

Abstract
Following subcellular fractionation, the complexity of proteins derived from a particular cellular compartment is often evaluated by gel electrophoretic analysis. For the proteomic cataloguing of these distinct protein populations and their biochemical characterization, gel electrophoretic protein separation can be conveniently combined with liquid chromatography mass spectrometry. Here we describe a gel-enhanced liquid chromatography mass spectrometry (GeLC-MS)/MS approach with a new bioanalytical focus on the proteomic profiling of mitochondrial contact sites from rat liver using the highly sensitive Orbitrap Fusion Tribrid mass spectrometer for optimum protein identification following extraction from dried and long-term stored gels. Mass spectrometric analysis identified 964 protein species in the mitochondrial contact site fraction, whereby 459 proteins were identified by ≥3 unique peptides. This included mitochondrial components of the supramolecular complexes that form the ATP synthase, the respiratory chain, ribosomal subunits and the cytochrome P450 system, as well as crucial components of the translocase complexes translocase of the inner membrane (TIM) and translocase of the outer membrane (TOM) of the two mitochondrial membranes. Proteomics also identified contact site markers, such as glutathione transferase, monoamine oxidase and the pore protein voltage dependent anion channel (VDAC)-1. Hence, this report demonstrates that the GeLC-MS/MS method can be used to study complex mixtures of proteins that have been embedded and stored in dried polyacrylamide gels for a long period of time. Careful re-swelling and standard in-gel digestion is suitable to produce peptide profiles from old gels that can be used to extract sophisticated proteomic maps and enable the subsequent bioinformatics analysis of the distribution of protein function and the determination of potential protein clustering within the contact site system.
Keywords: contact sites, MICOS, mitochondria, TIM, TOM, VDAC
Introduction
Differences in size, charge and solubility are critical physicochemical parameters that can be conveniently used for the biochemical separation of individual protein species [1]. Complex mixtures of proteins can be swiftly separated by a variety of one-dimensional polyacrylamide gel electrophoretic techniques [2–4] or two-dimensional gel electrophoresis using isoelectric focusing in the first dimension [5–7]. Gel electrophoretically separated proteins can then be stored for very long periods of time at room temperature following gel drying [8]. A great variety of labelling and staining methods for the detection of proteins in polyacrylamide gels are available to visualize separated protein bands or spots [9]. Following storage, gel-embedded polypeptides can be efficiently extracted from individual lanes or gel zones and recovered for subsequent biochemical analyses [10]. This opens up the possibility of re-analysing experiments that were carried out many years prior to the proteomic era of modern biochemistry [11]. The usage of highly sensitive mass spectrometry to characterize protein populations in long-term stored polyacrylamide gels promises a refined profiling to both confirm and extend the scope of previous gel-based protein biochemical studies.
A select number of previous attempts to study dried and stored gel samples by mass spectrometry have mostly focused on isolated proteins or relatively small numbers of electrophoretically separated protein species [12–16]. The analytical continuation of the proteomic profiling of dried and long-term stored protein gels, as outlined in this report, is based on the mass spectrometric screening of an isolated fraction of mostly soluble muscle proteins [17]. We have extended and refined this approach here to the comparative subproteomic identification of protein species in a critical membrane system, the contact sites between the outer and inner membranes of mitochondria. Rat liver mitochondrial fractions enriched with the outer membrane, inner membrane and contact sites were originally separated by one-dimensional gel electrophoresis over 30 years ago and then stored after drying in a lab book at room temperature [18].
For the systematic analysis of protein mixtures, one-dimensional sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) can be conveniently coupled to sensitive liquid chromatography tandem mass spectrometry. This biochemical approach, usually referred to as GeLC-MS/MS, represents a robust and well-established technique of modern mass spectrometry-based proteomics [19–21]. Here we outline a GeLC-MS/MS approach with a specific bioanalytical focus on the subproteomic profiling of rat liver mitochondria using the highly sensitive Orbitrap Fusion Tribrid mass spectrometer for optimum protein identification following extraction from dried and long-term stored gels. During the retrospective analysis of a long-term stored polyacrylamide gel, a combination of extensive subcellular fractionation, liver protein extraction, one-dimensional gel electrophoretic separation, in-gel digestion, liquid chromatography and advanced mass spectrometry succeeded in the systematic cataloguing of the mitochondrial contact site fraction. This resulted in the identification of 964 protein species, including 459 proteins for which sequence was covered by ≥ 3 unique peptides. The proteomic documentation of a large number of mitochondrial liver proteins with a close association to the contact site region clearly outperformed the findings from previous biochemical studies prior to the development of mass spectrometry-based proteomics [22].
Mitochondrial contact sites are dynamic and crucial membrane systems involved in energy metabolism, organelle signalling and protein translocation [23–25]. Previous investigations on the identification and characterization of protein constituents of mitochondrial contact sites were performed with biochemical, immunochemical and focused mass spectrometric analyses [26–28]. Building on these findings, we have utilized long-term stored polyacrylamide gels for the systematic mass spectrometric profiling of the entire protein population that constitutes mitochondrial contact sites, as outlined in this report.
Materials and methods
Materials
For the systematic profiling of the gel electrophoretically separated protein constituents of rat liver contact sites, a variety of general analytical grade reagents and materials were obtained from GE Healthcare (Little Chalfont, Buckinghamshire, UK), Bio-Rad Laboratories (Hemel-Hempstead, Hertfordshire, UK) and Sigma Chemical Company (Dorset, UK). Sequencing grade-modified trypsin was purchased from Promega (Madison, WI, USA) and Whatman nitrocellulose transfer membranes came from Invitrogen (Carlsbad, CA, USA). The chemiluminescence substrate and protease inhibitors were obtained from Roche Diagnostics (Mannheim, Germany). A primary antibody to the porin protein VDAC-1 (Abcam Cat# ab14734, RRID: AB_443084) was from Abcam (Cambridge, UK) and Chemicon International (Temecula, CA, USA) provided peroxidase-conjugated secondary antibodies.
Isolation of mitochondrial contact sites
A fraction highly enriched in mitochondrial contact sites was isolated by an optimized method as previously described in detail [18]. The workflow is summarized in Fig. 1 and consists of the isolation of mitochondria by differential centrifugation, followed by mitochondrial swelling, mitochondrial shrinkage, sonication and sucrose density gradient centrifugation for the enrichment of individual mitochondrial membrane systems. The protein constituents from the inner membrane, contact sites and outer membranes were originally separated on 10% SDS-PAGE gels [2] followed by protein staining with Coomassie Brilliant Blue. Standard vacuum drying of the gel was carried out between one layer of acetate film and one layer of thick filter paper in a solution of 30% methanol and 5% glycerol. Transportation of samples to Maynooth University was carried out in accordance with the Department of Agriculture (animal by-product register number 2016/16 to the Department of Biology, National University of Ireland, Maynooth, Co. Kildare).
Figure 1:

Overview of the bioanalytical workflow for the proteomic analysis of enriched contact sites from rat liver mitochondria. Shown is the isolation scheme of mitochondrial contact sites consisting of differential centrifugation, organelle swelling, organelle shrinkage, sonication and sucrose density gradient centrifugation to enrich fractions with individual membrane systems. The protein population of isolated contact sites was separated by gel electrophoresis and then gels dried and stored at room temperature. Following re-swelling of gels and in-gel digestion, contact site proteins were identified by LC-MS.
Sample preparation for mass spectrometric analysis
Re-swelling of the long-term stored polyacrylamide gel with the mitochondrial contact site fraction was carried out through overnight incubation at room temperature with gentle agitation in 30% methanol, 5% acetic acid and 5% glycerol [17]. Gel strips were then placed in fresh plastic tubes and incubated with shaking at room temperature; first for 4 h in 5% glycerol and 1% acetic acid, and then with 1% glycerol and 1% acetic acid overnight. Following gentle washing of gels with distilled water, the covering acetate sheet and filter paper were removed. For the proteolytic digestion of mitochondrial proteins prior to mass spectrometric peptide analysis, an established in-gel digestion protocol was used [29]. Protein lanes with purified contact sites from liver mitochondria [18] were cut into five separate segments and processed separately [30]. Individual Coomassie Brilliant Blue-stained gel zones were de-stained by the addition of 100 µl of 100 mM ammonium bicarbonate: neat acetonitrile (1: 1) solution, and incubated at 37°C for 30 min with gentle agitation. The solution was removed and 500 µl neat acetonitrile was added to each gel zone and incubated at room temperature for 10 min with gentle agitation. The solution was removed and gel pieces then underwent in-gel trypsin digestion using 100 µl of re-suspended trypsin and incubated at 4°C for 30 min to allow slow diffusion of trypsin into the gel. A further 20 µl of trypsin buffer was added, and gel zones were incubated for 90 min at 4°C [30]. A 100 µl of a 50 mM ammonium bicarbonate solution was added and left to incubate overnight at 37°C. A 220 µl extraction buffer [5% formic acid/neat acetonitrile (1: 2)] was added to gel pieces and incubated at 37°C for 15 min with agitation. The supernatant, containing peptides, was transferred to fresh tubes, and dried down by vacuum centrifugation. Dried peptides were re-suspended in 0.5% trifluoroacetic acid (TFA)/5% acetonitrile (ACN), purified by C18 spin columns and dried by vacuum centrifugation and stored at -80°C prior to mass spectrometric analysis [31].
Liquid chromatography mass spectrometry
Dried peptides were re-suspended in loading buffer consisting of 2% ACN and 0.05% TFA in liquid chromatography mass spectrometry (LC-MS) grade water. Peptide suspensions were then vortexed, sonicated and centrifuged briefly at 14 000×g before being transferred to mass spectrometry vials [32]. Reverse-phased capillary high-pressure liquid chromatography was carried out using the UltiMate 3000 nano system (Thermo Scientific) coupled directly in-line with the Thermo Orbitrap Fusion Tribrid Mass Spectrometer (Thermo Scientific). The digested samples were loaded onto the trapping cartridge (µ-Precolumn 300 µm i.d. 5 mm C18 PepMap100 5 µm 100-Å) at a flow rate of 25 µl/min with 2% (v/v) ACN, 0.1% (v/v) TFA for 3 min before being resolved onto an analytical column (Easy-Spray C18 75 µm × 500 mm, 2 µm bead diameter column). Peptides were eluted using the following binary gradient: solvent A (0.1% (v/v) formic acid in LC-MS grade water) and 2–27.5% solvent B [80% (v/v) ACN, 0.08% (v/v) formic acid in LC-MS grade water] for 60 min at a flow rate of 300 nl/min [33]. For peptide ionization, a voltage of 1.9 kV was applied and a capillary temperature of 320°C was used. Data-dependent acquisition with full scans in the 375–1500 m/z range was performed using an Orbitrap mass analyser with a resolution of 120 000 (at m/z 200), a targeted automatic gain control (AGC) value of 4E + 05 and a maximum injection time of 50 ms. The number of selected precursor ions for fragmentation was determined by the top-speed acquisition algorithm. Selected precursor ions were isolated in the Quadrupole with an isolation width of 1.6 Da. Peptides with a charge state of 2+ to 6+ were analysed and a dynamic exclusion was applied after 60 s. Precursor ions were fragmented using higher energy collision-induced dissociation with a normalized collision energy of 28%, and resulting MS/MS ions were measured in the linear ion trap. The typical MS/MS scan conditions were as follows: a targeted AGC value of 2E + 04 and a maximum fill time of 35 ms.
Protein profiling by label-free LC-MS/MS analysis
Proteins present in the enriched contact site fraction from rat liver mitochondria were identified using Proteome Discoverer 1.4 against Sequest HT (SEQUEST HT algorithm, licence Thermo Scientific, registered trademark University of Washington, USA) using the UniProtKB/Swiss-Prot Rattus norvegicus database. The following search parameters were used for protein identification: (i) peptide mass tolerance set to 10 ppm, (ii) MS/MS mass tolerance set to 0.6 Da, (iii) an allowance of up to two missed cleavages, (iv) carbamidomethylation set as a fixed modification and (v) methionine oxidation set as a variable modification. The data was filtered for high-confidence peptides as determined by Percolator validation in Proteome Discoverer. Bioinformatics was carried out by standard analysis software [34,35].
Immunoblot analysis
Standard immunoblotting was used to characterize the separated fractions from rat liver mitochondria enriched in the outer membrane, contact sites and inner membrane [18]. Following gel electrophoretic separation, proteins were transferred to nitrocellulose membranes, blocked in a milk protein solution [2.5% (w/v) fat-free milk powder in 10% phosphate-buffered saline] and incubated with primary antibody overnight [36]. Following a number of wash steps, membranes were incubated with horseradish peroxidase-conjugated secondary antibodies, and detected using enhanced chemiluminescence [36]. Densitometric scanning was performed using a HP PSC-2355 scanner.
Results and discussion
The method outlined in this report describes the successful proteomic analysis of the extracted protein constituents from a gel lane in a long-term stored SDS-PAGE gel that was originally used to demonstrate the difference in protein composition between fractions from rat liver mitochondria that are enriched in the outer membrane, inner membrane and contact sites [18]. The application of the GeLC-MS/MS method and the usage of the highly sensitive Orbitrap Fusion Tribrid mass spectrometer identified 964 protein species in the mitochondrial contact site fraction, as listed in Supplementary Table 1. The data underlying this study is available from Open Science Framework (10.17605/OSF.IO/8KQPB). Figure 2 gives an overview of the number of identified protein species in relation to their sequence coverage by numbers of unique peptides. Previous mass spectrometric studies of isolated mitochondria have identified a considerable number of protein species, including the mitochondrial proteomes from various organs and species [37–39], as well as subcellular structures within mitochondria [40–42]. Proteomic profiling has classified 900 high-confidence mitochondrial proteins and in addition over 2000 proteins in mitochondria-associated fractions [43]. Efficient protein separation by gel electrophoretic methodologies or liquid chromatography is often a prerequisite for in-depth bioanalytical applications. For the systematic cataloguing of the mitochondrial proteome [44–46], crucial parameters are the purity of starting material, sample handling and the rigor of the extraction protocol [47–49].
Figure 2:

Graphical presentation of the number of identified protein species in the contact site fraction from rat liver in relation to their sequence coverage by numbers of unique peptides.
Mitochondria are double membranous organelles with an outer and inner membrane system that separates an intermembrane space, as outlined in the diagram of Fig. 3. The protein banding patterns of enriched fractions from the outer membrane, contact sites and the inner membrane show distinct differences in the staining intensity of individual gel zones (Fig. 3). Antibody labelling of the three different mitochondrial membrane fractions clearly illustrate a striking difference in abundance of the outer membrane pore protein VDAC-1, which is lowest in the inner membrane and highest in the outer membrane (Fig. 3). The contact sites contain an intermediate amount of this pore protein of the outer membrane. The findings from the immunoblot analysis correlate well with the mass spectrometric data in relation to the porin isoform VDAC-1, which was detected by 19 unique peptides and a 64% coverage in contact sites (Supplementary Table 1). In contrast, 10 peptides and 46% coverage and 26 peptides and 75% coverage identified VDAC-1 in the inner membrane versus the outer membrane (not shown).
Figure 3:
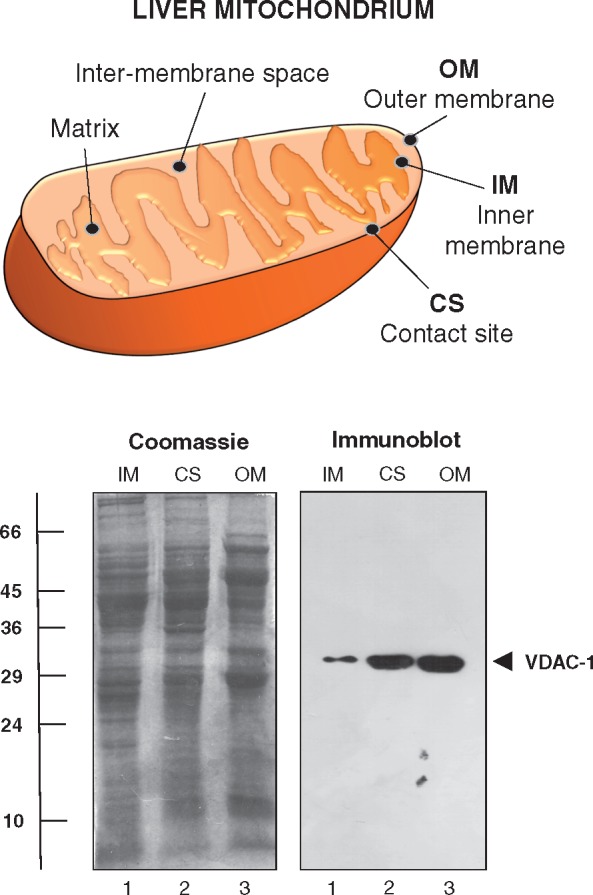
Gel electrophoretic and immunoblot analysis of isolated contact sites from rat liver mitochondria. Shown is a scheme of the double membranous system of mitochondria consisting of the outer membrane, contact sites and the inner membrane system that separates the intermembrane space. The Coomassie Brilliant Blue stained gel illustrates the differing protein band patterns of subcellular fractions enriched in the mitochondrial inner membrane (IM), contact sites (CS) and the mitochondrial outer membrane (OM). The immunoblot was labelled with antibodies to the outer membrane marker VDAC-1. The position of the immuno-reactive band is marked by an arrowhead. Molecular mass standards (in kDa) are indicated at the left of the gel image.
Following careful re-swelling of the dried and long-term stored gel [18], the proteomic profiling of the mitochondrial contact site fraction from rat liver was carried out by LC-MS/MS analysis. To illustrate the diversity of identified proteins and determine potential protein clusters, proteomic data were analysed by the bioinformatics programmes PANTHER [34] and STRING [35], respectively. Figure 4 summarizes the findings of the PANTHER analysis and shows that the contact site fraction contains a large number of oxidoreductases, transferases, hydrolases, nucleic acid binding proteins, enzyme modulators and transporters. This agrees with the fact that liver mitochondria are of central importance for a variety of hepatic functions, including oxidative bioenergetics, metabolic integration and protein translocation.
Figure 4:

Bioinformatic PANTHER analysis of the distribution of protein function in mitochondrial contact sites from rat liver. The classification system gives an overview of functionally related protein subfamilies in mitochondrial contact sites.
The systematic mapping and characterization of the liver proteome has included the identification of mitochondrial changes in health and disease [50]. In analogy, the detailed proteomic profiling of enriched contact sites reported here has identified mitochondrial components of the supramolecular complexes that form the respiratory chain, the ATP synthase, coenzyme biosynthesis clusters, ribosomal subunits and the cytochrome P450 system, as well as crucial components of the translocase complexes TIM and TOM of the two mitochondrial membranes. The STRING analysis shown in Fig. 5 summarizes potential protein–protein interaction patterns within the subproteome of mitochondrial contact sites. This presents a beneficial addition to the already existing proteomic listings and characterizations of subproteomic data sets of mitochondrial membrane systems [40–42]. The contact site fraction contains the marker enzyme glutathione transferase of this membrane system [18, 26–28], as well as the outer membrane markers monoamine oxidase and porin isoform VDAC-1 [23]. The ADP/ATP translocating protein complex and the glycolytic enzyme hexokinase were shown to be present in contact sites, which agree with previous biochemical studies [22, 27]. Mitochondria also form contact zones with the endoplasmic reticulum and interact with the cytoskeleton, which is confirmed by the presence of the SERCA-type Ca2+-pumping ATPase and tubulin isoforms, respectively. Key proteins linked to mitochondrial contact sites that have been identified by the GeLC-MS/MS study outlined here are summarized diagrammatically in Fig. 6. This includes the monoamine oxidase, the pore protein VDAC-1, MIC proteins belonging to the mitochondrial contact site and cristae organizing system MICOS, the ADP/ATP translocator and glutathione transferase GSTA, as well as various subunits of the translocase complexes TOM and TIM of the inner and outer mitochondrial membrane [23–25].
Figure 5:

Bioinformatic STRING analysis of potential interaction patterns between mass spectrometrically identified proteins in mitochondrial contact sites from rat liver. Shown are known and predicted protein–protein interactions between contact site proteins. Protein hubs, functional clusters and previously characterized marker proteins of contact sites are marked by circles.
Figure 6:

Overview of key components of mitochondrial contact sites from rat liver as determined by GeLC-MS/MS analysis of a long-term stored SDS-PAGE gel.
In eukaryotic cells, mitochondria are essential organelles that maintain cellular viability and generate the essential energy for the maintenance of basic biochemical and physiological processes. Mitochondria are the primary bioenergetic site for oxidative phosphorylation and are majorly involved in the integration of intermediate metabolism, protein translocation, cell cycle progression, calcium signalling, the biosynthesis of haem and iron–sulphur clusters and the regulation of apoptosis [51]. Altered expression levels within the mitochondrial proteome are critical factors for normal development and numerous diseases [52–54]. The method presented here is a suitable addition to the plethora of proteomic methodologies available to study the biochemistry of mitochondria and enables the re-evaluation of previous experiments on contact sites from liver mitochondria. This study has demonstrated that re-swelling of old polyacrylamide gels followed by in-gel digestion and highly sensitive mass spectrometry can be used for the advanced proteomic profiling of mitochondrial contact sites from rat liver and the systematic cataloguing of complex membrane subproteomes.
Supplementary data
Supplementary data are available at Biology Methods and Protocols online.
Conflict of interest statement. None declared.
Supplementary Material
Acknowledgements
The authors thank Prof. Dr. em. Dieter Brdiczka (Universität Konstanz) for the initiation of the mitochondrial contact sites project.
Funding
Research was supported by a Hume Scholarship from Maynooth University. The Orbitrap Fusion Tribrid mass spectrometer was funded under a Science Foundation Ireland Infrastructure Award to Dublin City University (SFI 16/RI/3701).
References
- 1. Righetti PG. Bioanalysis: its past, present, and some future. Electrophoresis 2004; 25:2111–27. [DOI] [PubMed] [Google Scholar]
- 2. Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970; 227:680–85. [DOI] [PubMed] [Google Scholar]
- 3. Brunelle JL, Green R.. One-dimensional SDS-polyacrylamide gel electrophoresis (1D SDS-PAGE). Methods Enzymol 2014; 541:151–59. [DOI] [PubMed] [Google Scholar]
- 4. Backman L, Persson K.. The no-nonsens SDS-PAGE. Methods Mol Biol 2018; 1721:89–94. [DOI] [PubMed] [Google Scholar]
- 5. Oliveira BM, Coorssen JR, Martins-de-Souza D.. 2DE: the phoenix of proteomics. J Proteomics 2014; 104:140–50. [DOI] [PubMed] [Google Scholar]
- 6. Murphy S, Dowling P, Ohlendieck K.. Comparative skeletal muscle proteomics using two-dimensional gel electrophoresis. Proteomes 2016; 4:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Meleady P. Two-dimensional gel electrophoresis and 2D-DIGE. Methods Mol Biol 2018; 1664:3–14. [DOI] [PubMed] [Google Scholar]
- 8. Stamova S, Michalk I, Bartsch H, Bachmann M.. Gel drying methods. Methods Mol Biol 2012; 869:433–36. [DOI] [PubMed] [Google Scholar]
- 9. Gauci VJ, Wright EP, Coorssen JR.. Quantitative proteomics: assessing the spectrum of in-gel protein detection methods. J Chem Biol 2011; 4:3–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kurien BT, Scofield RH.. Extraction of proteins from gels: a brief review. Methods Mol Biol 2012; 869:403–05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aebersold R, Mann M.. Mass-spectrometric exploration of proteome structure and function. Nature 2016; 537:347–55. [DOI] [PubMed] [Google Scholar]
- 12. Matsumoto H, Komori N.. Protein identification on two-dimensional gels archived nearly two decades ago by in-gel digestion and matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Anal Biochem 1999; 270:176–79. [DOI] [PubMed] [Google Scholar]
- 13. Dihazi H, Kessler R, Eschrich K.. In-gel digestion of proteins from long-term dried polyacrylamide gels: matrix-assisted laser desorption-ionization time of flight mass spectrometry identification of proteins and detection of their covalent modification. Anal Biochem 2001; 299:260–63. [DOI] [PubMed] [Google Scholar]
- 14. Panfilov O, Lanne B.. Peptide mass fingerprinting from wet and dry two-dimensional gels and its application in proteomics. Anal Biochem 2002; 307:393–95. [DOI] [PubMed] [Google Scholar]
- 15. Zhou J, Li J, Li J. et al. Dried polyacrylamide gel absorption: a method for efficient elimination of the interferences from SDS-solubilized protein samples in mass spectrometry-based proteome analysis. Electrophoresis 2010; 31:3816–22. [DOI] [PubMed] [Google Scholar]
- 16. Righetti PG, Lomonte B, Calvete JJ.. Resurrexit, sicut dixit, alleluia. Snake venomics from a 26-year old polyacrylamide focusing gel. J Proteomics 2012; 75:1074–78. [DOI] [PubMed] [Google Scholar]
- 17. Murphy S, Ohlendieck K.. Proteomic profiling of large myofibrillar proteins from dried and long-term stored polyacrylamide gels. Anal Biochem 2018; 543:8–11. [DOI] [PubMed] [Google Scholar]
- 18. Ohlendieck K, Riesinger I, Adams V. et al. Enrichment and biochemical characterization of boundary membrane contact sites from rat-liver mitochondria. Biochim Biophys Acta 1986; 860:672–89. [DOI] [PubMed] [Google Scholar]
- 19. Dzieciatkowska M, Hill R, Hansen KC.. GeLC-MS/MS analysis of complex protein mixtures. Methods Mol Biol 2014; 1156:53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piersma SR, Warmoes MO, de Wit M. et al. Whole gel processing procedure for GeLC-MS/MS based proteomics. Proteome Sci 2013; 11:17.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Paulo JA. Sample preparation for proteomic analysis using a GeLC-MS/MS strategy. J Biol Methods 2016; 3:45.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brdiczka DG, Zorov DB, Sheu SS.. Mitochondrial contact sites: their role in energy metabolism and apoptosis. Biochim Biophys Acta 2006; 1762:148–63. [DOI] [PubMed] [Google Scholar]
- 23. Brdiczka D, Beutner G, Rück A. et al. The molecular structure of mitochondrial contact sites. Their role in regulation of energy metabolism and permeability transition. Biofactors 1998; 8:235–42. [DOI] [PubMed] [Google Scholar]
- 24. Horvath SE, Rampelt H, Oeljeklaus S. et al. Role of membrane contact sites in protein import into mitochondria. Protein Sci 2015; 24:277–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wollweber F, von der Malsburg K, van der Laan M.. Mitochondrial contact site and cristae organizing system: a central player in membrane shaping and crosstalk. Biochim Biophys Acta 2017; 1864:1481–89. [DOI] [PubMed] [Google Scholar]
- 26. Ardail D, Privat JP, Egret-Charlier M. et al. Mitochondrial contact sites. Lipid composition and dynamics. J Biol Chem 1990; 265:18797–802. [PubMed] [Google Scholar]
- 27. Beutner G, Rück A, Riede B, Brdiczka D.. Complexes between porin, hexokinase, mitochondrial creatine kinase and adenylate translocator display properties of the permeability transition pore. Implication for regulation of permeability transition by the kinases. Biochim Biophys Acta 1998; 1368:7–18. [DOI] [PubMed] [Google Scholar]
- 28. Hoppel C, Kerner J, Turkaly P. et al. Isolation of hepatic mitochondrial contact sites: previously unrecognized inner membrane components. Anal Biochem 2002; 302:60–69. [DOI] [PubMed] [Google Scholar]
- 29. Shevchenko A, Tomas H, Havlis J. et al. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc 2006; 1:2856–60. [DOI] [PubMed] [Google Scholar]
- 30. Murphy S, Zweyer M, Mundegar RR. et al. Comparative gel-based proteomic analysis of chemically crosslinked complexes in dystrophic skeletal muscle. Electrophoresis 2018;39 1735–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Holland A, Henry M, Meleady P. et al. Comparative label-free mass spectrometric analysis of mildly versus severely affected mdx mouse skeletal muscles identifies annexin, lamin, and vimentin as universal dystrophic markers. Molecules 2015; 20:11317–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Murphy S, Zweyer M, Henry M. et al. Proteomic analysis of the sarcolemma-enriched fraction from dystrophic mdx-4cv skeletal muscle. J Proteomics 2018. doi: 10.1016/j.jprot.2018.01.015. [DOI] [PubMed] [Google Scholar]
- 33. O’Sullivan F, Keenan J, Aherne S. et al. Parallel mRNA, proteomics and miRNA expression analysis in cell line models of the intestine. World J Gastroenterol 2017; 23:7369–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mi H, Huang X, Muruganujan A. et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 2017; 45:D183–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Szklarczyk D, Morris JH, Cook H. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 2017; 45:D362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murphy S, Dowling P, Zweyer M. et al. Proteomic analysis of dystrophin deficiency and associated changes in the aged mdx-4cv heart model of dystrophinopathy-related cardiomyopathy. J Proteomics 2016;145:24–36. [DOI] [PubMed] [Google Scholar]
- 37. Pflieger D, Le Caer JP, Lemaire C. et al. Systematic identification of mitochondrial proteins by LC-MS/MS. Anal Chem 2002; 74:2400–06. [DOI] [PubMed] [Google Scholar]
- 38. Taylor SW, Fahy E, Zhang B. et al. Characterization of the human heart mitochondrial proteome. Nat Biotechnol 2003; 21:281–86. [DOI] [PubMed] [Google Scholar]
- 39. Rhee HW, Zou P, Udeshi ND. et al. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013; 339:1328–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Distler AM, Kerner J, Hoppel CL.. Proteomics of mitochondrial inner and outer membranes. Proteomics 2008; 8:4066–82. [DOI] [PubMed] [Google Scholar]
- 41. Zahedi RP, Sickmann A, Boehm AM. et al. Proteomic analysis of the yeast mitochondrial outer membrane reveals accumulation of a subclass of preproteins. Mol Biol Cell 2006; 17:1436–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hung V, Zou P, Rhee HW. et al. Proteomic mapping of the human mitochondrial intermembrane space in live cells via ratiometric APEX tagging. Mol Cell 2014; 55:332–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Morgenstern M, Stiller SB, Lübbert P. et al. Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep 2017; 19:2836–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ruiz-Romero C, Blanco FJ.. Mitochondrial proteomics and its application in biomedical research. Mol Biosyst 2009; 5:1130–42. [DOI] [PubMed] [Google Scholar]
- 45. Elstner M, Andreoli C, Klopstock T. et al. The mitochondrial proteome database. MitoP2. Methods Enzymol 2009; 457:3–20. [DOI] [PubMed] [Google Scholar]
- 46. Palmfeldt J, Bross P.. Proteomics of human mitochondria. Mitochondrion 2017; 33:2–14. [DOI] [PubMed] [Google Scholar]
- 47. Stimpson SE, Coorssen JR, Myers SJ.. Optimal isolation of mitochondria for proteomic analyses. Anal Biochem 2015; 475:1–3. [DOI] [PubMed] [Google Scholar]
- 48. Shibata T, Yamashita S, Hirusaki K. et al. Isolation of mitochondria by gentle cell membrane disruption, and their subsequent characterization. Biochem Biophys Res Commun 2015; 463:563–68. [DOI] [PubMed] [Google Scholar]
- 49. Lehr S, Hartwig S, Kotzka J.. Preparation of “functional” mitochondria: a challenging business. Methods Mol Biol 2015; 1264:1–8. [DOI] [PubMed] [Google Scholar]
- 50. Yu H, Wang F, Lin L. et al. Mapping and analyzing the human liver proteome: progress and potential. Expert Rev Proteomics 2016; 13:833–43. [DOI] [PubMed] [Google Scholar]
- 51. McBride HM, Neuspiel M, Wasiak S.. Mitochondria: more than just a powerhouse. Curr Biol 2006; 16:R551–60. [DOI] [PubMed] [Google Scholar]
- 52. Chan DC. Mitochondria: dynamic organelles in disease, aging, and development. Cell 2006; 125:1241–52. [DOI] [PubMed] [Google Scholar]
- 53. Monsalve M, Borniquel S, Valle I. et al. Mitochondrial dysfunction in human pathologies. Front Biosci 2007; 12:1131–53. [DOI] [PubMed] [Google Scholar]
- 54. Nunnari J, Suomalainen A.. Mitochondria: in sickness and in health. Cell 2012; 148:1145–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.