

Abstract
Targeted oncology therapies have revolutionized cancer treatment over the last decade and have resulted in improved prognosis for many patients. This advance has emanated from elucidation of pathways responsible for tumorigenesis followed by targeting of these pathways by specific molecules. Cardiovascular care has become an increasingly critical aspect of patient care in part because patients live longer, but moreover due to potential associated toxicities from these therapies. Because of the targeted nature of cancer therapies, cardiac and vascular side effects may additionally provide insights into the basic biology of vascular disease. We herein provide the example of tyrosine kinase inhibitors (TKI) utilized in chronic myelogenous leukemia (CML) to illustrate this medical transformation. We describe the vascular considerations for the clinical care of CML patients as well as the emerging literature on mechanisms of toxicities of the individual TKI. We additionally postulate that basic insights into toxicities of novel cancer therapies may serve as a new platform for investigation in vascular biology and a new translational research opportunity in vascular medicine.
Keywords: cardiovascular toxicity, cardio-oncology, peripheral arterial disease, tyrosine kinase inhibitors, chronic myelogenous leukemia
Graphical Abstract
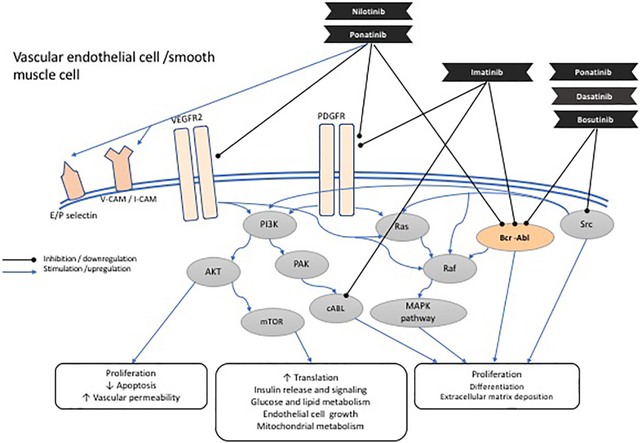
Kinase Inhibitors Used in Chronic Myelogenous Leukemia: Overview
Overactivation of kinases can lead to a number of pathological processes including cancer. In chronic myelogenous leukemia (CML), one such kinase (Abelson 1 kinase (ABL1)) becomes constitutively active as a result of a chromosomal translocation, juxtaposing the breakpoint cluster region (BCR) gene with the ABL kinase gene and genesis of the deregulated BCR-ABL1 kinase. Inhibitors of ABL1 kinase have resulted in remarkable efficacy in treating CML. In 2019, cardiovascular care has emerged as a crucial element of CML patient care for several reasons. First, CML patients are now expected to have near normal life expectancy and may succumb to other non-cancer morbidities and mortality. Second, numerous kinase inhibitors (KI) used for the treatment of CML are associated with vascular disease. Increasingly, cardiologists and vascular medicine physicians who specialize in oncology care (“cardio-oncologists”) play a critical role in CML care. In this review, we will discuss vascular aspects of care for CML patients. We will also discuss the emerging data on the mechanisms of vascular sequelae from CML TKI therapy and the implications for vascular medicine and vascular biology.
Kinase and Kinase Inhibitors (KI)
Kinases are enzymes that transfer phosphate groups from adenosine triphosphate (ATP) to specific protein or lipid substrates, leading to fundamental regulation of cell signaling. Over 500 kinases have been discovered, which are divided structurally into two groups: serine/threonine kinases and tyrosine/tyrosine-like kinases.1 KI inhibit either the ATP binding pocket (where the ATP binds) or the allosteric pocket (where the phosphorylated protein binds) and thus prevent kinase activity itself. (figure 1) While most KI are reversible, some KI (e.g., ibrutinib) bind to the proximal binding pocket with a reactive nucleophilic cysteine covalently and thus irreversibly. However, most KI are steady state competitive enzyme inhibitors with respect to ATP and either directly or indirectly interact with the ATP pocket.2
Figure 1 –

Mechanism of action of BCR-ABL tyrosine kinase in CML (Left). Mechanism of action of TKI in treatment of CML.(Right)
ATP: Adenosine triphosphate; ADP: Adenosine diphosphate; P: phosphate; CML: Chronic myelogenous leukemia; TKI: Tyrosine kinase inhibitor.
Because of the high similarity of the ATP binding sites across the entire kinome, small molecular KI can bind more than one kinase receptor, thus decreasing selectivity. For this reason, many KI occupy both the space where the ATP adenine group binds but also adjacent regions (including occasionally the adjacent allosteric pocket).3 Nevertheless, even with this approach the selectivity of approved KI is not absolute. Imatinib, the first approved KI for CML, inhibits not only the ABL1 tyrosine kinase relevant to CML, but also the tyrosine kinase receptor c-KIT, aberrantly activated in gastrointestinal stromal tumors (GIST), resulting in another drug indication for imatinib.4, 5
Kinases also play a critical role in cardiac, vascular and metabolic homeostasis; thus, altered activity of kinases in the cardiovascular systems can lead to pathology and various effects on the vasculature. For example, the kinase target of a specific TKI may have an oncogenic role if altered in a cancer cell but also a critical role, unaltered, in a myocyte or an endothelial cell. Inhibition of that kinase - as an unintended consequence – can lead to vascular disease. Such “on-target” toxicity is in contrast to “off-target” toxicity where the promiscuous nature of the TKI can lead to inhibition of structurally similar or related kinase receptor(s) which can cause disease. In addition, the effects of TKI on the cardiovascular system may not be entirely pathologic and may actually be beneficial. We will review these concepts in the case of CML and TKI used to treat CML below.
Tyrosine Kinase Inhibitors (TKI) – Inhibiting ABL1 kinase for the treatment of Chronic Myelogenous Leukemia (CML)
Five small molecule TKI have been approved to date for the treatment of CML.6 Imatinib was initially developed as a platelet-derived growth factor receptor (PDGFR) inhibitor, but was also found to inhibit other tyrosine kinases, such as ABL1 and c-KIT (the stem cell factor receptor).6 The International Randomized Interferon Versus STI571 (IRIS) trial demonstrated the unprecedented efficacy profile of imatinib in newly diagnosed CML. Remarkably, the longer term overall survival of patients treated with imatinib was close to 90%, establishing imatinib as a benchmark treatment for CML. In 2001, imatinib became the first small-molecule TKI approved by the FDA and has revolutionized the treatment of CML and other leukemias where the ABL1 tyrosine kinase is constitutively active.2
Since this initial historic approval, a number of later generation TKI were tested and approved for the treatment of CML. These included dasatinib, nilotinib, bosutinib and ponatinib. The impetus for introducing new TKI in this space was driven by several factors. First, although imatinib dramatically altered the natural history of CML, more than 20% of patients with CML were either unable to tolerate or developed resistance to imatinib. Second, the newer TKI were shown to be more potent inhibitors of ABL1 tyrosine kinase and could offer both salvage and augmented primary response. In head-to-head clinical trials with imatinib, dasatinib and nilotinib resulted in faster and greater proportion of molecular response as well as protection from early progression. Although this still has not resulted in better overall survival from trial data, both drugs were approved for front-line therapy. In 2017, bosutinib obtained FDA similar approval for frontline treatment of CML.
Despite the success of TKI therapy in CML, specific kinase domain mutations emerged in the leukemic clones driving resistance to TKI therapy. The most impactful and notorious is the example of the T315I mutation where threonine substitution for isoleucine at a key contact point severely affected TKI inhibition and was the major determinant of ABL1 tyrosine kinase activation within the leukemic clone.7 Ponatinib was developed as a multi-targeted KI with potent activity against the T315I mutation as well as the spectrum of other ABL1 kinase mutations. In a phase 2 trial (called the PACE study), ponatinib showed considerable response in patients who had failed other TKIs, leading to drug approval via the FDA accelerated approval program in 2012.8 However, one year later, ponatinib prescribing authorization was transiently suspended in the United States by the Food and Drug Administration due to emerging recognition of cardiovascular risks with the drug and need for updated label warnings. Not surprisingly the vascular/cardiovascular toxicities noted with ponatinib heralded closer assessment of cardiac, vascular and metabolic issues with all other TKI used for CML treatment.
Cardiovascular Risks Associated with TKI Used in CML – Clinical Studies
Despite considerable efficacy in patients who had failed other TKI, the early phase 2 trial with ponatinib showed considerable vascular toxicity. At a median follow-up of 12 months in the PACE trial, 6% of patients had coronary events, 3% had cerebrovascular events, and 4% had peripheral vascular events. At 28 months, cumulative events were 10%, 7%, and 7%, respectively. Indeed, retrospective analysis of the PACE database suggests a higher risk in patients with CV risk factors or CV disease, as well as a signal for CV events occurring in a ponatinib dose–dependent manner.9 In addition, at least a quarter of patients developed hypertension after initiating treatment. A cardiovascular signal for toxicity was also seen in a trial involving ponatinib as first-line therapy which led to trial suspension. The aggregate data strongly suggested that ponatinib is associated with higher risk of cardiovascular adverse events.
The cardiovascular risk profile of ponatinib brought into focus vascular and metabolic effects of other TKI used in CML. Certain cardiac signals become apparent early in drug approval for each therapy. For example, a small percentage of patients treated with both dasatinib and nilotinib were noted to have QT prolongation, although no clear risk of ventricular arrhythmias was identified. For this reason, QT assessment via an electrocardiogram was recommended for nilotinib. In the case of dasatinib, dyspnea observed in a fraction of patients led the US Food and Drug Administration (FDA) to issue a warning and recommend that patients be evaluated for signs and symptoms of cardiopulmonary disease before and during dasatinib treatment. The cause of dyspnea appears to be multi-factorial. In initial studies with dasatinib, a significant fraction of patients had pleural effusions. In addition to pleural effusion, dasatinib is also associated with pericardial effusion. The pathogenesis is unclear but is felt to be a “capillary leak” like syndrome.10 More concerning, reports of pulmonary hypertension were subsequently and additionally noted with dasatinib. In 2012, the French Pulmonary Hypertension Registry reported nine severe cases of dasatinib-associated pulmonary hypertension. Upon diagnosis, patients had moderate to severe precapillary pulmonary hypertension, with severe symptoms and hemodynamic compromise with two patients dying in follow-up.11 The incidence of dasatinib-associated pulmonary hypertension is felt to be at least 3% based on data from randomized trials. However, none of these trials systematically screened for pulmonary hypertension.
Vascular events – including cardiac, cerebral and peripheral events – have emerged as the biggest cardiovascular safety concern in CML patients. Before ponatinib-associated vascular complications became obvious late in 2013, initial case reports or case series of vascular events had been reported with nilotinib. A multicenter analysis of 179 patients revealed 11 patients (6.2%) who developed severe peripheral artery disease involving lower limbs. Eight patients required invasive therapy and four patients required amputation. More comprehensive appreciation of vascular events in CML patients was derived from clinical trials where newer TKI were compared to imatinib for efficacy. Although these trials were not designed to assess cardiovascular safety, an increased risk of vascular events was observed with newer TKI compared to imatinib. A 3-year follow-up of the ENESTnd trial (where nilotinib was compared to imatinib in front-line therapy) suggested a higher incidence of vascular events in patients treated with nilotinib compared with imatinib.12 These data were more striking at 5-year follow-up; twenty-eight out of 279 (10%) patients treated with nilotinib at 300 mg twice per day, 44 out 277 patients (15.9%) treated with nilotinib at 400 mg twice per day, and seven (2.5%) of 280 patients treated with imatinib 400 once per day had cardiovascular events.13 In these analyses cardiovascular events were defined as ischemic heart disease, ischemic cerebrovascular disease, and peripheral artery disease. These data suggest that nilotinib associated toxicity occurs in all arterial beds; fewer venous events were noted. 5 year follow-up report of the DASISION trial (where dasatinib was compared to imatinib in the front-line setting) noted a 5% risk of arterial ischemic events in patients on dasatinib compared with 2% risk for imatinib.14
In contrast, long term studies of patients treated with imatinib have general shown net neutral or possibly favorable effects on vascular disease risk. Arguably the best data come from head-to-head prospective studies where the efficacy of imatinib is compared to other TKI where cardiovascular event rates are lower in the imatinib arms of the studies. What remains unknown is baseline cardiovascular risk profile of CML patients irrespective of therapy or as they commence therapy. A current ongoing prospective study () is assessing the baseline cardiovascular risk factors and follow-up cardiovascular risks of a “real world” population of CML patients on chronic TKI therapy.
Cardiovascular Toxicities of TKI: Mechanistic Studies
The cardiovascular events observed in CML patients treated with TKI have led to mechanistic studies interrogating the vascular effects of each TKI, with some insight gained to date. Additional unanswered questions include the impact the CML diagnosis itself poses on risk of vascular events as well as the potential reversibility of any pathology stemming from either CML or TKI therapy as CML treatment paradigms shift towards a focus on finite therapy and treatment free remission (cessation of therapy in deep remission).
Nilotinib was found to upregulate pro-atherogenic adhesion-proteins on human endothelial cells, including intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion protein 1 (VCAM1), and E-selectin, which collectively can recruit inflammatory cells and platelets and promote vascular events. Increased expression of adhesion molecules is mediated through reduction in the level of miR-3121–3p, which additionally leads to upregulation IL-1β. It is proposed that the miR-3121–3p/IL-1β axis could be a potential target to prevent vascular events in CML patients determined to have high risk.15 Whole blood samples from nilotinib-treated CML patients demonstrated increased platelet adhesion. These patients also showed increased expression of markers of endothelial and platelet activation, including plasma soluble P- and E-selectin, sICAM-1, sVCAM-1, TNF-alpha, IL-6 levels and endogenous thrombin potential (ETP) levels in vivo, despite being on daily low-dose aspirin.16 Treatment of atherogenic (ApoE−/−) mice with nilotinib increases atherosclerotic build-up and blocked reperfusion and angiogenesis in a hind-limb-ischemia model of arterial occlusion.16 In mouse model studies, nilotinib remarkably enhanced thrombus growth and stability in damaged mesenteric arterioles and the carotid artery.17
Further interrogation of effects of nilotinib on other atherosclerosis risk factors have shown mixed results. A prospective clinical study showed that nilotinib doubles the levels of low-density lipoprotein cholesterol (“bad cholesterol”) but also raises high-density lipoprotein cholesterol levels.18 In atherosclerosis (APOE*3Leiden.CETP mouse) prone mice, nilotinib had no significant effect on cholesterol levels.19 On the other hand, early clinical trials with nilotinib showed hyperglycemia and development of ‘pre-diabetes’ or worsening of glycemic control in a subset of patients, an obvious risk factor for atherosclerosis.20, 21 From patient studies, there is no consensus with respect to the mechanisms of vascular toxicity associated with nilotinib. However, deep phenotyping in specific patients with vascular studies can be informative.22,23 Vessel wall magnetic resonance imaging in a case of nilotinib-associated cerebral stroke revealed diffuse concentric thickening of the intracranial artery walls, which is inconsistent with ordinary atherosclerosis (which often manifests with eccentric thickening).23 In addition, a study involving 159 patients on imatinib or nilotinib showed a higher incidence of abnormal ankle-brachial index (ABI) in patients treated with nilotinib. Abnormal ABI in patients treated with first- and second-line nilotinib was 26% and 35.7%, respectively, compared with 6.3% for first-line imatinib.24 These studies are informative because an abnormal ABI is a sensitive and specific test for not only peripheral artery disease but also systemic polyvascular atherosclerosis.25
The mechanisms of vascular toxicities associated with ponatinib may be different than nilotinib. Because of the promiscuity of ponatinib, multiple kinases (and thus pathways) in the vasculature can be affected. For example, unlike nilotinib, ponatinib is a potent inhibitor of VEGF receptors (e.g., kdr), leading to increased blood pressure, like other TKI with potent anti-VEGF properties.26, 5 Ponatinib has significant detrimental effects on cultured endothelial cells in vitro, both inhibiting proliferation and inducing apoptosis. Ponatinib also has direct pro-thrombotic effects increasing platelet activation and adhesion. A recent series of elegant experiments suggest that ponatinib vascular toxicity involves platelet adhesion followed by secondary microvascular angiopathy.27 Ultrasound molecular imaging demonstrates five to six-fold increased signal for glycoprotein-Ibα-mediated and von Willebrand factor (vWF)-mediated endothelial cell and platelet adhesion in ponatinib-treated mice compared to vehicle-treated controls. These effects were accentuated in atherosclerosis-prone (ApoE−/−) mice and were present in both large arteries as well as the microcirculation, the latter producing ventricular dysfunction.27 Given this elegant study, It is most likely that any degree of ventricular dysfunction seen with ponatinib is due to an indirect effect, rather than a direct effect on cardiomyocytes, as has been demonstrated in other models.28
Separate studies have been performed to interrogate the mechanisms of pulmonary hypertension associated with dasatinib. Treatment of rats with dasatinib led to a dose dependent endothelial cell apoptosis and dysfunction via increased mitochondrial reactive oxygen species (ROS) production. Over time dasatinib attenuated pulmonary vasoconstriction following hypoxia exposure and increased susceptibility to experimental pulmonary hypertension.29 In a separate study, a Rho-kinase inhibitor blunted the change in pulmonary pressures observed after dasatinib treatment, providing a potential for developing treatments.30 In both studies, the pulmonary effects were specific to dasatinib since they were not observed with other TKI used in CML treatment and consistent with clinical data.
Implications for Patient Care: Preventive and Treatment Strategies
In 2019, cardiovascular health has emerged as an important consideration in all CML patients irrespective of CML therapies. All CML patients should undergo general cardiovascular risk assessment and examination before starting of TKI therapy. Given the disparate effects of TKI on vascular disease, cardiovascular and metabolic factors (such as hypertension, hyperlipidemia and diabetes) should be assessed at various times following start of therapy (Figure 2). ABI is a sensitive and specific test for systemic atherosclerosis and offers an informative noninvasive diagnostic for patients staring specific TKI. A crucial point to clarify is whether baseline cardiovascular risk profile should definitively influence or dictate the choice of TKI selected for treatment. Patients considered high risk based on comorbidities or TKI selection may benefit from more frequent monitoring and from involvement of a cardiologist, vascular medicine specialist, or a cardio-oncologist to optimize primary and secondary prevention. This is particularly relevant for patients on ponatinib treatment. A simple ‘ABCDE’ algorithm is an established means to reduce cardiovascular events in the general population and maybe particularly relevant to the CML population (figure 3).31
Figure 2 –

Clinical recommendations for assessment of cardiovascular toxicity in individuals with chronic myelogenous leukemia receiving kinase inhibitors.
✔ Recommended; ✕ as clinically indicated;  Assess the QT prolongation.
Assess the QT prolongation.
 If there are any signs in favor of pulmonary arterial hypertension like dyspnea, consider echocardiogram as first line of screening.
If there are any signs in favor of pulmonary arterial hypertension like dyspnea, consider echocardiogram as first line of screening.
Figure 3 -.

“ABCDE” approach, which has been proposed to reduce CV disease in patients with CML receiving TKI treatment.
CML, chronic myeloid leukemia; CV, cardiovascular; TKI, tyrosine kinase inhibitor; ABI, ankle brachial index; BP, blood pressure; HTN, hypertension; ACEI, angiotensin-converting-enzyme inhibitors; ARB, angiotensin receptor blocker; BBs, beta blockers; dp-CCB, dihydropyridine calcium channel blockers; ndp-CCB, non-dihydropyridine calcium channel blockers.
Future Directions
In the future, better defining the various cardiovascular and metabolic effects of TKI will further advance the care and outcome of CML patients already dramatically improved by availability of TKI. Importantly, prospective, systematic cardiovascular toxicity assessment will be important in any future clinical trials in CML. Additionally, incorporation of adjudication of adverse events by independent experts (cardio-oncologists) could provide more reliable and clear safety data. Further study of real-world populations will allow better understanding of underlying cardiovascular risk factors and cardiovascular disease as well as the effects of CML itself and CML therapy. Finally, better delineation of mechanisms of underlying toxicities will be critical in terms of personalized preventive and treatment strategies for patients, knowing that the clear mechanisms by which TKI induce cardiovascular toxicity are still unknown and need to be subject to further investigation. These future directions are important because even today newer TKI are being introduced for CML therapy. For example, ABL001 (asciminib) is an allosteric inhibitor of BCR-ABL is currently being tested as a more effective therapy in CML. Indeed, the cardiovascular health of CML patients may be a more imperative topic in the future.32 Thus, it is strongly recommended to consider cardiovascular screening and follow up in the future clinical trials on new TKIs including ABI measurement, serial ECGs, baseline and periodic echocardiography.
Highlights:
The vascular considerations for the clinical care of CML patients have been addressed by mechanisms of toxicities of each TKI.
By defining the various cardiovascular and metabolic effects of TKI, the care and outcome of CML patients will be dramatically improved.
Basic insights into toxicities of novel cancer therapies may serve as a new platform for investigation in vascular biology and a new translational research opportunity in vascular medicine.
a). Acknowledgments
We thank Dr. Guido Zaman for providing the IC50 values for table 1.
Table 1-.
FDA approved tyrosine kinase inhibitors for treatment of CML by date, their molecular target, and their effect on the vasculature. (The IC50 values extracted from the website https://www.selleckchem.com/ and the article by Dr. Guido Zaman group.4)
| TKI | Year of approval | Target (IC50 in nM when available) | Vascular effect |
|---|---|---|---|
| Imatinib | 2001 | PDGFR α (0.1), KIT (0.1), ABL1(0.6) | Positive effect on blood glucose and lipid profile. |
| Bosutinib | 2014 | ABL1 (4.4), FGFR2, VEGFR2, PDGFRβ, SRC (1.2) | Hypertension |
| Dasatinib | 2006 | ABL1(0.27), KIT (79.0), PDGFR α, PDGFR β, SRC (0.8) | Platelet dysfunction, Pulmonary Hypertension |
| Nilotinib | 2007 | ABL1(18.5), KIT, PDGFR α | PAD, IHD/CVA, Hyperglycemia, Dyslipidemia |
| Ponatinib | 2012 | ABL1 (3.7), FGFR 1(2.2), FGFR 2 & 3, VEGFR 1& 2 & 3, PDGFR α (1.1), PDGFR β, KIT, SRC (5.4), TIE2 | PAD, Hypertension, IHD/CVA, VTE, Hyperglycemia |
b) Sources of Funding
JM is supported by NIH Grant R56HL141166 and R01HL141166. AWA receives funding from NIH K12 HL133117. AM is supported by a T32 training grant from National Institute of Health.
Abbreviations and Acronyms:
- KI
kinase inhibitors
- TKI
tyrosine kinase inhibitors
- CML
chronic myelogenous leukemia
- ABL1
Abelson 1 kinase
- BCR
breakpoint cluster region
- ATP
adenosine triphosphate
- GIST
gastrointestinal stromal tumors
- PDGFR
platelet-derived growth factor receptor
- IRIS
International Randomized Interferon Versus STI571 trial
- FDA
US Food and Drug Administration
- ICAM1
intercellular adhesion molecule 1
- VCAM1
vascular cell adhesion protein 1
- ETP
endogenous thrombin potential
- ABI
ankle-brachial index
- vWF
von Willebrand factor
- ROS
reactive oxygen species
Footnotes
Disclosure:
JM has served as a consultant/advisor for Novartis, Pfizer, Bristol-Myers Squibb, Takeda/Millennium, Ariad, Acceleron, Vertex, Incyte, Rgenix, Verastem, Pharmacyclics, StemCentRx, Heat Biologics, Daiichi-Sankyo, and Regeneron. MM has served as a consultant/advisor for Novartis, Pfizer, Bristol-Myers Squibb, Takeda/Millennium, Ariad, and receives institutional research support from Bristol Myers Squibb, Novartis, and Sun Pharma/SPARC. The other authors report no conflicts.
References:
- 1.Grimminger F, Schermuly RT, Ghofrani HA. Targeting non-malignant disorders with tyrosine kinase inhibitors. Nat Rev Drug Discov. 2010;9(12):956–970. doi: 10.1038/nrd3297. [DOI] [PubMed] [Google Scholar]
- 2.Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36(7):422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Wu P, Nielsen TE, Clausen MH. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov Today. 2016;21(1):5–10. doi: 10.1016/j.drudis.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 4.Uitdehaag JCM, De Roos JADM, Van Doornmalen AM, Prinsen MBW, De Man J, Tanizawa Y, Kawase Y, Yoshino K, Buijsman RC, Zaman GJR. Comparison of the cancer gene targeting and biochemical selectivities of all targeted kinase inhibitors approved for clinical use. PLoS One. 2014;9(3):1–13. doi: 10.1371/journal.pone.0092146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moslehi JJ, Deininger M. Tyrosine kinase inhibitor-associated cardiovascular toxicity in chronic myeloid leukemia. J Clin Oncol. 2015;33(35):4210–4218. doi: 10.1200/JCO.2015.62.4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roskoski R. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol Res. 2015;100:1–23. doi: 10.1016/j.phrs.2015.07.010. [DOI] [PubMed] [Google Scholar]
- 7.Lamontanara AJ, Gencer EB, Kuzyk O, Hantschel O. Mechanisms of resistance to BCR-ABL and other kinase inhibitors. Biochim Biophys Acta - Proteins Proteomics. 2013;1834(7):1449–1459. doi: 10.1016/j.bbapap.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 8.Mauro MJ, Flinn I, Ph D, Hare TO, Ph D, Hu S, Ph D, Narasimhan NI, Ph D, Rivera VM, Ph D, Clackson T, Ph D, Turner CD, Haluska FG, Ph D, Druker BJ, Deininger MWN, Ph D, Talpaz M. Ponatinib in Refractory Philadelphia Chromosome–Positive Leukemias. 2012. doi: 10.1056/NEJMoa1205127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cortes JE, Kim D, Pinilla-ibarz J, Coutre PD, Paquette R, Chuah C, Nicolini FE, Apperley JF, Khoury HJ, Talpaz M, Deangelo DJ, Abruzzese E, Rea D, Baccarani M. Ponatinib efficacy and safety in Philadelphia chromosome – positive leukemia : fi nal 5-year results of the phase 2 PACE trial. 2019;132(4):393–405. doi: 10.1182/blood-2016-09-739086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weatherald J, Chaumais MC, Montani D. Pulmonary arterial hypertension induced by tyrosine kinase inhibitors. Curr Opin Pulm Med. 2017;23(5):392–397. doi: 10.1097/MCP.0000000000000412. [DOI] [PubMed] [Google Scholar]
- 11.Montani D, Bergot E, Günther S, Savale L, Bergeron A, Bourdin A, Bouvaist H, Canuet M, Pison C, MacRo M, Poubeau P, Girerd B, Natali D, Guignabert C, Perros F, O’Callaghan DS, Jaïs X, Tubert-Bitter P, Zalcman G, Sitbon O, Simonneau G, Humbert M. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–2137. doi: 10.1161/CIRCULATIONAHA.111.079921. [DOI] [PubMed] [Google Scholar]
- 12.Larson RA, Hochhaus A, Hughes TP, Clark RE, Etienne G, Kim DW, Flinn IW, Kurokawa M, Moiraghi B, Yu R, Blakesley RE, Gallagher NJ, Saglio G, Kantarjian HM. Nilotinib vs imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia. 2012;26(10):2197–2203. doi: 10.1038/leu.2012.134. [DOI] [PubMed] [Google Scholar]
- 13.Hochhaus A, Saglio G, Hughes TP, Larson RA, Kim DW, Issaragrisil S, Le Coutre PD, Etienne G, Dorlhiac-Llacer PE, Clark RE, Flinn IW, Nakamae H, Donohue B, Deng W, Dalal D, Menssen HD, Kantarjian HM. Long-term benefits and risks of frontline nilotinib vs imatinib for chronic myeloid leukemia in chronic phase: 5-year update of the randomized ENESTnd trial. Leukemia. 2016;30(5):1044–1054. doi: 10.1038/leu.2016.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cortes JE, Saglio G, Kantarjian HM, Baccarani M, Mayer J, Boqué C, Shah NP, Chuah C, Casanova L, Bradley-Garelik B, Manos G, Hochhaus A. Final 5-year study results of DASISION: The dasatinib versus imatinib study in treatment-Naïve chronic myeloid leukemia patients trial. J Clin Oncol. 2016;34(20):2333–2340. doi: 10.1200/JCO.2015.64.8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sukegawa M, Wang X, Nishioka C, Pan B, Xu K, Ohkawara H, Hamasaki Y, Mita M, Nakamura K, Okamoto M, Shimura H, Ohta M, Ikezoe T. The BCR/ABL tyrosine kinase inhibitor, nilotinib, stimulates expression of IL-1β in vascular endothelium in association with downregulation of miR-3p. Leuk Res. 2017;58(May):83–90. doi: 10.1016/j.leukres.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 16.Hadzijusufovic E, Albrecht-Schgoer K, Huber K, Hoermann G, Grebien F, Eisenwort G, Schgoer W, Herndlhofer S, Kaun C, Theurl M, Sperr WR, Rix U, Sadovnik I, Jilma B, Schernthaner GH, Wojta J, Wolf D, Superti-Furga G, Kirchmair R, Valent P. Nilotinib-induced vasculopathy: Identification of vascular endothelial cells as a primary target site. Leukemia. 2017;31(11):2388–2397. doi: 10.1038/leu.2017.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alhawiti N, Burbury KL, Kwa FA, O’Malley CJ, Shuttleworth P, Grigg AP, Jackson DE. The tyrosine kinase inhibitor, nilotinib potentiates a prothrombotic state. Thromb Res. 2016;145:54–64. doi: 10.1016/j.thromres.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 18.Rea D, Mirault T, Cluzeau T, Gautier JF, Guilhot F, Dombret H, Messas E. Early onset hypercholesterolemia induced by the 2nd-generation tyrosine kinase inhibitor nilotinib in patients with chronic phase-chronic myeloid leukemia. Haematologica. 2014;99(7):1197–1203. doi: 10.3324/haematol.2014.104075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pouwer MG, Pieterman EJ, Verschuren L, Caspers MPM, Kluft C, Garcia RA, Aman J, Jukema JW, Princen HMG. The BCR-ABL1 Inhibitors Imatinib and Ponatinib Decrease Plasma Cholesterol and Atherosclerosis, and Nilotinib and Ponatinib Activate Coagulation in a Translational Mouse Model. Front Cardiovasc Med. 2018;5(June):1–13. doi: 10.3389/fcvm.2018.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ito Y, Miyamoto T, Chong Y, Maki T, Akashi K, Kamimura T. Nilotinib exacerbates diabetes mellitus by decreasing secretion of endogenous insulin. Int J Hematol. 2013;97(1):135–138. doi: 10.1007/s12185-012-1222-7. [DOI] [PubMed] [Google Scholar]
- 21.Beckman JA, Creager MA. Vascular complications of diabetes. Circ Res. 2016;118(11):1771–1785. doi: 10.1161/CIRCRESAHA.115.306884. [DOI] [PubMed] [Google Scholar]
- 22.Quintás-Cardama A, Kantarjian H, Cortes J. Nilotinib-associated vascular events. Clin Lymphoma, Myeloma Leuk. 2012;12(5):337–340. doi: 10.1016/j.clml.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki K, Yamamoto J, Kakeda S, Takamatsu S, Miyaoka R, Kitagawa T, Saito T, Nakano Y, Nishizawa S. Vessel wall magnetic resonance imaging findings and surgical treatment in nilotinib-associated cerebrovascular disease: A case report. Mol Clin Oncol. 2018:239–243. doi: 10.3892/mco.2018.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim TD, Rea D, Schwarz M, Grille P, Nicolini FE, Rosti G, Levato L, Giles FJ, Dombret H, Mirault T, Labussière H, Lindhorst R, Haverkamp W, Buschmann I, Dörken B, Le Coutre PD. Peripheral artery occlusive disease in chronic phase chronic myeloid leukemia patients treated with nilotinib or imatinib. Leukemia. 2013;27(6):1316–1321. doi: 10.1038/leu.2013.70. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch AT, Duval S. The global pandemic of peripheral artery disease. Lancet. 2013;382(9901):1312–1314. doi: 10.1016/s0140-6736(13)61576-7. [DOI] [PubMed] [Google Scholar]
- 26.Pandey AK, Singhi EK, Arroyo JP, Ikizler TA, Gould ER, Brown J, Beckman JA, Harrison DG, Moslehi J. Mechanisms of VEGF (Vascular Endothelial Growth Factor) Inhibitor–Associated Hypertension and Vascular Disease. Hypertension. 2017:HYPERTENSIONAHA.117.10271. doi: 10.1161/HYPERTENSIONAHA.117.10271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Latifi Y, Moccetti F, Wu M, Xie A, Packwood W, Qi Y, Ozawa K, Shentu W, Brown E, Shirai T, McCarty OJ, Ruggeri Z, Moslehi J, Chen J, Druker BJ, López JA, Lindner JR. Thrombotic microangiopathy as a cause of cardiovascular toxicity from the BCR-ABL1 tyrosine kinase inhibitor ponatinib. Blood. 2019;133(14):1597–1606. doi: 10.1182/blood-2018-10-881557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jacob F, Yonis AY, Cuello F, Luther P, Schulze T, Eder A, Streichert T, Mannhardt I, Hirt MN, Schaaf S, Stenzig J, Force T, Eschenhagen T, Hansen A. Analysis of tyrosine kinase inhibitor-mediated decline in contractile force in rat engineered heart tissue. PLoS One. 2016;11(2):1–18. doi: 10.1371/journal.pone.0145937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guignabert C, Phan C, Seferian A, Huertas A, Tu L, Thuillet R, Sattler C, Le Hiress M, Tamura Y, Jutant EM, Chaumais MC, Bouchet S, Manéglier B, Molimard M, Rousselot P, Sitbon O, Simonneau G, Montani DHM. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016;126(9):3207–3218. doi: 10.1172/jci86249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fazakas C, Nagaraj C, Zabini D, Végh AG, Marsh LM, Wilhelm I, Krizbai IA, Olschewski H, Olschewski A, Bálint Z. Rho-kinase inhibition ameliorates dasatinib-induced endothelial dysfunction and pulmonary hypertension. Front Physiol. 2018;9(MAY):1–14. doi: 10.3389/fphys.2018.00537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moslehi JJ. Cardiovascular Toxic Effects of Targeted Cancer Therapies. N Engl J Med. 2016;375(15):1457–1467. doi: 10.1056/NEJMra1100265. [DOI] [PubMed] [Google Scholar]
- 32.Wylie AA, Schoepfer J, Jahnke W, Sandra W, Loo A, Furet P, Marzinzik AL, Pelle X, Donovan J, Zhu W, Buonamici S, Hassan AQ, Lombardo F, Iyer V, Palmer M, Berellini G, Dodd S, Thohan S, Bitter H, Petruzzelli L, Vanasse KG, Warmuth M, Branford S, Ross DM, Timothy P, Hofmann F, Keen NJ, Sellers WR. The allosteric inhibitor ABL001 enables dual targeting of BCR–ABL1. Nat Publ Gr. 2017;543(7647):733–737. doi: 10.1038/nature21702. [DOI] [PubMed] [Google Scholar]