

Abstract
Purpose of Review:
An update is presented regarding neural tube defects (NTDs) including spina bifida and anencephaly, which are among the most common serious birth defects world-wide. Decades of research suggest that no single factor is responsible for neurulation failure, but rather NTDs arise from a complex interplay of disrupted gene regulatory networks, environmental influences and epigenetic regulation. A comprehensive understanding of these dynamics is critical to advance NTD research and prevention.
Recent Findings:
Next-generation sequencing (NGS) has ushered in a new era of genomic insight toward NTD pathophysiology, implicating novel gene associations with human NTD risk. Ongoing research is moving from a candidate gene approach toward genome-wide, systems-based investigations that are starting to uncover genetic and epigenetic complexities that underlie NTD manifestation.
Summary:
Neural tube closure is critical for the formation of the human brain and spinal cord. Broader, more all-inclusive perspectives are emerging to identify the genetic determinants of human NTDs.
Keywords: Neural tube, spina bifida, birth defects
Introduction
Neural tube defects (NTDs) refer to a group of often severe congenital malformations of the central nervous system that arise from a failure in neural tube closure in the first month of human embryonic development. This failure can occur during the processes of primary or secondary neurulation or postneurulation skeletogenesis. With a prevalence of 0.5 – 10 per 1000 live births and considerable variability among population geographic regions and ancestral background, NTDs are estimated to result in 300,000 to 500,000 new cases annually worldwide [1]. The most common NTDs, anencephaly and myelomeningocele, are often isolated and attributable to a multi-factorial causation with an enlarging list of genes potentially conferring risk in humans. Recent evidence suggests that digenic or polygenic mutations result in NTDs whereas post-neurulation defects (e.g. encephalocele) are more often either monogenic or associated with chromosomal rearrangement and tend to be syndromic, affecting multiple organs [2].
Several developmentally relevant signaling processes critical to neural tube closure include SHH, β-catenin / WNT (canonical WNT) and non-canonical WNT / PCP, pathways (Table I for nomenclature). These pathways direct cellular differentiation and convergent extension of the neural plate to elongate the rostral-caudal axis of the embryo. Disruption of these signaling processes have been observed in both mouse and human NTDs [3, 4]. Guided by over 300 gene associations with NTDs in mouse models to date [5], next generation sequencing (NGS) efforts in humans are starting to shed light on clinically relevant risk genes. Given the polygenic nature of many NTDs, various genome-wide approaches from a systems biology perspective are needed to unravel the genetic architecture of this complex disorder.
Table I.
Gene / Pathway Symbols and Corresponding Names
| APAF1 | Apoptotic Peptidase Activating Factor 1 |
| β−catenin / WNT | Canonical β−catenin-dependent Wnt Signaling Pathway |
| BIM | BCL2 Like 11 |
| CASP9 | Caspase 9 |
| CECR2 | CECR2 Histone Acetyl-Lysine Reader |
| CELSR1–3 | Cadherin EGF LAG Seven-Pass G-Type Receptor 1–3 |
| DAAM1 | Dishevelled Associated Activator of Morphogenesis 1 |
| DISP1 | Dispatched RND Transporter Family Member 1 |
| DLC1 | DLC1 Rho GTPase Activating Protein |
| DNMT3B | DNA Methyltransferase 3 Beta |
| DVL 1/2/3 | Dishevelled Segment Polarity Protein 1/2/3 |
| FOLR1/2/3 | Folate Receptor Alpha / Beta / Gamma |
| FRAS1 | Fraser Extracellular Matrix Complex Subunit 1 |
| FREM2 | FRAS1 Related Extracellular Matrix 2 |
| FZD 3/6 | Frizzled 3/6 |
| GPR161 | G Protein-Coupled Receptor 161 |
| ITGB1 | Integrin Subunit Beta 1 |
| JNK | c-Jun N-terminal kinase |
| MTHFD1 | Methylenetetrahydrofolate Dehydrogenase, Cyclohydrolase And Formyltetrahydrofolate Synthetase 1 |
| MTHFR | Methylenetetrahydrofolate Reductase |
| MTRR | 5-Methyltetrahydrofolate-Homocysteine Methyltransferase Reductase |
| MYO1E | Myosin IE |
| PAX3 | Paired Box 3 |
| PK | Prickle Planar Cell Polarity Protein |
| PTCH1 | Patched 1 |
| PTK7 | Protein Tyrosine Kinase 7 |
| RFC1 | Replication Factor C Subunit 1 |
| SCRIB | Scribble Planar Cell Polarity Protein |
| SHH | Sonic Hedgehog |
| SLC19A1 | Solute Carrier Family 19 Member 1 |
| SLC25A3 | Solute Carrier Family 25 Member 3 |
| SMARCA4 | SWI/SNF Related, Matrix Associated, Actin Dependent Regulator of Chromatin, Subfamily A, Member 4 |
| TRIM4 | Tripartite Motif Containing 4 |
| VANGL1/2 | Vangogh-Like Planar Cell Polarity Protein 1/2 |
| WDR63 | WD Repeat Domain 63 |
| WNT / PCP | Non-canonical WNT / PCP Signaling Pathway |
| XIST | X Inactive Specific Transcript |
Following the successes of maternal folic acid supplementation in reducing NTD incidence, considerable attention in research has been trained on genes involved in folate metabolism. This metabolic pathway supports a broader set of chemical steps involving the transfer of a single carbon unit, collectively known as one-carbon metabolism, which is directly involved in several significant physiologic processes of purine and thymidine biosynthesis, glycine, serine and methionine homeostasis and epigenetic balance. Therefore, deficits or dysfunction in the context of one-carbon metabolism can have a wide-ranging impact on NTD formation. Nevertheless, at least 30% of NTD cases are folate resistant, occurring despite prenatal folic acid supplementation. Here we review findings from recent studies that probe mechanistic aspects of NTD pathophysiology and highlight key gene regulatory networks as well as epigenetic processes that lead to failed neural tube closure.
NTD Risk Genes in Key Signaling Pathways
SHH signaling is critical for many aspects of early embryonic CNS development including dorsoventral patterning within the neural tube and regulation of neural plate bending. Numerous mouse models have shown that increased activation of SHH signaling activity promotes NTD formation, while other mutant models that display deficits in formation or function of cilia— a structure that promotes NT closure – exhibit decreased SHH signaling in the spinal cord. The mechanisms through which SHH pathway activity may influence NTDs include disrupted cell proliferation, differentiation or apoptosis [6]. Numerous mouse mutants that harbor genetic variants in Shh related genes display NTD phenotypes. In humans, variants in a few negative regulators of SHH (e.g. PTCH1 and PKA) have been linked with spina bifida [7, 8], although additional potential risk genes are anticipated.
Kim et al [9**] reported agreement between mouse and human data suggesting that deleterious GPR161 variants carry increased risk in humans, particularly for spina bifida. As a ciliary G-protein coupled receptor, GPR161 negatively regulates canonical WNT and retinoic acid signaling pathways during neurulation in mouse models, and also suppresses the SHH pathway via cAMP signaling [10, 11]. Kim and colleagues found two novel, rare GPR161 missense single nucleotide variants (SNVs) in their human NTD cohorts that were predicted in silico to be deleterious and studied the SNV impact by expressing mutant protein in 293T cells [9**]. They observed mislocalization of the mutant GPR161 receptors, increased SHH activity and reduced WNT activity, consistent with the transcriptional dysregulation that they found by RNA sequence analysis of Gpr161 knockout mice at embryonic day 9.5 (E9.5). These findings add further insight into the largely unresolved interplay of SHH and WNT signaling during neurulation.
Recent studies have interrogated candidate genes in other NTD implicated pathways including PCP, a pathway directly involved in aspects of neurulation and neural tube closure. This strongly conserved non-canonical WNT pathway has a well-established role in controlling vertebrate convergent extension to shape the neural plate for closure [12]. Initiating the pathway, ligand binding triggers complex formation and subcellular localization of several interacting transmembrane (FZD, VANGL, CELSR1–3) and cytoplasmic (DVL, DIVERSIN, PK, SCRIB) proteins. Downstream signaling involves RHO-GTPases and activation of JNK kinase and scaffold protein, DAAM1, which further leads to a variety of transcriptional and cytoskeletal changes in both actin and microtubule cytoskeletal components [FIGURE 1]. The WNT / PCP pathway has been implicated in axon guidance and regulating dendritic development among other functions [13, 14].
Figure 1. Non-canonical WNT / PCP and SHH signaling pathways.
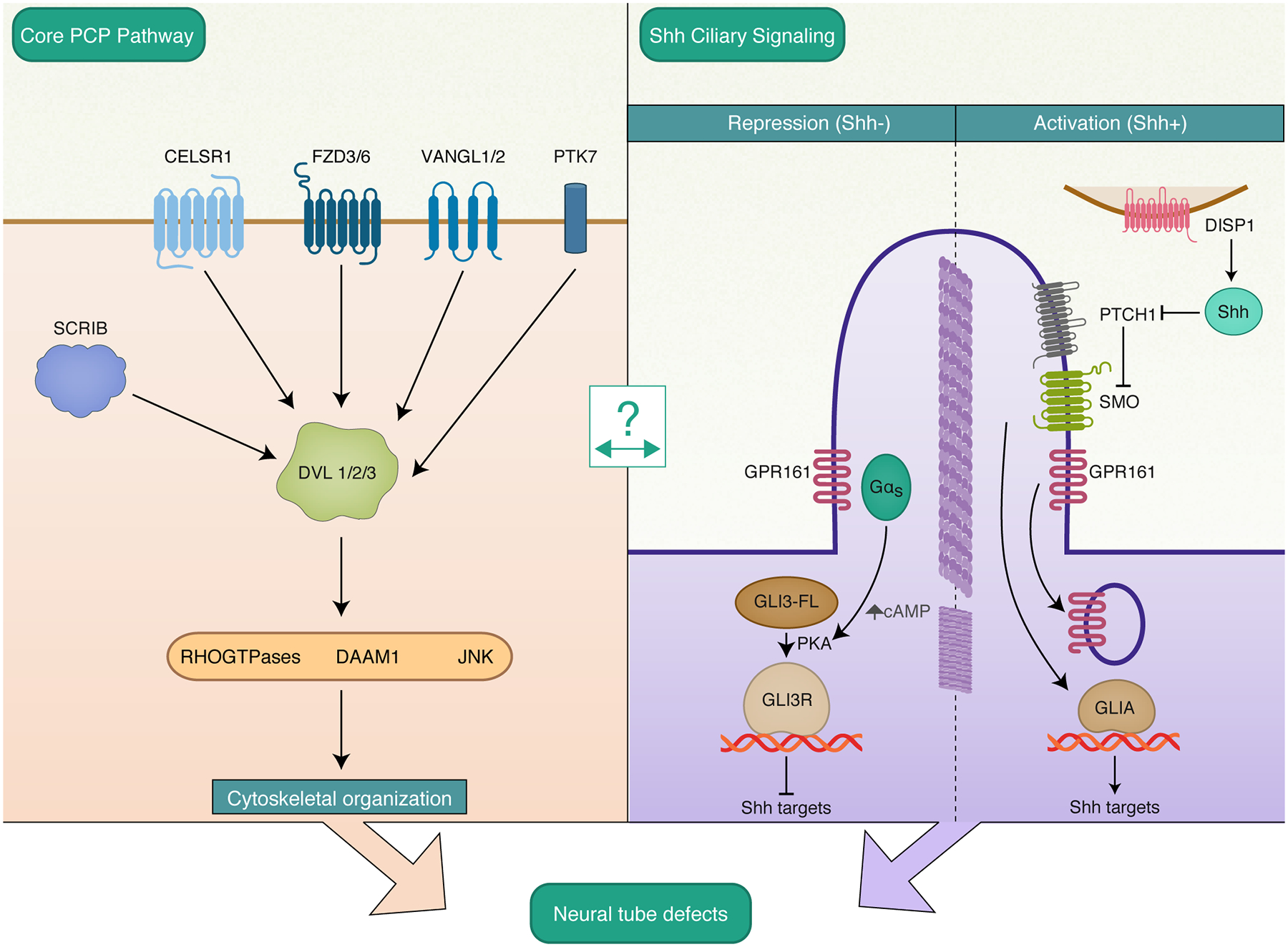
Digenic interactions in several core PCP genes are associated with NTD risk. Genetic variants in GPR161 may lead to NTDs associated with ciliary GPR161 mislocalization and dysregulated Wnt and Shh signaling. Molecular details of the cross-talk between PCP and Shh signaling remains to be elucidated. Abbreviations: CELSR1, Cadherin EGF LAG Seven-pass G-type Receptor 1; FZD3/6, Frizzled Class Receptor 3/6; VANGL1/2, Vangogh-Like Planar Cell Polarity Protein 1/2; PTK7, Protein Tyrosine Kinase 7; SCRIB, Scribble Planar Cell Polarity Protein; DVL 1/2/3, Dishevelled Segment Polarity Protein 1/2/3; DAAM1, Dishevelled Associated Activator of Morphogenesis 1/2/3; JNK, c-Jun N-terminal kinase; DISP1, Dispatched RND Transporter Family Member 1; PTCH1, Patched 1; SMO, Smoothened, Frizzled Class Receptor; GPR161, G Protein-Coupled Receptor 161.
Variants in PCP genes are increasingly implicated to confer risk for NTDs [15, 16]. Recent candidate gene approaches have interrogated PCP genes using specifically targeted DNA sequencing to find novel SNVs enriched in human NTD cases that are ripe for follow-up and functional analyses. At the same time, whole exome sequencing (WES) has begun unbiased surveys across the genome to detect the frequency of predicted deleterious mutations in NTD cases compared to unaffected controls, or search in child-parent trios, or multiplex families. One new study [17*] analyzed WES data in 8 families having two or more members affected by NTD and reanalyzed 43 trios, pooling 18 affected individual data from multiplex families and 43 affected singletons from trios. They screened for de novo and inherited mutations, using both candidate gene and genetic burden approaches. The authors found four novel loss-of-function (LOF) variants in three genes previously associated with NTDs (MTHFR, DLC1 and ITGB1), and also found enrichment in NTD cases of variants in MYO1E, a novel candidate gene involved in cytoskeletal remodeling [17*]. These results that further implicate cytoskeletal regulators like DLC1, ITGB1 an MYO1E underscore the need for further investigation into genes modulating actin-myosin dynamics and will surely involve crosstalk between cytoskeletal regulators and the WNT / PCP genes.
Another targeted panel sequencing approach [18] conducted in a cohort of 52 patients identified variants in novel genes FREM2 and DISP1 along with a relatively high prevalence of Wnt / PCP SNVs, compared to previous studies. FREM2 encodes an integral membrane protein and associates with FRAS1 forming a self-stabilizing complex in the extracellular matrix that is disrupted in Fraser syndrome and congenital diaphragmatic hernia [19, 20]. DISP1 is essential for vertebrate Hedgehog (Hh) signaling and has an important role in facilitating long-range Hh signaling [21]. Additional evidence implicating SHH in NTD suggests that SHH may help determine the temporal and spatial characteristics of cell polarity signaling via PCP as well as modulate gene expression through canonical, β-catenin / Wnt pathway activity [22].
An emerging hypothesis regarding NTD risk invokes a multi-hit or digenic interaction requirement for neurulation to fail, and while there are compelling data supporting this in mouse models, evidence is sparse in human genomic studies. The paucity of evidence for digenic and polygenic interactions in human studies is in part attributable to challenges posed by heterogeneous NTD subtypes, so that case phenotypic stratification reduces statistical power, as well as the exclusion of more common variants within genetic control databases that may nevertheless contribute to risk. A recent approach employed by Wang et al [23*] used targeted NGS to sequence Wnt / PCP genes in a cohort of 510 NTD patients, including spina bifida (232), anenecephaly (125), encephalocele (46) and multiple (99) NTD phenotypes. The authors found evidence of digenic interactions, with individual NTD cases containing rare variants (<0.01 mean allele frequency) found in CELSR1 and SCRIB, CELSR1 and DVL3 , or PTK7 and SCRIB genes. These findings indicate that NTD pathophysiology can result from digenically determined risk and suggest that grouping all NTD phenotypes together initially may be a fruitful approach to precede granular genotype-phenotype investigation.
In an elegant mouse genetic study, an insight into the recognized female predominance in NTD prevalence has further implicated digenic interactions. Delbridge et al [24**] made the surprising observation that p53 is required for Xist expression, it binds to enhancers in the X-inactivation centers, and p53 loss promotes stochastic biallelic expression of X-linked genes. Furthermore, digenic interactions were illustrated in that the 17%−60% penetrance of NTDs in p53−/− female mouse embryos increased to 100% when BIM – a pro-apoptotic gene – was also lost. These data should spark further investigation into X-linked gene interactions with autosomal variants. Intriguingly, variants in other apoptotic genes (APAF1 and CASP9) have been found in two families with folate-resistant NTDs [25], recapitulating phenotypes observed in knockout mouse models, and providing the first published report linking variants in apoptosis genes with human NTD risk. These recent contributions to the field provide strong genetic evidence supporting the hypothesis that loss or disruption of key programmed cell death genes is a significant risk factor for NTDs.
From Digenic to Polygenic and Genome-wide Modeling
A comprehensive interrogation of the genetic underpinning of NTDs must move from a candidate gene approach toward a genome-wide investigation of DNA variation, its impact on transcriptomics, as well as epigenetic modifications that underlie gene-environment interactions. Chen et al [26**] have made progress toward genome-wide investigations utilizing whole genome sequencing (WGS) toward a polygenic view of NTD risk. WGS covers not only the protein coding exons (exome) but also the other non-protein coding, 98% of the genome. In that study, computational evaluations using two different population cohorts, conservatively compared the distributions of rare, likely loss of function (LOF) variants by restricting analyses to protein-coding regions and so avoid the challenges of in silico pathogenicity prediction for non-coding variants. The authors found a significant statistical enrichment in rare singleton loss of function variants (SLoFVs) in NTD cases compared to controls from the 1000 Genomes Project and ExAC databases. The accumulation of SLoFVs in NTD affected individuals, compared to matched controls, was similar across two different ancestral populations thereby indicating that NTD risk in humans rises with the enrichment of deleterious variants in an individual. With polygenicity in mind, computational analyses utilizing genomic variant thresholds or polygenic risk scores in NTD biology will likely prove to be useful.
Protein encoding, exomes, though important, represent only around 3% of the genome. The range and diversity of genomic variation in NTD affected individuals must include structural variants (SVs) and SNVs in non-protein coding regions. SVs generally comprise at least 50 base pairs, and are investigated using both NGS and array-based platforms. SVs may arise as gains or losses in genetic material, e.g. copy number variants (CNVs), or as balanced rearrangements including inversions and translocations. Although there are more SNVs and small insertions/deletions (indels) compared to SVs in any given genome, SVs due to their range of sizes, account for more affected base pairs than SNVs and indels combined [27]. Moreover, SVs have recently been predicted to be responsible for 3.5–6.8% of expression quantitative trait loci (eQTL) sites [28], which are increasingly associated with complex genetic disorders including diabetes and Alzheimer’s disease. Despite the inherent challenges in SV detection from WGS data, recent approaches utilizing a combinatorial algorithmic strategy are showing great promise [29]. Accurate detection of this underexplored variation form can, in principle, help refine our understanding of the gene regulatory networks that are essential for proper neural tube closure.
Little is yet known about the contributions of SVs to human NTD risk. Array-based analyses have found enrichment of CNVs encompassing ciliogenic genes in a cohort of 85 NTD-affected embryos [30] and have highlighted haploinsufficiency of human PAX3 as a potential for a single gene cause of NTD [31]. More recently, Hofmeister et al [32*] described likely pathogenic CNVs in an NTD cohort using high-resolution copy number screening, including an intragenic deletion spanning exons 14–17 in the WDR63 gene. They supported the claim that this deletion is the likely cause of human occipital encephalocele by CRISPR/Cas9 mediated introduction of this deletion into a zebrafish model that also subsequently exhibited abnormal CNS development. In a neural tube-associated malformation, human congenital hydrocephalus, exome sequencing identified de novo variants, including CNV duplications at the SHH locus [33]. Overcoming the inherent technical challenges in SV detection from NGS data promises to enhance statistical power and reveal novel genes and networks relevant to NTD physiology.
One-Carbon Metabolism and Epigenetic Regulation
While NTDs can be resistant to folic acid supplementation, folate deficiency and variants in a growing number of genes involved in folate and one-carbon metabolism have an established risk [34]. Through coupled reactions that take place in the mitochondria and cytosol, folate metabolism supports one-carbon transfer events, which contribute purine and thymidine biosynthesis and the most ubiquitous methyl-donor, SAM, required for methylation of DNA, RNA, proteins and lipids [FIGURE 2]. The demand for one-carbon units is highest during fetal development. Disruption of DNA synthesis, required for cell proliferation, and DNA or histone methylation dependent epigenetic regulation of gene expression likely contribute to NTDs [35]. Human NTD-associated variants that impair folate one-carbon metabolism are coming to light. A recent study sequenced the exons and flanking introns from a cohort of 348 myelomeningocele subjects [36] and identified eight novel variants in a folate transporter (SLC19A1) as well as twenty novel variants in folate receptors (FOLR1, FOLR2, FOLR3), thus strengthening associations between folate intracellular transport and NTD risk. Another study [37] found additional polymorphisms in folate metabolic pathway genes (MTHFR, MTHFD1, MTRR, RFC1) as maternal NTD risk factors.
Figure 2. Key processes in folate one-carbon metabolism.
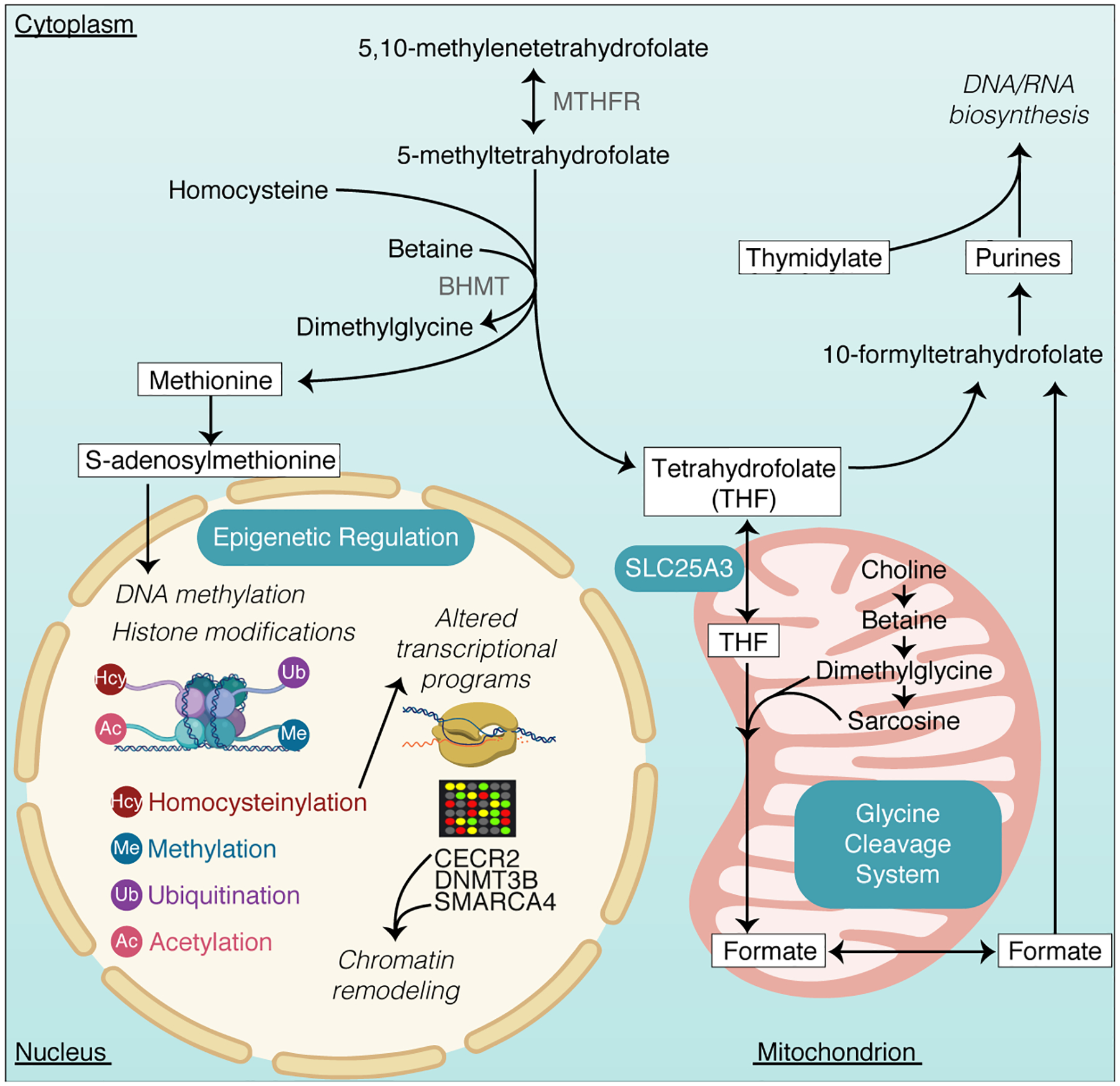
Compartmentalized reactions in the cytosol and mitochondrion are depicted. Variants in several genes regulating these steps have been linked with NTD risk and various epigenetic modifications - including DNA methylation requiring the primary methyl donor, S-adenosylmethionine (SAM) and histone homocysteinylation - have been shown to influence neural tube closure through aberrant expression of genes critical for neurulation. MTHFR, Methylenetetrahydrofolate Reductase; BHMT, Betaine-Homocysteine Methyltransferase; SLC25A3, Solute Carrier Family 25 Member 3; CECR2, CECR2 Histone Acetyl-Lysine Reader; DNMT3B, DNA Methyltransferase 3 Beta; SMARCA4, SWI/SNF Related, Matrix Associated, Actin Dependent Regulator of Chromatin, Subfamily A, Member 4.
Kim et al [38*] showed that inactivating the mitochondrial folate transporter Slc25a3 in mice induces folate-unresponsive cranial NTDs that can be mostly prevented with formate supplementation. These investigators corroborated their findings in mice by finding loss of function SLC25A3 variants in humans with NTDs. Further studies may enable determination of individual variant specificity that would indicate superiority of glycine or formate supplementation in certain individuals for prevention of folic acid resistant NTDs.
How one-carbon metabolism feeds into epigenetic regulation to further influence NTD formation deserves greater attention. As the major source of methyl donor for enzymatic reactions, dietary folate intake and metabolism comprise an important environmental influence on gene expression through DNA and/or histone methylation, but the mechanistic details relating to NTD risk are incompletely understood. Zhang et al [39] found that spinal cord tissues from human NTD fetuses had aberrant genome-wide methylation patterns compared to sex- and gestational week-matched controls. That study found hypomethylation patterns in TRIM4 associated with increased TRIM4 mRNA and protein expression that was postulated to influence NTD pathogenesis via immune-driven pathways.
Epigenetic modifications beyond methylation should be considered in NTD pathogenesis. A recent study [40*] illustrates histone homocysteinylation as influencing the expression of NTD critical genes. Homocysteine is an intermediate of methionine metabolism and has been associated with NTDs in a number of studies [41] though largely unknown mechanism(s). Investigators have observed that chromatin modulator, histone 3 (H3), is homocystenylated on lysine 79 (H3K79Hcy) when cellular homocysteine levels are elevated in human fetal brains. Chromatin immunoprecipitation followed by DNA sequencing (ChIP-seq) and RNA sequence analyses showed that elevated H3K79Kcy levels lead to decreased expression of several genes involved in NT closure (CECR2, SMARCA4, DNMT3B), suggesting that higher homocysteine levels contribute to NTD onset by this mechanism. SMARCA4 and CECR2 both function in chromatin remodeling and DNMT3B is a DNA methyltransferase, which further implicates crosstalk between homocysteine and methylation reactions. Other recently reported epigenetic mechanisms, such as histone ubiquitination and disruption of HOX gene expression, are emerging as potential NTD risk factors [42]. Such studies underscore the importance of genome-wide epigenetic investigations to illuminate the gene expression mechanisms underpinning of NTDs.
Genetic and epigenetic mechanisms influencing NTD pathophysiology have recently been investigated using three-dimensional neural cysts derived from human embryonic stem cells to recapitulate human NT formation in vitro. Valensisi et al [43] showed that 3-D neural cysts offer a distinct transcriptional and epigenomic landscapes that differ from two-dimensional neural stem cell cultures. These model systems can help bridge the gap between the environmentally influenced epigenetic responses to folate and their association with gene regulatory networks in human NTDs.
Conclusion
The advances in NGS technologies position us on the frontier of emerging reliable genetic tests with predictive value for NTD risk, insight into NTD polygenicity and opportunities for interrogating the diversity of genome variation. Studies are elucidating the complex roles that epigenetic modifications have in NTD causation. Increasing evidence for one-carbon transfer deficits implicate dysregulation of epigenetic mechanisms including, but not limited to, DNA methylation. Lastly, 3-D in vitro modeling using human cells can help disentangle the genetic and epigenetic landscape and further support computational genomic approaches toward understanding the complexities underlying NTD mechanisms.
Key Points.
Complex interactions between genetic, environmental and epigenetic phenomena give rise to NTDs.
The non-canonical WNT / PCP pathway contributes to NTD formation via cytoskeletal organization and potential crosstalk with SHH signaling.
Understanding the genetic basis of NTDs necessitates investigating both digenic and polygenic interactions as well as a broader range of genome variation.
Folate one-carbon metabolism carries a well-known NTD risk and bridges genetic and environmental processes through its critical regulation of epigenetic phenomena.
Acknowledgements
We would like to thank Christina Usher for assistance with the figure design
Financial Support and Sponsorship
Supported by P01HD067244 (MER) and T32HD060600 (PW)
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to report
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
* of special interest
** of outstanding interest
- 1.Copp AJ, Stanier P, Greene NDE Genetic Basis of Neural Tube Defects 2017. Textbook of Pediatric Neurosurgery. Springer, Cham, pp 1–28. [Google Scholar]
- 2.Rasmussen SA, Frias JL. Genetics of syndromic neural tube defects 2016. Neural tube defects: from origin to treatment. Oxford University Press, Oxford, pp 185–197. [Google Scholar]
- 3.López-Escobar B, Caro-Vega JM, Vijayraghavan DS, et al. The non-canonical Wnt-PCP pathway shapes the mouse caudal neural plate. Development. 2018. May 8;145(9). pii: dev157487. doi: 10.1242/dev.157487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Juriloff DM, Harris MJ. A consideration of the evidence that genetic defects in planar cell polarity contribute to the etiology of human neural tube defects. Birth Defects Res A Clin Mol Teratol. 2012. October;94(10):824–40. doi: 10.1002/bdra.23079. Epub 2012 Sep 28. Review. [DOI] [PubMed] [Google Scholar]
- 5.Harris MJ, Juriloff DM. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res A Clin Mol Teratol. 2010. August;88(8):653–69. doi: 10.1002/bdra.20676. Review. [DOI] [PubMed] [Google Scholar]
- 6.Murdoch JN, Copp AJ. The relationship between sonic Hedgehog signaling, cilia, and neural tube defects. Birth Defects Res A Clin Mol Teratol. 2010. August;88(8):633–52. doi: 10.1002/bdra.20686. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Z, Wang L, Shangguan S, et al. Association between PTCH1 polymorphisms and risk of neural tube defects in a Chinese population. Birth Defects Res A Clin Mol Teratol. 2013. June;97(6):409–15. doi: 10.1002/bdra.23152. Epub 2013 Jun 13. [DOI] [PubMed] [Google Scholar]
- 8.Wu J, Lu X, Wang Z, et al. Association between PKA gene polymorphism and NTDs in high risk Chinese population in Shanxi. Int J Clin Exp Pathol. 2013. November 15;6(12):2968–74. eCollection 2013. [PMC free article] [PubMed] [Google Scholar]
- 9.**. Kim SE, Lei Y, Hwang SH, et al. Dominant negative GPR161 rare variants are risk factors of human spina bifida. Hum Mol Genet. 2019. January 15;28(2):200–208. doi: 10.1093/hmg/ddy339. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper corroborates sound mouse data with human sequencing data in studying the impact variants in GPR161 have on NTD risk through dysregulated Shh and Wnt signaling.
- 10.Li BI, Matteson PG, Ababon MF, et al. The orphan GPCR, Gpr161, regulates the retinoic acid and canonical Wnt pathways during neurulation. Dev Biol. 2015. June 1;402(1):17–31. doi: 10.1016/j.ydbio.2015.02.007. Epub 2015 Mar 6. [DOI] [PubMed] [Google Scholar]
- 11.Mukhopadhyay S, Wen X, Ratti N, et al. The ciliary G-protein-coupled receptor Gpr161 negatively regulates the Sonic hedgehog pathway via cAMP signaling. Cell. 2013. January 17;152(1–2):210–23. doi: 10.1016/j.cell.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 12.Nikolopoulou E, Galea GL, Rolo A, et al. Neural tube closure: cellular, molecular and biomechanical mechanisms. Development. 2017. February 15;144(4):552–566. doi: 10.1242/dev.145904. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosso SB, Sussman D, Wynshaw-Boris A, et al. Wnt signaling through Dishevelled, Rac and JNK regulates dendritic development. Nat Neurosci. 2005. January;8(1):34–42. Epub 2004 Dec 19. [DOI] [PubMed] [Google Scholar]
- 14.Shafer B, Onishi K, Lo C, et al. Vangl2 promotes Wnt/planar cell polarity-like signaling by antagonizing Dvl1-mediated feedback inhibition in growth cone guidance. Dev Cell. 2011. February 15;20(2):177–91. doi: 10.1016/j.devcel.2011.01.002. Erratum in: Dev Cell. 2011 Mar 15;20(3):407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi Z, Yang X, Li BB, et al. Novel Mutation of LRP6 Identified in Chinese Han Population Links Canonical WNT Signaling to Neural Tube Defects. Birth Defects Res. 2018. January 15;110(1):63–71. doi: 10.1002/bdr2.1122. Epub 2017 Sep 29. [DOI] [PubMed] [Google Scholar]
- 16.Lei Y, Kim SE, Chen Z, et al. Variants identified in PTK7 associated with neural tube defects. Mol Genet Genomic Med. 2019. April;7(4):e00584. doi: 10.1002/mgg3.584. Epub 2019 Jan 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.*.Lemay P, De Marco P, Traverso M, et al. Whole exome sequencing identifies novel predisposing genes in neural tube defects. Mol Genet Genomic Med. 2019. January;7(1):e00467. doi: 10.1002/mgg3.467. Epub 2018 Nov 10. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper illustrates the utility of WES in de novo and inherited genetic analyses and the discovery of novel NTD risk genes.
- 18.Beaumont M, Akloul L, Carré W, et al. Targeted panel sequencing establishes the implication of planar cell polarity pathway and involves new candidate genes in neural tube defect disorders. Hum Genet. 2019. April;138(4):363–374. doi: 10.1007/s00439-019-01993-y. Epub 2019 Mar 5. [DOI] [PubMed] [Google Scholar]
- 19.Yu Q, Lin B, Xie S, et al. A homozygous mutation p.Arg2167Trp in FREM2 causes isolated cryptophthalmos. Hum Mol Genet. 2018. July 1;27(13):2357–2366. doi: 10.1093/hmg/ddy144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jordan VK, Beck TF, Hernandez-Garcia A, et al. The role of FREM2 and FRAS1 in the development of congenital diaphragmatic hernia. Hum Mol Genet. 2018. June 15;27(12):2064–2075. doi: 10.1093/hmg/ddy110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tukachinsky H, Kuzmickas RP, Jao CY, et al. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2012. August 30;2(2):308–20. doi: 10.1016/j.celrep.2012.07.010. Epub 2012 Aug 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Onishi K, Zou Y. Sonic Hedgehog switches on Wnt/planar cell polarity signaling in commissural axon growth cones by reducing levels of Shisa2. Elife. 2017. September 8;6 pii: e25269. doi: 10.7554/eLife.25269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Xiao Y, et al. Digenic variants of planar cell polarity genes in human neural tube defect patients. Mol Genet Metab. 2018. May;124(1):94–100. doi: 10.1016/j.ymgme.2018.03.005. Epub 2018 Mar 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.**.Delbridge ARD, Kueh AJ, Ke F, et al. Loss of p53 Causes Stochastic Aberrant X-Chromosome Inactivation and Female-Specific Neural Tube Defects. Cell Rep. 2019. April 9;27(2):442–454.e5. doi: 10.1016/j.celrep.2019.03.048. [DOI] [PubMed] [Google Scholar]; This study provides robust data suggesting partial X inactivation as a mechanism in female-specific NTDs in mice and propose that loss of p53-induced apoptosis predisposes to NTDs.
- 25.Spellicy CJ, Norris J, Bend R, et al. Key apoptotic genes APAF1 and CASP9 implicated in recurrent folate-resistant neural tube defects. Eur J Hum Genet. 2018. March;26(3):420–427. doi: 10.1038/s41431-017-0025-y. Epub 2018 Jan 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.**.Chen Z, Lei Y, Zheng Y, et al. Threshold for neural tube defect risk by accumulated singleton loss-of-function variants. Cell Res. 2018. October; 28(10):1039–1041. doi: 10.1038/s41422-018-0061-3. Epub 2018 Jul 5. [DOI] [PMC free article] [PubMed] [Google Scholar]; The computational approaches in this study are comprised of unbiased genomic analyses and present a strong framework for a polygenic view of NTD risk.
- 27.Spielmann M, Lupiáñez DG, Mundlos S. Structural variation in the 3D genome. Nat Rev Genet. 2018. July;19(7):453–467. doi: 10.1038/s41576-018-0007-0. Review. [DOI] [PubMed] [Google Scholar]
- 28.Chiang C, Scott AJ, Davis JR, et al. The impact of structural variation on human gene expression. Nat Genet. 2017. May;49(5):692–699. doi: 10.1038/ng.3834. Epub 2017 Apr 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kosugi S, Momozawa Y, Liu X, et al. Comprehensive evaluation of structural variation detection algorithms for whole genome sequencing. Genome Biol. 2019. June 3;20(1):117. doi: 10.1186/s13059-019-1720-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Shen Y, Gao Y, et al. Detection of copy number variants reveals association of cilia genes with neural tube defects. PLoS One. 2013;8(1):e54492. doi: 10.1371/journal.pone.0054492. Epub 2013 Jan 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goumy C, Gay-Bellile M, Eymard-Pierre E, et al. De novo 2q36.1q36.3 interstitial deletion involving the PAX3 and EPHA4 genes in a fetus with spina bifida and cleft palate. Birth Defects Res A Clin Mol Teratol. 2014. June;100(6):507–11. doi: 10.1002/bdra.23246. Epub 2014 Apr 18. [DOI] [PubMed] [Google Scholar]
- 32.Hofmeister W, Pettersson M, Kurtoglu D, et al. Targeted copy number screening highlights an intragenic deletion of WDR63 as the likely cause of human occipital encephalocele and abnormal CNS development in zebrafish. Hum Mutat. 2018. April;39(4):495–505. doi: 10.1002/humu.23388. Epub 2018 Jan 11. [DOI] [PubMed] [Google Scholar]
- 33.Furey CG, Choi J, Jin SC, et al. De Novo Mutation in Genes Regulating Neural Stem Cell Fate in Human Congenital Hydrocephalus. Neuron. 2018. July 25;99(2):302–314.e4. doi: 10.1016/j.neuron.2018.06.019. Epub 2018 Jul 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Au KS, Findley TO, Northrup H. Finding the genetic mechanisms of folate deficiency and neural tube defects-Leaving no stone unturned. Am J Med Genet A. 2017. November;173(11):3042–3057. doi: 10.1002/ajmg.a.38478. Epub 2017 Sep 25. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martiniova L, Field MS, Finkelstein JL, et al. Maternal dietary uridine causes, and deoxyuridine prevents, neural tube closure defects in a mouse model of folate-responsive neural tube defects. Am J Clin Nutr. 2015. April;101(4):860–9. doi: 10.3945/ajcn.114.097279. Epub 2015 Jan 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Findley TO, Tenpenny JC, O’Byrne MR, et al. Mutations in folate transporter genes and risk for human myelomeningocele. Am J Med Genet A. 2017. November;173(11):2973–2984. doi: 10.1002/ajmg.a.38472. Epub 2017 Sep 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai CQ, Fang YL, Shu JB, et al. Association of neural tube defects with maternal alterations and genetic polymorphisms in one-carbon metabolic pathway. Ital J Pediatr. 2019. March 14;45(1):37. doi: 10.1186/s13052-019-0630-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.*.Kim J, Lei Y, Guo J, et al. Formate rescues neural tube defects caused by mutations in Slc25a32. Proc Natl Acad Sci U S A. 2018. May 1;115(18):4690–4695. doi: 10.1073/pnas.1800138115. Epub 2018 Apr 16. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides interesting data on mitochondrial folate transporter variants in mice and partial rescue with formate supplementation.
- 39.Zhang H, Guo Y, Gu H, et al. TRIM4 is associated with neural tube defects based on genome-wide DNA methylation analysis. Clin Epigenetics. 2019. February 1;11(1):17. doi: 10.1186/s13148-018-0603-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.*.Zhang Q, Bai B, Mei X, et al. Elevated H3K79 homocysteinylation causes abnormal gene expression during neural development and subsequent neural tube defects. Nat Commun. 2018. August 24;9(1):3436. doi: 10.1038/s41467-018-05451-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows strong evidence for homocysteinylation as a histone modification and its subsequent potential influence on NTD risk.
- 41.Yang M, Li W, Wan Z, et al. Elevated homocysteine levels in mothers with neural tube defects: a systematic review and meta-analysis. J Matern Fetal Neonatal Med. 2017. September;30(17):2051–2057. doi: 10.1080/14767058.2016.1236248. Epub 2016 Oct 3. Review. [DOI] [PubMed] [Google Scholar]
- 42.Lin Y, Yu J, Wu J, et al. Abnormal level of CUL4B-mediated histone H2A ubiquitination causes disruptive HOX gene expression. Epigenetics Chromatin. 2019. April 16;12(1):22. doi: 10.1186/s13072-019-0268-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valensisi C, Andrus C, Buckberry S, et al. Epigenomic Landscapes of hESC-Derived Neural Rosettes: Modeling Neural Tube Formation and Diseases. Cell Rep. 2017. August 8;20(6):1448–1462. doi: 10.1016/j.celrep.2017.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]