

Abstract
Synthetic cannabinoids (SCBs), designer drugs marketed as legal alternatives to marijuana, act as ligands to cannabinoid receptors; however, they have increased binding affinity and potency, resulting in toxicity symptoms such as cardiovascular incidents, seizures, and potentially death. N‐(adamantan‐1‐yl)‐1‐(5‐fluoropentyl)‐1H‐indole‐3‐carboxamide (STS‐135) is a third generation SCB. When incubated with hepatocytes, it undergoes oxidation, hydrolysis, and glucuronidation, resulting in 29 metabolites, with monohydroxy STS‐135 (M25) and dihydroxy STS‐135 (M21) being the predominant metabolites. The enzymes responsible for this oxidative metabolism were unknown. Thus, the aim of this study was to identify the cytochrome P450 (P450s or CYPs) enzymes involved in the oxidative metabolism of STS‐135. In this study, STS‐135 was incubated with liver, intestinal, and brain microsomes and recombinant P450s to determine the enzymes involved in its metabolism. Metabolite quantification was carried out using ultra‐performance liquid chromatography. STS‐135 was extensively metabolized in HLMs and HIMs. Screening assays indicated CYP3A4 and CYP3A5 could be responsible for STS‐135’s oxidation. Through incubations with genotyped HLMs, CYP3A4 was identified as the primary oxidative enzyme. Interestingly, CYP2J2, a P450 isoform expressed in cardiovascular tissues, showed high activity towards the formation of M25 with a K m value of 11.4 μmol/L. Thus, it was concluded that STS‐135 was primarily metabolized by CYP3A4 but may have extrahepatic metabolic pathways as well. Upon exposure to STS‐135, individuals with low CYP3A4 activity could retain elevated blood concentration, resulting in toxicity. Additionally, CYP2J2 may aid in protecting against STS‐135‐induced cardiovascular toxicity.
Keywords: cytochrome P‐450 CYP3A, oxidation, STS‐135, synthetic cannabinoids, synthetic drugs
Abbreviations
- CBRs
cannabinoid receptors
- HBMs
human brain microsomes
- HLMs
human liver microsomes
- SCBs
synthetic cannabinoids
- UGT
UDP‐glucuronosyltransferase
1. INTRODUCTION
Synthetic cannabinoids (SCBs) are designer drugs present in products branded as “K2” or “Spice,” which are frequently marketed to consumers as safer, legal alternatives to marijuana. Originally, SCBs were synthesized as a means to investigate their potential as therapeutic ligands for cannabinoid receptors (CBRs); however, their predominant use has become recreational.1, 2 Often produced in clandestine labs, there are currently over 150 SCBs, with new analogs emerging as soon as existing compounds are made illegal. The greater danger of SCBs, as compared to endogenous cannabinoids and phytocannabinoids, results from the fact that SCBs have been observed to have a higher affinity for CBRs, and many of the SCB metabolites remain biologically active and exhibit an increased affinity and activity for CBRs as compared to the parent compounds.3, 4 Due to their high affinity to CBRs, exposure to SCBs can result in a number of effects similar to those elicited by the phytocannabinoids found in marijuana, including euphoria, analgesia, and appetite enhancement; however, in addition, SCBs frequently cause much more serious adverse effects including seizures, severe tachycardia, psychosis, hemorrhaging, and even death.5, 6, 7, 8, 9 While acute and chronic SCB abuse and its effects have been examined in various studies, information on the metabolism of SCBs is lacking, particularly for newer generations,this information is pertinent to understanding toxicity cases because accumulation of toxicants due to insufficient metabolism is a major mechanism of development of adverse toxic effects.3, 4, 10
STS‐135, also called N‐(adamantan‐1‐yl)‐1‐(5‐fluoropentyl)‐1H‐indole‐3‐carboxamide or 5F‐APICA, is the terminally‐fluorinated analog of SDB‐001 and APICA (Figure 1). It possesses a core with an indole structure and a substituted indole base. The base is substituted with a fluoropentyl chain at R1, suggesting the structure‐activity relationships found in the indole class of cannabimimetics, and at the terminal amine, R3, in the adamantane cage with a carboxamide group. This adamantane group is composed of four fused cyclohexane rings arranged in a unique structure called a diamondoid. STS‐135 is a potent agonist for both CB1 and CB2 receptors, producing reported, subjective effects similar to that of cannabis with a short duration and an emphasis on intense physical sensations.11, 12 Heart palpitations, vertigo, and sedation have been reported at doses that are lower than previously considered dangerous amounts, and users tend to suffer extreme anxiety or lose consciousness after consumption.12 Due to the strength of these effects generated at such low doses, at higher doses, there could be an increased risk of adverse effects, potentially resulting in death.11
Figure 1.
Structures of STS‐135 and its metabolites. Chemical structures of the third generation SCB STS‐135 and its main metabolites M25 and M21 are shown
Hepatic biotransformation contributes to the detoxification of many administered SCBs.3, 10, 13 Among the several xenobiotic‐detoxifying processes, oxidation, often performed by cytochrome P450s (P450s or CYPs), plays a significant role in their hepatic metabolism.10 P450s are a superfamily of microsomal hemoproteins that catalyze the hydroxylation of a wide range of xenobiotics, including SCBs, drugs, and environmental pollutants, as well as endogenous substances.10 In the human liver, at least 12 distinct P450 enzymes, including CYP1A2, −2C9, and −3A4, are functionally expressed and extensively contribute to the oxidative metabolism of hydrophobic compounds.14 The inhibition of detoxifying enzymes could result in elevated blood concentrations of ingested compounds and, thus, toxic effects.14, 15 As the extensive metabolism of SCBs often results in numerous biologically active metabolites, genetic polymorphisms of P450 and UDP‐glucuronosyltransferase (UGT) enzymes could potentially contribute to the idiosyncratic toxicity often experienced after SCB exposure. Individuals with genetic polymorphisms in these genes may be at a higher risk of developing toxic effects due to an inability to properly metabolize SCBs, depending on the availability or lack thereof of other metabolic pathways.15 Additionally, these polymorphisms are affected by race, resulting in entire groups of people being more susceptible to the toxic effects of these drugs.15 For example, a previous studied reported that one‐half of the surveyed African Americans expressed CYP3A5, while only one‐third of Caucasians did.16 Therefore, identifying the enzymes that are involved in the metabolism of toxic compounds is important in the prevention and treatment of potential adverse drug reactions.
It has been previously demonstrated that STS‐135 incubated with human hepatocytes undergoes oxidation, hydrolysis, and glucuronidation, resulting in the formation of 29 metabolites.17 Among them, monohydroxy STS‐135 (M25) and dihydroxy STS‐135 (M21) appear to be the primary metabolites; however, the enzymes responsible for the oxidative metabolism of this SCB have not yet been identified. In this study, our aim was to identify the P450s responsible for the formation of M25 and M21 by utilizing human liver microsomes (HLMs), genotyped HLMs, and recombinant P450s along with isoform specific inhibitors.
2. MATERIALS AND METHODS
2.1. Materials
All chemicals used for this study were of at least reagent grade. STS‐135 was obtained from Cayman Chemical. Pooled HLMs, genotyped HLMs, recombinant P450s, human brain microsomes (HBMs), and NADPH Regenerating System Solutions A and B were purchased from Corning. Human intestinal microsomes (HIMs) were purchased from Xenotech. LC/MS water, methanol, and acetic acid grade were purchased from Thermo Fisher Scientific. All other chemicals and reagents were purchased from Sigma‐Aldrich or Thermo Fisher Scientific, unless specified otherwise.
2.2. Screening of STS‐135 with HLMs, HIMs, HBMs, and recombinant P450s
The metabolism of STS‐135 was examined by analyzing the activity of HLMs, HBMs, and HIMs from 50‐donor pools along with assorted human recombinant P450 enzymes (50 µg CYP1A2, −2A6, −2B6, −2C9, −2C19, −2D6, −2E1, −2J2, −3A4, and −3A5) toward it. The final substrate concentration ranged 5‐200 µmol/L with 2% DMSO. We conducted a preliminary experiment and confirmed that the STS‐135 oxidizing activity was statistically the same between 1% and 2% organic solvent concentration in our enzyme assays. The substrate was added to each tube along with protein, water, and buffer (final concentration 0.1 mol/L KPO4, pH 7.4); the reactions were started with the addition of an NADPH‐regenerating system (1 mmol/L NADP+, 3 mmol/L glucose 6‐phosphate, 3 mmol/L MgCl2; 1 U/mL glucose 6‐phosphate dehydrogenase) to ensure the saturation of NADPH thus enabling cytochrome P450‐mediated reactions. Controls omitting the substrate, protein, and NADPH were included with each assay. Reactions were incubated at 37°C for 90 minutes or indicated time, and terminated by the addition of equal volumes of ethanol. Protein and other particulates were precipitated by centrifugation at 12 000g for 8 minutes and subsequently analyzed by an ultra‐performance liquid chromatography (UPLC) as described below. All reactions were performed in triplicate.
2.3. Inhibition of oxidative metabolism
Reactions were prepared following the protocol used in the screening assay, and inhibitors (α‐napthyflavone (α‐NF), quinidine (QND), N‐3‐benzylnirvanol (NBZ), and ketoconazole (KCZ) using vehicle as a control) were added to the reactions at concentrations of 10 and 100 µmol/L except NBZ, which was 10 and 64 µmol/L due to its insolubility. The substrate (STS‐135) concentration used in the inhibition assay was 50 µmol/L in order to obtain reliable substrate and metabolite peaks in the UPLC analysis. The inhibitors used in the present study were dissolved in either DMSO or methanol. The reaction mixtures were incubated for 90 min at 37°C, and the reactions were terminated by the addition of 30 μL of ethanol. After removal of the protein by centrifugation at 12 000g for 10 minutes, a 5 μL portion of the sample was subjected to analysis by UPLC. The final concentrations of the organic solvents in the incubation mixtures were less than 2%, and all reactions were performed in triplicate.
2.4. Effect of polymorphisms on STS‐135 metabolism
Assays were prepared as was described for the screening experiment, except that genotyped microsomes, CYP3A5*1/*1 and CYP3A5*3/*3, were used (Corning). While CYP3A5*1 exhibit normal CYP3A5 enzyme activity, CYP3A5*3 lacks the enzyme activity. However, the CYP3A5*3/*3 individual microsomes used in the current study exhibit higher CYP3A4 activity compared to the CYP3A5*3/*3 microsomes due to an inter‐individual variability in the CYP3A4 activity. Samples of the reactions were taken at the indicated times intervals (0, 10, 20, 30, 45, 60, 90, and 120 minutes), and the reactions were analyzed using UPLC.
2.5. UPLC analysis
The parent compound and its oxidized metabolites were identified by the ACQUITY UPLC System with a UV detector (Waters, Milford, MA,). The mobile phases were 0.1% acetic acid (A) and 100% methanol (B), and the flow rate was 0.5 mL/min with an elution gradient of 100% A (0‐0.2 minutes), a linear gradient from 100% A to 25% A‐75% B (0.2‐5 minutes), and 100% B (5‐7 minutes). The column was re‐equilibrated at initial conditions for 2.5 minutes between runs. The elution was monitored at 300 nm, and the results were analyzed with the Empower software (Waters).
2.6. Steady‐state enzyme kinetic assays
Incubation conditions were optimized for time and protein concentration, and all reactions were performed within the linear range of metabolite formation. Other than substrate concentrations and incubation times, the reaction mixture composition and analytical methods were identical to those described for the above screening assays. Incubations were carried out with recombinant CYP2J2 (50 µg protein) in the presence of various concentrations of the substrate (5‐200 µmol/L) for 90 minutes at 37°C.
2.7. Data analysis
Kinetic parameters were estimated from fitted curves using a program (http://www.ic50.tk) designed for non‐linear regression analysis. The Michaelis‐Menten equation, V = V max·[S]/(K m + [S]), was used to calculate the K m and V max values, where V is the velocity of the reaction, S is the substrate concentration, K m is the Michaelis constant and V max is the maximum velocity. The intrinsic clearance (CL int) is defined as V max/K m. The fitted curves were drawn using an online program (http://www.physiologyweb.com). Kinetic constants are reported as the mean ± SD of triplicate experiments. SDs were from three independent experiments. The half‐life was directly determined from linear regression. Statistical analysis for inhibitor studies was performed using the Analysis of Variance model and the Holm‐Bonferroni post hoc test.
3. RESULTS
3.1. Oxidative metabolism of STS‐135 by human microsomes
After screening human microsomes from the liver (HLMs), intestines (HIMs), and brain (HBMs) for their ability to metabolize STS‐135 at 50 µmol/L, it was determined that this compound was predominantly metabolized within the liver; the two predominant peaks observed here correspond with the mono‐ and dihydroxy metabolites, M25 and M21, as reported by 17 (Figure 1). Through comparison with the negative control, it was determined that the peak at retention time of 4.54 minutes was M21, the one at 4.89 minutes was M25, and the peak at 5.94 minutes was identified as the parent compound (STS‐135) (Figure 2). Specific activity for M25 was 0.0400 nmol/mg protein/min and for M21, 0.0460 nmol/mg protein/min in HLMs (Figure 3). Additionally, a significant amount of oxidative metabolism occurred in the intestines, with activity toward M25 at 0.0300 and M21 at 0.0110 nmol/mg protein/min in HIMs (Figure 3). However, there were no detectable metabolites produced by the HBMs, confirming the liver as the primary location of metabolic activity for STS‐135.
Figure 2.
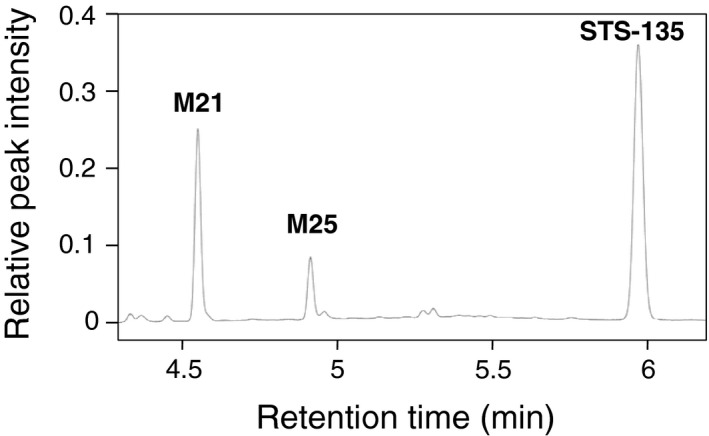
Representative chromatogram of STS‐135 and its main metabolites. STS‐135 was incubated with HLM and the reaction mixture was separated by UPLC. Retention times of STS‐135 and its primary metabolites, M21 and M25 were 4.54 (M21), 4.90 minutes (M25), and 5.94 minutes (STS‐135)
Figure 3.
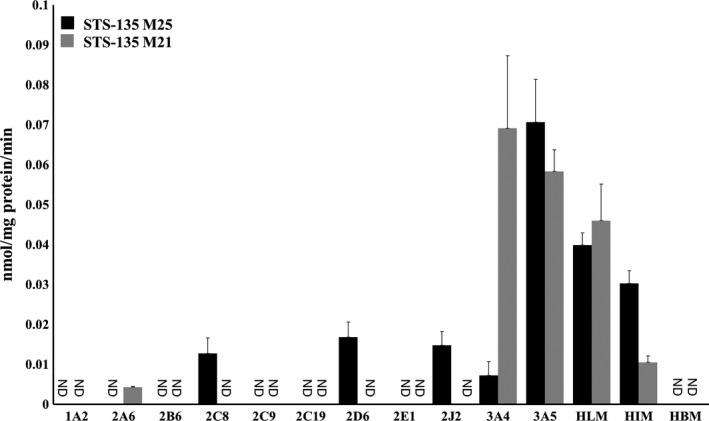
Hydroxylation activities of human recombinant P450s, HLMs, HIMs, and HBMs toward STS‐135. HLMs, HIMs, and HBMs along with 11 recombinant P450s (CYP1A2, −2A6, −2B6, −2C8, −2C9, −2C19, −2D6, −2E1, −2J2, −3A4, and −3A5) (50 µg) were used to screen for their ability to metabolize STS‐135 (50 µmol/L) and produce hydroxylated metabolites. At least one of two predominant metabolites (M25 and M21) was observed in reactions containing HLMs, HIMs, CYP2A6, −2C8, −2D6, −2J2, −3A4, and −3A5. Specific activities are expressed in nmol/mg protein/min
3.2. Identification of major oxidized metabolites of STS‐135 by recombinant proteins
As shown in Figure 3, HLMs, HIMs, and HBMs along with eleven human recombinant cytochrome P450 enzymes, CYP1A2, −2A6, −2B6, −2C8, −2C9, −2C19, −2D6, −2E1, −2J2, −3A4, and −3A5 were used to screen for their respective abilities to metabolize STS‐135 and produce the hydroxylated metabolites, M21 and M25. After incubation, the reactions were analyzed via UPLC following the protocol described above. Of all the recombinant proteins that were screened, CYP3A5 showed the greatest activity for M25 at 0.071 nmol/mg protein/min, and CYP3A4 and −3A5 showed highest activity for M21, exhibiting activities of 0.069 and 0.058 nmol/mg protein/min, respectively (Figure 3). CYP2C8, −2D6, and −2J2 produced only the monohydroxy M25 metabolite with activities of 0.013, 0.017, and 0.015 nmol/mg protein/min, respectively; in reactions containing CYP2A6, only the dihydroxy metabolite, M21, was present with a lower activity of 0.0042 nmol/mg protein/min. Figure 3 indicates that the majority of this metabolism is carried out by CYP3A4 and −3A5, which have previously been identified as two of the most abundant P450s in the liver.18
3.3. Inhibition of oxidative metabolism
To assist in the confirmation of the involvement of specific P450s in the metabolism of STS‐135 in the human liver, enzyme assays were performed in the presence of compounds, at 2 different concentrations, that are known to act as inhibitors towards specific families of P450s; these compounds included α‐NF, QND, NBZ, and KCZ. The specificity of the targets of the utilized inhibitors can be viewed in Table 1. The results were reported as a percentage of the specific activity shown using untreated reactions as the standard (Figure 4). α‐NF at 10 and 100 µmol/L did not show a significant inhibitory effect on the production of either M25 or M21, nor did 10 µmol/L QND or NBZ per Figure 3, where CYP1A2 was shown to not play a large role in the metabolism of this SCB. While 100 µmol/L QND and NBZ had little inhibitory effect on the formation of M21 (Figure 4B), they did inhibit the production of the M25 metabolite (Figure 4A); QND particularly showed a great magnitude of inhibitory power. As M21 is believed to be metabolized sequentially into M25, both concentrations of KCZ inhibited the formation of both the M25 and M21 metabolites, indicating that the CYP3A enzymes were prevented from metabolizing STS‐135 into its primary metabolites (Figure 4A and B). Thus, the data obtained from these inhibition assays supported the hypothesis that CYP3As were largely involved in the oxidative metabolism of STS‐135 in human livers.
Table 1.
Inhibitors used in the inhibition assay
Figure 4.
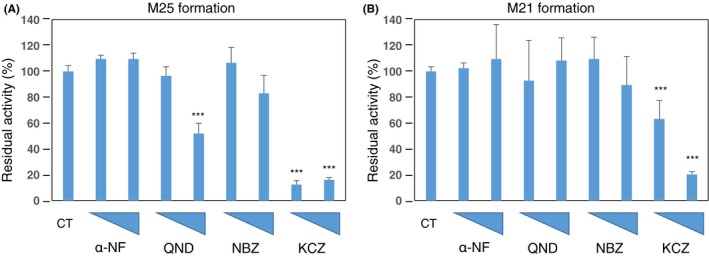
Enzymatic inhibition was performed to confirm the hydroxylation by various P450s. P450 inhibitors (α‐napthyflavone (α‐NF), quinidine (QND), N‐3‐benzylnirvanol (NBZ), and ketoconazole (KCZ) using water as a control) were added at 10 and 100 µmol/L concentrations, as denoted by the increasing height of the triangles on the x‐axis, to confirm the ability of P450s to oxidize STS‐135. (A) Both concentrations of KCZ inhibited the formation of the M25 metabolite by over 80%; 100 µmol/L QND inhibited its formation by approximately 50%. (B) 100 µmol/L KCZ inhibited the formation of M21 metabolites by decreasing activity by 70%. However, 10 µmol/L only reduced its formation by 30%. ***P < .001 compared to the control
3.4. Time‐dependent metabolism of STS‐135 in genotyped HLMs
While KCZ is effective at inhibiting the activities of CYP3A enzymes, it lacks the ability to discriminate between CYP3A4 and −3A5; therefore, to determine which enzyme played a predominant role in the metabolism of STS‐135 in the human liver, genotyped HLMs for CYP3A5 were used, and the samples were analyzed at the before mentioned time points. As mentioned previously, CYP3A5*1 possesses high activity for CYP3A5; in contrast, CYP3A5*3 shows no activity for CYP3A5; however, both microsomes possess CYP3A4 activity. Importantly, CYP3A4 activity was higher in the CYP3A5*3*3 microsomes that were used in the current study compared to microsomes genotyped as CYP3A5*1*1. The parent compound, STS‐135, decreased in a time dependent manner in reactions containing both the high and no CYP3A5 activity HLMs, CYP3A5*1*1 and CYP3A5*3*3, respectively (Figure 5A and B). Furthermore, it was observed that, as the amount of STS‐135 decreases, the amount of M25, the monohydroxylated metabolite, increases (Figure 5A and B). However, once it hits a plateau in its formation, M25 begins to decrease, and an increase in the amount of the dihydroxylated M21 metabolite follows. The half‐time (t1/2) for the disappearance of STS‐135 was 8.5 minutes when incubated in CYP3A5*3/*3 HLMs (Figure 5B) as opposed to the slower t1/2 observed in CYP3A5*1/*1 HLMs (21.1 min) (Figure 5A), indicating that the participation of CYP3A5 was irrelevant in the metabolism of STS‐135 and that the majority of the metabolism was performed by CYP3A4. The higher CYP3A4 activity in the HLMs genotyped CYP3A5 *3/*3 as compared to CYP3A5*1/*1 can explain the faster metabolic rate in the CYP3A5 *3/*3 microsomes.
Figure 5.
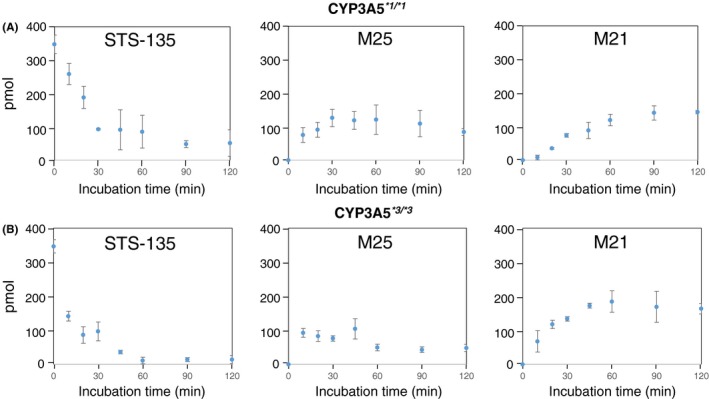
Time‐dependent metabolism of STS‐135 in genotyped HLMs. While CYP3A4 is not highly polymorphic, CYP3A5 is. Genotyped HLMs possessing (A) high (CYP3A5*1*1) and (B) low activity (CYP3A5*3*3) were used to determine whether CYP3A4 or CYP3A5 played a predominant role in the oxidative metabolism of STS‐135. The activity level of CYP3A5 was not shown to affect the production of M25 or M21
3.5. Steady‐state kinetic analysis using CYP2J2
As the recombinant CYP2J2 showed moderate activity towards STS‐135 and produced only a single metabolite, kinetic constants were analyzed. Figure 6 shows the kinetic profiles for the recombinant P450, which followed the classical Michaelis‐Menten kinetics curve. Steady‐state kinetic analysis of CYP2J2 resulted in a Vmax value of 17.2 ± 0.5 pmol/min/mg protein and a Km of 11.2 ± 1.7 µmol/L.
Figure 6.
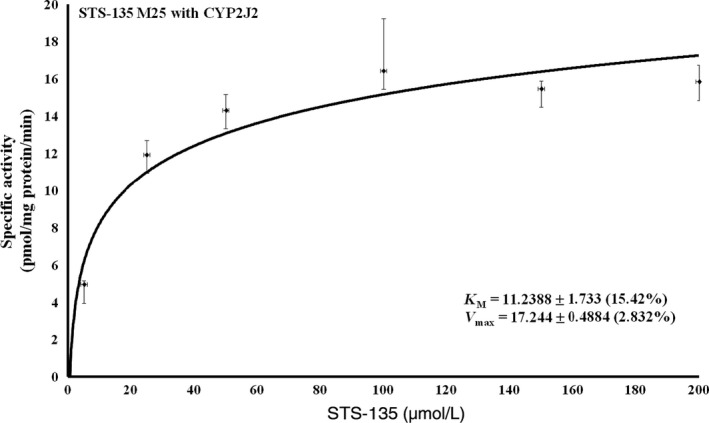
Steady‐state kinetic analysis of CYP2J2‐mediated hydroxylation of STS‐135. Kinetic constants for CYP2J2 and its production of M25 were analyzed, and its kinetic profile, which followed classical Michaelis‐Menten kinetics, resulted in a V max value of 17.244 ± 0.4884 and a K m of 11.2389 ± 1.733
4. DISCUSSION
SCBs were once marketed as a harmless, over the counter novelty items to consumers; however, the dangerous nature of these drugs of abuse has come to light. Like natural cannabinoids, including Δ9‐THC, SCBs bind to and act as ligands for CBRs, frequently having affinity for both receptors CB1 and CB2; however, these powerful cannabimimetics generate more varied clinical effects as compared to those generated by natural cannabinoids, and the severities of these effects are often of much greater magnitude.5, 6, 7, 8, 9 Furthermore, SCBs’ metabolism can result in biologically active metabolites that exhibit an even greater affinity for CBRs, increasing the risk of toxicity.3, 4 Unfortunately, the treatment for cases of toxicity induced by SCBs is palliative at best as there are no developed treatments. With abuse of SCBs on the rise, and clandestine labs manufacturing more and more analogs, understanding their mechanisms and the involved metabolic pathways is essential to the development of a practical and effective treatment for cases of toxicity.
STS‐135 is an extremely potent, third generation SCB with anecdotal evidence indicating that very little of the compound is required to generate a physiological response.12, 19 Currently, there is only one study that investigated and reported metabolism of STS‐135. According to the publication, two major hepatic metabolites of STS‐135 are M25 and M21, which are mono‐ and di‐hydroxilated metabolites, which suggests that CYPs are the major class of enzyme responsible for the oxidative metabolism of this compound. Although STS‐135 is metabolized by other phase I and II enzymes, contribution of these enzymes to overall hepatic metabolism of STS135 seemed minimum.17 The study also revealed that a significant proportion of STS‐135 can be metabolized in hepatocytes. Unfortunately, quantitative investigation of STS‐135 and its metabolites in blood or urine has not been reported. However, it is speculated that oxidation to M25 and M21 would be the main metabolic and clearance pathway of STS‐135 due to the general importance of liver in metabolic clearance of xenobiotics. However, the specific P450 enzymes involved in its metabolism remain unknown. There are a wide variety of P450 enzymes placed in several subfamilies that are present in the human body. Although there were numerous isoforms that exhibited the ability to produce the metabolites of these compounds, CYP3A4 and −3A5 appeared to be the enzymes predominantly responsible for the metabolism of this SCB (Figure 3). Furthermore, the data obtained with the genotyped liver microsomes showed that CYP3A4 was actually the predominant metabolizer. In our microsomal screening assay, no metabolism was detected in the HBMs (Figure 3), indicating that the brain lacks the enzymes necessary to metabolize or neutralize the effects of the parent SCB or its metabolites, increasing the risk for toxicity and potentially explaining the intensity of the reported symptoms. CYP2J2 is primarily expressed in the cardiovascular system, especially cardiomyocytes and endothelial cells.20, 21 This isoform plays a role in metabolizing endogenous polyunsaturated fatty acids into signaling molecules, and it also metabolizes arachidonic acid into various eicosatrienoic acid epoxides. Overexpressed CYP2J2 is found in numerous cancers where it can accelerate proliferation and protect the cell from apoptosis.22, 23 Due to its abundance in the heart, loss of function for this enzyme can result in an increased susceptibility to cardiac toxicity and, potentially, in damage to cardiac tissue. Another study has shown the importance of CYP2J2 in the protection of cardiac tissue from toxicity.24 In the current study, it was shown that STS‐135 was extensively metabolized by CYP2J2. Therefore, because metabolic inhibition of CYP2J2 by STS‐135 can result in disruption of heart function, this might explain the reported cases of heart toxicity in individuals who consumed STS‐135. Also, if the oxidative metabolite of STS‐135 whose production is mediated by CYP2J2 is still pharmacologically active, the metabolite itself may be involved in the heart toxicity. The toxicological mechanism of cardiotoxicity by STS‐135 and M25 needs to be further investigated in the future.
SCBs are posing a public health threat to consumers; during a five‐month period in 2015, there were over 3500 cases of SCB induced toxicity within the United States alone.25, 26 Additionally, it should be noted that these cases of toxicity are disproportionally occurring in teens and young adults, with one study reporting that of the deaths caused by SCBs, over one third occurred in individuals ages 13‐19.9 Therefore, understanding the mechanism behind the metabolism of these compounds and understanding how toxicities occur from this compound is pertinent. Thus, our study succeeded, with us identifying the primary enzymes involved in the metabolism of STS‐135, a highly potent, dangerous, third generation SCB. As discussed above, the STS‐135‐metabolizing enzymes are highly polymorphic. It should be noted that even without genetic polymorphisms, there is a significantly wide inter‐individual variability in the hepatic CYP3A4 activity in humans, suggesting that users possessing isoforms with reduced activity could potentially put themselves at risk for increased toxic effects, specifically in the heart and intestines. Furthermore, co‐administered drugs, food, herbs, and other SCBs could also be the factors increasing the risk of the STS‐135 toxicity due the facts that many of those can act as potent CYP3A inhibitors.
DISCLOSURES
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACKNOWLEDGEMENTS
This work was supported in part by a grant from the National Institute on Drug Abuse [NIH/NIDA DA039143 to ARP, WEF and PLP].
Jones S, Yarbrough AL, Fantegrossi WE, et al. Identifying cytochrome P450s involved in oxidative metabolism of synthetic cannabinoid N‐(adamantan‐1‐yl)‐1‐(5‐fluoropentyl)‐1H‐indole‐3‐carboxamide (STS‐135). Pharmacol Res Perspect. 2020;00:e00561 10.1002/prp2.561
Sabrina Jones and Azure L. Yarbrough are contributed equally.
REFERENCES
- 1. Castaneto MS, Gorelick DA, Desrosiers NA, et al. Synthetic cannabinoids: epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend. 2014;144:12‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hasin DS, Saha TD, Kerridge BT, et al. Prevalence of marijuana 787 use disorders in the united states between 2001–2002 and 2012–2013. JAMA Psychiatry. 2015;72:1235‐1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chimalakonda KC, Bratton SM, Le VH, et al. Conjugation of synthetic cannabinoids JWH‐018 and JWH‐073, metabolites by human UDP‐glucuronosyltransferases. Drug Metab Dispos. 2011;39:1967‐1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fantegrossi WE, Moran JH, Radominska‐Pandya A, et al. Distinct pharmacology and metabolism of K2 synthetic cannabinoids compared to Δ(9)‐THC: mechanism underlying greater toxicity? Life Sci. 2014;97:45‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brents LK, Reichard EE, Zimmerman SM, et al. Phase I hydroxylated metabolites of the K2 synthetic cannabinoid JWH‐018 retain in vitro and in vivo cannabinoid 1 receptor affinity and activity. PLoS ONE. 2011;6:e21917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lapoint J, James LP, Moran CL, et al. Severe toxicity following synthetic cannabinoid ingestion. Clin Toxicol. 2011;49:760‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shanks KG, Clark W, Behonick G. Death associated with the use of the synthetic cannabinoid ADB‐FUBINACA. J Anal Toxicol. 2016;40:236‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Stout SM, Cimino NM. Exogenous cannabinoids as substrates, inhibitors, and inducers of human drug metabolizing enzymes: a systematic review. Drug Metab Rev. 2014;46:86‐95. [DOI] [PubMed] [Google Scholar]
- 9. Trecki J, Gerona RR, Schwartz MD. Synthetic cannabinoid‐related illnesses and deaths. N Engl J Med. 2015;373:103‐107. [DOI] [PubMed] [Google Scholar]
- 10. Chimalakonda KC, Seely KA, Bratton SM, et al. Cytochrome P450‐mediated oxidative metabolism of abused synthetic cannabinoids found in K2/Spice: identification of novel cannabinoid receptor ligands. Drug Metab Dispos. 2012;40:2174‐2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Banister SD, Stuart J, Kevin RC, et al. Effects of bioisosteric fluorine in synthetic cannabinoid designer drugs JWH‐018, AM‐2201, UR‐144, XLR‐11, PB‐22, 5F‐PB‐22, APICA, and STS‐135. ACS Chem Neurosci. 2015;6:1445‐1458. [DOI] [PubMed] [Google Scholar]
- 12. Wilkinson SM, Banister SD, Kassiou M. Bioisosteric fluorine in the clandestine design of synthetic cannabinoids. Aust J Chem. 2015;68:4‐8. [Google Scholar]
- 13. Jones S, Yarbrough AL, Shoeib A, et al. Enzymatic analysis of glucuronidation of synthetic cannabinoid 1‐naphthyl 1‐(4‐fluorobenzyl)‐1H‐indole‐3‐carboxylate (FDU‐PB‐22). Xenobiotica. 2019;49:1388‐1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nebert DW, Russell DW. Clinical importance of the cytochromes P450. Lancet. 2002;360:1155‐1162. [DOI] [PubMed] [Google Scholar]
- 15. Patton AL, Seely KA, Yarbrough AL, et al. Altered metabolism of synthetic cannabinoid JWH‐018 by human cytochrome P450 2C9 and variants. Biochem Biophys Res Commun. 2018;498:597‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuehl P, Zhang J, Lin Y, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001;27:383‐391. [DOI] [PubMed] [Google Scholar]
- 17. Gandhi AS, Wohlfarth A, Zhu M, et al. High‐resolution mass spectrometric metabolite profiling of a novel synthetic designer drug, N‐(adamantan‐1‐yl)‐1‐(5‐fluoropentyl)‐1H‐indole‐3‐carboxamide (STS‐135), using cryopreserved human hepatocytes and assessment of metabolic stability with human liver microsomes. Drug Test Anal 2015;7:187‐198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paine MF, Hart HL, Ludington SS, et al. The human intestinal cytochrome P450 “Pie”. Drug Metab Dispos. 2006;34:880‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Basavarajappa BS, Subbanna S. Potential mechanisms underlying the deleterious effects of synthetic cannabinoids found in Spice/K2 products. Brain Sci. 2019;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. DeLozier TC, Kissling GE, Coulter SJ, et al. Detection of human CYP2C8, CYP2C9 and CYP2J2 in cardiovascular tissues. Drug Metab Dispos. 2007;35:682‐688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Evangelista EA, Kaspera R, Mokadam NA, et al. Activity, inhibition, and induction of cytochrome P450 2J2 in adult human primary cardiomyocytes. Drug Metab Dispos. 2013;41:2087‐2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen JK, Capdevilla J, Harris RC. Cytochrome p450 epoxygenase metabolism of arachidonic acid inhibits apoptosis. Mol Cell Biol. 2001;21:6231‐6233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chen C, Wei X, Rao X, et al. Cytochrome P450 2J2 is highly expressed in hematologic malignant diseases and promotes tumor cell growth. J Pharmacol Exp Ther. 2011;336:344‐355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang Y, El‐Sikhry H, Chaudhary KR, et al. Overexpression of CYP2J2 provides protection against doxorubicin‐induced cardiotoxicity. Am J Physiol Herat Circ Physiol. 2009;297:H37‐H46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hermanns‐Clausen M, Kneisel S, Szabo B, et al. Acute toxicity due to the confirmed consumption of synthetic cannabinoids: clinical and laboratory findings. Addiction. 2013;108:534‐544. [DOI] [PubMed] [Google Scholar]
- 26. Law R, Schier J, Martin C, et al. Centers for disease control (CDC). Notes from the field: increase in reported adverse health effects related to synthetic cannabinoid use‐united states, January‐May 2015. MMWR Morb Mortal Wkly Rep. 2015;64:618‐619. [PMC free article] [PubMed] [Google Scholar]
- 27. Cho US, Park EY, Dong MS, et al. Tight‐binding inhibition by α‐naphthoflavone of human cytochrome P450 1A2. Biochim Biophys Acta. 2003;1648:195‐202. [DOI] [PubMed] [Google Scholar]
- 28. Ching MS, Blake CL, Ghabrial H, et al. Potent inhibition of yeast‐expressed CYP2D6 by dihydroquinidine, quinidine, and its metabolites. Biochem Pharm. 1995;50:833‐837. [DOI] [PubMed] [Google Scholar]
- 29. Nirogi R, Palacharla RC, Uthukam V, et al. Chemical inhibitors of CYP450 enzymes in liver microsomes: combining selectivity and unbound fractions to guide selection of appropriate concentration in phenotyping assays. Xenobiotica. 2015;45:95‐106. [DOI] [PubMed] [Google Scholar]
- 30. Moltke LL, Greenblatt DJ, Duan SX, et al. In vitro prediction of the terfenadine‐ketoconazole pharmacokinetic interaction. J Clin Pharmacol. 1994;34:1222‐1227. [DOI] [PubMed] [Google Scholar]