

Abstract
Various antioxidants are being used to neutralize the harmful effects of reactive oxygen species (ROS) overproduced in the diseased tissue and contaminated environment. Polymer-directed crystallization of antioxidants has attracted attention as a way to control drug efficacy through molecular dissolution. However, most recrystallized antioxidants undertake the continuous dissolution independent of the ROS level, thus causing side-effects. This study demonstrates a unique method to assemble antioxidant crystals that modulate their dissolution rate in response to the ROS level. We hypothesized that antioxidants recrystallized using a ROS-labile polymer would be triggered to dissolve when ROS level increases. We examined this hypothesis by using catechin as a model antioxidant. Catechin was recrystallized using polyethylenimine cross-linked with ROS-labile diselanediylbis-(ethane-2,1-diyl)-diacrylate. Catechin crystallized with ROS-labile polymer displayed accelerated dissolution proportional to the H2O2 concentration. The ROS-responsive catechin crystals protected vascular cells from oxidative insults by activating intracellular glutathione peroxidase expression and, in turn, inhibiting an increase in the intracellular oxidative stress. In addition, the ROS-responsive catechin crystals alleviated changes in the heart rate of Daphnia magna in the oxidative media. We propose that the results of this study would be broadly useful to improving the therapeutic efficacy of a broad array of drug compounds.
Keywords: catechin, drug crystallization, Daphnia, cardioprotective effect, oxidative stress
Graphical Abstract
H2O2-responsive, anti-oxidizing drug crystals are assembled by polymer-directed recrystallization of catechin, for the first time. The catechin crystals exhibit the constant dissolution for 7 days and also display the accelerated dissolution in the oxidative media. The crystals can lower the intracellular oxidative stress of cells and protect heart of Daphnia from the oxidative stress.
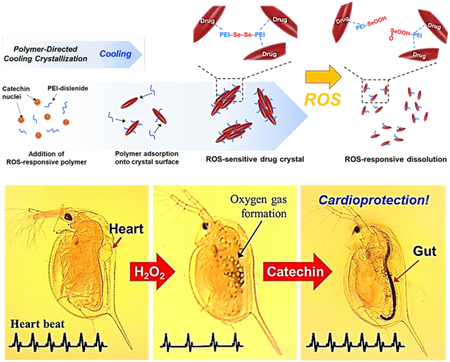
1. Introduction
Reactive oxygen species (ROS) including the superoxide anion, hydroxyl radical, and hydrogen peroxide (H2O2) are by-products of cellular metabolites. Under normal physiological conditions, ROS function as signaling molecules that regulate cell adhesion, proliferation, differentiation, senescence, and apoptosis.[1–5] ROS are also responsible for enhanced wound healing by stimulating tissue-forming cells and immune cells in a balanced manner.[6] However, abnormal overproduction of ROS and/or prolonged free radical actions increases oxidative stress and overwhelm the ability of living cells to maintain homeostasis. This abnormally increased oxidative stress denatures proteins, DNA, and cells.[7–9] In addition, the endogenous antioxidants such as catalase fail to neutralize the ROS, thus leading to chronic inflammatory disorders and impaired wound healing.[10–12]
Exogenous antioxidants including ascorbic acid, N-acetylcysteine, and catechin are administered to either scavenge ROS in tissue or stimulate cells to produce anti-oxidizing enzymes.[13–17] However, fast dissolution rate and rapid metabolism of the antioxidants stimulate cells to overproduce antioxidant enzymes and cause undesirable antioxidative stress. To this end, efforts are increasingly made to control the dissolution rate of antioxidants.[18–20] In particular, the polymer-directed drug crystallization garners attention because it can control polymorphs and size of drug molecules, both of which modulate the dissolution rate.[21–23]
Drug crystallization is a crucial purification process of drugs in the pharmaceutical manufacturing. It significantly influences drug properties including solubility, bioavailability, and stability. Various salts and polymers added during the crystallization induce different nucleation and growth behavior at different degrees of supersaturation. These molecules adsorb onto the surface of drug crystals during growth and disrupt the regular and repeating arrangements of the pure drug crystals. The resulting drug crystals possess dramatically different dissolution profiles, stability, and efficacy from those of the pure drug crystals.[23–25] However, most recrystallized drugs in physiological media exhibit a passive release profile characterized with zero or first-order dissolution kinetics. In case the dissolved amount of antioxidant is higher than needed, antioxidants may increase intracellular antioxidative stress level, thus limiting cell growth.[26] No antioxidant crystal can sense the ROS level and tune the dissolution rate actively.
To this end, this study demonstrates a simple and scalable process to manufacture ROS-responsive antioxidant crystals. We hypothesized that crystallization of the antioxidants with ROS-labile polymers would lead to ROS-responsive antioxidant crystals (Figure 1). We examined this hypothesis by using catechin, a polyphenolic flavonoid-type natural product that is mostly isolated from the green tea, red wine, and other fruits.[27] Catechin can scavenge free radicals and, in turn, reduce the risk of cancer and cardiovascular diseases.[28–30] The catechin was recrystallized by polyethylenimine (PEI) cross-linked by the diselanediylbis-(ethane-2,1-diyl)-diacrylate, denoted as PEI-diselenide. The molecular weight of PEI used herein is 800 g/mol, which is known to be minimally toxic both in vitro and in vivo.[31,32] The PEI forms hydrogen bonds with catechin to generate micro-sized catechin crystals. Small molecules containing selenium or sulfur have been increasingly used as stimuli-responsive linkers because of their redox properties. In particular, diselenide compounds can be reduced or oxidized easily due to the lower bonding energy of Se-Se (172 kJ mol−1) than S-S bond (240 kJ mol−1). Therefore, diselenide compounds are more sensitive to ROS than sulfur compounds.[33–38] Accordingly, catechin crystals complexed with PEI-diselenide can release catechin molecules actively when the intracellular oxidation level increases.
Figure 1.

Schematic illustration of the polymer-directed cooling crystallization process and the ROS-triggered disassembly of the catechin recrystallized with PEI-diselenides.
First, this study examined the interaction energy associated with the crystallization process and the morphology of resulting catechin crystals. Second, the H2O2-responsive dissolution rate of the catechin crystals was related to the intracellular oxidative stress and the metabolic activity of endothelial cells exposed to H2O2. Finally, the extent that H2O2-responsive catechin crystals protected living organisms from water contaminated with excessive H2O2 was evaluated by exposing Daphnia magna to H2O2 and examining their cardiac activity.
2. Results and Discussion
2.1. Synthesis of ROS-responsive PEI-diselenide
The diselanediylbis-(ethane-2,1-diyl)-diacrylate, a ROS-responsive cross-linker, was synthesized by substituting diol groups of 2,2´-diselanediylbis-(ethan-1-ol) with acrylate groups. First, the reaction between selenium and sodium borohydride led to the formation of (sodiodiselanyl)sodium (Na2Se2) and the further reaction between Na2Se2 and 2-bromoethanol resulted in 2,2´-diselanediylbis-(ethan-1-ol), as characterized by the methylene proton peaks (3.93 – 3.91 and 3.12 – 3.09 ppm) in the 1H NMR spectra (Step 1 in Figure S1 and Figure S2). Next, the nucleophilic substitution of the hydroxyl groups in the 2,2´-diselanediylbis-(ethan-1-ol) using acryloyl chloride yielded diselanediylbis-(ethane-2,1-diyl)-diacrylate, as characterized by the acrylate (6.44 – 5.81 ppm) and methylene proton peaks (4.47 – 3.10 ppm) of the 1H NMR spectra (Step 2 in Figure S1 and Figure S3). Finally, the PEI-diselenide was synthesized by the Michael-type cross-linking reaction between amino groups of PEI with acrylate groups of the diselanediylbis-(ethane-2,1-diyl)-diacrylate diacrylate (step 3 in Figure S1). The average molecular weight of PEI-diselenide was increased with the molar ratio between diselanediylbis-(ethane-2,1-diyl)-diacrylate and PEI according to the analysis made with the gel permeation chromatography (GPC) (Figure S4 and Table S1).
The extent to which PEI-diselenide obtained with 1:1 ratio degrades in response to H2O2 was examined by examining the change in the molecular weight with the GPC. The average molecular weight of PEI-diselenide was approximately 5,000 g/mol with a polydispersity index of 1.8. Interestingly, the PEI-diselenide incubated in the H2O2 solution exhibited a decrease in the molecular weight as shown by a shift in the elution time of the polymer (Figure S5). Increasing H2O2 concentration signified the reduction in the molecular weight of PEI-diselenide. In particular, the molecular weight of PEI-diselenide incubated in the 0.5 mM H2O2 solution for 24 hours was decreased from 5,000 to 890 g/mol along with an increased polydispersity index of 2.2 (Table S2).
2.2. Assembly of the polymer-directed crystal of catechin
We prepared the crystallized catechin by following the cooling crystallization process described in Figure S6. First, the catechin was dissolved in deionized water to make a solution with a concentration of 3 wt%. Next, the solution was cooled to 25 °C at a rate of 1 °C/min. The cooling process decreases the solubility of the catechin. The resulting supersaturated solution prompted the formation of catechin crystal nuclei. The resulting crystals continued to grow over 24 hours. According to the scanning electron microscopy images of the catechin crystals exhibited in Figure 2a-1, the recrystallized catechin formed micro-sized needles. Average diameter and length of the needle were 2.2 and 35.2 μm, respectively (Figure 2b).
Figure 2.

Scanning electron micrographs of (a) pure catechin crystals (a-1), catechin recrystallized with (a-2) hyaluronic acid (HA), (a-3) polyethylenimine (PEI), and (a-4) PEI-diselenide. (b) Average short-axis diameter and aspect ratio of the catechin recrystallized with different polymers. (c) DSC thermogram and (d) X-ray diffraction patterns of the recrystallized catechin. In (b), data points and error bars represent mean values and standard deviation of 100 samples per condition, respectively.
In parallel, the polymer-directed crystallization of catechin was conducted through the cooling crystallization of the mixture of polymer and catechin solution. We added poly(acrylic acid) (PAA, Mw ≈ 1,800 g/mol), hydroxypropyl cellulose (HPC, Mw ≈ 40,000 g/mol), polyethylene glycol (PEG, Mn ≈ 950 – 1,050 g/mol), and hyaluronic acid (HA, Mw = 620 – 1,200 kg/mol) to the aqueous solution of catechin (Figure S7). The resulting catechin crystals exhibited a similar morphology to catechin recrystallized without polymers (Figures 2a-2 and S7). PEI influenced size and shape of pure catechin crystals, while other polymers including hyaluronic acid, hydroxypropyl cellulose, poly(acrylic acid), and polyethylene glycol did not. In particular, the cooling crystallization of catechin in the presence of PEI resulted in acicular-shaped catechin crystals which are smaller than the crystals formed without polymers, denoted as pure catechin crystals (Figure 2a-3). The diameter and length of catechin recrystallized with PEI were 0.5 μm and 6.5 μm, respectively (Figure 2b). The PEI-diselenide also drove the catechin to form the crystals with similar morphology and size to that of catechin crystallized by the PEI (Figure 2a-4 and 2b).
2.3. Crystalline structure of polymer-directed catechin crystals
The polymorphs of catechin crystals were further characterized by measuring melting points of the recrystallized catechin using differential scanning calorimetry (DSC) (Figure 2c). The melting point of the recrystallized drug indicates the crystalline form. For instance, there are seven microcrystalline forms of catechin: two tetrahydrates, two monohydrates, and four anhydrates.[39–41] DSC thermogram of the recrystallized catechin exhibited three main peaks at 96, 143 and 177 °C. Melting points at 96 and 143 °C represent the tetrahydrate forms of catechin. Monohydrate forms of catechin are identified with a melting point of 177 °C.[40] The catechin recrystallized in the media dissolved with hyaluronic acid showed similar melting points to the polymer-free, pure catechin crystals consisting of a mixture of tetra- and monohydrates. In contrast, DSC thermograms of the catechin recrystallized with PEI or PEI-diselenide displayed only two melting points that correspond to the melting points of the tetrahydrate forms (i.e., 96 and 143 °C).
X-ray diffraction patterns of the recrystallized catechin were consistent with results of the DSC analysis. The catechin recrystallized in the pure water and that in the hyaluronic acid solution showed the peaks at 19.15° and 23.51°, both of which correspond to monohydrates (Figure 2d).[40,41] Both conditions also showed multiple peaks corresponding to tetrahydrates (16.65°, 20.88°, 25.95°, and 27.7°). In contrast, catechins recrystallized with PEI or PEI-diselenide exhibited the peaks corresponding to tetrahydrates.
Previous studies reported that recrystallization of catechin with pure water followed by air drying resulted in hydrate crystal forms due to the formation of hydrogen bonds between hydroxyl groups of catechin and water molecules.[40] Hyaluronic acid has hydroxyl groups that can also interact with catechin by hydrogen bonding. However, during the crystallization, it is likely that the interaction between catechin and water molecules is more energetically favorable than that between catechin and hyaluronic acid. In contrast, primary amine groups of PEI induced stronger hydrogen bonds with catechin than with water molecules, likely due to the stronger hydrogen bond energy of O–H···N than O–H···O. It is likely that hygroscopic PEI adsorbs onto the surface of growing catechin crystals and increase the number of water molecules associated with resulting crystals.
2.4. Thermodynamics analysis of the catechin crystal formation
We further examined the thermodynamic interaction between catechin and polymers using isothermal titration calorimetry (ITC) (Figure 3). In particular, we quantified the changes in the enthalpy (ΔH) and Gibbs free energy (ΔG) of the catechin solution when the polymers of interests were added to water at 40 °C. The changes in entropy (ΔS) was back-calculated from the values of ΔH and ΔG at a given temperature (T) using the equation.
Figure 3.

Representative calorimetric titrations of the catechin solution with (a) polyethylenimine (PEI), (b) PEI-diselenide, and (c) hyaluronic acid (HA). Upper graphs represent heat flow against time for the titration of catechin and lower graphs represent the integrated heats of each peak, normalized per mole of injectant (polymer).
| Eq.(1) |
The mixture of catechin and PEI showed the exothermic thermograms with ΔH and ΔG of −55.6 and −7.2 kcal/mol, respectively (Figure 3a and Table 1). This result affirms that the interaction between catechin and PEI was favorable thermodynamically. Also, the back-calculated ΔS (−1.2 kcal/mol) with negative value suggests that the exothermic interaction between catechin and PEI drove the thermodynamically favorable recrystallization process. In this analysis, the pH of 0.05 mM catechin solution used to prepare the catechin crystal was 8.2. Therefore, the catechin could be complexed easily with PEI (cationic polymer) during recrystallization. We also investigated the effects of pH on binding affinity between catechin and PEI-diselenide. Catechin becomes charged more positively with a decrease of pH because pKa of catechin is about 9. In this study, the pH of the catechin solution was from 8.2 to 7.0 by adding acetic acid. Catechin solutions with pH of 8.2, 7.4, and 7.0 led to the binding constant (K, 104 M−1) for PEI being 9.7, 7.1, and 2.3, respectively (Figure S8 and Table S3). This result suggests that the electrostatic interaction governs the interaction between PEI and catechin at a pH of 8.2.
Table 1.
Thermodynamic parameters quantified from the isothermal titration calorimetry analysis of the interaction between catechin and polymers. Titrations were run at 40 °C and units of the change in enthalpy (ΔH) and the change in Gibbs free energy (ΔG) are kcal/mol; the change in entropy (ΔS) is kcal/mol·°C; K is the binding constant (104 M−1).
| Polymer added | ΔH | ΔG | ΔS | K |
|---|---|---|---|---|
| PEI | −55.62 | −7.15 | −1.21 | 9.71 |
| PEI-diselenide | −25.69 | −6.58 | −0.48 | 3.88 |
| Hyaluronic acid | −0.30 | −2.87 | 0.06 | 0.01 |
The mixture of catechin and PEI-diselenide exhibited the same levels of ΔH and ΔG as the mixture of catechin and PEI (Figure 3b and Table 1). However, the binding constant (K) was almost 2.5-fold smaller than that for the mixture of catechin and PEI. The exothermic interaction between catechin and PEI-diselenide decreased with increasing the molar ratio of diselenide likely due to the decrease of amine groups available for binding with catechin (Figure S9). In contrast, no significant heat changes were observed with the addition of hyaluronic acid to the catechin solution (Figure 3c). Accordingly, the binding constant (K) was almost zero. These results indicated that there was little interaction between hyaluronic acid and catechin.
2.5. Analysis of H2O2-responsive dissolution of catechin crystals
We analyzed the dissolution profiles of recrystallized catechin in phosphate buffer saline (PBS) at 37 °C by measuring the amount of catechin dissolved and diffused from a dialysis membrane (Figure 4a). In the early stage, the pure catechin crystal was dissolved rapidly and released through the dialysis membrane. Catechin crystals recrystallized in the presence of HA did not exhibit a different hydrate form, morphology, and dissolution rate from pure catechin crystals. Therefore, the crystals recrystallized in the presence of non-interactive HA are not discussed further. In contrast, both catechin recrystallized with PEI and that with PEI-diselenide showed slower release profiles. Therefore, it took six days for the catechin crystals to be dissolved completely. There is a possibility that different hydrate forms of catechin crystals affect the dissolution rate. We prepared mono- and tetra-hydrate form of catechin crystals according to a previously reported method[40] and measured the dissolution rate. Mono- and tetra-hydrate catechin crystals showed a similar dissolution rate regardless of the hydrated form (Figure S10). These results state that the hydration state of recrystallized catechin crystals does not affect the dissolution rate of crystals. Therefore, we propose that the strong association between PEI and catechin is a major factor to decrease the dissolution rate of catechin in water.
Figure 4.

Cumulative release profiles of recrystallized catechin while being incubated in (a) PBS and (b) PBS containing 0.2 mM H2O2. (c) Release profiles and (d) The release rate of catechin recrystallized with PEI-diselenide in the PBS with different concentrations of H2O2. The dissolution rate of catechin crystals was calculated by fitting the cumulative amounts of catechin released between 1 and 48 h. (e) Cumulative release profiles of recrystallized catechin when exposed to 0.2 mM H2O2 solution after 1 day. All samples were incubated at 37 °C. Data points and error bars represent mean values and standard deviation of 3 samples per condition, respectively.
In parallel, catechin crystals were incubated in the PBS containing 0.2 mM of H2O2 (Figure 4b). The overall dissolution rates of the pure catechin crystals and the catechin crystals complexed with PEI decreased in the media containing H2O2 as compared with the crystals incubated in PBS free of H2O2. This result is due to the decreased pH of the media containing H2O2 (Figure S11). It is well known that the dissolution rate of catechin hydrate is dependent on pH.[41] Nevertheless, the PEI-free catechin crystal exhibited a faster dissolution rate than catechin complexed with PEI or PEI-diselenide, regardless of H2O2 concentration.
Interestingly, the catechin crystals prepared with the PEI-diselenide showed a faster dissolution rate than catechin crystals prepared with PEI only in the media containing 0.2 mM H2O2. 80% of the catechin complexed with PEI-diselenide was dissolved continuously over 6 days. During the same period, only 30% of the catechin was released from the catechin crystals complexed with PEI. This increased dissolution rate was attributed to the breakage of diselenide linkers triggered by H2O2. As confirmed with the GPC analysis shown in Figure S5, the average molecular weight of PEI-diselenide decreased in the media containing 0.2 mM H2O2. This molecular breakage is due to oxidation of hydrophobic selenides to water-soluble selenoxide or selenone.[33]
The dissolution profiles of the catechin complexed with PEI-diselenide was further characterized with media containing different H2O2 concentrations (Figure 4c). The concentration of H2O2 was varied from the normal physiological condition (0.1 μM) to the pathological range of H2O2 levels (0.1, 0.2, 0.3, and 0.5 mM).[42] Increasing H2O2 concentration led to the faster breakage of diselenide bonds in the catechin crystals as shown in Figure S5. As a consequence, the dissolution rate of catechin crystal was highly dependent on the H2O2 concentration. The amount of catechin released over 7 days increased with the H2O2 concentration. The release rate, quantified from the slope of the catechin release curve in Figure 4c, increased linearly with the H2O2 concentration (Figure 4d).
Also, the catechin crystals prepared with PEI-diselenide was incubated in the PBS over the first 24 hours followed by the addition of H2O2. By doing so, we examined whether the catechin crystals could adaptively expedite the dissolution rate. The final H2O2 concentration was 0.2 mM. Within the next 24 hours, the catechin crystals released the catechin molecules actively, as marked by the significant increase in the amount of catechin released (Figure 4e). This result confirms that the catechin crystals prepared using the PEI-diselenide can change their release profile in response to the external oxidative stimulus. Therefore, this H2O2-responsive drug crystal may be able to circumvent any side-effects resulting from continuous passive dissolution of drug molecules.
2.6. In vitro analysis of antioxidant effects of catechin crystals
The antioxidant activity of catechin crystals was tested by examining the metabolic activity of endothelial cells cultured in the media containing H2O2. First, the H2O2 concentrations above which most cells lost metabolic activity were determined. According to 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, which quantifies the fraction of metabolically active cells, 63 % of the cells lost their metabolic activity within 2 hours in the media containing 0.2 mM H2O2. Then, 98 % of cells lost their metabolic activity within 24 hours (Figure S12). Next, the concentration range at which catechin prevents the loss of cellular metabolic activity in 0.2 mM H2O2-containing media was determined by incubating cells with varying concentrations of catechin (Figure S13). 70 to 80 % of cells retained metabolic activity in the media dissolved with 1.6 and 3.2 mM catechin. Further increase of catechin concentration to 6.4 mM rather decreased the fraction of metabolically active cells, likely due to the increased intracellular antioxidative stress.
Separately, the PEI concentration which does not impact cellular metabolic activity was determined by incubating cells with varying concentrations of PEI. Cells were incubated in the media free of H2O2 for this analysis. More than 87 % of the cells remained metabolically active when PEI concentrations were lower than 0.12 mM (Figure S14). The PEI concentration in the recrystallized catechins was around 0.1 mM, which is lower than 0.12 mM.
Finally, antioxidative activity of catechin crystals was examined by incubating cells in the 0.2 mM H2O2-containing media (Figure 5a). Interestingly, after 24 h, 37 % of cells retained metabolic activity in the media suspended with catechin crystals complexed with PEI-diselenide. In contrast, only 7 % and 12 % of cells remained metabolically active in the media suspended with pure catechin crystals or catechin crystals complexed with PEI, respectively. The catechin crystals complexed with PEI made minimal difference from pure catechin crystals. The protective effect of catechin crystals decreased gradually over 7 days. However, a more significant fraction of cells remained metabolically active when cells were incubated with catechin crystals complexed with PEI-diselenide (Figure 5a).
Figure 5.

Analysis of the anti-oxidative effect of catechin crystals. Catechin crystals loaded in a dialysis bag were incubated with PBS containing 0.2 mM H2O2. Then, the media were collected and replaced with fresh media containing 0.2 mM H2O2 on Days 1, 3, 5 and 7. The collected media was used for the cell culture. (a) Quantification of the metabolically active C166 endothelial cells using the MTT assay kit. (b) Representative confocal images of the live-dead assay. Green color represents live cells, and red color represents dead cells. The scale bars represent 200 μm. (c) Quantification of the percentage of surface covered by live endothelial cells using the images in Figure S16a. (d) Intracellular oxidative stress of C166 cells incubated with different catechin concentrations. The values were normalized with respect to that of C166 cells without exposure to H₂O₂. (e) Representative fluorescence images of the oxidative stress of endothelial cells (C166) incubated in the media containing 0.2 mM H2O2 and different concentrations of catechin. The scale bars represent 100 μm. (f) Activity of glutathione peroxidase (GPx) in C166 cells. In (a), (b), (d), and (e) data points and error bars represent mean values and standard deviation of 5 samples per condition, respectively. * and ** represent the statistical significance of the difference of values between conditions indicated with line (*p < 0.05, **p < 0.01).
In addition, extents that selenium itself exerts antioxidative activity was examined by incubating cells in the 0.2 mM H2O2-containing media dissolved with varying concentrations of PEI-diselenide. According to MTT assay, PEI-diselenide solutions with concentrations varying from 0.01 to 0.16 mM did not increase the number of metabolically active cells (Figure S15).
The viability of cells was also assessed using the live-dead assay kit. A significant fraction of cells lost their viability when cells were cultured in media containing 0.2 mM H2O2 and catechin crystals for one day (Figures 5b and 5c). Additionally, cells treated with pure catechin crystals or catechin crystals complexed with PEI showed decreased surface coverage by live cells over 7 days (Figures 5c and S16). These results indicate that most of the dead cells were separated from the culture substrate. In contrast, cells treated with catechin crystals complexed PEI-diselenide showed insignificant changes in the surface coverage and number of live cells over 7 days compared with cells not exposed to H2O2 (Figures 5b, 5c, and S16).
The protective role of catechin crystals complexed with PEI-diselenide was attributed to the limited increase in the intracellular oxidative stress (Figure 5d). The intracellular oxidative stress level of cells was examined using CellROX® Green fluorogenic probe, which indicates the intracellular oxidative stress level with green fluorescence. Cells exposed to 0.2 mM H2O2 displayed the higher positive fluorescence around the nuclei than those not exposed to H2O2 (Figure 5e and S17). In contrast, the minimal level of fluorescence was found in cells incubated with the catechin crystals. According to the fluorescence images, the intracellular stress level decreased with increasing catechin concentration, thus confirming that catechin neutralized the oxidative effects of H2O2 within cells.
We further analyzed the DNA profiles of cells incubated with 0.2 mM H2O2 and catechin to examine the DNA protection potential of catechin from H2O2-induced oxidative stress. The extracted samples from cells incubated with 0.2 mM H2O2 showed fragmented DNAs (Figure S18). In contrast, the DNA from cells incubated with catechin and H2O2 exhibited a similar pattern to the DNA from cells not exposed to H2O2 (Figure S18). This result confirms that catechin prevents DNA fragmentation resulting from the H2O2-induced oxidative stress.
Furthermore, the activity of the glutathione peroxides, an antioxidant enzyme, in the cells was examined to evaluate the extent to which catechin crystals reduce oxidative stress by stimulating expression of glutathione peroxides (Figure 5f). In general, glutathione peroxides is involved in eliminating intra- and extracellular H2O2.[43] The high activity of glutathione peroxides implies that catechin molecules neutralize H2O2 molecules within cells, and subsequently lower the intracellular oxidative stress level. After 1 day of treatment, cells incubated with catechin crystals free of PEI showed a higher glutathione peroxides activity than those incubated with catechin complexed with PEI or PEI-diselenide. However, the cells treated with pure catechin crystals showed lower viability at day 1 (Figure 5b and 5c). It is likely that the uncontrollably dissolved catechin stimulates cells to overproduce GPx and eliminate H2O2 required for desired metabolic activity of cells. In contrast, catechin complexed with PEI-diselenide served to retain cell viability in an oxidative condition, because the catechin crystals released catechin molecules in response to the H2O2 level. Accordingly, after 7 days, cells incubated with catechin crystals complexed with PEI-diselenide showed a higher glutathione peroxides activity than those incubated with pure catechin crystals or catechin crystals complexed with PEI (Figure 5f). This result suggests that PEI-diselenide is advantageous to elevate the cellular glutathione peroxide activity in response to the increase of H2O2 level. In contrast, the catechin crystal free of PEI was consumed before Day 7. Also, the catechin crystals with PEI dissolved in the media slowly regardless of external ROS concentration, as shown in Figure 4, thus making catechin unavailable to effectively regulate the intracellular enzymatic activity.
2.7. Cardioprotective effects of catechin in Daphnia magna model.
The anti-oxidative efficacy of catechin crystal was further evaluated by exposing Daphnia magna to the oxidative media and examining the myocardial damage. Daphnia magna has been used to monitor not only the environmental impacts on the ecological system under guidelines by the Organization of Economic Cooperation and Development (OECD) but also evaluate the efficacy of cardioprotective drugs.[44,45]
First, Daphnia magna (Daphinids) was exposed to the media mixed with sub-lethal concentration of H2O2, such as 0.03 mM or 0.05 mM and catechin crystals for 48 h (Figure S19). H2O2 is often used to clean water fouled by excessive algae, thus raising a concern about impacts on living organism in water.[46] Then, we measured the heart rate of daphnids in accordance with OECD guidelines 202 (Figure 6a). The heart rate is an essential indication of the extent to which chemicals with potential toxicity influence physiology of daphnids.[44,45] Heart rates of daphnids also resemble the myogenic heart of vertebrates. Furthermore, daphnids have more genes similar to humans than other animal groups including insects and crustaceans.[47] According to the image of daphnids shown in Figure 6d, the gut of daphnids incubated in the media mixed with catechin crystals had turned black. As the catechin crystal is black, these images prove that daphnids took up the catechin crystals (Figure 6d-4 and 6d-5).
Figure 6.

Cardioprotective effects of catechin from H2O2-induced oxidative stress in Daphnia magna model. (a) Schematic illustration of toxicity test to assess effects of chemicals towards Daphnia magna. Young daphnids, aged less than 24 hours at the start of the test, were exposed to the test chemicals for a period of 48 hours. Then the mortality rate of daphnids was recorded at 48 hours and their heart rate was measured. (b) Heart rate of Daphnia magna subjected to H2O2 (0.03 or 0.05 mM). (c) Intracellular fluorescence of DCF converted from DCFDA by intracellular reactive oxygen species of Daphnia magna incubated with different conditions listed on the x-axis. The values were normalized to that of daphnids without exposure to catechin crystals and H₂O₂. The results are represented as means ± SD, n= 10. * and ** represent the statistical significance of the difference of values between conditions indicated with a line (*p < 0.05, **p < 0.01). (d) Representative optical microscope (OM) images of Daphnia magna: before treatment (d-1), after exposure to 0.05 mM H2O2 solution without catechin (d-2), with pure catechin crystals (d-3), catechin recrystallized with PEI (Catechin-PEI) (d-4), and catechin recrystallized with PEI-diselenide (Catechin-PEI-diselenide) (d-5), respectively.
The mean heart rates of daphnids exposed to 0.03 and 0.05 mM H2O2 decreased from 348 to 290 and 277 beats per minute (bpm), respectively (Figure 6b). H2O2 decreased the mean heart rates of daphnids in a dose-dependent manner while promoting the generation of bubbles within the body (Figure 6d and Movie S1). The intracellular oxidative stress level of daphnids was examined by 2,7-dichlorofluorescein diacetate (DCFDA) fluorogenic dye. Daphnids exposed to H2O2 showed the higher intracellular oxidative stress level than those not exposed to H2O2 (Figure 6c). This result implies that H2O2 increases intracellular oxidative stress level and, in turn, decreases the cardiac activity.
In contrast, daphnids not exposed to the H2O2 but incubated with catechin crystals displayed no decrease in heart rate. Daphnids exposed to 0.05 mM H2O2 with pure catechin crystals or catechin-PEI exhibited the decreased intracellular oxidative stress level and, in turn, recovered the heart rate imperfectly from 348 to 320 and 303 bpm, respectively. In contrast, catechin crystals complexed with PEI-diselenide served to increase the mean heart rate of daphnids incubated with 0.05 mM H2O2 solution from 348 to 340 bpm (Figure 6b). The recovered mean heart rate of daphnid was close to the normal heart rate (Movie S2). However, the heart rate of daphnids incubated with 0.03 mM H2O2 solution could not be fully recovered by catechin crystals complexed with PEI-diselenide, likely because of the limited breakage of Se-Se bonds of PEI-diselenide. The degree of recovery was comparable to that made with catechin crystals complexed with PEI.
We further examined the dissolution of catechin crystals in the media used for the culture of Daphnia magna using liquid chromatography-mass spectrometry (LC-MS) (Figure S20). When catechin crystallized with PEI-diselenide was incubated in the media mixed with sub-lethal concentration of H2O2 (i.e., 0.05 mM), concentration of dissolved catechin was significantly increased compared with the catechin crystallized with PEI. The result attests that catechin crystals formed with PEI-diselenide release catechin molecules continuously in the oxidative environment, similar to the in vitro test shown in Figure 4. The higher concentration of catechin with pure catechin crystals is attributed to the fast dissolution. In contrast, the lower concentration of catechin with catechin-PEI crystals is because of limited dissolution. We propose that catechin crystals complexed with PEI-diselenide in the Daphnia’s gut reduced the oxidative stress continuously due to the H2O2-triggered and sustained release of catechin. On the other hand, catechin crystals free of PEI were metabolized quickly as observed with faster disappearance of the positive color in the gut.
3. Conclusion
In conclusion, H2O2-responsive, antioxidant crystals were assembled by recrystallizing catechin with the ROS-labile polymer. The PEI crosslinked with diselanediylbis-(ethane-2,1-diyl)-diacrylate mitigated the catechin crystal growth during the crystallization, resulting in the micro-sized catechin crystal needles. Driven by the exothermic interaction between catechin and PEI-diselenide, the catechin crystallized with PEI-diselenide presented mainly a tetrahydrate form. The resulting catechin crystals exhibited an accelerated dissolution in response to the H2O2 concentration. The H2O2-responsive catechin crystals could lower the intracellular oxidative stress of cells exposed to the oxidative media over 7 days due to the constant expression of intracellular glutathione peroxidase. Therefore, the H2O2-responsive catechin crystals served to retain metabolic activity and viability of cells exposed to H2O2. In addition, H2O2-responsive catechin crystals were advantageous to alleviate the slowing down of the heart rate of Daphnia magna by H2O2 due to the continuous dissolution in the gut. Such protective effect could not be attained with pure catechin crystals nor catechin crystals complexed with PEI. Overall, the results of this study would be broadly useful in controlling polymorph, stimulus-responsiveness and therapeutic efficacy of various pharmaceutical ingredients.
4. Experimental Section
Materials:
Sodium borohydride (≥ 98.0%), selenium (≥ 99.5%), 2-bromoethanol (95%), triethylamine (≥ 99.0%), acryloyl chloride (97.0%, contains <210 ppm MEHQ as stabilizer), (+)–catechin hydrate (≥ 98.0%), poly(acrylic acid) (PAA, Mw ≈ 1,800 g/mol), hydroxypropyl cellulose (HPC, Mw ≈ 40,000 g/mol), polyethylene glycol (PEG, Mn ≈ 950 – 1,050 g/mol) and polyethylenimine (PEI, branched, Mw ≈ 800 g/mol) were purchased from Sigma-Aldrich. Hyaluronic acid (HA, Mw ≈ 620 – 1200 kg/mol) was purchased from Kikkoman. Tetrahydrofuran (THF, HPLG grade), deionized water, methylene chloride (99.6%), sodium chloride, magnesium sulfate hydrate (≥99.0%), ethyl acetate and n-hexane (anhydrous) were purchased from Fisher Scientific. H2O2 (30% solution) was purchased from Macron Fine Chemicals. A dialysis tube (MWCO: 3.5 kDa, Fisherbrand) was used to remove impurities in the chemical compounds. For drug release tests, a dialysis tube (MWCO: 500 – 1000 Da, Spectrum Labs) was used. C166 cells (mouse endothelial cell line from yolk sac) were obtained from American Type Cell Culture (ATCC CRL2581) and cultured according to the guidelines of ATCC. Daphnia eggs were obtained from Environmental Bio-Detection Products Incorporation (EBPI, Daphtoxkit F magna) and cultured according to the standard operational procedure.
Synthesis of PEI crosslinked with ROS-responsive diselenide:
Synthesis of 2,2`-diselanediylbis-(ethan-1-ol) Sodium borohydride (2.27 g, 0.06 mol) was dissolved in 50 mL of DI water under N2 atmosphere. Selenium (2.4 g, 0.03 mol) was added to the mixture in batches at 25 °C with stirring. After the mixture color was changed to milk white, another dosage of selenium (2.4 g, 0.03 mol) was added. Then, the mixture was kept at 80 °C for 3 h. The mixture was cooled down to room temperature. 2-bromoethanol (7.87 g, 0.03 mol) in 100 mL THF was added dropwise into the mixture. The subsequent reaction was refluxed for 24 h. The obtained solution was extracted with CH2Cl2 (100 mL × 3). The organic phase was combined, washed with 1 N NaCl and water, and then dried with MgSO4. The organic phase was filtered by vacuum filtration, and the organic phase was then concentrated by rotary evaporator. The crude product was purified by silica column chromatography with EtOAc/n-hexane (1/1 vol%) as an eluent. The product was obtained as yellow oil (yield: 60%); 1H NMR (500 MHz, CDCl3) δ (ppm): 4.01–3.98 (t, 1H), 3.93–3.91 (t, 4H), 3.31–3.28 (t, 1H), 3.12–3.09 (t, 4H), 13C NMR (500 MHz, CDCl3) δ (ppm): 61.73, 32.72 (Figure S1).
Synthesis of diselanediylbis-(ethane-2,1-diyl)-diacrylate.
2,2`-diselanediylbis-(ethan-1-ol) (2.5 g, 10 mmol, 1 equiv.) was dissolved in dichloromethane (20 mL) containing triethylamine (4.45 g, 50 mmol, 5 equiv.) at 0 °C. Acryloyl chloride (2.45 mL, 30 mmol, 3 equiv.) was added dropwise to the mixture which was stirred overnight. The chemical mixture was extracted with CH2Cl2. The organic phase was combined, washed with 1N NaCl and water, and then dried over anhydrous MgSO4. The crude product was purified by the silica gel column chromatography (EtOAc/n-hexane, 1/1). The product was obtained as yellow oil (yield: 48%); 1H NMR (500 MHz, CDCl3) δ (ppm): 6.44 – 6.38 (d, 4H), 6.15 – 6.07 (dd, 4H), 5.87 – 5.81 (d, 4H), 4.47 – 4.41 (t, 4H), 3.20 – 3.10 (t, 4H). 13C NMR (500 MHz, CDCl3) δ (ppm): 165.8, 131.2, 128.1, 64.1, 27.3 (Figure S2).
Synthesis of PEI crosslinked with diselenide.
The number of cross-links between PEI was controlled by changing the molar ratio between PEI and diselanediylbis-(ethane-2,1-diyl)-diacrylate from 1:1 to 1:25, and 1:5. PEI and diselanediylbis-(ethane-2,1-diyl)-diacrylate were dissolved in DMF and stirred at 60 °C for 48 h. The mixture was dialyzed against DI water using a dialysis tube (MWCO = 3.5 kDa) for 2 days and then lyophilized. The average molecular weight of resulting PEI-diselenide was examined by GPC.
Preparation of catechin crystals by polymer-directed recrystallization:
30 mg (10 wt% of catechin) of polymer additives (i.e., polyethylenimine, hyaluronic acid, poly(acrylic acid), poly(ethylene glycol), and hydroxypropyl cellulose) were dissolved in DI water (9.3 mL) for 1 day at 40 °C to ensure the complete dissolution. Then, 0.3g of catechin was added into the obtained polymer solution and stirred until all the solids completely were dissolved. The mixture was cooled to 20 °C with a controlled cooling rate of 1 °C/min, and the solution was further kept for 24 h until light orange-colored crystals formed. The obtained crystal solution was filtered through a Polyvinylidene Fluoride (PVDF) membrane (HVLP04700, pore size: 0.45 μm, Millipore) to collect catechin crystals. The crystals were gently washed 2 – 3 times with water to remove the unbound polymers. Then, the samples were dried under vacuum at room temperature for 24 h.
Characterization:
The morphology of catechin crystals was examined by an optical microscope (Leica DMIL) and an environmental scanning electron microscope (ESEM, Quanta FEG 450, FEI) at 10 kV acceleration voltage. One hundred particles from ten different SEM images of each sample were analyzed to measure the average particle size and aspect ratio. The polymorph of catechin crystals was investigated by differential scanning calorimetry (Perkin Elmer Diamond). Melting behavior was monitored under heating from 30 to 250 at 10 °C/mim. X-ray diffraction analysis (MiniFlex 600, Rigaku) was conducted to observe changes in the contents of crystal phases. The scans were performed in the 2-theta range of 10 – 80 at 10 °/min. The mean zeta-potential of the catechin crystals was characterized by the particle size analyzer (Litesizer 500™, Anton Paar). Thermodynamic analysis of the association between catechin and polymer additives was performed by Isothermal Titration Calorimetry (MicroCal). The 1.45 mL sample cell was filled with a 0.5 mM polymer solution. The cell was titrated with 28 injections of 10 μL catechin solution (0.05 mM). Each injection was performed over 17.1 s with a delay of 300 s between injections while stirring at 300 rpm. Thermodynamic binding parameters such as the binding constant, the change in enthalpy, and the change in entropy were obtained and calculated by fitting data to a single-site binding model. The first data point was not included in the analysis. The molecular weight and polydispersity of PEI-diselenide were determined by gel permeation chromatography (Breeze 2 GPC, Waters), with Styragel HT column (Waters). The mixture of acetic acid (0.1 mol/L)/sodium acetate (0.1 mol/L) (pH 2.8) was used as the eluent, with the elution rate of 1 mL/min. Polystyrene standards were used for calibration. Analytical LC-MS data for catechin crystals were acquired by high-performance liquid chromatography-tandem mass spectrometry using Agilent 1200 HPLC system with a 6460 electrospray triple-quadrupole mass spectrometer (Agilent Technologies). The mobile phases were 0.1% acetic acid in acetonitrile and 0.1% acetic acid in water. The mass spectrometer was operated in electrospray negative ionization mode, and the identification of MEHP in samples was achieved in SCAN mode by 50:50 ratio of mobile phase solvents.
Analysis of the release profile of catechin crystals:
25 mg of catechin crystals were placed in a dialysis tube (MWCO = 500 – 1000 Da) with phosphate-buffered saline (PBS, 1 mL). The dialysis tube was placed in the 499 mL of PBS media with different H2O2 concentrations (i.e., 0, 0.0001, 0.1, 0.2, 0.3, and 0.5 mM) and incubated at 37 °C under continuous shaking at 100 rpm. At the designated time points, dissolved catechin was collected from the incubation media and determined by reading the absorbance at the wavelength of 260 nm using the microplate spectrophotometer (Infinite 200 PRO, Tecan).
Analysis of the metabolic activity of cells:
A commercially available MTT assay kit (K299, Bio Vision) was used to measure the metabolic activity of cells in oxidative media. The assay was performed with several modifications to the manufacturer’s directions. Briefly, 160 mg of catechin crystals in the dialysis tube were incubated with 250 mL of Dulbecco’s Modified Eagle Medium (DMEM) containing 0.2 mM H2O2. Then, the media were collected and replaced with fresh DMEM containing 0.2 mM H2O2 after every designed time (1, 3, 5 and 7 days). The mean zeta potentials of catechin crystals, catechin complexed with PEI, and PEI-diselenide were 2.2, 18.0, and 5.9 mV, respectively. Catechin crystals complexed with PEI and PEI-diselenide showed a more positively charged surface than crystals free of PEI. To prevent charge-driven adsorption of catechin crystals on cellular membrane, catechin crystals were loaded in a dialysis bag (MWCO: 500 – 1000 Da) during incubation. Then, the incubation media were collected and used for the cell culture. Molecular weights of catechin and PEI used in this study are 290 Da and 800 Da, respectively. Therefore, it is unlikely that the catechin crystals complexed with PEI or PEI-diselenide pass through the dialysis bag. C166 cells were seeded onto 96-well plates at a density of 8,000 cells per well one day before treatment. By doing so, cells formed a thin endothelium. The cells were further incubated with the media as prepared above for 2 or 24 h. After that, 100 μL of growth media and 10 μL of MTT solution were then added to each well. The cells were incubated for 4 h at 37 °C. Resultant formazan crystals formed in each well were solubilized using 120 μL of DMSO upon removal of growth media. The absorbance of MTT was measured by using a microplate spectrophotometer (Infinite 200 PRO, Tecan) at the wavelength of 550 nm and 690 nm. The relative metabolic activity of cells was quantified as [(A550-A690)sample/(A550-A690)control].
Live-dead assay of cells:
A live/dead assay was performed using LIVE/DEAD® Viability/Cytotoxicity Assay Kit for mammalian cells (Invitrogen) according to the manufacturer’s instructions. The cultured cells were gently washed with 1× Dulbecco’s Phosphate Buffered Saline (DPBS) for 3 times. Calcein Acetoxymethyl (AM) and ethidium homodimer-1 (EthD-1) were diluted in 1× DPBS. 1 mL of diluted Calcein AM and EthD-1 solution was added to cultured cells and kept for 45 min at room temperature. The live cells are stained with Calcein-AM, and dead cells are stained with EthD-1. After staining, cells were gently washed with 1× DPBS for 3 times and imaged with a fluorescence microscope (Ti-U, Nikon).
Analysis of the intracellular oxidative stress:
C166 cells were seeded on 18 mm cover glass with the number of 10,000 cells for each. After one day incubation, cells were exposed to 0.2 mM H2O2 solution with catechin crystals. After 1.5 h, the intracellular oxidative stress was evaluated using the cellROX reagent (ThermoFisher), following the manufacturer’s instruction. Briefly, the C166 cells were incubated with 5 μM of cellROX reagent for 30 min, and the cells were washed with 1× DPBS two times. The cells were fixed with 3.7% formaldehyde solution for 15 min. For nuclear counterstain, cells were stained with 500 nM of 4′,6-diamidino-2-phenylindole (DAPI). Finally, the cells were imaged using a fluorescent microscope (Ti-U, Nikon), and the intensity was quantified using ImageJ software (NIH).
DNA fragmentation assay:
C166 cells were seeded in the T-75 flask with the number of 2,000,000 cells. After one day incubation, cells were exposed to 0.2 mM H2O2 solution with varying concentrations of catechin for 24h. Then, DNA of the cells was extracted using a DNA Extraction Kit (11835246001, Roche), following the manufacturer’s instruction. The extracted DNA from the cells was then separated by a standard 1% agarose gel. After electrophoresis, the gel was stained with ethidium bromide and photographed with gel documentation (BioRad).
Analysis of the glutathione peroxidase (GPx) activity of cells:
C166 cells were seeded onto 6-well plates at a density of 300,000 cells per well. The cells were further incubated with designed media for 12 h. The media was extracted from DMEM containing catechin crystals and 0.2 mM H2O2 after 1 or 7 days of incubation. The cells incubated in the oxidative media were collected and re-dispersed in 200 μL of cold PBS. Then, the mixture as centrifuged at 10 min at 14,000 rpm and the supernatant was used for the assay. All reagents and samples were equilibrated to 25 °C. The reaction progress was examined by monitoring the change in the absorbance at 340 nm resulting from consumption of NADPH. The unit of GPx activity (U/L) is the amount of GPx that produces 1 μmole of glutathione disulfide (GS-SG) per min at pH 7.6 and room temperature.
Daphnia manga culture:
During the experimental period, Daphnia magna neonates were cultured under a light cycle of 16 hours light and 8 hours darkness at 22.0 ± 0.5 °C water temperature in a climate incubator. All tests using Daphnia magna were conducted in six-well cell culture plates (Cellstar, Greiner Bio-one) filled with 10 mL of each test solution. The ISO standard freshwater (MicroBioTests Inc., Gent) was used as a control and a dilution solution. Concentration of H2O2 was kept constant at 0.03 or 0.05 mM, based on our preliminary experiments and toxicological data (Figure S13). The sub-lethal concentration (EC50) was 2 mg/L after 48 h. Heart rate and mortality of Daphnia were determined after 48 h incubation. The ISO medium mixed with catechin crystals or catechin crystals and H2O2 was added to the media in which Daphnia magna was cultured. 10 Daphnia magna was placed in 10 mL of media. After centrifugation of the media, the supernatants were filtered using 0.2 μm nylon filter and stored at –20 °C before analysis.
Analysis of the intracellular oxidative stress level in Daphnia magna:
The intracellular oxidative stress level of the daphnid was measured by 2,7-dichlorofluorescein diacetate (DCFDA) fluorogenic dye. Two-electron oxidation by intracellular reactive oxygen species converts DCFDA to fluorescent dichlorofluorescein (DCF). The final concentration of DCFDA was 0.01 mM with each 0.1 mL samples. Homogenized daphnids were used to determine the oxidative stress level. After 24h of exposure to 0.05 mM of the catechin crystals in the presence of H2O2 (0.03 mM or 0.05 mM), 8 daphnids were transferred to 0.1 mL of DCFDA solution for 4 h at 20 °C. The intracellular fluorescence level of DCF was monitored by a fluorescent microscope (Ti-U, Nikon), and the intensity was quantified using the ImageJ software (NIH).
Statistical analysis:
Averaged data is presented as mean ± standard deviation. Statistical significance between pairs of experimental populations is determined by Student’s t-tests.
Supplementary Material
Acknowledgments
B. S. Kim and J. Leong contributed equally to this work. The authors thank Oleg Davydovich and Prof. Jeffrey S. Moore (University of Illinois at Urbana-Champaign) for assistance in DSC and GPC analysis. This work was supported by Korea Institute of Science and Technology-Europe and partly by the National Science Foundation Research Training Grant (1735252), National Institutes of Health (1R21 HL109192), and the Department of Defense Vision Research Program under Award (W81XWH-17-1-022). J. Leong gratefully acknowledges the A*STAR Graduate Scholarship Overseas) from Agency for Science, Technology, and Research (A*STAR), Singapore.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or the author.
Contributor Information
Byoung Soo Kim, Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana–Champaign, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, Beckman Institute for Science and Technology, University of Illinois at Urbana–Champaign, Illinois 61801, United States.
Jiayu Leong, Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana–Champaign, Illinois 61801, United States; Institute of Bioengineering and Nanotechnology, 31 Biopolis Way, The Nanos, Singapore 138669, Singapore.
Seung Jung Yu, Department of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea.
Younghak Cho, Department of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea.
Chang Gyun Park, Environmental Safety Group, Korea Institute of Science and Technology (KIST–Europe), Saarbrucken 66123, Germany.
Da-Hye Kim, Environmental Safety Group, Korea Institute of Science and Technology (KIST–Europe), Saarbrucken 66123, Germany.
Eunkyung Ko, Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana–Champaign, Illinois 61801, United States; Carl R. Woese Institute for Genomic Biology, Beckman Institute for Science and Technology, University of Illinois at Urbana–Champaign, Illinois 61801, United States.
Sung Gap Im, Department of Chemical and Biomolecular Engineering, Korea Advanced Institute of Science and Technology, Daejeon 34141, Republic of Korea.
Jonghwi Lee, Department of Chemical Engineering and Materials Science, Chung-Ang University, Seoul 156-756, Republic of Korea jong@cau.ac.kr.
Young Jun Kim, Environmental Safety Group, Korea Institute of Science and Technology (KIST–Europe), Saarbrucken 66123, Germany youngjunkim@kist-europe.de.
Hyunjoon Kong, Department of Chemical and Biomolecular Engineering, University of Illinois at Urbana–Champaign, Illinois 61801, United States hjkong06@illinois.edu; Carl R. Woese Institute for Genomic Biology, Beckman Institute for Science and Technology, University of Illinois at Urbana–Champaign, Illinois 61801, United States.
References
- [1].Schieber M, Chandel NS, Curr. Biol 2014, 24, R453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, Symons M, Borrello S, Galeotti T, Ramponi G, J. Cell Biol 2003, 161, 933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].D’Autréaux B, Toledano MB, Nat. Rev. Mol. Cell Biol 2007, 8, 813. [DOI] [PubMed] [Google Scholar]
- [4].Redza-Dutordoir M, Averill-Bates DA, Biochim. Biophys. Acta - Mol. Cell Res 2016, 1863, 2977. [DOI] [PubMed] [Google Scholar]
- [5].Morimoto H, Iwata K, Ogonuki N, Inoue K, Atsuo O, Kanatsu-Shinohara M, Morimoto T, Yabe-Nishimura C, Shinohara T, Cell Stem Cell 2013, 12, 774. [DOI] [PubMed] [Google Scholar]
- [6].Dunnill C, Patton T, Brennan J, Barrett J, Dryden M, Cooke J, Leaper D, Georgopoulos NT, Int. Wound J 2017, 14, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sies H, Cadenas E, Philos. Trans. R. Soc. B Biol. Sci 1985, 311, 617. [DOI] [PubMed] [Google Scholar]
- [8].Slupphaug G, Kavli B, Krokan HE, Mutat. Res. Mol. Mech. Mutagen 2003, 531, 231. [DOI] [PubMed] [Google Scholar]
- [9].Kryston TB, Georgiev AB, Pissis P, Georgakilas AG, Mutat. Res. Mol. Mech. Mutagen 2011, 711, 193. [DOI] [PubMed] [Google Scholar]
- [10].Salganik RI, J. Am. Coll. Nutr 2001, 20, 464S. [DOI] [PubMed] [Google Scholar]
- [11].Miller ER, Pastor-Barriuso R, Dalal D, Riemersma RA, Appel LJ, Guallar E, Ann. Intern. Med 2005, 142, 37. [DOI] [PubMed] [Google Scholar]
- [12].Poljsak B, Milisav I, Oxid. Med. Cell. Longev 2012, 2012, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Patra RC, Swarup D, Dwivedi SK, Toxicology 2001, 162, 81. [DOI] [PubMed] [Google Scholar]
- [14].Halliwell B, Am. J. Med 1991, 91, S14. [DOI] [PubMed] [Google Scholar]
- [15].Zafarullah M, Li WQ, Sylvester J, Ahmad M, Cell. Mol. Life Sci 2003, 60, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bravo L, Nutr. Rev 1998, 56, 317. [DOI] [PubMed] [Google Scholar]
- [17].Oliver S, Vittorio O, Cirillo G, Boyer C, Polym. Chem 2016, 7, 1529. [Google Scholar]
- [18].Sansone F, Picerno P, Mencherini T, Villecco F, D’Ursi AM, Aquino RP, Lauro MR, J. Food Eng 2011, 103, 188. [Google Scholar]
- [19].Shao S, Li L, Yang G, Li J, Luo C, Gong T, Zhou S, Int. J. Pharm 2011, 421, 310. [DOI] [PubMed] [Google Scholar]
- [20].Liu F, Avena-Bustillos RJ, Chiou B-S, Li Y, Ma Y, Williams TG, Wood DF, McHugh TH, Zhong F, Food Hydrocoll. 2017, 62, 212. [Google Scholar]
- [21].Choi H, Lee H, Lee MK, Lee J, J. Pharm. Sci 2012, 101, 2941. [DOI] [PubMed] [Google Scholar]
- [22].Huang Y, Jiang Y, Yang X, Ren Y, Zhan D, Cölfen H, Hou Z, Liu XY, Cryst. Growth Des 2016, 16, 1428. [Google Scholar]
- [23].Zhong Z, Yang X, Guo B, Xu J, Huang Y, Cryst. Growth Des 2017, 17, 355. [Google Scholar]
- [24].Blagden N, de Matas M, Gavan PT, York P, Adv. Drug Deliv. Rev 2007, 59, 617. [DOI] [PubMed] [Google Scholar]
- [25].Lee MK, Lee H, Kim IW, Lee J, Pharmazie 2011, 66, 766. [PubMed] [Google Scholar]
- [26].Galati G, O’Brien PJ, Free Radic. Biol. Med 2004, 37, 287. [DOI] [PubMed] [Google Scholar]
- [27].Beecher GR, J. Nutr 2003, 133, 3248S. [DOI] [PubMed] [Google Scholar]
- [28].Ramos S, Mol. Nutr. Food Res 2008, 52, 507. [DOI] [PubMed] [Google Scholar]
- [29].Kleemann R, Verschuren L, Morrison M, Zadelaar S, van Erk MJ, Wielinga PY, Kooistra T, Atherosclerosis 2011, 218, 44. [DOI] [PubMed] [Google Scholar]
- [30].Gresele P, Cerletti C, Guglielmini G, Pignatelli P, de Gaetano G, Violi F, J. Nutr. Biochem 2011, 22, 201. [DOI] [PubMed] [Google Scholar]
- [31].Forrest ML, Koerber JT, Pack DW, Bioconjug. Chem 2003, 14, 934. [DOI] [PubMed] [Google Scholar]
- [32].Fischer D, Bieber T, Li Y, Elsässer H, Kissel T, Pharm. Res 1999, 16, 1273. [DOI] [PubMed] [Google Scholar]
- [33].Huo M, Yuan J, Tao L, Wei Y, Polym. Chem 2014, 5, 1519. [Google Scholar]
- [34].Zhang W, Zhou Y, Li X, Xu X, Chen Y, Zhu R, Yin L, Biomater. Sci 2018, 6, 1986. [DOI] [PubMed] [Google Scholar]
- [35].He Y, Nie Y, Cheng G, Xie L, Shen Y, Gu Z, Adv. Mater 2014, 26, 1534. [DOI] [PubMed] [Google Scholar]
- [36].Ma N, Li Y, Xu H, Wang Z, Zhang X, J. Am. Chem. Soc 2010, 132, 442. [DOI] [PubMed] [Google Scholar]
- [37].Ji S, Cao W, Yu Y, Xu H, Adv. Mater 2015, 27, 7740. [DOI] [PubMed] [Google Scholar]
- [38].Deng Q, Li X, Zhu L, He H, Chen D, Chen Y, Yin L, Biomater. Sci 2017, 5, 1174. [DOI] [PubMed] [Google Scholar]
- [39].Harper JK, Doebbler JA, Jacques E, Grant DM, Von Dreele RB, J. Am. Chem. Soc 2010, 132, 2928. [DOI] [PubMed] [Google Scholar]
- [40].Marti E, Heiber O, Gumma A, Huber G, Utsumi I, Nakagawa H, Miyata T, Akimoto K, Crystal modifications of (+)-catechin and pharmaceutical preparations containing them. 1985, 4,515,804. [Google Scholar]
- [41].Hergert HL, Kurth EF, J. Org. Chem 1953, 18, 521. [Google Scholar]
- [42].Halliwell B, Clement MV, Long LH, FEBS Lett. 2000, 486, 10. [DOI] [PubMed] [Google Scholar]
- [43].Hayes JD, McLellan LI, Free Radic. Res 1999, 31, 273. [DOI] [PubMed] [Google Scholar]
- [44].Villegas-Navarro A, Rosas-L E, Reyes JL, Comp. Biochem. Physiol. Part C Toxicol. Pharmacol 2003, 136, 127. [DOI] [PubMed] [Google Scholar]
- [45].Corotto F, Ceballos D, Lee A, Vinson L, Am. Biol. Teach 2010, 72, 176. [Google Scholar]
- [46].Matthijs HCP, Visser PM, Reeze B, Meeuse J, Slot PC, Wijn G, Talens R, Huisman J, Water Res. 2012, 46, 1460. [DOI] [PubMed] [Google Scholar]
- [47].Dzialowski EM, Turner PK, Brooks BW, Arch. Environ. Contam. Toxicol 2006, 50, 503. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.