

Abstract
The recognition and repair of DNA lesions occurs within a chromatin environment. Genetically tagging fluorescent proteins to DNA damage response proteins has provided spatial and temporal details concerning the establishment of biochemical subnuclear regions geared toward metabolizing genomic lesions. A specific marker for chromatin regions containing DNA breaks is required to study the initial dynamic structural changes in chromatin when DNA breaks occur. Here we present the experimental protocols used to investigate the dynamics of chromatin structure immediately after the simultaneous photoactivation of PAGFP-tagged core histone H2B and introduction of DNA breaks using UVA laser microirradiation on a laser scanning confocal microscope.
Keywords: DNA breaks, histones, chromatin, PAGFP, photoactivation, confocal microscopy, UVA laser microirradiation
1. Introduction
The appearance of DNA breaks presents severe implications for the integrity of the genome. A double-strand break is thought to be defining initial event that leads to a chromosomal translocation, which has the potential to transform a normal cell into the unstable tumorigenic state. Studying the cell biology of DNA breaks has focused largely on the proteins involved in sensing and metabolizing the genomic lesions. Physically, DNA breaks occur in a chromatin environment, therefore, it is of interest to determine how this environment is modulated upon the introduction of DNA breaks. It is known that the biochemical environment is altered as specific proteins are recruited (1) and post-translational modifications occur on a multitude of proteins, as in the phosphorylation of the core histone variant H2AX (2, 3), now a hallmark indicator of the presence of DNA breaks. This modification of a chromatin structural component also emphasizes the need to understand how the chromatin environment is modified upon the introduction of DNA breaks. Biochemical studies have provided valuable evidence for how large scale chromatin structure is influenced by DNA lesions (4, 5). However, the spatio-temporal context of these changes can only be provided by cell biology approaches, in particular, live-cell imaging.
Tagging DNA damage response proteins with genetically encoded fluorescent proteins has allowed researchers to monitor the redistribution and temporal recruitment of response proteins to sites of DNA breaks (6, 7). However, there is a lag period from the initial introduction of the experimentally induced DNA break until the steady-state accumulation of the protein at the DNA damage site. Furthermore, the multitude of response proteins display different recruitment kinetics and localize to distinct sub-compartments in the DNA lesion environment (8) and, therefore, not all proteins are representative of the chromatin structure surrounding the lesion.
Expression of GFP-tagged proteins has also allowed the application of photobleaching techniques to measure the kinetics of proteins in the spatial and biochemical context of the DNA breaks (6, 9). Photobleaching of fluorescent proteins in living cells involves directed exposure of a subset of fluorescent proteins to a short high intensity pulse of excitation light that photochemically alters the fluorophore resulting in the irreversible loss of emission light from the fluorescent protein, but does not physically disrupt the structural or biochemical activity of the tagged protein (10, 11). Photobleaching a subset of GFP-tagged proteins allows one to monitor the steady-state dynamics of the protein of interest by observing the redistribution of photobleached and non-photobleached proteins within a specific region of the cell. The inverse and complementary approach is to optically highlight a subset of fluorescent proteins and monitor the dynamics of the highlighted proteins (10). The advent of the photoactivatable variant of GFP, PAGFP (12) is a good example, where the non-activated PAGFP does not emit green fluorescence when exposed to 488 nm light, whereas upon a short high intensity pulse of ~400 nm light the protein is covalently photoconverted to the photoactivated state that now absorbs 488 nm light to successfully emit green fluorescence. Only those PAGFP molecules exposed to the pulse of ~400 nm light are photoactivated and emit green fluorescence when excited by 488 nm light. Thus PAGFP can be used as a highlighter protein in living cells (13).
Multiple methods have been employed to experimentally introduce DNA breaks within living mammalian cells to study the cell biological response to such genomic insults, including, but not limited to, ionizing radiation and laser microirradiation (2, 14–16). Laser scanning confocal microscopes equipped with continuous wave UVA lasers have been used to introduce DNA breaks in living cells (16, 17). However, most LSCMs equipped with continuous wave UVA lasers provide wavelengths that fall outside the efficient absorbance spectrum of the DNA molecule (18). Therefore, it is necessary to pre-sensitize cells with biochemical agents that either bind to the DNA molecule, such as the cell permeant Hoechst 33342 minor groove DNA-binding dye that causes minimal disruption to the structure of the DNA molecule (19), or those that are incorporated into the DNA molecule during DNA replication, such as the halogenated analog of deoxyuridine, BrdU (20–22). The presence of the DNA sensitizing agent facilitates the introduction of DNA breaks by UVA laser microirradiation using laser lines commonly found on confocal microscopes (351 and 364 nm) or laser microdissection microscopes (337 or 355 nm). The use of a laser scanning confocal microscope to introduce DNA breaks provides the distinct advantage of being able to subsequently image the specimen with high spatial and temporal resolution immediately after the introduction of DNA breaks (17). However, laser microdissection microscopes, which employ a pulsed UVA laser, are not optimally set up for high-resolution fluorescence imaging.
DNA is organized into chromatin structure with the nucleosome being the fundamental repeating unit, consisting of two molecules each of the core histones H2A, H2B, H3, and H4. The core histones are conserved stable structural components of the nucleosome that exhibit slow steady-state exchange (23, 24), under normal growth conditions.
Here we describe a method used to optically highlight and subsequently monitor chromatin in specific subnuclear regions containing DNA breaks in living cells with high spatial and temporal resolution. Development of the method required the ability to introduce DNA breaks in desired subnuclear regions; a specific stable marker for chromatin; that the chromatin marker highlight particular regions containing DNA breaks; and capability to visualize the subnuclear regions in real time in living cells. Thus, we combined UVA (364 nm) laser microirradiation and simultaneously photoactivation of H2B-PAGFP expressing cells pre-sensitized with the DNA-binding dye Hoechst 33342 with laser scanning confocal microscopy to achieve our goals. Here we will describe in detail how the experiments are set up and performed.
2. Materials
2.1. Cell Culture
Wild-type mouse embryo fibroblasts established from day 13.5 C57BL/6 embryos.
Dulbecco’s-modified Eagle’s medium (DMEM; Invitrogen) supplemented with 10% fetal calf serum, 100 U/ml penicillin (Gibco/BRL), 100 μg/ml streptomycin (Gibco/BRL), and 2 mM l-glutamine (Gibco/BRL).
Gridded chamber coverglass (cat# P35G-1.5–7-C-grid; MatTek).
Fugene 6 liposome transfection reagent (Roche).
Plasmid DNA used for transfection, H2B-PAGFP.
Microscopy medium: 25 mM HEPES in phenol red free DMEM (Invitrogen) supplemented with 10% fetal calf serum, 100 U/ml penicillin (Gibco/BRL), 100 μg/ml streptomycin (Gibco/BRL), and 2 mM l-glutamine (Gibco/BRL).
Hoechst 33342 (Sigma) 10 mg/ml working concentration in DMSO.
2.2. Immunolabeling
1 × PBS (Gibco/BRL).
16% paraformaldehyde solution, EM grade (Electron Microscopy Sciences): the day of the experiment freshly prepare 2% paraformaldehyde by diluting in 1 × PBS.
0.5% Triton X-100 in 1 × PBS.
1% bovine serum albumin (Sigma) in 1 × PBS; 0.05% goat serum (Sigma) in 1 × PBS.
Primary antibody mouse monoclonal anti-phospho-histone H2A.X (Ser139, clone JBW301, Upstate Biotech), 1 μg/μl stock solution.
Secondary antibody goat anti-mouse IgG highly cross-adsorbed AlexaFluor 546 (Molecular Probes/Invitrogen), stock 2 mg/ml solution.
2.3. Laser Scanning Confocal Microscopy
Laser scanning confocal microscope equipped with UVA (364 nm) and two visible (488 and 543 nm) laser lines, and an acousto-optical tunable filter (AOTF) (see Note 1).
40 × C-apochromat (N.A. 1.2) water immersion lens (see Note 2).
Temperature-controlled stage heater (Carl Zeiss MicroImaging) (see Note 3).
Objective lens heater (Bioptechs) (see Note 4).
Microscope software with photobleaching and time series acquisition modules (see Note 5).
Laser power meter capable of measuring UV and visible wave-lengths (the Ophir Nova by Ophir Optronics).
3. Methods
The method described below is dependent on two main contributing experimental factors. First, the transfection and pre-sensitization of the cells must be performed appropriately as to minimize alterations to the growth efficiency and biochemical stability of the cells. Overexpression of the PAGFP-tagged histone H2B will cause artifacts in the distribution and mobility of the protein, whereas inappropriate use of the Hoechst DNA-binding dye will disrupt DNA metabolic events, such as transcription, and detrimentally alter the growth state of the cells. Judicial use of both reagents will minimize both artifacts. Second, the appropriate amount of 364 nm UVA laser intensity is necessary for introducing DNA breaks. A calibration experiment is required to establish the most appropriate laser intensity needed to achieve a reasonable number of DNA breaks using UVA laser microirradiation. Artifacts will arise if the UVA laser intensity used to introduce DNA breaks is too high. Once these two experimental factors are established, running the experiments is straightforward.
3.1. Cell Culture, Transfection, and Pre-sensitization
Grow cells to 70% confluency in gridded chamber coverglass dishes in 2 ml of DMEM growth medium.
Transfect cells with H2B-PAGFP plasmid DNA using Fugene 6, as per the protocol suggested by the manufacturer, and incubate for 24 h at 37°C, 5% CO2.
Warm microscopy medium to 37°C. Add 0.75 μl of 10mg/ml Hoechst 33342 solution for each milliliter of medium used, for a final concentration of 7.5 μg/ml of Hoechst 33342 dye.
Replace growth medium in chamber dishes with Hoechst dye containing microscopy medium and incubate cells for 30 min at 37°C and 5% CO2 (see Note 6). Rinse the cells once with microscopy medium not containing Hoechst dye and add 2 ml per chamber dish of microscopy medium without Hoechst dye. The cells are now ready to be placed on the microscope.
3.2. Calibration of Laser Microirradiation Intensity for Introducing DNA Breaks
The purpose of this section is to record the microscope parameters and appropriate laser intensity needed to introduce DNA breaks in pre-sensitized cells. The aim is to determine the appropriate level of 364 nm laser intensity needed to introduce DNA breaks without overly damaging the cells. Exposure to laser intensity is governed by two factors: the actual output intensity of the laser available at the objective lens and the dwell time of the laser as it scans across the region of interest. There are a number of settings in the microscope software that influence these parameters; they will be mentioned in detail below. Once the parameters are established they should be maintained consistently throughout future experiments employing this method. Transfected cells are not absolutely necessary for the calibration step, but using them will help determine the detector settings required when running future photoactivation experiments.
Attach the objective heater to the 40 × C-apochromat water immersion objective lens and set to 37°C.
Attach the heated stage insert and set to 37° C.
Add a small drop (~20 μl) of distilled water on the objective lens and place the chamber coverglass with pre-sensitized cells in the heated stage and find best focus using brightfield illumination (see Note 7). Align and focus the condenser lens to establish Kohler illumination for subsequent acquisition of differential interference contrast (DIC) images.
Again using brightfield illumination, find a group of cells that are roughly in the same plane of focus. Record the position of the cells in relation to the grid imprinted on the coverslip.
Collect an initial image using a configuration suitable for collecting green fluorescence from PAGFP and corresponding DIC image. In establishing the optical configuration, a main dichroic mirror suitable for UV and 488 nm laser light must be used, at a minimum, since the 364 nm laser line will subsequently be used for laser microirradiation. Also, at this point the H2B-PAGFP will not emit green fluorescence, but it is important that the configuration be set up to allow acquisition of such fluorescence after photoactivation. The configuration that we consistently use includes a HFT UV/488 main dichroic mirror, a NFT 490 secondary beam splitter, and band-pass 505–550 nm emission filter. The pinhole diameter is opened slightly for live-cell imaging to provide an optical slice thickness of 1.5 μm. The DIC image is not confocal.
The laser power output of the Argon ion multi-line (458, 477, 488, and 514 nm) visible laser should be set to 25% maximum output with the 488 nm laser line used to image the photoactivated PAGFP. For imaging, the 488 nm laser intensity should be modulated to 3% transmission of the 25% output. The scan parameters for a 512 × 512 pixel frame size include a pixel dwell time of 3.2 μs (scan speed of 7 in the Zeiss LSM510 software) and optical zoom factor providing 0.22 μm X–γ pixel dimensions (Zoom factor 2 in the Zeiss LSM510 software).
Use the DIC image to place a ROI in the nucleus of each cell in the group. Remember to use ROIs of consistent dimensions (see Note 8).
Next set up the so-called “bleaching” parameters that will be used to introduce DNA breaks and photoactivate the H2B-PAGFP using the 364 nm UVA laser. We set the power output of the 80 mW Argon ion multi-line (351 nm and 364 nm) UV laser to 50% of maximum with the 364 nm laser line being used for UVA laser microirradiation. The AOTF-modulated transmission of the 364 nm laser line should be set to 20% of the 50% maximum output. The same scan speed (pixel dwell time of 3.2 μs) is used (see Note 9), with 10 scan iterations. The number of iterations includes the number of times the laser will scan through a given ROI (see Note 10).
Choose the option to scan once before exposure to the UVA laser, which will provide an image of the cells prior to introducing DNA breaks and photoactivating the H2B-PAGFP. A single post-UVA laser irradiation image is sufficient during the calibration step.
For the calibration procedure, move to different groups of cells, adjusting the positions of the ROIs and changing the AOTF-modulated transmission of the 364 nm laser line, with all other settings kept the same. Start by using the scale 2, 5, 10, 15, 20, 30, 40, 50, 75, and 100% transmission, with a different setting used for each new group of cells (see Note 11). Five minutes after the last group of cells has been exposed to UVA laser microirradiation, fix the cells in freshly prepared 2% paraformaldehyde in 1 × PBS at room temperature. Remember to record the position of each group of cells on the gridded coverslip.
With the chamber coverglass dish removed and the remaining distilled water cleaned from the objective lens, use the laser power meter to measure the 364 nm laser intensity at the objective for each AOTF-modulated transmission value and record the values (Fig. 9.1) (see Note 12).
Fig. 9.1.
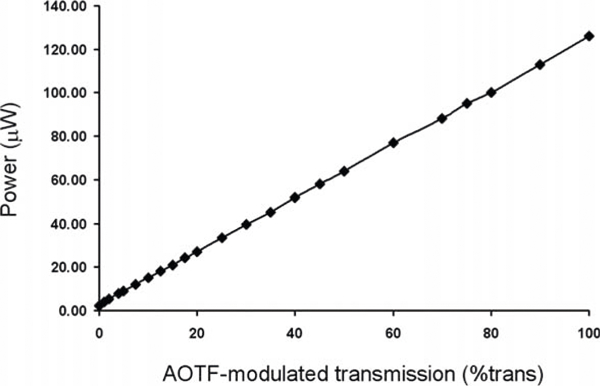
UVA (364 nm) laser intensity measurements. The laser power, in μW, measured at the 40 × C-apochromat objective lens in relation to the various AOTF-modulated percent transmission amounts of the laser light.
3.3. Immunolabeling of Laser Microirradiated Cells
This step is necessary to support the UVA laser intensity calibration of the microscope. The phosphorylated histone variant H2AX is a sensitive marker for the presence of DNA breaks and is used to determine whether the amount of UVA laser intensity is appropriate for introducing DNA breaks. Immuno-labeling of the fixed cells can be done in the chamber cover-glass dish.
After 5 min of fixation in 2% paraformaldehyde, rinse the cells with three successive changes of1 × PBS.
Permeabilize the cells by incubating with 0.5% Triton X-100 in 1 × PBS for 5 min at room temperature.
Rinse the cells three times with 1 × PBS.
Incubate with blocking buffer for 60 min at room temperature.
Remove blocking buffer.
Add primary anti-phosphorylated H2AX antibody at a final concentration of 1 μg/ml in 1 × PBS and incubate in a humidified chamber for 60 min at room temperature.
Rinse three times with 1 × PBS.
Add secondary goat anti-mouse AlexaFluor 546 (see Note 13) antibody at a final concentration of 10 μg/ml and incubate in a humidified chamber in the dark for 60 min at room temperature.
Rinse three times with 1 × PBS.
Leave cells in 1 × PBS.
3.4. Imaging the Immunolabeled Cells
Measuring the 364 nm UVA laser intensity during the calibration step provides a quantitative measure of the amount of laser microirradiation applied to the cells. The degree of phosphorylated H2AX present will be compared to the amount of laser intensity used. Since the appearance of phosphorylated H2AX is a sensitive indicator of the presence of DNA breaks, it will be used to help empirically determine the most suitable amount of laser microirradiation. The aim is to determine the minimal amount of UVA laser microirradiation that provides a consistent phosphorylated H2AX signal.
Relocate the cells on the gridded chamber coverglass using the 40× C-apochromat objective lens.
Choose an optical configuration suitable for exciting and collecting fluorescence for two channels, the green fluorescence from the H2B-PAGFP and the red fluorescence from the AlexaFluor 546, marking the presence of phosphorylated H2AX. A multi-track configuration (the two laser lines are actively switched on and off, but are not on simultaneously) with 488 and 543 nm laser line excitation. The power output of the 488 nm laser should be set to 30% maximum output and modulated to 10% transmission of the 30% output, whereas the HeNe 543 nm laser line should be set to maximum output and modulated at 50% transmission. The scan parameters for a 512 × 512 pixel frame size include a pixel dwell time of 2.56 μs (scan speed 8 in the Zeiss LSM510 software) and optical zoom factor providing 0.22 μm X–γ pixel dimensions (Zoom factor 2 in the Zeiss LSM510 software). For example, the multi-track configuration used with our microscope consistently includes a HFT UV/488/543/633 main dichroic mirror, a NFT 545 secondary beam splitter, and 505–530 nm band-pass emission filter in front of PMT detector accepting the H2B-PAGFP green fluorescence and a 560–615 nm band-pass emission filter in front of a second PMT detector accepting the AlexaFluor 546 red fluorescence. The pinhole diameter is set to one Airy unit which provides an optical slice thickness of 1.0 μm. A DIC image is also collected.
Move to the distinct groups of cells exposed to different amounts of UVA laser microirradiation while noting the presence of phosphorylated H2AX (Fig. 9.2). If the nucleus displays phosphorylated H2AX throughout the entire nucleus, beyond the ROI exposed to UVA laser microirradiation, that is a clear indication that the amount of UVA laser microirradiation used was too high and needs to be reduced.
Fig. 9.2.
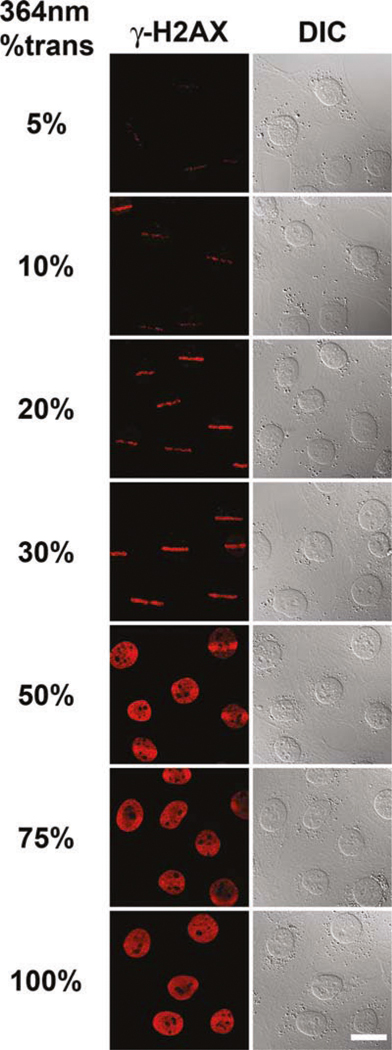
Calibration of the degree of phosphorylated H2AX present in relation to the amount of UVA laser intensity used for microirradiation. Several groups of wild-type MEFs were exposed to different amounts of 364 nm laser intensity. A single rectangular ROI was placed on each cell in a group and the cells subsequently microirradiated in that ROI. Confocal images of the resulting phosphorylated H2AX (γ-H2AX, left column) are shown. The corresponding DIC images are also shown (right column). Scale bar, 20 μm.
3.5. Imaging Photoactivated Chromatin Containing DNA Breaks
Now that the appropriate microscope parameters suitable for UVA laser microirradiation have been determined, the actual time-lapse experiments can be performed (Fig. 9.3).
Fig. 9.3.
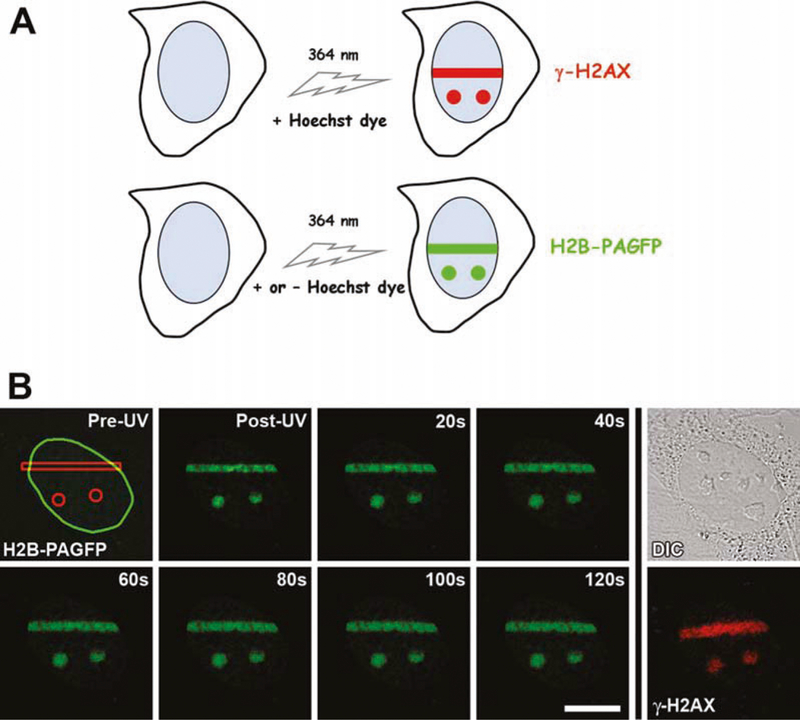
Simultaneous photoactivation of H2B-PAGFP and introduction of DNA breaks using UVA (364 nm) laser microirradiation. (A) a schematic indicating that DNA breaks are introduced upon exposure to 364 nm laser microirradiation in specific ROIs when cells are pre-sensitized with Hoechst DNA-binding dye, whereas H2B-PAGFP is photoactivated with the same 364 nm laser intensity in the presence or absence of the Hoechst dye. (B) Time-lapse series of a single pre-sensitized wild-type mouse embryo fibroblast expressing H2B-PAGFP. In this example, three ROIs were placed in the cell nucleus, one rectangle and two circles (shown in the Pre-UV image; with the oval outline denoting the boundary of the nucleus). After fixation and immunolabeling, the same cell was relocated and confocal image collected of the resultant DNA breaks, marked by the presence of phosphorylated H2AX (γ-H2AX, far right bottom panel). A corresponding DIC image is also shown. Scale bar, 20 μm.
Place the transfected H2B-PAGFP expressing cells in the gridded chamber coverglass dish on the heated 40 × C-apochromat water immersion lens.
The heated stage insert should be set to 37°C.
Set up the 364 nm laser microirradiation and photoactivation microscope parameters determined to be the most suitable from the calibration step. For our microscope configuration we use 50% UVA laser output, 20% AOTF-modulated transmission, pixel time of 3.2 μs (scan speed 7), optical zoom set to 0.22 μm pixel size, and 10 iterations of the ROI.
Find a group of cells using brightfield illumination, as described above (see Note 14).
Position the ROIs.
Acquire a time series of images including the pre-UVA laser exposure image and multiple images post-UVA laser exposure. The frequency of image acquisition is to be determined by the temporal information desired. Be careful that the sample is not imaged too frequently that overexposure to the 488 nm laser during imaging of the photoactivated H2B-PAGFP inadvertently disrupts the cells.
Acknowledgments
We thank George Patterson (NICHD/NIH) for the generous gift of the PAGFP construct and helpful technical assistance. We are grateful to Tom Misteli (NCI/NIH) for providing the H2B-GFP construct.
4. Notes
We used a Zeiss LSM510 META laser scanning confocal microscope equipped with a 80 mW Argon ion multi-line (351 and 364 nm) continuous wave UVA laser (Coherent Enterprise II 653, Coherent Laser Group, Santa Clara, CA), a 45 mW Argon ion multi-line (458, 477, 488, and 514 nm) visible laser (Lasos 60), and a 1.0 mW 543 nm HeNe laser. On this model of microscope, the UV and visible laser lines enter the scanhead from separate optical fibers and, therefore, to ensure that both UV and visible lasers are aligned the UV/V is collimator setting needs to be optimized for the objective lens used.
We used a Zeiss 40× C-apochromat (N.A. 1.2) lens for several of reasons: First, the use of a water immersion lens reduces refractive index mismatch between the specimen and the objective lens, thereby reducing the degree of spherical aberration in the resultant image. Second, the C-apochromat lens is corrected for multiple wavelengths helping to ensure that the 364 and 488 nm laser wavelengths are focused at the same plane in the specimen (see also Note 1). Last, the transmission efficiency of our C-apochromat lens is greater at 364 nm than for our 63 × oil immersion Plan-apochromat (N.A. 1.4) lens. Overall, the optical advantages gained by using the 40× C-apochromat lens outweighed the disadvantage of using an objective lens of lower numerical aperture (N.A. 1.2 versus N.A. 1.4).
We used a Zeiss heated stage insert that controls only the temperature of the stage and not the humidity or CO2 levels. For short-time course experiments such as described in this article, HEPES-buffered microscopy medium is sufficient to regulate the CO2 levels.
We used an objective heater from Bioptechs Inc., the large size to fit over the larger diameter collar of our 40 × C-apochromat lens. It is necessary to use an objective heater in conjunction with the heated stage if an environmental chamber is not used. The objective lens acts as a heat sink and the use of an objective heater helps stabilize the temperature and prevent temperature-related focal drift.
The software controlling the microscope and image acquisition parameters must have an accompanying module that allows the adaptive placement of ROIs. The software must also have a “bleaching” option in the module so that scanning of the 364 nm UVA laser can be limited to a short high intensity exposure in the confined area of the ROI. The Zeiss LSM510 software (version 3.2) used in our experiments has a bleaching option where the ROI can be established (both size and position in the image or specimen). It also allows the user to choose the desired wavelength and AOTF-modulated transmission (laser intensity) used to scan within the ROI. Although the “bleaching” option is used, we actually use the parameters to photoactivate the PAGFP rather than photobleach the PAGFP. An AOTF that modulates the laser intensity during the scanning process is necessary to restrict the high intensity UVA laser pulse to the desired ROI. The bleaching option in the Zeiss software is coupled to the time series acquisition, and thereby the bleach parameters can be run immediately before the time series is acquired.
Various cell types take up the cell permeant Hoechst 33342 dye at different rates and exhibit different degrees of sensitivity in the presence of the dye (25). The final concentration of Hoechst dye and the length of the incubation period in the presence of the dye may need to be adjusted for certain cell types. Overall, the aim is to add sufficient dye to sensitize the cells, but not too much to compromise the biochemical stability of the cells. Generally, after the addition of the dye the cells should continue to grow in culture through multiple cell cycles. If the majority of the cells exhibit poor survival or replication defects, then the concentration of the Hoechst dye used is too great and should be reduced. Once the appropriate dye concentration and incubation time have been established for a given cell type, those conditions must be used consistently for future experiments.
It is imperative that exposure to UV excitation by wide-field fluorescence illumination, such as through a DAPI filter, be strictly avoided. Such exposure will lead to the inadvertent introduction of DNA breaks and will compromise the experimental results. However, since the non-photoactivated H2B-PAGFP does not emit green fluorescence it is difficult to initially determine the transfection efficiency of the cells and whether it is worth continuing with further steps in the experiment. An extra chamber coverglass dish of transfected cells, not to be used for laser microirradiation, can be exposed to UV light using a DAPI filter since this exposure will also photo-activate the H2B-PAGFP and will provide some indication of the transfection efficiency of the cells. Again, this chamber dish should not be used in the laser microirradiation experiments.
Given the mechanics of scanning the laser across the sample, horizontal rectangular ROIs are preferable over vertical ROIs. The length (number of pixels in the X-axis) of the ROI may vary depending on the dimensions of the nucleus within individual cells, however, the thickness (number of pixels in the γ-axis) should be kept consistent.
Version 3.2 of the Zeiss LSM510 software forces one to use the same scan speed in the bleaching settings as that used in the image acquisition scan settings. Newer versions of the Zeiss software provide more flexibility in choosing different scan speeds for bleaching (photoactivating) and imaging.
The laser power output, the AOTF-modulated transmission of the laser allowed to reach the objective lens, the speed at which the laser is scanned across the sample (pixel dwell time), the distance that the laser covers over the particular time (optical zoom), and the number of scan iterations all influence the amount of laser intensity directed at the region of the nucleus. It is important to establish a good balance between all these parameters to ensure that the appropriate amount of laser intensity is used. The parameters mentioned above were found to be suitable for our Zeiss LSM510 META confocal microscope by running the calibration described above, but they will most definitely need to be adjusted for other similar microscopes.
Inherently there is an unavoidable time factor involved in performing the calibration procedure and this is why it is important to start at the lower laser intensity and progress to greater laser intensity.
Measuring the laser intensity at the objective will provide accurate values of the laser intensity suitable for running the UVA laser microirradiation experiments. As the UVA laser ages the output decreases and so recording periodic measurements of the laser intensity at the objective will allow adjustments to be made to the parameters to ensure the appropriate amount of laser intensity is used in future experiments.
The UVA microirradiated cells have H2B-PAGFP, and the red fluorescence emitting AlexaFluor 546 fluorophore is used for detecting the presence of phosphorylated H2AX. Our Zeiss LSM510 META microscope has a HeNe 543 laser line, and the excitation spectrum of the AlexaFluor 546 fluorophore is suited to this laser line.
Since the non-photoactivated PAGFP signal is very low, it is difficult to determine which cells are expressing H2B-PAGFP. To help overcome this, temporarily adjust the brightness and contrast of the displayed image. The presence of a faint green signal indicates that the cell may be expressing the protein. Return the contrast settings to normal and run the experiment. If the cell is indeed expressing the H2B-PAGFP, one will find out in the first image immediately after UVA laser microirradiation. If the activated PAGFP signal does not appear, most likely that cell is not expressing the H2B-PAGFP. Simply move on to a different cell and try again.
References
- 1.Downs JA, Nussenzweig MC, and Nussenzweig A (2007). Chromatin dynamics and the preservation of genetic information. Nature 447, 951–8. [DOI] [PubMed] [Google Scholar]
- 2.Rogakou EP, Boon C, Redon C, and Bonner WM (1999). DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Cell Biol 146, 905–16. [DOI] [PubMed] [Google Scholar]
- 3.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, and Bonner WM (1998) J Biol Chem 273, 5858–68. [DOI] [PubMed] [Google Scholar]
- 4.Smerdon MJ, Kastan MB, and Lieberman MW (1979). Distribution of repair-incorporated nucleotides and nucleosome rearrangement in the chromatin of normal and xeroderma pigmentosum human fibroblasts. Biochemistry 18, 3732–9. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi K, and Kaneko I (1985). Changes in nuclease sensitivity of mammalian cells after irradiation with 60Co gamma-rays. Int J Radiat Biol Relat Stud Phys Chem Med 48, 389–95. [DOI] [PubMed] [Google Scholar]
- 6.Houtsmuller AB, and Vermeulen W (2001). Macromolecular dynamics in living cell nuclei revealed by fluorescence redistribution after photobleaching. Histochem Cell Biol 115, 13–21. [DOI] [PubMed] [Google Scholar]
- 7.Lukas C, Falck J, Bartkova J, Bartek J, and Lukas J (2003). Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol 5, 255–60. [DOI] [PubMed] [Google Scholar]
- 8.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, and Lukas J (2006). Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 173, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Essers J, Houtsmuller AB, van Veelen L, Paulusma C, Nigg AL, Pastink A, Vermeulen W, Hoeijmakers JH, and Kanaar R (2002). Nuclear dynamics of RAD52 group homologous recombination proteins in response to DNA damage. Embo J 21, 2030–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lippincott-Schwartz J, Altan-Bonnet N, and Patterson GH (2003). Photobleaching and photoactivation: following protein dynamics in living cells. Nat Cell Biol Suppl, S7–14. [PubMed] [Google Scholar]
- 11.White J, and Stelzer E (1999). Photo-bleaching GFP reveals protein dynamics inside live cells. Trends Cell Biol 9, 61–5. [DOI] [PubMed] [Google Scholar]
- 12.Patterson GH, and Lippincott-Schwartz J (2002). A photoactivatable GFP for selective photolabeling of proteins and cells. Science 297, 1873–7. [DOI] [PubMed] [Google Scholar]
- 13.Patterson GH, and Lippincott-Schwartz J (2004). Selective photolabeling of proteins using photoactivatable GFP. Methods 32, 445–50. [DOI] [PubMed] [Google Scholar]
- 14.Cremer C, Cremer T, Fukuda M, and Nakanishi K (1980). Detection of laser – UV microirradiation-induced DNAphotolesions by immunofluorescent staining. Hum Genet 54, 107–10. [DOI] [PubMed] [Google Scholar]
- 15.Meldrum RA, Botchway SW, Wharton CW, and Hirst GJ (2003). Nanoscale spatial induction of ultraviolet photoproducts in cellular DNA by three-photon near-infrared absorption. EMBO Rep 4, 1144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Walter J, Cremer T, Miyagawa K, and Tashiro S (2003). A new system for laser-UVA-microirradiation of living cells. J Microsc 209, 71–5. [DOI] [PubMed] [Google Scholar]
- 17.Kruhlak MJ, Celeste A, Dellaire G, Fernandez-Capetillo O, Muller WG, McNally JG, Bazett-Jones DP, and Nussenzweig A (2006). Changes in chromatin structure and mobility in living cells at sites of DNA double-strand breaks. J Cell Biol 172, 823–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sutherland JC, and Griffin KP (1981). Absorption spectrum of DNA for wavelengths greater than 300 nm. Radiat Res 86, 399–409. [PubMed] [Google Scholar]
- 19.Geierstanger BH, and Wemmer DE (1995). Complexes of the minor groove of DNA. Annu Rev Biophys Biomol Struct 24, 463–93. [DOI] [PubMed] [Google Scholar]
- 20.Limoli CL, and Ward JF (1993). A new method for introducing double-strand breaks into cellular DNA. Radiat Res 134, 160–9. [PubMed] [Google Scholar]
- 21.Djordjevic B, and Djordjevic O (1965). Chromosomal aberrations in synchronized mammalian cells treated with 5-bromo-deoxyuridine and irradiated by ultra-violet light. Nature 206, 1165–6. [DOI] [PubMed] [Google Scholar]
- 22.Regan JD, Setlow RB, and Ley RD (1971). Normal and defective repair of damaged DNA in human cells: a sensitive assay utilizing the photolysis of bromodeoxyuridine. Proc Natl Acad Sci U S A 68, 708–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kimura H, and Cook PR (2001). Kinetics of core histones in living human cells: little exchange of H3 and H4 and some rapid exchange of H2B. J Cell Biol 153,1341–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siino JS, Nazarov IB, Svetlova MP, Solovjeva LV, Adamson RH, Zalenskaya IA, Yau PM, Bradbury EM, and Tomilin NV (2002) Photobleaching of GFP-labeled H2AX in chromatin: H2AX has low diffusional mobility in the nucleus. Biochem Biophys Res Commun 297, 1318–23. [DOI] [PubMed] [Google Scholar]
- 25.Fried J, Doblin J, Takamoto S, Perez A, Hansen H, and Clarkson B (1982) Effects of Hoechst 33342 on survival and growth of two tumor cell lines and on hematopoietically normal bone marrow cells. Cytometry 3, 42–7. [DOI] [PubMed] [Google Scholar]