

Abstract
Background
Post traumatic stress disorder (PTSD) is a prevalent and disabling disorder. Evidence that PTSD is characterised by specific psychobiological dysfunctions has contributed to a growing interest in the use of medication in its treatment.
Objectives
To assess the effects of medication for post traumatic stress disorder.
Search methods
We searched the Cochrane Depression, Anxiety and Neurosis Group specialised register (CCDANCTR‐Studies) on 18 August 2005, the Cochrane Central Register of Controlled Trials (CENTRAL) (The Cochrane Library issue 4, 2004), MEDLINE (January 1966 to December 2004), PsycINFO (1966 to 2004), and the National PTSD Center Pilots database. Reference lists of retrieved articles were searched for additional studies.
Selection criteria
All randomised controlled trials (RCTs) of pharmacotherapy for PTSD.
Data collection and analysis
Two raters independently assessed RCTs for inclusion in the review, collated trial data, and assessed trial quality. Investigators were contacted to obtain missing data. Summary statistics were stratified by medication class, and by medication agent for the selective serotonin reuptake inhibitors (SSRIs). Dichotomous and continuous measures were calculated using a random effects model, heterogeneity was assessed, and subgroup/sensitivity analyses were undertaken.
Main results
35 short‐term (14 weeks or less) RCTs were included in the analysis (4597 participants). Symptom severity for 17 trials was significantly reduced in the medication groups, relative to placebo (weighted mean difference ‐5.76, 95% confidence intervals (CI) ‐8.16 to ‐3.36, number of participants (N) = 2507). Similarly, summary statistics for responder status from 13 trials demonstrated overall superiority of a variety of medication agents to placebo (relative risk 1.49, 95% CI 1.28 to 1.73, number needed to treat = 4.85, 95% CI 3.85 to 6.25, N = 1272). Medication and placebo response occurred in 59.1% (N = 644) and 38.5% (628) of patients, respectively. Of the medication classes, evidence of treatment efficacy was most convincing for the SSRIs.
Medication was superior to placebo in reducing the severity of PTSD symptom clusters, comorbid depression and disability. Medication was also less well tolerated than placebo. A narrative review of 3 maintenance trials suggested that long term medication may be required in treating PTSD.
Authors' conclusions
Medication treatments can be effective in treating PTSD, acting to reduce its core symptoms, as well as associated depression and disability. The findings of this review support the status of SSRIs as first line agents in the pharmacotherapy of PTSD, as well as their value in long‐term treatment. However, there remain important gaps in the evidence base, and a continued need for more effective agents in the management of PTSD.
Plain language summary
Medication for post traumatic stress disorder
Post traumatic stress disorder (PTSD) occurs after exposure to significant trauma and results in enormous personal and societal costs. Although traditionally treated with psychotherapy, there is increasing recognition of a theoretical basis for medication treatments. This was a systematic review of 35 short‐term randomised controlled trials of pharmacotherapy for PTSD (4597 participants). A significantly larger proportion of patients responded to medication (59.1%) than to placebo (38.5%) (13 trials, 1272 participants). Symptom severity was significantly reduced in 17 trials (2507 participants). The largest trials showing efficacy were of the selective serotonin reuptake inhibitors, with long‐term efficacy also observed for these medications.
Background
Although the phenomenon of post traumatic stress disorder (PTSD) has long been recognised (for example as "shell shock" or "combat neurosis"), it is only relatively recently that this disorder has been officially recognised in the psychiatric nomenclature (APA 1980). Diagnostic criteria for PTSD provided by the 3rd edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM‐III) encouraged research on the epidemiology, psychobiology, and treatment of PTSD. Subsequent epidemiological research determined that the disorder is highly prevalent in a wide range of settings, particularly in those subjects who have been exposed to significant traumas (Breslau 1991; Davidson 1991; Kessler 1995). In addition, there is growing evidence that PTSD results in enormous personal and societal costs; this is based on chronicity of symptoms, high comorbidity of psychiatric and medical disorders, marked functional impairment, and estimations of economic costs (Solomon 1997; Brunello 2001).
By definition prior psychological trauma plays a causal role in PTSD, and psychotherapy has been widely employed in its management. Although psychodynamic psychotherapy has long been the mainstay of treatment, there have been few controlled studies of this modality (Brom 1989; Gersons 2000). Furthermore, the value of so‐called psychological debriefing in the immediate aftermath of trauma remains to be proven (Rose 1998; Rose 2002). Nevertheless, there is a growing body of evidence demonstrating that cognitive‐behavioural and similar psychotherapies are indeed effective in the treatment of PTSD (Keane 1989; Solomon 1992; Glynn 1995; Sherman 1998; van Etten 1998; Harvey 2003; Bisson 2005; Bradley 2005; NICE 2005).
There has also been increasing recognition, however, that PTSD is characterised by specific psychobiological dysfunctions (Yehuda 1995; Bonne 2004; Charney 2004), so providing a rationale for the use of medication treatments. PTSD is characterised by different symptom clusters, including intrusive/re‐experiencing, avoidant/numbing, and hyperarousal symptoms, and it is possible that each is mediated by different neurobiological mechanisms (Charney 1993), which may be normalised by specific pharmacological interventions. Certainly, there is growing evidence for rather specific dysregulations of neurotransmitter systems (including the serotonin, noradrenaline, and dopamine systems) and neuroendocrine systems (including the hypothalamus‐pituitary‐adrenal axis), as well as for structural and functional neuranatomical abnormalities in PTSD (Charney 1993; Yehuda 1995; Canive 1997; Connor 1998; Hull 2002; Bremner 2004).
Thus, whereas an older model was that medications might be valuable primarily as an adjunct to psychotherapy techniques in post‐traumatic reactions (Sargent 1940), contemporary psychobiological theory speculates that comorbid substance use in PTSD may represent an attempt at "self‐medication" and that prescribed medication may be able to play a primary role in preventing or reversing the dysfunctions of PTSD (Charney 1993; Charney 2004). Furthermore, several psychiatric disorders are often found comorbid with PTSD disorders (Kessler 1995), and certain of these are known to respond to medication. Indeed, the position that medication treatment may be useful in PTSD seems to have gained gradually increasing acceptance (van der Kolk 1983; Wise 1983; Friedman 1988; Friedman 1991; Faustman 1989; Walker 1989; Silver 1990; Allodi 1991; Davidson 1992; Davidson 1997a; Davidson 2000; Marshall 1996; Marshall 1998a; Shalev 1996; Connor 1998; Foa 1999; Cyr 2000; Marshall 2000; Asnis 2004; Ursano 2004).
Early reports of the pharmacotherapy of PTSD focused on the tricyclic antidepressants (TCAs) and the irreversible monoamine oxidase inhibitors (MAOIs) (Hogben 1981; White 1983; Burstein 1984; Milanes 1984; Falcon 1985; Bleich 1986; Davidson 1987; Lerer 1987; Frank 1988; Shestatzky 1988; Davidson 1989; Irwin 1989; Reist 1989; Olivera 1990; Chen 1991; Kosten 1991; Basoglu 1992; Rubin 1993; Demartino 1995). More recent work has focused on the selective serotonin reuptake inhibitors (SSRIs) (Davidson 1990;Burdon 1991; McDougle 1991; De Boer 1992; Dominiak 1992; Shay 1992; Nagy 1993; Fichtner 1994; Kline 1994; van der Kolk 1994; Brady 1995; Marmar 1996; Nagy 1996; Rothbaum 1996; Connor 1998; Davidson 1998a; Marshall 1998b; Marshall 2001; Brady 2000; Hertzberg 2000; Smajkic 2001; Tucker 2001; Martenyi 2002a; Zohar 2002; Tucker 2003; Brady 2004), and the serotonin antagonists and reuptake inhibitors (SARIs) (Liebowitz 1989; Hertzberg 1996; Hertzberg 1998; Hidalgo 1999).
Several other newly introduced antidepressants have also been studied (Katz 1994; Baker 1995 a; Neal 1997; Canive 1998; Davidson 1998a; Hamner 1998; Connor 1999; Davis 2000; Davis 2001; Davidson 2003). In addition, benzodiazepines (Dunner 1985; Lowenstein 1988; Braun 1990), beta‐blockers (Kolb 1984; Famularo 1988), buspirone (Simpson 1991; Wells 1991; Duffy 1992; Duffy 1994; LaPorta 1992; Fichtner 1994), clonidine (Kolb 1984; Kinzie 1989; Harmon 1996) and guanfacine (Horrigan 1996), cyproheptadine (Brophy 1991; Gupta 1998), d‐cycloserine (Heresco‐Levy 2002), inositol (Kaplan 1996), mood‐stabilizers (Kitchner 1985; Lipper 1986; Stewart 1986; van der Kolk 1987; Wolf 1988; Irwin 1989; Fichtner 1990; Fesler 1991; Szymanski 1991; Keck 1992; Forster 1994; Looff 1995; Ford 1996; Hertzberg 1999); typical (Bleich 1986; Dillard 1993) and atypical neuroleptics (Hamner 1996; Leyba 1998; Izrayelit 1998; Burton 1999; Butterfield 2001; Hamner 2003); and opioids (Glover 1993) have also received attention.
A systematic review of studies of pharmacotherapy for PTSD may be useful in tackling several questions for the field. Firstly, is pharmacotherapy in fact an effective form of treatment in PTSD? Given the preponderance of psychological models and evidence for the efficacy of certain forms of psychotherapy in PTSD (Bisson 2005), the role of pharmacotherapy remains debatable for many. In a recently published guideline for the treatment of PTSD, the National Institute of Clinical Evidence (NICE) recommended that preference be given to trauma‐focused psychological therapy over pharmacotherapy as a routine first line treatment for this disorder (NICE 2005). For those who accept a more dominant role for pharmacotherapy, questions about appropriate dose and duration arise, with current clinical recommendations suggesting that the SSRIs, for example, are prescribed at doses that increase to maximally effective/tolerated levels over a period of at least eight weeks (Foa 1999; Ballenger 2000; Ballenger 2004).
Secondly, are particular medication classes more effective in the treatment of symptoms and/or more acceptable to the patient in terms of adverse events than others? The use of novel agents (such as the serotonergic antidepressants) for PTSD in recent years raises the question of how these compare with older agents. Some recommendations, such as the expert consensus guideline series for the treatment of post traumatic stress disorder (Foa 1999), have suggested that the SSRIs, nefazodone, and venlafaxine are first‐line medications for the treatment of PTSD , with benzodiazepines and mood‐stabilisers having a role in patients with certain kinds of symptoms. Other recommendations have highlighted paroxetine and mirtazapine (NICE 2005). Support for such recommendations requires ongoing assessment of the literature on RCTs.
Thirdly, can a systematic review of RCTs provide information about the most important factors affecting pharmacotherapy response? Clinical factors, such as the kind of pre‐existing trauma (e.g. combat‐related or not), the duration of symptoms, and the presence of comorbid depression, early childhood trauma, and "secondary gain" for symptoms (for example, patients in ongoing litigation or receiving financial compensation) have all been suggested to play a role (Davidson 1993; van der Kolk 1994; Marshall 1998b; Davidson 2000). Methodological factors such as the duration of the trial or inclusion of patients with a minimal degree of symptom severity, may also affect treatment response. It is possible that the database of RCTs in PTSD may include information about some of these variables.
A number of reviews of the pharmacotherapy of PTSD have indeed been published in recent years (van der Kolk 1983; Friedman 1988; Friedman 1991; Davidson 1992; Davidson 1997a; Marshall 1996; Marshall 1998a; Solomon 1992; Shalev 1996; Otto 1996; Connor 1998; Albucher 2002; Asnis 2004). These reviews have been useful in summarising the existing research, pointing to methodological flaws, and outlining areas for future research. Nevertheless, few of these reviews have employed a systematic search strategy. It has recently been determined that even MEDLINE searches may miss over half of all RCTs in specialised health care journals (Hopewell 2002). Furthermore, few studies have estimated the effects of medication (Penava 1996; Davidson 1997a; van Etten 1998). Interestingly, in their meta‐analysis, Penava (Penava 1996) noted that effect size correlated with increased serotonergic specificity of the antidepressant. Further reviews in this area need to adhere to Cochrane Collaboration (Mulrow 1997) or similar (Moher 1999) guidelines for systematic identification of trials, investigation of sources of heterogeneity, measurement of methodological quality, and estimation of the effects of intervention.
The authors aimed to undertake a systematic review of randomised controlled (RCTs) of the pharmacotherapy of post traumatic stress disorder, following the guidelines and using the software of the Cochrane Collaboration.
Objectives
1. To identify and review all RCTs, including placebo controlled and comparative trials, of the pharmacotherapy of post traumatic stress disorder (PTSD), whether published or unpublished.
2. To provide an estimate of the effects of medication in reducing PTSD symptoms.
3. To determine whether particular classes of medication are more effective and/or acceptable than others in the treatment of PTSD.
4. To identify which factors (clinical, methodological) predict response to pharmacotherapy.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (placebo controlled and comparative trials) completed prior to the end of 2004 were considered for inclusion. Publication is not necessarily related to study quality and indeed publication may imply certain biases (Easterbrook 1991; Dickersin 1992; Scherer 1994), so unpublished abstracts and reports were also considered. Studies were not limited to any particular language. Differences between trials (for example, sample size, trial duration) were not used to exclude studies.
Types of participants
All studies of subjects with PTSD (as determined by the study authors) were included. There was no restriction on the basis of different diagnostic criteria for PTSD, duration and severity of PTSD symptoms, presence of comorbid disorders, or age and gender of subjects. However, these descriptors were tabulated in order to address the question of their possible impact on the effects of medication.
Types of interventions
The review focused only on medication treatments, in which the comparator was a placebo (active or non‐active) or other medication. A parallel review of the psychotherapy of PTSD has recently been completed by members of the Cochrane Collaboration (Bisson 2005). Trials in which ongoing pharmacotherapy is supplemented with augmentation medication (Hamner 1997;Heresco‐Levy 2002;Stein 2002; Hamner 2003;Monnelly 2003;Raskind 2003) will be included in a separate review of pharmacotherapy for treatment‐resistant anxiety disorders (Dhansay 2005). RCTs of medication prophylaxis for PTSD (Gelpin 1996;Pitman 2002; Schelling 2004) have also been reserved for a future Cochrane protocol.
Types of outcome measures
Primary outcomes PTSD symptom and symptom cluster response was determined from the total score on the Clinician Administered PTSD Scale (CAPS) (Blake 1990), a symptom severity measure that is increasingly used in RCTs of PTSD.
Treatment response (responders versus non‐responders) was determined from the Clinical Global Impressions scale ‐ improvement item (CGI‐I), or closely related measure such as the Duke Global Rating for PTSD scale (Davidson 1998b), or a closely related definition (Brady 2000). Responders are defined on the CGI‐I as those with a score of 1 = "very much" or 2 = "much" improved (Guy 1976); this is a widely used global outcome measure in RCTs of PTSD, where it appears robust (Davidson 1997b).
Secondary outcomes
PTSD symptom response was assessed for those trials which used other continuous measures of symptom severity besides the CAPS, as well as from summary statistics from self‐rated scales such as the Impact of Events Scale (IES) (Horowitz 1979), and the Davidson Trauma Scale (DTS) (Davidson 1997c). Self‐rated scales were frequently the only outcome measures used in older trials, and may continue to have a role in clinical practice. The efficacy of medication in alleviating symptoms within the three symptom clusters characteristic of PTSD (re‐experiencing/intrusion, avoidance/numbing, and hyperarousal) was determined using the CAPS‐B, CAPS‐C, and CAPS‐D subscales of the CAPS, as well as the relevant subscales of the self‐rated outcome measures.
The response of comorbid symptoms was measured by (a) depression scales, such as the Beck Depression Inventory (BDI) (Beck 1961), the Hamilton Depression scale (HAM‐D) (Hamilton 1959), and the Montgomery‐Asberg Depression Rating Scale (MADRS) (Montgomery 1979), and (b) anxiety scales, such as the Covi Anxiety Scale (CAS) (Covi 1984) and the Hamilton Anxiety scale (HAM‐A) (Hamilton 1960).
Quality of life measures, as well as measures of functional disability, such as the Sheehan Disability Scale (SDS), which includes subscales to assess work, social and family related impairment (Sheehan 1996), were also included when provided, to address the question of medication effectiveness.
The total proportion of participants who withdrew from the RCTs due to treatment emergent adverse events was included in the analysis as a surrogate measure of medication acceptability, in the absence of other more direct indicators of acceptability.
Search methods for identification of studies
Electronic Searches 1. The Cochrane Collaboration Depression, Anxiety and Neurosis Controlled Trials Register (CCDANCTR‐Studies) was searched using the following search strategy on 18 August 2005:
Diagnosis = Post‐Traumatic Stress Disorders
and
"Antidepressive Agents" OR "Monoamine Oxidase InhibitORs" OR "Selective Serotonin Reuptake InhibitORs" OR "Tricyclic Drugs" OR Acetylcarnitine OR Alaproclate OR Amersergide OR Amiflamine OR Amineptine OR Amitriptyline OR Amoxapine OR Befloxatone OR Benactyzine OR Brofaromine OR Bupropion OR Butriptyline OR Caroxazone OR ChlORpoxiten OR Cilosamine OR Cimoxatone OR Citalopram OR Clomipramine OR ClORgyline OR ClORimipramine OR Clovoxamine OR Deanol OR Demexiptiline OR Deprenyl OR Desipramine OR Dibenzipin OR Diclofensine OR Dothiepin OR Doxepin OR Duloxetine OR Escitalopram OR Etoperidone OR Femoxetine OR Fluotracen OR Fluoxetine OR Fluparoxan OR Fluvoxamine OR Idazoxan OR Imipramine OR Iprindole OR Iproniazid OR isocarboxazid OR Litoxetine OR Lofepramine OR Maprotiline OR Medifoxamine OR Melitracen OR Metapramine OR Mianserin OR Milnacipran OR Minaprine OR Mirtazapine OR Moclobemide OR Nefazodone OR Nialamide OR Nomifensine OR NORtriptyline OR Noxiptiline OR Opipramol OR Oxaflozane OR Oxaprotiline OR Pargyline OR Paroxetine OR Phenelzine OR Piribedil OR Pirlindole OR Pivagabine OR Prosulpride OR Protriptyline OR Quinupramine OR Reboxetine OR Rolipram OR Sertraline OR Setiptiline OR Teniloxine OR Tetrindole OR Thiazesim OR Thozalinone OR Tianeptine OR Toloxatone OR Tomoxetine OR Tranylcypromine OR Trazodone OR Trimipramine OR Venlafaxine OR Viloxazine OR Viqualine OR Zimeldine.
2. Additional searches were carried out on MEDLINE (via PubMed), PsycINFO, and The National PTSD Center Pilots database.
The MEDLINE search query, as derived from a highly sensitive search strategy developed by Robinson and Dickersin (Robinson 2002), was the following:
(randomized controlled trial [pt] OR controlled clinical trial [pt] OR randomized controlled trials [mh] OR random allocation [mh] OR double‐blind method [mh] OR single‐blind method [mh] OR clinical trial [pt] OR clinical trials [mh] OR ("clinical trial" [tw]) OR ((singl* [tw] OR doubl* [tw] OR trebl* [tw] OR tripl* [tw]) AND (mask* [tw] OR blind* [tw])) OR ("latin square" [tw]) OR placebos [mh] OR placebo* [tw] OR random* [tw] OR research design [mh:noexp] OR comparative study [mh] OR evaluation studies [mh] OR follow‐up studies [mh] OR prospective studies [mh] OR cross‐over studies [mh] OR control* [tw] OR prospectiv* [tw] OR volunteer* [tw]) NOT (animal [mh] NOT human [mh]) AND (Stress Disorders, Post‐Traumatic [mh:noexp] OR "posttraumatic stress disorder" [tw] OR "post traumatic stress disorder" [tw] OR PTSD [tw]) AND (pharmacother* [tw] OR medicat* [tw] OR drug* [tw] OR Drug Therapy [mh]). The PsycINFO search used the following search query: ("randomisation" OR "randomization") OR "controlled" AND ("post‐traumatic" OR posttraumatic) AND (medication OR pharmacotherapy OR treatment). PsycINFO includes the Dissertation Abstracts International database ‐ a database of unpublished dissertations.
The National PTSD Center Pilots database contains published and unpublished articles on PTSD. It was searched using the following search query: (randomisation or randomization) or controlled AND (post‐traumatic OR posttraumatic) AND (medication OR pharmacotherapy).
3. Unpublished trials were retrieved via the metaRegister module [mRCT] of the Controlled Trials database (http://www.controlled‐trials.com). The search terms used were "PTSD", "posttraumatic stress disorder", and "post traumatic stress disorder".
An initial broad strategy was undertaken to find not only RCTs, but also open‐label trials, as well as journal and chapter reviews of the pharmacotherapy of PTSD.
Reference Lists Additional RCTs were sought in reference lists of the retrieved articles and included studies in any language.
Data collection and analysis
Trial selection RCTs identified from the search were independently assessed for inclusion by two raters, based on information included in the abstract and/or main body of the trial report. RCTs which both raters regarded as satisfying the inclusion criteria specified in the "criteria for considering studies" section were collated. Studies for which additional information is required in order to determine their suitability for inclusion in the review have been listed in the "studies awaiting assessment" table in the Review Manager (RevMan) software, pending the availability of this information. Any disagreements in assessment and collation were resolved by discussion.
Data extraction Spreadsheet forms were designed for the purpose of recording descriptive information, summary statistics of the outcome measures, the quality scale ratings, and associated commentary. The data was subsequently exported to the RevMan software, which was used to conduct the meta‐analysis. Where information was missing, the reviewers contacted investigators by email in an attempt to obtain this information. In the case of one trial for which it was not possible to obtain exact treatment response figures (Marshall 2001), one of the reviewers used a ruler to estimate the mean number of responders within the comparison groups from a graph contained within the original trial report.
Data synthesis The following information was collated from each trial (additional information can be found in the "Characteristics of Included Studies" table):
(a) Description of the trials, including the primary researcher, the year of publication, and the source of funding.
(b) Characteristics of the interventions, including the number of participants randomised to the treatment and control groups, the number of total drop‐outs per group as well as the number that dropped out due to adverse effects, the dose of medication and the period over which it was administered, and the name and class of the medication (SSRIs, TCAs, MAOIs and "other medication").
(c) Characteristics of trial methodology, including the diagnostic (eg. DSM‐IV (APA 1994)) and exclusionary criteria employed, the screening instrument used (eg. the Structured Clinical Interview for DSM‐IV (SCID) (Spitzer 1996)) for both the primary and comorbid diagnoses, the presence of comorbid major depressive disorder (MDD), the use of a placebo run‐in or of a minimal severity criterion, the number of centres involved, and the trial's methodological quality (see below).
(d) Characteristics of participants, including gender distribution and mean and range of ages, mean length of time with PTSD symptoms, whether they have been treated with the medication in the past (treatment naivety), the number of participants in the sample with MDD, the number who experienced combat trauma, and the baseline severity of PTSD, as assessed by the trial's primary outcome measure or another commonly employed scale.
(e) Outcome measures employed (primary and secondary), and summary continuous (means and standard deviations (SD)) and dichotomous (number of responders) data. Additional information included whether data reflected the intent‐to‐treat (ITT) with last observation carried forward (LOCF) or mixed methods (MM) sample, or whether a completer/observed cases (OC) sample was reported.
Data analysis
Summary statistics for categorical and continuous measures were obtained from a random effects model (the random effects model includes both within‐study sampling error and between‐studies variation in determining the precision of the confidence interval (CI) around the overall effect size, whereas the fixed effects model takes only within‐study variation into account). The summary statistics were expressed in terms of an average effect size for each subgroup, as well as by means of 95% CIs.
Cross‐over trials were only included in the calculation of summary statistics when it was (a) possible to extract medication and placebo/comparator data from the first treatment period, or (b) when the inclusion of data from both treatment periods was justified through a wash‐out period of sufficient duration as to minimise the risk of carry‐over effects (a minimum of two weeks or longer in the case of trials assessing the efficacy of agents with extended half‐lives, such as the SSRI, fluoxetine (Gury 1999)). In the latter case, data from both periods were only included when it was possible to determine the correlation between participants' responses to the interventions in the different phases (Elbourne 2002).
In recognition of the possibility of differential effects for different types of medication, all of the comparisons were stratified by medication class. Medications which could not be classified as either SSRIs, TCAs or MAOIs were placed in a separate category, labelled "other medication". In addition, in the case of the SSRIs, in view of the large number of SSRI trials, comparisons between medication and placebo on the primary outcome measures and on the number of drop‐outs due to drug‐related adverse events were stratified by individual agents. Categorical data Relative risk (RR) of failure to respond to treatment was used as the summary statistic for the dichotomous outcome of interest (CGI‐I or related measure). RR was used instead of odds ratios, as odd ratios tend to underestimate the size of the treatment effect when the occurrence of the adverse outcome of interest is common (as was the case in this review, with an anticipated non‐response greater than 20%) (Deeks 2003), and because of the greater ease with which this statistic can be interpreted. Number needed to treat (NNT) was also included. The NNT is calculated as the inverse of the absolute risk reduction between the medication and control groups (McQuay 1997). It provides an indication of the number of patients who require treatment with medication before a single additional patient in the medication group responds to treatment, relative to the control group. The confidence intervals for the NNT were only calculated for significant treatment effects, given the difficulty of interpreting CIs which contain infinity (Altman 1998).
Continuous data Weighted mean differences (WMD) were calculated for continuous summary data obtained from studies that employed the CAPS. Alternatively, in cases in which a range of scales were employed, such as in the assessment of symptom severity on the self‐rated IES and DTS scales, or in the assessment of comorbid depression on the MADRS and HAM‐D, the standardised mean difference (SMD) was determined. This method of analysis standardises the differences between the means of the treatment and control groups in terms of the variability observed in the trial.
In the case of data from trials employing multiple fixed doses of medication, the bias introduced through comparing the summary statistics for multiple groups against the same placebo control was avoided by pooling the means and standard deviations across all of the treatment arms as a function of the number of participants in each arm. For the same reason, outcome comparisons were limited to those between only one medication and placebo in trials which compared several different medications with placebo. Selection of medication for analysis was done (prior to analysis, to avoid introducing bias) on the basis of which of the three major medication classes (TCAs, MAOIs, SSRIs) was least well represented. In addition, when including summary statistics from the self‐rated scales, preference was given to data from the DTS over the IES in trials which used both scales, given the late inclusion in the former of a subscale assessing the hyperarousal symptom cluster (Weiss 1997), and concerns regarding the psychometric properties of the IES subscales (Creamer 2003).
Quality assessment There has been some debate about how best to measure the quality of trials, and further work in this area remains necessary (Berlin 1999). In this review, one of the reviewers assessed the quality of the trials by means of the CCDAN Quality of Research Scale (CCDAN‐QRS) (Moncrieff 1999) (http://www.iop.kcl.ac.uk/IoP/ccdan/qrs.htm). This 23 item scale assesses a range of features such as sample size, the duration of the intervention, inclusion and exclusion criteria, and whether or not the power of the trial to detect a treatment effect was calculated. In addition, data for other trial characteristics which have been recognised as a potential sources of bias, such as the method used in generating the allocation sequence, the concealment of allocation, whether outcome assessment was blinded, and the number of participants lost to follow up, were also collated. This was regarded as necessary given doubts concerning the usefulness of an overall quality score from a scale composed of multiple items (Alderson 2003).
Heterogeneity Heterogeneity of treatment response, that is whether the differences between the results of trials were greater than would be expected by chance alone, was assessed visually from the forest plot of RR. It was also determined by means of the chi‐square test of heterogeneity, with a significance level of less than 0.10 interpreted as evidence of heterogeneity, given the low power of the chi squared statistic when the number of trials is small (Deeks 2003). In addition, the I‐square heterogeneity statistic reported by RevMan was used to test the robustness of the chi squared statistic to differences in the number of trials included in the groups being compared within each subgroup analysis (Higgins 2003). Differences on continuous measures in medication efficacy between these groups were assessed by means of Deeks' stratified test of heterogeneity (Deeks 2001). This method subtracts the sum of the chi squared statistics for each of the groups from the total chi squared for the subgroup analysis, to provide a measure (Qb) of heterogeneity between groups. Differences in treatment response on the CGI‐I was determined by whether the confidence intervals for the effect sizes of the subgroups overlap. This method was chosen in preference to the stratified test, due to inaccuracies in the calculation in RevMan of the chi squared statistic for dichotomous measures (Deeks 2003).
Subgroup analyses Subgroup analyses (Thomson 1994) were undertaken in order to assess the degree to which methodological differences between trials might have systematically influenced differences observed in the primary treatment outcomes.
The trials were grouped according to the following methodological sources of heterogeneity:
The involvement of participants from a single centre or multiple centres. Single‐centre trials are more likely to be associated with lower sample size but less variability in clinician ratings.
Whether or not trials were industry funded. In general, published trials which are sponsored by pharmaceutical companies appear more likely to report positive findings than trials which are not supported by for‐profit companies (Als‐Nielsen 2003; Baker 2003).
In addition, the following criteria were used to assess the extent of clinical sources of heterogeneity:
Whether or not the sample included combat veterans (this subgroup has been regarded as more resistant to treatment, and is arguably more likely to have more chronic and severe symptoms, to have comorbid depression, and to be male). For the purpose of this review, those trials for which 10 percent or fewer of the sample consisted of war veterans were classified as non‐combat veteran RCTs.
Whether or not the sample included patients diagnosed with major depression. Such an analysis might assist in determining the extent to which the efficacy of a medication agent in treating PTSD is independent of its ability to reduce symptoms of depression, an important consideration given the classification of many of these medications as antidepressants.
Sensitivity analysis Sensitivity analyses, which determine the robustness of the reviewers' conclusion to methodological assumptions made in conducting the meta‐analysis, were also performed. Sensitivity analyses were conducted to determine whether treatment response on the CGI‐I differed as a result of:
Treatment response versus non‐response as the unit of comparison in determining medication efficacy. This comparison is regarded as necessary given concerns that the former may result in less consistent summary statistics than the latter (Deeks 2002).
The exclusion of participants who were lost to follow up (LTF). This was determined through a "worst case/best case" scenario (Deeks 2003). In the worst case, all the missing data for the treatment group were recorded as non‐responders, whereas in the best case, all missing data in the control group were treated as non‐responders. (In the case of the one SSRI (Marshall 2004) and MAOI trial (Baker 1995 a) which only reported total LTF, the ratio of participants who dropped out in the medication and placebo group was determined from the average ratio between these groups for those RCTs in the respective classes which did provide this information). Should the conclusions regarding treatment efficacy not differ between these two comparisons, it can be assumed that missing data in trial reports do not have a significant influence on outcome.
Publication bias Publication bias was determined by visual inspection of a funnel plot of treatment response.
Results
Description of studies
The review included 35 short term RCTs of PTSD (4597 participants), three of which contained a maintenance component (Davidson 2001a, Marshall 2004, Martenyi 2002a)(see Comparison 06). Of the 35 trials, 30 were published, and all of these publications were in English. A placebo comparison group was employed in all but four of the trials (McRae 2004 and Saygin 2002 compared nefazodone with the SSRI sertraline, while Smajkic 2001 compared the efficacy of the SSRIs sertraline, paroxetine and the serotonin ‐ noradrenaline reuptake inhibitor (SNRI) venlafaxine. Chung 2004 assessed the efficacy and tolerability of mirtrazapine against that of sertraline). Of the remaining 31 short term RCTs, 17 of the trials included a SSRI treatment arm (one citalopramine, six fluoxetine, four paroxetine, seven sertraline), two trials a TCA intervention (one amitriptyline, one desipramine), four a MAOI intervention (two brofaromine, two phenelzine), and seven studies employed an intervention classified as "other medication". This last category included one benzodiazepine (alprazolam), two antipsychotics (olanzapine and risperidone), one anticonvulsant (lamotrigine), one second messenger system precursor (inositol), one SNRI (venlafaxine) and two novel antidepressants (mirtazapine and nefazodone). The SSRI trials can be distinguished from the non‐SSRI trials on a number of different study characteristics. The SSRI trials were significantly larger (mean = 184 participants) on average than the non‐SSRI trials (mean = 41 participants) (one‐sided Wilcoxon Mann‐Whitney test: W = 208, P = < 0.01), even when adjusting for the smaller number of participants in the non‐SSRI crossover trials (through doubling the sample size). The majority of the 17 placebo controlled short term trials published since 2000 have included SSRIs (N = 13), with only two SSRI trials being published prior to 2000 (Conner 1999; van der Kolk 1994).
None of the four acute RCTs which employed a cross‐over design (Braun 1990; Kaplan 1996; Reist 1989; Shestatzky 1988) provided sufficient information for inclusion in the calculation of summary statistics. These trials were all small and of poor quality (mean CCDAN‐QRS score of 12), however, so their exclusion is unlikely to have had a significant effect on the primary outcomes of the meta‐analysis. (In brief: Braun 1990 compared an intervention of alprazolam with placebo using a sample of 16 outpatients over a period of 12 weeks. Kaplan 1996 investigated the efficacy of inositol for a sample of 13 patients from two different outpatient clinics. Reist 1989 conducted a crossover trial of the TCA, desipramine, in treating 27 combat veterans over a period of 10 weeks. Shestatzky 1988 assessed the efficacy of the MAOI phenelzine in a 12 week trial of 13 PTSD outpatients).
In determining the long‐term effects of medication, Marshall (2004) assessed whether 17 responders in the medication arm of a 10 week trial of paroxetine continued to respond after an additional maintenance period of 12 weeks. In a relapse‐prevention trial of sertraline, Davidson (2001) set out to determine whether 50 patients who were randomised to a placebo control for 28 weeks were more likely to experience clinical deterioration or relapse than those 46 patients randomised to a sertraline intervention for the same period. Participants in the relapse prevention phase of this trial had completed a 12 week acute RCT of sertraline, and had also met responder criteria following the subsequent open‐label administration of this medication for a period of six months. In another relapse‐prevention trial, Martenyi (2002) re‐randomised 131 responders to a 12 week short‐term RCT of fluoxetine to an additional 24 weeks of placebo or medication.
A great deal of clinical heterogeneity was observed across patients in the RCTs included this review. Of the 34 acute trials that provided information on the nature of the index trauma for PTSD, six were composed exclusively of war veterans (Chung 2004; Davidson 1990; Hertzberg 2000; Kosten 1991; Pfizer589; Reist 1989), 9 contained individuals exposed to "civilian" traumas, such as earthquakes and child molestation (Brady 2000; Brady 2004; Conner 1999; Marshall 2004; McRae 2004; Pfizer588; Reich 2004; Saygin 2002; Smajkic 2001) and 19 contained patients exposed to both combat‐related and civilian traumas (Baker 1995 a; Braun 1990; Butterfield 2001; Davidson 2001a; Davidson 2003; Davidson 2004; Davis 2001; Eli Lilly 2006; Hertzberg 1999; Kaplan 1996; Katz 1994; Marshall 2001; Martenyi 2002a; Shestatzky 1988; Tucker 2001; Tucker 2003; van der Kolk 1994; van der Kolk 2004; Zohar 2002).Of the 24 short‐term RCTs that providing information on comorbid psychopathology, 18 reported that the patients in the trials were diagnosed with other anxiety disorders besides PTSD, as classified according to DSM III, DSM‐IV or DSM‐IV‐TR criteria (Brady 2000; Brady 2004; Braun 1990; Butterfield 2001; Davidson 1990; Davidson 2001a; Davidson 2003; Davis 2001; Hertzberg 2000; Kaplan 1996; Marshall 2001; Marshall 2004; Pfizer589; Reich 2004; Saygin 2002; Shestatzky 1988; Tucker 2001; Tucker 2003).The most commonly reported comorbid anxiety was panic disorder with or without agoraphobia, which was reported for 8 of the 12 studies that distinguished between individual anxiety disorder diagnostic categories (Braun 1990; Butterfield 2001; Davidson 1990; Davis 2001; Marshall 2001; Marshall 2004; Saygin 2002; Tucker 2003),
Summary statistics for the sertraline arm of the Tucker 2003 trial were excluded from the analysis, in favour of including the data from the less well represented citalopram arm. Data from the venlafaxine group in the unpublished Davidson trial (Davidson 2004) was given preference to that from the sertraline arm, for the same reason. Summary statistics from the phenelzine arm of the Kosten 1991 trial were chosen above those from the imipramine arm in order to equalise the number of MAOI and TCA trials.
Risk of bias in included studies
Generation of Allocation Sequence The randomisation procedure employed was described in four trials. Computer generated random codes were employed in three of these trials (Conner 1999; Davidson 2001a; Martenyi 2002a), while an urn randomisation procedure was followed in Brady 2004.
Allocation Concealment Of the 35 short term RCTs, only five described the allocation sequence which was used in assigning participants to the treatment and comparison groups. Adequate allocation concealment (randomisation log kept by each trial's pharmacy division) was practised in three of these trials (Conner 1999; Davis 2001; McRae 2004). The remaining two trials did not provide sufficient information to determine the adequacy of the concealment used (Kaplan 1996; Martenyi 2002a).
Blinding of Outcome Assessment Although the majority of the trials were described as "double‐blinded", only six of the short term trials explicitly described the assessment of outcome as blinded (Braun 1990; Davidson 1990; Davis 2001; Marshall 2004; Shestatzky 1988; van der Kolk 2004). The extent to which blinding was preserved in those flexible dose trials which adjusted dosage on the basis of tolerability is unclear, however. Indeed, outcome was assessed independently of medication administration and side‐effect evaluation in only one RCT (Marshall 2004). Two comparative RCTs did not employ any form of blinding (Smajkic 2001; Chung 2004), whereas it is not clear whether blinding was undertaken in another (Saygin 2002).
Loss to follow up On average, about 31.3 percent (718 out of 2291) of the participants in the 24 short term RCTs that provided drop‐out data did not reach study endpoint, with the majority (N = 13) of these trials excluding over a quarter of the sample. Of the 13 trials, six did not attempt to include the withdrawals in the summary statistics through estimating outcomes by means of either LOCF or MM analyses (Braun 1990;Chung 2004;Davidson 1990;Eli Lilly 2006;Saygin 2002;Smajkic 2001).
Quality Score The average quality score on the CCDAN‐QRS for the published short term trials was 22.8 points (range: 11 to 31) out of a maximum of 46 points. On this scale, 19 trials failed either to provide a record of the exclusion criteria used, or to report the number of people excluded by these criteria, 16 provided inadequate details of the side effects experienced by group, six RCTs did not provide information about funding, and three RCTs (excluding crossover trials) did not provide information about the comparability of the medication and control groups. Quality ratings were not calculated for the unpublished trials, due to the lack of sufficient descriptive data for these studies.
Effects of interventions
Primary outcome measures
Significant reductions in symptom severity were observed for patients who received medication in 17 short term trials from which it was possible to retrieve data for this outcome. The mean total CAPS score for the medication group was 5.76 points lower (95% CI ‐8.16 to ‐3.36, N = 2507) than that for the placebo group. Evidence for the efficacy of the SSRIs (N = 12) was once again observed (WMD = ‐5.95, 95% CI ‐8.9 to ‐3, N = 1907), with this class of medication making the largest contribution to the overall effect size observed (weight = 82.4%).
The comparison of the efficacy of particular SSRIs in reducing PTSD symptom severity provided evidence for the efficacy of both paroxetine (N = 4, WMD ‐10.49, 95% CI ‐13.87 to ‐7.11, N = 940) and to a lesser extent, sertraline (N = 6, WMD ‐3.78, 95% CI ‐6.9 to ‐0.65, N = 875), but not citalopram (N = 1, WMD ‐13.41, 95% CI ‐35 to 8.18, N = 33) or fluoxetine (N = 1, WMD ‐0.9, 95% CI ‐12.31 to 10.51, N = 59). The failure to detect a treatment effect for citalopram or fluoxetine presumably reflects the small samples, and hence low power, of these comparisons. There was no indication that brofaromine was more effective than placebo (N = 2, WMD ‐5.06, 95% CI ‐15.93 to 5.81, N = 178). Neither the single trials of the novel antidepressant nefazodone or the antipsychotic risperidone provided evidence of efficacy in reducing symptom severity (WMD ‐5.6, 95% CI ‐21.26 to 10.06, N = 41 and WMD ‐11, 95% CI ‐30.55 to 8.55, N = 21, respectively). This was also true of the single SNRI trial of venlafaxine (N = 1, WMD ‐4.8, 95% CI ‐11.73 to 2.13, N = 358).
With regards to direct comparisons, no difference in the reduction of symptom severity was observed in the two head‐to‐head comparisons of nefazodone and sertraline (SMD ‐0.19, 95% CI ‐0.63 to 0.25, N = 80), or in the single unpublished comparison of venlafaxine and sertraline (SMD ‐0.01, 95% CI ‐0.22 to 0.20, N = 352). Although the only trial to directly compare mirtazapine and sertraline (Chung 2004) reported that treatment with mirtazapine resulted in a larger number of responders on the CAPS than sertraline after six weeks of treatment (treatment response was defined as a reduction of over 30% on the total score of this scale), the authors were unable to detect a difference in efficacy when comparing these groups on the total CAPS score. Patients who received medication were significantly more likely to be responders than those who received placebo in the 13 trials that provided data on this outcome (RR 1.49, 95% CI 1.28 to 1.73; random effects model, N = 1272), as determined by response rates on the Clinical Global Impressions scale change item (or close equivalent). Response to medication occurred in 59.1% of subjects (N = 644), while response to placebo was seen in 38.5% of subjects (N = 628). The short term efficacy of medication treatment was observed for the SSRIs as a group (N = 7, RR 1.59, 95% CI 1.39 to 1.82, N = 999), to which the significant overall effect of medication on treatment response can once again primarily be attributed (weight = 69.1%).
The pattern of treatment response on the CGI‐I for the separate SSRI medications was similar to that observed for symptom severity, with both paroxetine (N = 3, RR 1.62, 95% CI 1.38 to 1.9, N = 719) and sertraline (N = 2, RR 1.71, 95% CI 1.22 to 2.4, N = 215) demonstrating efficacy. There was once again insufficient evidence to determine whether fluoxetine was effective in increasing the number of responders, relative to placebo (N = 2, RR 1.35, 95% CI 0.96 to 1.89, N =65). None of the individual trials of the TCA amitryptyline (RR 3.00, 95% CI 0.98 to 9.14, N = 40), the novel antidepressant mirtazapine (RR 2.91, 95% CI 0.82 to 10.39, N = 26), the antipsychotic olanzapine (N = 1, RR 1.00, 95% CI 0.42 to 2.4, N = 15), the anticonvulsant lamotrigine (N = 1, RR 2.00, 95% CI 0.33 to 12.18, N = 14), or the two trials of MAOI brofaromine (RR 1.16, 95% CI 0.79 to 1.72, N = 178) was significantly more effective than placebo in increasing treatment response.
The NNT analysis revealed that each patient who was treated with medication was approximately 21% more likely to become a responder as a result of being treated with medication over an average of 11 weeks than if they had been given placebo (NNT = 4.85, 95% CI 3.85 to 6.25). The equivalent percentages for the individual SSRIs was 23% for paroxetine (NNT = 4.31, 95% CI 3.33 to 6.25), 22% for sertraline (NNT = 4.49, 95% CI 2.86 to 10), and 16.5% for fluoxetine (NNT = 6.07). By way of comparison, a person diagnosed with PTSD was only 7% more likely to respond to treatment with brofaromine than with placebo (NNT = 13.94, 95% CI 4.76 to 14.29).
Continued reduction of symptom severity on the CAPS was observed in the 10 week extension phase of the 12 week placebo‐controlled RCT of paroxetine (Marshall 2004). An increased rate of relapse (defined as a >= 40% increase on the eight item Treatment Outcome PTSD scale (TOP‐8) and an increase in CGI‐S score of >= 2) was observed in those patients who were randomised to placebo after responding to a 12 week trial of fluoxetine (Martenyi 2002a). Davidson (2001) additionally found that over half of the 96 outpatients who had initially responded to six months of treatment with sertraline experienced worsening of symptoms once switched over to placebo, with patients in this group being 6.35 times more likely to relapse than those participants who remained on medication. A patient was considered to have relapsed in this trial if they met all of the following criteria: an increase in CGI‐I score of at least three points, an increase in CAPS score of at least 30% and 15 points, and if the patient experienced significant clinical deterioration (as determined by the clinician).
Secondary outcome measures
The overall symptom severity effect size for trials which did not employ the CAPS confirmed the presence of a treatment effect of medication relative to placebo (N =6, SMD ‐0.44, 95% CI ‐0.87 to ‐0.01, N = 147). Consistent results were also observed for the total scores of the self‐rated scales (IES and DTS), in which medication reduced symptom severity by ‐0.31 standard deviation units compared to placebo (N = 9, 95% CI ‐0.51 to ‐0.11, N = 882). A significant effect of medication treatment was observed for the SSRIs (SMD ‐0.20, 95% CI ‐0.34 to ‐0.06, N = 769), the RCT of the MAOI phenelzine (SMD ‐1.06, 95% CI ‐1.75 to ‐0.36, N = 37), as well as for the TCA amitryptiline (SMD ‐0.9, 95% CI ‐1.62 to ‐0.18, N = 33), while no such effect was found for either of the single olanzapine and nefazodone trials.
The significantly reduced scores on the re‐experiencing/intrusion (N = 9, WMD ‐2.06, 95% CI ‐3.02 to ‐1.1, N = 1304), avoidance/numbing (N = 9, WMD ‐4.06, 95% CI ‐5.41 to ‐2.7, N = 1304), and hyperarousal (N = 9, WMD ‐3.1, 95% CI ‐4.1 to ‐2.1, N = 1304) subscales of the CAPS indicates that the efficacy of medication is not limited to particular symptom clusters. The positive findings for the nine trials which provided summary statistics on these subscales was primarily attributable to the seven SSRI trials, which together contributed an average of 95% to the magnitude of the overall effect sizes on these subscales. The remaining trials of nefazodone and risperidone provided no evidence of efficacy on any of the symptom clusters measured by the CAPS. Symptoms of avoidance/numbing (N = 4, SMD ‐0.49, 95% CI ‐0.9 to ‐0.08, N = 268) but not re‐experiencing/intrusion (N = 4, SMD ‐0.43, 95% CI ‐0.98 to 0.13, N = 268) were reduced after treatment according to the subscales of the self‐rated scales.
With regards to comorbidity, medication demonstrated greater efficacy in alleviating the symptoms of depression than placebo, as assessed by a range of depression scales. This was true for both trials which reported mean endpoint scale ratings (N = 7, SMD ‐0.34, 95% CI ‐0.57 to ‐0.10, N = 459), as well as for the RCTs which only reported change scores (N= 3, SMD ‐0.33, 95% CI ‐0.6 tp ‐0.07, N = 887). The finding that SSRIs were more effective than placebo in the trials reporting change scores, but not in those which provided endpoint summary scores (N = 4, SMD ‐0.19, 95% CI ‐0.39 to 0.02, N = 364) could be attributed to less variability in the former comparison, as it only included a single medication agent (paroxetine). The increased precision of effect size estimates when change scores are used, and the greater sample size and associated power of this comparison are also likely causes of this discrepancy.
The observation that anxiety symptoms were not noticeably reduced by medication interventions, as assessed by the HAM‐A (N =3, WMD ‐2.17, 95% CI ‐7.22 to 2.88, N = 287), is largely a reflection of the lack of evidence for the efficacy of the SSRIs (N = 2, WMD 0.58, 95% CI ‐1.97 to 3.14, N = 254), with single trials of other medications demonstrating efficacy on the HAM‐A (amitryptiline) and on other anxiety scales (phenelzine). The only head‐to‐head comparison of nefazodone with sertraline for which comorbidity summary statistics were available demonstrated that these medications were equally effective in reducing symptoms of depression (WMD ‐0.84, 95% CI ‐7.88 to 6.20, N = 26) and of anxiety (WMD ‐3.23, 95% CI ‐10.9 to 4.44, N =26). No difference in the efficacy of mirtazapine and sertraline in reducing symptoms of depression on the HAM‐D was reported for the one trial which compared these medications.
Quality of life was significantly improved by pharmacotherapy (N = 5, WMD ‐2.54, 95% CI ‐3.68 to ‐1.41, N = 752), according to summary statistics on the Sheehan Disability Scale (SDS). This was primarily due to the SSRI interventions (WMD ‐2.56, 95% CI ‐3.7 to ‐1.41, N = 737), with only one of the four trials in this class not demonstrably superior to placebo in improving functioning according to this measure of social, work and family‐related functioning. Patients receiving medication were more likely to withdraw from treatment due to side‐effects experienced than those who received placebo (N = 21, RR 1.44, 95% CI 1.04 to 2, N = 2116). Nevertheless, the overall effect sizes for each of the medication classes reveals that the administration of medication in the 10 SSRI (N = 1649), three MAOI (N = 196) and two TCA (N = 100) trials did not result in a significantly greater number of withdrawals due to side effects than administering placebo (although the SSRIs come close (RR = 1.42, 95%CI = 0.99, 2.05 )). The same observation was made with respect to the individual SSRI agents. In addition, the overlap between confidence intervals reveals no differences in tolerability for either the medication classes or the SSRIs. Although Saygin (2002) found that nefazodone resulted in significantly higher side‐effect scores than sertraline on the CGI, no differences in tolerability between these medications was observed by McRae (2004). A significantly larger proportion of patients (8 out of 13) who were randomised to the venlafaxine arm of a small unblinded multi‐arm RCT dropped out due to adverse events (Smajkic 2001) than those patients randomised to either the sertraline or paroxetine arms of this trial.
Heterogeneity With regards to the overall heterogeneity of trial results, the chi squared test revealed a similar degree of variation between the outcomes of trials for both treatment response and symptom severity (Chi = 16.3, P = 0.18, df = 12 and Chi = 22.04, P = 0.14, df = 16, respectively). The same finding was made across all of the secondary outcome measures employed, with the exception of the scores on the HAM‐A (Chi = 10, P = 0.007), where amitryptiline demonstrated superiority over the two SSRI trials in reducing anxiety on this scale, relative to placebo. No differences were observed in the reduction of symptom severity between the SSRI and MAOIs trials (Qb = 0.06, P =0.81, df = 14), while extensive overlap between the confidence intervals for all of the medication classes on the CGI‐I indicated little difference in terms of the proportion of non‐responders in these groups. The separation of the effects of the SSRIs by agent revealed that paroxetine was more effective in reducing symptom severity than sertraline (Qb = 8.86, P < 0.01). Indeed, the reduction of symptom severity was nearly twice as large for paroxetine as for all the medications combined (WMD ‐10.49 versus ‐5.76). Nevertheless, no such differences in the efficacy of any of the SSRI medications was observed for treatment response. There were too few trials on the secondary outcomes to determine relative efficacy of different medication classes.
Subgroup analyses Symptom severity in the six trials which took place across multiple centres for which the CAPS total score was available was reduced to a greater extent than the eight trials conducted within single centres (Qb = 2.8, P = 0.09, df = 13). However, this effect was not observed in the analysis of treatment response (N = 13; 95% CI for single centre trials 1.2 to 2.6, N =188; 95% CI for multi‐centre trials 1.2 to 1.72, N = 1084). No difference in treatment response was evident in the comparison of industry funded trials versus non‐industry funded trials either, as the confidence interval of the effect size for the former (N = 9, 95% CI 1.41 to 1.84, N = 1053) was contained within the confidence interval of the latter (N = 3, 95% CI 0.81 to 2.87, N = 105).
Symptom severity decreased to an equivalent extent (Qb = 0.5, P = 0.48) in trials which included depressed participants (N = 9, SMD ‐0.35, 95% CI ‐0.46 to ‐0.23, N = 1304) as in those which did not (N = 2, SMD ‐0.48, 95% CI ‐1.52 to 0.56, N = 151). RCTs which contained few combat veterans (N = 8, average proportion of war veterans = 3%) demonstrated a significantly greater reduction in symptom severity following medication treatment (Qb = 4.12, P = 0.04) than trials with a large percentage of participants with combat‐related trauma (N = 4, average proportion = 61.1%). The difference between these groups was not detected with respect to treatment response, however, as evidenced by the inclusion of the confidence interval for the latter group (N = 5, average proportion = 3.8%) within the confidence interval of the former (N = 8, average proportion = 56%).
The finding of a difference in the reduction of symptom severity between trials with few war veterans versus those with many was not surprising, given the general characterisation of the war trauma subgroup of PTSD sufferers as more treatment resistant than other subgroups. War veteran samples are typically predominantly male (86.7% versus 32.8% in the groups compared in this review), and have more severe (84.9 versus 74.6 points on the CAPS) and chronic (21.1 years versus 10.9 years) PTSD than those trials composed of patients with other types of trauma. In addition, although it was not possible to observe differences in treatment response for these two group, four of the five trials with fewer war veterans demonstrated superior treatment response amongst participants given medication, as compared to none of the eight trials with a substantial proportion of veterans.
Sensitivity analyses
The comparison of the analysis of treatment efficacy in terms of treatment non‐response as opposed to response on the CGI‐I (or equivalent) revealed similar outcomes for both the overall short‐term efficacy of medication (N = 13, RR 0.66, 95% CI 0.6 to 0.74, N = 1272), as well as the efficacy of the SSRIs in treating PTSD (N = 7, RR 0.65, 95% CI 0.58 to 0.74, N = 999). However, whereas the use of treatment response as a summary statistic indicated that both the TCA amitryptyline and the novel antidepressant mirtazapine were no more effective than placebo, relative risk of non‐response to treatment provides evidence that both of these medications are more effective over the short‐term than placebo (mirtazapine: RR 0.45, 95% CI 0.22 to 0.94, N = 26; amitryptyline: RR 0.6, 95% CI 0.38 to 0.96, N = 40). It seems probable that the failure to find a significant effect for these two agents on treatment response reflects the lack of sensitivity of the relative risk of benefit for therapeutic trials to the effects of interventions when the placebo response rate is low (Deeks 2002), as was the case for these two trials. The number of participants responding to medication was significantly higher relative to the placebo control in both the worst case scenario (N = 12, RR 1.37, 95% CI 1.12 to 1.69, N = 1275), where those participants from the medication group who were not included in the analysis were regarded as non‐responders, and in the best case scenario (N = 12, RR 1.53, 95% CI 1.22 to 1.9, N = 1262), in which those participants excluded from the placebo control were regarded as non‐responders. The overlap in the confidence intervals for these two outcomes indicates that loss to follow up is unlikely to have influenced assumptions made about the overall efficacy of medication.
Publication bias
The distribution of trials on a funnel plot for treatment response (see Additional Figures: Figure 1) provides no evidence of substantial publication bias. A slight skewness in the distribution of trials on the funnel plot for the CAPS (see Additional Figures: Figure 2) could be interpreted as evidence of a tendency for trials with larger standard errors and smaller effect sizes to go unreported.
1.
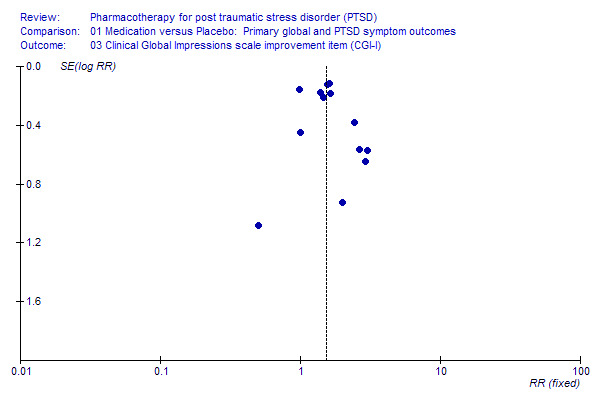
Publication Bias 2.
Funnel plot of publication bias on Clinical Global Impression Scale ‐ Improvement item (CGI‐S)
2.
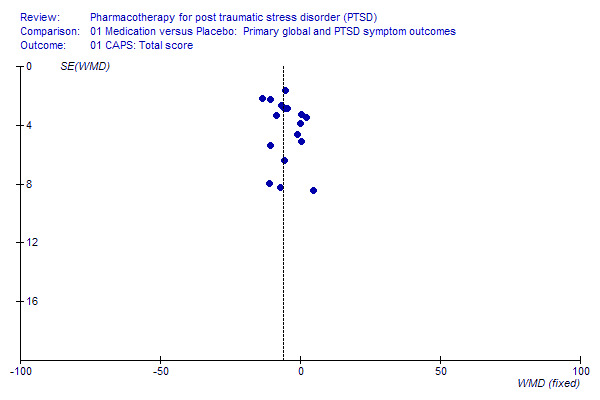
Publication Bias 1.
Funnel Plot of publication bias on Clinician Administered PTSD Scale (CAPS)
Discussion
This review provides evidence of the efficacy of medication in the short‐term treatment of PTSD, as assessed on the primary outcome measures of responder status and symptom severity. Medication was significantly more effective than placebo across the three symptom clusters which characterize PTSD (re‐experiencing/intrusion, avoidance/numbing, and hyperarousal) as assessed by the CAPS subscales. Scores on the self‐rated symptom severity scales confirmed medications' effectiveness in reducing overall symptom severity, as well as PTSD avoidance symptoms. In addition, the administration of medication resulted in a reduction in comorbid symptoms, and the improvement in quality of life measures. These findings hold despite the clinical heterogeneity of PTSD subjects included in the reviewed trials (see Table 1 ‐ Characteristics of Included Studies). Although a recent guideline noted that, with few exceptions, the overall effect size for medication trials of PTSD failed to exceed the limit of 0.5 defined as indicative of clinical effectiveness (NICE 2005), we would caution that there is no direct translation between the effect size statistic and assessments of clinical effectiveness. The findings here that the CGI‐I response rate was 59.1% on medication and 38.5% on placebo, and of a relatively low NNT of 4.85, support the growing clinical consensus that medication does have an important role in the treatment of PTSD.
The current evidence base of RCTs is unable to demonstrate superior efficacy or acceptability for any particular medication class. Although some have suggested that the SSRIs are more effective than older antidepressants (Dow 1997; Penava 1996), class membership did not contribute significantly to the variation observed in symptom severity outcomes between trials, while the confidence intervals for the summary statistic of responder status on the seven SSRI trials overlapped with that of the MAOI and TCA trials. Similarly, direct comparisons of sertraline and nefazodone demonstrated that these medications were equally effective in reducing PTSD symptom severity. Although the SSRIs have often been said to have superior tolerability in comparison to older medication classes, this was not readily apparent on analysis of drop‐out rates due to treatment emergent side‐effects in medication versus placebo groups. However, it should be emphasized that drop‐out rates due to adverse events may not always provide an accurate measure of medication tolerability (Loke 2005). In addition, it is important to be aware of the need for careful monitoring after initiation of SSRIs (CSM 2004). Nevertheless, the SSRI trials constitute the bulk of the evidence for the efficacy of medication in treating PTSD, both in terms of the number of studies and their size. The finding of the effectiveness of the SSRIs were also more robust to differences in the particular summary statistic employed than was the case for either the amitryptiline or mirtazapine trials. It is therefore reasonable to support the expert consensus (Foa 1999; Ballenger 2000; Ballenger 2004) that SSRIs constitute the first line medication choice in PTSD.
Nevertheless, the efficacy of medication in PTSD is unlikely to extend to all medications. While there is preliminary evidence that paroxetine is more effective than sertraline in reducing the severity of PTSD symptoms, and although the two mirtazapine trials provide some support for the efficacy of this agent, neither of the brofaromine, olanzapine and lamotrigine trials demonstrated efficacy with regards to treatment response or symptom reduction. Given the lack of a placebo control in two of the RCTs of nefazadone, and the negative finding with regards to symptom reduction in the third, evidence of the efficacy of this medication must be regarded as inconclusive. The failure to detect an effect of medication in the olanzapine and lamotrigine trials, despite other open‐label evidence that olanzapine is effective in combating PTSD (Petty 2001;Pivac 2004), may, however, be attributable to the small samples employed (average number = 15). Similarly, the failure of amitriptyline to alleviate the symptoms of PTSD in the only trial of this medication can perhaps be explained by the short duration of the trial (four weeks). Methodological limitations in the form of small sample size and short duration might also account for the negative findings of the crossover trials of alprazolam, inositol, desipramine, and phenelzine. This possibility is supported by the finding of reduced symptom severity in the only other controlled trial of phenelzine (Kosten 1991). Nevertheless, the lack of efficacy of desipramine could arguably support the (at this stage speculative) hypothesis that more noradrenergic agents (such as desipramine) are less useful than more serotonergic agents in PTSD (Dow 1997; Penava 1996). The question of whether benzodiazepines are useful immediately after trauma (Gelpin 1996; Mellman 1998) or in PTSD remains debated, although recent expert consensus panels have suggested caution (Foa 1999; Ballenger 2000; Ballenger 2004) in the use of these agents. Neither the potential clinical (presence of combat trauma, comorbid depression) or methodological (single versus multi‐centre trials, industry versus non‐industry funding) predictors of medication response tested in this review can account for the substantial proportion (41%) of patients who do not appear to respond to medication. The finding that symptom severity is reduced to a greater extent in the multi‐centre than the single centre trials should be interpreted with caution, not only due to the marginal significance of this finding, but also because it was not possible to replicate this finding with regards to treatment response. The failure to detect an association between the presence of participants with comorbid major depression and treatment efficacy indicates that medications are unlikely to exert their effects in PTSD indirectly via a reduction in depressive symptoms.
This review found some evidence that war veterans are more resistant to pharmacotherapy than other patient groups, at least with regards to the reduction of symptom severity. This was despite the fact that a number of RCTs with war veteran samples were excluded from this review (Hamner 1997; Hamner 2003; Stein 2002; Monnelly 2003; Raskind 2003; Bartzokis 2005) (see Table 2 ‐ Characteristics of Excluded Studies), and that it was not possible to classify certain large‐scale unpublished trials according to trauma type (Davidson 2004; Eli Lilly 2006; SKB627). Further research is therefore required to determine conclusively whether being a war veteran is a significant predictor of treatment response, and to distinguish the effects of this trauma subtype from other potential predictors of treatment response with which it is associated (such as being male, and having more chronic and severe PTSD). It is possible that the crucial factor is not so much being a war veteran, but rather being a veteran of particular wars (Zohar 2002). Given the heterogeneous phenomenology of PTSD, it remains crucial to determine the factors which do predict response to medication, and also to delineate whether certain medications are more effective for particular symptom sets (including symptoms such as psychosis (Hamner 1996), dissociation (Fichtner 1990; Marshall 1998b) and vulnerability to stress (Connor 1999)). Future RCTs and meta‐analyses should attempt to address such questions in greater detail.
The importance of long‐term treatment of PTSD is indicated by the observation of a continued improvement of PTSD symptoms following acute treatment with paroxetine but not placebo (Marshall 2004). The conclusion that a short‐term course of treatment with SSRIs may be inadequate is supported by increased relapse rates in trials of both fluoxetine (Martenyi 2002b) and sertraline (Davidson 2001b). The findings of these trials are consistent with consensus recommendations of six to twelve months medication treatment for acute PTSD (Foa 1999), and interventions of at least 12 months to prevent relapse in the treatment of chronic PTSD (Foa 1999; Ballenger 2000; Ballenger 2004). Although the current review does not directly address the question of whether medication or psychotherapy exerts a larger effect in PTSD (van Etten 1998), combined treatment is often used in clinical settings, and this may contribute to the effects of psychotherapy in published studies. Conversely, it has been speculated that the high placebo response rate observed in some of the trials included in this review (Brady 2000; van der Kolk 2004) might be the consequence of psychological support provided to the patients as an inadvertent consequence of participating in the rigorous assessment procedures implemented within the trials (Krakow 2000; van der Kolk 2004). Nevertheless, in one of the trials for which this suggestion was made (van der Kolk 2004), the authors still discovered psychotherapy (Eye Movement Desensitization and Reprocessing (EMDR)) to be more effective than fluoxetine in maintaining the complete remission of PTSD symptoms six months after the end of a eight week placebo‐controlled RCT. There are, however, few direct comparisons of the efficacy of pharmacotherapy and psychotherapy for PTSD, and given that few psychotherapy RCTs have masked treatment assignment (NICE 2005) indirect comparisons of effect sizes may also have limited utility. Theoretically, modern understanding of PTSD as involving psychobiological dysfunctions would indicate that it is unnecessary to institute false dichotomies between brain and mind, and that both kinds of intervention might be useful (Southwick 1993; Khouzam 1997). Certain medications (e.g. benzodiazepines) have, however, been argued to diminish the effects of psychotherapy, so rigorous comparative studies are needed.
Additional questions for future pharmacological research in the area of PTSD include the effectiveness of medication in clinical settings, and the precise effects of medication on quality of life measures (Fossey 1994; Rapaport 2002). Further research on medication in PTSD in different age samples (Famularo 1988; Looff 1995; Harmon 1996; Horrigan 1996; Seedat 2001), on patients with comorbid substance use disorders (Liebowitz 1989; Brady 1995; Brady 2004), and on more treatment‐refractory (Braun 1990; Demartino 1995; Hamner 1997; Hamner 1998; Hidalgo 1999; Stein 2002; Raskind 2003; Bartzokis 2005) or non‐compliant (Kroll 1990) patients is also needed. In addition, randomised controlled trials are needed to determine the efficacy of promising medications, such as tiagabine (Taylor 2003), tianeptine (Onder 2005) and topirimate (Berlant 2002; Berlant 2004), for which only open‐label trials have been conducted thus far.
Given the high prevalence and enormous personal and societal costs of PTSD, there are still relatively few RCTs of pharmacotherapy for PTSD. No controlled trials were found in paediatric or geriatric subjects. With few exceptions (Baker 1995 a; Reist 1989; Braun 1990; Davidson 1990; Hertzberg 1999; Hertzberg 2000; Brady 2004), trials have excluded patients with comorbid substance use. One such exception to be included in this review was a RCT of sertraline in treating concurrent PTSD and alcoholism (Brady 2004), in which little difference was found in the efficacy of medication versus placebo in reducing symptom severity. Also, although outside the scope of the current review, there seem to be few RCTS of the treatment of traumatised patients prior to their meeting criteria for PTSD (Robert 1999; Pitman 2002; Mellman 2002; Schelling 2004), a potentially important clinical area.
Finally, the inherent problems of meta‐analysis should also be borne in mind (Bailar 1997); certainly these are by no means a substitute for clinical research. This is especially the case when there is evidence of the possibility that smaller trials with negative outcomes are not being published. Furthermore, the context of clinical practice differs from controlled trials in many respects, not the least being the inclusion of more complex patients (including patients with possible "secondary gain" from symptoms, a group that has been specifically excluded from more recent RCTs (Brady 2000)) and the possible need for polypharmacy in a subgroup of PTSD patients (Kolb 1984; Kinzie 1989; Burdon 1991; Hargrave 1993; Leyba 1998). Moreover, methodological shortcomings, such as failure to employ independent outcome assessors in trials which adjust medication dosage on the basis of side‐effect severity, and inadequate statistical methods of compensating for patient withdrawals, risk undermining the best efforts to minimise bias in the findings of systematic reviews of health care interventions. However, an advantage of the Cochrane Collaboration is that it encourages regular updating of reviews in the light of new data, and hopefully additional data from new randomised controlled trials of medication in PTSD will become available for inclusion in a revised systematic review in the future. Given the high prevalence of PTSD, its chronicity and morbidity, and its enormous personal and societal costs, additional prospective research on the pharmacological prevention and treatment of this disorder is clearly required, and systematic reviews and retrospective analyses may be useful in integrating the findings of such work as well as in suggesting areas for further investigation.
Authors' conclusions
Implications for practice.
Medication treatments can be effective in PTSD, acting to reduce its core symptoms, and should be considered as part of the treatment of this disorder. The existing evidence base of RCTs includes a heterogenous sample of participants with a range of different traumas, trauma duration and severity, and comorbidity. Although there is also no clear evidence to show that any particular class of medication is more effective or better tolerated than any other, the greatest number of trials showing efficacy to date, as well as the largest, have been with the SSRIs. In contrast, there have been negative studies of benzodiazepines, MAOIs, anti‐psychotics, lamotrigine and inositol. In addition, although there was only one RCT in which psychotherapy was compared with pharmacotherapy, from a clinical perspective it is as well to remember the possible value of this modality alone or in combination with pharmacotherapy. The findings of maintenance trials support the value of long‐term interventions in increasing the efficacy of medication and preventing relapse.
Implications for research.
Given the prevalence and costs of PTSD, there is a need for further controlled clinical trials in the treatment of this disorder. The differential efficacy and acceptability of different classes of medication, including newer agents potentially useful in this disorder (e.g. escitalopram, tiagabine, tianeptine, tropirimate), requires study. Questions for future research also include the precise effects of medication on quality of life measures, appropriate dose and duration of medication, and determining factors which predict response to medication. Further research on the value of medication in PTSD in different trauma groups, in paediatric and geriatric subjects, in patients with comorbid substance use, and in treatment‐refractory patients is needed. Clinical trials to determine the possible benefits of early (prophylactic), combined (with psychotherapy), and long‐term (maintenance) intervention in PTSD may also be valuable.
Feedback
Comments submitted by Danielle Stacey, 8 December 2015
Summary
In 2009, Stein DJ et al. published a Cochrane review which provided us with a very thorough explanation of the literature surrounding pharmacotherapy for post‐traumatic stress disorder (PTSD).1 After a thorough review of the publication, we have a few enquiries regarding the patient population represented in the meta‐analysis and the statistical analysis conducted. In addition, we have provided some potential recommendations for future updates.
Clinical heterogeneity
Within a systematic review we test for statistical heterogeneity, as did Stein DJ et al, however we often over look clinical heterogeneity or diversity.2 We agree with the authors’ conclusions that “the existing evidence base of RCTs includes a heterogeneous sample of participants with a range of different traumas, trauma duration and severity, and comorbidity.” As an example we can look at analysis 1.1 which includes the following patient populations: Brady 2004, patients suffering from alcoholism; Brady 2000 and Davidson 2001, patients with anxiety and MDD; Pfizer 589, predominantly war veterans; and some trials that have a combination of these various characteristics. The significantly greater reduction in symptom severity in trials with less veteran participants highlights how important clinical diversity can be when interpreting results and when analyzing comparisons for treatment within a meta‐analysis. Given this, one could argue whether meta‐analysis of trials with high clinical variability is even appropriate. At the very least it warrants a section to inform clinicians which patient populations are encompassed in each analysis. We also suggest completing an additional subgroup analysis for trials including participants with concurrent anxiety disorders. After a random selection of three trials (Brady 2000, Davidson 2001, Marshall 2004) from analysis 1.1, we found that all three included participants with concurrent anxiety disorders at various rates. Together, these three trials comprise a weight of 16.2% of the random effects model. This additional subgroup analysis will aid in further defining the treatment populations within the review and the potential impact on the pooled efficacy results.
Inclusion of studies in meta‐analysis 1.3
Focusing again on the statistical analysis, at first glance, analysis 1.3 for clinical global impression scale improvement item (CGI‐I) seemed to include a low number of RCTs for SSRIs. After scanning the included trial characteristics tables, we noticed 12 trials that had CGI‐I (or similar scale) as a primary or secondary outcome (Davidson, Davidson 2001, Brady 2000, Pfizer588, Pfizer589, Zohar 2002, Marshall 2004, Tucker 2001, Marshall 2001, Herzberg 2000, Eli Lilly, and Connor 1999). Analysis 1.3 included only 7 trials in the forest plot (Brady 2000, Connor 1999, Hertzberg 2000, Marshall 2001, Marshall 2004, Tucker 2001, and Zohar 2002). It was mentioned in the description of studies section that Davidson would not be used for statistical analysis, however this does not explain why the remainder of the trials were not included. In addition, a similar statement claimed that data from Tucker 2003 would be excluded from statistical analysis, however this trial was included in other forest plots (eg. Analysis 1.1). A brief explanation to help understand the reasoning for inclusion of trials into each analysis would help in interpreting the results.
References
As previously mentioned, we took the time to randomly pull some of the trials that were cited in the review. Our first choice was Davidson 2001. From the included studies citations, we chose the second of four trials under Davidson 2001 given the lead author and year of publication.3 It was very apparent that this trial was in fact a maintenance trial, and answering a very different question than the review. This particular Davidson publication cited another Davidson publication, which was in fact the acute treatment trial. Upon further inspection, data from the original trial was included in the analysis, which is great. However, it is confusing for readers because wherever Davidson 2001 is cited in the review, may lead them to look at the maintenance trial and not the main trial of interest. We suggest, for trials that have the same lead author and publication year (eg. Davidson 2001), adding a distinguishing mark for each will help readers.
Additional points
The combination of clinical variability, unclear trial methodology and very thorough inclusion of information can lead to confusion and misinterpretation without a clear, concise and easy to analyze presentation. To make it easier for readers to gain a quick and accurate interpretation of the included data and results, we suggest the following changes:
1. Include a risk of bias graph or a risk of bias summary figure for the included trials.2 It is apparent that a bias assessment was completed, however each risk of bias section did not include all pertinent design factors to make an accurate assessment. Furthermore, placing these factors in the characteristic tables does not help viewers get an overall idea of the included trial quality, nor is it efficient. In addition, although it seems Cochrane is not a full supporter of quality scales, this information could also be displayed with or immediately after the risk of bias tool(s).
2. Conduct a subgroup analysis for trials including patients with concurrent anxiety disorders as previously suggested in an effort to address clinical heterogeneity.
3. Include reasoning for including or excluding trials in each analysis where applicable, as previously suggested.
4. For different trials that have the same lead author and publication year (eg. Davidson 2001), add a distinguishing mark for each.
5. On page 14, the following statement contains an error, “the conclusion that a short‐term course of treatment with SSRIs may be inadequate is supported by increased relapse rates in trials of both fluoxetine (Davidson 2001) and sertraline (Martenyi 2002).” Davidson 2001 and Martenyi 2002 should be switched.
References:
1. Stein DJ, Ipser JC, Seedat S. Pharmacotherapy for post traumatic stress disorder (PTSD). Cochrane Database of Systematic Reviews 2006, Issue 1. Art. No.: CD002795. DOI: 10.1002/14651858.CD002795.pub2
2. Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org
3. Davidson J, Pearlstein T, Londborg P, Brady KT, Rothbaum B, Bell J, et al.Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: results of a 28‐week double‐blind, placebo‐ controlled study. American Journal of Psychiatry 2001;158(12): 1974–81.
Reply
We would like to thank the reviewers for their close reading of our Cochrane review ‘Pharmacotherapy for posttraumatic stress disorder’. We would note that there have been considerable advances in methodological and reporting standards since this review was published, a decade ago in 2006, and that adherence to these standards in the imminent update of the review should help to address many of the reviewer’s concerns. Specifically, new Cochrane standards require a more thorough description of important aspects of studies in the description of included studies section, under the following subheadings – design, participant characteristics, diagnosis, co‐morbidities, setting, interventions, and outcomes. The updated review will incorporate these headings and this information will make it easier for the reader to assess important aspects of studies included in the review. Nevertheless, we have responded as best we can to the individual comments kindly provided by the reviewers below.
Clinical heterogeneity
The reviewers are correct in highlighting the importance of taking clinical variability into account when interpreting the results of this meta‐analysis. It was in recognition of this fact that we originally decided to investigate whether effect sizes differed in trials of civilian trauma versus war veterans. That said, trials of very homogenous clinical populations may have limited generalizability. We have opted, in response to the reviewer’s comments, to present a general descriptive overview of the patients included in the eligible studies in the Description of studies section of the review. This was done instead of providing the suggested breakdown of the clinical background of trial participants for each analysis, in the interests of readability. As evident in this paragraph, the majority (18/22 or 82%) of RCTs included in the meta‐analysis that described comorbid diagnoses in their patients documented comorbid anxiety disorders in addition to PTSD. The exceptions in this regards were Katz 1995, Kosten 1991, Chung 2004, and Reist 1989, with only Katz 1995 providing data for inclusion in the analyses of the review’s primary outcomes. Accordingly, we feel that there is not not much utility in conducting additional subgroup analyses to assess the effects of comorbid anxiety disorders in this review.
Inclusion of studies in meta‐analysis 1.3
With regards to the observation that the analysis of the effect of medication on the CGI‐I outcome (Analysis 1.3) does not appear to contain all included studies, when considered across all medication classes, Analysis 1.3 contains a total of 13 trials, including the majority of the trials that the reviewers listed as including the CGI‐I as a primary or secondary outcome. The discrepancy noted by the reviewers is due to the fact that only those trials for which it was possible to extract data on the particular outcomes of interest were included in the analyses (after requesting data from the authors, where these were not reported in the study publication). In order to pre‐empt confusion on this point, we have altered the text describing the primary outcomes in the Effects of interventions section, to make this observation explicit.
As far as the inclusion of data from Tucker 2003 in analyses conducted within the review is concerned, it was noted in paragraph 5 (now paragraph 6) of the Description of included studies section that “Summary statistics for the sertraline arm of the Tucker 2003 trial were excluded from the analysis, in favour of including the data from the less well represented citalopram arm.” Nevertheless, we determined that data from the sertraline arm of Tucker 2003 had inadvertently been included in the review in place of data from the citalopram arm from this trial (Analysis 1.2 and 2.2). This error has now been corrected, and the results of relevant analyses updated accordingly. This correction has not changed the findings of the review.
References
Additional entries have been added under the Included Studies category in RevMan (version 5.2) to accommodate the maintenance phases of both the Davidson 2001 and Martenyi 2002 short‐term trials. Study IDs have been distinguished by appending them with alphabetical characters.
Additional points
1. We thank the reviewers for this suggestion. Cochrane methodologies have improved in the 10 years since this review was published. Inclusion of two risk of bias summary figures ‐ one that presents judgements about each risk of bias item as percentages across all included studies; and another that presents judgements about each risk of bias item for each included study is now a standard requirement for Cochrane reviews, and accordingly both will be included in the update to this review. Additionally Cochrane now supports the assessment of quality of included studies using the GRADE approach and this methodology will also be applied and incorporated in the interpretation of results in the update of the review.
2. This is not a viable option, given the fact that the vast majority of included trials that provide information on comorbid diagnoses include patients diagnosed with other anxiety disorders (please refer to the response to the first comment above).
3. We trust that we have sufficiently addressed the reviewer’s concerns regarding the rational for including or excluding RCTs in the review in our response to this particular query above.
4. We have distinguished between the acute and maintenance components of trials included in the review by assigning different labels to these study components, as described above. We have also updated the Characteristics of included studies section of the review accordingly.
5. We have made the suggested switch, and linked the text to the appropriate references for the maintenance components of the sertraline (Davidson 2001b) and fluoxetine (Martenyi 2002b) trials.
Contributors
Feedback submitted by Danielle Stacey, BSc(Pharm), PharmD Student Aaron Tejani, BSc(Pharm), PharmD
Response submitted by Jonathan Ipser
What's new
| Date | Event | Description |
|---|---|---|
| 12 April 2016 | Feedback has been incorporated | Feedback incorporated and small amendments made to the description of studies and effects of intervention sections. |
| 4 November 2008 | Amended | Converted to new review format. |
History
Protocol first published: Issue 1, 1998 Review first published: Issue 4, 2000
| Date | Event | Description |
|---|---|---|
| 14 October 2005 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
The authors are supported by the MRC Research Unit on Anxiety and Stress Disorders (Cape Town, South Africa). The authors wish to thank Cochrane Collaboration internal and external reviewers for comments on an earlier draft, and for providing advice on the process of a Cochrane Collaboration review. In particular, Dianne O'Connell provided statistical advice for the first version of this review. We would also like to thank George Bartzokis, Susan Brady and Phebe Tucker for responding to requests for additional data for the update to the review, and Bessel van der Kolk and Randall Marshall for letting us have access to unpublished trials. At CCDAN, Hugh McGuire provided help with literature searches. Joy Oliver of the South African Cochrane Centre help retrieve a number of trial reports in updating the review. Finally, we are especially indebted to the UK Cochrane collaboration for providing funding to update this review, and Elizabeth Waters and Nandi Siegfried in particular for their part in organising the funding. Nandi Siegfried also arranged for one of the reviewers (JI) to attend a 5 day Cochrane workshop in preparation for updating this review.
Data and analyses
Comparison 1. Medication versus Placebo: Primary global and PTSD symptom outcomes.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 CAPS: Total score | 17 | 2507 | Mean Difference (IV, Random, 95% CI) | ‐5.76 [‐8.16, ‐3.36] |
| 1.1 SSRIs | 12 | 1909 | Mean Difference (IV, Random, 95% CI) | ‐5.59 [‐8.60, ‐2.58] |
| 1.2 MAOIs | 2 | 178 | Mean Difference (IV, Random, 95% CI) | ‐5.06 [‐15.93, 5.81] |
| 1.4 Other medication | 3 | 420 | Mean Difference (IV, Random, 95% CI) | ‐5.51 [‐11.53, 0.52] |
| 2 CAPS: Individual SSRI agents | 12 | 1909 | Mean Difference (IV, Random, 95% CI) | ‐5.59 [‐8.60, ‐2.58] |
| 2.1 Citalopram versus placebo | 1 | 35 | Mean Difference (IV, Random, 95% CI) | 4.78 [‐15.95, 25.51] |
| 2.2 Fluoxetine versus placebo | 1 | 59 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐12.31, 10.51] |
| 2.3 Paroxetine versus placebo | 4 | 940 | Mean Difference (IV, Random, 95% CI) | ‐10.49 [‐13.87, ‐7.11] |
| 2.4 Sertraline versus placebo | 6 | 875 | Mean Difference (IV, Random, 95% CI) | ‐3.78 [‐6.90, ‐0.65] |
| 3 Clinical Global Impressions scale improvement item (CGI‐I) | 13 | 1272 | Risk Ratio (M‐H, Random, 95% CI) | 1.49 [1.28, 1.73] |
| 3.1 SSRIs | 7 | 999 | Risk Ratio (M‐H, Random, 95% CI) | 1.59 [1.39, 1.82] |
| 3.2 MAOIs | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 1.16 [0.79, 1.72] |
| 3.3 TCAs | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 3.0 [0.98, 9.14] |
| 3.4 Other medication | 3 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.73, 3.20] |
| 4 CGI‐I: Individual SSRI agents | 7 | 999 | Risk Ratio (M‐H, Random, 95% CI) | 1.59 [1.39, 1.82] |
| 4.1 fluoxetine versus placebo | 2 | 65 | Risk Ratio (M‐H, Random, 95% CI) | 1.35 [0.96, 1.89] |
| 4.2 paroxetine versus placebo | 3 | 719 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [1.38, 1.90] |
| 4.3 sertraline versus placebo | 2 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 1.71 [1.22, 2.40] |
1.1. Analysis.
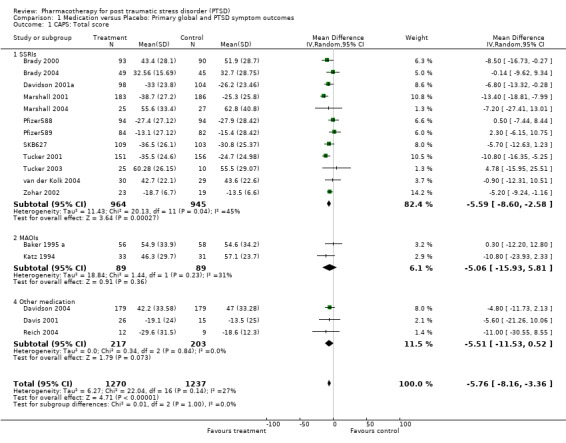
Comparison 1 Medication versus Placebo: Primary global and PTSD symptom outcomes, Outcome 1 CAPS: Total score.
1.2. Analysis.
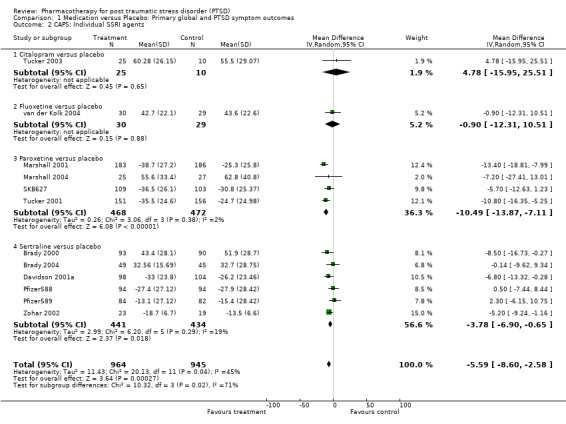
Comparison 1 Medication versus Placebo: Primary global and PTSD symptom outcomes, Outcome 2 CAPS: Individual SSRI agents.
1.3. Analysis.
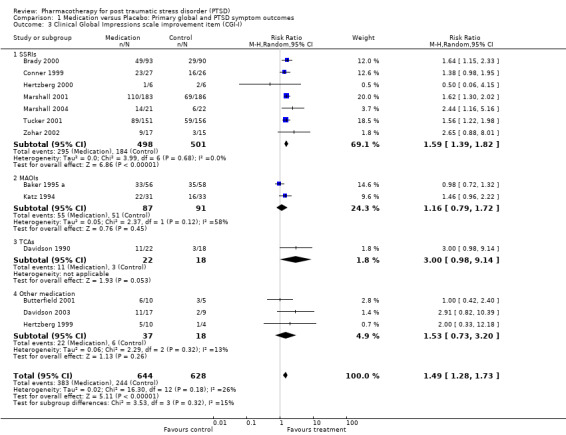
Comparison 1 Medication versus Placebo: Primary global and PTSD symptom outcomes, Outcome 3 Clinical Global Impressions scale improvement item (CGI‐I).
1.4. Analysis.
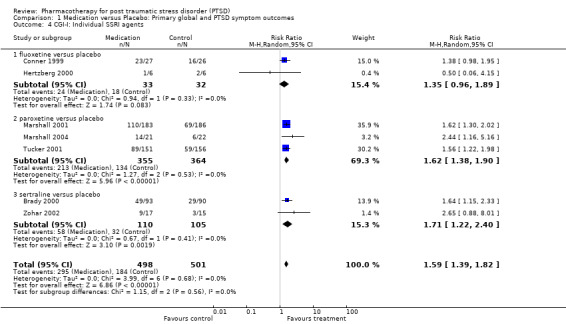
Comparison 1 Medication versus Placebo: Primary global and PTSD symptom outcomes, Outcome 4 CGI‐I: Individual SSRI agents.
Comparison 2. Medication versus Placebo: Secondary measures of PTSD.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Symptom severity: Other measures | 6 | 147 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.44 [‐0.87, ‐0.01] |
| 1.1 SSRIs | 2 | 59 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐1.66, 1.05] |
| 1.2 MAOIs | 0 | 0 | Std. Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 1.3 TCAs | 1 | 33 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.63 [‐1.33, 0.07] |
| 1.4 Other medications | 3 | 55 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.92, 0.32] |
| 2 Self‐rated scales: Total score | 9 | 882 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.51, ‐0.11] |
| 2.1 SSRIs | 5 | 771 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.34, ‐0.06] |
| 2.2 MAOIs | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.06 [‐1.75, ‐0.36] |
| 2.3 TCAs | 1 | 33 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐1.62, ‐0.18] |
| 2.4 Other medication | 2 | 41 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐1.20, 0.33] |
| 3 CAPS subscale: Re‐experiencing/intrusion | 9 | 1304 | Mean Difference (IV, Random, 95% CI) | ‐2.06 [‐3.02, ‐1.10] |
| 3.1 SSRIs | 7 | 1242 | Mean Difference (IV, Random, 95% CI) | ‐1.97 [‐3.11, ‐0.83] |
| 3.2 MAOIs | 0 | 0 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.3 TCAs | 0 | 0 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3.4 Other medication | 2 | 62 | Mean Difference (IV, Random, 95% CI) | ‐2.38 [‐6.52, 1.76] |
| 4 CAPS subscale: Avoidance/numbing | 9 | 1304 | Mean Difference (IV, Random, 95% CI) | ‐4.06 [‐5.41, ‐2.70] |
| 4.1 SSRIs | 7 | 1242 | Mean Difference (IV, Random, 95% CI) | ‐4.19 [‐5.58, ‐2.80] |
| 4.2 MAOIs | 0 | 0 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.3 TCAs | 0 | 0 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4.4 Other medication | 2 | 62 | Mean Difference (IV, Random, 95% CI) | ‐1.56 [‐7.56, 4.44] |
| 5 CAPS subscale: Hyperarousal | 9 | 1304 | Mean Difference (IV, Random, 95% CI) | ‐3.10 [‐4.10, ‐2.10] |
| 5.1 SSRIs | 7 | 1242 | Mean Difference (IV, Random, 95% CI) | ‐3.07 [‐4.09, ‐2.04] |
| 5.2 MAOIs | 0 | 0 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.3 TCAs | 0 | 0 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 Other medication | 2 | 62 | Mean Difference (IV, Random, 95% CI) | ‐3.77 [‐8.37, 0.84] |
| 6 Self‐rated subscale: Re‐experiencing/Intrusion | 4 | 268 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.43 [‐0.98, 0.13] |
| 6.1 SSRIs | 1 | 183 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.18 [‐0.47, 0.11] |
| 6.2 MAOIs | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.07 [‐1.77, ‐0.38] |
| 6.3 TCAs | 1 | 33 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐1.46, ‐0.04] |
| 6.4 Other medication | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | 0.51 [‐0.58, 1.61] |
| 7 Self‐rated subscale: Avoidance/numbing | 4 | 268 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.49 [‐0.90, ‐0.08] |
| 7.1 SSRIs | 1 | 183 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.19 [‐0.48, 0.10] |
| 7.2 MAOIs | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.81 [‐1.49, ‐0.14] |
| 7.3 TCAs | 1 | 33 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐1.62, ‐0.18] |
| 7.4 Other medication | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐1.38, 0.78] |
| 8 Self‐rated subscale: Hyperarousal | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐1.13, 1.01] |
| 8.4 Other medication | 1 | 15 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.06 [‐1.13, 1.01] |
2.1. Analysis.
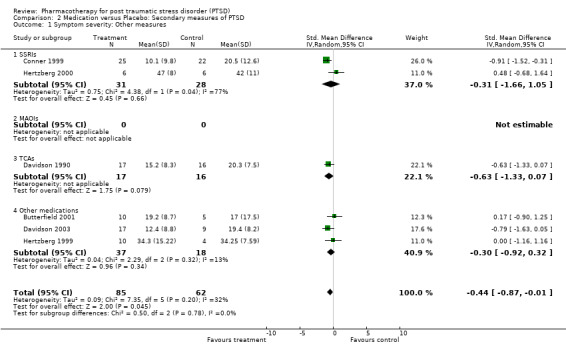
Comparison 2 Medication versus Placebo: Secondary measures of PTSD, Outcome 1 Symptom severity: Other measures.
2.2. Analysis.
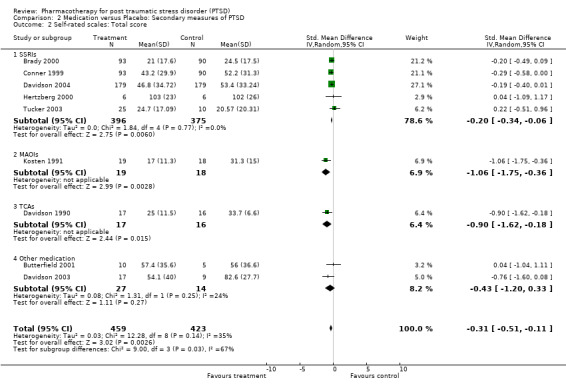
Comparison 2 Medication versus Placebo: Secondary measures of PTSD, Outcome 2 Self‐rated scales: Total score.
2.3. Analysis.
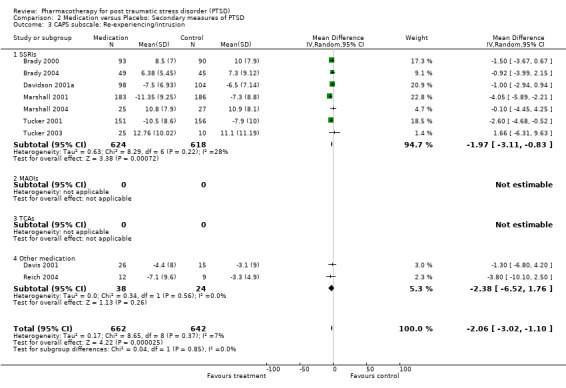
Comparison 2 Medication versus Placebo: Secondary measures of PTSD, Outcome 3 CAPS subscale: Re‐experiencing/intrusion.
2.4. Analysis.
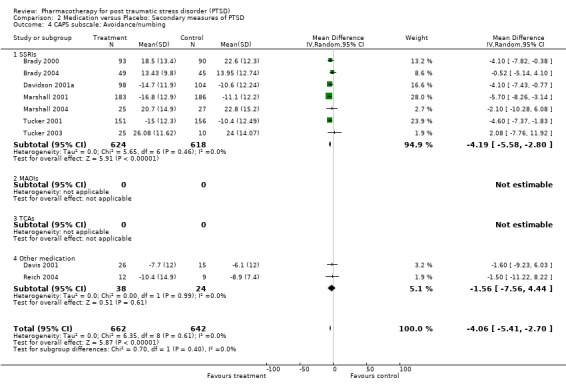
Comparison 2 Medication versus Placebo: Secondary measures of PTSD, Outcome 4 CAPS subscale: Avoidance/numbing.
2.5. Analysis.
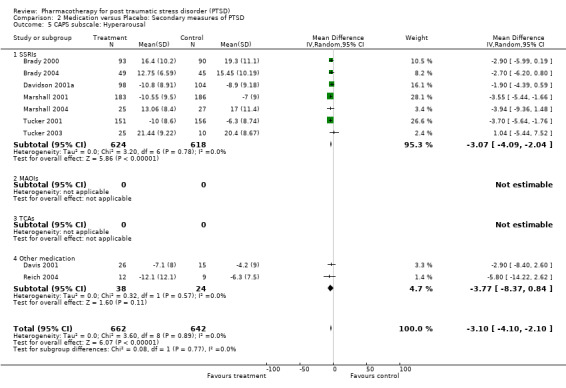
Comparison 2 Medication versus Placebo: Secondary measures of PTSD, Outcome 5 CAPS subscale: Hyperarousal.
2.6. Analysis.
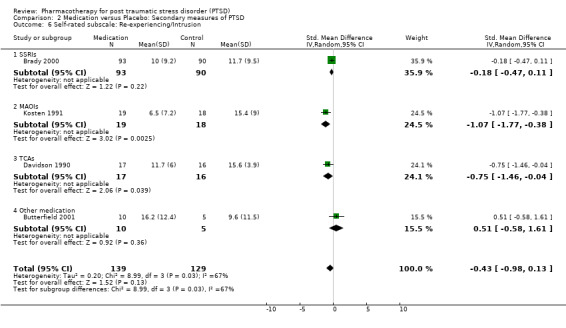
Comparison 2 Medication versus Placebo: Secondary measures of PTSD, Outcome 6 Self‐rated subscale: Re‐experiencing/Intrusion.
2.7. Analysis.
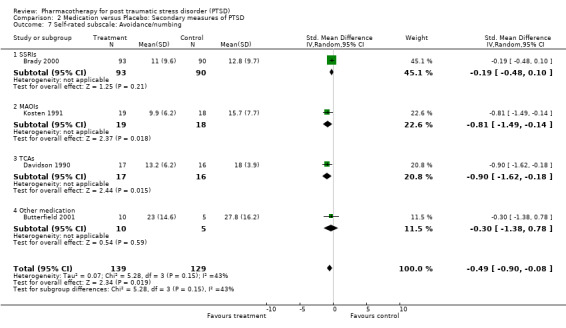
Comparison 2 Medication versus Placebo: Secondary measures of PTSD, Outcome 7 Self‐rated subscale: Avoidance/numbing.
2.8. Analysis.
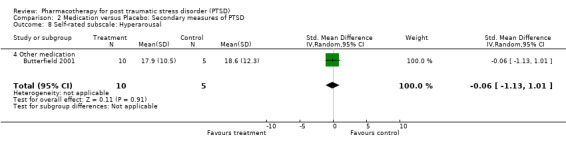
Comparison 2 Medication versus Placebo: Secondary measures of PTSD, Outcome 8 Self‐rated subscale: Hyperarousal.
Comparison 3. Medication versus Placebo: Comorbid symptoms.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Depression Scale (typically Hamilton Depression) | 7 | 459 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.34 [‐0.57, ‐0.10] |
| 1.1 SSRIs | 4 | 364 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.19 [‐0.39, 0.02] |
| 1.2 MAOIs | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐1.06, 0.25] |
| 1.3 TCAs | 1 | 33 | Std. Mean Difference (IV, Random, 95% CI) | ‐1.16 [‐1.90, ‐0.41] |
| 1.4 Other medication | 1 | 25 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.89 [‐1.78, ‐0.01] |
| 2 Depression Scale ‐ Change scores | 3 | 887 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.33 [‐0.60, ‐0.07] |
| 2.1 SSRIs | 3 | 887 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.33 [‐0.60, ‐0.07] |
| 2.2 MAOIs | 0 | 0 | Std. Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.3 TCAs | 0 | 0 | Std. Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.4 Other medication | 0 | 0 | Std. Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Anxiety ‐ Hamilton Anxiety Scale | 3 | 287 | Mean Difference (IV, Random, 95% CI) | ‐2.17 [‐7.22, 2.88] |
| 3.1 SSRIs | 2 | 254 | Mean Difference (IV, Random, 95% CI) | 0.58 [‐1.97, 3.14] |
| 3.3 TCAs | 1 | 33 | Mean Difference (IV, Random, 95% CI) | ‐7.60 [‐12.74, ‐2.46] |
| 3.4 Other medication | 0 | 0 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 4 Anxiety ‐ Other scales | 2 | 62 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.75 [‐1.28, ‐0.22] |
| 4.2 MAOIs | 1 | 37 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.67 [‐1.34, ‐0.01] |
| 4.4 Other medication | 1 | 25 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.88 [‐1.77, ‐0.00] |
3.1. Analysis.
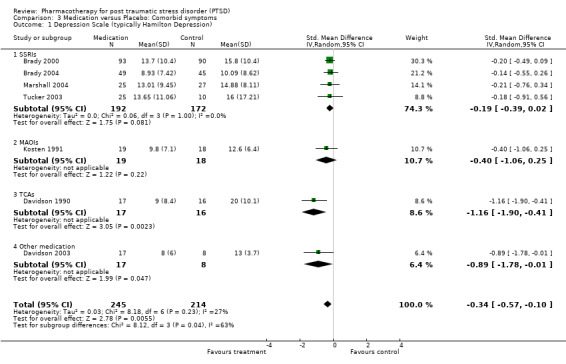
Comparison 3 Medication versus Placebo: Comorbid symptoms, Outcome 1 Depression Scale (typically Hamilton Depression).
3.2. Analysis.
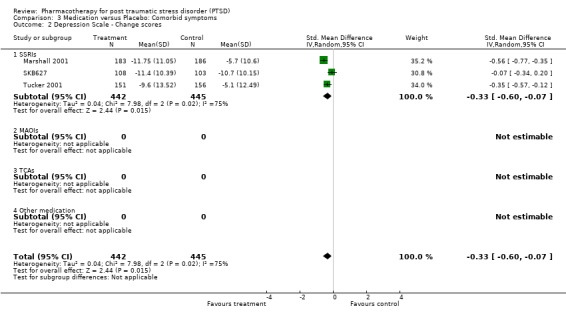
Comparison 3 Medication versus Placebo: Comorbid symptoms, Outcome 2 Depression Scale ‐ Change scores.
3.3. Analysis.
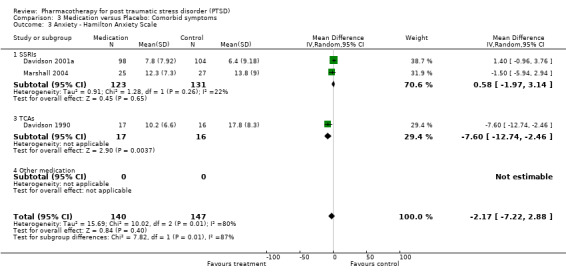
Comparison 3 Medication versus Placebo: Comorbid symptoms, Outcome 3 Anxiety ‐ Hamilton Anxiety Scale.
3.4. Analysis.
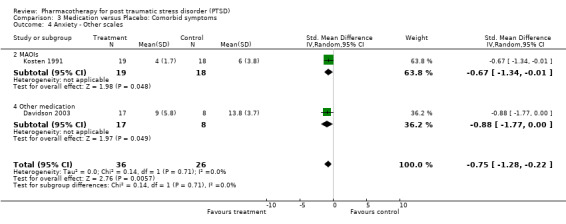
Comparison 3 Medication versus Placebo: Comorbid symptoms, Outcome 4 Anxiety ‐ Other scales.
Comparison 4. Medication versus Placebo: Quality of Life Scales.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Sheehan Disability Scale | 5 | 752 | Mean Difference (IV, Random, 95% CI) | ‐2.54 [‐3.68, ‐1.41] |
| 1.1 SSRIs | 4 | 737 | Mean Difference (IV, Random, 95% CI) | ‐2.56 [‐3.70, ‐1.41] |
| 1.4 Other medications | 1 | 15 | Mean Difference (IV, Random, 95% CI) | ‐1.5 [‐10.53, 7.53] |
4.1. Analysis.
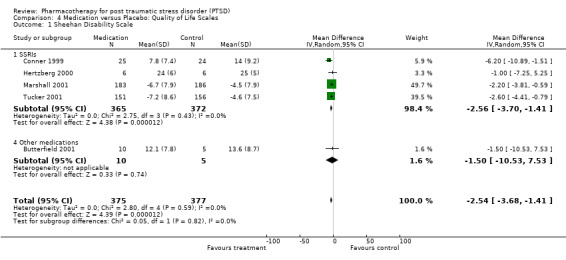
Comparison 4 Medication versus Placebo: Quality of Life Scales, Outcome 1 Sheehan Disability Scale.
Comparison 5. Medication versus Placebo: Drop‐out Rate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Drop‐out rate due to treatment emergent adverse effects | 21 | 2116 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [1.04, 2.00] |
| 1.1 SSRIs | 10 | 1649 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.99, 2.05] |
| 1.2 MAOIs | 3 | 196 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.29, 3.57] |
| 1.3 TCAs | 2 | 100 | Risk Ratio (M‐H, Random, 95% CI) | 5.45 [0.68, 43.88] |
| 1.4 Other medication | 6 | 171 | Risk Ratio (M‐H, Random, 95% CI) | 1.69 [0.53, 5.43] |
| 2 Drop‐out rate due to treatment emergent adverse effect: SSRI medications | 10 | 1649 | Risk Ratio (M‐H, Random, 95% CI) | 1.42 [0.99, 2.05] |
| 2.1 Fluoxetine | 3 | 367 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.25, 2.96] |
| 2.2 Paroxetine | 3 | 751 | Risk Ratio (M‐H, Random, 95% CI) | 1.48 [0.94, 2.31] |
| 2.3 Sertraline | 4 | 531 | Risk Ratio (M‐H, Random, 95% CI) | 1.54 [0.73, 3.26] |
5.1. Analysis.
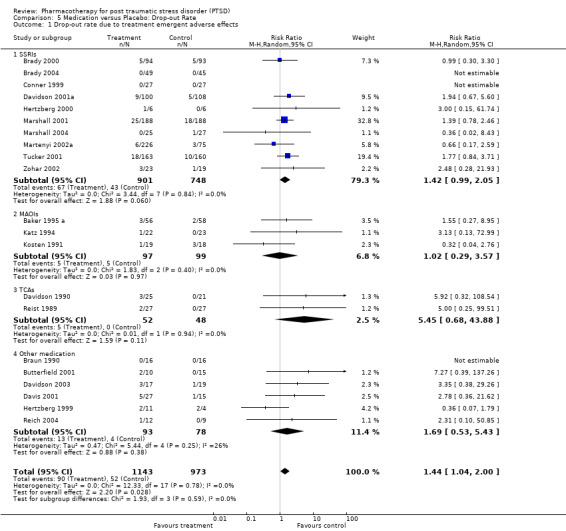
Comparison 5 Medication versus Placebo: Drop‐out Rate, Outcome 1 Drop‐out rate due to treatment emergent adverse effects.
5.2. Analysis.
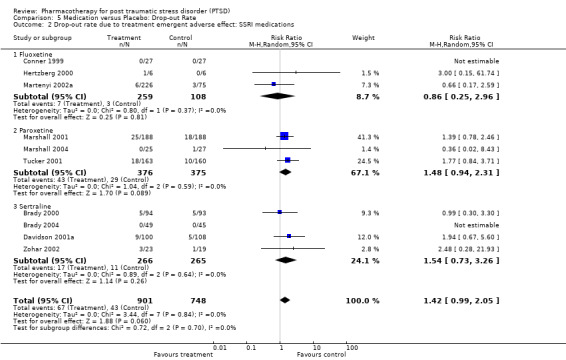
Comparison 5 Medication versus Placebo: Drop‐out Rate, Outcome 2 Drop‐out rate due to treatment emergent adverse effect: SSRI medications.
Comparison 6. Medication versus Placebo: Extension data.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2 Relapse data: Number of participants to relapse | 2 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.12, 0.70] |
| 2.1 SSRIs | 2 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 0.29 [0.12, 0.70] |
| 3 Continuation trials: Symptom severity on the CAPS | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 9.62 [‐3.53, 22.77] |
| 3.1 SSRIs | 1 | 16 | Mean Difference (IV, Random, 95% CI) | 9.62 [‐3.53, 22.77] |
6.2. Analysis.
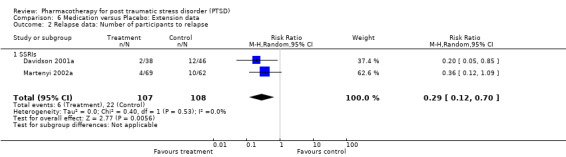
Comparison 6 Medication versus Placebo: Extension data, Outcome 2 Relapse data: Number of participants to relapse.
6.3. Analysis.
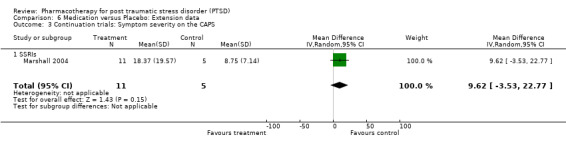
Comparison 6 Medication versus Placebo: Extension data, Outcome 3 Continuation trials: Symptom severity on the CAPS.
Comparison 7. Subgroup analyses ‐ Methodological criteria.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Single versus multi‐centre trials | 14 | 1895 | Mean Difference (IV, Random, 95% CI) | ‐5.62 [‐8.95, ‐2.29] |
| 1.1 Single centre trials | 6 | 302 | Mean Difference (IV, Random, 95% CI) | ‐2.20 [‐7.95, 3.55] |
| 1.2 Multi‐centre trials | 8 | 1593 | Mean Difference (IV, Random, 95% CI) | ‐6.49 [‐10.75, ‐2.22] |
| 2 Single versus multi‐centre trials | 13 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Single centre trials | 6 | 188 | Risk Ratio (M‐H, Random, 95% CI) | 1.77 [1.20, 2.60] |
| 2.2 Multi‐centre trials | 7 | 1084 | Risk Ratio (M‐H, Random, 95% CI) | 1.44 [1.20, 1.72] |
| 3 Industry versus non‐industry funded trials | 13 | 1899 | Mean Difference (IV, Random, 95% CI) | ‐6.49 [‐9.64, ‐3.34] |
| 3.1 Industry funded trials | 12 | 1840 | Mean Difference (IV, Random, 95% CI) | ‐6.83 [‐10.09, ‐3.57] |
| 3.2 Non‐industry funded trials | 1 | 59 | Mean Difference (IV, Random, 95% CI) | ‐0.90 [‐12.31, 10.51] |
| 4 Industry versus non‐industry funded trials | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Industry funded trials | 9 | 1053 | Risk Ratio (M‐H, Random, 95% CI) | 1.61 [1.41, 1.84] |
| 4.2 Non‐industry funded trials | 3 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.53 [0.81, 2.87] |
7.1. Analysis.
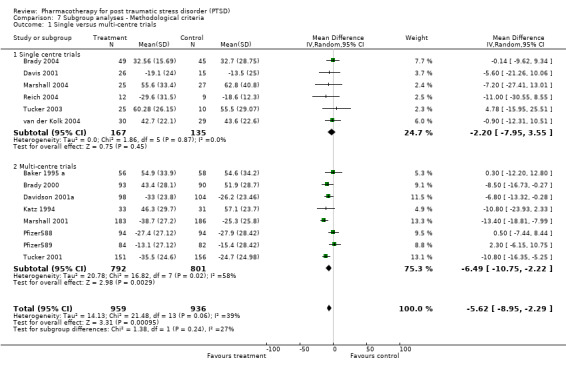
Comparison 7 Subgroup analyses ‐ Methodological criteria, Outcome 1 Single versus multi‐centre trials.
7.2. Analysis.
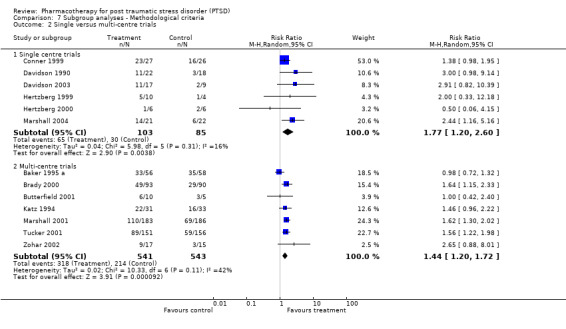
Comparison 7 Subgroup analyses ‐ Methodological criteria, Outcome 2 Single versus multi‐centre trials.
7.3. Analysis.
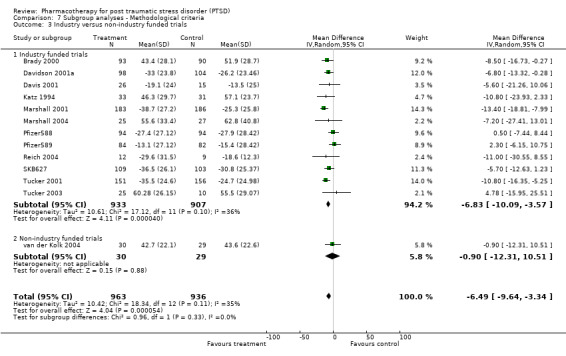
Comparison 7 Subgroup analyses ‐ Methodological criteria, Outcome 3 Industry versus non‐industry funded trials.
7.4. Analysis.
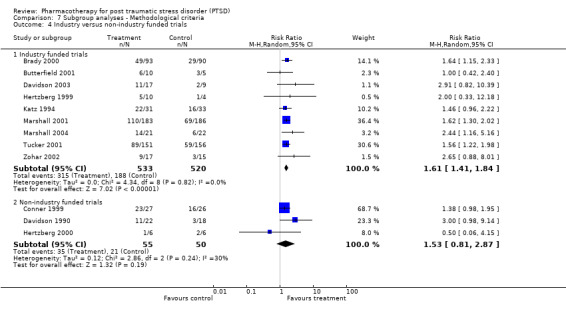
Comparison 7 Subgroup analyses ‐ Methodological criteria, Outcome 4 Industry versus non‐industry funded trials.
Comparison 8. Subgroup analyses ‐ Clinical criteria.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Inclusion of major depression vs. non‐inclusion: CAPS | 11 | 1455 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.45, ‐0.16] |
| 1.1 Trials including patients with major depressive disorder | 9 | 1304 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.35 [‐0.46, ‐0.23] |
| 1.2 Trials excluding patients with major depressive disorder | 2 | 151 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.48 [‐1.52, 0.56] |
| 2 Inclusion of major depression vs. non‐inclusion: Clinical Global Impressions scale improvement item (CGI‐I) | 7 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Trials including patients with major depressive disorder | 7 | 812 | Risk Ratio (M‐H, Random, 95% CI) | 1.62 [1.39, 1.89] |
| 2.2 Trials excluding patients with major depressive disorder | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 3 Inclusion of war veterans versus non‐inclusion: CAPS | 12 | 1801 | Mean Difference (IV, Random, 95% CI) | ‐6.18 [‐9.69, ‐2.66] |
| 3.1 Trials including war veterans | 4 | 385 | Mean Difference (IV, Random, 95% CI) | ‐1.73 [‐7.54, 4.07] |
| 3.2 Trials without war veterans | 8 | 1416 | Mean Difference (IV, Random, 95% CI) | ‐7.54 [‐11.37, ‐3.71] |
| 4 Inclusion of war veterans versus non‐inclusion: CGI‐I | 13 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Trials including war veterans | 8 | 317 | Risk Ratio (M‐H, Random, 95% CI) | 1.40 [0.99, 2.00] |
| 4.2 Trials without war veterans | 5 | 955 | Risk Ratio (M‐H, Random, 95% CI) | 1.59 [1.39, 1.81] |
8.1. Analysis.
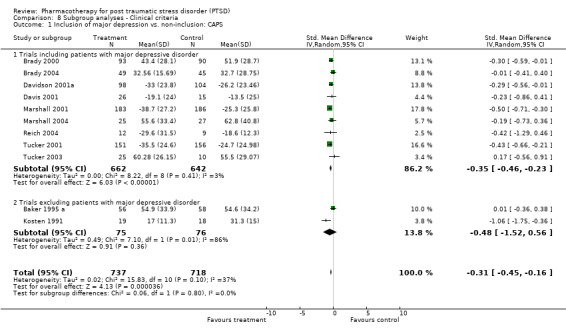
Comparison 8 Subgroup analyses ‐ Clinical criteria, Outcome 1 Inclusion of major depression vs. non‐inclusion: CAPS.
8.2. Analysis.
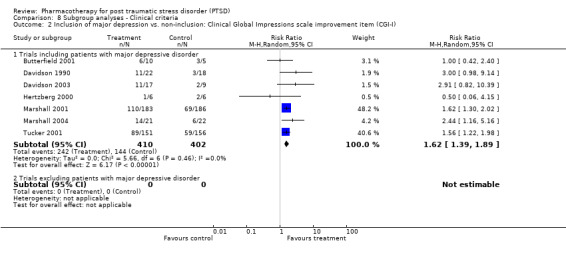
Comparison 8 Subgroup analyses ‐ Clinical criteria, Outcome 2 Inclusion of major depression vs. non‐inclusion: Clinical Global Impressions scale improvement item (CGI‐I).
8.3. Analysis.
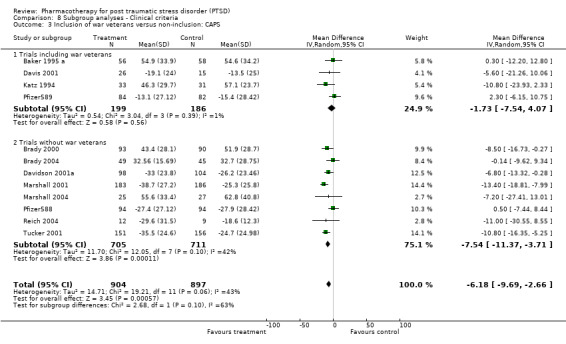
Comparison 8 Subgroup analyses ‐ Clinical criteria, Outcome 3 Inclusion of war veterans versus non‐inclusion: CAPS.
8.4. Analysis.
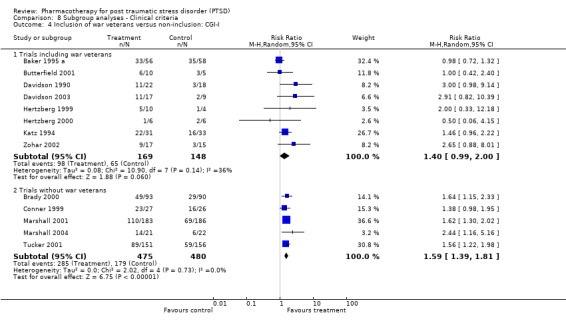
Comparison 8 Subgroup analyses ‐ Clinical criteria, Outcome 4 Inclusion of war veterans versus non‐inclusion: CGI‐I.
Comparison 9. Sensitivity analyses.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinical Global Impressions scale improvement item (CGI‐I) : non‐response | 13 | 1272 | Risk Ratio (M‐H, Random, 95% CI) | 0.66 [0.60, 0.74] |
| 1.1 SSRIs | 7 | 999 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.58, 0.74] |
| 1.2 MAOIs | 2 | 178 | Risk Ratio (M‐H, Random, 95% CI) | 0.80 [0.44, 1.44] |
| 1.3 TCAs | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.6 [0.38, 0.96] |
| 1.4 Other medication | 3 | 55 | Risk Ratio (M‐H, Random, 95% CI) | 0.59 [0.35, 0.98] |
| 2 "Worst case" loss to follow up analysis | 12 | 1275 | Risk Ratio (M‐H, Random, 95% CI) | 1.37 [1.12, 1.69] |
| 2.1 SSRIs | 7 | 1027 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [1.33, 1.74] |
| 2.2 MAOIs | 2 | 175 | Risk Ratio (M‐H, Random, 95% CI) | 0.28 [0.01, 5.42] |
| 2.3 TCAs | 1 | 43 | Risk Ratio (M‐H, Random, 95% CI) | 2.64 [0.86, 8.12] |
| 2.4 Other medication | 2 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.12 [0.51, 2.47] |
| 3 "Best case" loss to follow up analysis | 12 | 1262 | Risk Ratio (M‐H, Random, 95% CI) | 1.52 [1.22, 1.90] |
| 3.1 SSRIs | 7 | 1018 | Risk Ratio (M‐H, Random, 95% CI) | 1.64 [1.44, 1.87] |
| 3.2 MAOIs | 2 | 172 | Risk Ratio (M‐H, Random, 95% CI) | 0.48 [0.06, 3.86] |
| 3.3 TCAs | 1 | 43 | Risk Ratio (M‐H, Random, 95% CI) | 3.5 [1.13, 10.81] |
| 3.4 Other medication | 2 | 29 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.52, 2.51] |
9.1. Analysis.
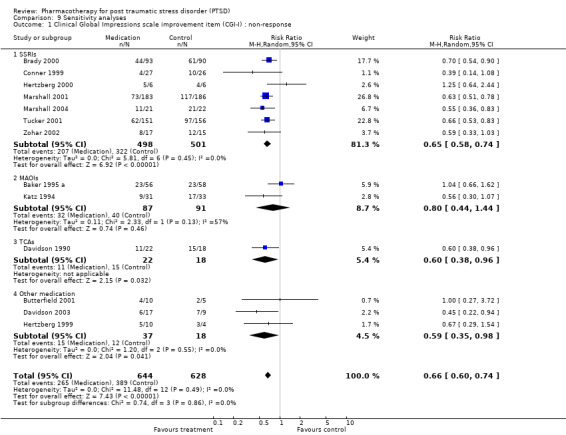
Comparison 9 Sensitivity analyses, Outcome 1 Clinical Global Impressions scale improvement item (CGI‐I) : non‐response.
9.2. Analysis.
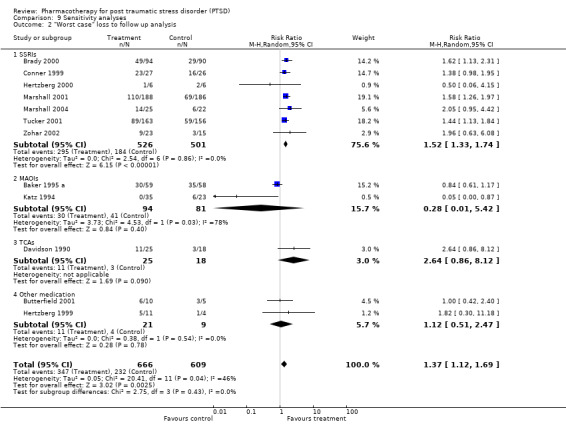
Comparison 9 Sensitivity analyses, Outcome 2 "Worst case" loss to follow up analysis.
9.3. Analysis.
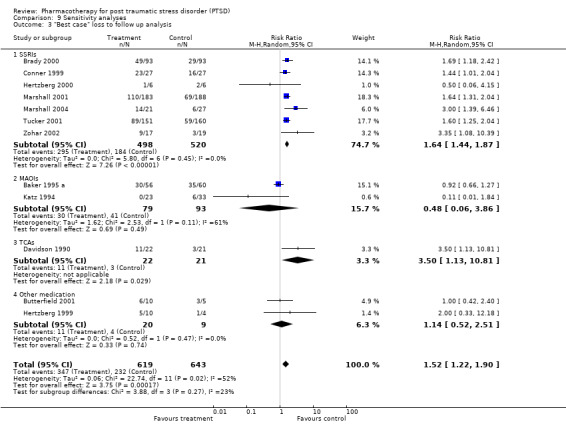
Comparison 9 Sensitivity analyses, Outcome 3 "Best case" loss to follow up analysis.
Comparison 10. Head‐to‐head comparisons.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Clinician administered scales: Symptom severity | 3 | 432 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.04 [‐0.23, 0.14] |
| 1.1 Nefazodone versus sertraline | 2 | 80 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.19 [‐0.63, 0.25] |
| 1.2 Venlafaxine versus sertraline | 1 | 352 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.01 [‐0.22, 0.20] |
| 2 Comorbid symptoms: Depression (MADRS) | 1 | 26 | Mean Difference (IV, Random, 95% CI) | ‐0.84 [‐7.88, 6.20] |
| 3 Comorbid symptoms: Anxiety (Hamilton Anxiety Scale) | 1 | 26 | Mean Difference (IV, Random, 95% CI) | ‐3.23 [‐10.90, 4.44] |
10.1. Analysis.
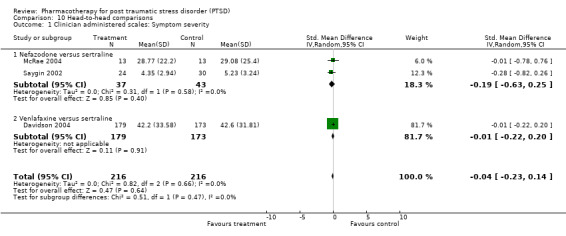
Comparison 10 Head‐to‐head comparisons, Outcome 1 Clinician administered scales: Symptom severity.
10.2. Analysis.
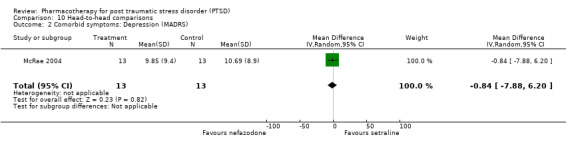
Comparison 10 Head‐to‐head comparisons, Outcome 2 Comorbid symptoms: Depression (MADRS).
10.3. Analysis.
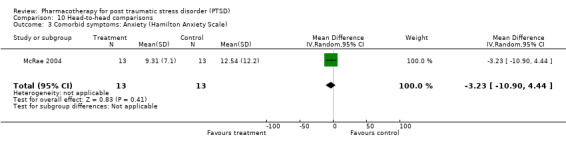
Comparison 10 Head‐to‐head comparisons, Outcome 3 Comorbid symptoms: Anxiety (Hamilton Anxiety Scale).
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Baker 1995 a.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, single‐blind placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 146 DSM‐III‐R PTSD, average age: 44 years (23‐73), 81% male, mean duration of diagnosis: 12.8 years, MDD not present, 60% combat‐related SCREENING Primary diagnosis: symptoms at least 6 months, CAPS >= 45, MADRS <= 22 Comorbidity: MADRS |
|
| Interventions | Description: brofaromine up to 150 mg/d vs placebo x 12 weeks | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: IES, DTS, CGI NOTES: CGI (values not reported, CGI‐C obtained from Davidson et al, 1997), ITT (LOCF 1 post‐baseline assessment) values provided. |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Not mentioned
Medication provided by industry: No
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: total of 35 Quality rating score: 15 Single‐blind placebo run‐in excluded 28 placebo‐responders Obtained additional HAM‐D and CAPS‐2 summary statistics from Sudie Back |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Brady 2000.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, single blind 2 week placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 187 DSM‐III‐R PTSD, 6% combat‐related, mean age: 39.9 years (18‐69), 27% male, 33% (62/187) with current major depression SCREENING Primary diagnosis: CAPS‐1, 6‐month duration, CAPS >= 50 at end of placebo run‐in Comorbidity: No information |
|
| Interventions | Description: sertraline 50‐200 mg/d (mean endpoint dose: 133.3 mg/d) versus placebo x 12 weeks | |
| Outcomes | Primary outcomes: CGI‐C, CGI‐S, CAPS‐2, IES. Responders defined as 30% or greater decrease on CAPS‐2 and CGI‐C of 1 or 2.
Secondary outcomes: DTS, HAM‐D, Q‐LES‐Q Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 30/94(32%) on sertraline, 28/93(30%) on placebo. Quality rating score: 31 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Brady 2004.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, fixed dose, double‐blind, 4 day tapering of medication at end of trial, single blind 1 week placebo run‐in BLINDING Participants: unclear Assessors: unclear Administrators: unclear ALLOCATION CONCEALMENT Method: unclear RANDOMISATION Method: urn randomisation by gender, depressive disorder, trauma type, and age of index trauma |
|
| Participants | SAMPLE
Description: 94 DSM‐IV PTSD with concurrent alcoholism (LOCF sample), no war veterans, mean age: 36.7 years, 54% male, SCREENING Primary diagnosis: CAPS, SCID, CAPS >= 30% decrease after placebo run‐in grounds for exclusion Comorbidity: TLFB |
|
| Interventions | Description: sertraline (50 mg/d ‐ 150 mg/d) versus placebo (50 mg/d ‐ 150 mg/d) x 12 weeks | |
| Outcomes | Outcomes: OCDS, CAPS, HAM‐D, SID, IES (not clear whether primary or secondary) Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: unclear
Medication provided by industry: Yes
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: information not provided, 3 patients excluded as responders during placebo run‐in Quality rating score: 22 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Braun 1990.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, crossover, 2 week titrated placebo washout, flexible dose, double‐blind, single centre BLINDING Participants: Unclear Assessors: Yes Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 16 DSM‐III PTSD, 25% combat‐veterans, mean age: 37.7 years (19‐56), 13% (2/16) MDD, described as chronic and treatment‐refractory SCREENING Primary diagnosis: Unclear Comorbidity: None |
|
| Interventions | Description: alprazolam 1.5mg to 6 mg/d (max avg: 4.65 mg/d) in divided doses vs placebo x 5 weeks each phase of crossover | |
| Outcomes | Outcomes: DSM based PTSD scale, IES, HAM‐D, HAM‐A, Visual Analogue (not clear which outcomes are secondary or primary). Data estimation: Observed cases for patients who completed 5 weeks of both phases |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Unclear
Medication provided by industry: Unclear
Any of the authors work for industry: Unclear ADDITIONAL INFORMATION Drop‐out rates: 3/7 (43%) on alprazolam, 3/9 (33%) on placebo. Quality Rating Scale score: 14 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Butterfield 2001.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 15 DSM‐IV PTSD, mean age: 43.2 years (26‐73), 14 women, 53.3% (8/15) MDD, most common comorbid diagnosis: GAD ‐ 64.3% (9/15), baseline severity on TOP‐8: olanzapine (19.3), placebo (21.8) SCREENING Primary diagnosis: SIP Comorbidity: MINI |
|
| Interventions | Description: olanzapine 5 mg/d ‐ 20mg/d (max. mean 14.1 mg/d) versys placebo (max. mean: 13.9mg/d) x 10 weeks | |
| Outcomes | Primary outcomes: TOP‐8, SPRINT,
Secondary outcomes: IES, DTS, CGI‐I, SDS, BAS, AIMS Data estimation: ITT (General linear model) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 3/15 (20%) on olanzapine and 1/5 (20%) on placebo. Quality rating score: 24 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Chung 2004.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, non‐blinded, single centre BLINDING Participants: No Assessors: No Administrators: No ALLOCATION CONCEALMENT Method: None RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 113 DSM‐IV Korean war veterans, mean age: 59.8 , mean duration of diagnosis:34.5, 17% (17/100) MDD, baseline severity on CAPS‐2: mirtazapine (103.2), sertaline (88.8) SCREENING Primary diagnosis: Unclear Comorbidity: None |
|
| Interventions | Description: mirtazapine 15 mg/d ‐ 34.1 mg/d (mean daily dose) versus sertraline 50mg/d ‐ 101.5mg/d (mean daily dose) x 6 weeks | |
| Outcomes | CAPS‐2, HAM‐D (17 item), CGI‐I, CGI‐S (no distinction made between primary and secondary outcomes) Data estimation: completer values |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 12.1% (7/58) on mirtazapine, 10.9% (6/55) on sertraline Quality rating score: 27 Patients taking antidepressants were placed on 7 day washout priot to entering trial |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Conner 1999.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, single centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: randomisation only known to hospital pharmacy RANDOMISATION Method: computer generated randomisation into groups |
|
| Participants | SAMPLE
Description: 54 DSM‐III PTSD, none combat‐related, duration of diagnosis: 6 years (median), median age: 37 years (18‐55), 91% female, baseline severity on DGRP: fluoxetine (4.2) placebo (4.6) SCREENING Primary diagnosis: SCID Comorbidity: None |
|
| Interventions | Description: fluoxetine (20‐60 mg/d; median: 30 mg/d) vs placebo (20‐60 mg/d; median: 40 mg/d) x 12 weeks | |
| Outcomes | Primary outcomes: DGRP(change, severity)
Secondary outcomes: SIP, DTS, SDS. VS. Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No
Medication provided by industry: Yes
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 6/27(22%) on fluoxetine and 12/27(44%) on placebo. Quality rating score: 28 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Davidson 1990.
| Methods | DESIGN
Description: balanced randomization, placebo‐controlled, parallel arms, flexible dose, double‐blind, single centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 46 DSM‐III PTSD, chronic, combat veterans, presumably all males, inpatients included, 20% (9/46) MDD, most common comorbid diagnosis: GAD (16), baseline severity on IES: amitriptyline (33.1) placebo (36.8) SCREENING Primary diagnosis: SI‐PTSD Comorbidity: None |
|
| Interventions | Description: amitriptyline (50‐300 mg/d) vs placebo x 8 weeks, ongoing supportive psychotherapy | |
| Outcomes | Primary outcomes: CGI‐I , SI‐PTSD, CGI‐S
Secondary outcomes: HAM‐D, HAM‐A, IES, NI, EPI Data estimation: completer values |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No
Medication provided by industry: No
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 8/25 (32%) on amitriptyline, 5/21 (24%) on placebo. Quality rating score: 24 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Davidson 2001a.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, multicentre, 1 week single‐blind placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: computer generated |
|
| Participants | SAMPLE
Description: 208 DSM‐III‐R PTSD, 5% war trauma, mean age: 37.1 (18‐69), 78% male, duration of diagnosis: 12.3 years, 40% (83/208) MDD, baseline severity on CAPS‐2: sertraline (73.9) placebo (73.5) SCREENING Primary diagnosis: CAPS‐1, CAPS‐2 >= 50, minimum 6 months duration Comorbidity: None |
|
| Interventions | Description: sertraline 25 mg/d to between 50‐200 mg/d (avg: 146.3 mg/d) vs. placebo x 12 weeks | |
| Outcomes | Primary outcomes: CAPS‐2, IES, CGI‐S, CGI‐I
Secondary outcomes: DTS, HAM‐D, HAM‐A, PSQI Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 30% on sertraline and 27% on placebo (not clear which sample was used as denominator) Quality rating score: 33 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Davidson 2001b.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, relapse prevention study for participants undergoing acute and continuation treatment in Brady 2000 and Davidson 2001a BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: unclear |
|
| Participants | SAMPLE 96 DSM‐III‐R PTSD, mean age=43.4 years, SD=10.3, 30% male SCREENING Responders (CGI ≤ 2, ≥30% improvement on total severity score for CAPS) in last two sessions of 28 week continuation treatment with sertraline |
|
| Interventions | Sertraline 50‐200 mg/d (avg 137 mg/day) vs. placebo (avg 145 mg/day) x 28 weeks | |
| Outcomes | Primary outcomes: Rates and times to event for 1) relapse, 2) relapse or discontinuation because of clinical deterioration (self‐
rated), and 3) acute exacerbation of PTSD.
Secondary outcomes: CAPS, HAM‐D, QLES Data estimation: Not applicable for primary outcomes (Kaplan Meier survival analysis) |
|
| Notes | INDUSTRY SUPPORT Industry funded: Yes (as reported in acute and continuation phase studies) Medication provided by industry: No (as reported in acute and continuation phase studies) Any of the authors work for industry: Yes | |
Davidson 2003.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, fixed dose, double‐blind, single centre, 1 week single‐blind placeb run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 29 DSM‐IV PTSD, 15% (4/26) war trauma, mean age: 46.5 years, 73% (19/26) MDD, baseline severity on SPRINT: mirtrazapine (21.7), placebo (25) SCREENING Primary diagnosis: SIP =< 20 Comorbidity: MINI |
|
| Interventions | Description: mirtrazapine 15 mg/d ‐ 45 mg/d (mean end dose: 38.8 mg/d) versus placebo (mean end dose: 43.3 mg/d) x 8 weeks | |
| Outcomes | Primary outcomes: SPRINT(Clinical Global Improvement & Total Scores)
Secondary outcomes: DTS, HADS, SIP, ASEX Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 18% (3/17) on mitrazapine and 33% (3/9) on placebo Quality rating score: 27 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Davidson 2004.
| Methods | DESIGN
Description: Randomised, double‐blind, placebo‐controlled, parallel flexible dose trial BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 538 DSM‐IV PTSD, 65.4% females, average age: 32 years SCREENING Primary diagnosis: Unclear Comorbidity: Unclear |
|
| Interventions | Description: sertraline (25 mg/d ‐200 mg/d), venlafaxine (37.5 mg/d ‐ 300 mg/d) versus placebo x mean duration of 84 days | |
| Outcomes | CAPS, CGI‐S, DTS, GAF (no distinction made between primary and secondary outcomes) Data estimation: Unclear |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Unclear
Medication provided by industry: Unclear
Any of the authors work for industry: Unclear ADDITIONAL INFORMATION Drop‐out rates: 40.5% (218/538) dropout rate ? Quality rating score: Not calculated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Davis 2001.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, multi‐centre BLINDING Participants: Yes Assessors: Yes Administrators: Yes (self‐administered) ALLOCATION CONCEALMENT Method: randomisation log kept by pharmacist RANDOMISATION Method: No information |
|
| Participants | SAMPLE
Description: 42 DSM‐IV PTSD, 98% (40/41) combat veterans, mean age: 53.8 years (32‐75), 98% (40/41) male, average duration of illness: 29.9 years, 39% (16/41) MDD, baseline severity on CAPS: nefazodone (81) and placebo (83.2) SCREENING Primary diagnosis: SCID Comorbidity: SCID |
|
| Interventions | Description: nefazodone 200 mg/d ‐ 600 mg/d (avg final dose: 435mg/d) versus placebo x 12 weeks | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: HAM‐A, HAM‐D, PTSD checklist, CADSS, GAFS, CGI Data estimation: LOCF (excluded 1 patient due to compromise of blind) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 46% (12/26) on nefazodone and 40% (6/15) on placebo Quality rating score: 25 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Eli Lilly 2006.
| Methods | DESIGN
Description: Randomised, double blind, placebo‐controlled, parallel fixed dose trial BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 411 PTSD SCREENING Primary diagnosis: Unclear Comorbidity: Unclear |
|
| Interventions | Description: fluoxetine 20 mg/d (N = 163) and 40 mg/d (N = 160) versus placebo (N = 88) x mean of 84 days | |
| Outcomes | TOP‐8,CGI‐S (no distinction made between primary and secondary outcomes) Data estimation: ITT |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Yes
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 259 (63%) dropouts in total Quality rating score: Not calculated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Hertzberg 1999.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arms, flexible dose, double blind, multi‐centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 15 DSM‐IV PTSD, 71% (10/14) war combat, mean age: 43.4 years (29‐53), 64% (9/14) male, baseline severity on SI‐PTSD: lamotrigine (44.8) and placebo (43) SCREENING Primary diagnosis: SIP Comorbidity: MINI |
|
| Interventions | Description: lamotrigine 25 mg/d ‐500mg/d (avg. max. dose: 380 mg/d) versus placebo x 8 weeks | |
| Outcomes | ITT(LOCF) values provided.
Primary outcomes: SIP, DGRP
Secondary outcomes: None Data estimation: LOCF (excluded 1 patient) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 27% (3/11) on lamotrigine and 75% (3/4) on placebo Quality rating score: 23 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Hertzberg 2000.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arms, flexible dose, double blind, single centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 12 DSM‐IV PTSD, male Vietnam combat veterans, mean age: 46 years (44‐48), 66% (8/12) MDD, baseline severity on DTS: fluoxetine (106) and placebo (111) SCREENING Primary diagnosis: SIP Comorbidity: SCID ‐ DSM‐III‐R |
|
| Interventions | Description: fluoxetine 10 mg/d ‐ 60 mg/d (mean endpoint dose: 48 mg/d) vs placebo x 12 weeks | |
| Outcomes | DTS, SDS, SIP, DGRP (no distinction between primary and secondary outcomes) Data estimation: Presumably LOCF (not clear in text) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No
Medication provided by industry: Yes
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 1/6 (17%) on fluoxetine and 0/6 (0%) on placebo Quality rating score: 21 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Kaplan 1996.
| Methods | Description: random‐assignment, placebo‐controlled, crossover, fixed dose, double‐blind, 2 week washout period BLINDING Participants: Yes Assessors: Unclear Administrators: Yes (self administered) ALLOCATION CONCEALMENT Method: Identical capsules used for medication and placebo RANDOMISATION Method: pre‐arranged random code |
|
| Participants | SAMPLE
Description: 17 DSM‐III‐R PTSD, 23% (3/13) combat‐related, mean age for OC: 39.7 year (25‐56), 62% male, duration of diagnosis for OC: 0.5‐28 years, no comorbid major depression, baseline severity on IES for OC: inositol (35.8) and placebo (34.9) SCREENING Primary diagnosis: No information Comorbidity: No information |
|
| Interventions | Description: inositol 12 g/d versus placebo x 4 weeks | |
| Outcomes | IES , SCL‐90 (1 centre), HAM‐A, HAM‐D (other centre)
(no distinction between primary and secondary outcomes) Data estimation: Completer values |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No information
Medication provided by industry: No
Any of the authors work for industry: No information ADDITIONAL INFORMATION Drop‐out rates: 4/17 (24%) Quality rating score: 13 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Katz 1994.
| Methods | Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, multi‐centre, 2 week single‐blind placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 64 DSM‐III‐R PTSD, 17% (8/45) combat‐related, median age: 39 (22‐62), 76% male, duration of diagnosis: 2.8 years, baseline severity on CAPS: brofaromine (80.6) and placebo (82.9) SCREENING Primary diagnosis: CAPS > 36, response of >= 20% decrease on CAPS during placebo run‐in Comorbidity: HAM‐D > 21 |
|
| Interventions | Description: brofaromine 50 mg/d ‐150 mg/d versus placebo x 14 weeks | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: CGI Data estimation: LOCF |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Unclear
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 30% (10/33) on brofaromine and 29% (9/31) on placebo Quality rating score: 24 CGI‐C values obtained from Davidson et al, 1997 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Kosten 1991.
| Methods | DESIGN
Description: random‐assignment, comparator and placebo‐controlled, parallel arm, flexible dose, double‐blind, multi‐centre BLINDING Participants: Yes Assessors: No Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 60 DSM‐III‐PTSD, all combat veterans, mean age: 39 years, all males, no subjects with comorbid major depression, baseline severity on IES: imipramine (36.5), phenelzine (30.6) and placebo (33) SCREENING Primary diagnosis: SCID Comorbidity: SADS |
|
| Interventions | Description: imipramine 50 mg/d ‐ 300 mg/d (avg max. dose: 225 mg/d) versus phenelzine 15 mg/d ‐ 75 mg/d (avg max. dose: 68 mg/d) versus placebo x 8 weeks | |
| Outcomes | Primary outcomes: IES
Secondary outcomes: Combat Scale; CAS; HAM‐D; HAM‐A; RSD Data estimation: LOCF (completers of 3 or more weeks) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No
Medication provided by industry: Yes
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 52% (12/23) on impramine, 21% (4/19) on phenelzine and 12/18 (67%) on placebo Quality rating score: 26 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Marshall 2001.
| Methods | DESIGN
Description: random‐assignment, comparator and placebo‐controlled, parallel arm, fixed dose, double‐blind, 1 week placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 563 DSM‐IV PTSD, mean age: 41.8 years, 57% (315 /551) female, average duration of diagnosis: 15.7 years, approximately 45% (248/551) comorbid MDD, baseline severity on CAPS‐2:: paroxetine 20mg/d (75.3), paroxetine 40mg/d (74.3), and placebo ( 74.4) SCREENING Primary diagnosis: MINI, CAPS‐1, CAPS‐2 >= 50 Comorbidity: No information |
|
| Interventions | Description: paroxetine (25 mg/d) or paroxetine (50 mg/d) versus placebo x 12 weeks | |
| Outcomes | Primary outcomes: CAPS‐2, CGI‐I
Secondary outcomes: DTS, TOP‐8, SDS, MADRS Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No information
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 35% (66/188) on 20mg/d paroxetine, 40% (74/187) on 40mg/d paroxetine, and 36% (68/188) placebo Quality rating score: 26 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Marshall 2004.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, 1 week single‐blind placebo run‐in, single centre, with maintenance phase BLINDING Participants: No Assessors: Yes Administrators: Yes ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 63 DSM‐IV PTSD, no combat‐related trauma, mean age: 39.8 years, 67% (35/52) female, 62.5% (30/48) MDD, baseline severity on CAPS: paroxetine (82.8) and placebo (84.2) SCREENING Primary diagnosis: SCID, minimum duration of 3 months PTSD Comorbidity: SCID I |
|
| Interventions | Description: paroxetine 10 mg/d ‐ 6 mg/d versus placebo x 10 weeks, followed by 12 week double‐blind maintenance phase for responders to paroxetine or placebo | |
| Outcomes | Primary outcomes: CGI, CAPS
Secondary outcomes: CAPS‐2, IPP, DES, HAM‐A, HAM‐D, CAP‐2 (items 22 & 23) Data estimation: Mixed‐effects model regression (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Unclear
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 32% (8/25) on paroxetine and 52% (14/27) on placebo Quality rating score: 21 7 patients removed as responders during placebo run‐in Assessments made in either Spanish or English |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Martenyi 2002a.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, fixed dose, double‐blind, multi‐centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: computer drug‐labelled emergency codes RANDOMISATION Method: computer generated random sequence |
|
| Participants | SAMPLE
Description: 301 DSM‐IV PTSD, acute, 46% combat‐related, mean age: 37.9 years, 81% male, baseline severity on CAPS: fluoxetine ( 80.5) and placebo (81.3) SCREENING Primary diagnosis: SCID‐I [modified], CAPS‐DX => 50, CGI‐S >= 4 Comorbidity: MADRS > 20 |
|
| Interventions | Description: fluoxetine 20 mg/d ‐ 80 mg/d, (avg dose: 57.8 mg/d) versus placebo x 12 weeks | |
| Outcomes | Primary outcomes: TOPS‐8 Secondary outcomes: CAPS‐2, CGI‐S, CGI‐I, DTS, MADRS, HAM‐A, SCL‐90‐R, DES | |
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Not mentioned
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: not provided Quality rating score: 31 The patient‐rated scales, the DTS and SCL‐90‐R, were translated into other languages |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Martenyi 2002b.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, relapse prevention component of Martenyi 2002b BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: unclear RANDOMISATION Method: unclear |
|
| Participants | 131 DSM‐IV PTSD, 81% male, avg age = 38.2 years, 47% trauma combat related. Patients required to present with 50% decrease after 12 weeks of acute treatment with fluoxetine on the TOP‐8 score, a CGI–S score <= 2, and failed diagnostic criteria for PTSD | |
| Interventions | Fluoxetine max avg dose at endpoint: 53 mg/d vs placebo x 24 weeks. Responders to 12 weeks of acute treatment with placebo maintained on placebo. No discontinuation taper of fluoxetine from the acute to relapse prevention phase | |
| Outcomes | Primary outcomes: Time to relapse on TOPS‐8 (>= 40% increase for 12 week acute baseline) and CGI‐S (>= 2 on CGI‐S from 12 week acute baseline) Secondary outcomes: CAPS‐2, CGI‐I, DTS, MADRS, HAM‐A, SCL‐90‐R | |
| Notes | INDUSTRY SUPPORT Industry funded: Yes Medication provided by industry: Not mentioned Any of the authors work for industry: No | |
McRae 2004.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, multi‐centre, 1 week single‐blind placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: randomisation sequence kept by research pharmacist RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 37 DSM‐IV PTSD, mean age: 40.3 years (18‐65), 77% (26/26) female, average duration of diagnosis: 22 years, baseline severity on CAPS: nefazedone (68.85) and placebo (73.77) SCREENING Primary diagnosis: MINI; CAPS‐1, minimal 3 month duration, CAPS‐2 >= 50 after placebo run‐in + CAPS not >30% decrease during run‐in Comorbidity: MINI |
|
| Interventions | Description: nafazedone 100 mg/d‐600 mg/d (avg: 463 mg/d) versus sertraline 50 mg/d‐200 mg/d (avg: 153 mg/d) x 12 weeks | |
| Outcomes | Primary outcomes: CAPS‐2, CGI‐I
Secondary outcomes: DTS, MADRS, HAM‐A, TOP‐8, PSQI, SDS Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: No
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 14 dropouts in total Quality rating score: 33 3 patients excluded during run‐in |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Pfizer588.
| Methods | DESIGN
Description: Randomised placebo‐controlled, double‐blind, parallel, flexible dose trial BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 193 DSM‐III‐R PTSD, physical/sexual assault, 74.6% female, average age: 37 years, average duration of illness: 10.5 years, baseline severity on CPS‐2: < 50 (except for one participant) SCREENING Primary diagnosis: Unclear Comorbidity: Unclear |
|
| Interventions | Description: sertraline (mean completer dose: 156 mg/d) versus placebo x mean of 74 days | |
| Outcomes | CAPS‐2, CGI‐I, IES, DTS, CGI‐S (no distinction made between primary and secondary outcomes) Data estimation: Unclear |
|
| Notes | INDUSTRY SUPPORT
Industry funded:
Medication provided by industry:
Any of the authors work for industry: ADDITIONAL INFORMATION Drop‐out rates: 50 (25.9%) dropped out in total Quality rating score: Not calculated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Pfizer589.
| Methods | DESIGN
Description: Randomised, double‐blind, placebo‐controlled, parallel flexible dose trial BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 169 DSM‐III‐R PTSD, predominantly war‐trauma sample (71%), 79.9% female, average age: 45 years, average duration of illness: 18 years SCREENING Primary diagnosis: Unclear Comorbidity: Unclear |
|
| Interventions | Description: sertraline (mean completer dose: 156 mg/d) versus placebo x mean duration of 72 days | |
| Outcomes | CAPS‐2, CGI‐I, CGI‐S, IES, DTS (no distinction made between primary and secondary outcomes) Data estimation: Unclear |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Yes
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 26 on sertraline, 14 on placebo Quality rating score: Not calculated Prevalence of patients with drug abuse history higher at baseline in medication than placebo group |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Reich 2004.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, single‐centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 21 DSM‐III‐R PTSD, mean age: 27.9 years (18‐56), 100% female, all suffered childhood physical, sexual, emotion or verbal abuse, 9 participants on concurrent medication, baseline severity on CAPS‐2: risperidone (63.5) and placebo (65.6) SCREENING Primary diagnosis: SCID, CAPS‐1 Comorbidity: None |
|
| Interventions | Description: risperidone mean dose of 0.5 mg q.h.s. (avg dose: 1.4 mg/d) at start to 8 mg/d versus placebo x 8 weeks | |
| Outcomes | Primary outcomes: CAPS‐1, CAPS‐2
Secondary outcomes: None Data estimation: LOCF and random effects time series modeling methods |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Unclear
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 75% (9/12) on risperidone and 78% (7/9) on placebo Quality rating score: 26 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Reist 1989.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, crossover, flexible dose, double‐blind, 4 day switched cross‐over, multi‐centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 27 DSM‐III PTSD, combat veterans, mean age 38.4 years (28‐64), all males, 33% (6/18) major depression, most prevalent comorbidity: 50% (9/18) dysthymic disorder, baseline severity on IES: desipramine (55.2) and placebo (56.2) SCREENING Primary diagnosis: Not mentioned Comorbidity: SCID, SCID ‐ DSM‐III‐R for personality disorders |
|
| Interventions | Description: desipramine 50 mg/d ‐200 mg/d (max avg: 165 mg/d) versus placebo x 8 weeks | |
| Outcomes | HAM‐D, HAM‐A, BDI, IES
(no distinction between primary and secondary outcomes) Data estimation: Completer values |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No information
Medication provided by industry: Unclear
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 33% (9/27) Quality rating score: 12 ongoing recreational and group therapies |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Saygin 2002.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible‐dose, single centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 60 DSM‐IV PTSD, earthquake survivors, mean age: 41.5 years, 76% (41/54) male, 9% (5/54) MDD, baseline severity on TOP‐8: nefazadone (15.75) and sertraline (19.27) SCREENING Primary diagnosis: SCID‐1 Comorbidity: Not mentioned |
|
| Interventions | Description: nefazadone 200 mg/d ‐ 400 mg/d (avg: 332.4 mg/d) versus sertraline 50 mg/d‐100 mg/d (avg: 68.3 mg/d) x 6 months | |
| Outcomes | PDS, TOP‐8, CGI (no distinction between primary and secondary outcomes) Data estimation: Completer values |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No
Medication provided by industry: No
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 20% (6/30) on nefazadone and 0% on sertraline Quality rating score: 25 Not clear whether study was blinded |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Shestatzky 1988.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, crossover, flexible‐dose, double‐blind, single centre, 2 week placebo washout BLINDING Participants: Unclear Assessors: Yes Administrators: Unclear ALLOCATION CONCEALMENT Method: Claimed, but method not mentioned RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 13 DSM‐III PTSD, mean age: 38.5 years (31‐50), average duration of diagnosis: 5.6 years, 2% (2/10) MDD, baseline severity on IES: phenelzine (34) and placebo (36) SCREENING Primary diagnosis: Not mentioned Comorbidity: Not mentioned |
|
| Interventions | Description: phenelzine 30 mg/d ‐ 75 mg/d (avg. max dose: 66 mg/d) versus placebo x 5 weeks | |
| Outcomes | CGI‐I, CGI‐S, CAPS‐2. ITT(LOCF) values provided.
Primary outcomes: CAPS‐2, CGI‐I, CGI‐S
Secondary outcomes: MADRS Data estimation: |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No
Medication provided by industry: No
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 54% (7/13) on phenelzine and 0% (0/13) on placebo Quality rating score: 14 much of descriptive data for 4 week completers in both phases; supportive psychotherapy provided to patients |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
SKB627.
| Methods | DESIGN
Description: Randomised, double‐blind, placebo‐controlled, parallel flexible dose trial, placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 322 DSM‐IV PTSD, 53.7% female SCREENING Primary diagnosis: Unclear Comorbidity: Unclear |
|
| Interventions | Description: paroxetine 20 mg/d ‐ 50 mg/d versus placebo x mean duration of 84 days | |
| Outcomes | CAPS‐2, CGI‐I (no distinction made between primary and secondary outcomes) Data estimation: ITT |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Yes
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: Not provided Quality rating score: Not calculated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Smajkic 2001.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, single centre BLINDING Participants: No Assessors: No Administrators: No ALLOCATION CONCEALMENT Method: None RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 32 DSM‐IV PTSD, bosnian refugees, no combat‐related trauma, mean age: 51.34 years (24‐63), 56% female, baseline severity on PSS‐Sev: sertraline (41.9), venlaxafine (37.8) and paroxetine (37.3) SCREENING Primary diagnosis: PSS Comorbidity: SCID, BDI |
|
| Interventions | Description: sertraline 50 mg/d ‐ 100 mg/d versus venlafaxine 75 mg/d ‐ 150 mg/d versus paroxetine (max dose 20mg/d) x 6 weeks | |
| Outcomes | Primary outcomes: PSS
Secondary outcomes: GAF, BDI Data estimation: Completer values |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No
Medication provided by industry: No
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 0% sertraline, 62% (8/13) venlafaxine and 0% paroxetine Quality rating score: 17 Scales translated into Bosnian. Participants received concurrent case management and supportive therapy |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | D ‐ Not used |
Tucker 2001.
| Methods | Description: random‐assignment, placebo‐controlled, parallel arm, flexible‐dose, double‐blind, multi‐centre, 1 week placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 323 DSM‐IV PTSD, 7% (21/307) combat‐related, mean age: 40.8 years, 66% female, average duration of diagnosis: 14.9 years, 35% (108/307) with comorbid major depression, baseline severity on CAPS‐2: paroxetine (73.2) and placebo (74.3) SCREENING Primary diagnosis: CAPS‐1; MINI, CAPS‐2 < 50 after placebo run‐in Comorbidity: Not mentioned |
|
| Interventions | Description: paroxetine 20 mg/d ‐50 mg/d (mean dose: 27.6 mg/d) versus placebo x 12 weeks | |
| Outcomes | Primary outcomes: CAPS‐2 (score < 20 = 'remission'), CGI‐I
Secondary outcomes: DTS, TOP‐8, SDS, MADRS Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Unclear
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 46% (70/151) on paroxetine and 42% (66/156) on placebo Quality rating score: 27 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Tucker 2003.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible‐dose, double‐blind, single centre, 1‐2 week taper at end BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: Unclear RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 59 DSM‐IV PTSD, mean age: 38.5 years, 72% (42/58) female, 3% (2/58) combat related, 78% (45/58) major depression, baseline severity on CAPS: citalopramine (91), sertraline (83.9) and placebo (94.2) SCREENING Primary diagnosis: SCID‐IV, CAPS‐I >=50 Comorbidity: Unclear |
|
| Interventions | Description: citalopram 20 mg/d ‐ 50 mg/d (final avg: 36.2 mg/d)) versus sertraline 50 mg/d ‐ 200 mg/d (final avg: 134.1mg/d)) versus placebo x 10 weeks | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: IES (revised), BDI (revised) Data estimation: LOCF (2 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Unclear
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: +‐20% (5/58) on citalopramine, +‐ 26% (6/23) on sertraline and +‐ 30% (3/10) on placebo Quality rating score: 26 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
van der Kolk 1994.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible‐dose, double‐blind, multi‐centre BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 64 DSM‐II‐R PTSD, 44% (28/64) combat‐related trauma, mean age: 40.4 years (22‐55), 66% (42/64) male, 54.8% (34/64) MDD SCREENING Primary diagnosis: CAPS >= 45 Comorbidity: Not mentioned |
|
| Interventions | Description: fluoxetine 20 mg/d ‐ 60 mg/d (avg. dose: 40 mg/d) versus placebo x 5 weeks | |
| Outcomes | CAPS, BDHI, HAM‐D, DES, DESI, acoustic startle response, rorschach inkblot test (no distinction between primary and secondary outcomes) Data estimation: Completer values |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Unclear
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 36% (12/33) on fluoxetine and 13% (4/31) on placebo Quality rating score: 22 Supportive psychotherapy was permitted |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
van der Kolk 2004.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible‐dose, double‐blind, multi‐centre, 6 month follow‐up BLINDING Participants: Unclear Assessors: Yes Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 59 DSM‐IV PTSD, mean age: 34.9 years (18‐65), 86% (51/59) female, average duration of diagnosis: 13.3 years, baseline severity on CAPS: fluoxetine (73.7) and placebo (70.3) SCREENING Primary diagnosis: Trauma incident >= 1 year ago, GAF < 40 Comorbidity: Not mentioned |
|
| Interventions | Description: fluoxetine 10 mg/d ‐ 60 mg/d versus Eye Movement Desensitization and Reprocessing (EMDR) versus placebo x 5 weeks | |
| Outcomes | Primary outcomes: CAPS
Secondary outcomes: BDI Data estimation: LOCF |
|
| Notes | INDUSTRY SUPPORT
Industry funded: No
Medication provided by industry: Unclear
Any of the authors work for industry: No ADDITIONAL INFORMATION Drop‐out rates: 13% (4/30) on fluoxetine and 10% (3/29) on placebo Quality rating score: 28 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Zohar 2002.
| Methods | DESIGN
Description: random‐assignment, placebo‐controlled, parallel arm, flexible dose, double‐blind, mutli‐centre, 1 week single blind placebo run‐in BLINDING Participants: Unclear Assessors: Unclear Administrators: Unclear ALLOCATION CONCEALMENT Method: No information RANDOMISATION Method: Unclear |
|
| Participants | SAMPLE
Description: 51 PTSD, 76% (32/42) combat‐related trauma, mean age: 40 years, 88% (37/42) male, baseline severity on CAPS‐2: sertraline (91.2) and placebo (93.3) SCREENING Primary diagnosis: minimum 6 month PTSD, CAPS‐1, CGI‐S >=4, CAPS‐2 >=50 required after placebo run‐in Comorbidity: Unclear |
|
| Interventions | Description: sertraline 50 mg/d ‐ 200 mg/d (120 mg/d) versus placebo (avg:147 mg/d) x 10 weeks | |
| Outcomes | Primary outcomes: CAPS‐2, CGI‐I, CGI‐S
Secondary outcomes: MADRS Data estimation: LOCF (1 post‐baseline assessment) |
|
| Notes | INDUSTRY SUPPORT
Industry funded: Yes
Medication provided by industry: Unclear
Any of the authors work for industry: Yes ADDITIONAL INFORMATION Drop‐out rates: 26% (6/23) on fluoxetine and 26% (5/19) on placebo Quality rating score: 30 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Acronyms for scales: AIMS: Abnormal Involuntary Movement Scale; ASEX: Arizona Sexual Experience Scale; ASI: Addiction Severity Index; BAS: Barnes Akathisia Scale; BDHI: Buss‐Durkee Hostility Inventory; BDI: Beck Depression Inventory; CADSS: Clinician Administered Dissociative States Scale; CAPS‐2: Clinician Administered PTSD Scale ‐ part 2; CAS: Covi Anxiety Scale; CES: Combat Exposure Scale; CES‐D: Center for Epidemiologic Studies Depression Scale (self‐rated); CGI‐I: Clinical Impression ‐ Improvement Scale; CS: Columbia Scale; DESI: Disorders of Extreme Stress Inventory; DES: Dissociative Experiences Scale; DGRP: Duke Global Rating for PTSD; DTS: Davidson Trauma Scale; EPI: Eyesenck Personality Inventory; HADS: Hospital Anxiety and Depression Scale; HAM‐A: Hamilton Anxeity Scale; IPP: Inventory of Personal Problems; MADRS: Montgomery‐Asberg Depression Rating Scale; MISS: Mississippi Scale for Combat‐Related PTSD; NI: Newcastle Index; OCDS: Obsessive Compulsive Drinking Scale; PDS: Posttraumatic Stress Disorder Scale PSQI: Pittsburgh Sleep Quality Index; PSS: PTSD Symptoms Scale; RSD: Raskin Scale for Depression; SADS: Schedule for Affective Disorders and Schizophrenia; SAS: Simpson‐Angus Scale; SCID: Structured Clinical Interview for DSM; SCID‐P: Structured Clinical Interview for DSM, Psychotic screen; SCL‐90‐R: Hopkins 90‐item Symptom Checklist‐Revised; SDS: Sheehan Disability Scale; SI‐PTSD: DSM based PTSD scale; SIP: Structured Interview for PTSD; SPRINT: Short PTSD Rating Interview scale; TLFB: Time‐Line Follow‐Back; VS: Vulnerability to the effects of stress scale; VAS: Visual Analog Scale; UKUSERS: UKU Side Effect Rating Scale; SPRINT: Short PTSD Rating Interview; TOP‐8: Treatment Outcome PTSD Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Aerni 2004 | All 3 patients received concurrent medication. In addition, one of the patients was treated with concurrent psychotherapy |
| Bartzokis 2005 | Augmentation trial of adjunctive risperidone for chronic, combat‐related PTSD. To be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders. |
| Bremner 1997 | Pharmacotherapy impact assessment trial |
| Coupland 1997 | Pharmacotherapy impact assessment trial |
| Eftekhari 2004 | Comparison of treatment of chronic PTSD with prolonged exposure or sertraline. No placebo or other medication used as control. |
| Frank 1988 | An extension of this database was subsequently published (Kosten et al, 1991). |
| Frommberger 1998 | Open label comparison of paroxetine and CBT. No placebo or alternative medication control |
| Gelpin 1996 | Trial of prophlactic treatment of recent trauma survivors with benzodiazepines. To be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Hamner 1997 | Buspirone potentiation of antidepressants for combat‐veteran PTSD sufferers. May be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders. |
| Hamner 1999 | Pharmacotherapy impact assessment trial |
| Hamner 2003 | Risperidone augmentation of medication for war‐veteran PTSD sample with psychotic symptoms. May be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Heresco‐Levy 2002 | Augmentation trial of d‐cycloserine for PTSD. May be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders. |
| Jacobs‐Rebhun 2000 | Trial of treatment for sleep disorders associated with PTSD |
| Kanter 2001 | Pharmacotherapy impact assessment trial |
| Kellner 2000 | Pharmacotherapy impact assessment trial. Not a treatment study |
| Kline 1994 | Open uncontrolled trial of sertraline for treatment‐resistant combat veteran PTSD patients |
| Monnelly 2003 | Augmentation of risperidone for irritable aggression symptoms in combat‐veteran PTSD sufferers. May be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders. |
| Morgan 1995 | Pharmacotherapy impact assessment trial |
| Nagy 1996 | The poster presentation provided neither response rates, nor means/standard deviations before and after medication/placebo. |
| Otto 2003 | Augmentation trial and comparison group receives medication and cognitive‐behaviour therapy |
| Pitman 1990 | Pharmacotherapy impact assessment trial |
| Pitman 2002 | Participants were allowed to continue with concommitant psychiatric treatments |
| Pivac 2004 | Open, non‐randomised trial of olanzapine versus fluphenazine for psychotic combat‐related PTSD |
| Randall 1995 | Pharmacotherapy impact assessment trial |
| Raskind 2003 | Trial classified as an augmentation trial for treatment resistant PTSD. To be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Reist 1995 | Pharmacotherapy impact assessment trial |
| Reist 2001 | Pharmacotherapy impact assessment trial |
| Schelling 2004 | Trial of hydrocortisone for post‐operative complications in cardiac surgery. As trials is concerned with the development of PTSD following surgery, and diagnosis with PTSD does not fall within its inclusion criteria, this is better considered a prophylaxis trial, and will be included in an upcoming protocol |
| Southwick 1997 | Pharmacotherapy impact assessment trial |
| Stein 2002 | Augmentation trial of adjunctive olanzapine for SSRI resistant combated‐related PTSD. To be included in an upcoming review of pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders |
| Vaiva 2003 | Pharmacotherapy impact assessment trial |
| van der Kolk 1989 | Pharmacotherapy impact assessment trial |
| Zatzick 2004 | Pharmacotherapy component of collaborative care intervention non‐randomised as only offered to patients with persistent PTSD symptoms |
Contributions of authors
Dan Stein co‐ordinated the intitial work of this review, including literature search, trial assessment, and data analysis. Jonathan Ipser assisted with all aspects of updating the review, including literature search, trial assessment, and data analysis. Soraya Seedat provided help with critical interpretation of studies.
Sources of support
Internal sources
No sources of support supplied
External sources
MRC Research Unit on Anxiety and Stress Disorders, Cape Town, South Africa.
Cochrane Collaboration, UK.
Declarations of interest
The MRC Anxiety and Stress Disorders Research Unit has received funding from almost all pharmaceutical companies involved with psychiatry in South Africa.
Potential conflicts of interest for individual reviewers Dan Stein has received research grants and/or consultancy honoraria from Astrazeneca, Eli‐Lilly, GlaxoSmithKline, Lundbeck, Orion, Pfizer, Pharmacia, Roche, Servier, Solvay, Sumitomo, and Wyeth. He has participated in a number of ongoing trials, and has presented data from some of these trials on behalf of the sponsoring companies.
Jonathan Ipser has no known conflicts of interest outside of his employment by the MRC Unit on Anxiety Disorders.
Soraya Seedat has received support from several companies (including GlaxoSmithKline, Eli‐Lilly, Pfizer, Cephalon) and has participated in several clinical trials.
Edited (no change to conclusions), comment added to review
References
References to studies included in this review
Baker 1995 a {published data only}
- Baker DG, Diamond BI, Gillette G, Hamner M, Katzelnick D, Keller T, et al. A double‐blind randomized placebo‐controlled multi‐center study of brofaromine in the treatment of post‐traumatic stress disorder. Psychopharmacology 1995;122(4):386‐9. [DOI] [PubMed] [Google Scholar]
- Connor KM, Hidalgo RB, Crockett B, Malik M, Katz RJ, Davidson JR. Predictors of treatment response in patients with posttraumatic stress disorder. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2001;25(2):337‐45. [DOI] [PubMed] [Google Scholar]
Brady 2000 {published data only}
- Brady K, Pearlstein T, Asnis GM, Baker D, Rothbaum B, Sikes CR, et al. Efficacy and safety of sertraline treatment of posttraumatic stress disorder. Archives of General Psychiatry 2000;283(14):1837‐44. [DOI] [PubMed] [Google Scholar]
- Davidson J, Pearlstein T, Londborg P, Brady KT, Rothbaum B, Bell J, et al. Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: Results of a 28‐week double‐blind, placebo‐controlled study. American Journal of Psychiatry 2001;158(12):1974‐81. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Landerman LR, Farfel GM, Clary CM. Characterizing the effects of sertraline in post‐traumatic stress disorder. Psychological Medicine 2002;32(4):661‐70. [DOI] [PubMed] [Google Scholar]
- Gaffney M. Factor analysis of treatment response in posttraumatic stress disorder. Journal of Traumatic Stress 2003;16(1):77‐80. [DOI] [PubMed] [Google Scholar]
- Rapaport MH, Endicott J, Clary CM. Posttraumatic stress disorder and quality of life: Results across 64 weeks of sertraline treatment. Journal of Clinical Psychiatry 2002;63(1):59‐65. [DOI] [PubMed] [Google Scholar]
- Rapaport MH, Farfel G, Clary CM. Quality of life improvement in PTSD with sertraline treatment: Results of a multicenter, placebo‐controlled trial. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):294. [Google Scholar]
Brady 2004 {unpublished data only}
- Brady KT, Sonne S, Anton RF, Randall CL, Back SE, Simpson K. Sertraline in the treatment of co‐occuring alcohol dependence and PTSD. Draft manuscript. [DOI] [PubMed]
- Sonne SC, Back SE, Diaz Zuniga C, Randall CL, Brady KT. Gender differences in individuals with comorbid alcohol dependence and post‐traumatic stress disorder. American Journal on Addictions 2003;12(5):412‐23. [PubMed] [Google Scholar]
Braun 1990 {published data only}
- Braun P, Greenberg D, Dasberg H, Lerer B. Core symptoms of posttraumatic stress disorder unimproved by alprazolam treatment. Journal of Clinical Psychiatry 1990;51(6):236‐8. [PubMed] [Google Scholar]
Butterfield 2001 {published data only}
- Butterfield MI, Becker ME, Connor KM, Sutherland S, Churchill LE, Davidson JR. Olanzapine in the treatment of post‐traumatic stress disorder: A pilot study. International Clinical Psychopharmacology 2001;16(4):197‐203. [DOI] [PubMed] [Google Scholar]
Chung 2004 {published data only}
- Chung MY, Min KH, Jun YJ, Kim SS, Kim WC, Jun EM. Efficacy and tolerability of mirtazapine and sertraline in Korean veterans with posttraumatic stress disorder: A randomized open label trial. Human Psychopharmacology 2004;19(7):489‐94. [DOI] [PubMed] [Google Scholar]
Conner 1999 {published data only}
- Barnett SD, Tharwani HM, Hertzberg MA, Sutherland SM, Connor KM, Davidson JR. Tolerability of fluoxetine in posttraumatic stress disorder. Progress in Neuro‐Psychopharmacology & Biological Psychiatry 2002;26(2):363‐7. [DOI] [PubMed] [Google Scholar]
- Connor KM, Sutherland SM, Tupler LA, Malik ML, Davidson JR. Fluoxetine in post‐traumatic stress disorder: Randomized, double‐blind study. British Journal of Psychiatry 1999;175:17‐22. [DOI] [PubMed] [Google Scholar]
- Malik ML, Connor KM, Sutherland SM, Smith RD, Davison RM, Davidson JR. Quality‐of‐life and posttraumatic stress disorder: A pilot study assessing changes in SF‐36 scores before and after treatment in a placebo‐ controlled trial of fluoxetine. Journal of Traumatic Stress 1999;12(2):387‐93. [DOI] [PubMed] [Google Scholar]
Davidson 1990 {published data only}
- Davidson JR, Kudler H, Smith R, Mahorney SL, Lipper S, Hammett E, et al. Treatment of posttraumatic stress disorder with amitriptyline and placebo. Archives of General Psychiatry 1990;47(3):259‐66. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Kudler HS, Saunders WB, Erickson L, Smith RD, Stein RM, et al. Predicting response to amitriptyline in posttraumatic stress disorder. American Journal of Psychiatry 1993;150(7):1024‐9. [DOI] [PubMed] [Google Scholar]
Davidson 2001a {published data only}
- Davidson JR, Landerman LR, Farfel GM, Clary CM. Characterizing the effects of sertraline in post‐traumatic stress disorder. Psychological Medicine 2002;32(4):661‐70. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Rothbaum BO, Kolk BA, Sikes CR, Farfel GM. Multicenter, double‐blind comparison of sertraline and placebo in the treatment of posttraumatic stress disorder. Archives of General Psychiatry 2001;58(5):485‐92. [DOI] [PubMed] [Google Scholar]
Davidson 2001b {published data only}
- Davidson J, Londborg P, Pearlstein T, Rothbaum B, Brady K, Farfel G. Double‐blind, randomized, 28‐week continuation study of sertraline and placebo in posttraumatic stress disorder. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):292. [Google Scholar]
- Davidson J, Pearlstein T, Londborg P, Brady KT, Rothbaum B, Bell J, et al. Efficacy of sertraline in preventing relapse of posttraumatic stress disorder: results of a 28‐week double‐blind, placebo‐controlled study. American Journal of Psychiatry 2001;158(12):1974‐81. [DOI] [PubMed] [Google Scholar]
- Rapaport MH, Endicott J, Clary CM. Posttraumatic stress disorder and quality of life: Results across 64 weeks of sertraline treatment. Journal of Clinical Psychiatry 2002;63(1):59‐65. [DOI] [PubMed] [Google Scholar]
Davidson 2003 {published data only}
- Davidson JR, Weisler RH, Butterfield MI, Casat CD, Connor KM, Barnett S, et al. Mirtzapine vs. placebo in posttraumatic stress disorder: A pilot trial. Biological Psychiatry 2003;53(2):188‐91. [DOI] [PubMed] [Google Scholar]
Davidson 2004 {unpublished data only}
- Davidson J, Rothbaum BO, Tucker P, Asnis Gr, Benattia I, Musgnung JJ. Venlafaxine extended release in posttraumatic stress disorder: A sertraline‐ and placebo‐controlled study . Journal of clinical psychopharmacology 2006 ; 26 ( 3 ):259‐267. [DOI] [PubMed] [Google Scholar]
- Davidson JR, Lipschitz A, Musgnung J. Treatment of PTSD: A comparison of venlafaxine extended release, sertraline, and placebo [abstract NR521] . 157th Annual Meeting of the American Psychiatric Association; 2004 May 1‐6; New York, NY 2004 :195. [Google Scholar]
- Davidson J, Lipschitz A, Musgnung JJ. Venlafaxine XR and sertraline in posttraumatic stress disorder: A placebo‐controlled trial. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
Davis 2001 {published data only}
- Davis LL, Ambrose S, English B, Dafore ME, Farley J, Bartolucci Al, et al. A placebo‐controlled study of nefazodone for the treatment of chronic posttraumatic stress disorder. 20th Annual Meeting, International Society for Traumatic Stress Studies, December 6 ‐ 9, New Orleans, LA. 2001.
- Davis LL, Jewell ME, Ambrose S, Farley J, English B, Bartolucci A, et al. A placebo‐controlled study of nefazadone for the treatment of chronic posttraumatic stress disorder. Journal of Clinical Psychopharmacology 2004;24(3):291‐7. [DOI] [PubMed] [Google Scholar]
Eli Lilly 2006 {unpublished data only}
- Martenyi F, Brown EB, Caldwell CD. Failed efficacy of fluoxetine in the treatment of posttraumatic stress disorder: results of a fixed‐dose, placebo‐controlled study . Journal of clinical psychopharmacology 2007 ; 27 ( 2 ):166‐170. [DOI] [PubMed] [Google Scholar]
- Eli Lilly. Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
Hertzberg 1999 {published data only}
- Hertzberg MA, Butterfield MI, Feldman ME, Beckham JC, Sutherland SM, Connor KM, et al. A preliminary study of lamotrigine for the treatment of posttraumatic stress disorder. Biological Psychiatry 1999;45(9):1226‐9. [DOI] [PubMed] [Google Scholar]
Hertzberg 2000 {published data only}
- Hertzberg MA, Feldman ME, Beckham JC, Kudler HS, Davidson JR. Lack of efficacy for fluoxetine in PTSD: A placebo controlled trial in combat veterans. Annals of Clinical Psychiatry 2000;12(2):101‐5. [DOI] [PubMed] [Google Scholar]
Kaplan 1996 {published data only}
- Kaplan Z, Amir M, Swartz M, Levine J. Inositol treatment of post‐traumatic stress disorder. Anxiety 1996;2(1):51‐2. [DOI] [PubMed] [Google Scholar]
Katz 1994 {published data only}
- Connor KM, Hidalgo RB, Crockett B, Malik M, Katz RJ, Davidson JR. Predictors of treatment response in patients with posttraumatic stress disorder. Progress in Neuro‐Psychopharmacology and Biological Psychiatry 2001;25(2):337‐45. [DOI] [PubMed] [Google Scholar]
- Katz RJ, Lott MH, Arbus P, Crocq L, Herlobsen P, Lingjaerde O, et al. Pharmacotherapy of post‐traumatic stress disorder with a novel psychotropic. Anxiety 1994;1(4):169‐74. [DOI] [PubMed] [Google Scholar]
Kosten 1991 {published data only}
- Kosten TR, Frank JB, Dan E, McDougle CJ, Giller EL Jr. Pharmacotherapy for posttraumatic stress disorder using phenelzine or imipramine. Journal of Nervous and Mental Disease 1991;179(6):366‐70. [DOI] [PubMed] [Google Scholar]
Marshall 2001 {published data only}
- Food, Drugs Administration (FDA). [ NDA 20‐031/S‐029 Paxil (Paroxetine Hydrochloride) Tablets; GlaxoSmithKline; Post‐traumatic Stress Disorder ]. Drugs@FDA [http://www.accessdata.fda.gov] Vol. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/20‐031S029.pdf (accessed 17 December 2013) .
- GlaxoSmithKline. A 12 week, double‐blind, fixed dose comparison of 20 and 40 mg daily of paroxetine and placebo in Patients suffering from Posttraumatic Stress Disorder (PTSD) . GSK ‐ clinical study register [www.gsk‐clinicalstudyregister.com] Study ID: PAR 29060 651 (BRL‐02960).
- Marshall RD, Beebe KL, Oldham M, Zaninelli R. Efficacy and safety of paroxetine treatment for chronic PTSD: A fixed‐dose, placebo‐controlled study. American Journal of Psychiatry 2001;158(12):1982‐8. [DOI] [PubMed] [Google Scholar]
Marshall 2004 {unpublished data only}
- Marshall R, Blanco C, Lewis‐Fernandez R, Simpson B, Lin SH, Garcia W, et al. Randomized controlled trial of paroxetine in adults with chronic PTSD. 18th Annual Meeting, International Society for Traumatic Stress Studies, November 7 ‐ 10, Baltimore, MD. 2002.
- Marshall RD, Lewis‐Fernandez R, Blanco C, Simpson HB, Lin S, Garcia W, et al. A controlled trial of paroxetine for chronic PTSD, dissociation and interpersonal problems in mostly minority adults. draft manuscript. [DOI] [PubMed]
Martenyi 2002a {published data only}
- Martenyi F, Brown EB, Zhang H, Prakash A, Koke SC. Fluoxetine versus placebo in posttraumatic stress disorder. Journal of Clinical Psychiatry 2002;63(3):199‐206. [DOI] [PubMed] [Google Scholar]
Martenyi 2002b {published data only}
- Martenyi F, Brown EB, Zhang H, Koke SC, Prakash A. Fluoxetine v. placebo in prevention of relapse in post‐traumatic stress disorder.. British Journal of Psychiatry 2002;181:315‐20. [DOI] [PubMed] [Google Scholar]
McRae 2004 {published data only}
- McRae AL, Brady KT, Mellman TA, Sonn SC, Killeen TK, Timmerman MA, et al. Comparison of nefazodone and sertraline for the treatment of posttraumatic stress disorder. Depression and Anxiety 2004;19(3):190‐6. [DOI] [PubMed] [Google Scholar]
Pfizer588 {unpublished data only}
- Food, Drugs Administration (FDA). Review and evaluation of clinical data [ Pfizer NDA application: Sertraline (Zoloft) for PTSD] . Drugs@FDA [www.accessdata.fda.gov] 199 9 ; Vol. http://www.accessdata.fda.gov/drugsatfda_docs/nda/99/19‐839S026_Zoloft_Clinr_P1.pdf ( accessed 31 January 2012 ) .
- Pfizer. Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
Pfizer589 {unpublished data only}
- Food, Drugs Adminstration (FDA). Review and evaluation of clinical data [Pfizer NDA application: Sertraline (Zoloft) for PTSD] . Drugs@FDA [www.accessdata.fda.gov] 1999 ; Vol. http://www.accessdata.fda.gov/drugsatfda_docs/nda/99/19‐839S026_Zoloft_Clinr_P1.pdf (accessed 31 January 2012) .
- Friedman MJ, Marmar CR, Baker DG, Sikes CR, Farfel GM. Randomized, double‐blind comparison of sertraline and placebo for posttraumatic stress disorder in a Department of Veterans Affairs setting . Journal of clinical psychiatry 2007 ; 68 ( 5 ):711‐20. [DOI] [PubMed] [Google Scholar]
- Pfizer. Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
Reich 2004 {published data only}
- Reich DB, Winternitz S, Hennen J, Watts T, Stanculescu C. A preliminary study of risperidone in the treatment of postraumatic stress disorder related to childhood abuse in women. Journal of Clinical Psychiatry 2004;65(12):1601‐5. [DOI] [PubMed] [Google Scholar]
Reist 1989 {published data only}
- Reist C, Kauffmann CD, Chicz Demet A, Chen CC, Demet EM. REM latency, dexamethasone suppression test, and thyroid releasing hormone stimulation test in posttraumatic stress disorder. Progress in Neuropsychopharmacology and Biological Psychiatry 1995;19(3):433‐43. [DOI] [PubMed] [Google Scholar]
- Reist C, Kauffmann CD, Haier RJ, Sangdahl C, DeMet EM, Chicz‐DeMet A, et al. A controlled trial of desipramine in 18 men with posttraumatic stress disorder. American Journal of Psychiatry 1989;146(4):513‐6. [DOI] [PubMed] [Google Scholar]
Saygin 2002 {published data only}
- Saygin MZ, Sungur MZ, Sabol EU, Cetinkaya P. Nefazodone versus sertraline in treatment of posttraumatic stress disorder. Bulletin of Clinical Psychopharmacology 2002;12(1):1‐5. [Google Scholar]
Shestatzky 1988 {published data only}
- Shestatzky M, Greenberg D, Lerer B. A controlled trial of phenelzine in posttraumatic stress disorder. Psychiatric Research 1988;24(2):149‐55. [DOI] [PubMed] [Google Scholar]
SKB627 {unpublished data only}
- Food, Drugs Administration (FDA). NDA 20‐031/S‐029 Paxil (Paroxetine Hydrochloride) Tablets; GlaxoSmithKline; Post‐traumatic Stress Disorder . Drugs@FDA [http://www.accessdata.fda.gov] Vol. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2001/20‐031S029.pdf (accessed 17 December 2013) .
- GlaxoSmithKline. A 12‐Week, Double‐Blind, Placebo‐Controlled, Parallel Group Study to Assess the Efficacy and Tolerability of Paroxetine in Patients Suffering from Posttraumatic Stress Disorder (PTSD) . GSK ‐ clinical study register [www.gsk‐clinicalstudyregister.com] Study ID: 29060/627.
- Stein DJ, Davidson J, Seedat S, Beebe K. Paroxetine in the treatment of post‐traumatic stress disorder: pooled analysis of placebo‐controlled studies . Expert Opin i on on Pharmacother apy 2003 ; 4 ( 10 ):1829‐38. [DOI] [PubMed] [Google Scholar]
- SmithKline Beecham. Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
Smajkic 2001 {published data only}
- Smajkic A, Weine S, Djuric‐Bijedic Z, Boskailo E, Lewis J, Pavkovic I. Sertraline, paroxetine, and venlafaxine in refugee posttraumatic stress disorder with depression symptoms. Journal of Traumatic Stress 2001;14(3):445‐52. [DOI] [PubMed] [Google Scholar]
Tucker 2001 {published data only}
- GlaxoSmithKline. A 12 Week, Double‐blind, Placebo‐controlled, parallel group study to assess the efficacy and tolerability of Paroxetine in Patients suffering from Posttraumatic Stress Disorder (PTSD) . GSK ‐ Clinical Study Register [www.gsk‐clinicalstudyregister.com] 2000 :Study ID: GSK 29060/648.
- Tucker P, Zaninelli R, Yehuda R, Ruggiero L, Dillingham K, Pitts CD. Paroxetine in the treatment of chronic posttraumatic stress disorder: Results of a placebo‐controlled, flexible‐dosage trial. Journal of Clinical Psychiatry 2001;62(11):860‐8. [DOI] [PubMed] [Google Scholar]
Tucker 2003 {published data only}
- Tucker P, Potter‐Kimball R, Wyatt DB, Parker DE, Burgin C, Jones DE, et al. Can physiologic assessment and side effects tease out differences in PTSD trials? A double‐blind comparison of citalopram, sertraline, and placebo. General Psychopharmacology 2003;37(3):135‐49. [PubMed] [Google Scholar]
van der Kolk 1994 {published data only}
- Kolk BA, Dreyfuss D, Michaels M, Shera D, Berkowitz R, Fisler R, et al. Fluoxetine treatment in posttraumatic stress disorder. Journal of Clinical Psychiatry 1994;55(12):517‐22. [PubMed] [Google Scholar]
van der Kolk 2004 {unpublished data only}
Zohar 2002 {published data only}
- Amital D, Zohar J, Kotler M, Bleich A, Miodovnik H, Nevo A ET AL. A placebo‐controlled study of sertraline in post traumatic stress disorder. International Journal of Neuropsychopharmacology 2000;3(Suppl 1):269. [Google Scholar]
- Amital D, Zohar J, Kotler M, Bleich A, nrodovnik H, Nevo A, et al. A placebo‐controlled trial pilot study of sertraline in PTSD. Annual Meeting of the American Psychiatric Association. American Psychiatric Association, 1999:157.
- Zohar J, Amital D, Miodownik C, Kotler M, Bleich A, Lane RM. Double‐blind placebo‐controlled pilot study of sertraline in military veterans with posttraumatic stress disorder. Journal of Clinical Psychopharmacology 2002;22(2):190‐5. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Aerni 2004 {published data only}
- Aerni A, Traber R, Hock C, Roozendaal B, Schelling G, Papassotiropoulos A, et al. Low‐dose cortisol for symptoms of posttraumatic stress disorder. American Journal of Psychiatry 2004;161(8):1488‐90. [DOI] [PubMed] [Google Scholar]
Bartzokis 2005 {published data only}
- Bartzokis G, Lu PH, Turner J, Mintz J, Saunders CS. Adjunctive risperidone in the treatment of chronic combat‐related posttraumatic stress disorder. Biological Psychiatry 2005;57(5):474‐9. [DOI] [PubMed] [Google Scholar]
Bremner 1997 {published data only}
- Bremner JD, Innis RB, Ng CK, Staib LH, Salomon RM, Bronen RA, et al. Positron emission tomography measurement of cerebral metabolic correlates of yohimbine administration in combat‐related posttraumatic stress disorder. Archives of General Psychiatry 1997;54(3):246‐54. [DOI] [PubMed] [Google Scholar]
Coupland 1997 {published data only}
- Coupland NJ, Lillywhite A, Bell CE, Potokar JP, Nutt DJ. A pilot controlled study of the effects of flumazenil in posttraumatic stress disorder. Biological Psychiatry 1997;41(9):988‐90. [DOI] [PubMed] [Google Scholar]
Eftekhari 2004 {unpublished data only}
- Eftekhari A, Miller H, Zoellner L, Feeny N. Prolonged exposure vs. sertraline: Secondary outcomes in PTSD treatment. 20th Annual Meeting, International Society for Traumatic Stress Studies, November 14 ‐ November 18, New Orleans, LA. 2004.
Frank 1988 {published data only}
- Frank JB, Kosten TR, Giller EL Jr, Dan E. A randomized clinical trial of phenelzine and imipramine for post‐traumatic stress disorder. American Journal of Psychiatry 1988;145(10):1289‐91. [DOI] [PubMed] [Google Scholar]
Frommberger 1998 {published data only}
- Frommberger U, Nyberg E, Richter H, Stieglitz RD, Berger M. Paroxetine vs. cognitive‐behavioral therapy in the treatment of depression in PTSD patients. XXIst Collegium Internationale Neuro psychopharmacologicum, Glasgow, Scotland. 12th 16th July. 1998.
Gelpin 1996 {published data only}
- Gelpin E, Bonne O, Peri T, Brandes D, Shalev AY. Treatment of recent trauma survivors with benzodiazepines: A prospective study. Journal of Clinical Psychiatry 1996;57(9):390‐4. [PubMed] [Google Scholar]
Hamner 1997 {published data only}
- Hamner M, Ulmer H, Horne D. Buspirone potentiation of antidepressants in the treatment of PTSD. Depression and Anxiety 1997;5(3):137‐9. [PubMed] [Google Scholar]
Hamner 1999 {published data only}
- Hamner MB, Ulmer HG, Horne DF, George MS, Arana GW. Procaine administration and behavioral responsivity in post‐traumatic stress disorder: a pilot study of tolerability. Human Psychopharmacology 1999;14(2):105‐11. [Google Scholar]
Hamner 2003 {published data only}
- Hamner MB, Faldowski RA, Ulmer HG, Frueh BC, Huber MG, Arana GW. Adjunctive risperidone treatment in post‐traumatic stress disorder: a preliminary controlled trial of effects on comorbid psychotic symptoms. International Clinical Psychopharmacology 2003;18(1):1‐8. [DOI] [PubMed] [Google Scholar]
Heresco‐Levy 2002 {published data only}
- Heresco‐Levy U, Kremer I, Javitt DC, Goichman R, Reshef A, Blanaru M, et al. Pilot‐controlled trial of D‐cycloserine for the treatment of post‐traumatic stress disorder. International Journal of Neuropsychopharmacology 2002;5(4):301‐7. [DOI] [PubMed] [Google Scholar]
Jacobs‐Rebhun 2000 {published data only}
- Jacobs‐Rebhun S, Schnurr PP, Friedman MJ, Peck R, Brophy M, Fuller D. Posttraumatic stress disorder and sleep difficulty. American Journal of Psychiatry 2000;157(9):1525‐6. [DOI] [PubMed] [Google Scholar]
Kanter 2001 {published data only}
- Kanter ED, Wilkinson CW, Radant AD, Petrie EC, Dobie DJ, McFall ME, et al. Glucocorticoid feedback sensitivity and adrenocortical responsiveness in posttraumatic stress disorder. Biological Psychiatry 2001;50(4):238‐45. [DOI] [PubMed] [Google Scholar]
Kellner 2000 {published data only}
- Kellner M, Wiedemann K, Yassouridis A, Levengood R, Guo LS, Holsboer F, et al. Behavioral and endocrine response to cholecystokinin tetrapeptide in patients with posttraumatic stress disorder. Biological Psychiatry 2000;47(2):107‐11. [DOI] [PubMed] [Google Scholar]
Kline 1994 {published data only}
- Kline NA, Dow BM, Brown SA, Matloff JL. Sertraline efficacy in depressed combat veterans with posttraumatic stress disorder. American Journal of Psychiatry 1994;151(4):621. [DOI] [PubMed] [Google Scholar]
Monnelly 2003 {published data only}
- Monnelly EP, Ciraulo DA, Knapp C, Keane T. Low‐dose risperidone as adjunctive therapy for irritable aggression in posttraumatic stress disorder. Journal of Clinical Psychopharmacology 2003;23(2):193‐6. [DOI] [PubMed] [Google Scholar]
Morgan 1995 {published data only}
- Morgan CA 3rd, Grillon C, Southwick SM, Nagy LM, Davis M, Krystal JH, et al. Yohimbine facilitated acoustic startle in combat veterans with post‐traumatic stress disorder. Psychopharmacology 1995;117(4):466‐71. [DOI] [PubMed] [Google Scholar]
Nagy 1996 {unpublished data only}
- Nagy LM, Southwick SM, Charney DS. Placebo‐controlled trial of fluoxetine in PTSD. International Society for Traumatic Stress Studies 12th Annual Meeting, San Francisco, CA, November 11. 1996.
Otto 2003 {published data only}
- Otto MW, Hinton D, Korbly NB, Chea A, Ba P, Gershuny BS, et al. Treatment of pharmacotherapy‐refractory posttraumatic stress disorder among Cambodian refugees: a pilot study of combination treatment with cognitive‐behavior therapy vs sertraline alone. Behaviour Research and Therapy 2003;41:1271‐6. [DOI] [PubMed] [Google Scholar]
Pitman 1990 {published data only}
- Pitman RK, Kolk BA, Orr SP, Greenberg MS. Naloxone‐reversible analgesic response to combat‐related stimuli in posttraumatic stress disorder. Archives of General Psychiatry 1990;47(6):541‐4. [DOI] [PubMed] [Google Scholar]
Pitman 2002 {published data only}
- Pitman RK, Sanders KM, Zusman RM, Healy AR, Cheema F, Lasko NB, et al. Pilot study of secondary prevention of posttraumatic stress disorder with propranolol. Biological Psychiatry 2002;51(2):189‐92. [DOI] [PubMed] [Google Scholar]
Pivac 2004 {published data only}
- Pivac N, Kozaric‐Kovacic D, Muck‐Seler D. Olanzapine versus fluphenazine in an open trial in patients with psychotic combat‐related post‐traumatic stress disorder. Psychopharmacology 2004;175(4):451‐6. [DOI] [PubMed] [Google Scholar]
Randall 1995 {published data only}
- Randall PK, Bremner JD, Krystal JH, Nagy LM, Heninger GR, Nicolaou AL, et al. Effects of the benzodiazepine antagonist flumazenil in PTSD. Biological Psychiatry 1995;38(5):319‐24. [DOI] [PubMed] [Google Scholar]
Raskind 2003 {published data only}
- Raskind MA, Peskind ER, Kanter ED, Petrie EC, Radant A, Thompson CE, et al. Reducation of nightmares and other PTSD symptoms in combat veterans by prazosin: A placebo‐controlled study. American Journal of Psychiatry 2003;160(2):371‐3. [DOI] [PubMed] [Google Scholar]
Reist 1995 {published data only}
- Reist C, Kauffmann CD, Chicz Demet A, Chen CC, Demet EM. REM latency, dexamethasone suppression test, and thyroid releasing hormone stimulation test in posttraumatic stress disorder. Progress in Neuropsychopharmacology and Biological Psychiatry 1995;19(3):433‐43. [DOI] [PubMed] [Google Scholar]
Reist 2001 {published data only}
- Reist C, Duffy JG, Fujimoto K, Cahill L. beta‐Adrenergic blockade and emotional memory in PTSD. International Journal of Neuropsychopharmacology 2001;4(4):377‐83. [DOI] [PubMed] [Google Scholar]
Schelling 2004 {published data only}
- Schelling G, Kilger E, Roozendaal B, Quervain DJ, Briegel J, Dagge A, et al. Stress doses of hydrocortisone, traumatic memories, and symptoms of posttraumatic stress disorder in patients after cardiac surgery: A randomized study. Biological Psychiatry 2004;55(6):627‐33. [DOI] [PubMed] [Google Scholar]
Southwick 1997 {published data only}
- Southwick SM, Krystal JH, Bremner JD, Morgan CA 3rd, Nicolaou AL, Nagy LM, et al. Noradrenergic and serotonergic function in posttraumatic stress disorder. Archives of General Psychiatry 1997;54(8):749‐58. [DOI] [PubMed] [Google Scholar]
Stein 2002 {published data only}
- Stein MB, Kline NA, Matloff JL. Adjunctive olanzapine for SSRI‐resistant combat‐related PTSD: A double‐blind, placebo‐controlled study. American Journal of Psychiatry 2002;159(10):1777‐9. [DOI] [PubMed] [Google Scholar]
Vaiva 2003 {published data only}
- Vaiva G, Ducrocq F, Jezequel K, Averland B, Lestavel P, Brunet A, et al. Immediate Treatment with propranolol decreases posttraumatic stress disorder two months after trauma. Biological Psychiatry 2003;54(9):947‐9. [DOI] [PubMed] [Google Scholar]
van der Kolk 1989 {published data only}
- Kolk BA, Greenberg MS, Orr SP, Pitman RK. Endogenous opioids, stress induced analgesia, and posttraumatic stress disorder. Psychopharmacology Bulletin 1989;25(3):417‐21. [PubMed] [Google Scholar]
Zatzick 2004 {published data only}
- Zatzick D, Roy‐Byrne P, Russo J, Rivara F, Droesch R, Wagner A, et al. A randomized effectiveness trial of stepped collaborative care for acutely injured trauma survivors. Archives of General Psychiatry 2004;61(5):498‐506. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Fluoxetine, Davidson {published data only (unpublished sought but not used)}
Fluoxetine, Lilly {published data only (unpublished sought but not used)}
Nefazodone 1, BMS {published data only (unpublished sought but not used)}
Nefazodone 2, BMS {published data only (unpublished sought but not used)}
Olanzapine, Davidson {published data only}
Paroxetine 2 ‐ SB {published data only (unpublished sought but not used)}
Paroxetine 3 ‐ SB {published data only (unpublished sought but not used)}
Sertraline 2, Pfizer {published data only (unpublished sought but not used)}
Sertraline 3, Pfizer {unpublished data only}
Sertraline 4, Pfizer {published data only}
SKB650 {unpublished data only}
- GlaxoSmithKline. A Study of the Maintained Efficacy and Safety of Paroxetine Versus Placebo in the Long‐Term Treatment of Posttraumatic Stress Disorder . GSK ‐ clinical study register [www.gsk‐clinicalstudyregister.com] 2001 :Study ID: 29060/627.
- SmithKline Beecham. Unpublished data. Appendix 14 of 2nd consultation draft for the NICE (2005) PTSD guidelines.
References to ongoing studies
Davis 2003 {published data only (unpublished sought but not used)}
- Davis L, Petty F. Gabaergic modulation in PTSD and treatment with anticonvulsants. 19th Annual Meeting, International Society for Traumatic Stress Studies, October 29 ‐ November 1, Chicago, IL. 2003.
DVA (unknown) {unpublished data only}
- Department of Veterans Affairs (sponsors). CSP #504 ‐ Risperidone Treatment for Military Service Related Chronic Post‐Traumatic Stress Disorder. http://clinicaltrials.gov/show/NCT00099983.
Mechoulam (THC) {unpublished data only}
- Raphael Mechoulam. delta‐9 tetrahydrocannabino (THC) for combat‐related post traumatic stress disorder. http://www.medscape.com/. Reuters Ltd.
Mithoefer 2005 {unpublished data only}
- Mithoefer, M. A Test of MDMA‐assisted Psychotherapy in People with Posttraumatic Stress Disorder. Current controlled trials (http://clinicaltrials.gov/show/NCT00090064).
NIAAA 2005 {unpublished data only}
- National Institute on Alcohol Abuse and Alcoholism (sponsors). Treatment for Alcoholism and Post‐Traumatic Stress Disorder (naltrexone). Current controlled trials (http://clinicaltrials.gov/show/NCT00006489).
Tucker 2004 {published data only (unpublished sought but not used)}
- Tucker P, Davis L, Cohen J. Pharmacotherapy of PTSD: Trauma and neurotransmitters across the lifespan. 20th Annual Meeting, International Society for Traumatic Stress Studies, November 14 ‐ November 18, New Orleans, LA. 2004.
Additional references
Albucher 2002
- Albucher RC, Liberzon I. Psychopharmacological treatment in PTSD: a critical review. Journal of Psychiatric Research 2002;36(6):355‐67. [DOI] [PubMed] [Google Scholar]
Alderson 2003
- Alderson P, Green S, Higgins JP. Assessment of study quality. Cochrane Reviewers’ Handbook 4.2.1 [updated December] Section 8;. Chichester, UK: John Wiley & Sons, Ltd., 2004. [Google Scholar]
Allodi 1991
- Allodi FA. Assessment and treatment of torture victims: A critical review. Journal of Nervous and Mental Disease 1991;179(1):4‐11. [DOI] [PubMed] [Google Scholar]
Als‐Nielsen 2003
- Als‐Nielsen B, Chen W, Gluud C, Kjaergard LL. Association of funding and conclusions in randomized drug trials ‐ A reflection of treatment effect or adverse events?. JAMA 2003;290(7):921‐8. [DOI] [PubMed] [Google Scholar]
Altman 1998
- Altman DG. Confidence intervals for the number needed to treat. BMJ 1998;317(7):1309‐1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐III). 3rd Edition. Washington, DC: American Psychiatric Association, 1980. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders (DSM‐IV). 4th Edition. Washington, DC: American Psychiatric Association, 1994. [Google Scholar]
Asnis 2004
- Asnis GM, Kohn SR, Henderson M, Brown NL. SSRIs versus non‐SSRIs in post‐traumatic stress disorder: an update with recommendations. Drugs 2004;64(4):383‐404. [DOI] [PubMed] [Google Scholar]
Bailar 1997
- Bailar JC 3rd. The promise and problems of meta‐analysis. New England Journal of Medicine 1999;337(8):559‐61. [DOI] [PubMed] [Google Scholar]
Baker 2003
- Baker CB, Johnsrud MT, Crismon ML, Rosenheck RA, Woods SW. Quantitative analysis of sponsorship bias in economic studies of antidepressants. British Journal of Psychiatry 2003;183:498‐506. [DOI] [PubMed] [Google Scholar]
Ballenger 2000
- Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ, Foa EB, Kessler RC, et al. Consensus statement on posttraumatic stress disorder from the International Consensus Group on Depression and Anxiety. Journal of Clinical Psychiatry 2000;61(Suppl 5):60‐6. [PubMed] [Google Scholar]
Ballenger 2004
- Ballenger JC, Davidson JR, Lecrubier Y, Nutt DJ, Marshall RD, Nemeroff CB, et al. Consensus statement update on posttraumatic stress disorder from the international consensus group on depression and anxiety. Jouornal of Clinical Psychiatry 2004;65(Suppl 1):55‐62. [PubMed] [Google Scholar]
Basoglu 1992
- Basoglu M, Marks IM, Senguen S. Amitriptyline for PTSD in a torture survivor: A case study. Journal of Traumatic Stress 1992;5:77‐83. [Google Scholar]
Beck 1961
- Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventory for measuring depression. Archives of General Psychiatry 1961;4:561‐71. [DOI] [PubMed] [Google Scholar]
Berlant 2002
- Berlant J, Kammen DP. Open‐label topiramate as primary or adjunctive therapy in chronic civilian posttraumatic stress disorder. Journal of Clinical Psychiatry 2002;63(1):15‐20. [DOI] [PubMed] [Google Scholar]
Berlant 2004
- Berlant JL. Prospective open‐label study of add‐on and monotherapy topiramate in civilians with chronic nonhallucinatory posttraumatic stress disorder. BMC Psychiatry 2004;4(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Berlin 1999
- Berlin JA, Rennie D. Measuring the quality of trials: The quality of quality scales. JAMA 1999;282(11):1083‐5. [DOI] [PubMed] [Google Scholar]
Bisson 2005
- Bisson JI, Andrew M. Psychological treatment of post‐traumatic stress disorder (PTSD). The Cochrane Database of Systematic Reviews 2005, Issue 3. [CD003388] [DOI] [PubMed] [Google Scholar]
Blake 1990
- Blake DD, Weathers FW, Nagy LM, et al. A clinician rating scale for assessing current and lifetime PTSD: The CAPS‐1. Behavior Therapy 1990;21:187‐8. [Google Scholar]
Bleich 1986
- Bleich A, Siegel B, Garb R, Lerer B. Post‐traumatic stress disorder following combat exposure: clinical features and psychopharmacological treatment. British Journal of Psychiatry 1986;149:365‐9. [DOI] [PubMed] [Google Scholar]
Bonne 2004
- Bonne O, Grillon C, Vythilingam M, Neumeister A, Charney DS. Adaptive and maladaptive psychobiological responses to severe psychological stress: implications for the discovery of novel pharmacotherapy. Neuroscience Biobehavioral Review 2004;28(1):65‐94. [DOI] [PubMed] [Google Scholar]
Bradley 2005
- Bradley R, Greene J, Russ E, Dutra L, Westen D. A multidimensional meta‐analysis of psychotherapy for PTSD. American Journal of Psychiatry 2005;162(2):214‐27. [DOI] [PubMed] [Google Scholar]
Brady 1995
- Brady KT, Sonne SC, Roberts JM. Sertraline treatment of comorbid posttraumatic stress disorder and alcohol dependence. Journal of Clinical Psychiatry 1995;56(11):502‐5. [PubMed] [Google Scholar]
Bremner 2004
- Bremner JD, Vermetten E. Neuroanatomical changes associated with pharmacotherapy in posttraumatic stress disorder. Annals of the New York Academy of Science 2004;1032:154‐7. [DOI] [PubMed] [Google Scholar]
Breslau 1991
- Breslau N, Davis GC, Andreski P. Traumatic events and post traumatic stress disorder in an urban population of young adults. Archives of General Psychiatry 1991;48(3):216‐22. [DOI] [PubMed] [Google Scholar]
Brom 1989
- Brom D, Kleber RJ, Defares PB. Brief psychotherapy for posttraumatic stress disorders. Journal of Consulting and Clinial Psychology 1989;57(5):607‐12. [DOI] [PubMed] [Google Scholar]
Brophy 1991
- Brophy MH. Cyproheptadine for combat nightmares in post‐traumatic stress disorder and dream anxiety disorder. Military Medicine 1991;156:100‐1. [PubMed] [Google Scholar]
Brunello 2001
- Brunello N, Davidson JR, Deahl M, Kessler RC, Mendlewicz J, Racagni G, et al. Posttraumatic stress disorder: diagnosis and epidemiology, comorbidity and social consequences, biology and treatment. Neuropsychobiology 2001;43(3):150‐62. [DOI] [PubMed] [Google Scholar]
Burdon 1991
- Burdon AP, Sutker PB, Foulks EF, Crane MU. Pilot program of treatment for PTSD. American Journal of Psychiatry 1991;148(9):1269‐70. [DOI] [PubMed] [Google Scholar]
Burstein 1984
- Burstein A. Treatment of posttraumatic stress disorder with imipramine. Psychosomatics 1984;25(9):681‐7. [DOI] [PubMed] [Google Scholar]
Burton 1999
- Burton JK, Marshall RD. Categorizing fear: the role of trauma in a clinical formulation. American Journal of Psychiatry 1999;156(5):761‐6. [DOI] [PubMed] [Google Scholar]
Canive 1997
- Canive JM, Lewine JD, Orrison WW Jr, Edgar CJ, Provencal SL, Davis JT, et al. MRI reveals gross structural abnormalities in PTSD. Annals of the New York Academy of Sciences 1997;821:512‐5. [DOI] [PubMed] [Google Scholar]
Canive 1998
- Canive JM, Clark RD, Calais LA, Qualls C, Tuason VB. Bupropion treatment in veterans with posttraumatic stress disorder: an open study. Journal of Clinical Psychopharmacology 1998;18(5):379‐83. [DOI] [PubMed] [Google Scholar]
Charney 1993
- Charney DS, Deutch AY, Krystal JH, Southwick SM, Davis M. Psychobiologic mechanisms of posttraumatic stress disiorder. Archives of General Psychiatry 1993;50(4):294‐306. [DOI] [PubMed] [Google Scholar]
Charney 2004
- Charney D. Psychobiological mechanisms of resilience and vulnerability: Implications for successful adaptation to extreme stress. American Journal of Psychiatry 2004;161(2):195‐216. [DOI] [PubMed] [Google Scholar]
Chen 1991
- Chen C. The obsessive quality and clomipramine treatment of PTSD. American Journal of Psychiatry 1991;148(8):1087‐8. [DOI] [PubMed] [Google Scholar]
Connor 1998
- Connor KM, Davidson JR. The role of serotonin in posttraumatic stress disorder: Neurobiology and pharmacotherapy. CNS Spectrums 1998;3(7S2):43‐51. [Google Scholar]
Connor 1999
- Connor KM, Davidson JR, Weisler RH, Ahearn E. A pilot study of mirtazapine in post‐traumatic stress disorder. International Clinical Psychopharmacology 1999;14(1):29‐31. [DOI] [PubMed] [Google Scholar]
Covi 1984
- Covi L, Lipman RS. Primary depression or primary anxiety A possible psychometric approach to a diagnostic dillema. Clinical Neuropharmacology 1984;7:S502‐503. [Google Scholar]
Creamer 2003
- Creamer M, Bell R, Failla S. Psychometric properties of the Impact of Event Scale— RPPsychometric properties of the Impact of Event Scale—Revised. Behaviour Research and Therapy 2003;41(12):1489–96. [DOI] [PubMed] [Google Scholar]
CSM 2004
- Committee on Safety of Medicines. Report of the CSM expert working group on the safety of selective serotonin reuptake inhibitor antridepressants. http://www.mhra.gov.uk/news/2004/SSRIfinal.pdf 2004.
Cyr 2000
- Cyr M, Farrar MK. Treatment for posttraumatic stress disorder. Annals of Pharmacotherapy 2000;34(3):366‐76. [DOI] [PubMed] [Google Scholar]
Davidson 1987
- Davidson J, Walker I, Kilts C. A pilot study of phenelzine in the treatment of post‐traumatic stress disorder. British Journal of Psychiatry 1987;150:252‐5. [DOI] [PubMed] [Google Scholar]
Davidson 1989
- Davidson JR, Nemeroff CB. Pharmacotherapy in posttraumatic stress disorder: historical and clinical considerations and future directions. Psychopharmacology Bulletin 1989;25(3):422‐5. [PubMed] [Google Scholar]
Davidson 1990
- Davidson JR, Roth S, Newman E. Fluoxetine in posttraumatic stress disorder. Journal of Traumatic Stress 1990;4:419‐23. [Google Scholar]
Davidson 1991
- Davidson JR, Hughes D, Blazer DG, George LK. Posttraumatic stress disorder in the community : an epidemiological study. Psychological Medicine 1991;21(3):713‐21. [DOI] [PubMed] [Google Scholar]
Davidson 1992
- Davidson JR. Drug therapy of posttraumatic stress disorder. British Journal of Psychiatry 1992;160:309‐14. [DOI] [PubMed] [Google Scholar]
Davidson 1993
- Davidson JR, Kudler HS, Saunders WB, Erickson L, Smith RD, Stein RM, et al. Predicting response to amitriptyline in posttraumatic stress disorder. American Journal of Psychiatry 1993;150(7):1024‐9. [DOI] [PubMed] [Google Scholar]
Davidson 1997a
- Davidson JR, Malik ML, Sutherland SN. Response characteristics to antidepressants and placebo in post‐traumatic stress disorder. International Clinical Psychopharmacology 1997;12(6):291‐6. [DOI] [PubMed] [Google Scholar]
Davidson 1997b
- Davidson JR. Biological therapies for posttraumatic stress disorder: an overview. Joournal of Clinical Psychiatry 1997;58(Suppl 9):33‐6. [PubMed] [Google Scholar]
Davidson 1997c
- Davidson JR, Book SW, Colket L, Tupler LA, Roth S, David D, et al. Assessment of a new self‐rating scale for posttraumatic stress disorder. Psychological Medicine 1997;27:153‐60. [DOI] [PubMed] [Google Scholar]
Davidson 1998a
- Davidson JR, Weisler RH, Malik M, Tupler LA. Fluvoxamine in civilians with posttraumatic stress disorder. Journal of Clinical Psychopharmacology 1998;18(1):93‐5. [DOI] [PubMed] [Google Scholar]
Davidson 1998b
- Davidson JR, Weisler RH, Malik ML, Connor MK. Treatment of posttraumatic stress disorder with nefazodone. International Clinical Psychopharmacology 1998;13(3):111‐3. [DOI] [PubMed] [Google Scholar]
Davidson 2000
- Davidson JR. Pharmacotherapy of posttraumatic stress disorder: Treatment options, long term follow‐up, and predictors of outcome. Journal of Clinical Psychiatry 2000;61(Suppl 5):52‐6. [PubMed] [Google Scholar]
Davis 2000
- Davis LL, Nugent AL, Murray J, Kramer GL, Petty F. Nefazodone treatment for chronic posttraumatic stress disorder: an open trial. Journal of Clinical Psychopharmacology 2000;20(2):159‐64. [DOI] [PubMed] [Google Scholar]
De Boer 1992
- Boer M, Op van Velde W, Falger PJ, Hovens JE, Groen JH, Duijn H. Fluvoxamine treatment for chronic PTSD: A pilot study. Psychotherapy & Psychosomatics 1992;57(4):158‐63. [DOI] [PubMed] [Google Scholar]
Deeks 2001
- Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in meta‐analysis.. In: Egger M, Davey SG, Altman DG editor(s). Systematic Reviews in Health Care: Meta‐Analysis in Context. 2nd Edition. London: BMJ Publication Group, 2001. [Google Scholar]
Deeks 2002
- Deeks JJ. Issues in the selection of a summary statistic for meta‐analysis of clinical trials with binary outcomes. Statistics in medicine 2002;21(11):1575‐600. [DOI] [PubMed] [Google Scholar]
Deeks 2003
- Deeks J, Higgins J, Altman D. Cochrane Reviewers’ Handbook 4.2.1 [updated December 2003]. In: Alderson P, Green S, Higgins J editor(s). The Cochrane Library. Chichester, UK: John Wiley & Sons, Ltd., 2003. [Google Scholar]
Demartino 1995
- Demartino R, Mollica RF, Wilk V. Monoamine oxidase inhibitors in posttraumatic stress disorder: promise and problems in Indochinese survivors of trauma. Journal of Nervous and Mental Disease 1995;183(8):510‐5. [DOI] [PubMed] [Google Scholar]
Dhansay 2005
- Dhansay Y, Ipser JC, Stein DJ. Pharmacotherapy augmentation strategies in treatment‐resistant anxiety disorders. The Cochrane Database of Systematic 2005, Issue 5. [Art. No.: CD005473. DOI: 10.1002/14651858.CD005473.] [DOI] [PubMed] [Google Scholar]
Dickersin 1992
- Dickersin K, Min YI, Meinert CL. Factors influencing the publication of research results : follow‐up of applications submitted to two substantial review boards. JAMA 1992;267(3):374‐8. [PubMed] [Google Scholar]
Dillard 1993
- Dillard ML, Bendfeldt F, Jernigan P. Use of thioridazine in post‐traumatic stress disorder. Southern Medical Journal 1993;86(11):1276‐8. [DOI] [PubMed] [Google Scholar]
Dominiak 1992
- Dominiak GM. Psychopharmacology of the abused. In: Shapiro S, Dominiak GM, et al. editor(s). Sexual trauma and psychopathology: Clinical intervention with adult survivors. New York, NY: Lexington Books/Macmillan, 1992:81‐111. [Google Scholar]
Dow 1997
- Dow B, Kline N. Antidepressant treatment of posttraumatic stress disorder and major depression in veterans. Annals of Clinical Psychiatry 1997;9(1):1‐5. [DOI] [PubMed] [Google Scholar]
Duffy 1992
- Duffy JD. Rapid response to buspirone in a case of post‐traumatic stress disorder. Annals of Clinical Psychiatry 1992;4:193‐6. [Google Scholar]
Duffy 1994
- Duffy JD, Malloy PF. Efficacy of buspirone in the treatment of posttraumatic stress disorder: An open trial. Annals of Clinical Psychiatry 1994;6(1):33‐7. [DOI] [PubMed] [Google Scholar]
Dunner 1985
- Dunner FJ, Edwards WP, Copeland PC. Clinical efficacy of alprazolam in PTSD patients. 138th Annual Meeting of the American Psychiatric Association, Los Angeles, CA. 1985.
Easterbrook 1991
- Eaterbrook PJ, Berline JA, Gopaian R, Matthews DR. Publication bias in clinical research. Lancet 1991;337(8746):867‐72. [DOI] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne DR, Altman DG, Higgins JPT, Curtin F, Worthington HV, Vail A. Meta‐analyses involving cross‐over trials: Methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Falcon 1985
- Falcon S, Ryan C, Chamberlain K, Curtis G. Tricyclics: possible treatment for post‐traumatic stress disorder. Journal of Clinical Psychiatry 1985;46(9):385‐9. [PubMed] [Google Scholar]
Famularo 1988
- Famularo R, Kinscherff R, Fenton T. Propranolol treatment for childhood posttraumatic stress disorder, acute type. A pilot study. American Journal of Diseases of Children Am J Dis Childhood 1988;142(11):1244‐7. [DOI] [PubMed] [Google Scholar]
Faustman 1989
- Faustman WO, White PA. Diagnostic and psychopharmacological treatment characteristics of 536 inpatients with posttraumatic stress disorder. Journal of Nervous and Mental Disease 1989;177(3):154‐9. [DOI] [PubMed] [Google Scholar]
Fesler 1991
- Fesler FA. Valproate in combat‐related posttraumatic stress disorder. Journal of Clinical Psychiatry 1991;52(9):361‐4. [PubMed] [Google Scholar]
Fichtner 1990
- Fichtner CG, Kuhlman DT, Gruenfeld MJ, Hughes JR. Decreased episodic violence and increased control of dissociation in a carbamazepine‐treated case of multiple personality. Biological Psychiatry 1990;27(9):1045‐52. [DOI] [PubMed] [Google Scholar]
Fichtner 1994
- Fichtner CG, Arora RC, O'Connor FL, Crayton JW. Platelet paroxetine binding and fluoxetine pharmacotherapy in posttraumatic stress disorder: Preliminary observations on a possible predictor of clinical treatment response. Life Sciences 1994;54(3):39‐44. [DOI] [PubMed] [Google Scholar]
Foa 1999
- Foa EB, Davidson JR, Frances A. The expert consensus guideline series: treatment of posttraumatic stress disorder: The Expert Consensus Panel for PTSD. Journal of Clinical Psychiatry 1999;60(Suppl. 16):1‐75. [PubMed] [Google Scholar]
Ford 1996
- Ford N. The use of anticonvulsants in posttraumatic stress disorder: case study and overview. Journal of Traumatic Stress 1996;9(4):857‐63. [DOI] [PubMed] [Google Scholar]
Forster 1994
- Forster PL, Schoenfield FB, Marmar CR, Lang AJ. Lithium for irritability in post‐traumatic stress disorder. Journal of Traumatic Stress 1994;8(1):143‐9. [DOI] [PubMed] [Google Scholar]
Fossey 1994
- Fossey MD, Hamner MB. Clonazepam‐related sexual dysfunction in male veterans with PTSD. Anxiety 1994;1(5):233‐6. [PubMed] [Google Scholar]
Friedman 1988
- Friedman MJ. Toward rational pharmacotherapy for posttraumatic stress disorder: an interim report. American Journal of Psychiatry 1988;145(3):281‐5. [DOI] [PubMed] [Google Scholar]
Friedman 1991
- Friedman MJ. Biological approaches to the diagnosis and treatment of post traumatic stress disorder. Journal of Traumatic Stress 1991;4:67‐91. [Google Scholar]
Gersons 2000
- Gersons BP, Carlier IV, Lamberts RD, Kolk BA. Randomized clinical trial of brief eclectic psychotherapy for police officers with posttraumatic stress disorder. Journal of Trauma Stress 2000;13(2):333‐47. [DOI] [PubMed] [Google Scholar]
Glover 1993
- Glover H. A preliminary trial of nalmefene for the treatment of emotional numbing in combat veterans with post‐traumatic stress disorder. Israel Journal of Psychiatry and Related Sciences 1993;30(4):255‐63. [PubMed] [Google Scholar]
Glynn 1995
- Glynn SM, Eth S, Randolph ET, et al. Behavioral family therapy for Vietnam combat veterans with posttraumatic stress disorder. Journal of Psychotherapy Practice and Research 1995;4:214‐23. [PMC free article] [PubMed] [Google Scholar]
Gupta 1998
- Gupta S, Popli A, Bathurst E, Hennig L, Droney T, Keller P. Efficacy of cyproheptadine for nightmares associated with posttraumatic stress disorder. Comprehensive Psychiatry 1998;39(3):160‐4. [DOI] [PubMed] [Google Scholar]
Gury 1999
- Gury C, Cousin F. Pharmacokinetics of SSRI antidepressants: half‐life and clinical applicability. Encephale 1999;25(5):470‐6. [PubMed] [Google Scholar]
Guy 1976
- Guy W. ECDEU assessment manual for psychopharmacology. Washington, DC: U.S. National Institute of Health, 1976. [Google Scholar]
Hamilton 1959
- Hamilton M. The assessment of anxiety states by rating. British Journal of Medical Psychology 1959;32:50‐5. [DOI] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery & Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamner 1996
- Hamner MB. Clozapine treatment for a veteran with comorbid psychosis and PTSD (let). American Journal of Psychiatry 1996;153(6):841. [DOI] [PubMed] [Google Scholar]
Hamner 1997
- Hamner M, Ulmer H, Horne D. Buspirone potentiation of antidepressants in the treatment of PTSD. Depression and Anxiety 1997;5(3):137‐9. [PubMed] [Google Scholar]
Hamner 1998
- Hamner MB, Frueh BC. Response to venlafaxine in a previously antidepressant treatment‐resistant combat veteran with post‐traumatic stress disorder. International Clinical Psychopharmacology 1998;13(5):233‐4. [DOI] [PubMed] [Google Scholar]
Hamner 2003
- Hamner MB, Faldowski RA, Ulmer HG, Frueh BC, Huber MG, Arana GW. Adjunctive risperidone treatment in post‐traumatic stress disorder: a preliminary controlled trial of effects on comorbid psychotic symptoms. International Journal of Clinical Psychopharmacology 2003;18(1):1‐8. [DOI] [PubMed] [Google Scholar]
Hargrave 1993
- Hargrave R. Serotonergic agents in the management of dementia and posttraumatic stress disorder. Psychosomatics 1993;34(5):461‐2. [DOI] [PubMed] [Google Scholar]
Harmon 1996
- Harmon RJ, Riggs PD. Clonidine for posttraumatic stress disorder in preschool children. Journal of the American Academy of Child & Adolescent Psychiatry 1996;35(9):1247‐9. [DOI] [PubMed] [Google Scholar]
Harvey 2003
- Harvey AG, Bryant RA, Tarrier N. Cognitive behaviour therapy for posttraumatic stress disorder. Clinical Psychology Review 2003;23(3):501‐22. [DOI] [PubMed] [Google Scholar]
Hertzberg 1996
- Hertzberg MA, Feldman ME, Beckham JC, Davidson JR. Trial of trazodone for posttraumatic stress disorder using a multiple baseline group design. Journal of Clinical Psychopharmacology 1996;16(4):294‐8. [DOI] [PubMed] [Google Scholar]
Hertzberg 1998
- Hertzberg MA, Feldman ME, Beckham JC, Moore SD, Davidson JR. Open trial of nefazodone for combat‐related posttraumatic stress disorder. Journal of Clinical Psychiatry 1998;59(9):460‐4. [DOI] [PubMed] [Google Scholar]
Hertzberg 1999
- Hertzberg MA, Butterfield MI, Feldman ME, Beckham JC, Sutherland SM, Connor KM, et al. A preliminary study of lamotrigine for the treatment of posttraumatic stress disorder. Biological Psychiatry 1999;45(9):1226‐9. [DOI] [PubMed] [Google Scholar]
Hidalgo 1999
- Hidalgo R. Nefazodone in posttraumatic stress: results from six open‐label trials. International Clinical Psychopharmacology 1999;14(2):61‐8. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JTP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(6):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hogben 1981
- Hogben GL, Cornfield RB. Treatment of traumatic war neurosis with phenelzine. Archives of General Psychiatry 1981;38(4):440‐5. [DOI] [PubMed] [Google Scholar]
Hopewell 2002
- Hopewell S, Clarke M, Lusher A, Lefebvre C, Westby M. A comparison of handsearching versus MEDLINE searching to identify reports of randomized controlled trials. Statistics in Medicine 2002;21(11):1625‐34. [DOI] [PubMed] [Google Scholar]
Horowitz 1979
- Horowitz M, Wilner N, Alvarez W. Impact of Event Scale: a measure of subjective stress. Psychosomatic Medicine 1979;41(3):209‐18. [DOI] [PubMed] [Google Scholar]
Horrigan 1996
Hull 2002
- Hull A. Neuroimaging findings in post‐traumatic stress disorder. British Journal of Psychiatry 2002;181:102‐10. [PubMed] [Google Scholar]
Irwin 1989
- Irwin M, Van‐Putten T, Guze B, Marder SR. Pharmacologic treatment of veterans with posttraumatic stress disorder and concomitant affective disiorder. Annals of Clinical Psychiatry 1989;1(2):127‐30. [Google Scholar]
Izrayelit 1998
- Izrayelit L. Schizoaffective disorder and PTSD successfully treated with olanzapine and supportive psychotherapy. Psychiatric Annals 1998;28(8):424‐6. [Google Scholar]
Keane 1989
- Keane T. Post‐traumatic stress disorder: current status and future directions. Behavior Therapy 1989;20:149‐53. [Google Scholar]
Keck 1992
- Keck PE, McElroy SL, Friredman LM. Valproate and carbamazepine in the treatment of panic and posttraumatic stress disorders. Journal of Clinical Psychopharmacology 1992;12(1S):36‐41. [DOI] [PubMed] [Google Scholar]
Kessler 1995
- Kessler RC, Sonnega A, Bromet E, Hughes M, Nelson CB. Posttraumatic stress disorder in the National Comorbidity Survey. Archives of General Psychiatry 1995;52(12):1048‐60. [DOI] [PubMed] [Google Scholar]
Khouzam 1997
- Khouzam HR, Kissmeyer P. Antidepressant treatment, posttraumatic stress disorder, survivor guilt, and spiritual awakening. Journal of Traumatic Stress 1997;10(4):691‐6. [DOI] [PubMed] [Google Scholar]
Kinzie 1989
- Kinzie JD, Leung P. Clonidine in Cambodian patients with post traumatic stress disorder. Journal of Nervous and Mental Disease 1989;177(9):546‐50. [DOI] [PubMed] [Google Scholar]
Kitchner 1985
- Kitchner I, Greenstein R. Low dose lithium carbonate in the treatment of post traumatic stress disorder: brief communication. Military Medicine 1985;150(7):378‐81. [PubMed] [Google Scholar]
Kolb 1984
- Kolb LC, Burris BC, Griffiths S. Propranolol and clonidine in the treatment of the chronic post‐traumatic stress disorders of war. In: Kolk BA editor(s). Post‐traumatic Stress Disorder: Psychological and Biological Sequelae. Washington, DC: American Psychiatric Press, 1984:98‐105. [Google Scholar]
Krakow 2000
- Krakow B, Hollifield M, Warner TD. Placebo effect in posttraumatic stress disorders. Journal of Clinical Psychiatry 2000;284(5):563. [DOI] [PubMed] [Google Scholar]
Kroll 1990
- Kroll J, Paul L, Habenicht M, Chan S, Yang M, Vang T, et al. Medication compliance, antidepressant blood levels, and side effects in Southeast Asian patients. Journal of Clinical Psychopharmacology 1990;10(4):279‐83. [PubMed] [Google Scholar]
LaPorta 1992
- LaPorta LD, Ware MR. Buspirone in the treatment of posttraumatic stress disorder. Journal of Clinical Psychopharmacology 1992;12(2):133‐4. [DOI] [PubMed] [Google Scholar]
Lerer 1987
- Lerer B, Bleich A, Kotler M, Garb R, Hertzberg M, Levin B. Posttraumatic stress disorder in Israeli combat veterans: effect of phenelzine treatment. Archives of General Psychiatry 1987;44(11):976‐81. [DOI] [PubMed] [Google Scholar]
Leyba 1998
- Leyba CM, Wampler TP. Risperidone in PTSD. Psychiatric Services 1998;49(2):245‐6. [DOI] [PubMed] [Google Scholar]
Liebowitz 1989
- Liebowitz NR, Mallakh RS. Trazodone for the treatment of anxiety symptoms in substance abusers. Journal of Clinical Psychopharmacology 1989;9(8):449‐51. [PubMed] [Google Scholar]
Lipper 1986
- Lipper S, Davidson JR, Grady TA, Edinger JD, Hammett EB, Mahorney SL, et al. Preliminary study of carbamazepine in post traumatic stress disorder. Psychosomatics 1986;27(12):849‐54. [DOI] [PubMed] [Google Scholar]
Loke 2005
- Higgins JPT, Green S. Appendix 6b. Including adverse effects. In: Loke YK, Price D, Herxheimer A editor(s). Cochrane Handbook for Systematic Reviews of Interventions 4.2.5 [Updated May 2005]. Vol. Issue 3, Chichester, UK: John Wiley & Sons, Ltd., 2005. [Google Scholar]
Looff 1995
- Looff D, Grimley P, Kuller F, Martin A, Shonfield L. Carbamazepine for PTSD. Journal of the American Academy of Child & Adolescent Psychiatry 1995;34(6):703‐4. [DOI] [PubMed] [Google Scholar]
Lowenstein 1988
- Lowenstein RJ, Hornstein N, Farber B. Open trial of clonazepam in the treatment of post traumatic stress symptoms in multiple personality disorder. Dissociation 1988;1:3‐12. [Google Scholar]
Marmar 1996
- Marmar CR, Schoenfeld F, Weiss DS, Metzler T, Zatzick D, Wu R, et al. Open trial of fluvoxamine treatment for combat‐related posttraumatic stress disorder. Journal of Clinical Psychiatry 1996;57(Suppl 8):66‐72. [PubMed] [Google Scholar]
Marshall 1996
- Marshall RD, Stein DJ, Liebowitz MR, Yehuda R. Psychopharmacology of posttraumatic stress disorder. Psychiatric Annals 1996;26:217‐26. [Google Scholar]
Marshall 1998a
- Marshall RD, Davidson JR, Yehuda R. Pharmacotherapy in the treatment of posttraumatic stress disorder and other trauma‐related disorders. In: Yehuda R editor(s). Psychological Trauma. Washington, DC: American Psychiatric Press, 1998:133‐77. [Google Scholar]
Marshall 1998b
- Marshall RD, Schneier FR, Fallon BA, Knight CB, Abbate LA, Goetz D, et al. An open trial of paroxetine in patients with noncombat‐related posttraumatic stress disorder. Journal of Clinical Psychopharmacology 1998;18(1):10‐8. [DOI] [PubMed] [Google Scholar]
Marshall 2000
- Marshall RD, Pierce D. Implications of recent findings in posttraumatic stress disorder and the role of pharmacotherapy. Harvard Review of Psychiatry 2000;7(5):247‐56. [PubMed] [Google Scholar]
McDougle 1991
- McDougle CJ, Southwick SM, Charney DS, James RL. An open trial of fluoxetine in the treatment of posttraumatic stress disorder. Journal of Clinical Psychopharmacology 1991;11(5):325‐7. [PubMed] [Google Scholar]
McQuay 1997
- McQuay HJ, Moore RA. Using numerical results from systematic reviews in clinical practice. Annals of Internal Medicine 1997;126(9):712‐20. [DOI] [PubMed] [Google Scholar]
Mellman 1998
- Mellman TA, Byers PM, Augenstein JS. Pilot evaluation of hypnotic medication during acute traumatic stress response. Journal of Traumatic Stress 1998;11(3):563‐9. [DOI] [PubMed] [Google Scholar]
Mellman 2002
- Mellman TA, Bustamante V, David D, Fins AI. Hypnotic medication in the aftermath of trauma. Journal of Clinical Psychiatry 2002;63(12):1183‐4. [DOI] [PubMed] [Google Scholar]
Milanes 1984
- Milanes F, Mack C. Phenelzine treatment of post‐Vietnam stress syndrome. VA Practitioner 1984;1:40‐9. [Google Scholar]
Moher 1999
- Moher D, Cook DJ, Eastwood S, Olkin I, Rennie D, Stroup DF. Improving the quality of reports of meta‐analyses of randomized controlled trials: the QUOROM statement. Lancet 1999;354(9193):1896‐900. [DOI] [PubMed] [Google Scholar]
Moncrieff 1999
- Moncrieff J, Churchill R, Drummond DC, McGuire H. Development of a quality assessment instrument for trials of treatments for depression and neurosis. International Journal of Methods in Psychiatric Research 1999;10(3):126‐33. [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382‐9. [DOI] [PubMed] [Google Scholar]
Mulrow 1997
- Mulrow CD, Oxman AD. Cochrane Collaboration Handbook. Oxford: Update Software, 1997. [Google Scholar]
Nagy 1993
- Nagy LM, Morgan CA, Southwick SM, Charney DS. Open prospective trial of fluoxetine for posttraumatic stress disorder. Journal of Clinical Psychopharmacology 1993;13(2):107‐13. [PubMed] [Google Scholar]
Neal 1997
- Neal LA, Shapland W, Fox C. An open trial of moclobemide in the treatment of post‐traumatic stress disorder. International Clinical Psychopharmacology 1997;12(4):231‐7. [DOI] [PubMed] [Google Scholar]
NICE 2005
- Guideline Development Group. Post‐traumatic stress disorder: The management of PTSD in adults and children in primary and secondary care. Vol. 26, London: Gaskell and the British Psychological Society, 2005. [PubMed] [Google Scholar]
Olivera 1990
- Olivera AT, Fero D. Affective disorders, DST, and treatment in PTSD patients: Clinical observations. Journal of Traumatic Stress 1990;3:407‐14. [Google Scholar]
Onder 2005
Otto 1996
- Otto MW, Penava SJ, Pollock RA, Smoller JW. Cognitive‐behavioral and pharmacological perspectives on the treatment of post traumatic stress disorder. In: Pollack MH, Otto MW, Rosenbaum JR editor(s). Challenges in clinical practice: Pharmacologic and psychosocial strategies. New York, NY: Guilford Press, 1996:219‐60. [Google Scholar]
Penava 1996
- Penava SJ, Otto MW, Pollack MH, Rosenbaum JF. Current status of pharmacotherapy for PTSD: An effect size analysis of controlled studies. Depression and Anxiety 1996;4(5):240‐2. [DOI] [PubMed] [Google Scholar]
Petty 2001
- Petty F, Brannan S, Casada J, Davis LL, Gajewski V, Kramer GL, et al. Olanzapine treatment for post‐traumatic stress disorder: an open‐label study. International Journal of Clinical Psychopharmacology 2001;16(6):331‐7. [DOI] [PubMed] [Google Scholar]
Rapaport 2002
- Rapaport MH, Endicott J, Clary CM. Posttraumatic stress disorder and quality of life: Results across 64 weeks of sertraline treatment. Journal of Clinical Psychiatry 2002;63(1):59‐65. [DOI] [PubMed] [Google Scholar]
Robert 1999
- Robert R, Blakeney PE, Villarreal C, Rosenberg L, Meyer WJ 3rd. Imipramine treatment in pediatric burn patients with symptoms of acute stress disorder: a pilot study. Journal of the American Academy of Child and Adolescent Psychiatry 1999;38(7):873‐82. [DOI] [PubMed] [Google Scholar]
Robinson 2002
- Robinson KA, Dickersin K. Development of a highly sensitive search strategy for the retrieval of reports of controlled trials using PubMed. International Journal of Epidemiology 2002;31(1):150‐3. [DOI] [PubMed] [Google Scholar]
Rose 1998
- Rose S, Bisson J. Brief early psychological interventions following trauma: A systematic review of the literature. Journal of Traumatic Stress 1998;11(4):697‐710. [DOI] [PubMed] [Google Scholar]
Rose 2002
- Rose S, Bisson J, Wessely S. Psychological debriefng for preventing post traumatic stress disorder (PTSD) (Review). The Cochrane Database of Systematic Reviews 2002, Issue 2. [DOI] [PubMed] [Google Scholar]
Rothbaum 1996
- Rothbaum BO, Ninan PT, Tomas L. Sertraline in the treatment of rape victims with posttraumatic stress disorder. Journal of Traumatic Stress 1996;9(4):865‐71. [DOI] [PubMed] [Google Scholar]
Rubin 1993
- Rubin MR, Solt V, Chen C‐J, et al. Anafranil's effect on the obsessions and sleep in PTSD. Proceedings and Abstracts of the 146th Annual Meeting of the American Psychiatric Association, San Francisco, CA. 1993:102.
Sargent 1940
- Sargent WW, Slater E. Acute war neuroses. Lancet 1940;140:1‐2. [Google Scholar]
Scherer 1994
- Scherer RW, Dickersin K, Langenberg P. Full publication of results initially presented in abstracts. A meta‐analysis. JAMA 1994;272(2):158‐62. [PubMed] [Google Scholar]
Seedat 2001
- Seedat S, Lockhat R, Kaminer D, Zungu‐Dirwayi N, Stein DJ. An open trial of citalopram in adolescents with post‐traumatic stress disorder. International Journal of Clinical Psychopharmacology 2001;16(1):21‐5. [DOI] [PubMed] [Google Scholar]
Shalev 1996
- Shalev A, Bonne E, Eth S. Treatment of posttraumatic stress disorder: A review. Psychosomatic Medicine 1996;58(2):162‐82. [DOI] [PubMed] [Google Scholar]
Shay 1992
- Shay J. Fluoxetine reduces explosiveness and elevates mood of Vietnam vets with PTSD. Journal of Traumatic Stress 1992;5:97‐101. [Google Scholar]
Sheehan 1996
- Sheehan DV, Harnett‐Sheehan K, Raj BA. The measurement of disability. International Clinical Psychopharmacology 1996;11 Suppl 3:89‐95. [DOI] [PubMed] [Google Scholar]
Sherman 1998
- Sherman JJ. Effects of psychotherapeutic treatments for PTSD: A meta‐analysis of controlled clinical trials. Journal of Traumatic Stress 1998;11(3):413‐35. [DOI] [PubMed] [Google Scholar]
Silver 1990
- Silver JM, Sandberg DP, Hales RP. New approaches in the pharmacotherapy of posttraumatic stress disorder. Journal of Clinical Psychiatry 1990;51(10 Suppl):33‐8. [PubMed] [Google Scholar]
Simpson 1991
- Simpson MA. Buspirone, benzodiazepines, and post‐traumatic stress disorder. Journal of Traumatic Stress 1991;4:305‐8. [Google Scholar]
Solomon 1992
- Solomon SD, Gerrity ET, Muff AM. Efficacy of treatments for posttraumatic stress disorder: An empirical review. JAMA 1992;268(5):633‐8. [PubMed] [Google Scholar]
Solomon 1997
- Solomon SD, Davidson JR. Trauma: Prevalence, impairment, service use, and cost. Journal of Clinial Psychiatry 1997;58(Suppl 5):1‐11. [PubMed] [Google Scholar]
Southwick 1993
- Southwick SM, Yehuda R. The interaction between pharmacotherapy and psychotherapy in the treatment of posttraumatic stress disorder. American Journal of Psychotherapy 1993;47(3):404‐10. [DOI] [PubMed] [Google Scholar]
Spitzer 1996
- Spitzer R, Williams J, Gibbons M. Instructional manual for the structured clinical interview for DSM‐IV.. New York. NY: New York State Psychiatric Institute., 1996. [Google Scholar]
Stewart 1986
- Stewart JT, Bartucci RJ. Posttraumatic stress disorder and partial complex seizures (let). American Journal of Psychiatry 1986;143(1):113‐4. [DOI] [PubMed] [Google Scholar]
Szymanski 1991
- Szymanski HV, Olympia J. Divalproex in posttraumatic stress disorder. American Journal of Psychiatry 1991;148(8):1086‐7. [DOI] [PubMed] [Google Scholar]
Taylor 2003
- Taylor FB. Tiagabine for posttraumatic stress disorder: a case series of 7 women. Journal of Clinical Psychiatry 2003;64(12):1421‐5. [DOI] [PubMed] [Google Scholar]
Thomson 1994
- Thomson SG. Why sources of heterogeneity in meta‐analysis should be investigated. BMJ 1994;309(6965):1351‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ursano 2004
- Ursano RJ, Bell C, Eth S, Friedman M, Norwood A, Pfefferbaum B, et al. Practice guideline for the treatment of patients with acute stress disorder and posttraumatic stress disorder. American Journal of Psychiatry 2004;161(11 Suppl):3‐31. [PubMed] [Google Scholar]
van der Kolk 1983
- Kolk BA. Psychopharmacological issues in post traumatic stress disorder. Hospital and Community Psychiatry 1983;34(8):683‐91. [DOI] [PubMed] [Google Scholar]
van der Kolk 1987
- Kolk BA. The drug treatment of post traumatic stress disorder. Journal of Affective Disorders 1987;13(2):203‐13. [DOI] [PubMed] [Google Scholar]
van Etten 1998
- Etten ML, Taylor S. Comparative efficacy of treatments for post‐traumatic stress disorder: a meta‐analysis. Clinical Psychology and Psychotherapy 1998;5:126‐44. [Google Scholar]
Walker 1989
- Walker JI. Chemotherapy of traumatic war stress. In: Miller TW, et al. editor(s). Stressful life events.. International Universities Press Stress and Health Series, Monograph 4. Madison, CT: International Universities Press, 1989:587‐601. [Google Scholar]
Weiss 1997
- Weiss DS, Marmar CR. The Impact of Event Scale—Revised. In: Wilson JP, Keane TM editor(s). Assessing psychological trauma and PTSD: A handbook for practitioners. New York, NY: Guilford Press, 1997:399‐411. [Google Scholar]
Wells 1991
- Wells GB, Chu C, Johnson R, et al. Buspirone in the treatment of post‐traumatic stress disorder and dream anxiety disorder. Military Medicine 1991;11:340‐3. [Google Scholar]
White 1983
- White NS. Post‐traumatic stress disorder. Hospital and Community Psychiatry 1983;34(11):1061‐2. [DOI] [PubMed] [Google Scholar]
Wise 1983
- Wise M. Post traumatic stress disorder: The human reaction to catastrophe. Drug Therapy 1983;3:97‐105. [Google Scholar]
Wolf 1988
- Wolf ME, Alavi A, Mosnaim AD. Posttraumatic stress disorder in Vietnam veterans: clinical and EEG findings; possible therapeutic effects of carbamazepine. Biological Psychiatry 1988;23(6):642‐4. [DOI] [PubMed] [Google Scholar]
Yehuda 1995
- Yehuda R, McFarlane AC. Conflict between current knowledge about posttraumatic stress disorder and its original conceptual basis. American Journal of Psychiatry 1995;152(12):1705‐13. [DOI] [PubMed] [Google Scholar]