

 This work is licensed under a
This work is licensed under a Abstract
Summary
Adrenocortical carcinoma (ACC) is an aggressive cancer that originates in the cortex of the adrenal gland and generally has a poor prognosis. ACC is rare but can be more commonly seen in those with cancer predisposition syndromes (e.g. Li-Fraumeni and Lynch Syndrome). The diagnosis of ACC is sometimes uncertain and it requires the use of precise molecular pathology; the differential diagnosis includes pheochromocytoma, adrenal adenoma, renal carcinoma, or hepatocellular carcinoma. We describe a case of a 57-year-old woman with Lynch Syndrome and metastatic ACC who was initially diagnosed as having pheochromocytoma. The tumor was first identified at 51 years of age by ultrasound followed by a CT scan. She underwent a left adrenalectomy, and the histopathology identified pheochromocytoma. Two years later, she had tumor recurrence with imaging studies showing multiple lung nodules. Following a wedge resection by video-assisted thoracoscopic surgery (VATS), histopathology was read as metastatic pheochromocytoma at one institution and metastatic ACC at another institution. She later presented to the National Institutes of Health (NIH) where the diagnosis of ACC was confirmed. Following her ACC diagnosis, she was treated with mitotane and pembrolizumab which were stopped due to side effects and progression of disease. She is currently receiving etoposide, doxorubicin, and cisplatin (EDP). This case highlights the importance of using a multi-disciplinary approach in patient care. Thorough evaluation of the tumor’s pathology and analysis of the patient’s genetic profile are necessary to obtain the correct diagnosis for the patient and can significantly influence the course of treatment.
Learning points:
Making the diagnosis of ACC can be difficult as the differential diagnosis includes pheochromocytoma, adrenal adenoma, renal carcinoma, or hepatocellular carcinoma.
Patients with Lynch Syndrome should undergo surveillance for ACC as there is evidence of an association between Lynch Syndrome and ACC.
Conducting a complete tumor immunoprofile and obtaining a second opinion is very important in cases of suspected ACC in order to confirm the proper diagnosis.
A multi-disciplinary approach including genetic testing and a thorough evaluation of the tumor’s pathology is imperative to ensuring that the patient receives an accurate diagnosis and the appropriate treatment.
Patient Demographics: Adult, Female, White, United States
Clinical Overview: Adrenal, Adrenal, Adrenocortical carcinoma, Lynch Syndrome*
Diagnosis and Treatment: Hypertension, Flushing, Headache, Asthenia, Histopathology, Ultrasound scan, CT scan, Endoscopy, MRI, PET scan, Gallium scan, Immunohistochemistry, Synaptophysin, Vimentin*, Calretinin*, SF-1*, KI-67*, Haematoxylin and eosin staining, Adrenalectomy, Resection of tumour, Thorascopic surgery*, Mitotane, Pembrolizumab*, Etoposide, Doxorubicin, Anthracyclines, Cisplatin, Statins, Sandostatin*, Cyclophosphamide*, Dacarbazine, Vincristine*
Related Disciplines: Oncology, Genetics, Pathology
Publication Details: Unique/unexpected symptoms or presentations of a disease, January, 2020
Background
Adrenocortical carcinoma (ACC) is a rare and aggressive cancer with an incidence of 0.7–2 cases per million persons per year. ACC generally has a poor prognosis with a 5-year survival rate of 20–25% (1, 2). ACC is a challenging disease with a broad range of clinical presentations (e.g. Cushing’s syndrome), often presenting in an advanced stage with a large, locally invasive primary tumor. Reaching the diagnosis of ACC is a multi-disciplinary effort, with elements provided by the clinical history, laboratory/hormonal evaluations, imaging modalities, and pathology review. Clinical presentation varies based on the functionality of the tumor, with a functional tumor characterized by secretion of hormones such as cortisol, aldosterone, or steroid hormone precursors which lead to symptoms such as Cushing’s syndrome and virilization. In addition, patients with functional tumors can experience poorly controlled hypertension, weight gain, and acne (3). Imaging modalities, particularly CT, are essential for the localization of adrenal tumors.
Most adrenocortical tumors appear sporadically. However, there are some hereditary cancer syndromes that are associated with ACCs. For example, Li-Fraumeni syndrome results from dominantly inherited germline mutations in the TP53 gene and has an increased risk of ACC. Mutations in the TP53 gene are more frequently identified in children with ACC (~50–80%) than adults with ACC. Lynch syndrome also predisposes patients to certain cancers due to mutations in the DNA mismatch repair genes MSH2, MSH6, MLH1, and PMS2. Lynch syndrome is associated with a high risk of colorectal cancer (80% lifetime) and other non-colonic cancers such as small bowel, stomach, hepatobiliary, urinary tract, and endometrial cancer (4). There have been a few case reports on the association of ACC and Lynch syndrome (4, 5) with some studies estimating the incidence at 3.2% (4). Additional cancer predisposition syndromes that are associated with ACC are multiple endocrine neoplasia type 1 and familial adenomatous polyposis (6).
Once ACC is suspected, surgical resection is recommended as an alternative to biopsy when feasible (7, 8). A post-surgical pathologic confirmation of disease is important as certain features, such as the mitotic rate, have been shown to predict outcomes and may be used to further guide treatment (9). Mitotane can be given as a type of adjuvant therapy after surgical resection as treatment for patients with metastatic ACC or as treatment in cases in which the tumor is unresectable. For more advanced cases, a combination of mitotane with etoposide, doxorubicin, and cisplatin (EDP) is given as standard treatment. Several clinical trials are currently assessing the use of immune checkpoint inhibitors to treat ACC (10).
The differential diagnosis of ACC includes pheochromocytoma, adrenal adenoma, renal carcinoma, or hepatocellular carcinoma; the ultimate diagnosis often rests on the pathologist. Here we present a case of one patient’s turbulent diagnostic journey to adrenocortical carcinoma given her atypical pathologic tumor features. Her case illustrates the importance of obtaining an accurate pathology diagnosis, while demonstrating how difficult this can be when faced with conflicting stains and tumor features.
Case presentation
A 57-year-old Caucasian female presented to the National Institutes of Health (NIH) with a complicated diagnostic journey with differential diagnosis of pheochromocytoma vs adrenocortical carcinoma (ACC). At the age of 47, she developed symptoms of flushing and hypertension for which she was treated symptomatically. At that time, the patient believed that these symptoms were caused by multiple sclerosis, diagnosed the year earlier. A pathogenic mutation (p.Q46X) in the MSH2 gene was identified and she was subsequently referred for genetic counseling and diagnosed with Lynch syndrome. In February 2013, an endoscopy and ultrasound (US) showed an incidental adrenal mass. MRI of the abdomen showed a 1.4 × 0.9 cm left adrenal mass that lacked T2 hyperintensity (Fig. 1A). A CT scan of the abdomen performed in July 2013 showed that the left adrenal mass measured 2.3 × 2.9 × 1.9 cm with 91.7 Hounsfield Units on post-contrast imaging (Fig. 1B). At that time, biochemical evaluation reported that 24-h urine catecholamines, metanephrines, free cortisol, and vanillylmandelic acid were normal.
Figure 1.

(A) MRI of the abdomen showed a 1.4 × 0.9 cm left adrenal mass. (B) CT (computed tomography) scan of the abdomen showed a left adrenal mass that measured 2.3 × 2.9 × 1.9 cm with 91.7 Hounsfield Units on post-contrast imaging. (C) Subsequently a CT scan of the chest showed no evidence of disease (D) while a follow-up CT showed multiple lung nodules.
Investigation and treatment
The patient subsequently underwent a left adrenalectomy and pathology showed pheochromocytoma with a PASS (Pheochromocytoma of the Adrenal gland Scaled Score) of 11 out of 20. She had resolution of her symptoms following surgery and underwent surveillance scans every 3–6 months. However, she had recurrence of symptoms in 2014 reported as asthenia and migraine headaches, but imaging showed no evidence of tumor recurrence (Fig. 1C). In September 2015, MRI of the abdomen and CT of the chest/abdomen/pelvis showed multiple lung nodules (Fig. 1D) and the patient subsequently underwent a wedge resection by video-assisted thoracoscopic surgery (VATS). Pathology from this surgery was reported as ACC at one institution but recorded as pheochromocytoma at another institution. The decision was made to treat her as pheochromocytoma, and she began treatment with monthly Sandostatin injections. In 2016, it was recommended to stop Sandostatin and start on cyclophosphamide, vincristine, and dacarbazine (CVD) for presumed metastatic pheochromocytoma. She completed eight cycles of CVD from May to October 2016. However, CVD was stopped due to progression of disease and side effects including grade 3 peripheral neuropathy. In November 2016, she was started on pembrolizumab (100 mg, i.v.) every 3 weeks based on her history of Lynch syndrome and loss of expression for MSH-2 and MSH-6 in her tumor cells.
The patient presented to the NIH in December 2017. Imaging studies at that time revealed a para-splenic mass and multiple metastatic bilateral lung masses (Fig. 2) also seen on 18F-Fluorodeoxyglucose (18FDG) PET/CT scan and negative 68Ga-dotatate PET/CT scan (Fig. 3). In addition, histological slides from her left adrenalectomy in 2013 and wedge resection in September 2015 were re-read at our institution and reported as metastatic ACC (Fig. 4). Specifically, two tissue fragments from the tumor nodule measured at 5.0 × 2.9 × 2.1 cm and 2.5 × 2.0 × 2.0 cm and appeared to arise from the adrenal gland. The tumor from the left adrenal gland predominantly involved the adrenal cortex. The tumor was composed of some cells with clear lipoid cytoplasm growing in a diffuse pattern, and the cells were reported as showing a large hyperchromatic nucleus. Mitotic figures were numerous with some having an atypical configuration. Vascular invasion was also present. Immunohistochemistry (IHC) performed reported positive staining for vimentin, synaptophysin, and focally calretinin. Negative staining was observed for chromogranin, desmin, inhibin, CD34, CK7, K20, CAM5.2, D2-40, and GATA 3. Further IHC performed at another institution reported positive staining for SF-1 and negative staining for chromogranin and TTF1. The Ki-67 was reported as 70–80%. Slides from the patient’s lung wedge resection showed tumor morphologically similar to the primary ACC (Fig. 4D). Based on the pattern of staining, pathology from our institution reported metastatic ACC. In March 2018, a biopsy of the para-splenic mass reported histopathologic findings consistent with ACC. This finding was confirmed at another institution.
Figure 2.

CT (computed tomography) of the chest, abdomen, and pelvis demonstrates tumor progression of multiple lung metastases and a metastatic splenic mass.
Figure 3.
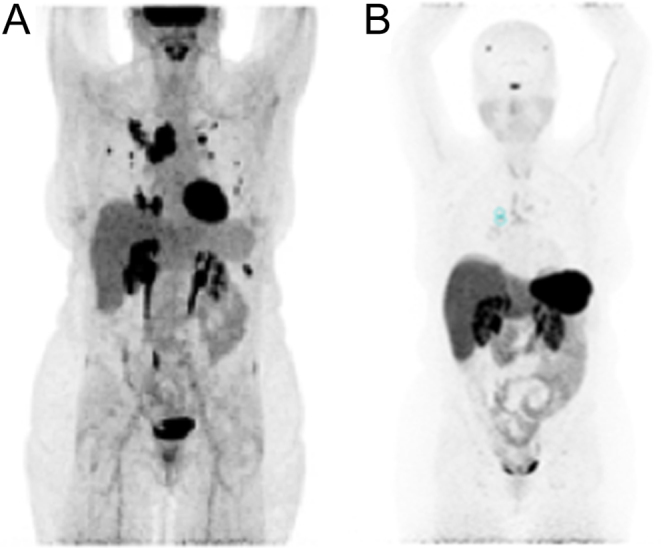
(A) Anterior MIP (maximal intensity projection) images with intense uptake on 18F-Fluorodeoxyglucose (18FDG) PET/CT scan (B) and negative uptake on 68Ga-dotatate PET/CT scan.
Figure 4.
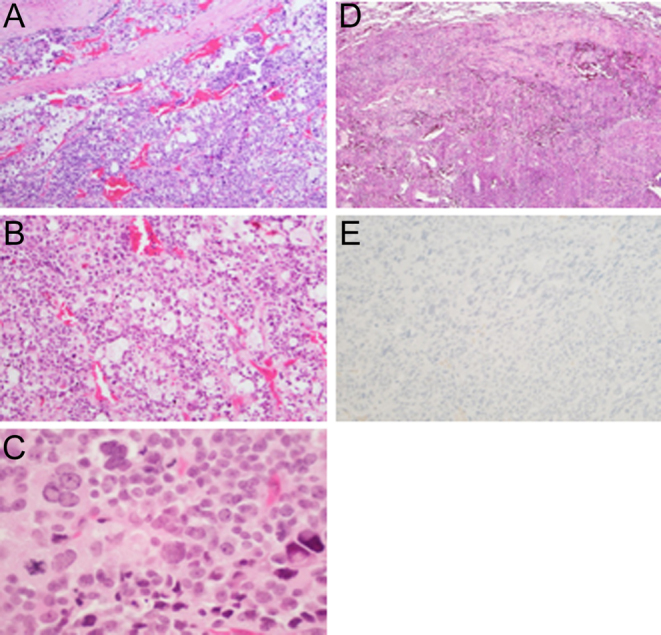
(A) Low power of the adrenocortical carcinoma involving the fibrous capsule (150× magnification, haemotoxylin and eosin staining). (B) Higher magnification of the tumor shows small cells with dark nucleus and scant cytoplasm intermixed with lipid-rich cells with prominent clear cytoplasm (200× magnification, haemotoxylin and eosin staining). (C) The tumor cells show variation in nuclear size and nuclear pleomorphism. Mitotic figures were present (250× magnification, haemotoxylin and eosin staining). (D) Metastatic tumor in the lung. The tumor showed the same characteristics of the primary adrenal lesion (150× magnification, haemotoxylin and eosin staining). (E) Immunohistochemical staining of the tumor was negative for chromogranin (150× magnification).
Outcome and follow-up
Following her ACC diagnosis, the patient started treatment with mitotane in July 2018. Mitotane was discontinued due to a severe skin reaction, which was treated with steroids. Mitotane was then re-attempted, but the skin reaction recurred and mitotane was discontinued. After mitotane was taken off her regimen, the patient re-started pembrolizumab in November 2018 which was later discontinued due to progression of disease. She is currently receiving etoposide, doxorubicin, and cisplatin (EDP).
Discussion
Adrenocortical carcinoma is an aggressive disease with relatively few treatment options. The chance of survival decreases dramatically as ACC progresses. Five-year survival drops from 66–82% for stage I to 0–17% for stage IV (1). As a result, getting an accurate and prompt diagnosis is imperative to effectively manage the patient’s disease and prolong survival. In the case of this patient, the correct diagnosis of ACC and the appropriate standard treatment were unfortunately delayed by a diagnostic work-up that concluded pheochromocytoma.
Pheochromocytoma and ACC have similarities that make obtaining an accurate diagnosis difficult. Both pheochromocytoma and cortisol or aldosterone-producing ACCs are associated with hypertension. In addition, they have similar average sizes, are both commonly found as incidentalomas, and can appear as a compact growth of large pleomorphic cells on a microscopic level. Tumor cells with large nuclei can be seen in both ACC and pheochromocytoma (11). In addition, hyaline globules are usually present in pheochromocytoma, but they have also been seen in cases of ACC. Nuclear pleomorphism can be seen in both cancers but is generally higher in pheochromocytoma (12). However, in pheochromocytoma, cells contain abundant basophilic cytoplasm, and mitotic figures are uncommon (11). Some cases of oncocytic ACC have reported that these tumor cells show prominent nuclei with a low mitotic rate. In addition, these cells lack the abundant smooth endoplasmic reticulum and tubular cristae commonly seen in conventional ACC. Staining was also negative for synaptophysin, chromogranin, and S-100 protein (13).
A review of 300 ACC cases revealed that the most common obstacle in diagnosis was discriminating between pheochromocytoma and ACC (14). One distinguishable difference is that pheochromocytomas secrete catecholamines while ACCs can secrete steroid hormones such as cortisol and aldosterone (3, 15). However, there have been few cases reported on pheochromocytoma with ectopic ACTH dependent Cushing’s syndrome (15). Immunohistochemical staining can help distinguish between ACC and pheochromocytoma; ACCs are positive for melan A, weakly positive for keratin, and negative for chromogranin A, while pheochromocytomas are positive for chromogranin A and negative for melan A and keratins (11).
The Weiss diagnostic criteria for the diagnosis of ACC includes three or more of the following features: high nuclear grade, mitotic rate >5 per 50 high-power fields, atypical mitotic figures, clear cells comprising 25% or less of the tumor, diffuse architecture, tissue necrosis, and venous, sinusoidal, and capsular invasion (16). Other criteria systems have been developed to differentiate between benign and malignant adrenocortical tumors. One system is a modification to the Weiss system that attaches numerical values to various Weiss criteria (17). Another system is based on an index that is calculated by assigning a numeric value to seven histopathologic parameters that include regressive changes, preservation of normal structure, nuclear atypia, nuclear hyperchromasia, structure of nucleoli, mitotic activity, and invasion of the capsule and/or blood vessel wall (18). Older systems involving histological and clinical parameters include a system that identified several criteria that were statistically significant in predicting metastasis such as weight loss, vascular invasion, tumor cell necrosis, a diffuse growth pattern, tumor mass, and broad fibrous bands traversing the tumor (19).
Additional studies may be useful in confirming malignant tissue such as ultrastructural studies and immunohistochemistry tests. In ultrastructural studies, ACC is evidenced by abundant smooth endoplasmic reticulum and mitochondria with prominent tubular or vesicular cristae. In immunohistochemistry studies, melan A, inhibin-alpha, and calretinin are tested. Cytokeratin is usually weakly positive and vimentin is strongly positive. In addition, synaptophysin is weakly positive and chromogranin is consistently negative. A marker for ACC cells includes Ad4BP/SP-1 and is relatively restricted in its distribution (20). A pathologic diagnosis for pheochromocytoma can be designated using the PASS system which uses growth pattern, necrosis, tumor cell spindling, mitotic count, atypical mitosis, invasion, nuclear pleomorphism, and hyperchromasia to separate tumors. The maximum PASS score is 20. Tumors with a PASS score of 4 or greater have a potential to be malignant while tumors with a PASS score of less than 4 are benign (21).
This case also provides further evidence on the association of Lynch syndrome and ACC. We concur with previous discussions that advocate adding ACC to the list of Lynch-related cancers as well as increasing surveillance of ACC in Lynch Syndrome patients (4). Mismatch repair genes are well known to increase risk of radiation-associated damage. As a result, this patient was advised against radiation treatment because of the presence of a pathogenic mutation in MSH2. This underscores the importance of identifying these defects in ACC patients, since a patient’s genetic profile can affect their treatment options and management. It is also important to regularly follow-up with these patients as they are at an increased risk for other cancers. ACC is very rare and aggressive, so increased screening of ACC in Lynch syndrome patients would be crucial in obtaining an early diagnosis and better prognosis.
This case highlights the importance of using a multi-disciplinary approach in patient care. Conducting a thorough pathology evaluation with complete tumor immunoprofiling is essential to receiving an accurate diagnosis. This case also underscores the significance of seeking multiple second opinions in cases of suspected ACC. Taking the proper measures to obtain the right diagnosis and conducting genetic testing will lead to the most appropriate and effective treatment for the patient.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of this case report.
Funding
This work was supported by the National Institutes of Health (NIH).
Patient consent
Written informed consent for publication of their clinical details and/or clinical images was obtained from the patient.
Author contribution statement
I S and S F drafted the manuscript. M R provided genetic counseling to the patient and was also involved in editing the manuscript. M M was the pathologist who interpreted the tumor pathology and provided the microscopic images for publication. B J T, K M R, and B W reviewed and edited the manuscript prior to submission. K P and J D R were the physicians who contributed to the care of the patient. J D R also performed final editing of the manuscript and gave the approval to submit the manuscript for publication.
References
- 1.Libé R. Adrenocortical carcinoma (ACC): diagnosis, prognosis, and treatment. Frontiers in Cell and Developmental Biology 2015. 45 ( 10.3389/fcell.2015.00045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Plager JE. Carcinoma of the adrenal cortex: clinical description, diagnosis, and treatment. International Advances in Surgical Oncology 1984. 329–353. [PubMed] [Google Scholar]
- 3.Ng L, Libertino JM. Adrenocortical carcinoma: diagnosis, evaluation and treatment. Journal of Urology 2003. 5–11. ( 10.1097/01.ju.0000030148.59051.35) [DOI] [PubMed] [Google Scholar]
- 4.Challis BG, Kandasamy N, Powlson AS, Koulouri O, Annamalai AK, Happerfield L, Marker AJ, Arends MJ, Nik-Zainal S, Gurnell M. Familial adrenocortical carcinoma in association with Lynch syndrome. Journal of Clinical Endocrinology and Metabolism 2016. 2269–2272. ( 10.1210/jc.2016-1460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Raymond VM, Everett JN, Furtado LV, Gustafson SL, Jungbluth CR, Gruber SB, Hammer GD, Stoffel EM, Greenson JK, Giordano TJ, et al Adrenocortical carcinoma is a lynch syndrome-associated cancer. Journal of Clinical Oncology 2013. 3012–3018. ( 10.1200/JCO.2012.48.0988) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petr EJ, Else T. Adrenocortical carcinoma (ACC): when and why should we consider germline testing? Presse Médicale 2018. e119–e125. ( 10.1016/j.lpm.2018.07.004) [DOI] [PubMed] [Google Scholar]
- 7.Gaujoux S, Brennan MF. Recommendation for standardized surgical management of primary adrenocortical carcinoma. Surgery 2012. 123–132. ( 10.1016/j.surg.2011.09.030) [DOI] [PubMed] [Google Scholar]
- 8.Mazzaglia PJ, Monchik JM. Limited value of adrenal biopsy in the evaluation of adrenal neoplasm: a decade of experience. Archives of Surgery 2009. 465–470. ( 10.1001/archsurg.2009.59) [DOI] [PubMed] [Google Scholar]
- 9.Assie G, Antoni G, Tissier F, Caillou B, Abiven G, Gicquel C, Leboulleux S, Travagli JP, Dromain C, Bertagna X, et al Prognostic parameters of metastatic adrenocortical carcinoma. Journal of Clinical Endocrinology and Metabolism 2007. 148–154. ( 10.1210/jc.2006-0706) [DOI] [PubMed] [Google Scholar]
- 10.Jasim S, Habra MA. Management of adrenocortical carcinoma. Current Oncology Reports 2019. 20 ( 10.1007/s11912-019-0773-7) [DOI] [PubMed] [Google Scholar]
- 11.Lloyd RV. Adrenal cortical tumors, pheochromocytomas and paragangliomas. Modern Pathology 2011. S58–S65. ( 10.1038/modpathol.2010.126) [DOI] [PubMed] [Google Scholar]
- 12.Gogoi G, Baruah MP, Borah P, Borgohain M. Adrenocortical carcinoma: an extremely uncommon entity and the role of immunohistochemistry in its diagnosis. Indian Journal of Endocrinology and Metabolism 2012. (Supplement 2) S373–S375. ( 10.4103/2230-8210.104098) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoang MP, Ayala AG, Albores-Saavedra J. Oncocytic adrenocortical carcinoma: a morphologic, immunohistochemical and ultrastructural study of four cases. Modern Pathology 2002. 973–978. ( 10.1038/modpathol.3880638) [DOI] [PubMed] [Google Scholar]
- 14.Duregon E, Volante M, Bollito E, Goia M, Buttigliero C, Zaggia B, Berruti A, Scagliotti GV, Papotti M. Pitfalls in the diagnosis of adrenocortical tumors: a lesson from 300 consultation cases. Human Pathology 2015. 1799–1807. ( 10.1016/j.humpath.2015.08.012) [DOI] [PubMed] [Google Scholar]
- 15.Sakuma I, Higuchi S, Fujimoto M, Takiguchi T, Nakayama A, Tamura A, Kohno T, Komai E, Shiga A, Nagano H, et al Cushing syndrome due to ACTH-secreting pheochromocytoma, aggravated by glucocorticoid-driven positive-feedback loop. Journal of Clinical Endocrinology and Metabolism 2016. 841–846. ( 10.1210/jc.2015-2855) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiss LM. Comparative histologic study of 43 metastasizing and nonmetastasizing adrenocortical tumors. American Journal of Surgical Pathology 1984. 163–169. ( 10.1097/00000478-198403000-00001) [DOI] [PubMed] [Google Scholar]
- 17.Aubert S, Wacrenier A, Leroy X, Devos P, Carnaille B, Proye C, Wemeau JL, Lecomte-Houcke M, Leteurtre E. Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. American Journal of Surgical Pathology 2002. 1612–1619. ( 10.1097/00000478-200212000-00009) [DOI] [PubMed] [Google Scholar]
- 18.Slooten HV, Schaberg A, Smeenk D, Moolenaar AJ. Morphologic characteristics of benign and malignant adrenocortical tumors. Cancer 1985. 766–773. () [DOI] [PubMed] [Google Scholar]
- 19.Hough AJ, Hollifield JW, Page DL, Hartmann WH. Prognostic factors in adrenal cortical tumors: a mathematical analysis of clinical and morphologic data. American Journal of Clinical Pathology 1979. 390–399. ( 10.1093/ajcp/72.3.390) [DOI] [PubMed] [Google Scholar]
- 20.Sasano H, Suzuki T, Nagura H, Nishikawa T. Steroidogenesis in human adrenocortical carcinoma: biochemical activities, immunohistochemistry, and in situ hybridization of steroidogenic enzymes and histopathologic study in nine cases. Human Pathology 1993. 397–404. ( 10.1016/0046-8177(93)90088-x) [DOI] [PubMed] [Google Scholar]
- 21.Thompson LD. Pheochromocytoma of the Adrenal gland Scaled Score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. American Journal of Surgical Pathology 2002. 551–566. ( 10.1097/00000478-200205000-00002) [DOI] [PubMed] [Google Scholar]