

SUMMARY
Autism spectrum disorder (ASD) manifests as alterations in complex human behaviors including social communication and stereotypies. In addition to genetic risks, the gut microbiome differs between typically-developing (TD) and ASD individuals, though it remains unclear whether the microbiome contributes to symptoms. We transplanted gut microbiota from human donors with ASD or TD controls into germ-free mice, and reveal that colonization with ASD microbiota is sufficient to induce hallmark autistic behaviors. The brains of mice colonized with ASD microbiota display alternative splicing of ASD-relevant genes. Microbiome and metabolome profiles of mice harboring human microbiota predict that specific bacterial taxa and their metabolites modulate ASD behaviors. Indeed, treatment of an ASD mouse model with candidate microbial metabolites improves behavioral abnormalities and modulates neuronal excitability in the brain. We propose that the gut microbiota regulates behaviors in mice via production of neuroactive metabolites, suggesting that gut-brain connections contribute to the pathophysiology of ASD.
Graphical Abstract
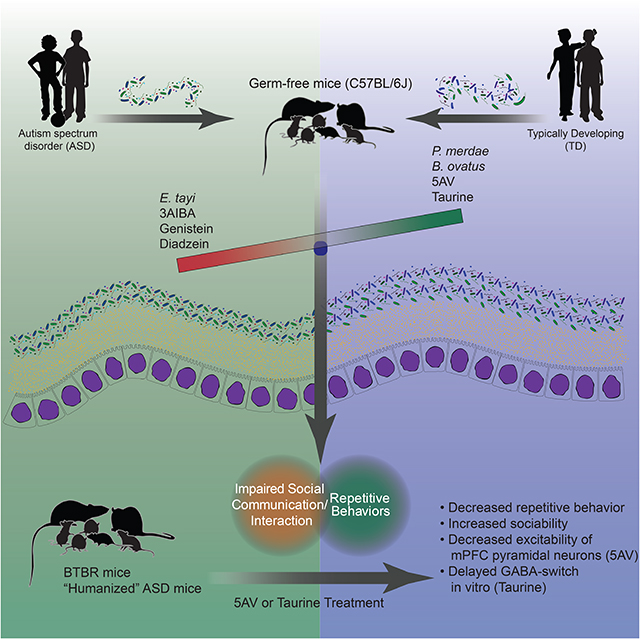
Repetitive and social behavioral abnormalities in mice with microbiomes from patients with autism spectrum disorder can be corrected by the administration of specific metabolites
INTRODUCTION
Autism spectrum disorder (ASD) is a group of neurodevelopmental conditions involving altered social communication and interaction, as well as repetitive, stereotyped behaviors. The prevalence of ASD in children and adolescents is 1 in 59 individuals in the US (Baio et al., 2018), although higher rates have also been reported (Xu et al., 2018), with males affected more frequently than females. Considerable genetic research has highlighted the importance of de novo mutations, copy number variations, and rare and common variants in ASD etiology (An et al., 2018; Gandal et al., 2018; Sanders et al., 2015; de la Torre-Ubieta et al., 2016), many of which have been modeled in animals (Patel et al., 2018). In genetically predisposed individuals, various environmental factors may modify and/or trigger psychiatric conditions (Mazina et al., 2015; Schaafsma et al., 2017). A non-genetic, yet heritable (Moeller et al., 2018) contributor to human behaviors may be the gut microbiota, a community of microorganisms humans harbor in our gastrointestinal (GI) tract that impacts development and function of the immune, metabolic, and nervous systems (Cho and Blaser, 2012; Sandoval-Motta et al., 2017). Diet and other interventions that reconfigure a microbiome can alter its function, either within a host or when inherited by its offspring (Sonnenburg et al., 2016), and may modify disease symptoms in individuals when coupled with a genetic risk (Luca et al., 2017; Sampson et al., 2016). Alternatively, a “pathogenic” microbiome itself may be sufficient to trigger disease even in otherwise non-predisposed individuals (Kim et al., 2017). For example, transplant of microbiota from patients with depression, irritable bowel syndrome-associated anxiety, or schizophrenia into wild-type mice promoted indicationspecific behavioral symptoms (De Palma et al., 2017; Zheng et al., 2016, 2019).
The gut microbiota has been suggested to play a role in ASD. Gut bacterial communities differ between individuals with ASD and typically-developing (TD) controls (Coretti et al., 2018; De Angelis et al., 2013; Finegold et al., 2010; Gondalia et al., 2012; Kang et al., 2013, 2017a; Kushak et al., 2016; Liu et al., 2019; Son et al., 2015; Strati et al., 2017; Wang et al., 2019; Williams et al., 2011), as well as in mouse models of ASD (Buffington et al., 2016; Coretti et al., 2017; Hsiao et al., 2013; Kim et al., 2017; Liu et al., 2018; de Theije et al., 2014). Fecal microbiome profiles are most divergent in ASD subjects with GI dysfunction, a common comorbidity of autism (Chaidez et al., 2014; Gorrindo et al., 2012). In addition, microbial-based interventions, including fecal transplantation, antibiotics, and probiotics, have shown promise in a limited number of human trials (Grimaldi et al., 2018; Kang et al., 2017b; Sandler et al., 2000). Specific gut microbes have demonstrated therapeutic potential in animal models of ASD (Buffington et al., 2016; Hsiao et al., 2013; Sgritta et al., 2018; Tabouy et al., 2018). Furthermore, changes in the microbiome often result in altered metabolic profiles, impacting the availability and diversity of nutrients and microbial metabolites (Dodd et al., 2017; Melnik et al., 2017; Sharon et al., 2014; Wikoff et al., 2009). Indeed, metabolomic analyses of serum, feces, and urine from ASD subjects have uncovered differences in various molecules compared to TD individuals, with many dysregulated compounds being of microbial origin (De Angelis et al., 2013; Kang et al., 2017a; Ming et al., 2012; Mussap et al., 2016; Wang et al., 2019). Notably, amino-acid transport and degradation differ between TD and ASD individuals (Aldred et al., 2003; Evans et al., 2008), intriguing since amino acids serve as precursors for many potent neuroactive molecules, such as classic neurotransmitters. In order to explore the contribution of the microbiota to ASD etiology, we sought to define functional contributions by the gut microbiota to behaviors associated with ASD. Herein, we report that colonization of germ-free (GF) wild-type mice with fecal microbiota from individuals with ASD was sufficient to promote core behavioral symptoms in their offspring, relative to those colonized with TD microbiota. We suggest a mechanism of action involving the production of microbial metabolites in the gut that affect brain function and regulate behavior.
RESULTS
Gut Microbiota from Individuals with ASD is Sufficient to Promote Altered Behaviors in Mice
Microbiome profiles of individuals with ASD, especially those of individuals with ASD with intestinal symptoms, are different from TD persons (De Angelis et al., 2013; Gondalia et al., 2012; Kang et al., 2013, 2017a; Son et al., 2015; Strati et al., 2017). While human studies are powerful, cross-sectional sampling after diagnosis yields associations that are unable to resolve cause-and-effect relationships, and do not inform whether gut microbiome changes are a consequence of disease (e.g., shaped by genetics, diet, GI symptoms, etc.) or if they contribute to symptoms. Accordingly, we tested whether altered human microbiota functionally promote ASD behaviors in mice. Fecal samples from human TD and ASD donors were selected based on Autism Diagnostic Observation Schedule (ADOS) (Gotham et al., 2007) and GI severity index (GSI) scores (Schneider et al., 2006) (Figure 1A–B, Table S1), and transplanted into GF wild-type mice, i.e., “humanized microbiome” mice. Donor fecal samples exhibited differences between TD and ASD samples in both α- and β-diversity within bacterial communities, as surveyed by high-throughput amplicon sequencing of the 16S rRNA gene (Figure 1C–D, Figure S1A–B). As ASD is a developmental disorder with evidence for prenatal effects (Hallmayer et al., 2011; Lyall et al., 2014), we colonized GF male and female C57BL/6J mice with TD or ASD donor samples (recipient TD and ASD, annotated rTD and rASD, respectively), and subsequently bred these animals (Figure 1A). Adult offspring mice (offspring TD and ASD, annotated oTD and oASD) that inherited human donor microbiota were either sampled (feces, serum, brains) or behavior tested (Figure 1A). We stratified donors using the standardized ADOS (StdADOS) score for disease severity (Gotham et al., 2009) into three groups: TD, Mild-ASD (StdADOS = 4–5), or ASD (StdADOS = 6–10) (Figure 1B, Table S1). oASD mice, colonized with samples from ASD donors, displayed behavioral deficits relevant to ASD, while oMild-ASD mice behaved similar to oTD controls (Figure S2A–E). Specifically, we observed increased repetitive behavior, decreased locomotion, and decreased communication in oASD mice compared to mice colonized with samples from TD controls (oTD), as tested by marble burying (MB), open-field testing (OFT), and ultrasonic vocalization (USV), respectively. No differences in sociability were observed by a three-chamber sociability test (Figure S2B). These core features of ASD were not the result of anxiety or other locomotion deficits, as colonized mice spent the same amount of time in the center of an open-field arena and male mice traveled similar distances (Figures S2C–D).
Figure 1. Colonization of mice with ASD microbiomes reproduces human behaviors.

(A) Experimental design: germ-free mice were colonized with fecal samples from TD or ASD donors at weaning and bred at 7–8 weeks of age. Offspring were behaviorally tested starting at 6 weeks of age, and various tissues and samples were collected at P45.
(B) Donor metadata. Metadata for sixteen donors used for mouse colonization, of which eight were followed up on with downstream analysis (in bold). See Table S1.
(C) α-diversity in human TD (circles) and ASD (mild ASD-blue squares, ASD-red squares) donors as measured by observed amplicon sequence variants (ASVs) from 16S rRNA gene sequencing of human TD and ASD donors. Eight samples used downstream are in dark-grey. Differences in medians tested by Kruskal-Wallis.
(D) First two axes of a principal coordinate analysis (PCoA) of unweighted UniFrac distances from 16S rRNA gene sequencing of human TD and ASD donors. NTD=5, Nmild ASD=3, NASD=8. Darker symbols denote samples in bold in Figure 1B. Group differences were tested by pairwise PERMANOVA.
(E-G) Behavioral phenotypes in humanized mice: (E) Number of marbles buried in Marble burying (MB) test (Cohen’s doTD-oASD= 0.64), (F) time socializing in direct social interaction (DSI)(Cohen’s doTD-oASD= −0.45), and (G) distance traveled in open field testing (OFT)(Cohen’s doTD-oASD= −0.58); in colonized offspring colored by donor. Hypothesis testing for differences of the means were done by a mixed effects analysis using donor diagnosis and mouse sex as fixed effects and donor ID as a random effect. p-values were derived from a chi-square test. NoASD= 121, NoTD= 85 (8–23 mice per donor, per gender). Data presented is the aggregate of all experiments.
(H) Spearman’s rank correlation between mouse behavior and donor metadata (see Table S1). Benjamini-Hochberg adjusted p-values for significant (α≤0.05) correlations are noted. Color scale denotes Spearman’s ρ from purple (positive correlation) to green (negative correlation). GSI: Gastrointestinal Severity Index; StdADOS: standardized Autism Diagnostic Observation Schedule score; PDDBI: Pervasive Developmental Disorder Behavior Inventory.
(I) α-diversity in humanized oTD and oASD mice as measured by observed amplicon sequence variants (ASVs) from 16S rRNA gene sequencing of human TD and ASD donors. Differences in medians tested by Kruskal-Wallis. Data are colored by donor. N=4–7 male offspring per donor.
(J) First three axes of a PCoA of unweighted UniFrac distances from oTD (circles) and oASD (squares) male offspring mice (colored by donor) from 16S rRNA gene sequencing of human TD and ASD donors. Group differences were tested by pairwise PERMANOVA. N=4–7 male offspring per donor.
(K) GraPhLan plot of LefSe linear discriminant analysis of microbiome profiles up to the genus level from 16S rRNA gene sequencing of human TD and ASD donors. Highlights denote significant taxonomic differences between oTD and oASD mice. N=4–7 male offspring per donor.
See also Figures S1–S3, Table S1.
Further in-depth studies were performed on a representative subset of fecal samples from three TD and five ASD donors (Figure 1B, in bold). Mice colonized with fecal samples from these donors were also behaviorally examined by the direct social interaction test (DSI), a more sensitive sociability assay. oASD mice “humanized” with fecal microbiota from ASD donors showed increased repetitive behavior, decreased sociability, and decreased locomotion compared to mice colonized with samples from TD controls (Figures 1E–G). Behavioral changes induced by ASD microbiota were more pronounced in male mice than in females (Figures S2A’–D’, S2F–G). Conversely, female oASD mice traveled shorter distances than oTD females in OFT (Figures S2C’, S2H). However, it is unknown whether this male bias was a true effect or an inherent result of our experimental design, using only human male donor samples in this study. Future studies comparing effects of samples from both male and female donors will help resolve this question. We find that the distance traveled in OFT negatively correlates with the donor age, indicating that some donor age-related microbiota changes may affect locomotion (Figure 1H). Additionally, increased repetitive behavior was highly correlated with ADOS and GSI scores (Figure 1H). It is possible that GI co-morbidities in the ASD donors and their correlation with some behaviors (Figure 1B, Table S1) may impact outcomes in experimental mice after microbiota transplant. However, we found no differences in mouse weight and intestinal barrier function between oTD and oASD mice (Figure S3A–B, Figure S3C–H). Further, there were no significant differences in a large panel of cytokines and chemokines from either the terminal ileum or the proximal colon (Figure S3I–J), suggesting no local inflammation in the GI tract of oASD mice as a result of “humanization” with ASD microbiota. Thus, microbiota transplantation from human ASD donors into mice can transfer ASD relevant behavioral deficits.
TD and ASD Colonized Mice Harbor Different Bacterial Species that Correlate with Behaviors
To validate the fidelity of microbiota transplantation, we collected feces from mice colonized with human samples (recipient TD and ASD, annotated rTD and rASD), and colon contents of their oTD and oASD offspring. As expected, the recipient microbiomes shifted away from that of donors, as seen three weeks after transplantation into mice (Figures S1C–E). We observe a decrease in α-diversity upon colonization of mice (donor to recipients), indicating loss of bacterial species due to sample processing and/or host-incompatibility (Figure S1D). Moreover, specific taxa, such as Actinobacteria and some Firmicutes, failed to engraft in mice, while others such as some Bacteriodetes thrived (Figure S1F–G). Sixty percent of the taxa in donor samples engrafted on average in recipient mice with no differences in engraftment or bacterial loads between oTD and oASD mice (Figure S1H–I, S1J–L). We observed a slight shift in α- and β-diversity in offspring compared to recipients (Figures S1C–E). While oASD mice did not exhibit the same decrease in α-diversity as the human donors (Figure 1I), we did observe a clear separation between oTD and oASD, similar to that in the human donor population, except for recipients of a single ASD-donor (oASD-1) (Figure 1J). oASD offspring displayed different bacterial profiles from oTD controls, driven by various taxa such as Bacteriodetes, β-Proteobacteria, Lactobacillales, Clostridiaceae, and Enterobacteriaceae (Figure 1K). Collectively, fecal transplantation from human donors into GF recipient mice maintained differences between TD and ASD microbiome profiles, which are vertically transmitted to offspring with fidelity.
We identified differentiating bacterial taxa between oTD and oASD microbiota using DESeq2 analysis. In total, 31 amplicon sequence variants (ASVs) were differentially abundant between groups (Figures 2A–2B). Those ASVs belong predominantly to the Clostridia and Bacteroidia classes, with single representatives from the Verrucomicrobia, α- and β-Proteobacteria phyla. ASVs belonging to Bacteroidia were associated with most TD controls. Specifically, Bacteroides ovatus (970ed_Bacteroides ovatus and 054dc_Bacteroides ovatus) and Parabacteroides merdae (4ae7e_Parabacteroides) were prevalent in all oTD samples, and absent from most or all oASD samples. Conversely, Eisenbergiela tayi (02b40_Lachnospiraceae and 29857_Lachnospiraceae) was prevalent among all oASD mice, and absent from oTD groups (Figure 2B). These observations were further corroborated by an unsupervised RandomForest classification analysis. The trained classifier assigned all offspring samples correctly by group. The model indicates 13 ASVs contributing >1% to discrimination between oTD and oASD samples (Figure 2C), including E. tayi, B. thetaiotaomicron, and P. merdae (Figure 2D). Thus, we found that differences in bacterial community composition between oTD and oASD mice were specific to certain taxa. Similar to human studies, β-diversity of bacterial communities differed between oASD and oTD mice (Liu et al., 2019). Specifically, Bacteroidetes, Bacteroides and Parabacteroides were decreased in oASD mice (De Angelis et al., 2015; Strati et al., 2017; Williams et al., 2011), with an increase in Akkermansia, Sutterella and Lachnospiraceae, as has been reported in humans (Kang et al., 2013; Williams et al., 2011).
Figure 2. Lachnospiraceae, Bacteroides, and Parabacteroides are differentially abundant in the oTD and oASD microbiomes.

(A) Volcano plot of differential bacterial abundance analysis as calculated by DESeq2 from 16S rRNA gene sequencing. Fold change as a factor Benjamini-Hochberg corrected p-values are plotted for each taxon. Significantly different taxa (α≤0.001) are colored according to their phylum.
(B) Heat map of differentially abundant amplicon sequence variants (ASVs) by DESeq2 (α≤0.001) from 16S rRNA gene sequencing. Features are named by their taxonomy with a unique feature identifier. Samples are clustered by Bray-Curtis distances.
(C) Microbiome features (ASVs) contributing >1% to classification between oTD and oASD samples by RandomForest. Taxon abundances from offspring mice were used to train a supervised Random Forest classifier based on donor diagnosis (oTD/oASD; accuracy ratio over baseline: 1.75). Taxa are ordered by their contribution to correct classification of microbiomes by diagnosis.
(D) Relative abundance of select taxa in the microbiome of male offspring, colored by donor. Hypothesis testing for differences of the means were tested by a linear mixed effects analysis with diagnosis as a fixed effect and donor ID as a random effect. NoTD= 15, NoASD= 20 (4–7 mice per donor).
(E) Abundance of select taxa in the offspring microbiome is correlated with behavior of male offspring. Spearman’s rank correlation between the microbiome and mouse behavior, by donor (as in Figure 1). Benjamini-Hochberg adjusted p-values (α≤0.05) for significant correlations are noted. Color scale denotes Spearman’s ρ from purple (positive correlation) to green (negative correlation).
(F-G) Relative abundance of P. merdae and E. tayi in the original human cohorts. Hypothesis testing by one-tailed Mann-Whitney U test. NTD=32, NASD=42.
We next explored whether specific ASVs positively or negatively co-vary with behavioral outcomes. The abundance of three of the four bacterial ASVs significantly correlated with both repetitive and social behaviors in male mice using Spearman’s rank correlation (Figure 2E). The Bacteroides (b20cd_Bacteroides) species and P. merdae (4ae7e_Parabacteroides) both correlated with reduced repetitive behavior and increased social behavior. Conversely, E. tayi (02b40_Lachnospiraceae) showed the opposite effects, as it correlated with increased repetitive behavior and social interaction deficits (Figure 2E). Accordingly, we validated that 16S rRNA gene ASVs that map to E. tayi, but not those that map to P. merdae, were more abundant in ASD individuals from large human cohort studies (Figure 2F–G). The association of specific bacterial species with oTD or oASD samples, which are also highly correlated with ASD-relevant behaviors, supports the hypothesis that specific bacteria may contribute to various symptoms of ASD.
ASD Microbiota Promote Extensive Alternative Splicing of ASD-relevant Genes in the Brain
Social behavior is mediated by multiple brain regions including the prefrontal cortex (PFC) and the striatum (STR) (Barak and Feng, 2016). Synaptic dysfunction and aberrant developmental trajectories in the STR may result in increased repetitive behaviors (Langen et al., 2014; Rothwell et al., 2014). Large scale analyses, comparing expression in brains of TD and ASD individuals highlight differential expression patterns of various genes and non-coding RNAs (Parikshak et al., 2016; Voineagu et al., 2011). RNA-seq analysis revealed few genes with significant differential expression profiles between oTD and oASD mice at P45 (Figures S4A–C). Three long-noncoding RNAs of unknown function from the PFC, and two additional genes from the combined PFC and STR dataset, displayed differential expression between groups of mice (Figures S4A–C, Figure S4D). One protein coding gene of known function, namely Daglb, was down-regulated in ASD-colonized mice. Diacylglycerol Lipase Beta (Daglb) is involved in endocannabanoid production and affects axonal growth during development (Bisogno et al., 2003; Powell et al., 2015) (Figure S4D). A gene set enrichment analysis (GSEA) highlighted several KEGG pathways that were significantly different in oASD brains (Figure 3A–B). Pathways involving transcription, translation, and protein quality control and export were upregulated in oASD brains (Figure 3A). A KEGG pathway involving RNA processing by the spliceosome was significantly upregulated in oASD mice (Figure 3A).
Figure 3. The microbiome impacts gene expression and alternative splicing of high confidence ASD genes in the mouse brain.

(A–B) KEGG pathways upregulated (A) and downregulated (B) in the brains of oASD mice by Gene Set Enrichment Analysis (GSEA).
(C) Venn diagram of differentially spliced genes in the STA and/or PFC between oTD and oASD mice (ASD microbiome Spliced Genes; FDR≤0.05), and their overlap with known ASD genes as curated by SPARK (odds ratio: 4.12 (95% CI 2.16–7.88), p < 0.0001) and SFARI Gene dataset (4 syndromic genes, 21 genes categories 1–3, and 13 genes category 4 and above; odds ratio: 1.39 (95% CI 1.01–1.92), p = 0.0401). Differential splicing events were identified by rMATS.
(D) Cell-type enrichment analysis of differentially-splicing events in brains of oASD mice. Odds ratio and 95% confidence intervals are presented.
(E) Enrichment of differentially-spliced events amongst previously reported targets of specific RNA-binding proteins and activity-dependent events in the brain.
(F) Examples of differential splicing events (FDR ≤ 0.05) in genes present in both SPARK and SFARI Gene. Data points colored by donor. PFC: NoASD= 19, NoTD= 14; STR: NoASD= 20, NoTD= 14 (3–6 mouse samples per donor/tissue). Benjamini-Hochberg corrected p-values were calculated by rMATS.
Given evidence for spliceosome alterations, we investigated whether oASD brains exhibited differential patterns of alternative splicing (AS), a feature found in human ASD brains (Gandal et al., 2018; Irimia et al., 2014; Parikshak et al., 2016; Parras et al., 2018). rMATS analysis (Shen et al., 2014) identified 560 genes that showed different AS events between oASD and oTD mice in either brain region (PFC: 265 events, STR: 257 events, Full dataset: 243 events): 123 with 3’ or 5’ alternative start sites (A3SS/A5SS) events, 78 with mutually exclusive exon (MXE) events, 381 with skipped exon (SE) events, and eight with retained introns (RI). We examined curated lists of high-confidence ASD risk genes, namely SPARK for Autism and the SFARI Gene dataset (Abrahams et al., 2013) (Figure 3C, Table S2). Fifty two of the 560 AS events occurred within genes associated with autism, with 11 present in both the SPARK gene list and SFARI Gene dataset, and 52 present in the SFARI gene list (Figure 3C). Differential splicing events were enriched for genes highly expressed in neurons, and under-enriched for in microglia in oASD mice compared to oTD animals (Figure 3D). Interestingly, we found that this dataset of differentially spliced genes was highly enriched for various ribosome binding protein (RBP) targets, but under-enriched in activity dependent transcripts (Figure 3E), suggesting that the gut microbiome can alter splicing programs at the RBP level.
We detected differential splicing events in several ASD-related genes, predominantly in the STR of oASD mice (Figure 3F). Amongst these is the Fragile X Mental Retardation 1 gene (FMR1), required for normal cognitive development (Iossifov et al., 2012; Parikshak et al., 2013; Sittler et al., 1996). oASD mice show decreased inclusion rates of a skipped exon in FMR1 (Figure 3F). We observed an increased inclusion rate of a mutually exclusive exon (MXE) in Neurexin-2 (Nrxn2), a presynaptic adhesion protein strongly linked to ASD (Dachtler et al., 2014; Südhof, 2008) (Figure 3F). Ank2, necessary for neuronal migration (Kordeli and Bennett, 1991; Willsey et al., 2013), exhibited decreased inclusion of a MXE in the STR of oASD mice, compared to oTD controls (Figure 3F). Other genes that show aberrant splicing in the PFC of mice include the calcium voltage-gated channel subunit Cacna1c, mutations in which cause Timothy syndrome (Splawski et al., 2004); adnylsuccinate lyase Adsl which is associated with infantile autism (Sivendran et al., 2004); Pogz, associated with ASD and intellectual disability (Stessman et al., 2016); as well as Ube3A, aberrant splicing of which can result in Angelman syndrome (Sartori et al., 2008). These data reveal a major shift in alternative splicing patterns of genes in the brains of oASD mice relative to oTD mice, with an enrichment for ASD-related genes in the differentially spliced subset that mirror findings from human brains.
Microbiota from TD and ASD-colonized Mice Produce Discrete Metabolite Profiles
Gene expression, splicing, and neuronal function in the brain can be regulated by small molecule metabolites (Nankova et al., 2014). An altered metabolome has been associated with ASD (Aldred et al., 2003; De Angelis et al., 2013; Evans et al., 2008; Kang et al., 2017a; Naushad et al., 2013; Yap et al., 2010), and many small molecules differing between TD and ASD likely result from microbial metabolism (De Angelis et al., 2015; Krajmalnik-Brown et al., 2015). We performed untargeted metabolomics analyses of colon contents from oTD and oASD mice (Figure 4A–B), and corresponding serum (Figure 4C). Twenty-seven out of 313 detected metabolites were significantly different in the colon contents of oASD mice, compared to oTD mice (Figure 4; Table S3) (Figure 4A–F). There were higher concentrations of amino acids in colon contents of oASD mice, similar to reports studying individuals with ASD (De Angelis et al., 2013). Notably, we found differences in several agonists and antagonists of the inhibitory gamma-aminobutyric acid (GABA) and glycine receptors (Figure 4G–I). Specifically, 5-aminovaleric acid (5AV) was significantly lower in oASD mice (Figure 4G). 5AV is a weak GABAA receptor agonist (Callery and Geelhaar, 1985), and is significantly lower in individuals with ASD (Ming et al., 2012). Lower levels of another weak GABAA agonist (and a potent glycine receptor agonist), namely taurine, are found in a subset of individuals with ASD (Adams et al., 2011; Park et al., 2017; Tu et al., 2012). Intriguingly, oASD mice have ~50% less taurine compared to oTD mice (Figures 4H). Together, lower levels of 5AV and taurine suggest that gut microbes may impact inhibitory GABA signaling. Conversely, 3-aminoisobutyric acid (3AIBA), a degradation product of thymine and a weak glycine receptor agonist, was increased in oASD mice (Figure 4I). Additional bioactive molecules elevated in colons of oASD mice are the soy-derived isoflavones, genistein and daidzein (Figures 4J). While 21 serum metabolites were differentially abundant (8 identified) in oASD mice versus oTD mice, only genistein is significantly different in both colon contents and serum (Figure 4C, F, J). Various sexually-dimorphic effects on neurodevelopment and behavior are linked to genistein, daidzein and their degradation products (Ponti et al., 2017; Rodriguez-Gomez et al., 2014; Westmark, 2014). Elevated levels of these metabolites suggest reduced dietary nutrient metabolizing bacteria in the oASD microbiota (Matthies et al., 2008, 2009).
Figure 4. oTD and oASD microbiomes imprint the colonic and serum metabolome of mice.

(A–C) Volcano plots of differentially abundant metabolites identified by an untargeted metabolomics of (A) colon contents by GC-MS, (B) colon contents by 1H NMR, and (C) serum by GC-MS. Significantly different metabolites with more than 50% difference are marked in red, and those with modest effects (<50%) are marked in yellow. NoASD= 20, NoTD= 15 (4–7 mice per donor). p-values were calculated using the maximum likelihood test of a mixed effect linear model.
(D-F) Heat maps of differentially abundant metabolites identified by an untargeted metabolomics of (D) colon contents by GC-MS, (E) colon contents by 1H NMR, and (F) serum by GC-MS. NoASD= 20, NoTD= 15 (4–7 mice per donor). Metabolite abundances were median-normalized and plotted based on the Z-score from purple for highly abundant metabolites, to green, for metabolites detected in low levels.
(G–I) Median-normalized concentrations of (G) 5-aminovaleric acid, (H) taurine, (I) 3-aminoisobutyric acid, and (J) the isoflavones daidzein and genistein in colon contents. Data point color denotes donor. NoASD= 20, NoTD= 15 (4–7 mice per donor). Bar graphs denote mean and S.E.M. p-values were calculated using the maximum likelihood test of a mixed effect linear model. oTD and oASD are offspring of recipient mice.
We analyzed metagenomic data from colon contents of mice by HUMAnN2 (Franzosa et al., 2018), and coupled these data with MIMOSA (Noecker et al., 2016) – a metabolic model-based framework for inferring the contribution of bacterial species and genes to the production and degradation of metabolites measured by NMR and GC-MS analysis (Figures S5). Interestingly, MIMOSA analysis suggested that amino acids were predominantly degraded/utilized by the microbiota in contrast with other metabolites (by specific Akkermansia, Alistipes and Bacteroides species) (Figure 5A, Figure S5A–B, Figure S5C–D). Specific KEGG pathways differentially present in oTD and oASD gut microbiomes, with various chemical structure transformation maps highly represented in oASD mice (Figure S5E). We further found that the metabolism of various amino acids, and specifically that of proline, taurine, and glutamate and glutamine, were differentially represented in the metagenomes of oASD mice (Figure 5B). These data indicate that the oTD microbiota preferentially metabolizes specific dietary amino-acids.
Figure 5. Metagenomic analysis corroborates amino acid metabolism is deficient in oASD mice.

(A) Putative bacterial contributors to variation in amino acids and other metabolites identified by a MIMOSA analysis, separated by their mechanism of action (synthesis/production, degradation/utilization, or both), and based on GC-MS (top) and NMR (bottom) metabolomic analysis and metagenomic analysis of colon contents in oASD mice.
(B) Differentially abundant KEGG orthologs involved in amino acid metabolism from HUMAnN2 by limma-voom analysis. Fold change and p-values are presented for significantly different pathways.
(C–E) Taurine production in oASD mice is deficient. (C) possible sources of taurine and KEGG orthologs involved. (D) abundance of K01442 and (E) K01580 copies in oTD and oASD mice quantified by HUMAnN2. Differences in Means were analyzed by a Kruskal-Wallis test.
(F–I) Pathways providing substrates for Stickland fermentation to produce 5AV are deficient in 0ASD mice. (F) pathways upstream to 5AV production and KEGG orthologs involved. (G) abundance of hypD (by ShortBRED) and the abundance of (H) K13821 and (I) K00286 copies in oTD and oASD mice quantified by HUMAnN2. Differences in Means were analyzed by a Kruskal-Wallis test.
See also Figure S5.
HUMAnN2 and ShortBRED analyses (Franzosa et al., 2018; Kaminski et al., 2015) of metagenomic data enabled exploring possible pathways for taurine and 5AV metabolism. Taurine can be produced by either deconjugation of tauro-conjugated bile acids, such as taurocholic acid, or by decarboxylation of Lcysteate (Figure 5C). HUMAnN2 analysis identified that both bile-salt hydrolase (K01442) and glutamate decarboxylase (K01580) were present at lower abundance in oASD metagenomes. Glutamate decarboxylase catalyzes the conversion of glutamate to GABA, and can also decarboxylate cysteate to taurine (Kim et al., 2009; Wu, 1982). Interestingly, MIMOSA analysis predicted that taurine concentrations may result from differential synthesis potential by three specific species: Alistipes sp. HGB5, Alistipes finegoldii, and Bacteroides xylanisolvens (Figure S5A). 5AV is likely the product of Stickland fermentation of proline (Huang et al., 2018). However, we found no differences in the abundance of proline reductase (Figure S5F–G). Δ1-pyrrolline-5-carboxylate (P5C) is an intermediate of proline production from trans-4-hydroxyproline (Hyp) by HypD (Huang et al., 2018; Levin et al., 2017), which is subsequently reduced to L-proline by P5C reductase (K00286). Additionally, the oxidation of L-proline by proline dehydrogenase (K13821) produces L-glutamate, with P5C as an intermediate (Liu et al., 2017). Hydroxyproline concentrations varied consistently with K00286 abundance (Figure S5B), and hypD levels were not significantly decreased in oASD samples (Figure 5G). However, we found a significant increase in K13821 (Figure 5H) and a significant decrease in P5C reductase (K00286) abundance (Figure 5I) in oASD mice. These data suggest that, in oTD microbiota, L-proline is produced from Hyp via P5C and subsequently serves as an electron acceptor to produce 5AV (Figure 5F–I). Conversely, in oASD microbiota, the balance shifts towards production of P5C and glutamate, and away from 5AV (Figure 5F–I). Together, empiric data and computational predictions suggest that amino acid metabolism may be a key mechanism by which the gut microbiota modulates behavior.
Administration of Metabolites Depleted in ASD Improve Behaviors in a Mouse Model
Microbial metabolites in the gut can impact neurological outcomes (Wang et al., 2018). We next correlated metabolite profiles to human subject metadata (e.g., age, ADOS score, and GSI score) and behavioral outcomes in male mice. Spearman’s rank correlation suggests specific metabolites that distinguish oASD and oTD mice, including taurine and 5AV, may protect mice from development of ASD-like behaviors (Figure 6A). In addition, we predict that genistein and daidzein (or the absence of their degradation products) contribute to repetitive behavior effects while lysine, 3AIBA, and genistein are predicted to influence locomotion (Figure 6A). Integration of microbiome and metabolome profiles enables the testable hypothesis that protective or pathogenic microbial metabolites may regulate specific behaviors associated with ASD.
Figure 6. Microbial metabolites impact behaviors and brain physiology in the BTBR mouse model.

(A) Spearman’s rank correlation between discrete metabolites and male mouse behavior (see Figure 1). Benjamini-Hochberg adjusted p-values for significant (α≤0.05) correlations are noted. Color scale denotes Spearman’s ρ from purple (positive correlation) to green (negative correlation).
(B) Targeted metabolomics for 5AV and taurine in E18.5 dams orally administered metabolites at 10mM in drinking water from E0.5 and on. Normalized concentrations of 5AV and taurine in dam’s colon contents, serum, amniotic fluid, and fetal brains were measured. Group differences were tested by Kruskal-Wallis test, Dunn post-hoc, and Holm correction for multiple comparisons. N=3–4 dams per group.
(C–E) 5AV and taurine ameliorate ASD-related behavioral deficits in the BTBR mouse model for ASD. Groups of mice were orally administered with either 10 mM taurine or 5AV in drinking water (ad libitum) before mating, and throughout their lifetime. Offspring were tested by (C) marble burying, (D) direct social interaction, and (E) open field tests, and compared to untreated vehicle controls. Results are aggregated from three independent experiments. NControl= 42, N5AV= 52, NTaurine= 33. Hypothesis on differences in means were tested by one-way ANOVA on trimmed means (10%) and subsequent post-hoc tests.
(F) Amplitude and frequency of mEPSCs in pyramidal neurons in the L5 of the mPFC in acute slices from 8–12 week old BTBR mice treated with 5AV, Taurine, or control from pregnancy to adulthood. tested by 1-way ANOVA on trimmed means (10%) and subsequent post-hoc tests. NControl = 21 cells in 5 mice, N5AV = 18 cells in 3 mice, NTaurine = 20 cells in 4 mice.
(G) Amplitude and frequency of mIPSCs in pyramidal neurons in the L5 of the mPFC in acute slices from 8–12 week old BTBR mice treated with 5AV, Taurine, or control from pregnancy to adulthood. tested by one-way ANOVA on trimmed means (10%) and subsequent post-hoc tests. NControl = 21 cells in 4 mice, N5AV = 17 cells in 3 mice, NTaurine = 20 cells in 4 mice.
(H) Excitability of pyramidal neurons in the L5 of the mPFC in acute slices from BTBR mice treated with 5AV, Taurine, or control, in response to step-wise injection of current, as measured by the number of action potential spikes. Two-way ANOVA and Dunnett’s post-hoc. * P < 0.05, ** P<0.01.
See also Figure S6.
The presence of both 5AV and taurine in the mouse colon depends on the microbiota (Figure S6A–B) (Matsumoto et al., 2017). We hypothesized that the decreased levels of taurine and 5AV in colon contents of oASD mice (see Figure 4G–H) could have an effect during gestation as well as adulthood.
We orally administered high concentrations (10mM) of either 5AV or taurine to pregnant C57BL/6J mice, and measured the abundance of these metabolites in colon contents, amniotic fluid, and fetal brains (Figure 6B). While 5AV reached the fetal compartment and the fetal brain (Figure 6B), taurine concentrations did not change with administration suggesting it may act locally in the gut.
To test the behavioral effects of 5AV and taurine, we employed the BTBR T+ tf/J (BTBR) mouse model that displays ASD-related behavioral changes (McFarlane et al., 2008; Meyza and Blanchard, 2017; Moy et al., 2007; Silverman et al., 2010). BTBR mice have known microbiome and GI changes relative to C57BL/6 mice (Coretti et al., 2017; Golubeva et al., 2017; Newell et al., 2016), as well as known metabolic deficits (Golubeva et al., 2017; Klein et al., 2016). We orally administered taurine to BTBR mice from conception through adulthood to capture both prenatal and postnatal neurodevelopmental periods, and compared their behavior to untreated animals. Indeed, taurine significantly reduced repetitive behavior, as measured by marble burying, and increased social duration in the DSI test (Figure 6C–D). Additionally, offspring treated with taurine displayed reduced anxiety, as indicated by increased center duration in the OFT (Figure 6E). 5AV was predicted to increase social behavior, based on correlation (Figure 6A). Administration of 5AV from conception through adulthood reduced repetitive behavior in BTBR mice (Figure 6C), although this outcome was not predicted by correlation. Importantly, we validated the predicted positive effects of 5AV on social behavior (Figure 6D). Exposure of taurine and 5AV to developing BTBR mice during the prenatal and weaning periods is critical, as administration to juvenile mice starting at 4 weeks of age and through adulthood did not rescue behavioral alterations (Figure S6C–E). A similar effect was observed when we administered 5AV from weaning to offspring mice colonized with human microbiota from donor ASD3 (Figure S6F–H). Mice administered 5AV showed increased locomotion, decreased center duration (Figure S6F), and increased sociability (Figure S6H), with no significant difference in repetitive behavior (Figure S6G). These findings support the hypothesis that metabolites of gut bacteria can contribute to the manifestations of ASD-like behaviors in mice.
5AV Reduces Neuronal Excitability in the Prefrontal Cortex of BTBR Mice
Some forms of ASD are thought to result from an imbalance between excitation and inhibition (E-I) in synaptic transmission and cortical circuitry (Lee et al., 2017; Nelson and Valakh, 2015; Rubenstein and Merzenich, 2003), although a recent report suggests E-I imbalance is not causative in some mouse models (Antoine et al., 2019). E-I imbalance, and its modulation by various treatments, have been reported in BTBR mice (Burket et al., 2013; Lee et al., 2017; Silverman et al., 2012, 2013). Targeting GABAergic signaling has been suggested to be an avenue for intervention in ASD (Cellot and Cherubini, 2014). To test whether 5AV and taurine affect cortical circuitry and perturb E-I balance in BTBR mice, we recorded from pyramidal neurons in layer 5 (L5) of the medial prefrontal cortex (mPFC) by whole-cell patch clamp in acute slices. Specifically, we investigated whether treatment with taurine or 5AV (from conception to adulthood) affected the intrinsic membrane and synaptic [miniature inhibitory or excitatory postsynaptic currents (mIPSC and mEPSC)] properties of pyramidal neurons. Neither 5AV nor taurine affected the intrinsic membrane properties of neurons or the frequency or amplitude of mEPSCs and mIPSCs, compared to controls (Figure S6I, Figure 6F–G). However, L5 pyramidal neurons from mice treated with 5AV, but not taurine, showed significantly decreased excitability as measured by the number of spikes observed as a result of injection of stepwise increasing currents (Figure 6H). Another process hypothesized to affect GABAergic signaling is the GABA excitatory to inhibitory switch during development, although it is still somewhat controversial (Ben-Ari et al., 2012; Valeeva et al., 2016). Since taurine has previously been shown to inhibit the potassium-chloride cotransporter KCC2 (Inoue et al., 2012), it might affect the excitatory-to-inhibitory switch in response to GABA. Primary cortical neurons from embryonic Sprague Dawley rat brains were cultured with taurine, 5AV, or controls (over 17 days in vitro), and responses to GABA exposure by calcium imaging were measured using a calcium-specific dye (Fluo-4). Compared to controls, cortical neurons treated with taurine exhibited a delayed switch from excitatory to inhibitory response to GABA (Figure S6J), suggesting that taurine might affect the development of neurons and their response to inhibitory neurotransmitters. We propose that the human microbiota can modulate complex behaviors in mice via the production of neuroactive microbial metabolites, with mechanisms that include regulation of E-I balances in the brain.
DISCUSSION
A potential link between the microbiota and ASD was suggested almost two decades ago, emboldened by two foundational observations: first it was reported that oral vancomycin treatment resulted in a short-term benefit in a small group of children with ASD (Sandler et al., 2000), and subsequently, Finegold and colleagues surveyed gut bacteria in children with ASD and noted differences compared to controls (Finegold et al., 2002). Since then numerous studies from diverse cohorts have reported that microbiome profiles of ASD individuals, especially those with co-morbid GI dysfunction, differ from that of TD controls (Coretti et al., 2018; De Angelis et al., 2013; Finegold et al., 2010; Gondalia et al., 2012; Kang et al., 2013, 2017a; Kushak et al., 2016; Liu et al., 2019; Son et al., 2015; Strati et al., 2017; Wang et al., 2019; Williams et al., 2011). The high prevalence of GI dysfunction (Adams et al., 2011; Chaidez et al., 2014; McElhanon et al., 2014), along with evidence of immune imbalance (Onore et al., 2012), further suggest microbiota involvement in ASD. While these compelling association studies in humans suggest gut bacteria may impact behavioral symptoms, a direct contribution by the microbiota to the pathophysiology and behavioral outcomes of ASD has not been described previously. We report herein that colonization of mice with gut microbiota from human donors with ASD, but not from TD controls, is sufficient to promote behaviors in mice consistent with the core behavioral features of ASD. Notably, phenotypes were more pronounced in male offspring mice; future work will address gender differences in mice that the current experimental design cannot resolve since we exclusively utilized male donors. Our results demonstrate that changes to the human microbiota can promote ASD-like behaviors in wild-type mice, but do not conclude that gut bacteria are entirely causal for symptoms. Factors such as altered host genetics and perinatal events, coupled with an altered microbiota, may together influence the etiology of ASD by compounding risks that enhance symptom severity.
The transcriptional program in ASD brains is distinct from that of TD brains (Gandal et al., 2018; Parikshak et al., 2016; Voineagu et al., 2011). RNAseq analysis of “humanized microbiota” mice showed few transcripts differing in expression levels. However, several disease-related pathways were upregulated. Recent studies have highlighted the importance of aberrant alternative splicing of mRNA in the brains of subjects with an ASD diagnosis (Corominas et al., 2014; Gandal et al., 2018; Irimia et al., 2014; Parikshak et al., 2016; Voineagu et al., 2011). Among upregulated KEGG pathways in oASD mice were genes related to the spliceosome, suggesting that the microbiota contribute to splicing regulation. Analysis of gene splicing revealed extensive differences between oTD and oASD mice, with 11 alternative splicing events in validated high-risk ASD genes. These findings suggest that gut microbes not only regulate gene expression in the mouse brain (Hoban et al., 2016; Neufeld et al., 2011; Stilling et al., 2015), but also affect alternative splicing (Stilling et al., 2018), and can regulate the production of specific isoforms of genes that have been implicated in ASD.
Gut bacteria can affect gene expression and host behavior through various pathways, including the production of neuroactive small molecules (Hsiao et al., 2013). Concentrations of several metabolites in the colon and circulation were regulated by the gut microbiota in a donor-specific manner, and we observed impaired amino acid metabolism. Two examples are taurine, the metabolic product of cysteate or taurocholic acid, and 5AV, the fermentation product of proline—both GABAA receptor agonists that are increased in the colons of oTD mice. Taurine is essential for brain development (Kilb and Fukuda, 2017; Tochitani, 2017) and can be produced by neurons and astrocytes (Vitvitsky et al., 2011) or transported through the blood-brain-barrier via the Taurine transporter TauT (Benrabh et al., 1995). 5AV acts as an anticonvulsant in mice (Dhaher et al., 2014; Samuels et al., 1983). We hypothesized that taurine and 5AV are neuroactive metabolites produced by the microbiota. Indeed, administration of either taurine or 5AV to BTBR mice from conception attenuated ASD-relevant behavioral alterations, namely stereotypies and social impairments. These GABAA receptor agonists may act to perturb the E-I balance in the brain, specifically during development. Extensive evidence highlight the importance of the E-I balance in neurodevelopmental disorders (Lee et al., 2017; Nelson and Valakh, 2015; Rubenstein and Merzenich, 2003). Administration of 5AV to BTBR mice decreased the excitability of L5 pyramidal neurons in the mPFC. Consistent with these results, social deficits in the CNTNP2-deficient mouse model of autism could be rescued by modulating the excitability of pyramidal neurons (Selimbeyoglu et al., 2017). Our results indicate that taurine can delay the excitatory-to-inhibitory developmental switch in neurons, at least in vitro, and may also act on brain function via other mechanisms (Bellono et al., 2017; Kaelberer et al., 2018). Together, these discoveries provide experimental evidence that microbial metabolites can mediate communication between the gut and brain in the context of modeling ASD in mice.
Recent advances in DNA sequencing technologies have empowered numerous microbiome profiling studies, revealing changes to microbial community compositions across diets, geographies, and diseases. While hypothesis-generating, translating these correlations into actionable outcomes is challenging in humans. GF animals harboring a human microbiota offer an experimental tool to investigate the function of human gut bacteria in a highly controlled environment. This is critical, as ASD is a heterogeneous condition, displaying a wide spectrum of symptom severity among affected individuals, with diverse inherent predispositions and extensive environmental exposures (Lombardo et al., 2019). While ours is a limited study, with 16 donor samples from a pediatric cohort, the results support a hypothesis that the human gut microbiota contributes to ASD phenotypes. Analysis of fecal samples from additional well- characterized cohorts will enable the identification of subsets of ASD individuals whose symptoms may be microbiota-related from those with disparate etiologies. The high concordance rates observed between monozygotic twins implicate a strong genetic basis for ASD (Tick et al., 2016). However, it is important to consider that both human and microbial genes are vertically heritable across generations. Accordingly, we propose that the etiology of ASD may lie in the human genome, the human microbiota, or both via gene-environment interactions. While the exciting prospect of genome editing remains on the horizon, development of microbiota-based interventions such as probiotics, fecal microbiota transplantation, or metabolites may offer a timely and tractable approach to addressing the lifelong challenges of ASD.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Contacts, Gil Sharon (gsharon@caltech.edu) and Sarkis K. Mazmanian (sarkis@caltech.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human fecal samples
For all animal experiments, Arizona State University (ASU) shared human fecal samples with California Institute of Technology (Caltech) with a Material Transfer Agreement and approval to share deidentified data by the Institutional Review Board (IRB) at ASU (ASU IRB protocol #: 1206007979, Caltech IRB protocol # 15–0569). Human fecal samples were previously collected from typically developing children and children with autism spectrum disorders (ASD) at ASU (Kang et al., 2013, 2017). Briefly, parents collected and froze a single fecal sample from each subject. Frozen fecal samples were shipped overnight to ASD with a cold pack, and stored at −80 °C. All fecal samples and their metadata including gastrointestinal (GI) and ASD-relevant clinical data were de-identified before being shared with Caltech. DNA samples from Kang et al., 2013, 2017 were resequenced and analyzed as specified below.
Mouse husbandry
All animal husbandry and experiments were approved by the Caltech Institutional Animal Care and Use Committee (IACUC protocol #1645). Throughout the study, colonized animals were maintained in autoclaved microisolator cages with autoclaved bedding (Aspen Chip Bedding, Northeastern Products Corp, Warrensburg, NY), water, and chow (Laboratory Autoclavable Rodent Diet 5010, LabDiet; St. Louis, MO, USA). Mice were maintained at an ambient temperature of 71–75 °F, 30% 70% humidity, at a cycle of 13 hours light & 11 hours dark.
Mouse Colonization
Germ-free (GF) C57BL/6J weanlings (3–4 weeks of age) from the Mazmanian laboratory colony were colonized with fecal samples from human donors. Human fecal samples were collected by the Krajmalnik-Brown laboratory at the Arizona State University as part of a previous study (Kang et al., 2013, 2017), and stored at −80 °C. Aliquots of 16 donor samples were sent to Caltech and used for colonization into GF mice. To that end, frozen aliquots were thawed in an anaerobic chamber and resuspended in two volumes of reduced sodium bicarbonate solution (final concentration 5 %) (Life Technologies; Carlsbad, CA, USA). Subsequently, samples were vigorously vortexed and spun down. Supernatants were then used to colonize GF mice by a single oral gavage (100 μl/mouse; Instech, PA, USA). Colonized mice (4–6 females and 2–3 males per donor) were then allowed to rest for 3 weeks, and were subsequently mated according to donor. Pregnant dams were single-housed at E15.5–17.5, and offspring were weaned at 3 weeks of age. At weaning, different litters born within up to a week apart were combined and housed in groups of 4–5 male or female mice per cage and used for subsequent analyses. Cages were assigned to either behavior testing or for tissue collection. Behavior testing started at 6 weeks of age, while tissues were collected at P45. In the initial stage of this study, we colonized and subsequently behaviorally tested mice with fecal samples from 13 human male donors. Subsequently, we tested and analyzed a subset of representative samples (two TD and three ASD) along with additional previously untested samples (one TD and two ASD), to ensure there is no bias in sample selection.
METHOD DETAILS
Behavior testing
All mice were tested using the same battery of behavioral tests, starting at six weeks of age, in the following order: open field testing, marble burying, social behavior, and USV (male-female context). Mice were allowed to rest for at least two nights after cage changing before they were tested, and tests were performed 4–7 days apart to allow mice to rest between tests. Mice were acclimated to the behavior testing room overnight to reduce stress and anxiety. Mice were tested during the light phase of the light cycle; to control for time of day effects, cages of different groups were alternated. During the initial discovery phase, the experimenter was blinded to the donor but not to the group. In the subsequent validation phase, the experimenter was blinded. Each donor sample was tested 1–2 times and the aggregated data is presented.
Open field testing (OFT)
OFT was performed in 50 × 50 cm2 white plexiglas arenas, recorded using an overhead camera, and tracked and analyzed using the EthoVision XT 10 software package (Noldus Information Technology; Leesburg, VA, USA). Prior to testing, the arena was disinfected using Rescue disinfectant (Virox technologies), followed by 70% ethanol and finally water. Mice were then introduced to the arena and allowed to explore for 10 min while tracked. The total distance traveled, and the number of entries and time spent in a 30 × 30 cm2 center square, were analyzed by the EthoVision XT 10.0 software (Noldus Information Technology; Leesburg, VA, USA).
Marble burying (MB) -
MB was performed in a normal cage bottom (Lab Products; Seaford, DE, USA) with floor area of 75 in2 filled with 3–4 cm of fresh, autoclaved wood chip bedding (Aspen chip bedding, Northeastern Products Corp; Warrensburg, NY, USA). Mice were first habituated to the cage for 10 min, and subsequently transferred to a holding cage while the bedding was leveled and 20 glass marbles (4 × 5) were placed on top. Mice were then returned to their own cage and removed after 10 min. The number of buried marbles (50% or more covered) was then recorded and photographed for reference. Bedding was replaced for each mouse, and marbles were soaked in 70% ethanol and dried in bedding in between each test. When the experimenter was not blinded to the treatment or group, the number of buried marbles was scored by a second experimenter blinded to the treatment/group from overhead images taken after the second 10 min session.
Three-chamber sociability test -
the 3-chamber sociability test was performed in a 60 × 40 cm2 white plexiglass box divided into three chambers (20 × 40 cm2) by clear plexiglass dividers. Mice were first habituated to the full empty arena for 10 min. Subsequently, mice were confined to the center chamber and a stimulus mouse (sex-matched adult SPF C57BL/6J) was placed in a small cage in one chamber (social chamber) while a small object was placed in a cage on the other chamber (non-social chamber). Mice were then allowed to travel between chambers for 10 min, and the movement of mice was recorded by an overhead camera and tracked using the EthoVision XT 10 software package (Noldus Information Technology; Leesburg, VA, USA). A sociability index was calculated by the following: . Prior to each test, arenas were disinfected using Rescue (formerly Accel) disinfectant (Virox Technologies; Oakville, ON, Canada), followed by 70% ethanol and finally water.
Direct social interaction (DSI) test
As a more sensitive measure for sociability, we also used the direct social interaction test where a mouse is allowed to interact with a stimulus mouse while the interaction is recorded using an overhead video camera. Each mouse was introduced to a fresh empty cage and allowed to habituate for 10 min; grooming behavior was scored for the last five min of this period. Subsequently, a stimulus mouse (either juvenile or adult SPF C57BL/6J (or BTBR), depending on the experiment) was introduced to the cage for 6 additional min. A blinded experimenter scored videos for any social approach, aggression, or grooming behavior using the ETHOM software (Shih and Mok, 2000). A set of reference videos was used to ensure consistency over time.
Ultrasonic vocalization (USV) -
The male-female paradigm was used to test deficits in communication in male mice. Mice were single-housed and exposed to a new SPF C57BL/6J (or BTBR) female for 10 min every day in the three days prior to the test. On the fourth day, mice were habituated to an empty cage (no bedding) with a filter soaked with a drop of fresh female urine for 10 min. Subsequently, the filter was removed and a novel female was introduced to the cage. Ultrasonic vocalizations were recorded using Avisoft UltraSoundGate 116Hme microphone (Avisoft Bioacoustics) and the Avisoft Sas lab Recorder software (Avisoft Bioacoustics). Total vocalization and vocalization counts were recorded during 3minute sessions of male-female interaction.
Tissue Collection
On P45, offspring mice were sacrificed by first administering 5% isoflurane by inhalation for 30 sec followed by cervical dislocation. Subsequently, blood was collected by heart puncture into 1.1ml z-gel serum collection tubes (Sarstedt; Germany). Serum was then collected according to the manufacturer’s instructions and stored frozen in −80 °C until analysis. Brains were macro-dissected using a mouse brain slicer (1mm coronal section slice intervals; Zivic Instruments; Pittsburgh, PA, USA) and sections of the prefrontal cortex and the striatum were collected into RNALater (Thermo; Waltham, MA, USA) and kept frozen in −80 °C until analysis. Intestines were dissected, colon and cecal contents collected separately and flash frozen while the intestinal tissue (~2 cm of the proximal colon and ~2 cm of the terminal ileum) were rinsed in PBS and frozen in RNALater. To control for effects by the time of collection, mice from different groups were sacrificed in an alternated fashion. All samples were then assigned an identification number that prevented from direct identification of the groups to facilitate blinded analysis of samples downstream.
Mouse fecal sample collection and microbial DNA extraction
Frozen mouse fecal samples were shipped overnight on dry ice to ASU and stored in −80 °C until DNA extraction. Human feces that were used as donor samples for the mouse experiments were also shipped back to ASU in order to be processed for microbial DNA extraction and next-generation sequencing together with mouse fecal samples. At ASU, microbial genomic DNA was extracted from fecal samples using the PowerSoil® DNA Isolation Kit (Mobio; Carlsbad, CA, USA) with a modification based on the manufacturer protocol. Quality and quantity of genomic DNA was verified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technology; Rockland, DE, USA).
16S rRNA gene sequencing
Qualified genomic DNA samples were processed for 16S rRNA library preparation and next-generation sequencing at the Microbiome Analysis Laboratory in the Biodesign Swette Center for Environmental Biotechnology (http://krajmalnik.environmentalbiotechnology.org/microbiome-lab.html). The Earth Microbiome Project standard protocols (http://www.earthmicrobiome.org/emp-standard-protocols/) were employed with the barcoded primer set 515F-806R (515F:GTGCCAGCMGCCGCGGTAA, 806R:GGACTACHVGGGTWTCTAAT) that targets the V4 region of the bacterial (and archeal) 16S rRNA gene (Caporaso et al., 2012). Paired-end, 2×150bp, next-generation sequencing was performed using MiSeq Illumina platform (MiSeq Reagent Kit v2; Illumina Inc.; San Diego, CA, USA) and microbiome sequencing data were analyzed using the Quantitative Insights Into Microbial Ecology (QIIME2) software package (Bolyen et al., 2018; Caporaso et al., 2010).
Shotgun sequencing for metagenomics
A miniaturized version of the Kapa HyperPlus Illumina-compatible library prep kit (Kapa Biosystems; Wilmington, MA, USA) was used for library generation. DNA extracts were normalized to 5 ng total input per sample in an Echo 550 acoustic liquid handling robot (Labcyte Inc; San Jose, CA, USA). A Mosquito HTS liquid-handling robot (TTP Labtech Inc was used for 1/10 scale enzymatic fragmentation, end-repair, and adapter-ligation reactions carried out using). Sequencing adapters were based on the iTru protocol (Glenn et al., 2016), in which short universal adapter stubs are ligated first and then sample-specific barcoded sequences added in a subsequent PCR step. Amplified and barcoded libraries were then quantified by the PicoGreen assay and pooled in approximately equimolar ratios before being sequenced on an Illumina HiSeq 4000 instrument to >30X coverage.
Microbiome analysis
16S rRNA gene
Demultiplexed sequencing outputs were obtained from the ASU sequencing facility and analyzed using the QIIME 2 (versions 2017.9 and 2018.8) software package according to the suggested workflow (Caporaso et al., 2010; Janssen et al., 2018). Since there was little overlap between forward and reverse reads, only forward reads (~150 bp long) were used for subsequent analysis. On average, 51,729 ± 1,331 reads were obtained per sample (median: 50,134 reads). Low quality bases were first removed from the reads and amplicon sequence variants (ASVs) were obtained using the Deblur denoising plugin (Amir et al., 2017) on reads trimmed to 120 bp present in the 16S reference dataset. Subsequently, fragments were inserted to 16S rRNA gene context by q2-fragment-insertion (Janssen et al., 2018) and alignments were obtained using MAFFT (Yamada et al., 2016) and a phylogenetic tree was generated using FastTree (Price et al., 2009). Alpha and Beta diversities were analyzed using the core-metrics-phylogenetic for observed ASVs, Faith’s phylogenetic diversity, and Pielou’s evenness measures for alpha diversity and unweighted UniFrac and Bray-Curtis for beta diversity measures (Lozupone and Knight, 2005). PCoAs were visualized by EMPeror (Vázquez-Baeza et al., 2013). Taxonomic analysis was performed using the q2-feature-classifier trained on GreenGenes 13_8 99% OTU table (Bokulich et al., 2018; McDonald et al., 2012). Differential abundance analysis was performed using the Phyloseq (1.20.0) and DESeq2 (1.16.1) R packages (Love et al., 2014; McMurdie and Holmes, 2013). Additional differential abundance analyses were performed using LefSe (Segata et al., 2011). To further analyze ASVs that contribute to the discrimination between NT and ASD samples and to behavioral phenotypes, a Random Forest analysis (Liaw et al., 2002), as implemented in QIIME 2, was used.
Metagenomics
The metagenomic data was processed using the Oecophylla pipeline (https://github.com/biocore/oecophylla). A total of 10,170,588 ± 996,036 (Mean ± SEM; Median 9,791,965) reads per mouse sample were obtained. The raw reads were examined with FastQC and low quality sequences and unwanted reads (0.044 ± 0.008%, Mean ± SEM; Median 0.03%) were filtered with Bowtie2 (v0.1; Langmead and Salzberg, 2012). The taxonomic composition was profiled using the default parameters of MetaPhlAn2 (Truong et al., 2015) through the Oecophylla pipeline. The functional gene pathway was profiled using the default settings of HUMAnN2 (Franzosa et al., 2018) through the Oecophylla pipeline with 43.1 ± 1.51% (Mean ± SEM; Median 40.45%) of the reads unassigned. HUMAnN2 uses the UniRef90, MetCyc and MinPath databases along with MetaPhlAn2 and ChocoPhlAn pangenome databases to characterize the pathways and genes in sequences (Truong et al., 2015). Gene family abundance, pathway abundance, and pathway coverage of each sample were generated from HUMAnN2; we used the gene family abundance output biom table for analysis. Differences in relative abundance between the sample groups were calculated with LIMMA (Law et al., 2014) and considered significant if p < 0.05 (Kruksal-Wallis test) after multiple test correction by FDR adjustment.
RNA Extraction and cDNA synthesis
Total RNA was extracted from de-identified tubes of mouse tissues using the RNeasy Kit (Qiagen; Germany), according to the manufacturer’s instructions. RNA concentrations and the 260/280 nm absorbance ratio were determined using NanoDrop One (Thermo; Waltham, MA, USA). cDNA was reverse transcribed using iScript cDNA Synthesis Kit (Bio-Rad; Hercules, CA, USA), according to the manufacturer’s protocol using 1 μg total RNA in 20 μL reactions.
Quantitative Real-Time PCR
qRT-PCR reactions were carried out in a total reaction volume of 12 l containing: 6 l of 2X Power SYBR Green PCR Master Mix (Applied Bioscience; Foster City, CA, USA), 0.5 l of each 10 M primers, and 5 l cDNA (10 ng per reaction). qRT-PCR were performed in duplicates using 384-well plate format in the ABI PRISM 7900 HT (Thermo; Waltham, MA, USA) with the following conditions: 2 min at 50°Cand 10 min at 95 °C, followed by 40 cycles of 15 sec at 95 °C and 1 min at 60 °C, dissociation stage consisted of 15 sec at 95 °C, 15 sec at 60 °C, 15 sec at 95 °C. the house-keeping gene β-actin was used to normalize expression levels between samples, and no template controls (NTCs) were used as negative controls. The following primers were used: ZO1: Forward: 5’- AGGACACCAAAGCATGTGAG-3’, Reverse: 5’-GGCATTCCTGCTGGTTACA-3’; Ocldn: Forward: 5’-TTGAAAGTCCACCTCCTTACAGA-3’, Reverse: 5’- CCGGATAAAAAGAGTACGCTGG-3’; ZO2: Forward: 5’-ATGGGAGCAGTACACCGTGA-3’, Reverse: 5’-TGACCACCCTGTCATTTTCTTG-3’;bActin: Forward: 5’-GGCTGTATTCCCCTCCATCG-3’, Reverse: 5’-CCAGTTGGTAACAATGCCATGT-3’
Intestinal permeability assay
10–11 week old offspring “humanized” mice were fasted (water ad libitum) for 4 hours. Subsequently, mice were gavaged with 4 KDa FITC-dextran (100 mg/ml; Sigma; St. Louis, MO, USA) at 600 mg/kg body weight. Four hours after gavage, mice were sacrificed by asphyxiation with CO2 and blood was collected by cardiac puncture into 1.1ml z-gel serum collection tubes (Sarstedt; Germany) and stored on ice in the dark at 4°C until collection according to the manufacturer’s instructions. 25 L of serum samples were loaded in each well and diluted with an equal volume of 1X PBS. FITC-dextran concentrations were determined by spectrophotofluorometry using Cytation5 (Biotek; Winooski, VT, USA) using wavelength setting of: excitation (485/20), emission (528/20) against a calibration curve of known FITC-dextran concentrations. Throughout the procedure, the experimenter was blinded to mouse groups.
Viable and total bacterial counts in feces
Fecal samples were collected from offspring “humanized” mice and homogenized in 10 μL/mg sterile 1X PBS. Suspensions were serially diluted and plated on Tryptic-Soy Agar plates with 5% sheep blood (TSA; Teknova; Hollister, CA, USA) and Brucella Agar with 5% Sheep blood plates (Teknova; Hollister, CA, USA). Subsequently, plates were incubated aerobically (TSA plates) or anaerobically (Brucella plates) at 37 °C for 48–72 hours. For total 16S rRNA gene quantification, DNA was extracted from 200 l of fecal suspensions using Quick-DNA Fecal/Soil Microbe Miniprep Kit (Zymo; Irvine, CA, USA), following manufacturer’s protocol. Fecal DNA from samples were eluted in 100 μL EB buffer, and concentrations were quantified using NanoDrop One (Thermo; Waltham, MA, USA). Femto Bacterial DNA Quantification Kit (Zymo; Irvine, CA, USA) was subsequently used for absolute quantification of 16S copies in fecal samples, according to the manufacturer’s instructions. qPCR reactions were prepared in duplicates in 96-well plate format and performed on an Eppendorf RealPlex4 (Eppendorf; Germany) with the following conditions: 10 mins at 95 °C, followed by 40 cycles of 30 sec at 95 °C, 30 sec at 50 °C and 1 min at 72 °C, then final extension of 7 mins at 72 °C. 16S concentrations were determined using a calibration curve of known concentrations of E. coli.
Multiplexed determination of cytokines and chemokines in the terminal ileum and proximal colon
Tissues were collected (terminal ileum and proximal colon) from “humanized” offspring mice and stored at −80 °C until extraction and analysis. Total protein was extracted using the Thermo Tissue extraction buffer I (Thermo; Waltham, MA, USA) by bead-beating with ceramic beads (Lysing Matrix D; MP Biomedicals; Santa Ana, CA, USA) at 3,000 rpm for 2 × 30 sec, lysates were then centrifuged 3 min at 16,000 × g at 4 °C and supernatants were collected. Protein concentration in lysates was determined by Pierce- BCA Protein Assay Kit (Thermo; Waltham, MA, USA), according to the manufacturer’s instructions and lysates were adjusted to 200 μg/ml with extraction buffer. 50 l lysate were diluted 2X in sample dilution buffer and analyzed in duplicates by multiplex ELISA using a mouse cytokine 23-plex (Bio-Plex Pro Mouse Cytokine 23-plex Assay) and Bio-Plex® 200 Systems (BioRad; Hercules, CA, USA) according to the manufacturer’s instructions. Analytes were quantified in each sample against a calibration curve of known concentrations.
Brain gene expression analysis
Brain tissue from prefrontal cortex and striatum was macro-dissected and flash frozen on dry ice. Approximately 30 mg of frozen brain tissue was then pulverized and RNA was extracted using Qiagen miRNAeasy kits according to the manufacturer’s instructions. RNA sequencing libraries were prepared using TruSeq Stranded mRNA Library Prep kits using polyA selection (Illumina; San Diego, CA, USA), and subsequently sequenced twice on an Illumina HiSeq 4000 with standard chemistry and protocols for 69 base pair paired end reads (UCLA Neuroscience Genomics Core), to achieve an average depth of 56 million reads per sample. Demulitplexed fastq files were mapped to the mouse reference genome assembly (GRCm38/mm10) using STAR with Gencode M10 annotations. Quality control was performed using PicardTools to generate a matrix of sequencing-related metrics for each sample (CollectAlignmnetSummaryMetrics, CollectRnaSeqMetrics, CollectGcBiasMetrics, CollectInsertSizeMetrics, MarkDuplicates). Two sequencing statistics, seqPC1 and seqPC2, were calculated as the first and second principal components of this matrix and were used as covariates in downstream analyses as previously published (Parikshak et al., 2016). Gene expression was quantified using featureCounts. Genes were filtered to retain only those (n=15695) with a minimum of 10 counts in at least half of the samples. Outlier samples (n=3) were identified and removed. Count-level data then underwent TMM scale normalization, followed by voom transformation and differential gene expression (DGE) using the limma package (Law et al., 2014) in R using the following covariates: Group, Brain Region, RIN, seqPC1, and seqPC2. The limma:duplicateCorrelation function was used to account for non-independence of mice exposed to the same microbiome donor. Test statistics were calculated for the group comparison and local FDR correction was applied to account for multiple comparisons using the fdrtool package in R (Strimmer, 2008). Genes with FDR < 0.1 were identified as being differentially expressed. Gene-set enrichment analysis (GSEA; Subramanian, et al., 2007) was performed on genes ranked by their differential expression T-statistic using the clusterProfiler (Yu et al, 2012) package in R with GO, KEGG, and hallmark gene sets downloaded from http://software.broadinstitute.org/gsea/msigdb/genesets.jsp.
Analysis of event-level differential splicing was performed using rMATS (v3.2.5; Shen et al., 2014). BAM files from ASD and NT groups were first merged. Percent spliced in (PSI) values were calculated for several classes of alternative splicing events, including skipped exon (SE), alternative 5’ splice site (A5SS), alternative 3’ splice site (A3SS), mutually exclusive exons (MXE), and retained introns (RI). Events with FDR < 0.1 were considered differentially spliced across groups. Enrichment of known autism risk genes (SFARI: https://gene.sfari.org/, SPARK for Autism: http://spark-sf.s3.amazonaws.com/SPARK_gene_list.pdf) among those exhibiting DS was assessed using logistic regression, controlling for gene length.
Cell-type specific expression analysis of differentially spliced genes was performed using the pSI package (specificity index; http://genetics.wustl.edu/jdlab/psi_package/) in R (Dougherty et al., 2010; Xu et al., 2014). Cell-type specific gene expression data was obtained from an RNAseq study of purified populations of neurons, astrocytes, oligodendrocytes, microglia, and endothelial cells derived from mouse cortex (Zhang et al., 2014). Significance was assessed using Fisher’s exact test with a pSI threshold set to 0.05, followed by Bonferroni correction of p-values.
Finally, we assessed whether differentially expressed genes were enriched for known targets of several ribosomal-binding protein (RBP) splicing factors. We manually curated lists of splicing targets derived experimentally using CLIP-seq and/or RBP knockout experiments in mice for the following RBP families: MBNL (Weyn-Vanhentenryck et al., 2018), RBFOX (Lee et al., 2016; Weyn-Vanhentenryck et al., 2018), PTBP (Vuong et al., 2016; Weyn-Vanhentenryck et al., 2018), NOVA (WeynVanhentenryck et al., 2018; Zhang et al., 2010), SRRM4 (Raj et al., 2014). Fisher’s exact test was used to calculate enrichment with background set to those genes exhibiting any alternative splicing in our dataset.
Metabolomics analysis
De-identified colon contents and serum samples were collected and flash-frozen at P45 without any buffers, and were shipped to the Dept. of Energy Pacific Northwest National Laboratory for metabolomic analysis by NMR and GC-MS. In colon contents, a total of 122 metabolites were identified by GC-MS (out of a total of 246 detected), and 67 metabolites were detected and identified by NMR. In serum, a total of 130 metabolites were identified by GC-MS (out of a total of 255 detected).
GC-MS sample preparation and analysis
Metabolites were extracted from murine colon contents and plasma samples using methanol (Deroussent et al., 2011; Snijders et al., 2016). Feces were homogenized and weighed, and chilled methanol (−20 °C) was added proportionally to the colon content sample (1 mL to 100 mg). Glass beads were added and the suspension was agitated and sonicated to extract metabolites. Supernatant was collected after centrifugation (15,000 g × 5 min at 4 °C) and 100 μL of each methanol layer was transferred to a new clean vial and subsequently dried under a speed-vacuum concentrator. 50 μL of serum samples were thawed and 200 μL of chilled methanol was added to denature proteins. Supernatants were collected after centrifugation (15,000 g × 5 min at 4 °C). All the samples were then dried completely and stored at −70 °C freezer until the instrumental analysis. Prior to analysis, the stored extracts were completely dried under speed-vacuum to remove moisture and were subsequently derivatized chemically, by methoxyamination and trimethylsilyation (TMS), as reported previously (Snijders et al., 2016). Briefly, methoxyamine (20 μL of a 30 mg mL-1 stock in pyridine) was added to each sample, followed by incubation at 37 °C with shaking for 90 min. subsequently, N-methyl-N(trimethylsilyl)trifluoroacetamide (MSTFA) with 1% trimethylchlorosilane (TMCS) (80 μL) was added to each vial, and incubated at 37 °C with shaking for 30 min. Samples were then allowed to cool to room temperature and were analyzed on the same day. Metabolites were resolved by gas chromatography using a HP-5MS column (30 min × 0.25 mm × 0.25 μm; Agilent Technologies). Samples (1 μL) were injected in splitless mode, and the helium gas flow rate was determined by the Agilent Retention Time Locking function based on analysis of deuterated myristic acid (Agilent Technologies; Santa Clara, CA, USA). The injection port temperature was held at 250 °C throughout the analysis. The GC oven was held at 60 °C for 1 min after injection, and the temperature was then increased to 325 °C by 10 °C/min, followed by a 10 min hold at 325 °C. The transfer line between GC and MS was maintained at 280 °C. All the MS data were collected over the mass range of 50–550 m/z under standard electron impact (EI) ionization mode at 70 eV of ionization energy. GC-MS raw data files were processed using the Metabolite Detector software (v2.5 beta; Hiller et al., 2009). Retention indices (RI) of detected metabolites were calculated based on the analysis of the FAMEs mixture (C8–C28), followed by their chromatographic alignment across all analyses after deconvolution. Metabolites were initially identified by matching experimental spectra to a PNNL augmented version of Agilent GC-MS metabolomics Library, containing spectra and validated retention indices for over 850 metabolites. Subsequently, any unknown peaks were matched to the NIST14 GC-MS library. All metabolite identification and quantification ions were validated and confirmed to reduce deconvolution errors during automated dataprocessing and to eliminate false identifications.
Proton NMR Metabolomics
A global metabolomics approach was used to obtain assignment and quantitation of metabolites via nuclear magnetic resonance (1H NMR). The one-dimensional (1D) 1H NMR spectra of all samples were collected in accordance with standard Chenomx (Edmonton, Alberta, Canada) sample preparation and data collection guidelines (Weijle, 2006). Fecal extract samples were diluted by 10 % (v/v) spike of a National Institute of Standards and Technology calibrated reference solution (100 % D2O, 5 mM 2,2 dimethyl-2-silapentane-5-sulfonate-d6 (DSS), and 0.1 % sodium azide). All NMR spectra were collected using a Varian Direct Drive 600 MHz NMR spectrometer equipped with a 5 mm triple-resonance salt-tolerant cold probe. The 1D 1H spectra were collected following standard Chenomx data collection guidelines (Weljie et al., 2006), employing a 1D NOESY presaturation (TNNOESY) experiment with 65536 complex points and at least 512 scans at 298 K.A presaturation delay of 1.5 sec was used to optimize water suppression. The 1D 1H NMR spectra of all samples were processed, assigned, and analyzed by using Chenomx NMR Suite 8.1 (Chenomx Inc.; Edmonton, AB, Canada) with quantification based on spectral intensities relative to the internal standard. Candidate metabolites present in each of the complex mixture were determined by matching the chemical shift, J-coupling, and intensity information of experimental NMR signals against the NMR signals of standard metabolites in the Chenomx library which include metabolites from the HMDB database.
Metabolomics data analysis
Data collected by GC-MS and NMR were log2 transformed. Sample outliers were identified and removed based on the RMD-PAV algorithm (Matzke et al., 2011) and a p-value threshold of 0.0001; based on this criterion, three samples were removed. An ANOVA filter (Webb-Robertson et al., 2010) was implemented which removed peaks that have inadequate data to perform a simple ANOVA test for any of the comparisons of interest. Finally, a global median normalization was applied to the data to correct for total metabolite content.
A mixed effects linear model was fit to the data, using the R package lme4 (Bates et al., 2015), with diagnosis (TD, ASD) as a fixed effect and the donor included as a random effect. A maximum likelihood test was conducted to test the null hypothesis that the mean metabolite abundance was equal across treatment groups (Chambers and Hastie, 1992).
Targeted metabolomics for taurine and 5-aminovaleric acid
Tissues (colon contents, serum, amniotic fluid, and fetal brain) were collected from age-matched GF and SPF E16.5 C57BL6/J dams or from SPF E18.5 C57BL/6 dams administered 10 mM 5AV, 10 mM taurine, or vehicle in drinking water from E0.5. Throughout the extraction, samples were kept at 4 °C on ice. Colon contents and fetal brains were homogenized in MilliQ water (4 μl / mg) by bead-beating with ceramic beads (Lysing Matrix D; MP Biomedicals; Santa Ana, CA, USA) at 3,000 rpm for 2 × 30 sec. Subsequently, a volume of 0.4 N Perchloric Acid spiked with known amounts of heavy standards (Taurine 1,2-13C2, 98% and Lysine:2HCl 13C6, 99%;15N2, 99; Cambridge Isotope Laboratories; Tewksbury, MA, USA) in water was added to each volume of both water-soluble tissue extracts, serum, or amniotic fluid, to a final concentration of 0.2 N and samples were vigorously vortexed for 15 sec. Tubes were then centrifuged for 20 min, at 10,000 × g, at 4 °C, supernatants were collected onto an UltraFree MC-GV 0.22 m centrifuge PVDF filter (Millipore; Burlington, MA, USA) and centrifuged again for 5 min, at 11,500 × g, at room-temperature. Filtrates were collected and subsequently analyzed by LC-MS/MS.
Analysis of the samples was performed on the ABSciex QTRAP 6500 LC-MS/MS system (Framingham, MA, USA), equipped with an Eksigent ekspert nanoLC 425 pump, ekspert nanoLC400 autosampler and Analyst software. Samples were directly injected (2 min) onto an Intrada amino acid column 1mm × 30mm (Imtakt USA; Portland, OR, USA) kept at room temperature. Chromatographic separation was achieved using a 50 μl/min flow rate and a linear gradient of 0 to 30 % B within 1 min; 30–100 % B in 2 min, followed by 100 % B for 2 min and equilibration for 2 min. Solvent A is 0.3 % formic acid in Acetonitrile and solvent B is 100 mM ammonium formate in 20 % acetonitrile. For all MRM experiments, 6500 QTrap acquisition parameters were as follows: 5500 V Ion-spray voltage, curtain gas setting of 30 and nebulizer gas setting of 20, interface heater at 150° C, 2.5 × 10–5 torr base pressure, and Q1 and Q3 set to unit resolution (0.6–0.8 Da full width at peak half-height). MRM acquisition methods were constructed with metabolite-specific tuned de-clustering potential (DP) and collision energy (CE) voltages. A default collision cell exit potential of 15 V was used for all MRM ion pairs. Raw data were analyzed using Skyline (MacLean et al., 2010) and peak areas were normalized to heavy standards.
MIMOSA integrative analysis
We used a metabolic modeling-based framework, MIMOSA, to identify metabolites whose variation across samples can be explained by variation in the metabolic potential of the microbiome (Noecker et al., 2016). Specifically, we used HUMAnN2 output, regrouped to KEGG pathways. We then analyzed each metabolomics dataset separately using the R package MIMOSA version 1.0.1 (http://elbo.gs.washington.edu/software_MIMOSA.html). MIMOSA constructs a community-wide metabolic model based on the inferred gene content of each sample and calculates community metabolic potential (CMP) scores, representing the relative capacity of the predicted community enzyme content in that sample to synthesize or degrade each metabolite. It then compares variation in these scores across samples to variation in measured metabolite concentrations using a rank-based Mantel test, to identify metabolites for which variation in concentration across samples is positively associated (consistent) with variation in community metabolism (as predicted by the CMP scores), using a local FDR q-value < 0.01. We identified metabolites for which variation in concentration across samples is negatively associated (contrasting) with CMP scores, with the same significance threshold. To identify potential contributing taxa for each metabolite, we calculated the Pearson correlation between the CMP scores obtained for a given metabolite across samples using the entire community and the CMP scores generated based on each species by itself (that is, recalculating the metagenome content and CMP scores based solely on the abundance of this species). OTUs for which this correlation coefficient for a given metabolite was greater than 0.5 were classified as potential contributing OTUs for that metabolite. Potential contributing genes were identified by calculating the Pearson correlation between the vectors of CMP scores obtained with and without reactions linked to that gene. Metabolites for which all main potential contributing genes were exclusively involved in synthesis reactions were classified as “Primarily predicted by synthesis”, and as “Primarily predicted by degradation” for those whose main potential contributors were exclusively involved in degradation reactions.
Metabolite administration
To test the effects specific metabolites have on behavioral phenotypes, metabolites were administered ad libitum in drinking water at a final concentration of 10 mM starting either three weeks before mating (pre-pregnancy group), or at 3–4 weeks of age (post-weaning group). Adult offspring (pre-pregnancy group) or adult treated animals were then tested for ASD-relevant behavioral phenotypes. Treatment continued throughout behavioral testing. To test the effects of potentially therapeutic compounds, BTBR T+ Itpr3tf/J mice were treated and tested. Throughout these experiments, mice were housed under standard conditions, with autoclaved water and irradiated chow ad libitum (PicoLab Rodent Diet 20 5053, LabDiet; St. Louis, MO, USA).
Slice Electrophysiology
Adult BTBR mice (8–12 weeks old) were first deeply anesthetized by intraperitoneal injection of ketamine/xylazine and then perfused transcardially with ice-cold sucrose slicing solution (sucrose 213 mM, KCl 2.5 mM, NaH2PO4 1.2 mM, NaHCO3 25 mM, glucose 10 mM, MgSO4 7 mM, CaCl2 1 mM, pH 7.35). After decapitation, brain was removed and immersed in the same ice-cold slicing solution. Coronal slices (300 μm) of medial prefrontal cortex were cut using a vibratome (VT-1200s; Leica; Buffalo Grove, IL, USA). We first let the slices recover in artificial cerebrospinal fluid (ACSF; 124 mM NaCl, 2.5 mM KCl, 1.2 mM NaH2PO4, 24 mM NaHCO3, 25 mM glucose, 1 mM MgSO4, 2 mM CaCl2) at 33 °C for 30 min and then held them at room temperature (~22 °C) until use.
During recording, slices were perfused continuously (~2 mL/min) with ACSF at 25 °C. Neurons were visualized and targeted using an upright IR-DIC microscope (BX51WI; Olympus; Center Valley, PA, USA). Whole-cell recordings were achieved using glass pipettes with an impedance of 3 to 6 MΩ when filled with intracellular solution (for current clamp, 145 mM K-gluconate, 2 mM NaCl, 4 mM KCl, 10 mM HEPES, 0.2 mM EGTA, 4 mM Mg-ATP, 0.3 mM Na-GTP, pH 7.25; for miniature IPSC, 145 mM CsCl, 2 mM MgCl2, 10 mM HEPES, 0.2 mM EGTA, 5 mM QX-314-Br, 2 mM Na2ATP, Na-GTP 0.3 mM, pH 7.25; for miniature EPSC,145 mM Cs(CH3)SO3, 2 mM MgCl2, 10 mM HEPES, 0.2 mM EGTA, 5mM QX-314-Br, 2 mM Na2ATP, 0.3 mM Na-GTP, pH 7.25). Electrical signal was sampled at 20 kHz and filtered at 2.9 kHz using an EPC 10 system (HEKA Elektronik; Holliston, MA, USA). Liquid junction potential was not corrected. To isolate the miniature EPSC/IPSC, we include synaptic blockers in ACSF (for mEPSC, 0.5 μM TTX, 100 μM picrotoxin; for mIPSC, 0.5 μM TTX, 10 μM CNQX, 25 μM DL-APV). Membrane potential of pyramidal neurons was held at −70/−60 mV.
Calcium imaging
Primary E18.5 Sprague Dawley rat cortical neurons (BrainBits; Springfield, IL, USA) were dissociated in papain (2 mg/ml; BrainBits, Springfield, IL, USA), according to the manufacturer’s instructions. Dissociated cells were resuspended in Neurobasal media (Gibco; Waltham, MA, USA) supplemented with 2 % B27-Plus (Gibco, Waltham, MA, USA), 1 % Glutagro (Corning; Corning, NY USA), and 1 % penicillin and streptomycin (Corning; Corning, NY USA). Cells were plated on Poly-D-Lysine pre-coated glass coverslips (Neuvitro; Vancouver, WA, USA) at 60,000 cells / cm2. Cells were incubated in a humidified incubator at 37 °C and 5 % CO2 for 6–17 days, and half of the media volume in each well was changed every 3 days. Before imaging, cells were loaded with Fluo-4-AM for 15 min at 37 °C. Fluo-4-AM was prepared in HEPES-buffered solution (HBS) containing: 150 mM NaCl, 3 mM KCl, 3 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 5 mM glucose, pH 4. During imaging, cells were perfused continuously with HBS at 25 °C. CNQX (10 μM) was added to the bath to block glutamatergic excitation. GABA (100 μM in HBS) was puff applied through a glass pipette (~1 μm tip diameter), placed 10~300 μm away from imaged cells, and air pulses (4~6 psi, 50ms) were generated by a Picospritzer system (Parker Hannifin; Cleveland, OH, USA). Illumination was provided by a halogen lamp and images were taken every 1 sec using a Rolera-XR camera (QImaging; Surrey, BC, Canada) controlled by Micro-Manager software (exposure duration 50 ms).
Data analysis was carried out using Matlab software. To quantify the calcium elevation triggered by GABA, we calculated F/F0 of each cell and a F/F0 higher than 5 % was considered a positive response. The final GABA switch curve was the best fitting to a logistic function: y = .
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis for behavioral outcomes in fecal transplanted offspring. Comparison of behavioral outcomes between TD Controls and ASD donors were tested using longitudinal linear mixed effects analyses, with test cycles and donors treated as repeated factors. Analyses were performed in SPSS (v 24); a priori alpha = 0.05. All outcomes were tested for normality and transformed as required. Diagonal covariance matrices were used so that intra-cycle and intra-donor correlations were accounted for in the modeling. The donor type (TD vs ASD) was the primary fixed effect measured, and mouse sex was an a priori covariate. Raw outputs from these analyses are included as supplementary data. Additional statistical analysis was done using R (3.4.1) or Python (3.6.4), using various packages to test mixed effects testing diagnosis (TD or ASD) as a fixed effect and donor and testing round as random effects. Wilcoxon and robust statistics to test 1-way ANOVA and t-tests on trimmed means, based on a generalization of the Welch method (t1dway, lincon, yuen, t=0.1; WRS2 0.9–2 (Mair, Wilcox, and Schoenbrodt 2017). Spearman’s rank correlations were performed using donor metadata and mouse behavior data (average behavior for all males in each donor group). Benjamini-Hochberg correction was performed for multiple comparison corrections. p-values ≤ 0.05 were considered significantly different. Visualization and additional statistical analysis was performed using Python (3.6.3), using the Matplotlib (2.2.3), Seaborn (0.7.1), StatsModels (0.9.0), Scipy (0.19.1), Pandas (0.23.4), pymer4 (0.6.0; Jolly, 2018), rpy2 (2.9.5) and Numpy (1.13.3) packages.
Test statistics and p-values for data presented in all figures is included in Table S4.
DATA AND SOFTWARE AVAILABILITY
Raw and analyzed data used to produce all figures are available at the following links:
Mendeley Data http://dx.doi.org/10.17632/ngzmj4zkms Human donor microbiome raw data are available in SRA (16S: PRJNA533120).
Mouse recipient microbiome raw data are available in SRA and EBI (16S: PRJNA431279, Shotgun: ERP113632 or Qiita (Gonzalez et al., 2018) study ID 11809).
RNA-seq raw data The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE109827 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109827).
Metabolomics raw data Metabolights MTBLS726
Supplementary Material
Figure S1. Engraftment fidelity for colonization of mice with human microbiomes. Related to Figure 1. (A) α diversity as measured by observed amplicon sequence variants (ASVs) from 16S rRNA gene sequencing in TD and ASD individuals from which donor samples in this study were used. Differences in means tested by Kruskal-Wallis. Sixteen samples used downstream are in dark-grey. Black Bar represents the mean. NTD=32, NASD=40.
(B) First three axes of a principal coordinate analysis (PCoA) of unweighted UniFrac distances from TD (circles) and ASD (squares) donors from 16S rRNA gene sequencing. Group differences were tested by pairwise PERMANOVA. Dark symbols denote samples that were further studied in depth. NTD=32, NASD=40.
(C) Box plots of pairwise distances of donor, recipients, and offspring mice to donor samples by unweighted UniFrac distances from 16S rRNA gene sequencing. Differences between samples were tested by pairwise PERMANOVA test.
(D) Box plots of α-diversity, as measured by the number of observed species from 16S rRNA gene sequencing. Group differences tested by Kruskal-Wallis test.
(E) First two axes of a PCoA of unweighted UniFrac distances from 16S rRNA gene sequencing of human TD and ASD donor population. Group differences by experimental stage (as in Panel C; donors, recipients, and offspring) or by donor diagnosis were tested by pairwise PERMANOVA.
(F-G) Taxonomic profile in donors (D), recipients (P), and offspring (F1) at the phylum level by diagnosis (F) and by donor (G) from 16S rRNA gene sequencing.
(H-I) Taxa engraftment in mice at the species level from 16S rRNA gene sequencing. The fraction of taxa present in mice and respective donor, as well as the cumulative relative abundance of shared taxa in the donor are plotted. Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and experimental stage, donor ID, and mouse sex as random effects.
(J-L) Fecal bacterial load in offspring of colonized male mice. (J) Total 16S rRNA gene was measured by qPCR and quantified by a calibration curve with E. coli DNA. (K) Aerobic and (L) anaerobic viable counts by plating on Tryptic Soy blood agar or Brucella blood agar, respectively. Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and donor ID as a random effect.
Table S3. Metabolomic analysis of colon contents by GC-MS and NMR, and of serum by GC-MS. Related to Figure 4.
Table S4. Statistical tests for data presented in figures. Related to Figure 1, 2, 3, 4, 5, 6, S1, S2, S3, S4, S5, and S6.
Figure S2. Colonization with human ASD microbiomes reproduces behavioral deficits in mice. Related to Figure 1. (A-E, A’-D’) Behavioral outcomes in offspring of colonized mice from 16 donors:
(A, A’) Repetitive behavior by marble burying test
(B, B’) 3-chamber sociability index
(C-D, C’-D’) distance traveled and center duration by open field testing.
(E) communication by ultrasonic vocalization (USV) in offspring of mice colonized with human samples from 16 donors, colored by donor. ASD-Mild defined as StdADOS = 4–5, and ASD defined as StdADOS = 6–10 (Gotham et al. 2007). Hypothesis testing for differences of the means were done by a random effects analysis and p-values from a chi-square test. NoASD= 179, NoASD-Mild= 44, NoTD= 111 (4–23 mice per unique donor per sex). Data stratified by mouse sex is presented in (A’, B’, C’, and D’) where α = 0.025 for Bonferroni-corrected statistical significance. Data presented is the aggregate of all experiments.
(F-H) Behavioral outcomes in offspring of colonized mice from selected eight donors stratified by mouse sex: (F) Repetitive behavior by marble burying test, (G) time socializing in direct social interaction, and (H) distance traveled in open field testing in colonized offspring colored by donor. Hypothesis testing for differences of the means were done by a linear mixed effects analysis and p-values from a chi-square test. NoASD= 121, NoTD= 85 (8–23 mice per donor, per sex). α = 0.025 for Bonferroni-corrected statistical significance. Data presented are the aggregate of all experiments.
Figure S3. Gastrointestinal physiology and immunity are similar in offspring mice colonized with human ASD microbiomes, compared to TD microbiomes. Related to Figure 1. (A–B) Gastrointestinal physiology in offspring mice as measured by (A) Mouse weight at 12 weeks of age, (B) intestinal permeability as measured by FITC-Dextran (4 KDa) in serum following gavage. NoTD=10, NoASD=14 (2–4 male mice per donor). Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and donor ID, experimental stage, and test date as random effects.
(C-H) Expression of the tight-junction genes Occludin (Ocldn), Zonula Occludens 1 (ZO1), and Zonula Occludens 2 (ZO2) in the distal ileum (C–E) and the proximal colon (F-H) of oTD and oASD mice, as measured by qRT-PCR. NoTD=15, NoASD=20 (3–7 male mice per donor). Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and donor ID as a random effect.
(I-J) Quantification of cytokines and chemokines in the distal ileum (I) and the proximal colon (J) of oTD and oASD mice, as measured by Bioplex 23-plex assay. NoTD=15, NoASD=20 (3–7 male mice per donor). Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and donor ID as a random effect.
Figure S4. Differential gene expression in the prefrontal cortex (PFC) and striatum (STR). Related to Figure 3. (A–C) Volcano plot of genes expressed in the aggregated data from both PFC and STR(A), the PFC (B), or the STR (C) of TD and ASD-colonized offspring (FDR ≤ 0.1, in red). Fold change as a factor Benjamini-Hochberg corrected p-values are plotted for each gene. STR: Striatum; PFC:Prefrontal cortex. p-values and false discovery rates were calculated using the limma and fdrtools packages.
(D) Relative expression levels (arbitrary units; A.U) of differentially expressed genes in brains of oTD and oASD mice (FDR ≤ 0.1). Protein coding genes (4930539E08Rik, Daglb) in aggregated data from both STR and PFC, and long non-coding RNAs (Gm26944, Gm13016, Gm17259) in PFC. Data points colored by donor. PFC: NoASD= 19, NoTD= 14, STR: NoASD= 20, NoTD= 14 (3–6 mouse samples, per tissue). p-values and false discovery rates were calculated using the limma and fdrtools packages.
Figure S5. Functional analysis by metagenomics and MIMOSA model-based analysis of microbial contributions to metabolite profiles. Related to Figure 4 and Figure 5. (A–B) MIMOSA-model prediction of strains involved in production or degradation of specific metabolites. Columns correspond to MetaPhlAn2-identified species in oTD and/or oASD metagenomes. Rows correspond to the metabolites across detected by NMR (A) or GC-MS (B) that were significantly consistent with metabolic potential at q-value ≤ 0.1. Blue squares indicate that the estimated metabolic potential of the strain in question is consistent with contributing to variation in that metabolite (production or utilization/degradation). The area of the colored points along the bottom indicates the relative abundances of each taxon in oTD and oASD samples. The segments along the bottom indicate the relative ratio of each taxon in oTD versus oASD samples. The segments along the left side show the average difference in metabolite concentration Z-scores between the control and ASD donor samples. The column of colored tiles indicates the MIMOSA correlation between metabolic potential scores and metabolite concentrations for each metabolite.
(C–D) Contributions of microbial strains identified by MIMOSA as putatively involved in the production and degradation of metabolites. The fraction of contributing strains with potential for synthesis, degradation, or neither (0) is shown for oTD and oASD samples, and for amino acid metabolites compared to other compound classes, measured by NMR (C) and GC-MS (D), illustrating a trend of reduced amino acid metabolism in oASD samples.
(E) limma-voom differential abundance of KEGG pathways between oTD and oASD metagenomes.
(F - G) 5-aminovaleric acid production by Stickland fermentation of D-proline. (F) abundance of prdA and (G) prdB copies in oTD and oASD mice quantified by ShortBRED. Differences in Means were analyzed by a Kruskal-Wallis non-parametric test.
Figure S6. Metabolite administration post weaning has no effects on ASD-related behaviors in BTBR mice. Related to Figure 5. (A–B) Targeted metabolomics for (A) 5AV and (B) taurine in GF and SPF E16.5 dams. Normalized concentrations of 5AV and taurine in dam’s colon contents, serum, amniotic fluid, and fetal brains were measured. Group differences were tested by Kruskal-Wallis test. N=3 dams per group.
(C–E) Mice were orally administered 10 mM 5AV or taurine in drinking water starting at 3–4 weeks of age, and throughout their lifetime. Offspring were tested by (C) marble burying, (D) direct social interaction, and (E) open field tests, and compared to untreated vehicle controls. Results are aggregated from 2 independent experiments. NControl= 20, N5AV= 18, NTaurine= 20. Hypothesis on differences in means were tested by one-way ANOVA on trimmed means (10%) and subsequent post-hoc tests.
(F-H) Offspring “humanized” oASD3 mice were orally administered 10 mM 5AV in sterile drinking water starting at 3–4 weeks of age, and throughout their lifetime. Offspring were tested by (F) open field tests, (G) marble burying, and (H) direct social interaction, and compared to untreated vehicle controls. NControl= 20, N5AV= 20. Hypothesis on differences in means were tested by one-tailed t-tests on trimmed means (10%).
(I) Basal properties of L5 pyramidal neurons in the mPFC: F-I Slope, rheobase, slow afterhyperpolarization (sAHP) amplitude, resting membrane potential, resting membrane resistance, and voltage response to a steady hyperpolarizing current recorded (Sag). Hypothesis on differences in means were tested by one-way ANOVA on trimmed means (10%). NControl = 20 cells in 4 mice, N5AV = 17 cells in 3 mice, NTaurine = 20 cells in 4 mice.
(J) Proportion of GABA-excitable cortical rat neurons as a function of days in culture (DIV), treated with either 5AV, Taurine, or control. NControl = 1,126, N5AV = 1,306, NTaurine = 1,471 (over 10 time points at DIV6–17).
Table S2. Microbiome-Induced, ASD-Relevant Splicing events. Related to Figure 3.
HIGHLIGHTS.
Mice harboring human ASD, but not TD, microbiomes exhibit ASD-like behaviors
ASD and TD microbiota produce differential metabolome profiles in mice
Extensive alternative splicing of risk genes in brains of mice with ASD microbiota
BTBR mice treated with 5AV or taurine improved repetitive and social behaviors
ACKNOWLEDGMENTS
The authors would like to thank Drs. H. Chu, G. Lenz, C. Schretter, and D. Dar, and members of the Mazmanian laboratory for critical discussions. We thank the staff at the Caltech Office of Laboratory Animal Resources. We also thank Y. Huang for hypD reference sequences. We thank Dr. J. Adams for critical review on the manuscript. We also thank Dr. J. Maldonado and M. Bennett for their support on 16S rRNA gene sequencing. We thank G. Humphrey, J. DeRight Goldasich, T. Schwartz, R. Salido Benitez, and G. Ackermann for their support in shotgun sequencing. Metabolomics analyses were supported by the Microbiomes in Transition (MinT) Initiative as part of the Laboratory Directed Research and Development Program at PNNL. Metabolomics measurements were performed in the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the U.S. Department of Energy Office of Biological and Environmental Research and located at PNNL in Richland Washington. PNNL is a multi-program national laboratory operated by Battelle for the DOE under contract DE-AC05-76RLO 1830. This work was supported by Autism Speaks Postdoctoral Fellowship in Translational Research 9718 and Human Frontiers Science Program Long-Term Fellowship 2012/65 (to G.S,), SFARI Bridge to Independence Award (to M.J.G), The San Diego Diversity Fellowship and the National Biomedical Computation Resource (to B.C.T). Funding includes grants from NIH (GM124312-01 to E.B., NS104925 to C.L., HD055784, MH100027 to D.H.G, and MH100556 to S.K.M.), Autism Research Institute, the Emch Foundation, the Brenen Hornstein Autism Research & Education Foundation (to D.W.K. and R.K.B.), Lynda and Blaine Fetter, the Simons Foundation, and the Heritage Medical Research Institute (to S.K.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTERESTS:
D.W.K. and R.K-B. have pending/approved patent applications related to the use of FMT and/or probiotics for various conditions including ASD. G.S. and S.K.M. have filed a pending patent application for the use of specific microbes and metabolites for various neurodevelopmental conditions. S.K.M is a co-founder of Axial Biotherapeutics and member of its scientific advisory board.
REFERENCES
- Abrahams BS, Arking DE, Campbell DB, Mefford HC, Morrow EM, Weiss LA, Menashe I, Wadkins T, Banerjee-Basu S, and Packer A (2013). SFARI Gene 2.0: a community-driven knowledgebase for the autism spectrum disorders (ASDs). Mol. Autism 4, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Zech Xu Z, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, et al. (2017). Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams JB, Audhya T, McDonough-Means S, Rubin RA, Quig D, Geis E, Gehn E, Loresto M, Mitchell J, Atwood S, et al. (2011). Nutritional and metabolic status of children with autism vs. neurotypical children, and the association with autism severity. Nutr. Metab 8, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred S, Moore KM, Fitzgerald M, and Waring RH (2003). Plasma amino acid levels in children with autism and their families. J. Autism Dev. Disord 33, 93–97. [DOI] [PubMed] [Google Scholar]
- An J-Y, Lin K, Zhu L, Werling DM, Dong S, Brand H, Wang HZ, Zhao X, Schwartz GB, Collins RL, et al. (2018). Genome-wide de novo risk score implicates promoter variation in autism spectrum disorder. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baio J, Wiggins L, Christensen DL, Maenner MJ, Daniels J, Warren Z, Kurzius-Spencer M, Zahorodny W, Robinson Rosenberg C, White T, et al. (2018). Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2014. MMWR Surveill. Summ 67, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak B, and Feng G (2016). Neurobiology of social behavior abnormalities in autism and Williams syndrome. Nat. Neurosci 19, 647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates D, Mächler M, Bolker B, and Walker S (2015). Fitting Linear Mixed-Effects Models Using lme4. Journal of Statistical Software, Articles 67, 1–48. [Google Scholar]
- Bellono NW, Bayrer JR, Leitch DB, Castro J, Zhang C, O’Donnell TA, Brierley SM, Ingraham HA, and Julius D (2017). Enterochromaffin Cells Are Gut Chemosensors that Couple to Sensory Neural Pathways. Cell 170, 185–198.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Kahle KT, and Cherubini E (2012). The GABA excitatory/inhibitory shift in brain maturation and neurological disorders. Neuroscientist 18, 467–486. [DOI] [PubMed] [Google Scholar]
- Benrabh H, Bourre JM, and Lefauconnier JM (1995). Taurine transport at the blood-brain barrier: an in vivo brain perfusion study. Brain Res 692, 57–65. [DOI] [PubMed] [Google Scholar]
- Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams E-J, et al. (2003). Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol 163, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich NA, Kaehler BD, Rideout JR, Dillon M, Bolyen E, Knight R, Huttley GA, and Gregory Caporaso J (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet C, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. (2018). QIIME 2: Reproducible, interactive, scalable, and extensible microbiome data science (PeerJ Preprints) [DOI] [PMC free article] [PubMed]
- Buffington SA, Di Prisco GV, Auchtung TA, Ajami NJ, Petrosino JF, and Costa-Mattioli M (2016). Microbial Reconstitution Reverses Maternal Diet-Induced Social and Synaptic Deficits in Offspring. Cell 165, 1762–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burket JA, Benson AD, Tang AH, and Deutsch SI (2013). D-Cycloserine improves sociability in the BTBR T+ Itpr3tf/J mouse model of autism spectrum disorders with altered Ras/Raf/ERK1/2 signaling. Brain Res. Bull 96, 62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callery PS, and Geelhaar LA (1985). 1-Piperideine as an in vivo precursor of the gamma-aminobutyric acid homologue 5-aminopentanoic acid. J. Neurochem 45, 946–948. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellot G, and Cherubini E (2014). GABAergic signaling as therapeutic target for autism spectrum disorders. Front Pediatr 2, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaidez V, Hansen RL, and Hertz-Picciotto I (2014). Gastrointestinal problems in children with autism, developmental delays or typical development. J. Autism Dev. Disord 44, 1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JM, and Hastie T (1992). Statistical Models in S (Wadsworth & Brooks/Cole Advanced Books & Software; ). [Google Scholar]
- Cho I, and Blaser MJ (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet 13, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coretti L, Cristiano C, Florio E, Scala G, Lama A, Keller S, Cuomo M, Russo R, Pero R, Paciello O, et al. (2017). Sex-related alterations of gut microbiota composition in the BTBR mouse model of autism spectrum disorder. Sci. Rep 7, 45356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coretti L, Paparo L, Riccio MP, Amato F, Cuomo M, Natale A, Borrelli L, Corrado G, Comegna M, Buommino E, et al. (2018). Gut Microbiota Features in Young Children With Autism Spectrum Disorders. Front. Microbiol 9, 3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corominas R, Yang X, Lin GN, Kang S, Shen Y, Ghamsari L, Broly M, Rodriguez M, Tam S, Trigg SA, et al. (2014). Protein interaction network of alternatively spliced isoforms from brain links genetic risk factors for autism. Nat. Commun 5, 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dachtler J, Glasper J, Cohen RN, Ivorra JL, Swiffen DJ, Jackson AJ, Harte MK, Rodgers RJ, and Clapcote SJ (2014). Deletion of α-neurexin II results in autism-related behaviors in mice. Transl. Psychiatry 4, e484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis M, Piccolo M, Vannini L, Siragusa S, De Giacomo A, Serrazzanetti DI, Cristofori F, Guerzoni ME, Gobbetti M, and Francavilla R (2013). Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS One 8, e76993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Angelis M, Francavilla R, Piccolo M, De Giacomo A, and Gobbetti M (2015). Autism spectrum disorders and intestinal microbiota. Gut Microbes 6, 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Palma G, Lynch MDJ, Lu J, Dang VT, Deng Y, Jury J, Umeh G, Miranda PM, Pigrau Pastor M, Sidani S, et al. (2017). Transplantation of fecal microbiota from patients with irritable bowel syndrome alters gut function and behavior in recipient mice. Sci. Transl. Med 9. [DOI] [PubMed] [Google Scholar]
- Deroussent A, Rodriguez S, Martelli S, Seck A, Dubus-Daudigeos E, Desmaële D, Vassal G, and Paci A (2011). Quantification of dimethyl-ifosfamide and its N-deschloropropylated metabolites in mouse plasma by liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci 879, 743–750. [DOI] [PubMed] [Google Scholar]
- Dhaher R, Damisah EC, Wang H, Gruenbaum SE, Ong C, Zaveri HP, Gruenbaum BF, and Eid T (2014). 5-aminovaleric acid suppresses the development of severe seizures in the methionine sulfoximine model of mesial temporal lobe epilepsy. Neurobiol. Dis 67, 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd D, Spitzer MH, Van Treuren W, Merrill BD, Hryckowian AJ, Higginbottom SK, Le A, Cowan TM, Nolan GP, Fischbach MA, et al. (2017). A gut bacterial pathway metabolizes aromatic amino acids into nine circulating metabolites. Nature 551, 648–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty JD, Schmidt EF, Nakajima M, and Heintz N (2010). Analytical approaches to RNA profiling data for the identification of genes enriched in specific cells. Nucleic Acids Res 38, 4218–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans C, Dunstan RH, Rothkirch T, Roberts TK, Reichelt KL, Cosford R, Deed G, Ellis LB, and Sparkes DL (2008). Altered amino acid excretion in children with autism. Nutr. Neurosci 11, 9–17. [DOI] [PubMed] [Google Scholar]
- Finegold SM, Molitoris D, Song Y, Liu C, Vaisanen M-L, Bolte E, McTeague M, Sandler R, Wexler H, Marlowe EM, et al. (2002). Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis 35, S6–S16. [DOI] [PubMed] [Google Scholar]
- Finegold SM, Dowd SE, Gontcharova V, Liu C, Henley KE, Wolcott RD, Youn E, Summanen PH, Granpeesheh D, Dixon D, et al. (2010). Pyrosequencing study of fecal microflora of autistic and control children. Anaerobe 16, 444–453. [DOI] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, et al. (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal MJ, Zhang P, Hadjimichael E, Walker RL, Chen C, Liu S, Won H, van Bakel H, Varghese M, Wang Y, et al. (2018). Transcriptome-wide isoform-level dysregulation in ASD, schizophrenia, and bipolar disorder. Science 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn TC, Nilsen R, Kieran TJ, Finger JW, Pierson TW, Bentley KE, Hoffberg S, Louha S, Garcia-De-Leon FJ, del Rio Portilla MA, et al. (2016). Adapterama I: Universal stubs and primers for thousands of dual-indexed Illumina libraries (iTru & iNext; ). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubeva AV, Joyce SA, Moloney G, Burokas A, Sherwin E, Arboleya S, Flynn I, Khochanskiy D, Moya-Pérez A, Peterson V, et al. (2017). Microbiota-related Changes in Bile Acid & Tryptophan Metabolism are Associated with Gastrointestinal Dysfunction in a Mouse Model of Autism. EBioMedicine 24, 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondalia SV, Palombo EA, Knowles SR, Cox SB, Meyer D, and Austin DW (2012). Molecular characterisation of gastrointestinal microbiota of children with autism (with and without gastrointestinal dysfunction) and their neurotypical siblings. Autism Res 5, 419–427. [DOI] [PubMed] [Google Scholar]
- Gonzalez A, Navas-Molina JA, Kosciolek T, McDonald D, Vázquez-Baeza Y, Ackermann G, DeReus J, Janssen S, Swafford AD, Orchanian SB, et al. (2018). Qiita: rapid, web-enabled microbiome meta-analysis. Nat. Methods 15, 796–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrindo P, Williams KC, Lee EB, Walker LS, McGrew SG, and Levitt P (2012). Gastrointestinal dysfunction in autism: parental report, clinical evaluation, and associated factors. Autism Res 5, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotham K, Risi S, Pickles A, and Lord C (2007). The Autism Diagnostic Observation Schedule: revised algorithms for improved diagnostic validity. J. Autism Dev. Disord 37, 613–627. [DOI] [PubMed] [Google Scholar]
- Gotham K, Pickles A, and Lord C (2009). Standardizing ADOS scores for a measure of severity in autism spectrum disorders. J. Autism Dev. Disord 39, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi R, Gibson GR, Vulevic J, Giallourou N, Castro-Mejía JL, Hansen LH, Leigh Gibson E, Nielsen DS, and Costabile A (2018). A prebiotic intervention study in children with autism spectrum disorders (ASDs). Microbiome 6, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallmayer J, Cleveland S, Torres A, Phillips J, Cohen B, Torigoe T, Miller J, Fedele A, Collins J, Smith K, et al. (2011). Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 68, 1095–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller K, Hangebrauk J, Jäger C, Spura J, Schreiber K, and Schomburg D (2009). MetaboliteDetector: comprehensive analysis tool for targeted and nontargeted GC/MS based metabolome analysis. Anal. Chem 81, 3429–3439. [DOI] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. (2013). Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Martínez-Del Campo A, and Balskus EP (2018). Anaerobic 4-hydroxyproline utilization: discovery of a new glycyl radical enzyme in the human gut microbiome uncovers a widespread microbial metabolic activity. Gut Microbes 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Furukawa T, Kumada T, Yamada J, Wang T, Inoue R, and Fukuda A (2012). Taurine inhibits K+-Cl-cotransporter KCC2 to regulate embryonic Cl-homeostasis via with-no-lysine (WNK) protein kinase signaling pathway. J. Biol. Chem 287, 20839–20850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee Y-H, Narzisi G, Leotta A, et al. (2012). De novo gene disruptions in children on the autistic spectrum. Neuron 74, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallières M, Tapial J, Raj B, O’Hanlon D, et al. (2014). A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159, 1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen S, McDonald D, Gonzalez A, Navas-Molina JA, Jiang L, Xu ZZ, Winker K, Kado DM, Orwoll E, Manary M, et al. (2018). Phylogenetic Placement of Exact Amplicon Sequences Improves Associations with Clinical Information. mSystems 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly E (2018). Pymer4: Connecting R and Python for Linear Mixed Modeling. The Journal of Open Source Software 3, 862. [Google Scholar]
- Kaelberer MM, Buchanan KL, Klein ME, Barth BB, Montoya MM, Shen X, and Bohórquez DV (2018). A gut-brain neural circuit for nutrient sensory transduction. Science 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, and Huttenhower C (2015). High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. PLoS Comput. Biol 11, e1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D-W, Park JG, Ilhan ZE, Wallstrom G, Labaer J, Adams JB, and Krajmalnik-Brown R (2013). Reduced incidence of Prevotella and other fermenters in intestinal microflora of autistic children. PLoS One 8, e68322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D-W, Ilhan ZE, Isern NG, Hoyt DW, Howsmon DP, Shaffer M, Lozupone CA, Hahn J, Adams JB, and Krajmalnik-Brown R (2017a). Differences in fecal microbial metabolites and microbiota of children with autism spectrum disorders. Anaerobe [DOI] [PubMed] [Google Scholar]
- Kang D-W, Adams JB, Gregory AC, Borody T, Chittick L, Fasano A, Khoruts A, Geis E, Maldonado J, McDonough-Means S, et al. (2017b). Microbiota Transfer Therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilb W, and Fukuda A (2017). Taurine as an Essential Neuromodulator during Perinatal Cortical Development. Front. Cell. Neurosci 11, 328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H-W, Kashima Y, Ishikawa K, and Yamano N (2009). Purification and characterization of the first archaeal glutamate decarboxylase from Pyrococcus horikoshii. Biosci. Biotechnol. Biochem 73, 224–227. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, Longman RS, Honda K, Littman DR, Choi GB, et al. (2017). Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MS, Newell C, Bomhof MR, Reimer RA, Hittel DS, Rho JM, Vogel HJ, and Shearer J (2016). Metabolomic Modeling To Monitor Host Responsiveness to Gut Microbiota Manipulation in the BTBRT+ tf/j Mouse. J. Proteome Res 15, 1143–1150. [DOI] [PubMed] [Google Scholar]
- Kordeli E, and Bennett V (1991). Distinct ankyrin isoforms at neuron cell bodies and nodes of Ranvier resolved using erythrocyte ankyrin-deficient mice. J. Cell Biol 114, 1243–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajmalnik-Brown R, Lozupone C, Kang D-W, and Adams JB (2015). Gut bacteria in children with autism spectrum disorders: challenges and promise of studying how a complex community influences a complex disease. Microb. Ecol. Health Dis 26, 26914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He Z-X, et al. (2015). Excess of rare, inherited truncating mutations in autism. Nat. Genet 47, 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushak RI, Winter HS, Buie TM, Cox SB, Phillips CD, and Ward NL (2016). Analysis of the Duodenal Microbiome in Autistic Individuals: Association with Carbohydrate Digestion. J. Pediatr. Gastroenterol. Nutr [DOI] [PubMed] [Google Scholar]
- Langen M, Bos D, Noordermeer SDS, Nederveen H, van Engeland H, and Durston S (2014). Changes in the development of striatum are involved in repetitive behavior in autism. Biol. Psychiatry 76, 405–411. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Lee J, and Kim E (2017). Excitation/Inhibition Imbalance in Animal Models of Autism Spectrum Disorders. Biol. Psychiatry 81, 838–847. [DOI] [PubMed] [Google Scholar]
- Lee J-A, Damianov A, Lin C-H, Fontes M, Parikshak NN, Anderson ES, Geschwind DH, Black DL, and Martin KC (2016). Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron 89, 113–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin BJ, Huang YY, Peck SC, Wei Y, Martínez-Del Campo A, Marks JA, Franzosa EA, Huttenhower C, and Balskus EP (2017). A prominent glycyl radical enzyme in human gut microbiomes metabolizes trans-4-hydroxy-l-proline. Science 355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liaw A, Wiener M, and et al. (2002). Classification and regression by randomForest. R News 2, 18–22. [Google Scholar]
- Liu F, Horton-Sparks K, Hull V, Li RW, and Martínez-Cerdeño V (2018). The valproic acid rat model of autism presents with gut bacterial dysbiosis similar to that in human autism. Mol. Autism 9, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Li J, Wu F, Zheng H, Peng Q, and Zhou H (2019). Altered composition and function of intestinal microbiota in autism spectrum disorders: a systematic review. Transl. Psychiatry 9, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L-K, Becker DF, and Tanner JJ (2017). Structure, function, and mechanism of proline utilization A (PutA). Arch. Biochem. Biophys 632, 142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo MV, Lai M-C, and Baron-Cohen S (2019). Big data approaches to decomposing heterogeneity across the autism spectrum. Mol. Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, and Knight R (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luca F, Kupfer SS, Knights D, Khoruts A, and Blekhman R (2017). Functional Genomics of Host–Microbiome Interactions in Humans. Trends Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyall K, Schmidt RJ, and Hertz-Picciotto I (2014). Maternal lifestyle and environmental risk factors for autism spectrum disorders. Int. J. Epidemiol 43, 443–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, and MacCoss MJ (2010). Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Ooga T, Kibe R, Aiba Y, Koga Y, and Benno Y (2017). Colonic Absorption of Low-Molecular-Weight Metabolites Influenced by the Intestinal Microbiome: A Pilot Study. PLoS One 12, e0169207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthies A, Clavel T, Gütschow M, Engst W, Haller D, Blaut M, and Braune A (2008). Conversion of daidzein and genistein by an anaerobic bacterium newly isolated from the mouse intestine. Appl. Environ. Microbiol 74, 4847–4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthies A, Blaut M, and Braune A (2009). Isolation of a human intestinal bacterium capable of daidzein and genistein conversion. Appl. Environ. Microbiol 75, 1740–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzke MM, Waters KM, Metz TO, Jacobs JM, Sims AC, Baric RS, Pounds JG, and Webb-Robertson B-JM (2011). Improved quality control processing of peptide-centric LC-MS proteomics data. Bioinformatics 27, 2866–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazina V, Gerdts J, Trinh S, Ankenman K, Ward T, Dennis MY, Girirajan S, Eichler EE, and Bernier R (2015). Epigenetics of autism-related impairment: copy number variation and maternal infection. J. Dev. Behav. Pediatr 36, 61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, and Hugenholtz P (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElhanon BO, McCracken C, Karpen S, and Sharp WG (2014). Gastrointestinal symptoms in autism spectrum disorder: a meta-analysis. Pediatrics 133, 872–883. [DOI] [PubMed] [Google Scholar]
- McFarlane HG, Kusek GK, Yang M, Phoenix JL, Bolivar VJ, and Crawley JN (2008). Autism-like behavioral phenotypes in BTBR T+ tf/J mice. Genes Brain Behav 7, 152–163. [DOI] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnik AV, da Silva RR, Hyde ER, Aksenov AA, Vargas F, Bouslimani A, Protsyuk I, Jarmusch AK, Tripathi A, Alexandrov T, et al. (2017). Coupling Targeted and Untargeted Mass Spectrometry for Metabolome-Microbiome-Wide Association Studies of Human Fecal Samples. Anal. Chem 89, 7549–7559. [DOI] [PubMed] [Google Scholar]
- Meyza KZ, and Blanchard DC (2017). The BTBR mouse model of idiopathic autism Current view on mechanisms. Neurosci. Biobehav. Rev 76, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming X, Stein TP, Barnes V, Rhodes N, and Guo L (2012). Metabolic perturbance in autism spectrum disorders: a metabolomics study. J. Proteome Res 11, 5856–5862. [DOI] [PubMed] [Google Scholar]
- Moeller AH, Suzuki TA, Phifer-Rixey M, and Nachman MW (2018). Transmission modes of the mammalian gut microbiota. Science 362, 453–457. [DOI] [PubMed] [Google Scholar]
- Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, Barbaro JR, Wilson LM, Threadgill DW, Lauder JM, et al. (2007). Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav. Brain Res 176, 4–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussap M, Noto A, and Fanos V (2016). Metabolomics of autism spectrum disorders: early insights regarding mammalian-microbial cometabolites. Expert Rev. Mol. Diagn 16, 869–881. [DOI] [PubMed] [Google Scholar]
- Nankova BB, Agarwal R, MacFabe DF, and La Gamma EF (2014). Enteric bacterial metabolites propionic and butyric acid modulate gene expression, including CREB-dependent catecholaminergic neurotransmission, in PC12 cells--possible relevance to autism spectrum disorders. PLoS One 9, e103740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naushad SM, Jain JMN, Prasad CK, Naik U, and Akella RRD (2013). Autistic children exhibit distinct plasma amino acid profile. Indian J. Biochem. Biophys 50, 474–478. [PubMed] [Google Scholar]
- Nelson SB, and Valakh V (2015). Excitatory/Inhibitory Balance and Circuit Homeostasis in Autism Spectrum Disorders. Neuron 87, 684–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell C, Bomhof MR, Reimer RA, Hittel DS, Rho JM, and Shearer J (2016). Ketogenic diet modifies the gut microbiota in a murine model of autism spectrum disorder. Mol. Autism 7, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noecker C, Eng A, Srinivasan S, Theriot CM, Young VB, Jansson JK, Fredricks DN, and Borenstein E (2016). Metabolic Model-Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links between Ecological and Metabolic Variation. mSystems 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onore C, Careaga M, and Ashwood P (2012). The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun 26, 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Luo R, Zhang A, Won H, Lowe JK, Chandran V, Horvath S, and Geschwind DH (2013). Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 155, 1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikshak NN, Swarup V, Belgard TG, Irimia M, Ramaswami G, Gandal MJ, Hartl C, Leppa V, Ubieta L de la T, Huang J, et al. (2016). Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 540, 423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Cohen I, Gonzalez M, Castellano MR, Flory M, Jenkins EC, Brown WT, and Schuller-Levis G (2017). Is Taurine a Biomarker in Autistic Spectrum Disorder? Adv. Exp. Med. Biol 975, 3–16. [DOI] [PubMed] [Google Scholar]
- Parras A, Anta H, Santos-Galindo M, Swarup V, Elorza A, Nieto-González JL, Picó S, Hernández IH, Díaz-Hernández JI, Belloc E, et al. (2018). Autism-like phenotype and risk gene mRNA deadenylation by CPEB4 mis-splicing. Nature [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel J, Lukkes JL, and Shekhar A (2018). Overview of genetic models of autism spectrum disorders. Prog. Brain Res 241, 1–36. [DOI] [PubMed] [Google Scholar]
- Ponti G, Rodriguez-Gomez A, Farinetti A, Marraudino M, Filice F, Foglio B, Sciacca G, Panzica GC, and Gotti S (2017). Early postnatal genistein administration permanently affects nitrergic and vasopressinergic systems in a sex-specific way. Neuroscience 346, 203–215. [DOI] [PubMed] [Google Scholar]
- Powell DR, Gay JP, Wilganowski N, Doree D, Savelieva KV, Lanthorn TH, Read R, Vogel P, Hansen GM, Brommage R, et al. (2015). Diacylglycerol Lipase α Knockout Mice Demonstrate Metabolic and Behavioral Phenotypes Similar to Those of Cannabinoid Receptor 1 Knockout Mice. Front. Endocrinol 6, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj B, Irimia M, Braunschweig U, Sterne-Weiler T, O’Hanlon D, Lin Z-Y, Chen GI, Easton LE, Ule J, Gingras A-C, et al. (2014). A global regulatory mechanism for activating an exon network required for neurogenesis. Mol. Cell 56, 90–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Gomez A, Filice F, Gotti S, and Panzica G (2014). Perinatal exposure to genistein affects the normal development of anxiety and aggressive behaviors and nitric oxide system in CD1 male mice. Physiol. Behav 133, 107–114. [DOI] [PubMed] [Google Scholar]
- Rognes T, Flouri T, Nichols B, Quince C, and Mahé F (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, and Südhof TC (2014). Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstein JLR, and Merzenich MM (2003). Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav 2, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, Challis C, Schretter CE, Rocha S, Gradinaru V, et al. (2016). Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 167, 1469–1480.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuels S, Fish I, and Schwartz SA (1983). Anticonvulsant activity of glycylglycine and delta-aminovaleric acid: evidence for glutamine exchange in amino acid transport. J. Neurochem 40, 1063–1068. [DOI] [PubMed] [Google Scholar]
- Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE, Murtha MT, Bal VH, Bishop SL, Dong S, et al. (2015). Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 87, 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Hultman C, Larsson H, and Reichenberg A (2017). The Heritability of Autism Spectrum Disorder. JAMA 318, 1182–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler RH, Finegold SM, Bolte ER, Buchanan CP, Maxwell AP, Väisänen ML, Nelson MN, and Wexler HM (2000). Short-term benefit from oral vancomycin treatment of regressive-onset autism. J. Child Neurol 15, 429–435. [DOI] [PubMed] [Google Scholar]
- Sandoval-Motta S, Aldana M, Martínez-Romero E, and Frank A (2017). The Human Microbiome and the Missing Heritability Problem. Front. Genet 8, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sartori S, Anesi L, Polli R, Toldo I, Casarin A, Drigo P, and Murgia A (2008). Angelman syndrome due to a novel splicing mutation of the UBE3A gene. J. Child Neurol 23, 912–915. [DOI] [PubMed] [Google Scholar]
- Schaafsma SM, Gagnidze K, Reyes A, Norstedt N, Månsson K, Francis K, and Pfaff DW (2017). Sex-specific gene-environment interactions underlying ASD-like behaviors. Proc. Natl. Acad. Sci. U. S. A 114, 1383–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CK, Melmed RD, Barstow LE, Enriquez FJ, Ranger-Moore J, and Ostrem JA (2006). Oral human immunoglobulin for children with autism and gastrointestinal dysfunction: a prospective, open-label study. J. Autism Dev. Disord 36, 1053–1064. [DOI] [PubMed] [Google Scholar]
- Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, Ramakrishnan C, Fenno LE, Davidson TJ, Wright M, et al. (2017). Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci. Transl. Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgritta M, Dooling SW, Buffington SA, Momin EN, Francis MB, Britton RA, and Costa-Mattioli M (2018). Mechanisms Underlying Microbial-Mediated Changes in Social Behavior in Mouse Models of Autism Spectrum Disorder. Neuron [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon G, Garg N, Debelius J, Knight R, Dorrestein PC, and Mazmanian SK (2014). Specialized metabolites from the microbiome in health and disease. Cell Metab 20, 719–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu Z-X, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc. Natl. Acad. Sci. U. S. A 111, E5593–E5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih H-T, and Mok H-K (2000). ETHOM: event-recording computer software for the study of animal behavior. Acta Zool. Taiwanica 11, 47–61. [Google Scholar]
- Silverman JL, Yang M, Lord C, and Crawley JN (2010). Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci 11, 490–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Smith DG, Rizzo SJS, Karras MN, Turner SM, Tolu SS, Bryce DK, Smith DL, Fonseca K, Ring RH, et al. (2012). Negative allosteric modulation of the mGluR5 receptor reduces repetitive behaviors and rescues social deficits in mouse models of autism. Sci. Transl. Med 4, 131ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Oliver CF, Karras MN, Gastrell PT, and Crawley JN (2013). AMPAKINE enhancement of social interaction in the BTBR mouse model of autism. Neuropharmacology 64, 268–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sittler A, Devys D, Weber C, and Mandel JL (1996). Alternative splicing of exon 14 determines nuclear or cytoplasmic localisation of fmr1 protein isoforms. Hum. Mol. Genet 5, 95–102. [DOI] [PubMed] [Google Scholar]
- Sivendran S, Patterson D, Spiegel E, McGown I, Cowley D, and Colman RF (2004). Two novel mutant human adenylosuccinate lyases (ASLs) associated with autism and characterization of the equivalent mutant Bacillus subtilis ASL. J. Biol. Chem 279, 53789–53797. [DOI] [PubMed] [Google Scholar]
- Snijders AM, Langley SA, Kim Y-M, Brislawn CJ, Noecker C, Zink EM, Fansler SJ, Casey CP, Miller DR, Huang Y, et al. (2016). Influence of early life exposure, host genetics and diet on the mouse gut microbiome and metabolome. Nat Microbiol 2, 16221. [DOI] [PubMed] [Google Scholar]
- Son JS, Zheng LJ, Rowehl LM, Tian X, Zhang Y, Zhu W, Litcher-Kelly L, Gadow KD, Gathungu G, Robertson CE, et al. (2015). Comparison of Fecal Microbiota in Children with Autism Spectrum Disorders and Neurotypical Siblings in the Simons Simplex Collection. PLoS One 10, e0137725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, and Sonnenburg JL (2016). Diet-induced extinctions in the gut microbiota compound over generations. Nature 529, 212–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Stessman HAF, Willemsen MH, Fenckova M, Penn O, Hoischen A, Xiong B, Wang T, Hoekzema K, Vives L, Vogel I, et al. (2016). Disruption of POGZ Is Associated with Intellectual Disability and Autism Spectrum Disorders. Am. J. Hum. Genet 98, 541–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilling RM, Moloney GM, Ryan FJ, Hoban AE, Bastiaanssen TF, Shanahan F, Clarke G, Claesson MJ, Dinan TG, and Cryan JF (2018). Social interaction-induced activation of RNA splicing in the amygdala of microbiome-deficient mice. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strati F, Cavalieri D, Albanese D, De Felice C, Donati C, Hayek J, Jousson O, Leoncini S, Renzi D, Calabrò A, et al. (2017). New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 5, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strimmer K (2008). fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Kuehn H, Gould J, Tamayo P, and Mesirov JP (2007). GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics 23, 3251–3253. [DOI] [PubMed] [Google Scholar]
- Südhof TC (2008). Neuroligins and neurexins link synaptic function to cognitive disease. Nature 455, 903–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabouy L, Getselter D, Ziv O, Karpuj M, Tabouy T, Lukic I, Maayouf R, Werbner N, Ben-Amram H, Nuriel-Ohayon M, et al. (2018). Dysbiosis of microbiome and probiotic treatment in a genetic model of autism spectrum disorders. Brain Behav. Immun 73, 310–319. [DOI] [PubMed] [Google Scholar]
- de Theije CGM, Wopereis H, Ramadan M, van Eijndthoven T, Lambert J, Knol J, Garssen J, Kraneveld AD, and Oozeer R (2014). Altered gut microbiota and activity in a murine model of autism spectrum disorders. Brain Behav. Immun 37, 197–206. [DOI] [PubMed] [Google Scholar]
- Tick B, Bolton P, Happé F, Rutter M, and Rijsdijk F (2016). Heritability of autism spectrum disorders: a meta-analysis of twin studies. J. Child Psychol. Psychiatry 57, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tochitani S (2017). Functions of Maternally-Derived Taurine in Fetal and Neonatal Brain Development In Taurine 10, (Springer, Dordrecht: ), pp. 17–25. [DOI] [PubMed] [Google Scholar]
- de la Torre-Ubieta L, Won H, Stein JL, and Geschwind DH (2016). Advancing the understanding of autism disease mechanisms through genetics. Nat. Med 22, 345–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903. [DOI] [PubMed] [Google Scholar]
- Tu W-J, Chen H, and He J (2012). Application of LC-MS/MS analysis of plasma amino acids profiles in children with autism. J. Clin. Biochem. Nutr 51, 248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valeeva G, Tressard T, Mukhtarov M, Baude A, and Khazipov R (2016). An Optogenetic Approach for Investigation of Excitatory and Inhibitory Network GABA Actions in Mice Expressing Channelrhodopsin-2 in GABAergic Neurons. J. Neurosci 36, 5961–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vázquez-Baeza Y, Pirrung M, Gonzalez A, and Knight R (2013). EMPeror: a tool for visualizing high-throughput microbial community data. Gigascience 2, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitvitsky V, Garg SK, and Banerjee R (2011). Taurine biosynthesis by neurons and astrocytes. J. Biol. Chem 286, 32002–32010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, and Geschwind DH (2011). Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong JK, Lin C-H, Zhang M, Chen L, Black DL, and Zheng S (2016). PTBP1 and PTBP2 Serve Both Specific and Redundant Functions in Neuronal Pre-mRNA Splicing. Cell Rep 17, 2766–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Wan J, Rong H, He F, Wang H, Zhou J, Cai C, Wang Y, Xu R, Yin Z, et al. (2019). Alterations in Gut Glutamate Metabolism Associated with Changes in Gut Microbiota Composition in Children with Autism Spectrum Disorder. mSystems 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Harvey L, Martin R, van der Beek EM, Knol J, Cryan JF, and Renes IB (2018). Targeting the gut microbiota to influence brain development and function in early life. Neurosci. Biobehav. Rev 95, 191–201. [DOI] [PubMed] [Google Scholar]
- Webb-Robertson B-JM, McCue LA, Waters KM, Matzke MM, Jacobs JM, Metz TO, Varnum SM, and Pounds JG (2010). Combined statistical analyses of peptide intensities and peptide occurrences improves identification of significant peptides from MS-based proteomics data. J. Proteome Res 9, 5748–5756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weljie AM, Newton J, Mercier P, Carlson E, and Slupsky CM (2006). Targeted profiling: quantitative analysis of 1H NMR metabolomics data. Anal. Chem 78, 4430–4442. [DOI] [PubMed] [Google Scholar]
- Westmark CJ (2014). A hypothesis regarding the molecular mechanism underlying dietary soy-induced effects on seizure propensity. Front. Neurol 5, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck SM, Feng H, Ustianenko D, Duffié R, Yan Q, Jacko M, Martinez JC, Goodwin M, Zhang X, Hengst U, et al. (2018). Precise temporal regulation of alternative splicing during neural development. Nat. Commun 9, 2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, and Siuzdak G (2009). Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc. Natl. Acad. Sci. U. S. A 106, 3698–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams BL, Hornig M, Buie T, Bauman ML, Cho Paik M, Wick I, Bennett A, Jabado O, Hirschberg DL, and Lipkin WI (2011). Impaired carbohydrate digestion and transport and mucosal dysbiosis in the intestines of children with autism and gastrointestinal disturbances. PLoS One 6, e24585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsey AJ, Sanders SJ, Li M, Dong S, Tebbenkamp AT, Muhle RA, Reilly SK, Lin L, Fertuzinhos S, Miller JA, et al. (2013). Coexpression networks implicate human midfetal deep cortical projection neurons in the pathogenesis of autism. Cell 155, 997–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY (1982). Purification and characterization of cysteic acid and cysteine sulfinic acid decarboxylase and L-glutamate decarboxylase from bovine brain. Proc. Natl. Acad. Sci. U. S. A 79, 4270–4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wells AB, O’Brien DR, Nehorai A, and Dougherty JD (2014). Cell type-specific expression analysis to identify putative cellular mechanisms for neurogenetic disorders. J. Neurosci 34, 1420–1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada KD, Tomii K, and Katoh K (2016). Application of the MAFFT sequence alignment program to large data—reexamination of the usefulness of chained guide trees. Bioinformatics 32, 3246–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap IKS, Angley M, Veselkov KA, Holmes E, Lindon JC, and Nicholson JK (2010). Urinary metabolic phenotyping differentiates children with autism from their unaffected siblings and age-matched controls. J. Proteome Res 9, 2996–3004. [DOI] [PubMed] [Google Scholar]
- Yu G, Wang L-G, Han Y, and He Q-Y (2012). clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Frias MA, Mele A, Ruggiu M, Eom T, Marney CB, Wang H, Licatalosi DD, Fak JJ, and Darnell RB (2010). Integrative modeling defines the Nova splicing-regulatory network and its combinatorial controls. Science 329, 439–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, Zeng L, Chen J, Fan S, Du X, et al. (2016). Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol. Psychiatry 21, 786–796. [DOI] [PubMed] [Google Scholar]
- Zheng P, Zeng B, Liu M, Chen J, Pan J, Han Y, Liu Y, Cheng K, Zhou C, Wang H, et al. (2019). The gut microbiome from patients with schizophrenia modulates the glutamate-glutamine-GABA cycle and schizophrenia-relevant behaviors in mice. Science Advances 5, eaau8317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Engraftment fidelity for colonization of mice with human microbiomes. Related to Figure 1. (A) α diversity as measured by observed amplicon sequence variants (ASVs) from 16S rRNA gene sequencing in TD and ASD individuals from which donor samples in this study were used. Differences in means tested by Kruskal-Wallis. Sixteen samples used downstream are in dark-grey. Black Bar represents the mean. NTD=32, NASD=40.
(B) First three axes of a principal coordinate analysis (PCoA) of unweighted UniFrac distances from TD (circles) and ASD (squares) donors from 16S rRNA gene sequencing. Group differences were tested by pairwise PERMANOVA. Dark symbols denote samples that were further studied in depth. NTD=32, NASD=40.
(C) Box plots of pairwise distances of donor, recipients, and offspring mice to donor samples by unweighted UniFrac distances from 16S rRNA gene sequencing. Differences between samples were tested by pairwise PERMANOVA test.
(D) Box plots of α-diversity, as measured by the number of observed species from 16S rRNA gene sequencing. Group differences tested by Kruskal-Wallis test.
(E) First two axes of a PCoA of unweighted UniFrac distances from 16S rRNA gene sequencing of human TD and ASD donor population. Group differences by experimental stage (as in Panel C; donors, recipients, and offspring) or by donor diagnosis were tested by pairwise PERMANOVA.
(F-G) Taxonomic profile in donors (D), recipients (P), and offspring (F1) at the phylum level by diagnosis (F) and by donor (G) from 16S rRNA gene sequencing.
(H-I) Taxa engraftment in mice at the species level from 16S rRNA gene sequencing. The fraction of taxa present in mice and respective donor, as well as the cumulative relative abundance of shared taxa in the donor are plotted. Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and experimental stage, donor ID, and mouse sex as random effects.
(J-L) Fecal bacterial load in offspring of colonized male mice. (J) Total 16S rRNA gene was measured by qPCR and quantified by a calibration curve with E. coli DNA. (K) Aerobic and (L) anaerobic viable counts by plating on Tryptic Soy blood agar or Brucella blood agar, respectively. Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and donor ID as a random effect.
Table S3. Metabolomic analysis of colon contents by GC-MS and NMR, and of serum by GC-MS. Related to Figure 4.
Table S4. Statistical tests for data presented in figures. Related to Figure 1, 2, 3, 4, 5, 6, S1, S2, S3, S4, S5, and S6.
Figure S2. Colonization with human ASD microbiomes reproduces behavioral deficits in mice. Related to Figure 1. (A-E, A’-D’) Behavioral outcomes in offspring of colonized mice from 16 donors:
(A, A’) Repetitive behavior by marble burying test
(B, B’) 3-chamber sociability index
(C-D, C’-D’) distance traveled and center duration by open field testing.
(E) communication by ultrasonic vocalization (USV) in offspring of mice colonized with human samples from 16 donors, colored by donor. ASD-Mild defined as StdADOS = 4–5, and ASD defined as StdADOS = 6–10 (Gotham et al. 2007). Hypothesis testing for differences of the means were done by a random effects analysis and p-values from a chi-square test. NoASD= 179, NoASD-Mild= 44, NoTD= 111 (4–23 mice per unique donor per sex). Data stratified by mouse sex is presented in (A’, B’, C’, and D’) where α = 0.025 for Bonferroni-corrected statistical significance. Data presented is the aggregate of all experiments.
(F-H) Behavioral outcomes in offspring of colonized mice from selected eight donors stratified by mouse sex: (F) Repetitive behavior by marble burying test, (G) time socializing in direct social interaction, and (H) distance traveled in open field testing in colonized offspring colored by donor. Hypothesis testing for differences of the means were done by a linear mixed effects analysis and p-values from a chi-square test. NoASD= 121, NoTD= 85 (8–23 mice per donor, per sex). α = 0.025 for Bonferroni-corrected statistical significance. Data presented are the aggregate of all experiments.
Figure S3. Gastrointestinal physiology and immunity are similar in offspring mice colonized with human ASD microbiomes, compared to TD microbiomes. Related to Figure 1. (A–B) Gastrointestinal physiology in offspring mice as measured by (A) Mouse weight at 12 weeks of age, (B) intestinal permeability as measured by FITC-Dextran (4 KDa) in serum following gavage. NoTD=10, NoASD=14 (2–4 male mice per donor). Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and donor ID, experimental stage, and test date as random effects.
(C-H) Expression of the tight-junction genes Occludin (Ocldn), Zonula Occludens 1 (ZO1), and Zonula Occludens 2 (ZO2) in the distal ileum (C–E) and the proximal colon (F-H) of oTD and oASD mice, as measured by qRT-PCR. NoTD=15, NoASD=20 (3–7 male mice per donor). Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and donor ID as a random effect.
(I-J) Quantification of cytokines and chemokines in the distal ileum (I) and the proximal colon (J) of oTD and oASD mice, as measured by Bioplex 23-plex assay. NoTD=15, NoASD=20 (3–7 male mice per donor). Differences in means were tested by linear mixed effects with donor diagnosis as a fixed effect and donor ID as a random effect.
Figure S4. Differential gene expression in the prefrontal cortex (PFC) and striatum (STR). Related to Figure 3. (A–C) Volcano plot of genes expressed in the aggregated data from both PFC and STR(A), the PFC (B), or the STR (C) of TD and ASD-colonized offspring (FDR ≤ 0.1, in red). Fold change as a factor Benjamini-Hochberg corrected p-values are plotted for each gene. STR: Striatum; PFC:Prefrontal cortex. p-values and false discovery rates were calculated using the limma and fdrtools packages.
(D) Relative expression levels (arbitrary units; A.U) of differentially expressed genes in brains of oTD and oASD mice (FDR ≤ 0.1). Protein coding genes (4930539E08Rik, Daglb) in aggregated data from both STR and PFC, and long non-coding RNAs (Gm26944, Gm13016, Gm17259) in PFC. Data points colored by donor. PFC: NoASD= 19, NoTD= 14, STR: NoASD= 20, NoTD= 14 (3–6 mouse samples, per tissue). p-values and false discovery rates were calculated using the limma and fdrtools packages.
Figure S5. Functional analysis by metagenomics and MIMOSA model-based analysis of microbial contributions to metabolite profiles. Related to Figure 4 and Figure 5. (A–B) MIMOSA-model prediction of strains involved in production or degradation of specific metabolites. Columns correspond to MetaPhlAn2-identified species in oTD and/or oASD metagenomes. Rows correspond to the metabolites across detected by NMR (A) or GC-MS (B) that were significantly consistent with metabolic potential at q-value ≤ 0.1. Blue squares indicate that the estimated metabolic potential of the strain in question is consistent with contributing to variation in that metabolite (production or utilization/degradation). The area of the colored points along the bottom indicates the relative abundances of each taxon in oTD and oASD samples. The segments along the bottom indicate the relative ratio of each taxon in oTD versus oASD samples. The segments along the left side show the average difference in metabolite concentration Z-scores between the control and ASD donor samples. The column of colored tiles indicates the MIMOSA correlation between metabolic potential scores and metabolite concentrations for each metabolite.
(C–D) Contributions of microbial strains identified by MIMOSA as putatively involved in the production and degradation of metabolites. The fraction of contributing strains with potential for synthesis, degradation, or neither (0) is shown for oTD and oASD samples, and for amino acid metabolites compared to other compound classes, measured by NMR (C) and GC-MS (D), illustrating a trend of reduced amino acid metabolism in oASD samples.
(E) limma-voom differential abundance of KEGG pathways between oTD and oASD metagenomes.
(F - G) 5-aminovaleric acid production by Stickland fermentation of D-proline. (F) abundance of prdA and (G) prdB copies in oTD and oASD mice quantified by ShortBRED. Differences in Means were analyzed by a Kruskal-Wallis non-parametric test.
Figure S6. Metabolite administration post weaning has no effects on ASD-related behaviors in BTBR mice. Related to Figure 5. (A–B) Targeted metabolomics for (A) 5AV and (B) taurine in GF and SPF E16.5 dams. Normalized concentrations of 5AV and taurine in dam’s colon contents, serum, amniotic fluid, and fetal brains were measured. Group differences were tested by Kruskal-Wallis test. N=3 dams per group.
(C–E) Mice were orally administered 10 mM 5AV or taurine in drinking water starting at 3–4 weeks of age, and throughout their lifetime. Offspring were tested by (C) marble burying, (D) direct social interaction, and (E) open field tests, and compared to untreated vehicle controls. Results are aggregated from 2 independent experiments. NControl= 20, N5AV= 18, NTaurine= 20. Hypothesis on differences in means were tested by one-way ANOVA on trimmed means (10%) and subsequent post-hoc tests.
(F-H) Offspring “humanized” oASD3 mice were orally administered 10 mM 5AV in sterile drinking water starting at 3–4 weeks of age, and throughout their lifetime. Offspring were tested by (F) open field tests, (G) marble burying, and (H) direct social interaction, and compared to untreated vehicle controls. NControl= 20, N5AV= 20. Hypothesis on differences in means were tested by one-tailed t-tests on trimmed means (10%).
(I) Basal properties of L5 pyramidal neurons in the mPFC: F-I Slope, rheobase, slow afterhyperpolarization (sAHP) amplitude, resting membrane potential, resting membrane resistance, and voltage response to a steady hyperpolarizing current recorded (Sag). Hypothesis on differences in means were tested by one-way ANOVA on trimmed means (10%). NControl = 20 cells in 4 mice, N5AV = 17 cells in 3 mice, NTaurine = 20 cells in 4 mice.
(J) Proportion of GABA-excitable cortical rat neurons as a function of days in culture (DIV), treated with either 5AV, Taurine, or control. NControl = 1,126, N5AV = 1,306, NTaurine = 1,471 (over 10 time points at DIV6–17).
Table S2. Microbiome-Induced, ASD-Relevant Splicing events. Related to Figure 3.
Data Availability Statement
Raw and analyzed data used to produce all figures are available at the following links:
Mendeley Data http://dx.doi.org/10.17632/ngzmj4zkms Human donor microbiome raw data are available in SRA (16S: PRJNA533120).
Mouse recipient microbiome raw data are available in SRA and EBI (16S: PRJNA431279, Shotgun: ERP113632 or Qiita (Gonzalez et al., 2018) study ID 11809).
RNA-seq raw data The data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE109827 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE109827).
Metabolomics raw data Metabolights MTBLS726