

Abstract
Leptomeningeal anastomoses or pial collateral vessels play a critical role in cerebral blood flow (CBF) restoration following ischemic stroke. The magnitude of this adaptive response is postulated to be controlled by the endothelium, although the underlying molecular mechanisms remain under investigation. Here we demonstrated that endothelial genetic deletion, using EphA4fl/fl/Tie2-Cre and EphA4fl/fl/VeCahderin-CreERT2 mice and vessel painting strategies, implicated EphA4 receptor tyrosine kinase as a major suppressor of pial collateral remodeling, CBF, and functional recovery following permanent middle cerebral artery occlusion. Pial collateral remodeling is limited by the crosstalk between EphA4-Tie2 signaling in vascular endothelial cells, which is mediated through p-Akt regulation. Furthermore, peptide inhibition of EphA4 resulted in acceleration of the pial arteriogenic response. Our findings demonstrate that EphA4 is a negative regulator of Tie2 receptor signaling, which limits pial collateral arteriogenesis following cerebrovascular occlusion. Therapeutic targeting of EphA4 and/or Tie2 represents an attractive new strategy for improving collateral function, neural tissue health, and functional recovery following ischemic stroke.
Keywords: Neuroscience, Vascular Biology
Keywords: Stroke
Introduction
Ischemic stroke results in significant cerebral blood flow (CBF) loss to brain regions predominately served by the middle cerebral artery, leading to cell death and neural tissue dysfunction. However, restoration of CBF may occur through developmentally regulated adaptations to the vascular network called leptomeningeal anastomoses or pial collateral vessels, which provide retrograde reperfusion to vulnerable tissue regions. This phenomenon is essential for preventing or reducing the consequences of an ischemic attack; however, the molecular mechanism(s) regulating the patient-specific pial collateral response remains understudied. While tissue plasminogen activator (tPA) and/or Alteplase remains the gold standard for stroke therapy, it is not effective in large vessel occlusions (LVOs) or when treatment is started prior to 4.5 hours after ischemic attack (1, 2) or 9 hours after ischemic attack in select patients (3). Recently, the use of endovascular thrombectomy has improved outcomes and extended the therapeutic window (4–7). In fact, it has been reported that endovascular thrombectomy has extended the treatment window up to at least 24 hours (8). Extension of this window due to pial collateral-mediated reperfusion enables CBF to maintain tissue protection in the penumbra region. Thus, improving our basic understanding of how collateral vessels remodel will allow us to better diagnose and treat patients with divergent responses.
The extent of collateral-mediated reperfusion has a major impact on preserving the penumbra and often dictates stroke outcome (9–13). Several studies have demonstrated that the outcome after thrombolysis and thrombectomy therapy is highly dependent on patient collateral scoring (14–19). Patients with high collateral function also have higher reperfusion rates after tPA therapy and a lower risk for symptomatic bleeding after reperfusion therapy (20). Pharmacological targeting of the pial collateral network therefore represents a viable therapeutic option to improve outcomes following embolic stroke, by sustaining penumbral blood flow and preserving neural tissue health. The middle cerebral arteries (MCAs), anterior cerebral arteries (ACAs), and posterior cerebral arteries (PCAs) anastomose at their distal ends to form the MCA-ACA and MCA-PCA collateral vessels (21). Collateral vessels do not show a proximal-to-distal axis with respect to blood flow, rather they connect 2 arterial branches with opposing flow. Therefore, collateral blood flow is bidirectional along the collateral vessel wall (22). Following an obstruction, collateral vessels undergo immense vascular restructuring and remodeling (enlargement), also called arteriogenesis, to allow for unidirectional retrograde blood reperfusion into the area of an occluded arterial branch (22, 23).
Although the benefits of targeting the collateral system are evident, further research is essential to elucidate the mechanism(s) underlying this remarkable tissue-saving adaptation. Collateral remodeling is initiated when mechanoreceptors on endothelial cells (ECs) detect fluid shear stress after an occlusion (24–26). Thus, the ECs comprised within the collateral niche may perform essential duties during the active remodeling process. Recently, we implicated EphA4 receptor tyrosine kinase in limiting collateral formation and recovery from hindlimb ischemia following femoral artery ligation (FAL) (27). The current study demonstrates that ECs are central to pial collateral remodeling after ischemic stroke and the presence of EphA4 suppresses their growth properties in vitro and in vivo by acting as an inhibitor of the Akt/Tie2 receptor-signaling pathway. These findings greatly improve our understanding of the cellular and molecular mechanism(s) involved in pial collateral remodeling following vascular obstruction.
Results
EphA4fl/fl/Tie2-Cre knockout mice show improved CBF, neural tissue, and behavioral recovery following permanent middle cerebral artery occlusion (pMCAO).
In the current study, we used EphA4fl/fl/Tie2-Cre knockout (KO) mice to evaluate changes in CBF following acute ischemic stroke and subsequent outcomes compared with EphA4fl/fl WT mice. Vascular recombination was confirmed using Tie2-Cre/ROSAmTmG reporter mice (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI131493DS1) and as previously described (27). CBF was measured by laser doppler prior to and at 5 minutes, then again 1–4 days after pMCAO in the ipsilateral hemisphere. The perfusion units (PFUs) were quantified and are represented relative to baseline preinjury CBF (Figure 1, A and B). No significant difference in CBF was observed at 5 minutes after pMCAO between WT and KO mice (relative PFUs: 0.532 ± 0.026 vs. 0.502 ± 0.034). However, we observed a significant increase in CBF at 1 day (0.87 ± 0.05 vs. 0.69 ± 0.05), 2 days (0.94 ± 0.06 vs. 0.73 ± 0.05), 3 days (0.95 ± 0.05 vs. 0.79 ± 0.06), and 4 days (0.97 ± 0.04 vs. 0.82 ± 0.04) in KO mice compared with WT mice. These findings correlated with reduced infarct volume in KO mice (15.57 ± 3.26 mm3) compared with WT mice (26.77 ± 3.13 mm3) at 4 days after pMCAO (Figure 1, C–E). Likewise, we found that KO mice showed improvements in behavioral recovery. Rotarod assessment demonstrated a significant increase in motor function in KO compared with WT mice at 7 days and increased trend at 3 days and 14 days after pMCAO (Figure 1F). While increased neurological severity scoring was observed following pMCAO, no significant difference was found between WT and KO mice (Figure 1G). However, assessment using novel object recognition (NOR) showed that pMCAO reduced the novel object preference index in WT but not KO mice at 3 days (62.75 ± 1.08 vs. 45.58 ± 2.61), 7 days (63.81 ± 2.57 vs. 50.01 ± 1.68), and 14 days (69.42 ± 3.15 vs. 50.16 ± 0.87) (Figure 1H). These findings demonstrate that EC-specific EphA4 is a mediator of functional deficits and neural tissue damage following pMCAO.
Figure 1. Increased CBF and reduced infarct volume in EC-specific KO mice following pMCAO.

(A) Laser doppler images before and after pMCAO. Panel shows representative images from EphA4fl/fl WT and EphA4fl/fl/Tie2::Cre KO mice before and after pMCAO. (B) Quantified analysis shows increased CBF in KO compared with WT mice; n = 7–10. (C) Representative serial Nissl images of 3 bregma levels in WT and (D) KO mice 1 day after pMCAO. (E) Quantified infarct volume shows a significant reduction in infarct volume in KO compared with WT mice; n = 6. (F) Rotarod assessment of WT and KO mice. KO mice performed significantly better than WT mice 3 and 7 days after stroke. (G) NSS and (H) NOR were analyzed 3–14 days after pMCAO. Two-way ANOVA with Bonferroni’s post hoc test; n = 9–17. *P < 0.05, ****P < 0.0001 compared with corresponding WT mice; ####P < 0.0001 compared with corresponding sham mice. White dotted lines in A indicate standardized ROI used for CBF quantification of each sample.
EphA4fl/fl/Tie2-Cre mice show enhanced pial collateral remodeling following pMCAO.
To evaluate whether improvements in CBF and behavioral recovery coincided with changes in pial collateral remodeling, we performed vessel painting 1 day and 4 days after pMCAO on EphA4fl/fl and EphA4fl/fl/Tie2-Cre mice. While we observed an increase in ipsilateral compared with contralateral pial collateral diameter in WT (Figure 2, A and B) and KO (Figure 2, C and D) mice, EC-specific EphA4 ablation significantly enhanced remodeling of MCA-ACA inter-collaterals 1 day (KO 41.08 ± 2.16 μm vs. WT 29.59 ± 1.79 μm) and 4 days (KO 53.29 ± 2.39 μm vs. WT 39.03 ± 1.84 μm) after pMCAO (Figure 2E). Given that the PCAs could also provide retrograde reperfusion into the territory of the occluded MCAs, we also evaluated those inter-collaterals. MCA-PCA also showed increased collateral diameter in KO compared with WT at 1 day (37.52 ± 1.88 μm vs. 26.90 ± 1.20 μm, respectively) and 4 days (50.43 ± 2.83 μm vs. 37.72 ± 2.15 μm, respectively) (Figure 2F). Diameter of all inter-collaterals (collaterals between the main branches of the MCA-ACA and MCA-PCA) combined showed an increase in KO mice compared with WT at 1 day (39.30 ± 1.45 μm vs. 28.31 ± 1.11 μm, respectively) and 4 days (51.75 ± 1.85 μm vs. 38.56 ± 1.36 μm, respectively) (Figure 2G). No significant difference was observed in contralateral collateral diameter between groups or in the total number of inter-collaterals (Figure 2H) after pMCAO, suggesting that EphA4 improved CBF through collateral remodeling rather than having greater numbers to begin with, which was confirmed by the total number of inter-collateral counts. Moreover, breakdown of collateral size shows that the largest increase in collateral diameter occurred in the ipsilateral KO mice, where 85% of collaterals were greater than 31 μm at 1 day and 91% at 4 days after pMCAO, compared with 10%–15% on the contralateral side. Conversely, 40% of the WT ipsilateral collaterals were greater than 31 μm at 1 day and 75% at 4 days after pMCAO. Additionally, a greater number of KO collaterals were larger than 50 μm: 52% compared with 18% in WT mice (Figure 2, I and J).
Figure 2. Increased collateral remodeling in EC-specific KO mice after pMCAO.

(A and B) Vessel painted WT brain 1 day after pMCAO showing pial collateral vessels (arrows) in the ipsilateral hemisphere. (C and D) KO vessel painted brain 1 day after pMCAO. (E) MCA-ACA collateral diameter analyses 1 and 4 days after pMCAO. KO mice show increased collateral size compared with WT mice (*). Ipsilateral collaterals are larger in diameter compared with contralateral at both time points (#). Ipsilateral vessels 4 days after injury are significantly larger than 1 day after injury in both genotypes ($). (F) MCA-PCA collateral analyses 1 and 4 days after stroke. (G) Average inter-collateral analyses and (H) inter-collateral counts show no significant difference between time points or between ipsilateral and contralateral hemispheres. Breakdown of collateral vessel size at 1 day (I) and 4 days (J) after pMCAO. One-way ANOVA with Bonferroni’s post hoc test; n = 7–10 per group. #,$P < 0.05; **,##,$$P < 0.01, ***,###,$$$P < 0.001; ****,####,$$$$P < 0.0001. Scale bars in A–D: 1 mm.
Given Tie2 may also be expressed by a subset of immune cells, we further validated the cell-autonomous role of EC-specific EphA4 in pial collateral remodeling by utilizing an inducible Cre system. We first tested the utility of Tie2-CreERT2 versus VeCadherin-CreERT2 mice using a reporter system and found that, 2 weeks after 5 consecutive injections of 2 mg/kg/day tamoxifen, VeCadherin-CreERT2/RosamTmG mice showed complete recombination, whereas Tie2-CreERT2/RosamTmG showed incomplete recombination (Supplemental Figure 2, A–F). This was true even at 3 mg/kg/day (data not shown). We then analyzed collateral diameters 1 day after pMCAO in tamoxifen-treated EphA4fl/fl (iWT) and EphA4fl/fl/VeCadherin-CreERT2 (iKO) mice and found a significant increase in ipsilateral pial size in iKO mice compared with iWT mice (Supplemental Figure 2, G–I). These findings demonstrate that loss of EC-specific EphA4 can accelerate pial arteriogenesis as early at 24 hours after pMCAO.
Evidence of early cellular remodeling in EphA4fl/fl/Tie2-Cre vessel painted MCA-ACA collaterals.
EphA4 suppresses EC proliferation in vitro (27) and the pial collateral niche undergoes active cellular remodeling following pMCAO (28). To address whether the loss of EC-specific EphA4 improved early remodeling of pial vessels by enhancing EC growth properties, we performed immunolabeling for cell division marker, PCNA on wild type and KO cortical tissue whole mounts 1 day after pMCAO and vessel painting. Maximum Z-projected confocal image analysis shows KO mice (Figure 3, A–C) displayed a greater number of PCNA+ cells in the MCA-ACA pial ipsilateral collateral vessel wall (Figure 3, I–K) compared with WT ipsilateral and KO contralateral vessels (Figure 3, D–H). These findings suggest EphA4 may limit early remodeling, in part, by suppressing EC proliferation in the collateral niche.
Figure 3. Increased cell proliferation within the MCA-ACA collateral niche of KO mice 1 day after pMCAO.

Vessel painted brains were immuno-labeled with anti-PCNA (green; white arrows) and imaged by high magnification confocal imaging. Maximum Z-projection analysis of the entire Z-stacked vessel was used to assess cell division for evidence of collateral remodeling as early as 1 day after pMCAO in the ipsilateral (A–E) and contralateral (F and G) hemispheres. (A) Representative 3D projected image from vessel painted (red) ipsilateral KO pial surface of an MCA-ACA collateral colabeled with antibodies against PCNA. KO collateral vessels show a greater number of PCNA+ cells aligned within (elongated compared with surrounding PCNA+ cells) the vessel wall compared with WT. (B and C) Representative single channel images of A, showing PCNA expression in the KO collateral niche compared with the ipsilateral WT collateral niche (D and E). No PCNA staining was seen in the contralateral pial collaterals of the same representative injured WT animal (F and G). (H) Quantified data showing the number of PCNA+ cells within the collateral vessel wall in increased in KO mice. (I–K) High-magnification confocal ortho view from inset in A, showing cell division in the KO collateral wall. Scale bars: 50 μm (B–G) and 10 μm (I–K). Two-way ANOVA with Bonferroni’s post hoc test; n = 5 mice per group; 4–5 MCA-ACA collaterals were analyzed per mouse. ****P < 0.0001.
EphA4fl/fl/Tie2-Cre mice display increased p-Akt and Angpt2 cortical expression after pMCAO.
To test whether Tie2-specific deletion of EphA4 could influence p-Akt/Angpt pathways after stroke, we assessed the protein lysates derived from the ipsilateral cortex 1 day and 4 days after pMCAO in EphA4fl/fl and EphA4fl/fl/Tie2-Cre mice. Using Western blot analysis, we observed a significant decrease in the expression of p-Akt in WT pMCAO-injured cortices compared with WT sham (0.06 ± 0.03 vs. 0.27 ± 0.05, relative to total Akt, respectively). However, the level of p-Akt was maintained in the KO injured cortex compared with sham at 1 day (0.23 ± 0.02 vs. 0.38 ± 0.14, relative to total Akt, respectively) (Figure 4, A and B). No significant change was seen 4 days after pMCAO (Figure 4, C and D). We also evaluated the expression of angiopoietin-1 and -2 (Angpt1, Angpt2) one day after pMCAO and found a significant reduction in Angpt2, exclusively expressed on ECs and a small population of immune cells, in the WT injured cortex compared with sham (0.61 ± 0.10 vs. 1.35 ± 0.13, respectively). This effect was attenuated in KO mice (1.19 ± 0.17 vs. 1.32 ± 0.10, respectively) (Figure 4, E and F). Angpt1 showed no significant difference between samples (Supplemental Figure 3C). These data reveal that EphA4 suppresses Angpt2 and p-Akt signaling following pMCAO.
Figure 4. Expression of p-Akt and Angpt2 is increased in KO mice 1 day after pMCAO.

(A) Western blot analysis 1 day after pMCAO. (B) Densitometric analysis shows a significant increase of p-Akt in KO mice cortical protein lysates compared with WT mice. (C) Western blot analysis from ipsilateral hemisphere lysates 4 days after pMCAO. (D) There was no significant difference in p-Akt expression. (E) Angpt1, Angpt2; Western blot analysis. (F) One day after stroke, there is significantly elevated Angpt2 protein in KO mice lysates compared with WT mice. One-way ANOVA with Bonferroni’s post hoc test; n = 4–7 mice per group. *P < 0.05, **P < 0.01.
Blocking Tie2 receptor prevents collateral remodeling, p-Akt expression, and neuroprotection in EphA4fl/fl/Tie2-Cre mice.
To investigate whether the genetic deletion of EC-specific EphA4 enhances collateral remodeling, p-Akt expression, and neuroprotection via Tie2 receptor signaling, we administered 5 mg/kg/day soluble Tie2-Fc or human Fc-control via tail vein injection immediately after pMCAO in EphA4fl/fl and EphA4fl/fl/Tie2-Cre mice. We observed a significant attenuation of ipsilateral collateral remodeling in KO mice receiving soluble Tie2-Fc (31.84 ± 1.06 μm) (Figure 5, B and C) compared with Fc-control (41.00 ± 4.80 μm) (Figure 5, A and C) 1 day after pMCAO. No effect on collateral diameter was found in the ipsilateral hemisphere of WT mice (Figure 5C). Similar changes were seen when comparing MCA-ACA (Figure 5D) and MCA-PCA (Figure 5E). These findings coincided with increased infarct volume in KO mice receiving Tie2-Fc (24.93 ± 3.60 mm3) compared with Fc-control (12.16 ± 3.52 mm3) (Figure 5, F and G). Furthermore, blocking Tie2 receptor signaling suppressed p-Akt expression in the cortex of KO mice treated with soluble Tie2-Fc (0.05 ± 0.03, relative to total Akt) compared with Fc-control (0.17 ± 0.02, relative to total Akt) (Figure 5, H and I).
Figure 5. Blocking Tie2 receptor attenuates neuroprotection and p-Akt in EC-specific EphA4 KO mice.

(A and B) Ipsilateral hemisphere of vessel painted pMCAO KO mice after Fc or Tie2-Fc treatment. (C) Quantitative analysis shows a reduction in the diameter of total inter-collateral KO mice treated with Tie2-Fc compared with Fc-treated mice; n = 5–9. WT mice showed no significant difference following Tie2-Fc; n = 4–5. (D) MCA-ACA collateral diameters. (E) MCA-PCA collateral diameters. (F) Representative Nissl staining of Fc and Tie2-Fc brains 1 day after pMCAO, demarcating the area of infarct. (G) Quantified graph shows that Tie2-Fc–treated KO mice have a significant increase in infarct volume compared with Fc-treated KO mice. (H) Western blot analysis of ipsilateral brain lysates from Fc- and Tie2-Fc–treated KO mice 1 day after pMCAO. (I) Quantified graph shows that Tie2-Fc–treated KO mice have reduced p-Akt expression compared with Fc-treated mice; n = 4 mice per group. Unpaired t test and 1-way ANOVA with Bonferroni’s post hoc test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Scale bars: 1 mm (A and B).
EphA4 suppresses Tie2/p-Akt expression to limit EC proliferation in vitro.
Collateral remodeling is initiated by the interplay between endothelial cell proliferation and inflammation (29–31). To test whether inflammatory stimuli regulates the expression of EphA4 and Tie2 on the endothelium and to determine whether EphA4 loss-of-function results in differential gene expression, we subjected brain-derived WT and KO ECs (27) to lipopolysaccharide (LPS) for 4 and 24 hours. We confirmed genetic knockout of Epha4 and showed that LPS stimulation results in a transient reduction in Epha4 and Tie2 at 4 hours and no change at 24 hours in WT ECs (Figure 6, A and B). Conversely, KO ECs show a significant increase in Tie2 expression in both vehicle and LPS-treated cells compared with WT ECs at both time points (Figure 6B). In addition, Angpt2, a known mediator of collateral remodeling in hindlimb ischemia (32), is reduced following 4- and 24-hour LPS stimulation in WT ECs, which is greatly enhanced in KO ECs (Figure 6C). To gain insight into the role of EphA4/Tie2 crosstalk on endothelial cell function, we blocked Tie2 signaling in WT and KO ECs using soluble Tie2-Fc and Fc-control, then evaluated proliferation and p-Akt expression. Compared with Fc-control, we found that Tie2-Fc treatment significantly attenuated the enhanced proliferation observed in KO ECs while having no effect on WT ECs in a dose-dependent manner (Figure 6, D–F). We previously showed that KO ECs have increased p-Akt expression compared with WT ECs (27). Here we show by Western blot analysis that p-Akt protein expression in KO ECs was reduced following Tie2-Fc treatment (Figure 6G). These data suggest that EphA4 suppresses EC proliferation and Akt signaling by regulating Tie2 expression and function. Last, we found that 87.5 and 106 dyne/cm2 shear stress for 4 hours significantly increased mRNA expression of Epha4 in WT ECs (Figure 6H). The WT and KO EC purity was also assessed by qPCR at passage 3 (Figure 3I). Taken together, our in vitro findings suggest that collateral shear stress may upregulate EphA4 expression, leading to suppression of Tie2 signaling and subsequent collateral remodeling (Figure 6I).
Figure 6. EphA4 suppresses Tie2/p-Akt expression to limit brain-derived EC proliferation.

(A–C) Primary cultured WT and KO brain-derived EC mRNA expression of Epha4, Tie2, and Angpt2 following 4 or 24 hours of 1 μg/mL LPS stimulation, respectively. Quantification is relative to vehicle control at each time point. (D–F) KO ECs treated with soluble Tie2-Fc for 24 hours show dose-dependent attenuation of proliferation compared with Fc-control. No effect was seen following treatment in WT ECs. (G) Representative Western blot image shows attenuation of p-Akt in KO ECs after 24 hours of treatment with 20 μg/mL Tie2-Fc compared with Fc-control. (H) Increased mRNA expression of Epha4 in WT ECs 4 hours after shear stress at pathological flow rates of 87.5 and 106 dyne/cm2 compared with control (ctl), no flow, or homeostatic flow at 50 dyne/cm2. (I) Fold change of gene expression in WT and KO ECs by qPCR at passage 3, demonstrating purity of cultured ECs. Biological triplicates were used and genes were represented relative to WT Tie2 expression. Four independent experiments were performed in triplicate for each experiment. One-way or 2-way ANOVA with Bonferroni’s post hoc test; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. Scale bars: 100 μm.
Pharmacological inhibition of EphA4 increases collateral remodeling after pMCAO.
Finally, we evaluated whether pharmacological inhibition of EphA4 could recapitulate the enhanced collateral remodeling seen in EphA4fl/fl/Tie2-Cre and EphA4fl/fl/VeCadherin-CreERT2 mice. To test this, we utilized sub-Q mini-osmotic pump infusion of the EphA4 peptide inhibitor, KYL, which prevents ligand binding and EphA4 activation (33–35). At 4 days after pMCAO, we observed a significant increase in the ipsilateral pial collateral diameter in KYL-treated mice (Figure 7E) compared with vehicle control (Figure 7D). This effect was observed in the MCA-ACA (50.61 ± 1.55 μm KYL vs. 42.35 ± 2.67 μm vehicle), MCA-PCA (49.15 ± 2.99 μm KYL vs. 35.90 ± 3.22 μm vehicle) and combined (49.88 ± 1.64 μm KYL vs. 39.13 ± 2.19 μm vehicle) arteriole branches (Figure 7, A–C). A breakdown of collateral size showed a significant increase in the percentage of collaterals greater than 50 μm compared with vehicle (47% vs. 21%, respectively) (Figure 7F). We also found cultured WT ECs treated with KYL and an additional EphA4 blocking peptide, VTM-EEKK (36), showed increased p-Akt levels compared with vehicle-treated WT ECs, similar to expression in KO cells (Supplemental Figure 4, A and B). EC proliferation was increased by KYL treatment, in a dose-dependent manner, in WT ECs (Supplemental Figure 4, C–H). These findings are similar to the effects seen following genetic deletion of EphA4 and provide evidence for pharmacological targeting of this pathway for collateral therapeutics.
Figure 7. Pharmacological inhibition of EphA4 increases pial collateral size 4 days after pMCAO.

(A) MCA-ACA collaterals (yellow arrows) show that KYL significantly increased the diameter of ipsilateral MCA-ACA inter-collaterals compared with those treated with vehicle (saline) (*). Both vehicle and KYL ipsilateral collaterals are significantly larger compared with contralateral (#). (B) MCA-PCA ipsilateral inter-collaterals of animals treated with KYL are also significantly larger compared with those of vehicle control. (C) Quantified graph showing that the average inter-collateral diameter after stroke is increase in KYL-treated mice. (D) Representative images of WT vessel painted ipsilateral hemisphere with vehicle treatment compared with (E) KYL treatment. (F) Breakdown of collateral sizes after pMCAO shows a significant increase in the percentage of collaterals whose diameters are greater than 51 μm, and a precipitous drop in the percentage of collaterals whose diameters are between 21–30 μm in KYL mice; n = 8 mice per group. One-way and 2-way ANOVA with Bonferroni’s post hoc test. *,#P < 0.05; **P < 0.01; ***P < 0.001; ****,####P < 0.0001. Scale bars: 500 μm.
Discussion
Since their discovery by Heubner in 1874, leptomeningeal anastomoses have been implicated as critical determinants of injury severity. However, the cellular and molecular underpinnings of the arteriogenic response remain under investigation following ischemic stroke. Findings from the current study describe a mechanism that restricts outward growth and remodeling. Our results implicate the Tie2-EphA4 axis as a critical regulator of pial collateral remodeling following stroke. The presence of EphA4 on the endothelium acts as a negative cue within the collateral niche to restrict Tie2 receptor function via the p-Akt pathway, thereby restricting collateral outgrowth and remodeling. EphA4fl/fl/Tie2-Cre mice show increased p-Akt signaling, collateral remodeling, and tissue protection, which were attenuated in the presence of soluble Tie2 inhibitor. Enhanced collateral growth correlated with improved CBF 1–4 days after pMCAO in the absence of Tie2-specific EphA4. Additional findings using inducible EphA4fl/fl/VeCadERT2 mice further demonstrate that loss of EphA4 on ECs can enhance collateral remodeling as early as 1 day after pMCAO. Finally, we show that inhibition of EphA4 using the KYL peptide inhibitor mimicked the enhanced collateral response we observed genetic deletion in EC-specific KO mice. Overall, these findings highlight EphA4 and the Angpt/Tie2 axis as an important target for collateral therapeutics in stroke.
Eph receptor tyrosine kinases are widely known to control cell migration, proliferation, and survival in the CNS. Although ephrin/Eph molecules play critical roles in numerous biological processes (37–39), the role of Eph/Ephrin signaling in cerebral arteriogenesis has not been investigated. Previous studies have demonstrated that ephrinB2, an arterial-specific marker involved in arteriovenous specification (38, 40, 41), is induced following cyclic stretch and limits the migration of smooth muscle cells and transmigration of monocytes in vitro (42). EphA4 displays remarkable ligand-binding promiscuity and its binding complex with ephrinB2 has been described (43). While we cannot rule out associations with other A- or B-class ligands, it is plausible that EphA4-ephrinB2 interaction is induced within the collateral vessel wall following MCA occlusion, and that this region-specific partnership acts to prevent outward growth and remodeling. EphrinB2 is located at the luminal and junctional endothelial cell surface where it associates with CD31 (44). Korff et al. demonstrated that quiescent smooth muscle–contacting ECs show uniform luminal expression of ephrinB2, which translocates to inter-endothelial cell junctions in a context-dependent manner. If this could occur under shear conditions within the collateral vessel wall, then association with and activation of EC-specific EphA4 forward receptor signaling may result in Tie2/p-Akt pathway suppression during remodeling. Further studies evaluating ephrinB2–/– or ephrinB2LacZ mice could provide additional insight. Last, we showed that cultured EC treatment and in vivo infusion with KYL can increase p-Akt EC proliferation and enhance collateral outward growth, respectively. While these findings suggest that inhibiting EC-specific EphA4 on the collateral vessel wall may mediate these effects, we cannot rule out the possibility that systemic delivery of KYL may also regulate EphA4 activation on peripheral immune cells, which could contribute to these effects.
Our findings show that EphA4-Tie2 receptor crosstalk plays a critical role in pial collateral remodeling after stroke. Angiopoietin ligands of the Tie2 receptor are known regulators of arteriogenesis, which have been shown to affect blood flow recovery in ischemia (32, 45, 46). Although previous studies demonstrate these effects in non-CNS ischemic conditions, no studies have been conducted to assess the functional relevance of Tie2 signaling in the cerebrovascular collateral network after stroke. In particular, Angpt2 has been found to improve blood flow and arteriogenesis in hindlimb ischemia (32). Our studies revealed a significant reduction in Angpt2 and no change in Angpt-1 protein expression in the ipsilateral cortex of WT mice 1 day after pMCAO. Angpt2 levels were maintained in KO mice alongside increased collateral growth, suggesting that heightened Tie2 activation in the absence of EphA4 can accelerate collateral remodeling at this early time point. Indeed, blockade of the Tie2 receptor restored the size of KO pial collaterals to WT levels while having no effect in WT mice 1 day after pMCAO. Although we did not evaluate Angpt2 after 4 days, we postulate that the levels in WT mice would return to normal, similar to the p-Akt expression, concomitant with a substantial increase in WT collateral size as compared with the evaluation done at 1 day. Tie2 is expressed exclusively on the vascular endothelium and a subset of immune cells (47–49). Given that immune cells, such as monocytes, can mediate collateral growth, we cannot rule out the possibility that Tie-specific deletion of EphA4 on immune cells contributes to these effects. Enhanced pial collateral growth in VeCadERT2/EphA4fl/fl mice, however, mimicked EphA4 deletion in Tie2-expresing cells, further confirming that the suppressive effects of EphA4 on pial collaterals is mediated by endothelial cell regulation. Future studies will begin to address the earliest changes that may occur within 24 hours in the absence of EphA4, including both active remodeling and immediate retrograde compensation. Together with extensive in vitro EC analysis of EphA4 expression and function, our data support the role of EC-derived EphA4 as a major inhibitor of pial collateral growth.
It remains unclear what mechanistic role Angpt2 plays in pial collateral growth in EphA4fl/fl/Tie2-Cre mice after pMCAO. Ang1 is an established agonist of Tie2 activation, whereas Ang2 is a competitive inhibitor, although this has been challenged to be cell-type and context dependent (49). For example, at high or sustained concentrations, Angpt2 can stimulate Tie2 on ECs (50, 51), and acts as an agonist alongside Ang1 in the presence of TNF (52). Moreover, Tie2 interaction with Tie1 or VE-PTP receptors may further regulate the sensitivity of Tie2 to its ligands (53–55). The expression of these receptors has yet to be evaluated in the collateral vessel niche following occlusion. Our findings reveal that pMCAO induces an acute loss of Angpt2 expression which is mediated by EphA4 signaling and may prevent early induction of pial collateral remodeling. While these findings were observed in whole cortex samples, Tie2 and Angpt2 are predominately expressed by ECs. We cannot rule out the possible contribution of Tie2 and Angpt2 expression from infiltrating immune cells, but our in vitro findings provide strong support for the role of EphA4 in suppressing Tie2/Angpt2 signaling via p-Akt signaling directly in ECs. Given the importance of collateral growth in maintaining blood flow to the vulnerable penumbra region prior to thrombectomy (56), strategies that target the EphA4/Angpt2 discord to accelerate this process may help alleviate neural tissue damage and dysfunction.
The current study expands our knowledge of the wide net that Eph receptors cast on cellular function and provides key mechanistic insight into the growth constraints that limit the unique adaptive response of cerebral collateral vessels. To date, it remains unclear why some stroke patients display high collateral function after occlusion, while others do not. Understanding the mechanism(s) that regulate and importantly restrict this process will be crucial for devising approaches to predict collateral engagement while improving collateral health and neurological outcome in patients with ischemic stroke.
Methods
Animals
All rodents were bred and housed in an AAALAC-accredited, virus/antigen-free facility with a 12-hour light-dark cycle. Food and water were provided ad libitum. EphA4fl/fl and Tie2-Cre mice (catalog 012916 and catalog 008863, respectively, Jackson Laboratory) as well as VeCadERT2 mice (a gift from John Chappell, Fralin Biomedical Research Institute, Virginia Tech, Roanoke, Virginia) were backcrossed 10 generations on the CD1 background and crossbred until the desired experimental male mice at 8–10 weeks of age were generated. Male 8- to 10-week-old CD1 mice (Charles River) were used for peptide studies. All mice were coded at the time of surgery for double-blinded experimentation.
Surgical procedures and treatments
Ischemic stroke was induced by pMCAO as previously described (28). Briefly, 8- to 12-week-old male mice were injected with analgesic Buprenorphine-SR (0.15 mg/kg, ZooPharm) followed by induction of anesthesia using 2% isoflurane-30% oxygen. An incision was made and skull thinned to expose and cauterize the main and 2 distal branches of the left MCA. Sham controls received identical procedures without ligation. For Tie2-Fc study, immediately following pMCAO, mice were administered either 5 mg/kg of soluble Tie2-Fc or 2.5 mg/kg soluble human Fc-control via tail vein injection, then euthanized at 24 hours. EphA4 blocking peptide, KYLPYWPVLSSL, was implanted in adult male CD1 mice subcutaneously via mini-osmotic pumps (Alzet, Inc; flow rate 0.5 μL/hour) at 10 mg/kg/day or saline vehicle immediately following pMCAO. Osmotic pumps were loaded and preincubated in sterile saline overnight at 37°C to prime the pumps. Mice were euthanized by vessel painting 4 days after pMCAO.
Vessel painting and collateral quantification
Vessel painting was performed as previously described (27, 57). Briefly, mice were injected with heparin (2000 U/kg) and sodium nitroprusside (SNP, 0.75 mg/kg) 5 minutes prior to euthanization, using an overdose of isoflurane. When breathing stopped, mice were perfused with 10 mL of 1× PBS containing 20 U/mL heparin to flush blood from the vascular system, then a mixture of 10 mL DiI (0.01 mg/mL, Invitrogen) and 4% sucrose-PBS-heparin was perfused to label the arteriole vasculature, using a flow rate of 2 mL/min, followed by 50 mL of 4% cold paraformaldehyde (PFA). Fixed brains were imaged at ×4 magnification on an upright fluorescence microscope (BX-51, Olympus America) or inverted 880 Zeiss confocal microscope. Images were imported into ImageJ (NIH) for quantification of the number and diameter of inter-collaterals, as described (28). Similarly, images were assessed using ImageJ to quantify the number of PCNA+/VP+ cells within the collateral vessel walls from maximum Z-projected images using the cell-counting tool.
Infarct volume
Fresh frozen brains were embedded in OCT, snap frozen, and serial cryo-sectioned. Infarct volume (mm3) was assessed by using the Cavalieri Estimator from nonbiased StereoInvestigator software (MicroBrightField) as previously described (58). Briefly, 6 serial coronal sections were then stained with 0.2% cresyl violet solution (Electron Microscopy Science) and the infarcted area was identified by loss of Nissl staining and pyknotic neurons. Volume analysis was performed by estimating the area of tissue loss in the ipsilateral cortical hemisphere using six 30-μm serial coronal sections. A 100 μm–spaced grid was placed over the ipsilateral hemisphere in the Cavalieri probe and the infarcted area was scored.
Immunohistochemistry of cortical whole mounts
Cortical whole mounts were dissected and placed in 1X PBS overnight as previously described (28). Briefly, whole mounts were blocked in 2% fish gelatin (Sigma-Aldrich) with 0.1% Triton X-100 and incubated overnight in mouse anti–smooth muscle actin (SMA) (1:1000; Abcam, ab7817) at 4°C. Whole mounts were washed then incubated with anti-mouse Alexa Fluor 488–conjugated secondary antibody (Thermo Fisher Scientific) washed then imaged on Zeiss 880 confocal microscope (Carl-Zeiss). For PCNA staining, whole mounts were treated as previously described (59).
Western blot analysis
Ipsilateral cortical tissue or cell lysate from culture was homogenized in RIPA buffer as previously described (27), then 20–100 μg protein was separated by 10% SDS-PAGE. Membranes were blocked in TBS/0.1% Tween20 (TBST)/5% bovine serum albumin (BSA), then incubated in the following primary antibodies: phospho-Akt (catalog 4051, Cell Signaling Technology), pan Akt (catalog 4691, Cell Signaling Technology), angiopoietin 1 (catalog AF923, R&D Systems), and angiopoietin 2 (ab155106, Abcam) in blocking solution overnight. Membranes were then washed and incubated with secondary antibodies (anti-rabbit IgG Dylight conjugate 680 or anti-mouse IgG Dylight conjugate 800; Cell Signaling Technology), then imaged using LI-COR Odyssey Imaging Systems (LI-COR, Inc.). Band intensities were quantified using LI-COR’s Image Studio software.
Endothelial cell cultures
Primary brain-derived ECs were isolated, cultured, and purity characterized as previously described (27). Briefly, cells were cultured in defined media then treated with human Fc-control or soluble Tie2-Fc (Sino Biologics) diluted in EC base media (Cell Biologics Inc.) without growth factors for 24 hours. Protein was isolated using RIPA buffer for Western blot analysis of p-Akt expression. Additionally, for proliferation assays, 10 μM BrdU was added for 1 hour in the Tie2-Fc or Fc-control wells. Cells were then fixed and subjected to immunohistochemistry using anti-BrdU (1:100, Abcam). Images were taken on an Olympus BX-51 at ×10. The number of total cells and the number of BrdU-positive cells were counted using DAPI counterstain and quantified using ImageJ. For stimulation experiments, 300,000 cells were plated in 6-well plates containing growth media in the presence or absence of 1 μg/mL LPS or following exposure to 30 and 50 dyne/cm2 using an orbital shaker. RNA was extracted 4 and 24 hours after stimulation. For peptide inhibitor studies, 15,000 cells were plated in each well of a 96-well plate containing serum-free media treated with either VTM-EEKK, VTA-EEKK, KYL, or water (vehicle control) at 75, 250, or 500 μM. BrdU (Sigma-Aldrich, 10 μM) was added, and 24 hours later cells were fixed for immunohistochemistry using 1:1000, rat anti-BrdU (MA1-82088; Thermo Fisher Scientific) in blocking solution overnight at 4°C. Donkey anti-rat Alexa-Flour 488–conjugated secondary antibody (A-21208; 1:500, Thermo Fisher Scientific) along with DAPI (catalog 4083; 1:2000, Cell Signaling Technology) were used. The images at ×4 objective were taken, then the number of cells (BrdU-positive and total DAPI-positive) were counted using NIH ImageJ software.
Quantitative real-time PCR
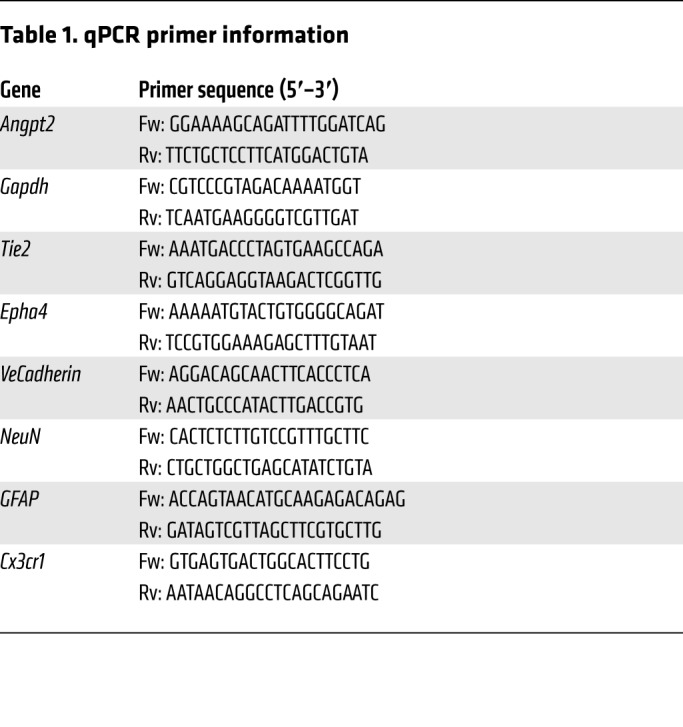
Total RNA from cells isolated according to manufactures instructions using TRIzol reagent (Ambion) per the manufacturer’s instructions. RNA was quantified by absorbance with spectrophotometer ND-1000 (NanoDrop). cDNA was made with the iScript cDNA synthesis kit (BioRad) per the manufacturer’s specifications. For qRT-PCR analysis, 50 ng cDNA per reaction was amplified with iTaq Universal SYBR Green Supermix (BioRad). Expression changes were calculated using ΔCq values with reference to Gapdh internal control. Relative expression was calculated then normalized to untreated WT samples. All primer efficiencies ranged from 87%–113% (Table 1).
Table 1. qPCR primer information.
Behavioral testing
All animal behaviors were performed as previously described for pMCAO (28).
Rotarod.
Mice were pretrained for 4 consecutive days prior to pMCAO or sham injury at 10 rpm and an acceleration of 0.1 rpm per second over 10 minutes. A baseline was collected on the fourth day, then tested 3, 7, and 14 days after injury.
Neurological severity scoring (NSS).
Sensorimotor deficits were tested before injury and 3 to 14 days after injury. NSS is a composite of motor, sensory (visual, tactile, and proprioceptive), reflex, and balance tests. Function was graded on a scale of 0 to 14 (normal = 0; maximal deficit = 14), where 1 point is awarded for the inability to perform the task.
Novel object recognition.
Cognitive and spatial deficits were tested using NOR. Briefly, mice were introduced to 2 identical objects on day 1 and then one of the objects was replaced with a new object the following day (test day) in a 40 cm3 arena. Time of exploration of the old and new object was recorded over 5 minutes. Preference of object was calculated as a ratio of exploration time of the specific object to the total time of exploration.
Cerebral blood flow
CBF was assessed as previously described (28). Briefly, CBF was assessed before, 5 minutes, and 1 to 4 days after injury using the Moor LDI2-HIR Laser Doppler and Moor Software Version 5.3 (Moor Instruments) Briefly, mice were anesthetized with 2% isoflurane-30% O2 and CBF, in PFUs, was scanned using a 2.5 cm × 2.5 cm scanning area. Tissue perfusion was quantified using the same standard region of interest (ROI) for every sample, representing the same area of the left hemisphere or side of injury and represented as a ratio of post-PFUs relative to each individual animal’s preinjury scan.
Statistical analysis
Data were graphed using GraphPad Prism, version 7 (GraphPad Software, Inc.). Student’s 2-tailed t test was used for comparison of 2 experimental groups. Multiple comparisons were done using 1-way or 2-way ANOVA and repeated measures where appropriate followed by Bonferroni’s post hoc test. Changes were identified as significant at P less than 0.05. Mean values were reported together with SEM. An experimenter blinded to the conditions performed all quantifications.
Study approval.
All procedures were conducted in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and were approved by the Virginia Tech IACUC (15-063 and 18-088).
Author contributions
BO, YQ, and XW performed research and analyzed data. BO, JBM, and MHT wrote and edited the paper, designed the research, and contributed reagents/analytic tools. WAM, MC, JC, and AH performed research and analyzed data.
Supplementary Material
Acknowledgments
We recognize the Institute for Critical Technology and Science, Regenerative Medicine Interdisciplinary graduate education program (IGEP), Center for Engineered Health, and Initiative for Maximizing Student Development (5R25GM072767-12). We thank Jennifer Munson and Chase Cornelison for their assistance with shear stress induction and John Chappell for the gift of VeCadherin-CreERT2 mice. This work was supported by the National Institute of Neurological Disorders and Stroke of the NIH through grants R15NS081623 (to MHT), R01NS112541 (to MHT), and F31NS095719-01A1 (to BO).
Version 1. 11/05/2019
In-Press Preview
Version 2. 01/21/2020
Electronic publication
Version 3. 02/03/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(2):1024–1035.https://doi.org/10.1172/JCI131493.
Contributor Information
Benjamin Okyere, Email: bokyere@vt.edu.
William A. Mills, III, Email: tre1@vt.edu.
Xia Wang, Email: wangx@vt.edu.
Michael Chen, Email: michaelwchen11@gmail.com.
Jiang Chen, Email: jicheng@vt.edu.
Amanda Hazy, Email: amanh15@vt.edu.
Yun Qian, Email: qyun8@vt.edu.
John B. Matson, Email: jmatson@vt.edu.
Michelle H. Theus, Email: mtheus@vt.edu.
References
- 1.del Zoppo GJ, et al. Recombinant tissue plasminogen activator in acute thrombotic and embolic stroke. Ann Neurol. 1992;32(1):78–86. doi: 10.1002/ana.410320113. [DOI] [PubMed] [Google Scholar]
- 2.Hacke W, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008;359(13):1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 3.Ma H, et al. Thrombolysis guided by perfusion imaging up to 9 hours after onset of stroke. N Engl J Med. 2019;380(19):1795–1803. doi: 10.1056/NEJMoa1813046. [DOI] [PubMed] [Google Scholar]
- 4.Berkhemer OA, et al. A randomized trial of intraarterial treatment for acute ischemic stroke. N Engl J Med. 2015;372(1):11–20. doi: 10.1056/NEJMoa1411587. [DOI] [PubMed] [Google Scholar]
- 5.Goyal M, et al. Randomized assessment of rapid endovascular treatment of ischemic stroke. N Engl J Med. 2015;372(11):1019–1030. doi: 10.1056/NEJMoa1414905. [DOI] [PubMed] [Google Scholar]
- 6.Jovin TG, et al. Thrombectomy within 8 hours after symptom onset in ischemic stroke. N Engl J Med. 2015;372(24):2296–2306. doi: 10.1056/NEJMoa1503780. [DOI] [PubMed] [Google Scholar]
- 7.Saver JL, et al. Stent-retriever thrombectomy after intravenous t-PA vs. t-PA alone in stroke. N Engl J Med. 2015;372(24):2285–2295. doi: 10.1056/NEJMoa1415061. [DOI] [PubMed] [Google Scholar]
- 8.Nogueira RG, et al. Trevo versus Merci retrievers for thrombectomy revascularisation of large vessel occlusions in acute ischaemic stroke (TREVO 2): a randomised trial. Lancet. 2012;380(9849):1231–1240. doi: 10.1016/S0140-6736(12)61299-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Srinivasan A, Goyal M, Al Azri F, Lum C. State-of-the-art imaging of acute stroke. Radiographics. 2006;26 Suppl 1:S75–S95. doi: 10.1148/rg.26si065501. [DOI] [PubMed] [Google Scholar]
- 10.Cheng-Ching E, et al. Degree of collaterals and not time is the determining factor of core infarct volume within 6 hours of stroke onset. AJNR Am J Neuroradiol. 2015;36(7):1272–1276. doi: 10.3174/ajnr.A4274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheripelli BK, Huang X, McVerry F, Muir KW. What is the relationship among penumbra volume, collaterals, and time since onset in the first 6 h after acute ischemic stroke? Int J Stroke. 2016;11(3):338–346. doi: 10.1177/1747493015620807. [DOI] [PubMed] [Google Scholar]
- 12.Christoforidis GA, Vakil P, Ansari SA, Dehkordi FH, Carroll TJ. Impact of pial collaterals on infarct growth rate in experimental acute ischemic stroke. AJNR Am J Neuroradiol. 2017;38(2):270–275. doi: 10.3174/ajnr.A5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jung S, et al. Factors that determine penumbral tissue loss in acute ischaemic stroke. Brain. 2013;136(Pt 12):3554–3560. doi: 10.1093/brain/awt246. [DOI] [PubMed] [Google Scholar]
- 14.Galimanis A, et al. Endovascular therapy of 623 patients with anterior circulation stroke. Stroke. 2012;43(4):1052–1057. doi: 10.1161/STROKEAHA.111.639112. [DOI] [PubMed] [Google Scholar]
- 15.Kucinski T, et al. Collateral circulation is an independent radiological predictor of outcome after thrombolysis in acute ischaemic stroke. Neuroradiology. 2003;45(1):11–18. doi: 10.1007/s00234-002-0881-0. [DOI] [PubMed] [Google Scholar]
- 16.Leng X, Lan L, Liu L, Leung TW, Wong KS. Good collateral circulation predicts favorable outcomes in intravenous thrombolysis: a systematic review and meta-analysis. Eur J Neurol. 2016;23(12):1738–1749. doi: 10.1111/ene.13111. [DOI] [PubMed] [Google Scholar]
- 17.Liebeskind DS, Jahan R, Nogueira RG, Zaidat OO, Saver JL, SWIFT Investigators Impact of collaterals on successful revascularization in Solitaire FR with the intention for thrombectomy. Stroke. 2014;45(7):2036–2040. doi: 10.1161/STROKEAHA.114.004781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nogales-Cadenas R, et al. GeneCodis: interpreting gene lists through enrichment analysis and integration of diverse biological information. Nucleic Acids Res. 2009;37(Web Server issue):W317–W322. doi: 10.1093/nar/gkp416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carmona-Saez P, Chagoyen M, Tirado F, Carazo JM, Pascual-Montano A. GENECODIS: a web-based tool for finding significant concurrent annotations in gene lists. Genome Biol. 2007;8(1):R3. doi: 10.1186/gb-2007-8-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bang OY, et al. Collateral flow averts hemorrhagic transformation after endovascular therapy for acute ischemic stroke. Stroke. 2011;42(8):2235–2239. doi: 10.1161/STROKEAHA.110.604603. [DOI] [PubMed] [Google Scholar]
- 21.Menon BK, et al. Assessment of leptomeningeal collaterals using dynamic CT angiography in patients with acute ischemic stroke. J Cereb Blood Flow Metab. 2013;33(3):365–371. doi: 10.1038/jcbfm.2012.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toriumi H, Tatarishvili J, Tomita M, Tomita Y, Unekawa M, Suzuki N. Dually supplied T-junctions in arteriolo-arteriolar anastomosis in mice: key to local hemodynamic homeostasis in normal and ischemic states? Stroke. 2009;40(10):3378–3383. doi: 10.1161/STROKEAHA.109.558577. [DOI] [PubMed] [Google Scholar]
- 23.Li Y, Choi WJ, Qin W, Baran U, Habenicht LM, Wang RK. Optical coherence tomography based microangiography provides an ability to longitudinally image arteriogenesis in vivo. J Neurosci Methods. 2016;274:164–171. doi: 10.1016/j.jneumeth.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamiya A, Bukhari R, Togawa T. Adaptive regulation of wall shear stress optimizing vascular tree function. Bull Math Biol. 1984;46(1):127–137. doi: 10.1007/BF02463726. [DOI] [PubMed] [Google Scholar]
- 25.Pipp F, et al. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler Thromb Vasc Biol. 2004;24(9):1664–1668. doi: 10.1161/01.ATV.0000138028.14390.e4. [DOI] [PubMed] [Google Scholar]
- 26.Zarins CK, Zatina MA, Giddens DP, Ku DN, Glagov S. Shear stress regulation of artery lumen diameter in experimental atherogenesis. J Vasc Surg. 1987;5(3):413–420. [PubMed] [Google Scholar]
- 27.Okyere B, et al. Endothelial-specific EphA4 negatively regulates native pial collateral formation and re-perfusion following hindlimb ischemia. PLoS ONE. 2016;11(7):e0159930. doi: 10.1371/journal.pone.0159930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okyere B, Creasey M, Lebovitz Y, Theus MH. Temporal remodeling of pial collaterals and functional deficits in a murine model of ischemic stroke. J Neurosci Methods. 2018;293:86–96. doi: 10.1016/j.jneumeth.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu H, et al. AMP-activated protein kinase α1 in macrophages promotes collateral remodeling and arteriogenesis in mice in vivo. Arterioscler Thromb Vasc Biol. 2016;36(9):1868–1878. doi: 10.1161/ATVBAHA.116.307743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fung E, Helisch A. Macrophages in collateral arteriogenesis. Front Physiol. 2012;3:353. doi: 10.3389/fphys.2012.00353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guan Y, et al. The formation of aberrant collateral vessels during coronary arteriogenesis in dog heart. Cells Tissues Organs (Print) 2016;201(2):118–129. doi: 10.1159/000442381. [DOI] [PubMed] [Google Scholar]
- 32.Tressel SL, et al. Angiopoietin-2 stimulates blood flow recovery after femoral artery occlusion by inducing inflammation and arteriogenesis. Arterioscler Thromb Vasc Biol. 2008;28(11):1989–1995. doi: 10.1161/ATVBAHA.108.175463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamberto I, et al. Distinctive binding of three antagonistic peptides to the ephrin-binding pocket of the EphA4 receptor. Biochem J. 2012;445(1):47–56. doi: 10.1042/BJ20120408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murai KK, Nguyen LN, Koolpe M, McLennan R, Krull CE, Pasquale EB. Targeting the EphA4 receptor in the nervous system with biologically active peptides. Mol Cell Neurosci. 2003;24(4):1000–1011. doi: 10.1016/j.mcn.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 35.Noberini R, et al. Small molecules can selectively inhibit ephrin binding to the EphA4 and EphA2 receptors. J Biol Chem. 2008;283(43):29461–29472. doi: 10.1074/jbc.M804103200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu B, et al. Potent and selective EphA4 agonists for the treatment of ALS. Cell Chem Biol. 2017;24(3):293–305. doi: 10.1016/j.chembiol.2017.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steinle JJ, Meininger CJ, Forough R, Wu G, Wu MH, Granger HJ. Eph B4 receptor signaling mediates endothelial cell migration and proliferation via the phosphatidylinositol 3-kinase pathway. J Biol Chem. 2002;277(46):43830–43835. doi: 10.1074/jbc.M207221200. [DOI] [PubMed] [Google Scholar]
- 38.Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell. 1998;93(5):741–753. doi: 10.1016/S0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- 39.Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3(7):475–486. doi: 10.1038/nrm856. [DOI] [PubMed] [Google Scholar]
- 40.Gale NW, et al. Ephrin-B2 selectively marks arterial vessels and neovascularization sites in the adult, with expression in both endothelial and smooth-muscle cells. Dev Biol. 2001;230(2):151–160. doi: 10.1006/dbio.2000.0112. [DOI] [PubMed] [Google Scholar]
- 41.Shin D, et al. Expression of ephrinB2 identifies a stable genetic difference between arterial and venous vascular smooth muscle as well as endothelial cells, and marks subsets of microvessels at sites of adult neovascularization. Dev Biol. 2001;230(2):139–150. doi: 10.1006/dbio.2000.9957. [DOI] [PubMed] [Google Scholar]
- 42.Korff T, Braun J, Pfaff D, Augustin HG, Hecker M. Role of ephrinB2 expression in endothelial cells during arteriogenesis: impact on smooth muscle cell migration and monocyte recruitment. Blood. 2008;112(1):73–81. doi: 10.1182/blood-2007-12-128835. [DOI] [PubMed] [Google Scholar]
- 43.Qin H, Noberini R, Huan X, Shi J, Pasquale EB, Song J. Structural characterization of the EphA4-Ephrin-B2 complex reveals new features enabling Eph-ephrin binding promiscuity. J Biol Chem. 2010;285(1):644–654. doi: 10.1074/jbc.M109.064824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Korff T, et al. Endothelial ephrinB2 is controlled by microenvironmental determinants and associates context-dependently with CD31. Arterioscler Thromb Vasc Biol. 2006;26(3):468–474. doi: 10.1161/01.ATV.0000200081.42064.e7. [DOI] [PubMed] [Google Scholar]
- 45.Chae JK, et al. Coadministration of angiopoietin-1 and vascular endothelial growth factor enhances collateral vascularization. Arterioscler Thromb Vasc Biol. 2000;20(12):2573–2578. doi: 10.1161/01.ATV.20.12.2573. [DOI] [PubMed] [Google Scholar]
- 46.Siddiqui AJ, et al. Combination of angiopoietin-1 and vascular endothelial growth factor gene therapy enhances arteriogenesis in the ischemic myocardium. Biochem Biophys Res Commun. 2003;310(3):1002–1009. doi: 10.1016/j.bbrc.2003.09.111. [DOI] [PubMed] [Google Scholar]
- 47.Fukuhara S, Sako K, Noda K, Nagao K, Miura K, Mochizuki N. Tie2 is tied at the cell-cell contacts and to extracellular matrix by angiopoietin-1. Exp Mol Med. 2009;41(3):133–139. doi: 10.3858/emm.2009.41.3.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fiedler U, Augustin HG. Angiopoietins: a link between angiogenesis and inflammation. Trends Immunol. 2006;27(12):552–558. doi: 10.1016/j.it.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 49.Brindle NP, Saharinen P, Alitalo K. Signaling and functions of angiopoietin-1 in vascular protection. Circ Res. 2006;98(8):1014–1023. doi: 10.1161/01.RES.0000218275.54089.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim I, Kim JH, Moon SO, Kwak HJ, Kim NG, Koh GY. Angiopoietin-2 at high concentration can enhance endothelial cell survival through the phosphatidylinositol 3’-kinase/Akt signal transduction pathway. Oncogene. 2000;19(39):4549–4552. doi: 10.1038/sj.onc.1203800. [DOI] [PubMed] [Google Scholar]
- 51.Teichert-Kuliszewska K, et al. Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of Tie2. Cardiovasc Res. 2001;49(3):659–670. doi: 10.1016/S0008-6363(00)00231-5. [DOI] [PubMed] [Google Scholar]
- 52.Hashimoto T, Wu Y, Boudreau N, Li J, Matsumoto M, Young W. Regulation of tie2 expression by angiopoietin--potential feedback system. Endothelium. 2004;11(3-4):207–210. doi: 10.1080/10623320490512417. [DOI] [PubMed] [Google Scholar]
- 53.Winderlich M, et al. VE-PTP controls blood vessel development by balancing Tie-2 activity. J Cell Biol. 2009;185(4):657–671. doi: 10.1083/jcb.200811159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seegar TC, et al. Tie1-Tie2 interactions mediate functional differences between angiopoietin ligands. Mol Cell. 2010;37(5):643–655. doi: 10.1016/j.molcel.2010.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eklund L, Kangas J, Saharinen P. Angiopoietin-Tie signalling in the cardiovascular and lymphatic systems. Clin Sci. 2017;131(1):87–103. doi: 10.1042/CS20160129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nogueira RG, et al. Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. N Engl J Med. 2018;378(1):11–21. doi: 10.1056/NEJMoa1706442. [DOI] [PubMed] [Google Scholar]
- 57.Defazio RA, et al. A protocol for characterizing the impact of collateral flow after distal middle cerebral artery occlusion. Transl Stroke Res. 2011;2(1):112–127. doi: 10.1007/s12975-010-0044-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brickler TR, et al. Angiopoietin/Tie2 axis regulates the age-at-injury cerebrovascular response to traumatic brain injury. J Neurosci. 2018;38(45):9618–9634. doi: 10.1523/JNEUROSCI.0914-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kennedy BK, Barbie DA, Classon M, Dyson N, Harlow E. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev. 2000;14(22):2855–2868. doi: 10.1101/gad.842600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.