

Abstract
Microbial species play important roles in different environments and the production of high-quality genomes from metagenome data sets represents a major obstacle to understanding their ecological and evolutionary dynamics. Metagenome-Assembled Genomes Orchestra (MAGO) is a computational framework that integrates and simplifies metagenome assembly, binning, bin improvement, bin quality (completeness and contamination), bin annotation, and evolutionary placement of bins via detailed maximum-likelihood phylogeny based on multiple marker genes using different amino acid substitution models, next to average nucleotide identity analysis of genomes for delineation of species boundaries and operational taxonomic units. MAGO offers streamlined execution of the entire metagenomics pipeline, error checking, computational resource distribution and compatibility of data formats, governed by user-tailored pipeline processing. MAGO is an open-source-software package released in three different ways, as a singularity image and a Docker container for HPC purposes as well as for running MAGO on a commodity hardware, and a virtual machine for gaining a full access to MAGO underlying structure and source code. MAGO is open to suggestions for extensions and is amenable for use in both research and teaching of genomics and molecular evolution of genomes assembled from small single-cell projects or large-scale and complex environmental metagenomes.
Keywords: metagenomics, evolutionary analyses, microbial draft genomes, species boundaries, FastANI, genome assembly and binning
Microbial species play important roles in different environments characterized by a span of organismal complexities. The shotgun sequencing coupled to metagenomic analyses are used to study microbial communities in these environments. The analysis and biological interpretation of sequence information derived from complex communities or single-amplified cell communities represented as metagenome or whole-genome sequencing data sets, respectively, is challenging and crucially depends on sophisticated computational resources and analyses. These include various pieces of software and steps (e.g., read assembly, binning, annotation, bin evaluation) next to program-specific settings, file format conversions and decision points that require and consume substantial time, computational resources and may introduce unintended bias (Sczyrba et al. 2017). Obtaining genomes from metagenomes is an emerging approach with the potential for large-scale recovery of high-quality near-complete genomes amenable for analyses of their evolutionary divergence, evolutionary dynamics, and abundance in original samples (Meyer et al. 2018).
Advances in computational tools have improved our ability to address relevant evolutionary questions. However, computational costs for hundreds of samples are measured in tenths of thousands of CPU hours. The development of highly successful tools such as FastQC (Andrews 2010), fastp (Chen et al. 2018), IDBA-UD (Peng et al. 2012), megaHIT (Li et al. 2015), metaSPAdes (Nurk et al. 2017), maxBin (Wu et al. 2016), MetaBAT (Kang et al. 2015), CONCOCT (Alneberg et al. 2014), BinSanity (Graham et al. 2017), Dereplication-Aggregation Scoring Tool (Sieber et al. 2018), CheckM (Parks et al. 2015), ezTree (Wu, 2018), and lessons learned through the Critical Assessment of Metagenomic Information (CAMI; Sczyrba et al. 2017; Meyer et al. 2018; Fritz et al. 2019) enabled the field of molecular evolution of Bacteria and Archaea domains to progress from being a descriptive to an experimental endeavor, providing insight into evolutionary wealth of novel metagenome-assembled genomes (MAGs), novel microbial lineages uncovered from the environment, hence substantially revising and expanding the tree of life (Parks et al. 2017; Parks et al. 2018) and evolutionary dynamic in complex environments and medicine (Lin and Kussell, 2019; Garud et al. 2019). Although the tools are widely used, a number of limitations (supplementary table S1, Supplementary Material online) and their dispersed and boutique nature is limiting their integration and presents an obstacle to their reproducible use within community, their further adoption alongside the ubiquitous increases in sequencing volumes, study complexity (Jain et al. 2018), emerging standards (Sczyrba et al. 2017; Bowers et al. 2017), and technology upgrades (e.g., Nanopores).
To date, no uniform piece of software exists that would integrate efficiently, scalable and reproducibly all the steps linking the raw outputs from the sequencing platform (i.e., sequence data sets) over the steps of sequence quality trimming, assembly, binning, bin improvement, bin quality control, bin annotation, to evolutionary and phylogenomic placement of bins based on multiple orthologous marker genes on protein level, provide core- and pan-genome analyses and species boundary delineation through fast average nucleotide identity (ANI) of resulting draft genomes. The field-wide analysis standards are emerging due to the ongoing efforts (Sczyrba et al. 2017; Meyer et al. 2018; Fritz et al. 2019); however, the lack of reproducible framework makes it difficult to embrace these standards, perform meta-analyses of existing data (Schloss et al. 2009; Parks et al. 2017) or simply remap and extend past analyses (Parks et al. 2018; Jain et al. 2018) to evolutionary dynamics (Garud et al. 2019).
A single software platform, Metagenome Assembled Genomes Orchestra (MAGO) (fig. 1; supplementary table S1, Supplementary Material online) was developed to fill this gap and to overcome the limitations (supplementary table S2, Supplementary Material online) by integrating an ensemble of previously developed tools, streamlining their performance and deliver compatibility of data formats, together with additional features for error checking, effective computational resource use, governed by user-tailored pipeline processing (as specified by a textual configuration file). MAGO currently makes use of the three most effective assemblers and six binners put forward by CAMISIM (Fritz et al. 2019) and AMBER (Meyer et al. 2018) studies, respectively. The resulting bins are further improved by additional (the seventh) binner, Dereplication-Aggregation Scoring Tool (Sieber et al. 2018) and evaluated by CheckM according to their quality (% completeness and % contamination; Parks et al. 2015) in line with MIMAG standard (Bowers et al. 2017). CheckM utilizes a broader set of orthologous protein marker genes specific to the position of each MAG within a reference genome tree and information about collocation of these genes, based on amino acid identity between marker genes. Finally, the produced collection of high quality MAGs can be used to extract protein-coding single-copy orthologous marker genes using functional annotation and build maximum likelihood trees from amino acid sequences with different amino acid substitution models within MAGO using ezTree (Wu, 2018). The resulting alignment file can be exported to build user specific trees in existing high-end software (e.g., MEGA, Kumar et al. 2018). To annotate and calculate core- and pan-genomes MAGO integrates Prokka (Seemann, 2014) and Roary (Page et al. 2015) and makes outputs (fasta, gbk) available for additional downstream analyses of genome rearrangements (e.g., Mauve, Darling et al. 2010). FastANI (Jain et al. 2018) is utilized for high-throughput ANI analysis of MAGs that is used to define species boundaries and Operational Taxonomic Unit (OTU) delineation at various thresholds of ANI. All outputs are readily made available in structured directories for additional inspection and inclusion in other types of analyses tools (e.g., MEGA-X, Kumar et al. 2018; GTDB-Tk, Parks et al. 2018; MAGpy, Stewart et al. 2019). In total, MAGO consists of a number (n = 53) of externally developed pieces of software (supplementary table S1, Supplementary Material online) and >9,000 lines of Python code integrated into seamless workflow to perform error checking of pipeline configuration and to prevent suboptimal utilization of computational resources.
Fig. 1.
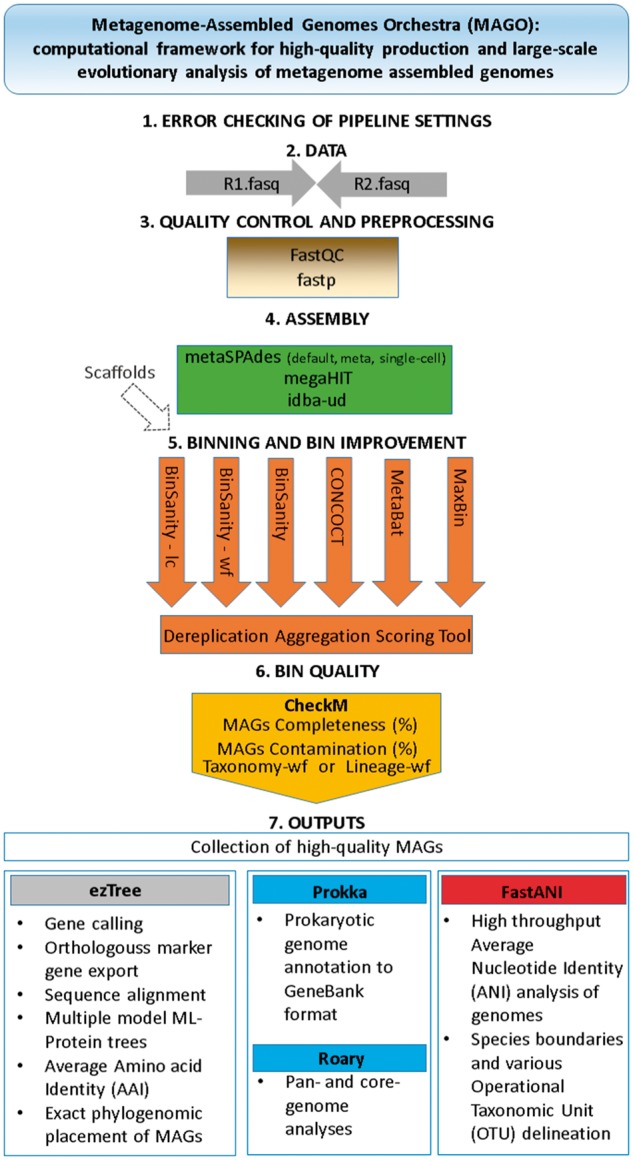
A schematic representation of steps integrated within MAGO starting from the input of raw sequencing data to MAGs, bin quality checking and the production of a collection of high-quality MAGs. These are further utilized in analysis of evolutionary relationships to produce maximum-likelihood (ML) phylogenomic placement, MAGs annotation, and core/pan genome calculations next to determination of species boundaries and operational taxonomic units at genomic level. The outputs are easily integrated into recently developed tools (e.g., MEGA-X, Kumar et al. 2018; GTDB-Tk, Parks et al. 2018; MAGpy, Stewart et al. 2019).
To overcome the constraints of web-based implementations of existing software and the known software limitations described above (supplementary table S2, Supplementary Material online) MAGO was made available as a singularity image (https://www.sylabs.io/singularity/; last accessed September 04, 2019) and a Docker container (https://www.docker.com; last accessed September 04, 2019) for high performance computing (HPC) purposes, and also as a VirtualBox (https://www.virtualbox.org/; last accessed September 04, 2019) virtual machine (as outlined in supplementary materials and methods, Supplementary Material online). By making MAGO an open-source-software package under the Commons Creative Attribution CC-BY License (https://creativecommons.org/licenses/; last accessed September 04, 2019) the software is free and open to modifications by other researchers. It is available for download at the project website (http://mago.fe.uni-lj.si; last accessed October 28, 2019). The accompanying preprepared example pipelines and test data set document necessary information about the use of MAGO, enhance reproducibility as the entire pipeline settings can now easily be shared as a single textual pipeline file between researchers, and results reproduced independently (supplementary figs. S1 and S2, Supplementary Material online).
The abilities of MAGO are attested by the quality of the underlying pieces of software (supplementary table S1, Supplementary Material online) and their respective publications. Increasingly complex model data sets spanning CAMI (Sczyrba et al. 2017) and EBI (https://www.ebi.ac.uk/ena/data/view/PRJEB8286; last accessed September 04, 2019) were used in benchmarking MAGO (supplementary table S3, Supplementary Material online; results not shown). The Genome Assembly Gold-standard Evaluations (GAGE) and single-cell amplified genome project (Salzberg et al. 2012; Kogawa et al. 2018) were used for realistic pure culture data analyses (supplementary table S3, Supplementary Material online; supplementary figs. S3–S7, Supplementary Material online). Finally, a number of real case metagenomics data sets (n = 106; s = 0.4 TB; supplementary table S3, Supplementary Material online) were analyzed: 1) the moose rumen microbiome (Svartström et al. 2017; figs. 2 and 3), and 2) longitudinal American pre/term delivery microbiomes (Goltsman et al. 2018; supplementary figs. S4–S9, Supplementary Material online).
Fig. 2.

Overview of the basic quality metrics of MAGs reconstructed from the moose rumen microbiome collection (samples S1–6) (supplementary table S3, Supplementary Material online; Svartström et al. 2017): (A) completeness (>50%); (B) contamination (<10%).
Fig. 3.

Genetic discontinuity observed in the wild moose rumen MAGs shown for the first 5,000 pairwise genome comparisons (supplementary table S3, Supplementary Material online). Values of FastANI estimates in the ANI range of 75–100% are shown. The 95% and 83% ANI thresholds of FastANI estimates serve to delineate comparisons belonging to the same species (>95% intraspecies ANI) or different species (<83% interspecies ANI).
Unless otherwise stated, in analyses of 280 GB data set of the moose rumen microbiome collection (supplementary table S3, Supplementary Material online; Svartström et al. 2017) all parameters used were the default for each subroutine. After initial sequence quality control (FastQC, fastp), each sample was assembled (MEGAHIT) and binned individually (MaxBin, metaBAT, and Concoct), aggregated and dereplicated (Dereplication-Aggregation Scoring Tool). CheckM was used to assess the quality of resulting MAGs (% completeness; % contamination). Single-sample binning produced a total of 3,012 bins. The distribution of the produced MAGs into high- and medium-quality MAGs was based on the criteria defined by the minimum information about a metagenome-assembled genome (MIMAG) standards (Bowers et al. 2017) (high: >90% completeness and <5% contamination, presence of 5S, 16S, and 23S rRNA genes, and at least 18 tRNAs; medium: ≥50% completeness and <10% contamination). Given that few of the MAGs with >90% completeness and <5% contamination in general pass the MIMAG thresholds regarding the presence of rRNA and tRNA genes due to known issues relating to the difficulties in assembly of rRNA regions, the MAGs of high quality are described as “near complete” in general (Bowers et al. 2017). Medium quality bins (n = 670) represented 22.2 ± 3.4% of all bins, whereas 75%, 80% complete bins (10% contamination) (Stewart et al. 2019) next to near complete bins represented 14.7 ± 3.4% (n = 443), 12.9 ± 2.9% (n = 389), and 6.5 ± 1.2% (197) of all recovered MAGs, respectively. In general, MAGO enabled to recover 13 MAGs (80% complete; 10% contamination; dereplicated) per each 10 GB of input sequence data.
The resulting MAGs obtained in this study were first used to explore the existence of genetic discontinuity among the microbial species as observed in large collections of complete genomes from unrelated studies (Jain et al. 2018). The bimodal distribution, with the vast majority (99.8%) of the total genome comparisons showing either > 95% intraspecies ANI or <83% interspecies ANI values, was observed also for the pairwise comparisons of MAGs recovered in this study (fig. 3). It is highly likely that the discontinuity represents a true biological signature, confirming the existence of sequence-discrete populations in natural environments. Although the exact biological mechanisms giving rise to this phenomenon were not explored in this study, the existence of genetic discontinuity in various environments provides opportunity to reconsider its potential origins: 1) decreased recombination frequency below 95% ANI; 2) dispersal limitations in habitats; 3) reduced diversity due to ongoing competition; 4) stochastic events over long periods of time, and provides opportunity to extend analyses from Bacterial and Archaeal domain toward plasmids (Nurk et al. 2017) and viruses (Sutton et al. 2019) for which MAGO can be adopted. In addition, the reconstructed MAGs were compared with a large and heterogeneous collection of characterized prokaryotic genomes (n = 91, 761; Jain et al. 2018). The majority of MAGs recovered in this study exhibited ANI < 83% (i.e., interspecies ANI values) with genomes in the collection. According to the species demarcation cut-off of ∼95% ANI the MAGs recovered from actively fermenting wild moose rumen represent potentially new species amenable for detailed genomic analyses.
MAGO efficiently alleviates the metagenome data analysis bottleneck and provides an important and straightforward-to-implement step toward making the future large-scale evolutionary analyses of MAGs efficient, flexible, scalable and reproducible, enforcing the MIMAG standard. Its outputs are easily integrated into downstream pipelines such as The Genome Taxonomy Database (GTDB) to establish a standardized microbial taxonomy based on genome phylogeny (http://gtdb.ecogenomic.org/; last accessed September 04, 2019). MAGO is open to suggestions for extensions and is amenable for use in both research and teaching of genomics and molecular evolution of genomes assembled from small single-cell projects or large-scale and complex environmental metagenomes.
Supplementary Material
Acknowledgments
B.M. was in part supported through Slovenian Research Agency Program (SRA/ARRS P0-0095). L.D. acknowledges the support of Slovenian Research Agency (SRA/ARRS R51867). This study was in part supported through SRA/ARRS projects J1-6732 (Community level transcriptomic de-novo assembly reveals microbial enzymes that effectively contribute to complex plant polymer degradation) and J1-6741 (Employing the recent advances in metagenomics to explore the karst groundwater microbiome) to B.S.
B.S. was in part supported through visiting professorships awarded by University of Innsbruck, Institute of Microbiology, Innsbruck, Republic of Austria, and CEEPUS Freemover Grant. The ongoing support of Heribert Insam, University of Innsbruck is gratefully acknowledged.
Michael Fink and Hermann Schwaerzler are acknowledged for their support with singularity on HPCC Leo3e, Leo4, and Mach2: The computational results presented have been achieved (in part) using the HPC infrastructure of the University of Innsbruck, and University of Ljubljana. The computational results presented have been achieved (in part) using the MACH2 Interuniversity Shared Memory Supercomputer.
Zala Prevoršek and Nejc Porenta are acknowledged for comments on previous versions of the manuscript.
The COST Actions CA15120 (Open Multiscale Systems Medicine), CA17118 (Identifying Biomarkers Through Translational Research for Prevention and Stratification of Colorectal Cancer), and CA18131 (Statistical and machine learning techniques in human microbiome studies) are acknowledged for discussions during the preparation of the manuscript.
We thank the Editor and Reviewers for their constructive reviews that helped improve the original manuscript.
References
- Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ, Lahti L, Loman NJ, Andersson AF, Quince C.. 2014. Binning metagenomics contigs by coverage and composition. Nat Methods. 11(11):1144–1146. [DOI] [PubMed] [Google Scholar]
- Andrews A. 2010. FastQC: a quality control tool for high throughput sequence data. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/; last accessed September 04, 2019.
- Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol. 35(8):725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J.. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34(17):i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling ACE, Mau BT, Perna NT.. 2010. Progressive mauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5(6):e11147.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritz A, Hofmann P, Majda S, Dahms E, Dröge J, Fiedler J, Lesker TR, Belmann P, DeMaere MZ, Darling AE, et al. 2019. CAMISIM: simulating metagenomes and microbial communities. Microbiome 7(1):17. DOI:10.1186/s40168-019-0633-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garud NR, Good BH, Hallatschek O, Pollard KS.. 2019. Evolutionary dynamics of bacteria in the gut microbiome within and across hosts. PLoS Biol. 17(1):e3000102.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goltsman DSA, Sun CL, Proctor DM, Digiulio DB, Robaczewska A, Thomas BC, Shaw GM, Stevenson DK, Holmes SP, Banfield JF, et al. 2018. Metagenomic analysis with strain-level resolution reveals fine-scale variation in the human pregnancy microbiome. Genome Res. 28(10):1467–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham ED, Heidelberg JF, Tully BJ.. 2017. BinSanity: unsupervised clustering of environmental microbial assemblies using coverage and affinity propagation. PeerJ 5:e3035.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AL, Castelle CJ, Dupont CL, Banfield JF.. 2019. Lateral gene transfer shapes the distribution of RuBisCO among Candidate Phyla Radiation bacteria and DPANN archaea. Mol Biol Evol. 36(3):435–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S.. 2018. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 9(1):5114.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DD, Froula J, Egan R, Wang Z.. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from microbial communities. PeerJ 3:e1165.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Li M, Knyaz C, Tamura K.. 2018. MEGA X: molecular Evolutionary Genetics Analysis across Computing Platforms. Mol Biol Evol. 35(6):1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogawa M, Hosokawa M, Nishikawa Y, Mori K, Takeyama H.. 2018. Obtaining high-quality draft genomes from uncultured microbes by cleaning and co-assembly of single-cell amplified genomes. Sci Rep. 8(1):2059.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Liu CM, Luo R, Sadakane K, Lam TW.. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31(10):1674–1676. [DOI] [PubMed] [Google Scholar]
- Lin M, Kussell E.. 2019. Inferring bacterial recombination rates from large-scale sequencing datasets. Nat Methods. 16(2):199–204. [DOI] [PubMed] [Google Scholar]
- Meyer F, Hofmann P, Belmann P, Garrido-Oter R, Fritz A, Sczyrba A, McHardy AC.. 2018. AMBER: assessment of Metagenome BinnERs. Giga Sci. 7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S, Meleshko D, Korobeynikov A, Pevzner PA.. 2017. metaSPAdes: a new versatile metagenomics assembler. Genome Res. 27(5):824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MT, Fookes M, Falush D, Keane JA, Parkhill J.. 2015. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31(22):3691–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW.. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25(7):1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks DH, Rinke C, Chuvochina M, Chaumeil PA, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson W.. 2017. Recovery of nearly 8, 000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol. 2(11):1533–1542. [DOI] [PubMed] [Google Scholar]
- Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil PA, Hugenholtz P.. 2018. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol. 36(10):996–1004. [DOI] [PubMed] [Google Scholar]
- Peng Y, Leung HCM, Yiu SM, Chin F.. 2012. IDBA-UD: a de novo assembler for single-cell and metagenomics sequencing data with highly uneven depth. Bioinformatics 28(11):1420–1428. [DOI] [PubMed] [Google Scholar]
- Salzberg SL, Phillippy AM, Zimin A, Puiu D, Magoc T, Koren S, Treangen TJ, Schatz MC, Delcher AL, Roberts M, et al. 2012. GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res. 22(3):557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 75(23):7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sczyrba A, Hofmann P, Belmann P, Koslicki D, Janssen S, Dröge J, Gregor I, Majda S, Fiedler J, Dahms E, et al. 2017. Critical Assessment of Metagenome Interpretation – a benchmark of metagenomics software. Nat Methods. 14(11):1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069.24642063 [Google Scholar]
- Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF.. 2018. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol. 3(7):836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RD, Auffret MD, Snelling TJ, Roehe R, Watson M.. 2019. MAGpy: a reproducible pipeline for the downstream analysis of metagenome-assembled genomes (MAGs). Bioinformatics 35(12):2150–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RD, Auffret MD, Warr A, Walker AW, Roehe R, Watson M.. 2019. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol. 37(8):953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton TDS, Clooney AG, Ryan FJ, Ross RP, Hill C. 2019. Choice of assembly software has a critical impact on virome characterisation. Microbiome 7:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svartström O, Alneberg J, Terrapon N, Lombard V, de Bruijn I, Malmsten J, Dalin A-M, Muller EEL, Shah P, Wilmes P, et al. 2017. Ninety-nine de novo assembled genomes from the moose (Alces alces) rumen microbiome provide new insights into microbial plant biomass degradation. ISME J. 11(11):2538–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YW, Simmons BA, Singer S.. 2016. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomics datasets. Bioinformatics 32(4):605–607. [DOI] [PubMed] [Google Scholar]
- Wu YW. 2018. ezTree: an automated pipeline for identifying phylogenetic marker genes and inferring evolutionary relationships among uncultivated prokaryotic draft genomes. BMC Genomics 19(S1):921.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.