

Abstract
Increased rates of locoregional recurrence are observed in patients with basal-like breast cancer (BC) despite the use of radiation therapy (RT); therefore, approaches that result in radiosensitization of basal-like BC are critically needed. Using patients’ tumor gene expression data from 4 independent data sets, we correlated gene expression with recurrence to find genes significantly correlated with early recurrence after RT. The highest-ranked gene, TTK, was most highly expressed in basal-like BC across multiple data sets. Inhibition of TTK by both genetic and pharmacologic methods enhanced radiosensitivity in multiple basal-like cell lines. Radiosensitivity was mediated, at least in part, through persistent DNA damage after treatment with TTK inhibition and RT. Inhibition of TTK impaired homologous recombination (HR) and repair efficiency, but not nonhomologous end-joining, and decreased the formation of Rad51 foci. Reintroduction of wild-type TTK rescued both radioresistance and HR repair efficiency after TTK knockdown; however, reintroduction of kinase-dead TTK did not. In vivo, TTK inhibition combined with RT led to a significant decrease in tumor growth in both heterotopic and orthotopic, including patient-derived xenograft, BC models. These data support the rationale for clinical development of TTK inhibition as a radiosensitizing strategy for patients with basal-like BC, and efforts toward this end are currently underway.
Keywords: Oncology, Therapeutics
Keywords: Breast cancer, DNA repair, Radiation therapy
Introduction
Breast cancer (BC) is the most commonly diagnosed invasive cancer in women in the United States and the second leading cause of cancer-related deaths (1). Radiation therapy (RT) remains a mainstay therapy for patients with BC and has been shown to not only reduce local recurrence (LR) but improve overall survival for these patients (2). Although RT is effective for many patients with BC, a significant proportion of them, especially those with basal-like BC, continue to have high rates of LR and poor overall survival, suggesting that RT is not as effective in those patient populations (3–7).
Although effective targeted treatment options are available for estrogen receptor–positive (ER+) and human epidermal growth factor receptor 2–positive (HER2+) BCs, fewer targeted treatment options exist for women with basal-like BCs. Recent advancements in BC subtyping have allowed for more advanced clinical risk stratification and treatment recommendations for patients; however, recommendations for radiation dose and treatment scheduling remain similar for all BC subtypes (8, 9). In an era of precision medicine and molecularly targeted therapy, understanding the molecular drivers of radiation resistance in basal-like BC remains a critical unmet clinical need.
To that end, previous studies have attempted to identify and characterize targets for the radiosensitization of BC, including basal-like BC. These studies have identified cell-cycle and DNA damage response proteins that may be implicated in RT resistance (10). As expected, targeting DNA damage response–related proteins is a promising strategy for radiosensitization (11). Recently, cell-cycle proteins have also been shown to be possible radiosensitization targets, often through modulation of the effectiveness of DNA damage repair genes (12, 13). To date, however, few therapies targeting these proteins have been clinically implemented, owing, in part, to the dose-limiting toxicities and off-target effects of these agents.
Here, we sought to identify novel radiosensitizing targets in aggressive basal-like BC. In this study, we used clinical data sets to correlate gene expression with recurrence-free survival after radiation in women with BC and nominate TTK as a potential mediator of radioresistance in aggressive subtypes of BC. Using clinical and preclinical data, we demonstrate that TTK was overexpressed in locally recurrent and basal-like BC. Preclinical studies show that inhibition of TTK, both genetically and pharmacologically, leads to increased radiosensitivity of basal-like BC cell lines and patient-derived xenograft) models. Our preclinical studies showed that radiosensitization is kinase function dependent and is mediated, at least in part, through impaired homologous recombination (HR) repair efficiency. Finally, we validated TTK inhibition–mediated radiosensitivity in vivo using a clinical-grade pharmacologic inhibitor.
Results
TTK is the top gene correlated to recurrence after radiation in BC across 4 independent data sets.
In an effort to identify genes that play a role in radioresistance and thus increase rates of LR in BC, we correlated gene expression to early (defined as 3 years or earlier) recurrence, including LR, across 4 independent data sets that included women treated with radiation according to the standard of care. We restricted our results to genes with an OR of 2.0 or greater and a multiple-testing–corrected P value of less than 1.0 × 10–6. Within these constraints, we found 10 genes that were significantly correlated with early recurrence across all 4 BC data sets (Figure 1A). These genes were ranked on the basis of their average differential log2 fold change across all 4 data sets, among patients with early (≤3 years) recurrence and those who did not have evidence of recurrence at 3 years. This nomination identified TTK, also known as monopolar spindle 1 (Mps1), as the top-ranked gene, with an average log2 fold change of 1.73 across the 4 independent data sets. To further refine our nomination, we focused on genes with a clinical-grade inhibitor currently in development. TTK was 1 of only 3 genes found to currently have a pharmacological agent in clinical trial according to ClinicalTrials.gov (Table 1). To confirm our findings, we performed Kaplan-Meier analyses of 2 independent data sets (Servant and Vande Vijver), as well as with 1 of the original 4 data sets (Wang). These data sets all had more carefully annotated LR-specific information and included women treated with RT. In all 3 data sets, TTK expression above the median was correlated to a decrease in local recurrence–free survival (LRFS) (Servant: hazard ratio = 1.70, P = 0.004; Vande Vijver: hazard ratio = 2.42, P = 0.005; Wang: hazard ratio = 2.23, P < 0.0001) (Figure 1, B–D). Furthermore, when divided into quartile expression, TTK expression was associated with a stepwise decrease in LRFS in these data sets (Supplemental Figure 1, A–C; supplemental material available online with this article; https://doi.org/10.1172/JCI130435DS1). Univariate analysis (UVA) showed that TTK expression was significantly correlated with LRFS in all 3 data sets (Tables 2, 3, 4, 5, 6, and 7). In multivariate analysis (MVA), using a stepwise logistic regression model, TTK remained the strongest predictor of LR (hazard ratio, 1.29–11.29), independent of all other clinicopathologic features (Tables 2–7).
Figure 1. TTK expression correlates with BC recurrence and independently predicts LRFS.

(A) Four BC data sets (Desmedt, van ‘t Veer, Wang, and Schmidt) were used to identify genes associated with early recurrence (within 3 years) (OR >2.0; P < 1.0 × 10–6). (B–D) Kaplan-Meier LRFS analysis of 3 independent data sets: Servant (B), Vande Vijver (C), and Wang (D) demonstrated that patients with higher-than-the-median expression of TTK had significantly higher rates of LR after radiation compared with patients with lower-than-the-median TTK expression. (E) TTK was overexpressed in basal-like BC compared with expression in other BC subtypes (P < 0.0001) and was overexpressed in BC compared with healthy tissue (P < 0.0001) in the METABRIC data set. (F) TTK was overexpressed in basal-like BC compared with non–basal-like BC, using transcripts per million (TPM) measurement, in the University of Michigan’s institutional data set (Met500) (P < 0.0001). A 2-sided Student’s t test and a 1-way ANOVA were used for statistical analyses. Error bars represent SD.
Table 1. Genes associated with locoregional recurrence after radiation.

Table 2. UVA of the Servant data set.
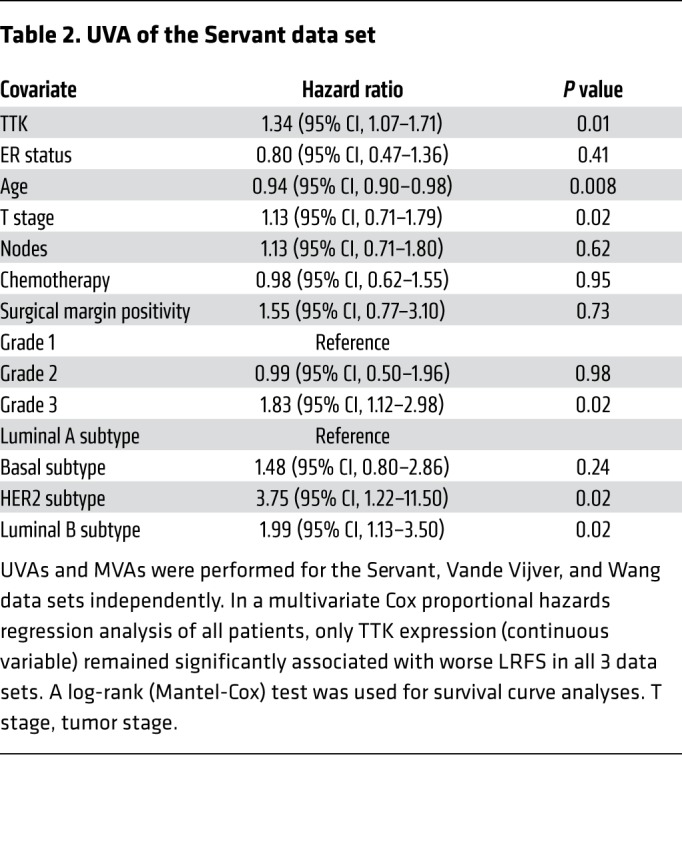
Table 3. MVA of the Servant data set.

Table 4. UVA of the Vande Vijver data set.
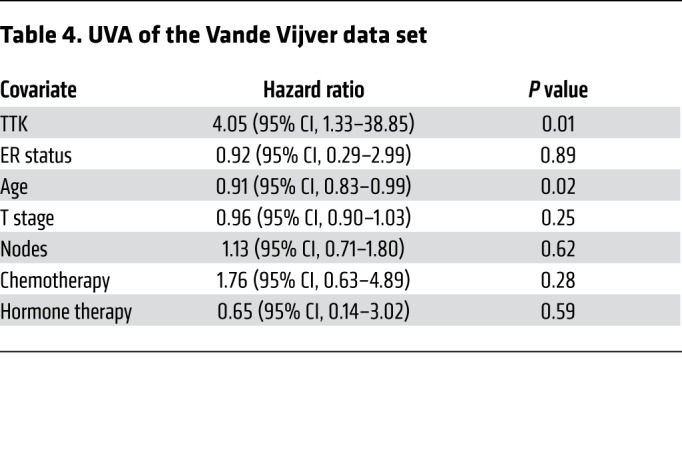
Table 5. MVA of the Vande Vijver data set.

Table 6. UVA of the Wang data set.
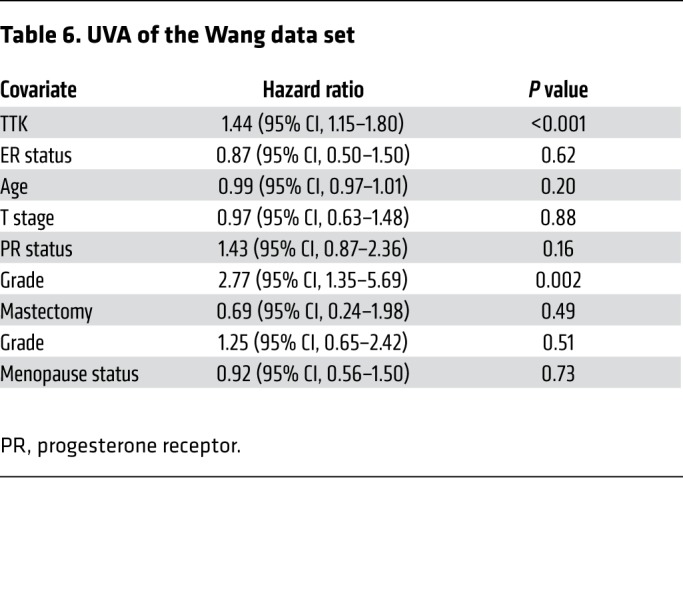
Table 7. MVA of the Wang data set.

We then evaluated TTK expression in multiple independent data sets to determine whether it is associated with any intrinsic subtype of BC. In each data set evaluated, TTK expression was significantly elevated in patients with ER– tumors compared with patients with ER+ tumors (P < 0.001; Supplemental Figure 1, D–F). Moreover, using the METABRIC (Molecular Taxonomy of Breast Cancer International Consortium) data set (14) (n = 1,986 patients) to evaluate TTK expression by BC-intrinsic subtype, we found that TTK expression was highest in the basal-like subtype and was significantly overexpressed in BC tissue versus healthy tissue (P < 0.0001; Figure 1E). Furthermore, in an institutionally assembled data set of BC metastatic tumors (MET500 patients; ref. 15), we found that TTK was significantly overexpressed in basal-like BC compared with other subtypes (Figure 1F). This association was also seen in The Cancer Genome Atlas (TCGA) BC data set (n = 945 patients) (Supplemental Figure 1G). Using RNA-Seq data for BC cell lines from the Cancer Cell Line Encyclopedia (CCLE), we found that TTK was overexpressed in ER– BC cell lines compared with expression in ER+ BC cell lines and was more highly expressed in basal-like cell lines than in HER2+ or luminal cell lines (P < 0.001; Supplemental Figure 1, H and I). We measured TTK protein expression in a panel of BC cell lines, which confirmed higher expression of TTK protein in basal-like BC cell lines, with MDA-MB-231 and BT-549 having the highest TTK protein expression levels (Supplemental Figure 1, J and K). Finally, we compiled the mutational landscapes of the cell lines used for further studies (Supplemental Figure 1L and ref. 16).
TTK inhibition radiosensitizes basal-like BC cell lines.
The correlation between TTK and early recurrence suggests that TTK may be involved in the RT response in BC. To further examine this, we performed gene set enrichment analysis (GSEA). Here, TTK expression was correlated with the expression of all other genes in the data set from TCGA and rank ordered by correlation coefficient. This gene list was then input for GSEA to identify pathways and networks associated with TTK expression. We found that cell-cycle genes in the ionizing radiation (IR) response at 6 and 24 hours were significantly enriched concepts at the top of the list (enrichment score >3.5 and P < 0.00001) (Supplemental Figure 2A). Negatively correlated concepts were related to ER+ and luminal BC, further validating our original nomination of TTK as being associated with basal-like BC (Supplemental Figure 2B). Together, these results indicate that TTK may be involved in the radiation response in BC and may function as a mediator of radiosensitivity.
To measure the effect of TTK perturbation on radiosensitivity in vitro, we used previously characterized radioresistant BC cell lines with high TTK expression (MDA-MB-231 and BT-549) (Supplemental Figure 1, J and K, and ref. 17). We performed clonogenic survival assays on stable, basal-like BC cell lines using doxycycline-inducible (Dox-inducible) shRNA to knock down TTK. Dox-induced TTK knockdown increased radiosensitivity in multiple shTTK stable clones in both MDA-MB-231 (radiation enhancement ratio [rER]: shTTK-1, 1.42–1.63; shTTK-2, 1.21–1.25) (Figure 2A) and BT-549 (rER for shTTK-1: 1.21–1.25; rER for shTTK-2: 1.21–1.26) (Figure 2C) cell lines. We also observed a significant decrease in the percentage of surviving cells after 2 Gy radiation (SF-2 Gy) in shTTK Dox+ compared with shTTK Dox– clones. We confirmed the knockdown of TTK protein with varying degrees of cytotoxicity in both cell lines (Figure 2, B and D, and Supplemental Figure 2, C and D). Additionally, we found that Dox treatment had no effect on radiosensitivity in shControl stable MDA-MB-231 or BT-549 cell lines (Figure 2, A–D).
Figure 2. Inhibition of TTK confers radiosensitivity in multiple basal-like BC cell lines with high baseline TTK expression.

(A and C) shRNA-induced TTK knockdown (shTTK) increased radiosensitivity in MDA-MB-231 (rER: 1.21–1.63) and BT-549 (rER: 1.21–1.26) cell lines. (B and D) The addition of Dox led to TTK knockdown in multiple stable clones in MDA-MB-231 and BT-549 cell lines. (E–G) Pharmacological inhibition of TTK induced radiosensitivity of MDA-MB-231 cells (rER: 25 nM 1.15–1.18, 37.5 nM 1.23–1.29, 50 nM 1.34–1.39), BT-549 cells (rER: 25 nM 1.10–1.17, 37.5 nM 1.11–1.30, 50 nM 1.23–1.39), and SUM-159 cells (rER: 25 nM 1.11–1.26, 37.5 nM 1.35–1.64, 50 nM 1.74–2.27) in a dose-dependent fashion. Data represent the mean of 3 independent experiments, and error bars represent SEM for clonogenic assays and SD for SF-2 Gy. A 2-sided Student’s t test was used for comparison of shRNA clonogenic assays, and a 1-way ANOVA with Dunnett’s multiple comparisons test was used for comparison of B909 clonogenic assays. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. shCon, shControl.
To confirm that TTK kinase function, and not just protein structural or scaffolding function, is important in mediating the response to RT in basal-like BC models, we performed clonogenic assays with the ATP-competitive TTK inhibitor Bayer 1161909 empesertib (hereafter referred to as B909), which is currently in clinical development (18). This drug was chosen, as it is currently the only TTK inhibitor in phase I/II clinical trials and because the target specificity, pharmacodynamics, and pharmacokinetics have already been well established (18). The drug doses used for radiosensitization studies were approximately half of the IC50 of proliferation in order to evaluate radiosensitization, and not single-agent antiproliferative effects (Supplemental Figure 2, E–G). We confirmed TTK inhibition by Western blot analysis of phospho–histone 3 (Ser10), a reported marker of functional TTK (Supplemental Figure 2, H and I, and ref. 19). A dose-dependent decrease in SF-2 Gy was also observed in all cell lines, and B909 caused varying degrees of cytotoxicity in all cell lines (Supplemental Figure 2, K–M). However, combination treatment of B909 and RT did not significantly decrease growth compared with B909 alone (Supplemental Figure 2J).
As a final, independent confirmation of radiosensitization, we performed clonogenic survival assays using an additional TTK inhibitor, NMS-P715 (19). TTK inhibition with NMS-P715 also increased radiosensitivity and significantly decreased the SF-2 Gy, further indicating that TTK kinase function is important for radioresistance (Supplemental Figure 2N). Cytotoxicity and rER with NMS-P715 are summarized in Supplemental Figure 2O.
TTK inhibition leads to persistent DNA damage after radiation.
Although radiosensitization can be induced through a number of mechanisms, we hypothesized that TTK-mediated radiosensitization may be due in part to decreased dsDNA damage repair efficiency. To evaluate the effect of TTK inhibition on dsDNA break repair, we measured γH2AX foci (more than 15 foci per cell), a marker for unresolved dsDNA damage, in cells treated with the TTK inhibitor, RT, or combination treatment over time (20). Using the MDA-MB-231 shTTK and BT-549 shTTK models, we measured γH2AX foci at various time points after treatment with DMSO, Dox (2 μg/mL) alone, RT (2 Gy) alone, or a combination of Dox and RT. Thirty minutes after RT, we detected equivalent levels of γH2AX+ cells in the cells treated with RT alone or with combined treatment (~70% in MDA-MB-231 shTTK and ~80% in BT-549 shTTK), whereas the nonirradiated cells had few γH2AX+ cells. Over time, the cells treated with RT alone repaired the RT-induced dsDNA damage more efficiently than did the combination treatment group at 4, 16, and 24 hours for MDA-MB-231 shTTK cells (Figure 3A) and at 4, 12, and 16 hours for BT-549 shTTK cells (Figure 3B), suggesting that TTK knockdown delayed dsDNA break repair efficiency. Representative images are shown of γH2AX, 24 hours after RT of MDA-MB-231 shTTK cells (Supplemental Figure 3A) and 16 hours after RT of BT-549 shTTK cells (Figure 3C).
Figure 3. TTK inhibition in combination with RT leads to persistent dsDNA damage over time.

(A and B) Combination treatment of Dox-inducible shRNA and RT led to persistent dsDNA damage over time in 2 basal-like BC cell lines: MDA-MB-231 (A) and BT-549 (B). (C) Representative images of BT-549 γH2AX foci at 16 hours. Original magnification, ×60. (D and E) Pharmacological inhibition of TTK kinase function, using B909, in combination with RT led to persistent dsDNA damage over time in 2 basal-like BC cell lines, MDA-MB-231 (D) and BT-549 (E). (F) Representative images of MDA-MB-231 γH2AX foci at 24 hours. Original magnification, ×60. Data represent the mean of 3 independent experiments repeated in triplicate, with approximately 100 cells counted for each experiment, and error bars represent SD. A 2-sided Student’s t test was used for comparison. *P < 0.05 and **P < 0.01
To assess the contribution of TTK kinase function in dsDNA break repair, we also measured unresolved dsDNA damage after treatment with B909 (75 nM) of MDA-MB-231 and BT-549 cell lines. Approximately 75% of cells treated with 2 Gy radiation were positive for γH2AX foci 30 minutes after radiation (Figure 3, D and E). As seen in the shTTK cell lines, combination treatment of B909 and RT resulted in persistent γH2AX foci+ cells over time in MDA-MB-231 cells (16 and 24 hours) (Figure 3D) and BT-549 (12 and 16 hours) (Figure 3E). Representative images are shown of γH2AX foci staining 24 hours after RT in MDA-MB-231 cells (Figure 3F) and 16 hours after RT in BT-549 cells (Supplemental Figure 4B).
Finally, we assessed dsDNA break repair using a second inhibitor, NMS-P715, in MDA-MB-231 cells. As previously observed, TTK inhibition in combination with RT, led to persistent γH2AX foci at 16 and 24 hours (Supplemental Figure 3B). Representative images of γH2AX foci at the 24-hour time point are shown in Supplemental Figure 3D. These results indicate that TTK inhibition may lead to radiosensitization of basal-like BC cell lines, at least in part as a result of impaired dsDNA damage repair.
TTK inhibition decreases HR-mediated DNA damage repair.
HR and nonhomologous end-joining (NHEJ) are the 2 prominent mechanisms in dsDNA repair. Although either may be involved in dsDNA break repair, previous reports suggested a potential correlation between TTK expression and HR (21). To further investigate these possible mechanisms of radiosensitization, we again performed GSEA by correlating gene expression with TTK expression. Here, we used the Kyoto Encyclopedia of Genes and Genomes (KEGG) to nominate cellular pathways related to TTK expression. In both the METABRIC and TCGA data sets, HR was significantly correlated with TTK gene expression and was listed among the top-8 positively correlated concepts (Figure 4, A and B). However, NHEJ was not significantly correlated with TTK expression in either data set. Thus, we hypothesized that TTK-mediated radiosensitization and persistent unresolved dsDNA breaks are due, at least in part, to decreased HR repair efficiency. Using stable cell lines with a well-characterized and validated HR-specific GFP reporter system, we tested the efficiency of HR after TTK knockdown (22–24). In both MDA-MB-231 and BT-549 cell lines, siRNA-mediated TTK knockdown significantly decreased HR efficiency compared with siNT (Figure 4, C and D). As controls, knockdown of Rad51, a key protein in the HR pathway, significantly decreased HR efficiency using this reporter system, whereas knockdown of XRCC6 (Ku70), a key protein in NHEJ, had no effect on HR efficiency (Figure 4, C and D, and ref. 25). To evaluate the dependence of this TTK-mediated HR repair on TTK kinase function, we used pharmacological TTK inhibition via B909 (50 nM and 75 nM). As with TTK knockdown, we found that treatment with B909 significantly decreased HR efficiency in MDA-MB-231 and BT-549 cells (Figure 4, E and F). Checkpoint kinases 1 and 2 (CHK1 and CHK2), which are critical proteins in the HR response, and DNA-dependent protein kinase (DNAPK), which is required for effective NHEJ repair, served as model system controls. As expected, pharmacologic inhibition of CHK1/-2 by AZD7762 (150 nM), an equipotent CHK1/-2 inhibitor, decreased HR efficiency, whereas the DNAPK inhibitor NU7441 (1.5 μM), which is not known to affect HR, had no effect on HR proficiency (Figure 4, E and F, and refs. 26–28). All experiments were repeated in a second clone to reduce clone-specific effects and confirm that TTK knockdown or inhibition decreases HR efficiency (Supplemental Figure 4, A–D).
Figure 4. TTK inhibition reduces HR efficiency.

(A and B) KEGG analysis through GSEA correlated the HR pathway with TTK expression in the METABRIC (A) and TCGA (B) data sets. (C and D) siRNA-induced TTK knockdown reduced HR efficiency in an HR-specific report system in MDA-MB-231 (C) and BT-549 (D) cells. (E and F) Inhibition of TTK kinase function, by B909 at 50 nM and 75 nM, reduced HR efficiency in MDA-MB-231 (E) and BT-549 (F) cells. TTKi, TTK inhibition; CHKi, CHK1/2 inhibition; DNAPKi, DNAPK inhibition. TTKi1, TTK inhibition with 50 nM B909; TTKi2, TTK inhibition with 75 nM B909. (G and I) TTK knockdown via a Dox-inducible shRNA reduced Rad51 foci formation after 4 Gy radiation in MDA-MB-231 (G) and BT-549 (I) cell lines. (H and J) Representative images of MDA-MB-231 (H) and BT-549 (J) Rad51 foci and Western blots showing no change in total Rad51 levels after Dox or RT treatment. Original magnification, ×60 (H and I). Data represent the mean of 3 independent experiments, and error bars represent SD. A 1-sided t test corrected for multiple comparisons was used for comparison of HR efficiency assays, and a 2-sided Student’s t test was used for comparisons of Rad51 foci experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. NES, normalized enrichment score.
To further corroborate our findings that TTK knockdown and inhibition decreased HR proficiency, we performed Rad51 foci formation assays after RT (4 Gy) in MDA-MB-231 shTTK and BT-549 shTTK stable cell lines. Rad51 foci formation is a marker for active HR repair; therefore, inhibition of Rad51 foci formation is indicative of impaired HR proficiency (25). In both cell lines, combination treatment with Dox (TTK knockdown) and RT resulted in a significant decrease in Rad51 foci formation at both early and late time points (MDA-MB-231: 6 and 24 hours, BT-549: 4 and 16 hours) compared with RT alone. We observed few Rad51 foci at either time point in cells treated with DMSO or Dox alone (Figure 4, G and I). Representative images at 24 hours (MDA-MB-231) and 16 hours (BT-549) are shown, and Western blot analyses showed that Rad51 protein expression was equal across all treatment groups, indicating that the decrease in Rad51 foci cannot be attributed to a more general decrease in Rad51 protein after TTK knockdown (Figure 4, H and J). These results indicate that RT induces Rad51 foci formation, whereas TTK knockdown inhibits this formation and likely leads to decreased HR efficiency.
Finally, we treated BT-549 cell lines with B909 alone, RT alone, or a combination of B909 and RT and found a decrease in both phosphorylated breast cancer 1 (p-BRCA1) and p-CHK1 after combined treatment of B909 and RT compared with RT alone (Supplemental Figure 4E). Decreased levels of p-BRCA1 and p-CHK1 are canonical markers of impaired HR efficiency. Additionally, combination treatment with B909 and RT decreased phosphorylated replication protein A (p-RPA) levels, indicating a disruption of the HR pathway and possibly a lack of Rad51 foci formation after TTK knockdown (Supplemental Figure 4F and ref. 29). This further indicates that TTK inhibition via B909 disrupts HR, which probably leads to increased radiosensitivity of basal-like BC cell lines.
TTK inhibition has no effect on NHEJ repair.
Although TTK inhibition decreased HR efficiency, we also tested the effect of TTK inhibition on NHEJ efficiency using a well-characterized and validated NHEJ-specific reporter plasmid system (30, 31). Dox-induced TTK knockdown in MDA-MB-231 and BT-549 shTTK cells did not decrease NHEJ efficiency, whereas treatment with the known DNAPK inhibitor NU7441 (1.5 μM) significantly decreased the efficiency of NHEJ in both cell lines (Figure 5, A and B). To evaluate the effect of TTK kinase function on NHEJ efficiency, we also treated MDA-MB-231 and BT-549 cells with B909 (50 nM and 75 nM). Neither B909 nor AZD7762 (CHK1/-2 inhibitor) affected NHEJ efficiency, whereas NU7441 (DNAPK inhibitor) significantly decreased the efficiency of NHEJ (Figure 5, C and D).
Figure 5. TTK knockdown has no effect on NHEJ repair efficiency.

(A and B) TTK knockdown via Dox-inducible shRNA had no effect on NHEJ efficiency in MDA-MB-231 (A) or BT-549 (B) cells, whereas DNAPK inhibition significantly reduced NHEJ efficiency. (C and D) Pharmacologic inhibition of TTK kinase, using B909 at 50 nM and 75 nM, had no effect on NHEJ, whereas DNAPK inhibition significantly reduced NHEJ efficiency in MDA-MB-231 (C) and BT-549 (D) cell lines. (E and F) Inhibition of TTK with B909 had no effect on p-Ku80 (Thr714) levels. Data represent the mean of 3 independent experiments, and error bars represent SD. A 2-sided Student’s t test was used for comparison of shTTK NHEJ assays, and a 1-way ANOVA with Dunnett’s multiple comparisons test was used for comparison of B909 NHEJ assays. *P < 0.05 and **P < 0.01.
Additionally, we treated MDA-MB-231 and BT-549 cells with B909 alone, RT alone, or a combination of B909 and RT to determine whether canonical NHEJ phosphorylated proteins were affected. In both MDA-MB-231 and BT-549 cells, we found no difference in p-Ku80 (Thr714) levels following combined treatment of B909 and RT compared with RT alone (Figure 5, E and F). Together, these results confirm that TTK inhibition has no effect on NHEJ repair efficiency.
Kinase-dead TTK does not rescue the radiosensitivity phenotype.
To validate that inhibition of TTK kinase function is responsible for the radiosensitization phenotype previously observed, we performed clonogenic survival assays using siRNAs to deplete endogenous TTK expression while reintroducing siRNA-resistant wild-type (WT) or kinase-dead (KD) TTK. As previously demonstrated, knockdown of TTK using siRNAs significantly (P < 0.01) increased radiosensitivity in both MDA-MB-231 and SUM-159 cell lines. Reexpression of WT TTK restored radioresistance in both cell lines; however, reexpression of KD TTK did not restore radioresistance in either cell line (Figure 6, A and B). Western blot analyses showed that the siRNA dramatically reduced endogenous TTK expression. It also confirmed robust expression of WT and KD TTK upon reintroduction using siRNA-resistant constructs (Supplemental Figure 6A). A summary of cytotoxicity and rER is provided in Supplemental Figure 5, B and C. Together, these results confirm that the kinase function of TTK is essential for the radioresistance phenotype observed in basal-like BC models.
Figure 6. After knockdown of TTK, WT TTK rescues the radiosensitization phenotype, but KD TTK does not.

(A and B) siRNA-induced TTK knockdown led to radiosensitization of MDA-MB-231 (A) and SUM-159 (B) cell lines. The addition of WT TTK rescued this phenotype, whereas KD TTK did not. (C and D) Knockdown of TTK using a siRNA decreased HR efficiency, whereas the reintroduction of WT TTK rescued HR efficiency. However, reintroduction of KD TTK did not rescue HR efficiency in MDA-MB-231 (C) or BT-549 (D) cell lines. (E) siRNA-induced TTK knockdown decreased Rad51 foci formation, however, reintroduction of WT TTK rescued Rad51 foci formation. Introduction of KD TTK was unable to rescue Rad51 foci formation. (F) Representative images of Rad51 foci at 4 hours. Original magnification, ×60.Data represent the mean of 3–4 independent experiments, and error bars represent SD. A 1-way ANOVA with Dunnett’s multiple comparisons test was applied. *P < 0.05, **P < 0.01, and ****P < 0.0001. Lipo, Lipofectamine.
We also wanted to confirm that inhibition of TTK kinase function was responsible for the impaired HR phenotype. Using the stable HR GFP reporter cell lines described above, we measured the impact of TTK knockdown using siRNA as well as the reintroduction of WT and KD TTK. In both MDA-MB-231 and BT-549 cell lines, we found that reexpression of WT TTK rescued HR efficiency, whereas expression of KD TTK was not able to rescue HR competency after siRNA-mediated TTK knockdown of TTK (Figure 6, C and D). To further confirm, the role of TTK kinase function on HR, we conducted Rad51 foci formation experiments. Forty-eight hours before RT (4 Gy), BT-549 cells were pretreated with one of the following: Lipofectamine alone, an siRNA targeting TTK, an siRNA plus WT TTK, or an siRNA plus KD TTK. TTK knockdown resulted in a significant decrease in Rad51 foci formation (Figure 6E). However, reexpression of WT TTK after knockdown rescued Rad51 foci formation, but reexpression of KD TTK was unable to rescue appropriate Rad51 foci formation (Figure 6E). Representative images of Rad51 foci 4 hours after RT are shown in Figure 6F.
TTK knockdown or inhibition reduces tumor growth in vivo.
Once we established that TTK knockdown or inhibition leads to radiosensitization of basal-like BC in vitro and that this effect is mediated, at least in part, by decreased dsDNA break repair efficiency through HR, we sought to determine whether inhibition of TTK in vivo similarly leads to radiosensitization. We initially used the MDA-MB-231 shTTK–knockdown model in heterotopic xenograft studies by injecting cells subcutaneously into the flanks of female mice. After the tumors were established and grew to approximately 100 mm3 in size, the mice received either no treatment, TTK knockdown with Dox, RT alone, or a combination of TTK knockdown with Dox and RT. RT was administered in 6 doses of 2 Gy over 6 days, beginning 72 hours after the initial Dox treatment to knock down TTK (Figure 7A). Combination treatment significantly reduced relative tumor growth compared with no treatment, TTK knockdown with Dox, or RT alone (Figure 7B). The time to tumor tripling significantly increased for combination treatment (undefined) compared with no treatment (14 days), TTK knockdown with Dox (19 days), or RT (17.5 days) (Figure 7C). Mouse weights were not significantly different between treatment groups in this study (Supplemental Figure 6A). To confirm that the addition of Dox reduced TTK expression, we performed immunohistochemical analyses of tumor samples and found that Dox-induced TTK knockdown significantly reduced TTK expression in the shTTK Dox+ tumors compared with the shTTK Dox– tumors (Figure 7, D and E). Finally, using the fractional tumor volume (FTV) method to measure synergy between treatments, we found that a combination of TTK knockdown and RT led to at least an additive, if not superadditive, effect (ratio [R] <1) (Supplemental Figure 6B).
Figure 7. Combination treatment of TTK inhibition and RT reduces basal-like BC tumor growth in vivo.

(A) Model of treatment schedule for in vivo studies. (B) Dox-inducible MDA-MB-231 shTTK cells had decreased tumor growth following a combination of Dox and RT (n = 16) compared with TTK knockdown (n = 15) or RT (n = 16) alone. (C) Combination treatment (RT plus Dox) leads to increased time to tumor tripling in vivo. (D) Immunohistochemistry from shTTK in vivo model depicts success knockdown of TTK after the addition of Dox. Original magnification, ×1 and ×15 (enlarged insets). (E) Average percentage of TTK+ cells across 4 tumors from the shTTK in vivo model, with and without Dox. (F) TTK inhibition using combination treatment of B909 (1 mg/kg) and RT led to decreased tumor growth and (G) increased time to tumor tripling in MDA-MB-231 BC cells (n = 16 tumors per group). (H) In an orthotopic PDX model, combined treatment of B909 (2.5 mg/kg) and RT decreased tumor growth compared with placebo, B909 only, or RT only. (I) Combination treatment with B909 and RT led to increased time to tumor tripling. A 1-way ANOVA with Dunnett’s multiple comparisons test and log-rank (Mantel-Cox) test were used for analyses. Error bars indicate the SEM. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
To establish that Dox has no effect on tumor growth and to determine that the decrease in tumor growth previously seen was not an artifact of a single shTTK clone, we performed a second study with 4 independent, stable MDA-MB-231 shRNA groups (shControl Dox–, shControl Dox+, shTTK-2 Dox+, and shTTK-2 Dox+ plus RT). We observed no difference in tumor growth between shControl with or without Dox, indicating that Dox alone had no effect on tumor growth (Supplemental Figure 6, C and D). As seen in our previous xenograft study, a combination of TTK knockdown and RT decreased tumor growth and increased time to tumor tripling (34 days) compared with shControl Dox– (10.5 days), shControl Dox+ (13 days), and shTTK-2 Dox+ (15 days) (Supplemental Figure 6, C and D). Again, mouse weights were similar in all groups (Supplemental Figure 6E).
To assess the role of TTK kinase function, and not just protein expression, in radiosensitization in vivo, we performed a xenograft study using the clinical-grade TTK inhibitor B909 to test whether TTK kinase inhibition would also decrease tumor growth and increase time to tumor tripling. Using a similar design scheme, we treated mice with either placebo (critical micellar concentration–Tween-80 [CMC–Tween-80]), RT alone, B909 (1 mg/kg) alone, or a combination of therapies (RT plus B909). Combination treatment significantly decreased relative tumor growth and significantly increased the time to tumor tripling (undefined number of days) compared with placebo (11 days), RT alone (22 days), or B909 alone (15 days) (Figure 7, F and G). Interestingly, 19% of tumors in the combination treatment group remained stable in size and were not growing even at the time of study completion (38 days), suggesting a sustained, durable response even weeks after the completion of therapy. As with TTK knockdown, combination treatment of B909 and RT had little effect on mouse weights (Supplemental Figure 6F). Furthermore, using the FTV method to measure synergy between treatments, we found that inhibition of TTK kinase function had a synergistic effect with RT (R >1) (Supplemental Figure 6G).
To confirm these findings independently, we used an orthotopic PDX model to test the efficacy of B909 plus RT. In this model, we implanted basal-like BC PDX (the PDX mutations are listed in Supplemental Figure 1L) tumors into the mammary fat pads of mice and allowed them to grow to approximately 100 mm3 in size. The mice received either placebo treatment (CMC–Tween-80), RT alone, B909 (2.5 mg/kg) alone, or combination therapy (RT plus B909). In agreement with the previous animal studies, combination treatment led to a significant decrease in tumor growth and increased time to tumor tripling (undefined number of days) compared with placebo (9 days), RT alone (22 days), or B909 alone (13 days) (Figure 7, H and I). Combination treatment of B909 and RT did not cause weight loss in mice, however, mice that received either placebo or B909 alone gained weight throughout the study, a finding that can be attributed to the growth of the PDX tumors (Supplemental Figure 6H). As seen in our previous study, combination treatment of B909 and RT was synergistic (R >1) and led to tumor regression in many mice (Supplemental Figure 6I). These results indicate that TTK inhibition, using B909 in combination with RT, inhibits tumor growth and delays the time to tumor tripling. Together, our findings in multiple nonoverlapping models suggest that the combination treatment of B909 and RT may be a feasible strategy for the treatment of patients with basal-like BC with a high risk of LR.
Discussion
In this study, we unbiasedly nominated TTK as the gene most strongly correlated with BC recurrence after radiation in 4 independent patient data sets (Figure 1). TTK expression is strongly associated with triple-negative breast cancer/basal-like (TNBC/basal-like) BC subtypes. Using both genetic (shRNA and siRNA) and pharmacologic (NMS-P715 and Bayer 1161909 empesertib) TTK inhibition, we induced radiosensitization in multiple basal-like BC cell lines (Figure 2). We found that TTK inhibition led to persistent, unresolved dsDNA damage over time (Figure 3). TTK knockdown or inhibition led to impaired HR with no effect on NHEJ, and impaired HR was responsible, at least in part, for increased radiosensitivity of basal-like BC cell lines (Figures 4 and 5). We also show that inhibition of TTK kinase function was responsible for increased radiosensitivity and loss of HR efficiency through the use of WT and KD TTK reexpression after endogenous TTK knockdown (Figure 6). In vivo, both genetic and pharmacologic TTK inhibition decreased tumor growth and increased the time to tumor tripling in both cell line and orthotopic PDX models (Figure 7). Together, these results demonstrate that TTK inhibition, in combination with RT, is a potentially effective strategy for the radiosensitization of basal-like BC that may ultimately lead to decreased rates of recurrence for patients.
In our nomination of novel targets for the radiosensitization of BC, we found TTK, also known as Mps1, to be the top target for radiosensitization of basal-like BC. TTK is overexpressed in various cancers and has previously been studied as a target for the treatment of BC, glioblastoma, ovarian cancer, colon cancer, and others (18, 19, 32–35). TTK has been well characterized for its role in the spindle assembly checkpoint (SAC) complex, which prevents progression from metaphase to anaphase in mitosis when problems occur in metaphase (36–40). Previously, TTK inhibition has been shown to cause irregular mitosis as well as increased aneuploidy, lagging chromosomes, and mitotic catastrophe (41, 42). Given these findings, previous groups have focused on TTK inhibition as a monotherapy, in combination with conventional chemotherapies, or in combination with anti–programmed cell death 1 (anti–PD-1) antibodies (18, 33, 41, 43). To date, however, few studies have characterized the role of TTK in the radiation response or as a possible combination therapy with RT (32). Additionally, previous studies have implicated TTK in HR- and NHEJ-mediated dsDNA break repair, although these studies suggest that TTK may be more strongly linked to the HR pathway than to the NHEJ pathway (21, 32). Our results add to this growing body of literature and are the first to our knowledge to suggest that TTK inhibition is a viable strategy for the radiosensitization of basal-like BC. We also demonstrate for the first time to our knowledge that this radiosensitivity in BC is mediated, at least in part, through impaired HR repair.
Our nomination process, in 4 distinct BC data sets, identified 10 genes that are correlated with recurrence in patients treated with radiation. In addition to the top nominated gene (TTK), multiple other identified genes (including EZH2 and KPNA2) have previously been associated with recurrence and radioresistance in various cancers (44, 45). This suggests that our unbiased approach to nominate novel mediators of recurrence was rational and effective. Although we only studied the effect of TTK inhibition on radiosensitization in this study, these additional genes may also be strong targets for the radiosensitization of BC and warrant further investigation.
Here, we show that TTK kinase function mediates HR competency; however, there are probably additional mechanisms for radiosensitization that are influenced by TTK. Increases in mitotic catastrophe, aneuploidy, and cell-cycle defects have previously been linked to TTK inhibition and are likely to contribute to the observed radiosensitization (32, 33, 40, 42, 46). In our study, we used 3 basal-like BC cell lines and a PDX model that were all BRCA1 WT in order to asses HR competency. We hypothesize that TTK inhibition may also have utility in BRCA1-mutant BC cell lines through its role in the SAC complex and not through impaired HR. However, further studies are necessary to validate this hypothesis.
We have demonstrated that TTK inhibition had no effect on NHEJ efficiency (Figure 5), despite other reports showing reduced NHEJ efficiency after TTK inhibition (32). This discrepancy may be explained by the different model systems and different pharmacological inhibitors used. For example, previous studies reporting that TTK inhibition led to impaired NHEJ used the TTK inhibitor NMS-P715 (32). However, at higher doses, NMS-P715 also inhibits maternal embryonic leucine zipper kinase (MELK), a kinase that our laboratory has shown to be critical to the NHEJ pathway (19). These off-target effects may have caused the decreased NHEJ efficiency seen in their study. However, further research needs to be performed in multiple nonoverlapping models to adequately address this concern. Furthermore, although we demonstrate that TTK inhibition led to a decrease in HR repair efficiency, we have not yet identified the mechanism by which this occurs. Additional studies are currently underway to understand how TTK interacts with proteins in the HR pathway and how TTK inhibition leads to decreased HR efficiency.
LRs after radiation remain a significant issue for women with basal-like BC, as the molecular drivers of these radioresistant recurrences are currently unclear. This study identifies TTK as a potential molecular mediator of radioresistance in basal-like BC. These data suggest that utilizing TTK inhibitors in combination with radiation may lead to improved rates of local control and disease cure for women with basal-like BC with high TTK expression. Future studies by our group and others will test this hypothesis in clinical trials, with the goal of improving local control and survival in women with these aggressive forms of BC.
Methods
Gene nomination.
Four independent data sets of primary tumor samples from women with BC with associated, curated recurrence data and gene expression were used for nomination (Wang [ref. 47], Desmedt [ref. 48], van ‘t Veer [ref. 49], and Schmidt [ref. 50]). Using Oncomine.org for analysis and “invasive ductal breast carcinoma – recurrence at 3 years – top 1% overexpressed” as a primary concept filter, we identified the genes whose expression was significantly correlated to a recurrence event within 3 years of diagnosis, with an OR of greater than 2 and a multiple testing corrected P value of less than 0.000001 in each data set as originally reported (51). The identified genes from each data set were then compared for overlap in all 4 data sets to generate the final list of genes for further investigation.
GSEA.
Gene expression was correlated to TTK expression in the TCGA BC data sets and ranked by correlation coefficient. The settings used in the GSEA were c2.all.v6.symbols.gmt (curated) as the gene sets database, 1000 permutations, and the minimum size was 15. For GSEA KEGG analysis, both METABRIC and TCGA were used with c2.cp.kegg.v7.0.symbols.gmt, 1000 permutations, and a minimum size of 10 (52, 53).
Cell culture.
The basal-like BC cell lines MDA-MB-231 and BT-549 were grown from frozen samples (American Type Culture Collection [ATCC]). MDA-MB-231 cells were grown in DMEM (Invitrogen, Thermo Fisher Scientific) supplemented with 10% FBS (Invitrogen) and penicillin/streptomycin (Invitrogen). BT-549 cells were grown in RPMI 1640 (Invitrogen) with 10% FBS (Invitrogen). SUM-159 cells were originally sourced from Steve P. Either (University of Michigan) and were obtained internally from Sofia Merajver (University of Michigan). SUM-159 cells were grown in HAMS F-12 media (Thermo Fisher Scientific) supplemented with 5% FBS (Invitrogen), 5 mg/mL human insulin (MilliporeSigma), 10 mmol/L HEPES (Thermo Fisher Scientific), 1 mg/kg hydrocortisone (MilliporeSigma), and an antibiotic-antimycotic. All cell lines were grown in a 5% CO2 incubator. Cells were passaged at approximately 70% confluence. Cell lines were authenticated at the University of Michigan DNA Sequencing core facility before use and tested for mycoplasma routinely (MycoAlert, Lonza).
Clonogenic survival assays.
Exponentially growing cells were plated in 6-well plates overnight before treatment with Dox, the drug, siRNA, or TTK plasmids. Cells were pretreated with, Dox (2 μg/mL) for 36 hours, with the drug for 2 hours, siRNA for approximately 8–24 hours, and TTK plasmids for 24 hours before RT. Cells were allowed to grow for 7–14 days before being fixed (7:1, methanol/acetic acid) and stained (crystal violet). Fifty or more cells constituted a colony. A linear-quadratic survival curve was fitted to each assay, as previously described (54).
Western blot analysis.
Cells were washed twice with ice-cold PBS and lysed with RIPA buffer (Thermo Fisher Scientific) supplemented with phosSTOP (Roche) and cOmplete Mini Protease Inhibitors (MilliporeSigma). Western blot analysis was performed as previously described (55). Specific antibody information and dilutions are provided in Supplemental Table 1.
Transfections, siRNAs, shRNAs, and plasmids.
siRNAs and shRNAs were ordered from Dharmacon and are listed in Supplemental Table 1. siRNAs were transfected using Lipofectamine RNAiMax (Invitrogen) and Opti-MEM media (Invitrogen). WT and KD TTK plasmids were provided by the Yu laboratory and contained a 6x Myc tag (56). Specific information on siRNAs, shRNAs, and plasmids is provided in Supplemental Table 1.
Irradiation.
Irradiation was performed using a Kimtron IC 225 (Kimtron Medical) at a dose rate of approximately 2 Gy/min at the University of Michigan Comprehensive Cancer Center Experimental Irradiation Core (Ann Arbor, Michigan, USA). Dosimetry was performed semiannually using an ionization chamber connected to an electrometer system that was directly traceable to a National Institute of Standards and Technology calibration. The beam was collimated with a 0.1-mm Cu inherent filter, and a 0.2-mm Cu filter was used for cell line irradiation. A 2-mm Cu filter was used for in vivo xenograft experiments.
γH2AX and Rad51 foci formation assay.
γH2AX and Rad51 foci were analyzed as previously described (57). Cells with more than 15 γH2AX foci or more than 10 Rad51 foci were scored as positive. Blinded analysis was performed when counting cells positive for foci. The list of antibodies used is provided in Supplemental Table 1.
HR reporter assay.
Cells were transfected with the HR reporter DR-GFP plasmid using Lipofectamine 2000 (Invitrogen) following the manufacturer’s recommendations, with Geneticin (Thermo Fisher Scientific) selection and validation by flow cytometry for GFP+ cells after I-SceI treatment (58). Validated clones were plated in a 6-well plate, pretreated with the indicated siRNA for 24 hours, drug for 1 hour, or TTK plasmids for 48 hours. The SceI adenovirus was added to the cells for 48 hours, and cells were harvested and sorted by flow cytometry for GFP+ cells, which indicated successful HR.
NHEJ reporter assay.
The pEYFP plasmid (gift from the Canman Laboratory, University of Michigan, Ann Arbor, Michigan, USA) was completely digested with 20 U NheI-HF and purified using a QIAquick PCR Purification Kit (QIAGEN, catalog 28104). Cells (1.0 × 105 cells per well) were plated in 6-well plates. The following day, 1 μg of the digested pEYFP plasmid was transfected per well. One hour before transfection, the cells were treated with either the TTK inhibitor or DMSO. Cells were harvested at the indicated time points, and the DNA plasmids were isolated with the QIAprep Spin Miniprep Kit (QIAGEN, catalog 27106). SYBR Green (Thermo Fisher Scientific) real-time quantitative PCR was conducted in triplicate in a 384-well PCR system, using DNA as a template. The specific primers used are listed in Supplemental Table 1. Relative DNA repair efficiency was calculated by the comparative method normalized to the Ct value of the internal control.
Mouse xenograft experiments.
Cells were injected subcutaneously into the bilateral flanks or orthotopically into the mammary fat pads of CB-17 SCID female mice that were originally sourced from Charles River Laboratories and maintained in a breeding colony at the University of Michigan. Tumors were allowed to grow to approximately 100 mm3 in size and were randomized before treatment began. Dox (1 μg/mL) was given through the drinking water to induce shTTK cells 72 hours prior to the beginning of RT. Bayer 1161909 (B909) was administered at a dose of 1 mg/kg twice a day for 2 days over a 4-week period, 1 day prior to the start of RT. RT was administered in 6 doses of 2 Gy. Tumor size was measured 3 times a week using a digital caliper, and tumor volume was calculated using the equation: volume = (length × width2) × π/6. Additive and synergistic effects were calculated using the FTV method as previously described (59, 60).
Study approval.
The protocols used in this study were approved by the IACUC of the University of Michigan.
Statistics.
Statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software). A log-rank (Mantel-Cox) test was used for survival curve analyses. One-way ANOVA was performed for BC subtype analysis. A 2-sided Student’s t test and 1-way ANOVA with Dunnett’s multiple comparisons test were used for in vitro statistical analyses. A 1-way ANOVA with Dunnett’s multiple comparisons test and a log-rank (Mantel-Cox) test were used for in vivo analyses. A P value of 0.05 or less was considered statistically significant.
Author contributions
BCC and CS conceived, designed, and developed the study and designed the methodology. BCC, LM, CLR, ML, MC, KWR, AZ, AMP, ARM, NH, MA, TW, EO, YSN, and DGT acquired data. BCC, LM, CLR, ML, MC, KWR, AMP, ARM, EO, YSN, DGT, and SM analyzed and interpreted data. BCC, LM, CLR, MC, KWR, AZ, AMP, ARM, NH, MA, EO, YSN, PB, TSL, SN, LJP, AC, and CS contributed to the writing, review, and/or revision of the manuscript. BCC, CS, LJP, and SM provided administrative, technical, or material support. BCC and CS supervised the study.
Supplementary Material
Acknowledgments
The authors thank Meredith Morgan for lending us the HR reporter construct and CHK1/-2 inhibitor (AZD7762), Christine Canman for the pEYFP plasmid, Hongtao Yu for the WT and KD TTK plasmids, and Steven Kronenberg for his assistance in creating figures and illustrations. This work was supported by a grant from the ASTRO-BCRF Career Development Award to End Cancer and the University of Michigan Rogel Cancer Center (P30CA046592, to CS). BCC, AMP, and ARM are supported by training grants through the NIH. BCC is supported by the National Cancer Institute (NCI) (T32-CA140044 and T32-CA009676); AMP is supported by the National Institute of General Medical Sciences (NIGMS) (T32-GM007767); and ARM is supported by the NIGMS (T-32 GM113900 and T32-GM007315).
Version 1. 01/21/2020
Electronic publication
Version 2. 02/03/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(2):958–973.https://doi.org/10.1172/JCI130435.
Contributor Information
Leah Moubadder, Email: leahmoubadder@gmail.com.
Cassandra L. Ritter, Email: cassanri@umich.edu.
Meilan Liu, Email: lmiu@med.umich.edu.
Meleah Cameron, Email: cameronmeleah@gmail.com.
Kari Wilder-Romans, Email: kariwi@med.umich.edu.
Amanda Zhang, Email: amanzhan@umich.edu.
Andrea M. Pesch, Email: ampesch@umich.edu.
Anna R. Michmerhuizen, Email: annamich@umich.edu.
Nicole Hirsh, Email: nhirsh@umich.edu.
Marlie Androsiglio, Email: marandro@umich.edu.
Eric Olsen, Email: olsene0101@gmail.com.
Powel H. Brown, Email: phbrown@mdanderson.org.
Shyam Nyati, Email: shyamnya@med.umich.edu.
Lori J. Pierce, Email: ljpierce@umich.edu.
Corey Speers, Email: cspeers@med.umich.edu.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68(1):7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- 2.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG) et al. Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet. 2011;378(9804):1707–1716. doi: 10.1016/S0140-6736(11)61629-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Caan BJ, et al. Intrinsic subtypes from the PAM50 gene expression assay in a population-based breast cancer survivor cohort: prognostication of short- and long-term outcomes. Cancer Epidemiol Biomarkers Prev. 2014;23(5):725–734. doi: 10.1158/1055-9965.EPI-13-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Horton JK, Jagsi R, Woodward WA, Ho A. Breast cancer biology: clinical implications for breast radiation therapy. Int J Radiat Oncol Biol Phys. 2018;100(1):23–37. doi: 10.1016/j.ijrobp.2017.08.025. [DOI] [PubMed] [Google Scholar]
- 5.Lowery AJ, Kell MR, Glynn RW, Kerin MJ, Sweeney KJ. Locoregional recurrence after breast cancer surgery: a systematic review by receptor phenotype. Breast Cancer Res Treat. 2012;133(3):831–841. doi: 10.1007/s10549-011-1891-6. [DOI] [PubMed] [Google Scholar]
- 6.Kyndi M, et al. Estrogen receptor, progesterone receptor, HER-2, and response to postmastectomy radiotherapy in high-risk breast cancer: the Danish Breast Cancer Cooperative Group. J Clin Oncol. 2008;26(9):1419–1426. doi: 10.1200/JCO.2007.14.5565. [DOI] [PubMed] [Google Scholar]
- 7.Sjöström M, et al. Response to radiotherapy after breast-conserving surgery in different breast cancer subtypes in the Swedish Breast Cancer Group 91 Radiotherapy Randomized Clinical Trial. J Clin Oncol. 2017;35(28):3222–3229. doi: 10.1200/JCO.2017.72.7263. [DOI] [PubMed] [Google Scholar]
- 8.Perou CM, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 9.Prat A, Parker JS, Fan C, Perou CM. PAM50 assay and the three-gene model for identifying the major and clinically relevant molecular subtypes of breast cancer. Breast Cancer Res Treat. 2012;135(1):301–306. doi: 10.1007/s10549-012-2143-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Speers C, et al. Identification of novel kinase targets for the treatment of estrogen receptor-negative breast cancer. Clin Cancer Res. 2009;15(20):6327–6340. doi: 10.1158/1078-0432.CCR-09-1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balbous A, et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC Cancer. 2016;16:604. doi: 10.1186/s12885-016-2647-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Speers C, et al. Maternal embryonic leucine zipper kinase (MELK) as a novel mediator and biomarker of radioresistance in human breast cancer. Clin Cancer Res. 2016;22(23):5864–5875. doi: 10.1158/1078-0432.CCR-15-2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei D, et al. Inhibition of protein phosphatase 2A radiosensitizes pancreatic cancers by modulating CDC25C/CDK1 and homologous recombination repair. Clin Cancer Res. 2013;19(16):4422–4432. doi: 10.1158/1078-0432.CCR-13-0788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curtis C, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486(7403):346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Robinson DR, et al. Integrative clinical genomics of metastatic cancer. Nature. 2017;548(7667):297–303. doi: 10.1038/nature23306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehmann BD, et al. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121(7):2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Speers C, Zhao S, Liu M, Bartelink H, Pierce LJ, Feng FY. Development and validation of a novel radiosensitivity signature in human breast cancer. Clin Cancer Res. 2015;21(16):3667–3677. doi: 10.1158/1078-0432.CCR-14-2898. [DOI] [PubMed] [Google Scholar]
- 18.Wengner AM, et al. Novel Mps1 kinase inhibitors with potent antitumor activity. Mol Cancer Ther. 2016;15(4):583–592. doi: 10.1158/1535-7163.MCT-15-0500. [DOI] [PubMed] [Google Scholar]
- 19.Colombo R, et al. Targeting the mitotic checkpoint for cancer therapy with NMS-P715, an inhibitor of MPS1 kinase. Cancer Res. 2010;70(24):10255–10264. doi: 10.1158/0008-5472.CAN-10-2101. [DOI] [PubMed] [Google Scholar]
- 20.Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 21.Peng G, et al. Genome-wide transcriptome profiling of homologous recombination DNA repair. Nat Commun. 2014;5:3361. doi: 10.1038/ncomms4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999;13(20):2633–2638. doi: 10.1101/gad.13.20.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci USA. 1994;91(13):6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weinstock DM, Nakanishi K, Helgadottir HR, Jasin M. Assaying double-strand break repair pathway choice in mammalian cells using a targeted endonuclease or the RAG recombinase. Methods Enzymol. 2006;409:524–540. doi: 10.1016/S0076-6879(05)09031-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cruz C, et al. RAD51 foci as a functional biomarker of homologous recombination repair and PARP inhibitor resistance in germline BRCA-mutated breast cancer. Ann Oncol. 2018;29(5):1203–1210. doi: 10.1093/annonc/mdy099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zabludoff SD, et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol Cancer Ther. 2008;7(9):2955–2966. doi: 10.1158/1535-7163.MCT-08-0492. [DOI] [PubMed] [Google Scholar]
- 27.Leahy JJ, et al. Identification of a highly potent and selective DNA-dependent protein kinase (DNA-PK) inhibitor (NU7441) by screening of chromenone libraries. Bioorg Med Chem Lett. 2004;14(24):6083–6087. doi: 10.1016/j.bmcl.2004.09.060. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y, et al. Preclinical evaluation of a potent novel DNA-dependent protein kinase inhibitor NU7441. Cancer Res. 2006;66(10):5354–5362. doi: 10.1158/0008-5472.CAN-05-4275. [DOI] [PubMed] [Google Scholar]
- 29.Shi W, et al. The role of RPA2 phosphorylation in homologous recombination in response to replication arrest. Carcinogenesis. 2010;31(6):994–1002. doi: 10.1093/carcin/bgq035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Q, Karnak D, Tan M, Lawrence TS, Morgan MA, Sun Y. FBXW7 facilitates nonhomologous end-joining via K63-linked polyubiquitylation of XRCC4. Mol Cell. 2016;61(3):419–433. doi: 10.1016/j.molcel.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deans AJ, Khanna KK, McNees CJ, Mercurio C, Heierhorst J, McArthur GA. Cyclin-dependent kinase 2 functions in normal DNA repair and is a therapeutic target in BRCA1-deficient cancers. Cancer Res. 2006;66(16):8219–8226. doi: 10.1158/0008-5472.CAN-05-3945. [DOI] [PubMed] [Google Scholar]
- 32.Maachani UB, et al. Targeting MPS1 enhances radiosensitization of human glioblastoma by modulating DNA repair proteins. Mol Cancer Res. 2015;13(5):852–862. doi: 10.1158/1541-7786.MCR-14-0462-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faisal A, et al. Characterisation of CCT271850, a selective, oral and potent MPS1 inhibitor, used to directly measure in vivo MPS1 inhibition vs therapeutic efficacy. Br J Cancer. 2017;116(9):1166–1176. doi: 10.1038/bjc.2017.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ling Y, et al. Overexpression of Mps1 in colon cancer cells attenuates the spindle assembly checkpoint and increases aneuploidy. Biochem Biophys Res Commun. 2014;450(4):1690–1695. doi: 10.1016/j.bbrc.2014.07.071. [DOI] [PubMed] [Google Scholar]
- 35.Xie Y, et al. Mps1/TTK: a novel target and biomarker for cancer. J Drug Target. 2017;25(2):112–118. doi: 10.1080/1061186X.2016.1258568. [DOI] [PubMed] [Google Scholar]
- 36.Lim G, Huh WK. Rad52 phosphorylation by Ipl1 and Mps1 contributes to Mps1 kinetochore localization and spindle assembly checkpoint regulation. Proc Natl Acad Sci USA. 2017;114(44):E9261–E9270. doi: 10.1073/pnas.1705261114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Isokane M, et al. ARHGEF17 is an essential spindle assembly checkpoint factor that targets Mps1 to kinetochores. J Cell Biol. 2016;212(6):647–659. doi: 10.1083/jcb.201408089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moura M, et al. Protein phosphatase 1 inactivates Mps1 to ensure efficient spindle assembly checkpoint silencing. Elife. 2017;6:e25366. doi: 10.7554/eLife.25366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Espert A, Uluocak P, Bastos RN, Mangat D, Graab P, Gruneberg U. PP2A-B56 opposes Mps1 phosphorylation of Knl1 and thereby promotes spindle assembly checkpoint silencing. J Cell Biol. 2014;206(7):833–842. doi: 10.1083/jcb.201406109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dou Z, et al. Dynamic localization of Mps1 kinase to kinetochores is essential for accurate spindle microtubule attachment. Proc Natl Acad Sci USA. 2015;112(33):E4546–E4555. doi: 10.1073/pnas.1508791112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mason JM, et al. Functional characterization of CFI-402257, a potent and selective Mps1/TTK kinase inhibitor, for the treatment of cancer. Proc Natl Acad Sci USA. 2017;114(12):3127–3132. doi: 10.1073/pnas.1700234114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Daniel J, Coulter J, Woo JH, Wilsbach K, Gabrielson E. High levels of the Mps1 checkpoint protein are protective of aneuploidy in breast cancer cells. Proc Natl Acad Sci USA. 2011;108(13):5384–5389. doi: 10.1073/pnas.1007645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maia AR, et al. Inhibition of the spindle assembly checkpoint kinase TTK enhances the efficacy of docetaxel in a triple-negative breast cancer model. Ann Oncol. 2015;26(10):2180–2192. doi: 10.1093/annonc/mdv293. [DOI] [PubMed] [Google Scholar]
- 44.Debeb BG, et al. EZH2 expression correlates with locoregional recurrence after radiation in inflammatory breast cancer. J Exp Clin Cancer Res. 2014;33:58. doi: 10.1186/s13046-014-0058-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dahl E, et al. Molecular profiling of laser-microdissected matched tumor and normal breast tissue identifies karyopherin alpha2 as a potential novel prognostic marker in breast cancer. Clin Cancer Res. 2006;12(13):3950–3960. doi: 10.1158/1078-0432.CCR-05-2090. [DOI] [PubMed] [Google Scholar]
- 46.Choi M, Min YH, Pyo J, Lee CW, Jang CY, Kim JE. TC Mps1 12, a novel Mps1 inhibitor, suppresses the growth of hepatocellular carcinoma cells via the accumulation of chromosomal instability. Br J Pharmacol. 2017;174(12):1810–1825. doi: 10.1111/bph.13782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365(9460):671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- 48.Desmedt C, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007;13(11):3207–3214. doi: 10.1158/1078-0432.CCR-06-2765. [DOI] [PubMed] [Google Scholar]
- 49.van ‘t Veer LJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt M, et al. The humoral immune system has a key prognostic impact in node-negative breast cancer. Cancer Res. 2008;68(13):5405–5413. doi: 10.1158/0008-5472.CAN-07-5206. [DOI] [PubMed] [Google Scholar]
- 51.Rhodes DR, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9(2):166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34(3):267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 53.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gill S, et al. Prognostic web-based models for stage II and III colon cancer: A population and clinical trials-based validation of numeracy and adjuvant! online. Cancer. 2011;117(18):4155–4165. doi: 10.1002/cncr.26003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu C, et al. Effect of epidermal growth factor receptor inhibitor on development of estrogen receptor-negative mammary tumors. J Natl Cancer Inst. 2003;95(24):1825–1833. doi: 10.1093/jnci/djg117. [DOI] [PubMed] [Google Scholar]
- 56.Kang J, Chen Y, Zhao Y, Yu H. Autophosphorylation-dependent activation of human Mps1 is required for the spindle checkpoint. Proc Natl Acad Sci USA. 2007;104(51):20232–20237. doi: 10.1073/pnas.0710519105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wei D, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72(1):282–293. doi: 10.1158/0008-5472.CAN-11-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mao Z, Seluanov A, Jiang Y, Gorbunova V. TRF2 is required for repair of nontelomeric DNA double-strand breaks by homologous recombination. Proc Natl Acad Sci USA. 2007;104(32):13068–13073. doi: 10.1073/pnas.0702410104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matar P, et al. Combined epidermal growth factor receptor targeting with the tyrosine kinase inhibitor gefitinib (ZD1839) and the monoclonal antibody cetuximab (IMC-C225): superiority over single-agent receptor targeting. Clin Cancer Res. 2004;10(19):6487–6501. doi: 10.1158/1078-0432.CCR-04-0870. [DOI] [PubMed] [Google Scholar]
- 60.Yokoyama Y, Dhanabal M, Griffioen AW, Sukhatme VP, Ramakrishnan S. Synergy between angiostatin and endostatin: inhibition of ovarian cancer growth. Cancer Res. 2000;60(8):2190–2196. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.