

Abstract
DNA base modifications and mutations are observed in all genomes throughout the kingdoms of life. Proteins involved in their establishment and removal were shown to use a base flipping mechanism to access their substrates. To better understand how proteins flip DNA bases to modify or remove them, we optimized and developed a pipeline of methods to step-by-step detect the process starting with protein–DNA interaction, base flipping itself and the ensuing DNA base modification or excision. As methylcytosine is the best-studied DNA modification, here we focus on the process of writing, modifying and reading this DNA base. Using multicolor electrophoretic mobility shift assays, we show that the methylcytosine modifier Tet1 exhibits little DNA sequence specificity with only a slight preference for methylated CpG containing DNA. A combination of chloroacetaldehyde treatment and high-resolution melting temperature analysis allowed us to detect base flipping induced by the methylcytosine modifier Tet1 as well as the methylcytosine writer M.HpaII. Finally, we show that high-resolution melting temperature analysis can be used to detect the activity of glycosylases, methyltransferases and dioxigenases on DNA substrates. Taken together, this DNA base flipping analytical pipeline (BaFAP) provide a complete toolbox for the fast and sensitive analysis of proteins that bind, flip and modify or excise DNA bases.
Keywords: DNA modifications, base flipping, electrophoretic mobility shift assay, high-resolution DNA melting analysis, base excision, methylcytosine
Introduction
DNA base modifications diversify the genome and regulate gene expression in a spatio-temporal manner. One of the best-studied DNA base modifications is 5-methylcytosine (5mC). Its establishment and maintenance are catalyzed by DNA methyltransferases (Dnmts), which transfer a methyl group from S-adenosylmethionine (SAM) to the fifth position of cytosine [1]. However, cytosine is sheltered inside the DNA double helix and stabilized by base pairing with the base guanine (G). Thus, to access and subsequently modify cytosine, a DNA base flipping mechanism is used by Dnmts. The base flipping phenomenon was first described in 1994 in a ternary complex containing HhaI methyltransferase (M.HhaI) [2], DNA and S-adenosylhomocysteine (SAH). In this complex, cytosine is rotated out from the DNA backbone by 180° and inserted into the catalytic pocket of the M.HhaI enzyme. Since then, more and more Dnmts were described to use a base flipping mechanism to achieve DNA base modifications, including M.HaeIII [3], M.TaqI [4], M.T4Dam [5], M.EcoDam [6], and the murine DNA methyltransferase Dnmt1 [7].
Moreover, 5mC can be modified by Ten-eleven translocation proteins (Tet) to 5-hydroxymethylcytosine [8, 9], 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) in an iterative iron and oxoglutarate dependent oxidation reaction [10, 11]. Similar to other DNA base modifiers, Tet proteins use a base flipping mechanism to modify 5mC to 5hmC [12, 13] and subsequently to 5fC and 5caC [14, 15].
In addition to DNA modifications, mutations of DNA bases that create mismatched base pairing are observed in vivo. Deamination of 5mC for instance [16], which either occurs spontaneously or is catalyzed by APOBEC3A, creates T:G mismatches [17]. To ensure proper genetic and epigenetic heritable information for subsequent cell generations, these mutated DNA bases must be repaired. One enzyme identified to be involved in T:G mismatch repair is Mbd4, a member of the MBD protein family that contains a glycosylase domain [18]. Following binding, the aberrant base is flipped out of the DNA helix and removed by Mbd4. The resulting abasic site is then repaired by enzymes of the base excision repair pathway [19].
To date, several methods are used to detect DNA base flipping, including X-ray crystallography [2] and nuclear magnetic resonance (NMR) spectroscopy [20]. Although X-ray crystallography provides the absolute proof of DNA base flipping, it is very demanding and time-consuming and depends on sophisticated equipment. The above makes it also not the method of choice to analyze the effect of mutations. The 2-aminopurine (2AP), an analog of the base adenine, has been used to detect base flipping in real time [21]. The fluorescence signal of 2AP is highly quenched when it is incorporated into DNA and located inside of the DNA helix. Conformational changes (bending/kinking) of the DNA increase 2AP fluorescence; however, the highest fluorescence intensities were observed when 2AP is flipped out of the DNA helix [22]. Moreover, several chemicals that react with flipped DNA bases have been identified. The resulting reaction products can be subsequently visualized by piperidine-induced strand cleavage and electrophoresis on denaturing gels [23–25]. Chloroacetaldehyde (CAA), for instance, reacts with the unpaired bases A and C to form 1,N6-ethenoadenine and 3,N4-ethenocytosine, respectively [26]. In combination with piperidine treatment, several Dnmts and endonucleases were successfully verified to use a base flipping mechanism to modify DNA bases [24]. Although M.HpaII belongs to the DNA methyltransferase family, several studies failed to detect that it induces base flipping [24], indicating that the current detection methods do not provide sufficient sensitivity and require optimization.
Since double-stranded DNA is mainly stabilized by base pairing between complementary strands, as well as base stacking interactions between adjacent bases [27], any DNA base modification would affect DNA double strand stability. To determine the thermostability of double-stranded DNA, the melting temperature measurement was developed. In particular, high-resolution melting (HRM) temperature analysis was shown to be a highly sensitive method that allows discrimination of a single base mutation within double-stranded DNA [28].
In this study, we developed a pipeline of methods to analyze every step of the DNA base flipping process. We apply these methods to the analysis of the cytosine base modifiers (M.HpaII and Tet1) and a methylcytosine reader and glycosylase (Mbd4). We show that our pipeline of methods is sensitive enough to analyze DNA binding, DNA stability, DNA base flipping and DNA base modification/excision induced by a variety of proteins involved in modifying and reading DNA bases.
Materials and methods
Step-by-step detailed protocols and materials for every assay are included in the Supplementary Data.
DNA preparation
DNA oligonucleotides were purchased from IBA (Germany) or IDT (Integrated DNA Technologies, Germany). To prepare fully-, hemi- or un-methylated double-stranded DNA, same molar of upper (CGup or MGup, 42-mer) and lower strand (Fill-In-ATTO550 or Fill-In-ATTO647N, 20-mer) were mixed in NEBuffer 2 (50 mM NaCl, 10 mM Tris–HCl, 10 mM MgCl2, 1 mM dithiothreitol; NEB, USA), denatured at 95 °C for 2 min and annealed by slowly cooling down to 37 °C. Then, the primer extension reactions were performed in the presence of 0.05 U/µl Klenow fragment, 1 mM dTTP, dGTP, dATP (PeqLab, Germany) and either 0.1 mM dCTP or dmCTP at 37 °C for 1 h. The methylation states of double-stranded DNA were confirmed by restriction enzyme MspI and HpaII digestion. To prepare double-stranded DNA containing T:G mismatch, same molar of upper (MGup, 42-mer) and lower strand (T:G mismatch DNA, 42-mer) were mixed in NEBuffer 2, denatured at 95 °C for 2 min and annealed by slowly cooling down to 37 °C.
PCR fragments were amplified from a plasmid containing a MINX sequence [29] using either dCTP or dmCTP as described [30].
Protein purification
GFP-, YFP-, mcherry- or His tagged proteins were purified from Sf9 insect cells as previously described [30]. For GFP and YFP purification, Ni-NTA beads coupled to GFP binding protein (GBP; [31]) were used. For mcherry purification, Ni-NTA beads coupled to RFP binding protein (RBP; [32]) were used. For His-tagged proteins, Talon beads (Clontech Laboratories, USA) were used. M.HpaII methyltransferase was purchased from NEB (cat. no. M0214S).
Electrophoretic mobility shift analysis
Purified proteins were incubated with the different fluorescently labeled oligonucleotides in binding buffer (20 mM HEPES, pH 7.9, 1 mM EDTA, 3 mM MgCl2, 2 mM DTT, 4% glycerol, and 0.1% Triton X-100) for 90 min at 37 °C for all proteins except Mbd4, which was incubated at 4 °C for 30 min. Complexes were analyzed on a nondenaturing 4.5% polyacrylamide gel (acrylamide/bis-acrylamide, 30% solution). Fluorescent signals (protein and DNA) were detected using a fluorescence imager (Amersham Imager 600 RGB) and also a fluorescence plate reader (TECAN Infinite® M200).
The fluorescent signals from the plate reader were plotted as a heatmap using a self-written R script (see Supplementary Data).
HRM temperature analysis
To detect DNA base flipping, we incubated the oligonucleotides with purified proteins at 37 °C for 2 h in the presence or absence of 2-CAA. After incubation, Platinum® SYBR® Green qPCR SuperMix-UDG (Invitrogen, cat. no. 11733046) was denatured and added to the sample. HRM temperature analysis was performed in a StepOnePlus™ Real-Time PCR machine (Applied Biosystems). Samples were first denatured at 95 °C for 30 s, then temperature was decreased to 50 °C with a decreasing rate of 2%, followed by increasing the temperature to 90 °C with 0.1 °C steps.
To detect base modification/excision, we incubated the oligonucleotides with purified proteins at 37 °C for 2 h. Then HRM temperature analysis was performed as just described.
Data normalization and visualization were performed using a self-written R script (see Supplementary Data).
Results and discussion
Protein–DNA binding assay
We hypothesized that three steps are involved in DNA base modification: (i) protein–DNA binding, (ii) base flipping, and (iii) base modification itself (Fig. 1). Therefore, we first aimed to understand how proteins interact with DNA, a process that is also critical for basic biological processes such as DNA replication, repair, and transcription in vivo. To better understand these processes, methods allowing accurate detection of protein–DNA interactions are essential. In the past few decades, several methods have been developed to detect protein–DNA interactions [33]. In particular, electrophoretic mobility shift assays (EMSAs) showed to be a robust and sensitive method [34]. Classical EMSAs use radioactively labeled DNA to visualize protein–DNA complexes, whereby proteins in the complex are detected only indirectly (Fig. 2A). However, to better understand the composition of protein–DNA complexes it is very important to directly detect both, protein and DNA signals. Therefore, we optimized multicolor EMSAs, which allows simultaneous detection of differently labeled DNA oligonucleotides and proteins. Because large complexes composed of multiple proteins bound to one DNA molecule cannot easily enter the gel, the fluorescence signals in the gel pockets were also included in our detection (Fig. 2A, right lane 5). Through detection of all complexes and complex components, new potential applications, such as protein competition assays and competitive binding to different DNA species can be implemented (Fig. 2A, right lane 5) [30]. Here, we use multicolor EMSA to directly test the specificity of Tet1 for differently modified DNA substrates in one reaction, which would have not been possible with classical radioactive EMSAs.
Figure 1:

Stepwise dissection of DNA base flipping. Three steps: (i) protein–DNA binding, (ii) base flipping, and (iii) base modification are involved in DNA base modification or repair. A: adenine, T: thymine, C: cytosine, G: guanine. Tet: Tet-eleven translocation protein. M.HpaII: HpaII methyltransferase. Mbd4: Methyl-CpG binding domain protein 4. The green shape represents protein, which binds, flips, and modifies DNA bases.
Figure 2:

Binding specificity of Tet1 to DNA. (A) Comparison of conventional EMSA (left, lanes 1–3) and multicolor EMSA (right lanes 1–5). Left lane 1: protein with isotope labeled DNA. Left lane 2: protein with isotope labeled DNA and competitor DNA. Left lane 3: isotope labeled DNA. Right lane 1: fluorescently tagged protein with fluorescently labeled DNA. Right lane 2: fluorescently tagged protein with fluorescently labeled DNA and competitor DNA. Right lane 3: fluorescently labeled DNA. Right lane 4: fluorescently tagged protein with two different fluorescently labeled DNAs. Right lane 5: Two different fluorescently tagged proteins with fluorescently labeled DNAs. (B) Binding specificity of Tet1CD (catalytic domain of Tet1) and MBD of Mecp2 (methyl-CpG binding protein 2) to methylated DNA. Purified mcherry-Tet1CD (2 µM) (magenta) was incubated with a 42-bp long oligonucleotide (1 µM) containing a symmetrically methylated CpG site (black) at 37 °C. After 90 min, free DNA and DNA-Tet1CD complexes were separated on a native polyacrylamide gel. As a control, the MBD protein (green, 2 µM) of Mecp2 was used. Minus and plus indicate cathode and anode, respectively. The fluorescence intensities were detected on a fluorescence microplate reader (see “Materials and methods” section) and intensities were plotted as line plots and heatmaps using RStudio. The upper part shows binding of Tet1CD (left) and the MBD of Mecp2 (right) to methylated DNA. The lower part shows binding of Tet1CD (left) or MBD (right) to methylated DNA in the presence of competitor DNA poly(dI:dC) (grey, 200 ng). Independent experiments were repeated twice. Shown is one representative result. A summary of the binding specificity of Tet1CD and MBD is shown at the bottom of the figure.
The preparation of fluorescently labeled DNA oligonucleotides, purification of fluorescently tagged proteins (Supplementary Fig. S1) as well as the detailed multicolor EMSAs protocol are described in the “Materials and methods” section. To detect signals of fluorescently labeled proteins and DNA oligonucleotides, we initially used a fluorescence imager with 460 nm, 520 nm, and 630 nm excitation wavelengths, as well as 525BP20, 605BP40, and 705BP40 emission filters. However, since the filter settings were not suitable to discriminate yellow fluorescent protein (YFP) and the mcherry protein signals (Supplementary Fig. S2A), we tested and developed a method to detect any combination of fluorescence signals on the gel using a fluorescence microplate reader, which gives full spectral excitation and emission flexibility. Moreover, this detection way immediately provides intensity values without the need for image analysis, which is a must when using fluorescence imagers. A detailed description on how to image polyacrylamide gels on a fluorescence microplate reader can be found in the “Materials and methods” section (Supplementary Fig. S2B-2F) [30].
Since the catalytic domain of Tet1 (Tet1CD) is sufficient for 5mC to 5hmC conversion [30,35], we were interested in how Tet1CD interacts with DNA. To this end, we incubated purified, mcherry-tagged Tet1CD proteins with ATTO-647N labeled DNA oligonucleotides containing a single fully methylated CpG site for 90 min at 37 °C. Protein–DNA complexes and free DNA were then separated on a native polyacrylamide gel. As a control, the methyl-CpG binding domain (MBD) of Mecp2, which was shown to have a preference for methylated CpG dinucleotides [30] was used. As shown in Fig. 2B, both proteins formed complexes with methylated DNA under the given reaction conditions (Fig. 2B, upper row). To further test the specificity of Tet1CD proteins to methylated DNA, we incubated both proteins with methylated DNA in the presence of unmethylated competitor DNA poly(dI:dC). In contrast to the MBD, Tet1CD did not form a complex with methylated DNA in the presence of poly(dI:dC) (Fig. 2B, lower row) indicating that Tet1CD interacts with DNA irrespective of its methylation status.
To further confirm and extend these data, we incubated His-tagged Tet1CD or YFP-tagged MBD proteins with DNA oligonucleotides containing a single fully methylated CpG (ATTO647N labeled) together with either a single unmethylated CpG (ATTO550 labeled) or a non-CpG (ATTO550 labeled). After 90 min of incubation at 37 °C, DNA–protein complexes were separated on a native polyacrylamide gel. As shown in Fig. 3, Tet1CD formed complexes with methylated and unmethylated oligonucleotides corroborating the previous results using poly(dI:dC) as competitor. When compared to MBD proteins, which have a preference for methylated DNA, Tet1CD showed only a slight preference for methylated oligonucleotides. Moreover, Tet1CD exhibited also slightly higher binding affinities to methylated CpG DNA compared to non-CpG DNA (Supplementary Fig. S3). These results indicate that although 5mC is a substrate of Tet, it does not significantly enhance Tet binding to DNA, whereas it does enhance the binding of MBD proteins.
Figure 3:

Binding specificity of Tet1CD and MBD of Mecp2 to methylated and unmethylated DNA. Purified His-Tet1CD or YFP-MBD was incubated with methylated CpG and unmethylated CpG DNA. After 90 min of incubation at 37 °C, free DNA and DNA–protein complexes were separated on a 4.5% native polyacrylamide gel. To quantify the binding, the amounts of unbound DNA (intensities of free DNA bands measured using Image J) were plotted below the gel. Independent experiments were repeated twice. Shown is one representative result. mC:mC, fully methylated CpG containing DNA; C:C, unmethylated CpG containing DNA. Free DNA and protein–DNA complexes are indicated on the gel.
Multicolor fluorescently tagged proteins and fluorescently labeled DNA have already been used to detect protein and DNA interactions. To detect fluorescence intensities, previous studies used fluorescence spectrophotometry analysis [37]. In addition, multicolor EMSAs were also used to measure fluorescence intensity and to determine the specificity of proteins to multiple DNA substrates [38]. However, the simultaneous detection of multiple fluorescence signals is often limited, since excitation and emission filters are not easily exchangeable in commercial fluorescence imagers. Our novel detection method, which is based on a fluorescence multiwell plate reader, however, allows detection of most commercially available fluorescent dyes without spectral bleed-through and offers full freedom to choose dyes for protein or DNA labeling.
DNA base flipping detection assay
After protein–DNA complex formation, most DNA base modifiers or excision proteins use a base flipping mechanism to access their DNA substrate (Fig. 1).
M.HhaI crystal structures showed that base flipping occurs in both, ternary complexes including DNA, the methyltransferase and its cofactor, as well as in binary complexes lacking its cofactor [20,22], indicating that the cofactor itself is not crucial for the base flipping reaction. Moreover, previous studies showed that M.HhaI induced base flipping can be detected only in the absence of its cofactor by CAA treatment [24], suggesting that the cofactor blocks the reaction of CAA with the flipped base. Until now, CAA has been used in combination with piperidine (which cleaves modified nucleotides such as 3,N4-ethenocytosine) to detect base flipping in various systems due to its high reactivity with unpaired cytosines. The product of the reaction, 3,N4-ethenocytosine, disrupts hydrogen bonds with guanine and decreases DNA double strand stability [39,40] (Fig. 4A). Since HRM analysis is used to measure DNA thermostability, we tested whether a novel combination of CAA treatment and HRM analysis could be used to detect DNA base flipping.
Figure 4:

Detection of HpaII methyltransferase (M.HpaII) mediated DNA base flipping. (A) 0.5 µM of hemimethylated or non-CCGG containing DNA was incubated with or without 1.25 pmol (4 units) of M.HpaII in the presence of 30 mM CAA. CAA reacts with cytosine when it is flipped out of the DNA helix. The reaction product 3,N4-ethenocytosine (εC) disrupts hydrogen bonds with the base guanine in the complementary strand. The presence of 3,N4-ethenocytosine (εC) can be detected with HRM analysis, due to its low Tm contribution to double-stranded DNA. (B) Normalized SYBR green fluorescence intensities (left) and the corresponding derivative of intensities (right) were plotted using RStudio. Independent experiments were repeated two times. Shown is one representative result with three technical replicates. The P-values of student’s t-test are indicated in the plot. Tm: melting temperature. C: cytosine; G: guanine; M:C: hemimethylated DNA; X:X: non-CpG containing DNA.
Since CAA reacts with both, adenosine and cytosine in single-stranded DNA, which is produced during HRM analysis, we first tested the effect of different CAA concentrations on the thermostability of double-stranded DNA under the HRM analysis conditions. Therefore, we incubated hemimethylated DNA with varying amounts of CAA and performed HRM analysis 1 h after incubation at 37 °C. As shown in Supplementary Fig. S4A, under our assay condition the highest CAA concentration, where most of the double-stranded DNA was still intact, was 50 mM. Next, we incubated hemimethylated DNA with CAA concentrations between 0 mM and 50 mM in the presence or absence of M.HpaII (without the cofactor SAM), respectively. Seventy minutes after incubation at 37 °C, the HRM analysis was performed (Supplementary Fig. S4B). We observed the largest melting temperature (Tm) differences between M.HpaII treated and untreated DNA in the presence of 30 mM CAA (Supplementary Fig. S4B). Furthermore, we observed no detectable Tm differences with high CAA concentrations (40 mM and 50 mM) (Supplementary Fig. S4B). However, with low CAA concentrations, the Tm differences correlated with CAA amounts (Supplementary Fig. S4B). Thus, we found 30 mM CAA to be the best condition for the detection of cytosine flipping. To further validate this result, we used a control oligonucleotide, which does not contain the M.HpaII recognition motif CCGG, for which M.HpaII showed a slightly lower binding preference than for CCGG containing DNA (Supplementary Fig. S4C). As shown in Fig. 4B, a similar melting temperature was observed for the non-CCGG containing oligonucleotide in the presence of M.HpaII and CAA. In contrast, hemimethylated CCGG containing oligonucleotide showed a decreased melting temperature with M.HpaII incubation. These results indicate that the CAA reaction only takes place when M.HpaII recognizes its substrate. Previous studies [24] using CAA concentrations >50 mM could not detect M.HpaII-induced DNA base flipping and accordingly claim that CAA disrupts protein–DNA complexes, which might already be the case at our highest tested concentrations (40 mM and 50 mM). In summary, we showed that the combination of CAA treatment and HRM analysis can be used to detect base flipping induced by Dnmts, such as M.HpaII, which until now could not be shown by any other methods.
Next, we assayed Tet1-mediated DNA base flipping using CAA treatment and HRM analysis. We first tested the complex formation ability of Tet1 and DNA in the presence of 30 mM CAA. As shown in Supplementary Fig. S5A, binding of Tet1 to DNA is not affected by the presence of CAA. Therefore, we next incubated hemimethylated DNA with or without Tet1 proteins (without cofactors Fe(II) and 2-oxoglutarate) in the presence of 30 mM or 0 mM of CAA. After 2 h of incubation, the HRM analysis was performed (Fig. 5A). Similar to M.HpaII, the Tm values decreased with Tet1 incubation in the presence of 30 mM CAA (Supplementary Fig. S5B). To further test whether the CAA reaction is specific for flipped 5mC, we incubated a hemimethylated CpG or non-CpG containing DNA with the Tet1 protein in the presence of 30 mM CAA. As shown in Fig. 5B, a decrease Tm value was also observed for non-CpG containing DNA raising the possibility that Tet flips other bases. However, the Tm decrease for non-CpG DNA is smaller than for CpG containing DNA. These results indicate that the combination of CAA and HRM can be used to detect Tet protein-induced base flipping. In summary, the combination of CAA treatment and HRM analysis can be used to detect DNA base flipping induced by M.HpaII and Tet1.
Figure 5:

Detection of Tet1 mediated DNA base flipping. (A) 0.5 µM of hemimethylated or non-CCGG containing DNA was incubated with or without 2 µM of the catalytic domain of Tet1 (Tet1CD) in the presence of 30 mM CAA. CAA reacts with 5mC when it is flipped out of the DNA helix. The reaction product 3,N4-ethenomethylcytosine (εmC) disrupts hydrogen bonds with base guanine in the complementary strand. The presence of 3,N4-ethenomethylcytosine (εmC) can be detected with HRM analysis, due to its low Tm contribution to double-stranded DNA. (B) Normalized fluorescent SYBR green intensities (left) and the corresponding derivative of intensities (right) were plotted using RStudio. Independent experiments were repeated two times. Shown is one representative result with three averaged technical replicates. The P-values of student’s t-test are indicated in the plot. Tm: melting temperature. C: cytosine; G: guanine; M:C: hemi methylated DNA; X:X: non-CpG containing DNA.
DNA base excision detection assay
As previously mentioned, the glycosylase Mbd4 also uses a DNA base flipping mechanism to excise mismatched DNA bases. The base 2-aminopurine (2AP) has previously been used to detect base flipping because it fluoresces when not base paired. Since Mbd4 excises mismatched DNA bases, we tested whether this simpler method could be used to measure base flipping or base excision. We first tested the fluorescence measurement of 2AP with 320 nm excitation and 370 nm emission wavelengths using a fluorescence micro plate reader. As shown in Supplementary Fig. S6A, 0.4 µM of single-stranded oligonucleotide containing 2AP showed higher fluorescence intensity than an oligonucleotide containing canonical DNA bases. Then, we further incubated Mbd4 proteins (Supplementary Fig. S6D) with a double-stranded DNA oligonucleotide containing a 2AP:G mismatch (Supplementary Fig. S6B and Fig. S6C) in the context of a methylated CpG site and measured the 2AP fluorescence intensity (Supplementary Fig. S7A). No increased fluorescence intensity for oligonucleotides containing 2AP was observed in the presence of Mbd4 (Supplementary Fig. S7A). Previous studies showed that Mbd4 preferentially binds to DNA containing a T:G mismatch (Supplementary Fig. S8) which serves as its substrate [18, 41]. To further test whether 2AP could be excised by Mbd4, we performed a Mbd4 glycosylase activity assay by running a denaturing gel using a T:G mismatch containing oligo as a control. As shown in Supplementary Fig. S7B, Mbd4 showed a glycosylase activity for the T:G mismatched oligo but not for the 2AP:G mismatched oligo. Since Mbd4 can excise the mismatched base T and thereby change the melting temperature of the DNA, we performed HRM analysis to test whether it could be used to detect Mbd4-induced base excision. As shown in Fig. 6, a small but significantly decreased Tm was observed for DNA containing mismatched T:G with Mbd4 incubation (Fig. 6A), but not for hemimethylated DNA (Fig. 6B). These results indicate that in addition to denaturing gel analysis, HRM can be used to detect Mbd4-induced base excision.
Figure 6:

Detection of Mbd4 mediated DNA base excision. Detection of Mbd4 mediated base excision using 42 bp oligonucleotides containing T:G mismatch in the context of a methylated CpG (A). As a control, hemimethylated oligonucleotides were used (B). In all, 1 µM of oligonucleotides was incubated with 2 µM of Mbd4 for 120 min at 37 °C. After incubation, the samples were incubated at 50 °C for 1 h in the presence of 20 µg proteinase K. Then, HRM analysis was performed. Solid and dashed lines indicate incubation of DNA with or without Mbd4, respectively. Independent experiments were repeated at least three times. Shown is one representative result with four technical replicates. The P-values of student’s t-test are indicated in the plot. T: thymine; C: cytosine.
DNA base modification detection assay
Tet proteins oxidize 5mC to 5hmC in the presence of their cofactors Fe(II) and 2-oxoglutarate [8, 9]. Recent studies have shown that similar to 5mC, 5hmC is a stable epigenetic mark [42]. To better understand the function of 5hmC, several methods have been developed to quantify the amount of 5hmC [43]. The classical method used to measure 5mC in the past few decades is bisulfite conversion sequencing; however, this method cannot discriminate 5mC from 5hmC [44]. Fortunately, several variants capable of discriminating 5mC from 5hmC [45, 46] and even from 5fC [47] and 5caC [48] have been developed and successfully used to map the different DNA modifications across the genome. Besides sophisticated sequencing methods, restriction enzymes, which specifically digest 5mC/5hmC modified DNA were discovered [49] and antibodies, that specifically recognize DNA base modifications, have been developed. However, all of the above mentioned methods are time-consuming and some of them, like antibody-based approaches, are restricted by their respective detection sensitivity [50].
Previous studies showed that 5mC increases DNA double strand stability by strengthening base stacking interactions [51]. Here, we tested whether modifications of 5mC affect DNA thermostability by HRM analysis. In accordance with previous studies, we found that 5hmC containing DNA exhibits lower Tm values compared to 5mC containing DNA (Supplementary Fig. S9A) [52–54]. As with 5hmC, 5fC, and 5caC containing DNA exhibited lower Tm values (Supplementary Fig. S9A) when compared to methylated DNA and, accordingly, decreased DNA double strand thermostability. The observation that the oxidized derivatives of 5mC decrease DNA thermostability implies that Tet-mediated 5mC oxidation can be detected indirectly by measuring DNA thermostability.
As we have shown above, the HRM analysis can be used to measure DNA double strand thermostability. We first tested the effect of oligonucleotide length (10, 20, and 42 bp) on the melting temperature of unmodified oligonucleotides. We found that 20 and 42 bp oligonucleotides gave melting temperatures >50 °C (Supplementary Fig. S9B). We next assayed Tet1 oxidation using the 42-bp oligonucleotide by HRM analysis. We incubated fully methylated 42 bp oligonucleotides with Tet1 proteins in the presence of their cofactors. As a control, reactions were prepared in duplicates, whereby one sample contained and the other one lacked Tet1. After 2 h of incubation at 37 °C, the HRM analysis was performed. We measured a decreased Tm value of the Tet1 modified versus the unmodified oligonucleotide (Supplementary Fig. S9C). This difference was very small (0.23 °C). As one strategy to enhance this Tm difference, we tested addition of a shorter 20-mer oligonucleotide with the same sequence as the middle part of the 42-bp oligonucleotide before running the HRM analysis (Supplementary Fig. S9D). As shown in Supplementary Fig. S9D, DNA incubated with Tet1 proteins exhibited 0.6 °C lower Tm values than DNA incubated without Tet1 enzymes. Since we showed that Tet oxidation products decrease Tm values of double-stranded DNA (Supplementary Fig. S9A), we conclude that Tet-mediated 5mC oxidation can be detected by HRM analysis. Considering that Tet proteins produce a mixture of 5mC modifications with variant Tm values (Supplementary Fig. S9A), the expected melting curve would have multiple peaks. To test the behavior of DNA mixtures with different Tm values, we performed HRM analysis with different ratios of unmethylated and fully methylated DNA. As depicted in Supplementary Fig. S9E, only one peak could be detected for each DNA mixture; however, Tm values increased with the amount of methylated DNA. This result indicates that the measured Tm values represent the Tm of a DNA mixture, where the amount of each DNA modification affects the observed Tm. Taken together, we show that HRM analysis is sensitive enough to detect Tet-mediated 5mC oxidation activity.
As mentioned in the introduction, deamination of one 5mC at symmetrically methylated CpG sites creates T:G mismatches in vivo. Up until now it is not known whether Tet proteins can act on the opposite non-deaminated 5mC. To test this, we incubated DNA oligonucleotides containing a T:G mismatch in the context of a methylated CpG site with Tet1 proteins and performed the HRM analysis. As shown in Supplementary Fig. S9F, we found a 0.3 °C decreased Tm value in the presence of Tet1, indicating that Tet1 proteins oxidize 5mC in mismatched CpG sites.
Tet-mediated 5mC oxidation is a Fe(II) and 2-oxoglutarate dependent process. Besides these two cofactors, DTT and ATP were reported to be crucial for Tet1 enzymatic activity [55]. To test whether our assay can be used to detect the effect of cofactors on Tet1 activity, we incubated fully methylated oligonucleotides with Tet1 proteins in oxidation buffer with or without ATP and DTT. After the oxidation reaction, the HRM analysis was performed. As shown in Supplementary Fig. S10A, Tet1 did not alter Tm values of DNA when incubated in oxidation buffer without DTT and ATP. In the presence of ATP and DTT, in contrast, the Tm of DNA was decreased (by 0.6 °C) in the presence of Tet1 (Supplementary Fig. S10B), indicating that DTT and ATP are crucial for Tet1-mediated oxidation and HRM analysis can be used to test Tet activity.
Since only one symmetrically methylated CpG is present in the 42-bp oligonucleotide and the decreased Tm after Tet1 incubation is, therefore, small, we next tested whether shorter oligonucleotides would lead to greater Tm differences. We chose a 20-mer long oligonucleotide as this still gave rise to a Tm >50 °C (Supplementary Fig. S9A). As shown in Fig. 7A, the Tm difference between the 20 bp DNA with or without Tet is 1 °C, which is higher than the 0.6 °C for the 42-bp DNA.
Figure 7:

Detection of Tet1-mediated 5mC oxidation. (A) Detection of Tet oxidation activity using 20 bp oligonucleotides. In all, 0.5 µM of fully methylated 20 bp DNA was incubated with 2 µM of Tet1CD. After 2 h incubation, DNA was used for HRM analysis. Independent experiments were repeated twice. Shown is one representative result with three technical replicates. The P-values of student’s t-test are indicated in the plot. (B) Detection of Tet oxidation activity using a PCR fragment containing multiple methylated cytosines. In all, 50 ng of 377 bp methylated PCR fragment was incubated with 2 µM of Tet1CD. After 2 h incubation, DNA was used for HRM analysis. Independent experiments were repeated twice. Shown is one representative result with four technical replicates. hmC: 5-hydroxymethylcytosine; fC: 5-Formylcytosine; caC: 5-carboxylcytosine.
To further assess whether the HRM could be used to detect Tet activity in multiple 5mC containing DNA, a 377-bp PCR fragment was incubated with Tet1 and subjected to HRM analysis. This amplicon contains 189 5mC and 42 mCpG sites. As shown in Fig. 7B, although the Tm values vary between reactions possibly due to the varying levels of 5mC oxidation, a large Tm shift was observed (around 3 °C) due to Tet activity.
In summary, these results indicate that HRM can be used to test Tet oxidation activity in vitro even with a single (5mC) base resolution.
Prior to 5mC oxidation by Tet enzymes, cytosine methylation is required and performed by Dnmts. The HpaII methyltransferase is one of these enzymes and methylates the internal cytosine in CCGG sequence (Supplementary Fig. S11A). As methylation of cytosine increases DNA Tm values (Supplementary Fig. S9E), we further tested whether the HRM analysis could be used to detect methyltransferase activity. To this end, we incubated M.HpaII and unmethylated CCGG containing oligonucleotide in the presence of its cofactor SAM. Two hours after incubation, the reactions were directly used for HRM analysis. As shown in Supplementary Fig. S11B, a significantly increased Tm (0.3 °C) was observed with M.HpaII incubation compared to non-M.HpaII incubated DNA. In addition, similar to Tet oxidation, the Tm difference (1.3 °C) is larger when a 20-bp oligonucleotide was used (Fig. 8, compare red line without enzyme with blue line with 2 units of enzyme). These results indicate that the HRM can be used to detect methyltransferase activity in vitro.
Figure 8:
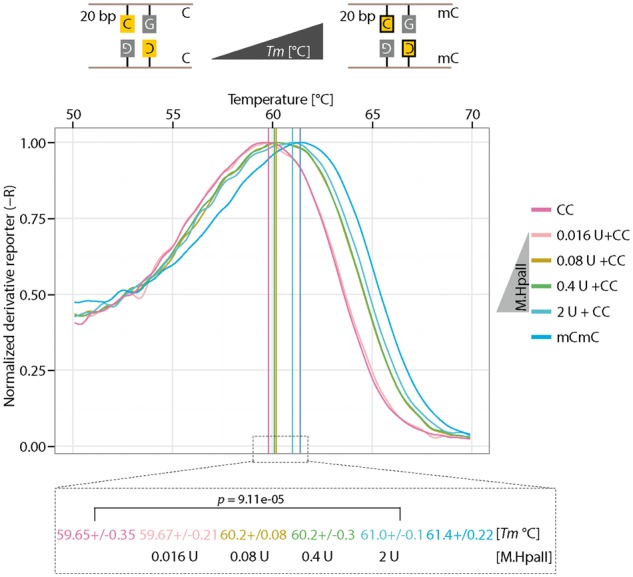
Detection of methyltransferase activity. Detection of M.HpaII activity using 20 bp oligonucleotides containing an unmethylated CpG. 0.1 µM of unmethylated DNA was incubated with varying amount of M.HpaII for 120 min at 37 °C. After incubation, HRM analysis was performed. Independent experiments were repeated at least two times. Shown is one representative result with three technical replicates. The P-values of student’s t-test are indicated in the plot. C: cytosine; U: M.HpaII units.
Compared to antibody-based assays, the HRM detection of either methyltransferase activity or Tet-mediated 5mC oxidation activity is time saving and highly reproducible. A single modification in DNA is usually difficult to detect by antibodies. HRM analysis, however, is sensitive enough to discriminate even single modifications in DNA oligonucleotides. Altogether, we show that HRM analysis can be used to test Tet-mediated 5mC oxidation and methyltransferase activity in vitro.
Conclusions
Accurate detection of DNA binding, base flipping and modification is crucial to understand the function and regulation of various proteins, such as methylcytosine writers, readers, and modifiers. Here, we present methods to step by step detect Tet1 binding, flipping, and oxidation of 5mC (Fig. 9). Multicolor EMSAs can be used to measure substrate preferences of multiple proteins to multiple oligonucleotides in one reaction. However, the analysis of DNA protein interactions is often limited, since conventional fluorescence imagers contain fixed excitation and emission filters that allow the detection of only some color combinations. Thus, we developed a method for the simultaneous detection of various commercially available fluorescent proteins/dyes on a fluorescence plate reader without spectral bleed-through. As a result, detection optimization allowed us to quantitate the binding of Tet1 proteins to differentially labeled DNA substrates by EMSAs and we found that Tet1CD proteins exhibited little DNA sequence specificity with only a slight preference for methylated CpG containing DNA.
Figure 9:

Summary of the different methods used for detection of DNA–protein interactions, DNA base flipping and DNA base modifications. A: adenine; T: thymine; C: cytosine; G: guanine. HRM temperature analysis. The green shape represents protein, which binds, flips, and modifies or excises DNA bases.
Currently used methods for the detection of base flipping are costly and time consuming. Here, we developed a rapid and sensitive assay, which combines CAA treatment and HRM temperature analysis to assay proteins that use a base flipping mechanism. With this combined method, we could show that Tet1 and also M.HpaII flip its substrate for subsequent modification. The base flipping activity of the latter had so far escaped detection with currently available methods.
For the detection of base excision by Mbd4 glycosylase, denaturing gel analysis has been thus far the method of choice. We show here that HRM analysis can be used to detect a single abasic site and, thus, is an alternative sensitive method to assay glycosylase activity.
Finally, we show that HRM analysis can be used to detect Tet activity under different buffer conditions and can further be extended to methyltransferase activity. Accordingly, this method is particularly suitable to screen different cofactors and inhibitors that facilitate or impede Tet or methyltransferase activity.
In conclusion, all of the above methods are easily performed, highly reproducible, and are suitable for the detection of DNA base modifications, their readers, and modifiers.
Supplementary Material
Acknowledgments
We thank Heinrich Leonhardt laboratory for providing DNA oligonucleotides containing 5mC, 5hmC, 5fC, and 5caC. We also thank Cathia Rausch for critical reading of the article and Henry D. Herce and Heinrich Leonhardt for helpful suggestions. Author contributions: P.Z., F.D.H., A.K.L., K.B., and M.H. performed experiments. P.Z. analyzed the data. P.Z. and M.C.C. conceived the project and P.Z., F.D.H., A.K.L., and M.C.C. wrote the article.
Supplementary data
Supplementary data is available at Biology Methods and Protocols online.
Conflict of interest statement. None declared.
Funding
P.Z. received a fellowship of the China Scholarship Council. This work was supported in part by grants of the Deutsche Forschungsgemeinschaft DFG CA 198/7 and DFG CA 198/10 to M.C.C.
References
- 1. Bestor T, Laudano A, Mattaliano R. et al. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol 1988;203:971–83. [DOI] [PubMed] [Google Scholar]
- 2. Klimasauskas S, Kumar S, Roberts RJ. et al. HhaI methyltransferase flips its target base out of the DNA helix. Cell 1994;76:357–69. [DOI] [PubMed] [Google Scholar]
- 3. Reinisch KM, Chen L, Verdine GL. et al. The crystal structure of HaeIII methyltransferase convalently complexed to DNA: an extrahelical cytosine and rearranged base pairing. Cell 1995;82:143–53. [DOI] [PubMed] [Google Scholar]
- 4. Goedecke K, Pignot M, Goody RS. et al. Structure of the N6-adenine DNA methyltransferase M.TaqI in complex with DNA and a cofactor analog. Nat Struct Biol 2001;8:121–5. [DOI] [PubMed] [Google Scholar]
- 5. Yang Z, Horton JR, Zhou L. et al. Structure of the bacteriophage T4 DNA adenine methyltransferase. Nat Struct Biol 2003;10:849–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horton JR, Liebert K, Bekes M. et al. Structure and substrate recognition of the Escherichia coli DNA adenine methyltransferase. J Mol Biol 2006;358:559–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Song J, Teplova M, Ishibe-Murakami S. et al. Structure-based mechanistic insights into DNMT1-mediated maintenance DNA methylation. Science 2012;335:709–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kriaucionis S, Heintz N.. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009;324:929–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tahiliani M, Koh KP, Shen Y. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009;324:930–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pfaffeneder T, Hackner B, Truss M. et al. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew Chem Int Ed Engl 2011;50:7008–12. [DOI] [PubMed] [Google Scholar]
- 11. Ito S, Shen L, Dai Q. et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011;333:1300–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hu L, Li Z, Cheng J. et al. Crystal structure of TET2-DNA complex: insight into TET-mediated 5mC oxidation. Cell 2013;155:1545–55. [DOI] [PubMed] [Google Scholar]
- 13. Hashimoto H, Pais JE, Zhang X. et al. Structure of a Naegleria Tet-like dioxygenase in complex with 5-methylcytosine DNA. Nature 2014;506:391–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hashimoto H, Pais JE, Dai N. et al. Structure of Naegleria Tet-like dioxygenase (NgTet1) in complexes with a reaction intermediate 5-hydroxymethylcytosine DNA. Nucleic Acids Res 2015;43:10713–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hu L, Lu J, Cheng J. et al. Structural insight into substrate preference for TET-mediated oxidation. Nature 2015;527:118–22. [DOI] [PubMed] [Google Scholar]
- 16. Duncan BK, Miller JH.. Mutagenic deamination of cytosine residues in DNA. Nature 1980;287:560–1. [DOI] [PubMed] [Google Scholar]
- 17. Wijesinghe P, Bhagwat AS.. Efficient deamination of 5-methylcytosines in DNA by human APOBEC3A, but not by AID or APOBEC3G. Nucleic Acids Res 2012;40:9206–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hendrich B, Hardeland U, Ng HH. et al. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature 1999;401:301–4. [DOI] [PubMed] [Google Scholar]
- 19. Krokan HE, Nilsen H, Skorpen F. et al. Base excision repair of DNA in mammalian cells. FEBS Lett 2000;476:73–7. [DOI] [PubMed] [Google Scholar]
- 20. Klimasauskas S, Szyperski T, Serva S. et al. Dynamic modes of the flipped-out cytosine during HhaI methyltransferase-DNA interactions in solution. EMBO J 1998;17:317–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Neely RK, Daujotyte D, Grazulis S. et al. Time-resolved fluorescence of 2-aminopurine as a probe of base flipping in M.HhaI-DNA complexes. Nucleic Acids Res 2005;33:6953–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Holz B, Klimasauskas S, Serva S. et al. 2-Aminopurine as a fluorescent probe for DNA base flipping by methyltransferases. Nucleic Acids Res 1998;26:1076–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Serva S, Weinhold E, Roberts RJ. et al. Chemical display of thymine residues flipped out by DNA methyltransferases. Nucleic Acids Res 1998;26:3473–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Daujotyte D, Liutkeviciute Z, Tamulaitis G. et al. Chemical mapping of cytosines enzymatically flipped out of the DNA helix. Nucleic Acids Res 2008;36:e57.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zagorskaite E, Sasnauskas G.. Chemical display of pyrimidine bases flipped out by modification-dependent restriction endonucleases of MspJI and PvuRts1I families. PLoS One 2014;9:e114580.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kusmierek JT, Singer B.. Chloroacetaldehyde-treated ribo- and deoxyribopolynucleotides. 1. Reaction products. Biochemistry 1982;21:5717–22. [DOI] [PubMed] [Google Scholar]
- 27. Yakovchuk P, Protozanova E, Frank-Kamenetskii MD.. Base-stacking and base-pairing contributions into thermal stability of the DNA double helix. Nucleic Acids Res 2006;34:564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Taylor CF. Mutation scanning using high-resolution melting. Biochem Soc Trans 2009;37:433–7. [DOI] [PubMed] [Google Scholar]
- 29. Zillmann M, Zapp ML, Berget SM.. Gel electrophoretic isolation of splicing complexes containing U1 small nuclear ribonucleoprotein particles. Mol Cell Biol 1988;8:814–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ludwig AK, Zhang P, Hastert FD. et al. Binding of MBD proteins to DNA blocks Tet1 function thereby modulating transcriptional noise. Nucleic Acids Res 2017;45:2438–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rothbauer U, Zolghadr K, Muyldermans S. et al. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 2008;7:282–9. [DOI] [PubMed] [Google Scholar]
- 32. Pichler G, Leonhardt H, Rothbauer U.. Fluorescent protein specific Nanotraps to study protein-protein interactions and histone-tail peptide binding. Methods Mol Biol 2012;911:475–83. [DOI] [PubMed] [Google Scholar]
- 33. Dey B, Thukral S, Krishnan S. et al. DNA-protein interactions: methods for detection and analysis. Mol Cell Biochem 2012;365:279–99. [DOI] [PubMed] [Google Scholar]
- 34. Hellman LM, Fried MG.. Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc 2007;2:1849–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang P, Rausch C, Hastert FD. et al. Methyl-CpG binding domain protein 1 regulates localization and activity of Tet1 in a CXXC3 domain-dependent manner. Nucleic Acid Res 2017;45:7178–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Muller U, Bauer C, Siegl M. et al. TET-mediated oxidation of methylcytosine causes TDG or NEIL glycosylase dependent gene reactivation. Nucleic Acids Res 2014;42:8592–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Frauer C, Leonhardt H.. A versatile non-radioactive assay for DNA methyltransferase activity and DNA binding. Nucleic Acids Res 2009;37:e22.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Spruijt CG, Gnerlich F, Smits AH. et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 2013;152:1146–59. [DOI] [PubMed] [Google Scholar]
- 39. Sági J, Perry A, Hang B. et al. Differential destabilization of the DNA oligonucleotide double helix by a T.G mismatch, 3,N(4)-ethenocytosine, 3,N(4)-ethanocytosine, or an 8-(hydroxymethyl)-3,N(4)-ethenocytosine adduct incorporated into the same sequence contexts. Chem Res Toxicol 2000;13:839–45. [DOI] [PubMed] [Google Scholar]
- 40. Wang G, Dunman PM, Humayun MZ.. Replication of M13 single-stranded DNA bearing a site-specific ethenocytosine lesion by Escherichia coli cell extracts. Cell Res 1997;7:1–12. [DOI] [PubMed] [Google Scholar]
- 41. Hashimoto H, Zhang X, Cheng X.. Excision of thymine and 5-hydroxymethyluracil by the MBD4 DNA glycosylase domain: structural basis and implications for active DNA demethylation. Nucleic Acids Res 2012;40:8276–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bachman M, Uribe-Lewis S, Yang X. et al. 5-Hydroxymethylcytosine is a predominantly stable DNA modification. Nat Chem 2014;6:1049–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Plongthongkum N, Diep DH, Zhang K.. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet 2014;15:647–61. [DOI] [PubMed] [Google Scholar]
- 44. Huang Y, Pastor WA, Shen Y. et al. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One 2010;5:e8888.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Booth MJ, Branco MR, Ficz G. et al. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012;336:934–7. [DOI] [PubMed] [Google Scholar]
- 46. Yu M, Hon GC, Szulwach KE. et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 2012;149:1368–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Booth MJ, Marsico G, Bachman M. et al. Quantitative sequencing of 5-formylcytosine in DNA at single-base resolution. Nat Chem 2014;6:435–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lu X, Song CX, Szulwach K. et al. Chemical modification-assisted bisulfite sequencing (CAB-Seq) for 5-carboxylcytosine detection in DNA. J Am Chem Soc 2013;135:9315–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Szwagierczak A, Brachmann A, Schmidt CS. et al. Characterization of PvuRts1I endonuclease as a tool to investigate genomic 5-hydroxymethylcytosine. Nucleic Acids Res 2011;39:5149–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang Y, Pastor WA, Zepeda-Martinez JA. et al. The anti-CMS technique for genome-wide mapping of 5-hydroxymethylcytosine. Nat Protoc 2012;7:1897–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Acosta-Silva C, Branchadell V, Bertran J. et al. Mutual relationship between stacking and hydrogen bonding in DNA. Theoretical study of guanine-cytosine, guanine-5-methylcytosine, and their dimers. J Phys Chem B 2010;114:10217–27. [DOI] [PubMed] [Google Scholar]
- 52. Thalhammer A, Hansen AS, El-Sagheer AH. et al. Hydroxylation of methylated CpG dinucleotides reverses stabilisation of DNA duplexes by cytosine 5-methylation. Chem Commun (Camb) 2011;47:5325–7. [DOI] [PubMed] [Google Scholar]
- 53. Lopez CM, Lloyd AJ, Leonard K. et al. Differential effect of three base modifications on DNA thermostability revealed by high resolution melting. Anal Chem 2012;84:7336–42. [DOI] [PubMed] [Google Scholar]
- 54. Severin PM, Zou X, Schulten K. et al. Effects of cytosine hydroxymethylation on DNA strand separation. Biophys J 2013;104:208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. He YF, Li BZ, Li Z. et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 2011;333:1303–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.