

Abstract
BACKGROUND
Adoptive transfer of donor-derived EBV-specific cytotoxic T-lymphocytes (EBV-CTLs) can eradicate EBV-associated lymphomas (EBV-PTLD) after transplantation of hematopoietic cell (HCT) or solid organ (SOT) but is unavailable for most patients.
METHODS
We developed a third-party, allogeneic, off-the-shelf bank of 330 GMP-grade EBV-CTL lines from specifically consented healthy HCT donors. We treated 46 recipients of HCT (n = 33) or SOT (n = 13) with established EBV-PTLD, who had failed rituximab therapy, with third-party EBV-CTLs. Treatment cycles consisted of 3 weekly infusions of EBV-CTLs and 3 weeks of observation.
RESULTS
EBV-CTLs did not induce significant toxicities. One patient developed grade I skin graft-versus-host disease. Complete remission (CR) or sustained partial remission (PR) was achieved in 68% of HCT recipients and 54% of SOT recipients. For patients who achieved CR/PR or stable disease after cycle 1, one year overall survival was 88.9% and 81.8%, respectively. In addition, 3 of 5 recipients with POD after a first cycle who received EBV-CTLs from a different donor achieved CR or durable PR (60%) and survived longer than 1 year. Maximal responses were achieved after a median of 2 cycles.
CONCLUSION
Third-party EBV-CTLs of defined HLA restriction provide safe, immediately accessible treatment for EBV-PTLD. Secondary treatment with EBV-CTLs restricted by a different HLA allele (switch therapy) can also induce remissions if initial EBV-CTLs are ineffective. These results suggest a promising potential therapy for patients with rituximab-refractory EBV-associated lymphoma after transplantation.
TRIAL REGISTRATION
Phase II protocols (NCT01498484 and NCT00002663) were approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center, the FDA, and the National Marrow Donor Program.
FUNDING
This work was supported by NIH grants CA23766 and R21CA162002, the Aubrey Fund, the Claire Tow Foundation, the Major Family Foundation, the Max Cure Foundation, the Richard “Rick” J. Eisemann Pediatric Research Fund, the Banbury Foundation, the Edith Robertson Foundation, and the Larry Smead Foundation. Atara Biotherapeutics licensed the bank of third-party EBV-CTLs from Memorial Sloan Kettering Cancer Center in June 2015.
Keywords: Immunology, Transplantation
Keywords: Cancer immunotherapy, T cells
Introduction
EBV-induced lymphomas are a significant cause of morbidity and mortality for recipients of allogeneic hematopoietic cell transplants (HCTs) and solid organ transplants (SOTs) (1–5). While decreasing immunosuppression can induce remissions of EBV-associated post-transplantation lymphoproliferative disorder (EBV-PTLD) in recipients of SOT with benign polyclonal lymphoid hyperplasias (5), it is not effective in HCT recipients or in SOT recipients who present with monoclonal/monomorphic lymphoma (5). Although combination chemotherapy can induce remissions in 40%–50% of SOT patients with monoclonal disease, relapses are common (6, 7). Furthermore, treatment-related mortality after R-CHOP ranges from 6% to 30%. In HCT patients, combination chemotherapy is associated with increased morbidity and mortality (8). Rituximab, administered preemptively, can induce sustained reversal of EBV viremia in up to 83% of HCT recipients (9); but only 50%–60% of patients with clinically and radiologically established disease achieve remissions (10–12).
In 1994, our group reported 5 HCT patients with monoclonal EBV-associated lymphomas who achieved complete remission (CR) after infusion of lymphocytes (donor lymphocyte infusion [DLI]) from their EBV-seropositive transplant donors and correlated response with emergence of donor-derived EBV-CTLs in the blood after transfer (13, 14). In 1995, Rooney et al. (15) first used HCT donor–derived EBV-specific cytotoxic T lymphocytes (EBV-CTLs) generated in vitro to treat or prevent EBV lymphomas following HCT without graft-versus-host disease (GvHD) (16). Subsequently, small case series have confirmed that HCT donor–derived EBV-CTLs can induce clearance of viremia and durable CRs of EBV+ lymphomas in 50%–70% of cases (17–21). In SOT patients, autologous EBV-CTLs can also induce CR or transient partial remissions (PRs) of EBV lymphomas (22–25). However, EBV viremia is rarely cleared (23, 25–27). Furthermore, autologous EBV-CTLs are difficult to generate if the SOT recipient (a) is seronegative or (b) has received rituximab. For both HCT and SOT recipients, logistics and culture times required to generate EBV-CTLs in time to treat these rapidly progressive lymphomas have been prohibitive.
To provide rapid access, we and others have explored partially HLA-matched EBV-CTLs derived from healthy donors other than the transplant donor (i.e., third-party donors). In 2002, Haque et al. (28) first reported the use of such cells in the treatment of 8 SOT recipients with EBV-PTLD, including one with lymphoma who achieved a PR. They then (29) treated 31 SOT and 2 HCT recipients with EBV-PTLD, of whom 14 (including 2 HCT patients) achieved CR and 3 PR. Subsequently, we reported 5 patients with EBV+ lymphomas complicating allogeneic cord blood or T cell–depleted HCT grafts whom we treated with partially HLA-matched third-party EBV-CTLs selected for restriction by an HLA allele shared by the allogenic HCT donor and/or the patient’s disease. Of these, 4 achieved durable CR (17, 30). Subsequently, limited case series have used third-party EBV-specific or multi-virus-specific T cells to treat EBV-associated lymphoproliferative disorder (EBV-LPD) or EBV viremia complicating cord blood or marrow HCTs, SOTs, or genetic immune deficiencies (29–39) (summarized in Table 1). However, the number of patients treated for EBV+ lymphoma is small.
Table 1. Summary of reported experience with adoptive therapy with third-party donor–derived EBV-CTLs.

Here, we report a single-center cohort of 46 patients with rituximab-refractory lymphomas developing after HCT or SOT who were treated with banked third-party EBV-CTL lines between October 2005 and May 2015, and describe attributes of the disease, its prior treatment, and the T cells used for adoptive therapy that are associated with clearance or continued progression of disease.
Results
Patient characteristics
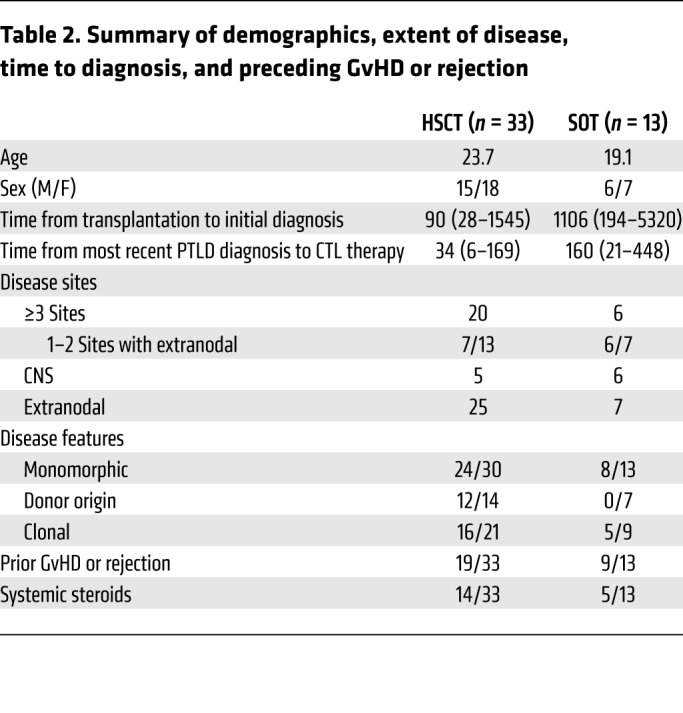
Characteristics of the 46 patients and their clinical and radiologic manifestations of EBV disease prior to treatment with EBV-CTLs are described in Supplemental Tables 1 and 2 (supplemental material available online with this article; https://doi.org/10.1172/JCI121127DS1) and summarized in Table 2. These patients had either progressed during rituximab treatment, failed to fully respond to it, or had a recurrence after a prior response. Prior therapy for 8 of 33 HCT patients and 12 of 13 SOT patients also included chemotherapy and/or radiation therapy. One of the HCT patients had failed to respond to EBV-CTLs from his HLA-disparate transplant donor. At time of referral, 27 of 33 HCT and 12 of 13 SOT recipients had high-risk disease based on location (≥3 of 7 anatomical sites of involvement and/or extranodal disease) (10).
Table 2. Summary of demographics, extent of disease, time to diagnosis, and preceding GvHD or rejection.
Pathologic characteristics of EBV malignancies
Histopathologic and genetic features of the EBV-associated lymphomas are described in Supplemental Table 3. The lymphomas were all of B cell type, and were monomorphic diffuse large B cell lymphomas in 24 of 30 HCT recipients (80%) and 8 of 13 SOT recipients (62%).
In HCT recipients the malignancy was monoclonal in 16 of 21 patients adequately tested and of transplant donor origin in 12 of 14 patients, including 1 with lymphoma cells from both cord blood units following double cord blood transplantation (CBT). In SOT recipients, 7 of 7 lymphomas tested were of host origin.
Characterization of EBV-CTLs infused
As shown in Figure 1, the EBV-CTLs contained more than 95% CD3+ T cells and fewer than 1% NK cells. Most EBV-CTL lines contained more than 90% CD8+ T cells. However, 7 CTL lines contained more than 50% CD4+ T cells. All T cell lines demonstrated EBV-specific cytotoxic activity. In limiting dilution assays, the EBV-CTLs contained a median of 6323.5 EBV-specific cytotoxic T cell precursors (EBV-CTLps) (range, 2.5–76,982 EBV-CTLps per 106 cells), and, in response to irradiated fully allogeneic PBMCs, generated low or undetectable alloreactive CTLps (median 1.2, range 0–27.4 allo-CTLps per 106 cells). EBV-CTLs administered to HCT patients did not differ significantly from those administered to SOT patients, either in types of T cells administered or in their frequencies of EBV-CTLps (data not shown).
Figure 1. Characterization of 55 EBV-specific cytotoxic T cells infused.

All T cell lines, including those predominantly containing CD4+ T cells, demonstrated EBV-specific cytotoxic activity against autologous EBV-BLCLs and did not kill NK cell–sensitive targets (K562), EBV-negative autologous or recipient-derived PHA blasts, or HLA-mismatched EBV-BLCLs. (A) Phenotype (CD3, CD8, CD4, and NK). (B) Cytotoxic activity of EBV-specific T cell lines against autologous BLCLs (circles), autologous PHA blasts (squares), mismatched targets (triangles), and NK-sensitive K562 targets (inverted triangles). (C) EBV-CTLp frequency and alloreactive CTL precursor (allo-CTLp) frequency in lines infused to treat patients. In limiting dilution assays, the EBV-CTLs contained a median of 6323.5 EBV-CTLps per 106 cells (range, 2.5–76,982 EBV-CTLps per 106 cells), and, in response to irradiated fully allogeneic PBMCs, generated low or undetectable alloreactive CTLps (median 1.2, range 0–27.4 allo-CTLps per 106 cells). Error bars indicate ± SD.
HLA restrictions were identified for each of the 55 EBV-CTL lines used; 19 (34%) were restricted by a single HLA-A (n = 15), HLA-B (n = 3), or HLA-DR (n = 1) allele, 26 lines (47%) by 2 (n = 13) or 3 (n = 13) alleles, and 10 (18%) by 4 or more alleles.
As might be expected, the EBV-CTL lines selected were most commonly restricted by class I HLA alleles prevalent in the ethnically diverse population of the New York area, such as HLA-A*0201, -B*0702, -A*0301, and -B*0801. On the basis of the HLA restrictions of the EBV-CTL lines in our bank, and the HLA alleles inherited by over 400 patients referred for transplantation, we estimate that a bank including EBV-CTLs restricted by 40 HLA alleles would be able to provide suitably restricted EBV-CTLs for over 95% of this population.
Treatment with third-party–derived EBV-CTLs is well tolerated
No immediate adverse reactions were observed due to infusion of EBV-CTLs. One patient developed de novo grade I acute GvHD of the skin, which resolved with topical therapy; none of the 19 patients with prior GvHD required additional therapy for GvHD after EBV-CTL therapy. No patient experienced CTL-related de novo suppression of neutrophil, red cell, or platelet counts or, in SOT patients, evidence of organ rejection.
Clinical responses of EBV-associated lymphomas to third-party EBV-CTL infusions
Responses to treatment with EBV-CTLs were classified as CR, partial remission (PR), stable disease (SD), or progression of disease (POD) using the International Workshop Criteria for assessing response to treatment in non-Hodgkin lymphoma (40). Only 8 of 33 HCT and 1 of 13 SOT patients achieved a CR after the first cycle of EBV-CTLs (Table 3 and Figure 2). An additional 9 patients (7 HCT, 2 SOT) achieved a PR. Thus, the response (CR + PR) after cycle 1 was 39% (18/46). However, as shown in Table 3 and Figure 2, response rates (CR + PR) increased with additional cycles, with maximal response achieved after a median of 2 cycles (range, 1–5). Of 33 HCT patients, 19 ultimately achieved a CR and 3 a stable PR (CR + PR = 68%). Of 13 SOT patients, 2 achieved a CR and 5 achieved durable PRs (CR + PR = 54%). In all, 29 of the 45 evaluable patients (64%) achieved a CR or sustained PR. The overall survival at 2 years was 57% for HCT and 54% for SOT recipients (Figure 2C). Both the complete and, strikingly, the partial remissions in the HCT and SOT groups have been durable (6–115 months).
Table 3. Response to first and ultimate cycle of EBV-CTLs.
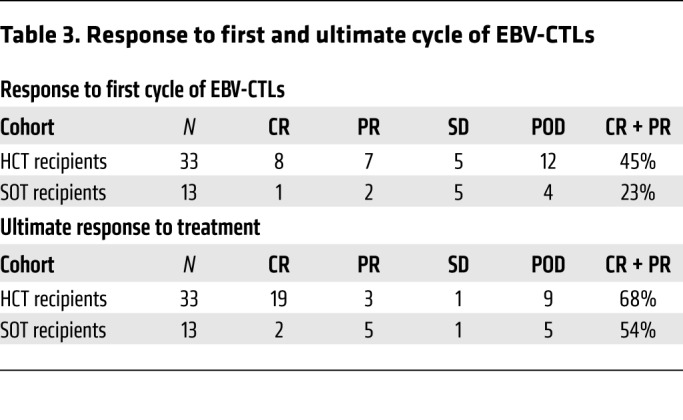
Figure 2. Number of cycles to best response (CR or PR), and survival of patients with HCT or SOT.

(A) Patients with EBV lymphoma after HCT. (B) Patients with EBV lymphoma after SOT. Patients achieving a CR (black) after the first cycle of EBV-CTLs included 8 of 33 HCT recipients and 1 of 13 SOT recipients. An additional 9 patients (7 HCT, 2 SOT) achieved a PR (gray), and 10 had SD. Thus, the overall response (CR + PR) after cycle 1 was 39% (18/46). With subsequent cycles of EBV-CTLs the response rate increased to 22 of 33 HCT (68%) and 7 of 13 SOT (54%) recipients. (C) Kaplan-Meier probabilities of survival for HCT and SOT patients at 2 years were 57% and 54%, respectively.
Outcomes based on EBV-CTLs administered are summarized in Figure 3. Twenty-one patients received a single cycle of EBV-CTLs, of whom 8 (7 HCT, 1 SOT) achieved a CR and 1 a durable PR (>2.5 years). However, 11 of 21 patients had POD through the first cycle, of whom 9 died early of EBV+ lymphoma. Median survival was 32 days (10–62 days) from initiation of EBV-CTLs. One additional patient with POD died of sepsis during the evaluation period; one patient received an alternate therapy and responded but died 12.1 months later of GvHD that predated EBV cell therapy. One other patient relapsed with his primary leukemia 1 day after his third dose of EBV-CTLs. This patient could be evaluated for toxicity and overall survival, but not for EBV lymphoma response because of chemotherapy introduced to treat the leukemia. This patient achieved remission of both EBV lymphoma and leukemia. However, leukemia relapsed 1 year after treatment; this patient died of leukemia 3 months later.
Figure 3. Flowchart of treatment and responses for patients treated for EBV-PTLD with third-party EBV-CTLs.

DOD, dead of disease.
Of the 25 patients who received more than 1 cycle of EBV-CTLs, 16 (1 CR, 6 PR, 8 SD, 1 nonevaluable) received EBV-CTLs from the same EBV-CTL line, with 9 ultimately achieving a CR and 5 a PR.
Three patients (1 PR, 1 SD, 1 POD) received subsequent cycles of EBV-CTLs from a different donor, but restricted by the same shared HLA allele as the primary cycle of cells; all achieved a CR. The patient with POD (UPN 4234) received a second HLA-B*07:02–restricted EBV-CTL line recognizing epitopes from both EBNA3C and EBNA3A rather than EBNA3A alone (data not shown).
Six patients (1 PR, 1 SD, 4 POD) received switch therapy with secondary cycles of EBV-CTLs restricted by a different HLA allele. Based on prior analyses of EBV+ lymphoma cells isolated from nonresponding patients (17, 41), we reasoned that switching to EBV-CTLs specific for an epitope presented by a different shared HLA allele might better treat an EBV lymphoma that was initially resistant. Of these 6, 1 with POD achieved a CR, 1 with POD achieved a PR, 1 in PR continued in PR until starting alternative therapy 4.9 months later, and 1 remained with SD. The other 2 patients continued with POD. However, progression was slowed, with survival extended to 215 and 266 days, respectively.
As shown in Figure 4A, most patients with continued progression of lymphoma died within a month of completing the first cycle, reflecting the rapid course of disease if not checked early in progression. Cumulative risk of death due to EBV lymphoma was 26% (Figure 4B). All deaths attributable to EBV lymphoma occurred within 8.8 months following initiation of T cell therapy in HCT and 5.8 months in SOT recipients.
Figure 4. Overall survival at 2 years.

(A) Survival of all patients based on evaluation of response to the first cycle of third-party EBV-CTLs. (B) Cumulative probability of death due to EBV lymphoma. (C) Survival of patients who received subsequent cycles of third-party EBV-CTLs, based on status of the EBV lymphoma immediately prior to initiation of the second cycle of EBV-CTLs. OS, overall survival.
For the 18 patients who achieved a CR or PR following the first cycle, survival at 2 years was 83%, and equivalent whether they did or did not receive a second cycle. As shown in Figure 4C, of those with SD after cycle 1 who received subsequent cycles, 72% were surviving at 2 years. In patients with POD following cycle 1 who received subsequent cycles of EBV-CTLs from a different donor, 3 of 5 (60%) responded and survived free of EBV+ lymphoma 2 years later.
In patients who achieved CR or PR, clinical improvements, including defervescence, shrinkage of palpable nodes, reduction of organomegaly, and resolution of pain or intestinal bleeding, were first detected 8–15 days after infusion of the effective T cells. Improvements in radiologic/endoscopic findings were documented by 28–35 days after the start of therapy. In patients with SD, symptoms including pain and fever plateaued or improved, but radiographic abnormalities did not improve. In contrast, patients who failed to respond showed persistence of fever and other clinical symptoms with continued clinical deterioration and/or worsening of radiologic findings.
In responding patients who had detectable EBV DNA levels in the blood before T cell infusion, 2 log10 reductions of EBV DNA levels were a useful initial indicator of response. However, because of prior treatment with rituximab, EBV DNA was not detectable in the blood of 7 HCT patients and 7 SOT patients despite evidence of POD.
Clinical and immunologic variables affecting outcome
Clinical characteristics of patients associated with response.
The patient characteristics examined for an association with response are summarized in Table 4. All sites of involvement, including the CNS, responded to EBV-CTL therapy. Of 11 patients with evidence of CNS involvement, 5 achieved CR and 4 durable PR. The proportion of patients with multiple sites of disease who achieved a CR or PR (52%) was significantly lower than that of patients with fewer than 3 sites of disease (80%) (P = 0.06). Patients with extranodal sites of lymphoma also had a lower response rate (P = 0.008). Patients treated with rituximab and chemotherapy before EBV-CTLs also fared worse than those previously treated with rituximab alone (45% vs. 80%, P = 0.03). In this series, no significant differences in response were seen when EBV-CTLs were administered to patients receiving concomitant steroid therapy. However, no patient was receiving a dose of ≥0.5 mg/kg prednisone or its equivalent. Of those receiving ≥0.2 mg/kg/d prednisone or its equivalent, 4 of 7 (57%) responded, compared with 7 of 12 (58%) patients receiving less than 0.2 mg/kg and 18 of 26 (69%) receiving no steroids.
Table 4. Comparison of clinical and treatment variables predicting response to third-party EBV-CTL therapy.

As shown in Table 5, the overall response rates (CR + PR) among recipients of unmodified and T cell–depleted HCT were similar (70% and 75%, respectively) and only slightly higher than that observed in cord blood graft recipients (60%). Complete response rates were similar (60%, 58%, and 60% for unmodified, T cell–depleted, and cord blood grafts). Notably, levels of CD3+ and CD3+CD4+ T cells and responses to phytohemagglutinin (PHA) were also similar in the 3 HCT groups.
Table 5. Baseline immune phenotype, function, and transplant type in responders and nonresponders.

Although the overall rate of durable responses (CR + PR) did not differ significantly between HCT and SOT recipients, the CR rate was higher in HCT patients than in SOT recipients (58% vs. 15%, P = 0.007). Since we would expect EBV-CTLs to be more susceptible to rejection in patients with residual T cell function, we examined whether differences in endogenous T cell populations in HCT and SOT recipients might be correlated with differences in CR rates observed. As shown in Table 5, the numbers of CD4+ and CD8+ T cells or T cell responses to PHA prior to adoptive therapy were not significantly higher in responders versus nonresponders in either the HCT or the SOT group. However, the CD4 and CD8 levels, as well as the PHA responses in the overall HCT group, were significantly lower than those of the SOT recipients, reflecting their greater degree of T cell deficiency prior to treatment.
Characteristics of EBV-CTLs and in vivo EBV-CTL proliferation after transfer associated with response.
As also shown in Table 4, lines used to treat patients who did or did not achieve a CR or PR did not differ significantly in the dose of EBV-specific CTLps per kilogram administered (P = 0.94). The distribution of CD4+ and CD8+ T cells among the EBV-CTLs administered was also similar, with a median of 11% CD4+ T cells among EBV-CTLs administered to patients achieving a CR or PR, compared with 9% in patients who failed to respond (P = 0.58).
The degree of HLA matching between EBV-CTLs administered and the HCT donor and patient or the SOT patient was not correlated with response (Table 4). Overall, the EBV-CTLs were matched with the patient and, for HCT, the transplant donor at a median of 4 of 10 alleles. A CR or PR was achieved in 12 of 19 (63%) patients matched for 1–3 HLA alleles and 17 of 26 (65%) matched for 4–8 alleles (P = 0.99). Results were similar for patient groups analyzed by transplant type. We also used the Cochran-Armitage test to identify any trends in compatibility associated with response. Again, no significant relationship was observed (P = 0.52). EBV lymphomas emerging in HCT recipients were of transplant donor type in 12 of 14 cases. However, we also found no significant trend in response between the number of HLA alleles shared by the EBV-CTLs and the HCT donor (P = 0.98).
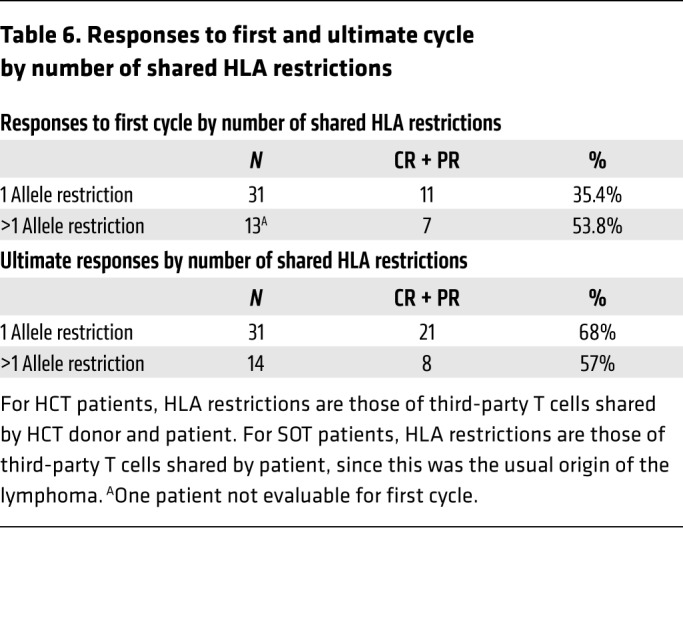
We also examined correlations between responses and the number of HLA restrictions shared by the patient (for SOT and HCT recipients) and/or the transplant donor (for HCT recipients). As shown in Table 6, in response to the EBV-CTLs initially transferred, 11 of 31 evaluable patients (35%) who received EBV-CTLs restricted by a single shared HLA allele achieved a CR or PR, and 6 patients achieved SD. In comparison, 7 of 13 patients (54%) treated with EBV-CTLs restricted by at least 2 shared alleles achieved a CR or PR, and 1 had SD. Ultimately, however, 21 of 31 patients treated with EBV-CTLs restricted by a single shared HLA allele (68%) achieved a CR or durable PR, compared with 8 of 14 patients (57%) treated with EBV-CTLs restricted by at least 2 shared alleles (P = 0.5).
Table 6. Responses to first and ultimate cycle by number of shared HLA restrictions.
We also examined the potential influence of HLA restrictions on the results observed in different patients receiving the same EBV-CTL line. In this series, EBV-CTL lines from 7 donors were used to treat more than 1 patient: 4 lines were used in 3 patients apiece, 2 in 2 patients, and 1 in 7 patients. The response rate among recipients of these lines did not differ significantly from the overall response rates in 10 of 14 HCT recipients (71%) and 5 of 8 SOT recipients (62%). Of the 7 donor lines, 5 had more than 1 restricting HLA allele. Among 12 recipients of these 5 lines, 3 shared only 1 of the EBV-CTLs’ restricting HLA alleles, of whom only 1 responded. In contrast, 8 of 9 recipients inheriting more than 1 of the EBV-CTLs’ restricting alleles responded.
Taken together, these data suggest that while restriction of the EBV-CTLs by a single shared HLA allele is sufficient, there may be an advantage to selection of EBV-CTLs restricted by more than 1 HLA allele shared by the patient’s disease. In this limited series, we did not detect an association between administration of EBV-CTLs restricted by any specific HLA allele and clinical response.
Although the characteristics of the EBV-CTLs analyzed did not predict response, EBV-CTLs that induced a CR or PR were regularly associated with a marked increase in blood levels of EBV-CTLps during the treatment cycle (Figure 5). Among responders, EBV-CTLp frequencies increased by a mean of 294-fold over preinfusion levels compared with a 3.4-fold increase in nonresponders (Table 4; P = 0.001). Increases in EBV-CTLp frequencies observed in patients responding to their first cycle were usually detected by 10–21 days after the initial infusion and coincided with clinical improvement. Increases in EBV-CTLps were also detected in 6 of 7 patients with SD after the first cycle who ultimately achieved a CR or PR.
Figure 5. EBV-CTLp frequency after first cycle of adoptive therapy with third-party EBV-CTLs.

Expansions could be detected in patients with responses as well as those with stable disease. Individual patients are demonstrated by distinctly colored lines. (A) EBV-CTLps tested in patients with complete response after first cycle of third-party EBV-CTLs. (B) EBV-CTLps tested in patients with partial response after first cycle of third-party EBV-CTLs. (C) EBV-CTLps tested in patients with stable disease after first cycle of third-party EBV-CTLs. (D) EBV-CTLps tested in patients with POD after first cycle of third-party EBV-CTLs.
The contrast between expansion of EBV-CTLps in responding patients and the lack thereof in patients with POD was also observed in the same patient among those who failed to respond to EBV-CTLs restricted by 1 HLA allele but subsequently responded to EBV-CTLs restricted by a different shared HLA allele. This is exemplified by patient UPN 5597 (Figure 6), who presented with a host-derived EBV+ lymphoma and had POD after treatment with 3 separate EBV-CTL lines restricted by HLA-A*11:01, including 2 lines that had previously induced a CR or PR in other patients (lines A and B). During these cycles, increases in CTLp frequencies were not observed (Figure 6B). He was then switched to an EBV-CTL line restricted by another shared allele, HLA-B*44:03 (line D). This line induced a CR, associated with a marked increment in CTLp frequencies.
Figure 6. Response to EBV-CTLs restricted by either HLA-A*1101 or HLA-B*4403.

(A) High-resolution typing of the patient and the origin of the EBV+ lymphoma and of the 4 EBV-CTL lines successively infused. Blue type indicates the restricting HLA allele of the EBV-CTL line. (B) Time course of EBV lymphoma and response to successive EBV-CTL lines (EBV PCR as an additional marker of disease). (C) Successive PET scans of disease progression and regression. (D) Distinctive pattern of STRs in EBV-CTLs. Top: Baseline host prior to CTL infusion. Middle: Third-party EBV-CTL donor. Bottom: EBV-specific T cells in blood 32 days after initial infusion and 16 days after third infusion in cycle 1 of EBV-CTLs from donor D, prior to cycle 2. Post-CTL-infusion specimen demonstrating the presence of CTL donor D cells (arrows indicate unique peaks corresponding to donor). s/p, status/post.
Subsequently, as shown in Table 7, we found that the HLA-A*11:01–restricted EBV-CTL lines failed to lyse the HLA-A*11:01+ EBV+ lymphoma cells grown from the patient’s lymphomatous tonsil. In contrast, the HLA-B*44:03–restricted EBV-CTL line was cytotoxic in vitro against both the patient’s HLA-B*44:03+ EBV+ lymphoma and HLA-B*44:03+ EBV B95.8–transformed B lymphoblastoid cell lines (EBV-BLCLs). These findings provide indirect but corroborating evidence supporting the hypothesis that the HLA-B*44:03–restricted EBV-CTLs recognized and were able to lyse the EBV+ lymphoma in vivo, while the HLA-A*11:01–restricted EBV-CTLs were not.
Table 7. Cytotoxic activity of third-party–derived EBV-CTLs used for treatment of patient UPN 5597.

To better understand the basis for the failure of the HLA-A*1101–restricted EBV-CTLs, we sequenced the latent proteins LMP1, LMP2, EBNA1, EBNA2, EBNA3A, EBNA3B, and EBNA3C from the EBV virus in the lymphoma cells grown from the patient and compared these sequences with those of EBV strain B95.8, focusing on sequences encoding all epitopes known to be presented by HLA-A*1101. Concurrently, we identified the peptide epitopes targeted by the HLA-A*1101–restricted EBV-CTL lines. Line A was specific for 2 EBNA3B peptides, 416IVTDFSVIK and 551DEPASTEPVHDQLL, known to be presented by HLA-A*1101. The DNA sequences for both of these EBNA3B peptides in the patient’s lymphoma cells and the B95.8 strains of EBV were identical. Line B was also specific for 551DEPASTEPVHDQLL. Unfortunately, we did not have sufficient cells from line C to establish this specificity.
We also evaluated the lymphoma cells for the presence of EBNA3B protein. By FACS analysis, EBNA3B was detected at fluorescence intensity levels comparable to those in EBV-BLCLs transformed by EBV strain 95.8. The lymphoma cells also expressed HLA-A*1101 protein, as assessed by FACS analysis of the cells using an HLA-A*1101–specific antibody (MyBioSource Inc.).
HLA-disparate third-party EBV-CTLs may persist for extended periods in organ allograft recipients maintained on nonsteroidal immunosuppressive drugs
Because of limitations in the number of lymphocytes left after sequential testing of EBV-CTLps, we were only able to assess the persistence of the third-party EBV-CTLs in 3 HCT and 2 SOT recipients. In 2 HCT patients tested before the second and third doses in a 3-week cycle, IFN-γ+ EBV-CTLs detected were exclusively from the third-party donor, as shown for patient UPN 5603 in Figure 7A. However, in the second patient, who had only achieved SD, these T cells could not be detected 5 days after the third dose in the cycle. In the third patient, UPN 5597 (depicted in Figure 6), who had failed treatment with HLA-A*1101–restricted EBV-CTLs but achieved a PR after an initial cycle of treatment with EBV-CTLs restricted by HLA-B*4403 (donor D), IFN-γ+ T cells bearing short tandem repeats (STRs) unique to donor D were detectable at least through 18 days after the last infusion of that first cycle of donor D EBV-CTLs. At the same time, EBV-CTLs from the prior donors were not detected (Figure 6D). Unfortunately, later samples were not available.
Figure 7. Detection of third-party donor EBV-CTLs using short tandem repeat analysis in 2 patients.

(A) HCT recipient. (B) SOT recipient. Baseline host prior to CTL infusion, third-party EBV-CTL donor, and post-CTL-infusion specimens demonstrating the presence of CTL donor cells at serial time points after infusion (arrows indicate unique peaks corresponding to donor). In the SOT patient, third-party donor EBV-CTLs were still detectable 23.7 months after the last infusion.
Of 2 SOT patients who were treated while on tacrolimus or sirolimus, one who failed to respond did not have IFN-γ+ EBV-CTLs bearing STRs unique to the third-party donor 24 days after completion of her second treatment cycle. In contrast, the other patient, a renal allograft recipient who achieved a durable PR after treatment with third-party EBV-CTLs matched for 6 of 10 HLA alleles, had T cells bearing STRs unique to the third-party donor 7 days after his first dose and again at 22 days after his first cycle. Strikingly, these third-party T cells still represented 16% of the IFN-γ+ T cells responding to the EBV peptide pool 23.7 months after his last dose of these cells (Figure 7B). In contrast, in a third patient, a heart allograft recipient, who achieved a PR of a host-origin EBV lymphoma after repeated courses of EBV-CTLs from 2 donors, including her HLA haplotype–mismatched mother, IFN-γ+ EBV-specific CTLs that were exclusively of host type were detected from 6 months to a year after completion of adoptive therapy.
Discussion
This study details the effects of partially HLA-matched, appropriately restricted, “off-the-shelf” third-party EBV-CTLs in the treatment of 46 recipients of allogeneic HCT or SOT with EBV+ lymphomas who had failed treatment with rituximab. The EBV-CTLs were derived from a bank of 330 cryopreserved EBV-CTL lines, generated under good manufacturing practice (GMP) and precharacterized as to HLA type, immune phenotype, lack of alloreactivity, EBV-specific cytotoxicity, and HLA restriction. This precharacterization permitted selection and treatment with appropriately HLA-restricted EBV-CTLs, within as few as 1–2 days of patient referral.
In vivo, the EBV-CTLs were well tolerated. No HCT or SOT recipient exhibited evidence of graft rejection or required treatment for a flare of GvHD; de novo GvHD requiring treatment was observed in only 1 patient who experienced a grade 1, transient skin rash. Furthermore, clinical responses were not associated with the cytokine release syndrome observed in patients responding to T cells expressing a chimeric antigen receptor (42). Thus, adoptive transfer of these EBV-CTLs has been safe.
The EBV+ lymphomas targeted by the third-party EBV-CTLs in this study uniformly presented as rapidly developing B cell malignancies that were monomorphic diffuse large B cell lymphomas in 80% of the cases. All 46 patients had rituximab-refractory disease, 20 had also failed to respond to chemotherapy and/or radiation therapy, 26 of 46 (56%) had disease in at least 3 anatomic sites, and 13 of the other 20 patients (65%) had extranodal disease, including 7 with disease in the CNS. These disease characteristics have been associated with a uniformly poor prognosis (10). Nevertheless, 68% of the HCT recipients and 54% of the SOT recipients treated with third-party EBV-CTLs achieved a CR or durable PR.
These results are similar to those we previously reported for HCT recipients with rituximab-refractory EBV lymphomas treated with HCT donor–derived EBV-CTLs (17). However, those patients usually responded by 3 weeks after a single cycle of EBV-CTLs. In contrast, responses to third-party EBV-CTLs were cumulative, requiring a median of 2 cycles to achieve CR or durable PR. This finding likely reflects the more limited potential of third-party T cells to persist after transfer, as may the initial SDs achieved by first cycles in ultimately responsive lymphomas and the lower rate of CRs in less immunocompromised SOT recipients.
We could not predict responders on the basis of doses of EBV-CTLps or the immunophenotype of the T cells administered. While Haque et al. (29) reported higher response rates in patients treated with more closely HLA-matched EBV-CTLs, we found no significant correlation between degree of HLA match and subsequent response. However, we selected EBV-CTLs primarily on the basis of restriction by an HLA allele that would be shared by the patient’s EBV-associated lymphoma. HLA restriction was prioritized because the EBV-CTLs in each line were usually restricted by only 1–3 HLA alleles and our prior studies, both in mice bearing multiple EBV-BLCL xenografts (43) and in patients receiving transplant donor–derived EBV-CTLs (18), had shown that such T cells selectively accumulate in and only induce remissions of tumors coexpressing EBV and the HLA allele by which the EBV-CTLs are restricted. Since third-party EBV-CTLs are rarely fully HLA-matched, they may be restricted by HLA alleles not shared by the patient’s disease. Thus, selection on the basis of HLA restriction may supersede the impact of matching for more HLA alleles.
Selection of EBV-CTL lines based on predetermined HLA restriction also provides particular advantages for treatment of EBV lymphomas of undefined origin in recipients of an HLA-disparate HCT, SOT, or cord blood graft, since an EBV-CTL restricted by an HLA allele shared by both the transplant donor and recipient addresses both possibilities.
Although we did not identify third-party EBV-CTL attributes that predicted responses, in this study, as in trials of transplant donor–derived EBV-CTLs (17, 30, 44), increases in the frequency of EBV-specific CTLps were regularly detected in patients who achieved CR or PR. They were also observed in patients with SD who subsequently attained a CR or PR, but not in patients with POD.
Multiple factors may contribute to POD despite treatment with appropriately HLA-restricted EBV-CTLs. Disease status affects outcome, since patients with extensively pretreated lymphomas or extranodal disease at time of referral had a poorer prognosis. Host factors (e.g., allospecific antibodies or residual alloresponsive T cells) could also induce rejection of EBV-CTLs early after infusion. Furthermore, EBV has developed multiple mechanisms to evade EBV-CTLs, ranging from epitope variation between different EBV strains (45, 46) to viral-encoded evasins that can usurp antigen processing and presentation, downregulate specific HLA alleles (47, 48), or directly inhibit T cell function (49). In our study, the consistent correlation observed between POD and failure of adoptively transferred EBV-CTLs to expand in vivo usually reflects the inability of EBV-CTLs to recognize the patient’s EBV lymphoma. Previously (17), we reported 3 patients who failed to respond to HCT donor–derived EBV-CTLs, even though the EBV lymphoma in each patient was of HCT donor origin. In each case, the EBV-CTLs lysed autologous B95.8 virus–transformed B cells used to sensitize the T cells but not autologous EBV+ cells grown from the patient’s lymphoma. However, T cells sensitized with the autologous lymphoma cells killed both donor-derived tumor and B95.8-transformed BLCLs, suggesting a lack of an antigen on the lymphoma recognized by B95.8-sensitized T cells rather than a functional impairment of the presentation of EBV antigens by the EBV lymphoma cells. In a similar case, Gottschalk et al. (41) demonstrated that spontaneously transformed EBV+ B cells isolated from a patient who failed to respond to EBV B95.8–sensitized T cells had a mutation resulting in deletion of the 2 epitopes of EBNA3B presented by HLA-A*11:01 targeted by those T cells.
In our patient UPN 5597, sequencing of EBV DNA encoding latent EBV proteins from the lymphomatous tonsil did not reveal mutations or deletions that would alter EBNA3B epitopes targeted by the HLA-A1101–restricted T cells. Furthermore, both EBNA3B and HLA-A1101 were normally expressed. These findings suggest that the presentation of targeted epitopes of EBNA3B by HLA-A1101 is otherwise impaired, potentially reflecting EBV-derived evasins capable of selectively interfering with antigen processing or transporter associated with antigen processing-mediated peptide transfer to HLA-A1101 for presentation (48, 49). These mechanisms are under study.
Based on our initial findings, we hypothesized that for patients with POD, selection of an alternate CTL line, particularly one specific for an epitope presented by another HLA allele shared by the patient’s disease, might prove effective. Our analyses of UPN 5597 (Figure 6) support this hypothesis. Furthermore, this “switch” therapy has induced durable complete or partial remissions in 3 of 5 patients who had POD after their first cycle. These responses were also closely correlated with expansions of EBV-CTLps in the blood. Thus, in patients with POD, switching to EBV-CTLs restricted by a different shared HLA allele can provide effector cells able to recognize the tumor, and induce remissions of disease. Furthermore, since 9 of 11 patients with POD who received only 1 cycle of EBV-CTLs died of lymphoma at a median of 28 days after initiation of EBV-CTL therapy, a trial of switch therapy in patients who continue to progress through the first 21 days of a cycle is warranted.
A striking, but yet poorly understood, finding has been the durability of both CRs and PRs observed following adoptive transfer of third-party EBV-CTLs. Because these T cells are allogeneic to both HCT and SOT recipients, we expected these T cells to be rejected. Indeed, the rationale for giving repeated 3-week cycles of cells was the hypothesis that such repeated doses would provide a more sustained exposure to the adoptively transferred EBV-CTLs. The cumulative nature of the responses observed following cycles of third-party EBV-CTLs also provides indirect evidence consistent with this hypothesis, as does the predominance of PRs observed in SOT patients, which contrasts with the preponderance of CRs achieved in the more T cell–deficient HCT recipients. That the increments in CTLp frequencies after each dose usually lasted for only 2–4 weeks also suggests their engraftment is brief. Thus, although the initial responses were closely related to increases in EBV-CTLps, with IFN-γ+ EBV-specific T cells of third-party donor origin by STR detected 10–21 days after infusion, the durability of responses observed more likely reflects activation of endogenous T cell responses. This is potentially stimulated by cross-presentation of antigens from EBV-associated lymphomas targeted by the third-party EBV-CTLs and potentially by recruitment of endogenous T cells responding to the allogeneic third-party effector cells. However, based on our findings and data from prior reports (29, 32), the possibility that persisting small populations of third-party EBV-CTLs also contribute to this durability must be considered. Previously, Leen et al. (32) and Haque et al. (29) have reported detection of third-party EBV-CTLs up to 94 days after infusion, in HCT and SOT recipients, respectively. While we found EBV-specific T cells of third-party origin (by STR) in 2 HCT recipients tested at 5 and 18 days following infusion, we did not have later samples to ascertain persistence. However, the finding of EBV-reactive third-party T cells in 1 SOT recipient at 23.7 months after infusion indicates that in some patients, these functional but HLA-disparate third-party EBV-CTLs can persist for extended periods. This persistence may be due to the ongoing immunosuppressive effects of tacrolimus. The possibility that the EBV-CTLs also contribute to their own survival through a veto function (50) also warrants evaluation. Ultimately, ongoing studies of the distribution and fate of genetically distinguishable third-party EBV-CTLs should clarify the relative contribution of the adoptively transferred EBV-CTLs and any endogenously generated T cells to the enduring responses observed.
In summary, third-party EBV-CTLs that are partially HLA-matched and appropriately HLA restricted can induce durable CRs or PRs in a high proportion of HCT and SOT patients with high-risk, rituximab-refractory EBV lymphomas without significant toxicity, graft injury, or GvHD. Maximal responses are cumulative, requiring, on average, two 3-week cycles of EBV-CTL infusions. Patients responding to a particular EBV-CTL line distinctively exhibit increases in the frequency of EBV-specific T cells in the blood within 10–21 days of the first infusion. Furthermore, patients with POD after 1 cycle can respond to treatment with an alternate EBV-CTL line specific for a different epitope presented by an alternate HLA allele shared by the lymphoma. Thus, off-the-shelf EBV-CTLs can provide multiple immediately accessible options for potentially curative treatment of high-risk EBV lymphomas complicating HCT or SOT.
Methods
Patients.
A total of 46 patients received third-party EBV-CTLs between October 2005 and May 2015 as treatment for EBV-associated B cell lymphomas that developed after an allogenic HCT (n = 33) or SOT (n = 13) and were refractory to or relapsed after therapy with rituximab.
Study approval.
All patients gave written consent and were treated on 1 of 2 consecutive single-arm phase II protocols (NCT01498484 and NCT00002663, www.ClinicalTrials.gov) approved by the IRB at the Memorial Sloan Kettering Cancer Center, by the FDA, and by the National Marrow Donor Program IRB. For completeness, we have included 4 patients previously reported (17, 30) who were the first to receive third-party EBV-CTLs following approval of the amendment of IRB 95-024 (NCT00002663) permitting their use. These patients are UPN 3520, UPN 5603, UPN 5628, and UPN 5629, identified by superscript “F” in Supplemental Table 1.
Trial treatment consisted of EBV-CTLs matched with the patient for at least 2 of 10 HLA alleles by high-resolution typing (HLA-A, -B, -C, -DR, or -DQ) and restricted by an HLA allele shared by the EBV lymphoma (when origin was known) and the HCT donor and patient for HCT recipients or the patient for SOT recipients. A treatment cycle consisted of 3 weekly i.v. infusions of 1 × 106 (protocol 1) or 2 × 106 (protocol 2) EBV-CTLs/kg, followed by a 3-week period of observation. Patients failing to achieve CR who had no therapy-related toxicity could receive additional cycles or be referred for alternate therapy. Primary study endpoints were (a) incidence of complete or partial responses as determined by clinical and radiographic criteria, and (b) incidence of infusion-related toxicities, alterations of HCT or SOT function, or GvHD. Secondary endpoints included (a) alterations in EBV DNA levels and (b) alterations in EBV-CTL precursor frequency measured in sequential blood specimens obtained before and after each infusion.
Diagnosis and characterization of EBV-LPD.
The EBV-associated lymphomas were classified according to the WHO criteria (51). Biopsy specimens were tested for EBV by in situ hybridization for EBV-encoded small RNAs and in some cases by immunohistology for LMP1. They were also tested for B and T cell markers. Whenever possible, we examined the EBV+ tumor cells for clonality of the B cells and their origin (host or donor). The genetic origin of the lymphoma was identified as donor or host, using FISH for XX versus XY in sex-mismatched transplants and using donor- or host-unique PCR-amplified STR polymorphisms (52, 53). Clonality of the tumors was identified by analysis of immunoglobulin rearrangements (54). Clonality of the EBV virus was determined by the method of Gulley and Raab-Traub (55).
In the case of UPN 5597, we were able to grow the clonal EBV lymphoma cells from the diseased tonsil and evaluate their sensitivity to lysis by the HLA-A1101– and HLA-B4403–restricted EBV-specific T cells sequentially infused for treatment. We also used next-generation sequencing to sequence the DNA of the EBV latent proteins from those lymphoma cells and compared the sequences of the latent EBV proteins and, particularly, the epitopes targeted by the EBV-specific T cells with those of the B95.8 strain of EBV.
For DNA sequencing, BLCL genomic libraries were constructed from 100 ng DNA using KAPA Hyper Prep Kit (KK8504, Kapa Biosystems) with 8 cycles of PCR. After sample barcoding, 130 ng–1.2 μg of each library was pooled and captured twice by hybridization with 112 EBV-specific biotinylated baits (Integrated DNA Technologies). Baits were designed based on complete sequence of B95.8 genome (GenBank V01555.2) and covered coding regions of 7 EBV latent genes (LMP1, LMP2, EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C). Capture pools were sequenced on a HiSeq 4000 System in a 100 bp/100 bp or 125 bp/125 bp paired-end run, using the HiSeq 3000/4000 SBS Kit (Illumina). An average of 2.7 million paired reads were generated per sample. Guided assembly of sequence reads was performed using Burrows-Wheeler Aligner (SourceForge) against reference B95.8 genome (GenBank V01555.2).
Generation and characterization of EBV-CTL lines.
EBV-CTLs were generated from a leukapheresis or unit of blood provided by healthy EBV-seropositive HCT donors who consented to these donations for the expressed purpose of generating EBV-CTLs for adoptive therapy in the recipient of their HCT or other patients with EBV-associated malignancies. The bank of 330 EBV-CTL lines was generated under FDA-compliant, good manufacturing practice (GMP) conditions as previously described (17). Briefly, T cells, enriched from PBMCs by depletion of monocytes by plastic adherence and of NK cells by adsorption to anti-CD56 immunomagnetic beads (Miltenyi Biotec), were sensitized in vitro at a 20:1 responder/stimulator (R/S) ratio with irradiated autologous EBV-transformed B cells (EBV-BLCLs) generated previously by transformation with the B95.8 strain of EBV (provided by C. Rooney, Baylor College of Medicine, Houston, Texas, USA). T cells were then cultured in Yssel’s medium (Gemini Bioproducts) supplemented with 5% heat-inactivated pooled normal human serum and restimulated with the same EBV-BLCLs weekly at a 4:1 R/S ratio.
Beginning day 16, IL-2 (Novartis) was added at 10–50 IU/mL 3 times per week. After 28–35 days of culture, T cells were characterized by flow cytometry and evaluated for EBV-specific cytotoxicity, lack of alloreactivity, and HLA restrictions (17). T cells meeting release criteria for adoptive therapy were aliquoted into labeled vials and cryopreserved.
The frequency of EBV-specific and alloreactive CTL precursors (CTLps) in EBV-CTL lines was measured by limiting dilution analysis (LDA) (14). When possible, the ability of EBV-CTLs to recognize endogenous EBV derived from a patient was assessed by measurement of cytotoxic activity against spontaneously transformed EBV+ B cells cultured from either a tumor biopsy or the PBMCs of the patient.
The epitope specificities of certain HLA-restricted third-party EBV-CTLs used for treatment were identified by assessment of their cytotoxicity against autologous PHA blasts loaded with different epitopes of each latent EBV protein known to be presented by an HLA allele of the T cell donor, as previously described (17).
Patient evaluations.
Patients were monitored for response by sequential clinical assessments, and imaging by CT, PET/CT, and/or MRI prior to and at the end of each cycle or as clinically indicated. EBV DNA copy numbers in the blood were monitored from 1995 to 2003 with a semiquantitative PCR-amplified assay and since 2003 with a quantitative real-time PCR assay. Patients were also monitored closely for serious adverse events using the National Cancer Institute (NCI) Common Toxicity Grading Criteria and for acute GvHD as graded by the NCI consensus criteria (https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/Archive/CTCAE_4.0_2009-05-29_QuickReference_8.5x11.pdf).
EBV-CTLp frequencies in the blood were quantitated by LDA (14) before EBV-CTL infusions and thereafter on days 1, 7, 14, 21, and 28, monthly for 4 months, and at 6- to 12-month intervals thereafter.
To test for the presence of third-party EBV-CTLs in the circulation after adoptive transfer, EBV-reactive T cells were examined for third-party donor–specific STRs as previously described to monitor marrow chimerism (52, 53). Briefly, Ficoll-Hypaque–separated PBMCs were sensitized for 12 hours with either a pool of EBV peptides (JPT Peptide Technologies), irradiated EBV-BLCLs from the HCT donor, or spontaneously EBV-transformed cells grown from the patient’s tumor. EBV-reactive T cell fractions were isolated by FACS sorting of CD3+ T cells coexpressing IFN-γ, or by immunoadsorption of activated CD3+ T cells coexpressing CD137. DNA from the CD137-enriched fractions as well as DNA from blood samples from the patient before transplantation, the HCT donor, and the third-party EBV-CTL donor was extracted using Qiagen EZ1 DNA blood kits. Samples were tested using primers from 2 autosomal STR kits (GenePrint Fluorescent STR multiplex systems: CTTv and GammaSTR kits) following the manufacturer’s protocol (Promega Corp.). Up to 8 loci were examined to identify the most informative alleles for quantitative assessment of host and donor proportions. Fluorescently labeled PCR products were detected using an ABI 3730 Genetic analyzer (Thermo Fisher Scientific) and analyzed with GeneMapper software (Applied Biosystems). The presence of third-party donor cells was determined by identification of unique donor peaks not present in the host, or the transplant donor.
Statistics.
The Kaplan-Meier method was used to estimate probability of survival over time. Comparisons of response rates between groups were assessed with Fisher’s exact test. The Cochran-Armitage test was used to determine whether there was a trend between the degree of HLA matching with the transplant donor or recipient and the patient’s response. The Wilcoxon rank sum test was used to evaluate whether CD4+ T cell or EBV-CTLp frequencies in the EBV-CTLs correlated with response.
Study approval.
The studies presented were approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center, (New York, New York, USA), the FDA, and the National Marrow Donor Program. All subjects or their legal proxy provided informed consent prior to their participation in the studies.
Author contributions
SP, ED, SS, GH, JB, PD, MAP, and EP all accrued patients to protocol and managed the care of the patients. GB evaluated radiographic assessments of patients. JRF assessed pathology of lesions. AH assisted with methodology for assessing durability of infused EBV-CTLs. SP served as investigator and then principal investigator of the enrolling protocols, enrolled patients, collected and analyzed data, and wrote the manuscript. ED managed the generation and characterization of the EBV-CTL bank and performed postinfusion in vitro assays, analyzed data, and contributed figures to the manuscript. SS collected clinical data. BG, KCH, AMH, HL, BS, FB, KJC, HCM, NAK, GK, RK, JWY, SG, AJ, EP, DP, and JB enrolled and managed patients on the protocol. GH was the biostatistician in design of the protocol and data analysis. MA, CM, and S. Hsu performed analysis of STRs discriminating EBV-CTL origins. S. Haque, JB, and MD reviewed studies for outcome assessments. RJO served as initial principal investigator, enrolled patients, analyzed data, and wrote the manuscript. CS was co-PI on the protocol, recruited and treated patients, and participated in writing the manuscript.
Supplementary Material
Acknowledgments
We acknowledge the use of the Integrated Genomics Operation Core, funded by an NCI Cancer Center Support Grant (P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. We thank members of the Center for Immune Cellular Therapy laboratory, Bone Marrow Transplantation Programs in Pediatrics and Medicine, Memorial Sloan Kettering Cancer Center, for their technical support. This work was supported through NIH grants CA23766 and R21CA162002, the Aubrey Fund, the Claire Tow Foundation, the Major Family Foundation, the Max Cure Foundation, the Richard “Rick” J. Eisemann Pediatric Research Fund, the Banbsury Foundation, the Edith Robertson Foundation, and the Larry Smead Foundation. Atara Biotherapeutics licensed the EBV-CTL bank in June 2015 and is developing this as ATA-129.
Version 1. 11/05/2019
In-Press Preview
Version 2. 01/06/2020
Electronic publication
Version 3. 02/03/2020
Print issue publication
Funding Statement
This work was supported through NIH grants CA23766, NIH R21CA162002, Aubrey Fund, The Claire Tow Foundation, Major Family Foundation, Max Cure Foundation, Richard “Rick” J. EIsemann Pediatric Research Fund, Banbury Foundation, Edith Robertson Foundation, Larry Smead Foundation.
NCI Cancer Center Support Grant
Footnotes
Conflict of interest: ED and ROR had consultancy agreements with Atara Biotherapeutics. ED and ROR are inventors on technology referenced in this work (SK2013-71 [patient ID], Proprietary Cell Banks for Use in 3rd-Party EBV-Specific T Cell Therapy; and SK2018-122 [patient ID], Methods of Selecting T Cell Lines for Adoptive Cellular Therapy). SP is an inventor on technology referenced in this work (SK2013-71 and SK2018-122) and has waived rights to revenue generated from these inventions. Memorial Sloan Kettering Cancer Center (MSK), which owns the technology, has licensed this technology to Atara, and MSK has interests in Atara through this licensing arrangement.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(2):733–747. https://doi.org/10.1172/JCI121127.
Contributor Information
Susan Prockop, Email: prockops@mskcc.org.
Ekaterina Doubrovina, Email: doubrove@mskcc.org.
Stephanie Suser, Email: susers@mskcc.org.
Glenn Heller, Email: hellerg@mskcc.org.
Juliet Barker, Email: barkerj@mskcc.org.
Parastoo Dahi, Email: dahip@mskcc.org.
Miguel A. Perales, Email: peralesm@mskcc.org.
Esperanza Papadopoulos, Email: papadope@mskcc.org.
Craig Sauter, Email: sauterc@mskcc.org.
Hugo Castro-Malaspina, Email: castromh@mskcc.org.
Farid Boulad, Email: bouladf@mskcc.org.
Kevin J. Curran, Email: currank@mskcc.org.
Sergio Giralt, Email: giralts@mskcc.org.
Boglarka Gyurkocza, Email: gyurkocb@mskcc.org.
Katharine C. Hsu, Email: hsuk@mskcc.org.
Ann Jakubowski, Email: jakubowa@mskcc.org.
Alan M. Hanash, Email: hanasha@mskcc.org.
Nancy A. Kernan, Email: kernann@mskcc.org.
Rachel Kobos, Email: kobosr@mskcc.org.
Guenther Koehne, Email: GuentherK@baptisthealth.net.
Heather Landau, Email: landauh@mskcc.org.
Doris Ponce, Email: ponced@mskcc.org.
Barbara Spitzer, Email: spitzerb@mskcc.org.
James W. Young, Email: youngj@mskcc.org.
Gerald Behr, Email: behrg@mskcc.org.
Mark Dunphy, Email: dunphym@mskcc.org.
Sofia Haque, Email: haques@mskcc.org.
Julie Teruya-Feldstein, Email: julie.teruya-feldstein@mountsinai.org.
Maria Arcila, Email: arcilam@MSKCC.ORG.
Christine Moung, Email: Moungc@mskcc.org.
Susan Hsu, Email: Susan.Hsu@redcross.org.
Aisha Hasan, Email: hasana@mskcc.org.
References
- 1.Singavi AK, Harrington AM, Fenske TS. Post-transplant lymphoproliferative disorders. Cancer Treat Res. 2015;165:305–327. doi: 10.1007/978-3-319-13150-4_13. [DOI] [PubMed] [Google Scholar]
- 2.Hoegh-Petersen M, et al. High incidence of post transplant lymphoproliferative disorder after antithymocyte globulin-based conditioning and ineffective prediction by day 28 EBV-specific T lymphocyte counts. Bone Marrow Transplant. 2011;46(8):1104–1112. doi: 10.1038/bmt.2010.272. [DOI] [PubMed] [Google Scholar]
- 3.San-Juan R, et al. Epstein-Barr virus-related post-transplant lymphoproliferative disorder in solid organ transplant recipients. Clin Microbiol Infect. 2014;20(suppl 7):109–118. doi: 10.1111/1469-0691.12534. [DOI] [PubMed] [Google Scholar]
- 4.Morscio J, Dierickx D, Tousseyn T. Molecular pathogenesis of B-cell posttransplant lymphoproliferative disorder: what do we know so far? Clin Dev Immunol. 2013;2013:150835. doi: 10.1155/2013/150835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petrara MR, Giunco S, Serraino D, Dolcetti R, De Rossi A. Post-transplant lymphoproliferative disorders: from epidemiology to pathogenesis-driven treatment. Cancer Lett. 2015;369(1):37–44. doi: 10.1016/j.canlet.2015.08.007. [DOI] [PubMed] [Google Scholar]
- 6.Glotz D, et al. The Seville expert workshop for progress in posttransplant lymphoproliferative disorders. Transplantation. 2012;94(8):784–793. doi: 10.1097/TP.0b013e318269e64f. [DOI] [PubMed] [Google Scholar]
- 7.Trappe R, et al. Sequential treatment with rituximab followed by CHOP chemotherapy in adult B-cell post-transplant lymphoproliferative disorder (PTLD): the prospective international multicentre phase 2 PTLD-1 trial. Lancet Oncol. 2012;13(2):196–206. doi: 10.1016/S1470-2045(11)70300-X. [DOI] [PubMed] [Google Scholar]
- 8.Davis CL. Interferon and cytotoxic chemotherapy for the treatment of post-transplant lymphoproliferative disorder. Transpl Infect Dis. 2001;3(2):108–118. doi: 10.1034/j.1399-3062.2001.003002108.x. [DOI] [PubMed] [Google Scholar]
- 9.Styczynski J, et al. Response to rituximab-based therapy and risk factor analysis in Epstein Barr Virus-related lymphoproliferative disorder after hematopoietic stem cell transplant in children and adults: a study from the Infectious Diseases Working Party of the European Group for Blood and Marrow Transplantation. Clin Infect Dis. 2013;57(6):794–802. doi: 10.1093/cid/cit391. [DOI] [PubMed] [Google Scholar]
- 10.Choquet S, et al. Rituximab in the management of post-transplantation lymphoproliferative disorder after solid organ transplantation: proceed with caution. Ann Hematol. 2007;86(8):599–607. doi: 10.1007/s00277-007-0298-2. [DOI] [PubMed] [Google Scholar]
- 11.Trappe RU, et al. Response to rituximab induction is a predictive marker in B-cell post-transplant lymphoproliferative disorder and allows successful stratification into rituximab or R-CHOP consolidation in an international, prospective, multicenter phase II trial. J Clin Oncol. 2017;35(5):536–543. doi: 10.1200/JCO.2016.69.3564. [DOI] [PubMed] [Google Scholar]
- 12.Fox CP, et al. EBV-associated post-transplant lymphoproliferative disorder following in vivo T-cell-depleted allogeneic transplantation: clinical features, viral load correlates and prognostic factors in the rituximab era. Bone Marrow Transplant. 2014;49(2):280–286. doi: 10.1038/bmt.2013.170. [DOI] [PubMed] [Google Scholar]
- 13.Papadopoulos EB, et al. Infusions of donor leukocytes to treat Epstein-Barr virus-associated lymphoproliferative disorders after allogeneic bone marrow transplantation. N Engl J Med. 1994;330(17):1185–1191. doi: 10.1056/NEJM199404283301703. [DOI] [PubMed] [Google Scholar]
- 14.Lucas KG, Small TN, Heller G, Dupont B, O’Reilly RJ. The development of cellular immunity to Epstein-Barr virus after allogeneic bone marrow transplantation. Blood. 1996;87(6):2594–2603. doi: 10.1182/blood.V87.6.2594.bloodjournal8762594. [DOI] [PubMed] [Google Scholar]
- 15.Rooney CM, et al. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet. 1995;345(8941):9–13. doi: 10.1016/S0140-6736(95)91150-2. [DOI] [PubMed] [Google Scholar]
- 16.Heslop HE, et al. Long-term restoration of immunity against Epstein-Barr virus infection by adoptive transfer of gene-modified virus-specific T lymphocytes. Nat Med. 1996;2(5):551–555. doi: 10.1038/nm0596-551. [DOI] [PubMed] [Google Scholar]
- 17.Doubrovina E, et al. Adoptive immunotherapy with unselected or EBV-specific T cells for biopsy-proven EBV+ lymphomas after allogeneic hematopoietic cell transplantation. Blood. 2012;119(11):2644–2656. doi: 10.1182/blood-2011-08-371971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gerdemann U, et al. Rapidly generated multivirus-specific cytotoxic T lymphocytes for the prophylaxis and treatment of viral infections. Mol Ther. 2012;20(8):1622–1632. doi: 10.1038/mt.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Icheva V, et al. Adoptive transfer of epstein-barr virus (EBV) nuclear antigen 1-specific t cells as treatment for EBV reactivation and lymphoproliferative disorders after allogeneic stem-cell transplantation. J Clin Oncol. 2013;31(1):39–48. doi: 10.1200/JCO.2011.39.8495. [DOI] [PubMed] [Google Scholar]
- 20.Comoli P, et al. Preemptive therapy of EBV-related lymphoproliferative disease after pediatric haploidentical stem cell transplantation. Am J Transplant. 2007;7(6):1648–1655. doi: 10.1111/j.1600-6143.2007.01823.x. [DOI] [PubMed] [Google Scholar]
- 21.Imashuku S, et al. Unsuccessful CTL transfusion in a case of post-BMT Epstein-Barr virus-associated lymphoproliferative disorder (EBV-LPD) Bone Marrow Transplant. 1997;20(4):337–340. doi: 10.1038/sj.bmt.1700883. [DOI] [PubMed] [Google Scholar]
- 22.Savoldo B, et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs) Blood. 2006;108(9):2942–2949. doi: 10.1182/blood-2006-05-021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khanna R, et al. Activation and adoptive transfer of Epstein-Barr virus-specific cytotoxic T cells in solid organ transplant patients with posttransplant lymphoproliferative disease. Proc Natl Acad Sci U S A. 1999;96(18):10391–10396. doi: 10.1073/pnas.96.18.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Comoli P, et al. Treatment of EBV-related post-renal transplant lymphoproliferative disease with a tailored regimen including EBV-specific T cells. Am J Transplant. 2005;5(6):1415–1422. doi: 10.1111/j.1600-6143.2005.00854.x. [DOI] [PubMed] [Google Scholar]
- 25.Sherritt MA, et al. Reconstitution of the latent T-lymphocyte response to Epstein-Barr virus is coincident with long-term recovery from posttransplant lymphoma after adoptive immunotherapy. Transplantation. 2003;75(9):1556–1560. doi: 10.1097/01.TP.0000058745.02123.6F. [DOI] [PubMed] [Google Scholar]
- 26.Haque T, et al. Reconstitution of EBV-specific T cell immunity in solid organ transplant recipients. J Immunol. 1998;160(12):6204–6209. [PubMed] [Google Scholar]
- 27.Comoli P, et al. Infusion of autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for prevention of EBV-related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood. 2002;99(7):2592–2598. doi: 10.1182/blood.V99.7.2592. [DOI] [PubMed] [Google Scholar]
- 28.Haque T, et al. Treatment of Epstein-Barr-virus-positive post-transplantation lymphoproliferative disease with partly HLA-matched allogeneic cytotoxic T cells. Lancet. 2002;360(9331):436–442. doi: 10.1016/S0140-6736(02)09672-1. [DOI] [PubMed] [Google Scholar]
- 29.Haque T, et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood. 2007;110(4):1123–1131. doi: 10.1182/blood-2006-12-063008. [DOI] [PubMed] [Google Scholar]
- 30.Barker JN, et al. Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood. 2010;116(23):5045–5049. doi: 10.1182/blood-2010-04-281873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Naik S, et al. Adoptive immunotherapy for primary immunodeficiency disorders with virus-specific T lymphocytes. J Allergy Clin Immunol. 2016;137(5):1498–1505.e1. doi: 10.1016/j.jaci.2015.12.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leen AM, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013;121(26):5113–5123. doi: 10.1182/blood-2013-02-486324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Q, Burton R, Reddy V, Lucas KG. Safety of allogeneic Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for patients with refractory EBV-related lymphoma. Br J Haematol. 2002;118(3):799–808. doi: 10.1046/j.1365-2141.2002.03683.x. [DOI] [PubMed] [Google Scholar]
- 34.Uhlin M, et al. Rapid salvage treatment with virus-specific T cells for therapy-resistant disease. Clin Infect Dis. 2012;55(8):1064–1073. doi: 10.1093/cid/cis625. [DOI] [PubMed] [Google Scholar]
- 35.Gallot G, et al. T-cell therapy using a bank of EBV-specific cytotoxic T cells: lessons from a phase I/II feasibility and safety study. J Immunother. 2014;37(3):170–179. doi: 10.1097/CJI.0000000000000031. [DOI] [PubMed] [Google Scholar]
- 36.Vickers MA, et al. Establishment and operation of a Good Manufacturing Practice-compliant allogeneic Epstein-Barr virus (EBV)-specific cytotoxic cell bank for the treatment of EBV-associated lymphoproliferative disease. Br J Haematol. 2014;167(3):402–410. doi: 10.1111/bjh.13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tzannou I, et al. Off-the-shelf virus-specific T cells to treat BK virus, human herpesvirus 6, cytomegalovirus, Epstein-Barr virus, and adenovirus infections after allogeneic hematopoietic stem-cell transplantation. J Clin Oncol. 2017;35(31):3547–3557. doi: 10.1200/JCO.2017.73.0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schultze-Florey RE, et al. Dissecting Epstein-Barr virus-specific T-cell responses after allogeneic EBV-specific T-cell transfer for central nervous system posttransplant lymphoproliferative disease. Front Immunol. 2018;9:1475. doi: 10.3389/fimmu.2018.01475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiou FK, Beath SV, Wilkie GM, Vickers MA, Morland B, Gupte GL. Cytotoxic T-lymphocyte therapy for post-transplant lymphoproliferative disorder after solid organ transplantation in children. Pediatr Transplant. 2018;22(2):e13133. doi: 10.1111/petr.13133. [DOI] [PubMed] [Google Scholar]
- 40.Cheson BD. Staging and response assessment in lymphomas: the new Lugano classification. Chin Clin Oncol. 2015;4(1):5. doi: 10.3978/j.issn.2304-3865.2014.11.03. [DOI] [PubMed] [Google Scholar]
- 41.Gottschalk S, et al. An Epstein-Barr virus deletion mutant associated with fatal lymphoproliferative disease unresponsive to therapy with virus-specific CTLs. Blood. 2001;97(4):835–843. doi: 10.1182/blood.V97.4.835. [DOI] [PubMed] [Google Scholar]
- 42.Lee DW, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124(2):188–195. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koehne G, et al. Serial in vivo imaging of the targeted migration of human HSV-TK-transduced antigen-specific lymphocytes. Nat Biotechnol. 2003;21(4):405–413. doi: 10.1038/nbt805. [DOI] [PubMed] [Google Scholar]
- 44.Rooney CM, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92(5):1549–1555. [PubMed] [Google Scholar]
- 45.Khanim F, Yao QY, Niedobitek G, Sihota S, Rickinson AB, Young LS. Analysis of Epstein-Barr virus gene polymorphisms in normal donors and in virus-associated tumors from different geographic locations. Blood. 1996;88(9):3491–3501. doi: 10.1182/blood.V88.9.3491.bloodjournal8893491. [DOI] [PubMed] [Google Scholar]
- 46.Rajcani J, Szenthe K, Banati F, Szathmary S. Survey of Epstein Barr virus (EBV) immunogenic proteins and their epitopes: implications for vaccine preparation. Recent Pat Antiinfect Drug Discov. 2014;9(1):62–76. doi: 10.2174/1574891X09666140828114812. [DOI] [PubMed] [Google Scholar]
- 47.Masucci MG, Stam NJ, Torsteinsdottir S, Neefjes JJ, Klein G, Ploegh HL. Allele-specific down-regulation of MHC class I antigens in Burkitt lymphoma lines. Cell Immunol. 1989;120(2):396–400. doi: 10.1016/0008-8749(89)90207-4. [DOI] [PubMed] [Google Scholar]
- 48.Albanese M, et al. Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc Natl Acad Sci U S A. 2016;113(42):E6467–E6475. doi: 10.1073/pnas.1605884113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ressing ME, et al. Epstein-Barr virus evasion of CD8(+) and CD4(+) T cell immunity via concerted actions of multiple gene products. Semin Cancer Biol. 2008;18(6):397–408. doi: 10.1016/j.semcancer.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 50.Or-Geva N, et al. Towards ‘off-the-shelf’ genetically modified T cells: prolonging functional engraftment in mice by CD8 veto T cells. Leukemia. 2018;32(4):1039–1041. doi: 10.1038/leu.2017.332. [DOI] [PubMed] [Google Scholar]
- 51.Swerdlow SH, et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood. 2016;127(20):2375–2390. doi: 10.1182/blood-2016-01-643569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Clark JR, et al. Monitoring of chimerism following allogeneic haematopoietic stem cell transplantation (HSCT): technical recommendations for the use of short tandem repeat (STR) based techniques, on behalf of the United Kingdom National External Quality Assessment Service for Leucocyte Immunophenotyping Chimerism Working Group. Br J Haematol. 2015;168(1):26–37. doi: 10.1111/bjh.13073. [DOI] [PubMed] [Google Scholar]
- 53.Kristt D, et al. Hematopoietic chimerism monitoring based on STRs: quantitative platform performance on sequential samples. J Biomol Tech. 2005;16(4):380–391. [PMC free article] [PubMed] [Google Scholar]
- 54.Inghirami G, Szabolcs MJ, Yee HT, Corradini P, Cesarman E, Knowles DM. Detection of immunoglobulin gene rearrangement of B cell non-Hodgkin’s lymphomas and leukemias in fresh, unfixed and formalin-fixed, paraffin-embedded tissue by polymerase chain reaction. Lab Invest. 1993;68(6):746–757. [PubMed] [Google Scholar]
- 55.Gulley ML, Raab-Traub N. Detection of Epstein-Barr virus in human tissues by molecular genetic techniques. Arch Pathol Lab Med. 1993;117(11):1115–1120. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.