

Abstract
ATP-sensitive potassium (KATP) channels are uniquely evolved protein complexes that couple cell energy levels to cell excitability. They govern a wide range of physiological processes including hormone secretion, neuronal transmission, vascular dilation, and cardiac and neuronal preconditioning against ischemic injuries. In pancreatic β-cells, KATP channels composed of Kir6.2 and SUR1, encoded by KCNJ11 and ABCC8, respectively, play a key role in coupling blood glucose concentration to insulin secretion. Mutations in ABCC8 or KCNJ11 that diminish channel function result in congenital hyperinsulinism. Many of these mutations principally hamper channel biogenesis and hence trafficking to the cell surface. Several small molecules have been shown to correct channel biogenesis and trafficking defects. Here, we review studies aimed at understanding how mutations impair channel biogenesis and trafficking and how pharmacological ligands overcome channel trafficking defects, particularly highlighting recent cryo-EM structural studies which have revealed the mechanisms of channel assembly and pharmacological chaperones.
Keywords: sulfonylurea receptor, sulfonylureas, carbamazepine, ABC transporter, Kir channel, protein assembly, protein trafficking, congenital hyperinsulinism
Introduction
In 1983, Noma discovered in cardiac myocytes a potassium channel that was activated by cyanide and inhibited by intracellular ATP, and named it the ATP-sensitive potassium (KATP) channel [1]. Soon after, studies into the electric mechanisms of insulin release from the pancreatic β-cell led to the identification of a similar KATP channel that serves as a key molecular link between glucose metabolism and insulin secretion [2, 3]. Additional KATP channels were subsequently identified in brain, skeletal muscle, and vascular smooth muscle [4–6]. By coupling cellular adenine nucleotide concentrations with membrane potential, these channels control a range of physiological activities, including hormone secretion, vasodilation, and cardiac contraction to enable adaptation to changing metabolic environments (reviewed in [7–11]). Molecular cloning and functional characterization revealed the proteins that make up the channel: an inwardly rectifying potassium (Kir) channel and a protein called sulfonylurea receptor (SUR) belonging to the ABC (ATP-Binding Cassette) transporter superfamily [12]. KATP channels found in different tissues have distinct functional and pharmacological properties, resulting from combinations of either Kir6.1 or Kir6.2 with SUR1 or SUR2, which occurs as two major splice variants SUR2A and SUR2B [13–15]. Kir6.2/SUR1 channels are expressed primarily in the pancreatic endocrine islets and the brain, Kir6.2/SUR2A channels in cardiac myocytes and skeletal muscle, and Kir6.1/SUR2B channels in vascular smooth muscle. Mutations in ABCC8 and KCNJ11, the genes encoding SUR1 and Kir6.2, respectively, are linked to a range of insulin secretion and neurological disorders (reviewed in [7, 16, 17]), Enhanced channel currents due to mutations in ABCC9 and KCNJ8, genes encoding SUR2 and Kir6.1, respectively, have recently been shown to underlie a cardiovascular disorder known as Cantú syndrome [18], while a frameshift mutation in ABCC9 that results in non-functional channels causes a disease named ABCC9-related Intellectual disability Myopathy Syndrome or AIMS [19].
Owing to their importance in health and disease, KATP channels have attracted much research attention. In particular, the pancreatic/neuronal Kir6.2/SUR1 subtype has been intensely studied and will be the focus of this review. In pancreatic β-cells, KATP conductance maintains the membrane potential in a hyperpolarized resting state at basal glucose concentrations. Upon glucose stimulation, KATP channels close causing membrane depolarization, which activates voltage-gated Ca2+ channels. Ca2+ influx then triggers insulin release. In this way, KATP channels couple glucose stimulation to insulin secretion. They are the major target of insulin secretagogues used to treat type 2 diabetes (T2D). For example, sulfonylureas, drugs which have been widely prescribed since the 1950s to treat T2D [20], stimulate insulin secretion and lower blood glucose by reducing KATP channel activity. Numerous channel mutations have been identified in patients with insulin secretion disorders [21, 22]. Mutations that reduce KATP currents by reducing the number of channels present in the plasma membrane and/or the channel’s ability to open at low glucose concentrations (referred to as loss-of-function mutations) lead to persistent β-cell depolarization. Consequently, insulin secretion persists despite severe hypoglycemia as seen in the disease congenital hyperinsulinism (CHI). By contrast, mutations that increase KATP currents (referred to as gain-of-function mutations) cause β-cell hyperpolarization and insufficient insulin secretion even when blood glucose levels are high, resulting in neonatal diabetes [16, 17]. In some cases, gain-of-function mutations cause developmental delay, epilepsy, in addition to neonatal diabetes, known as DEND syndrome. Reconstitution studies revealed that many CHI mutations diminish KATP currents by impairing channel folding, assembly and trafficking to the plasma membrane [23]. Patients with such mutations often require pancreatectomy to prevent life-threatening hypoglycemia caused by uncontrolled insulin secretion [24, 25]. This prompted efforts to identify small molecule chaperones that can correct channel expression defects caused by disease mutations [23].
In this article we discuss current understanding of the molecular mechanism of SUR1/Kir6.2 KATP channel assembly and trafficking, how disease mutations disrupt these processes to prevent surface expression, and how pharmacological ligands overcome channel assembly and trafficking defects caused by certain mutations. In particular, we will highlight insight gained from recent high resolution channel structures obtained using single-particle cryo-electron microscopy.
Overview of KATP channel biogenesis and functional regulation
Functional expression of KATP channels requires co-assembly of Kir6.2 and SUR1 into a heterooctameric complex in the endoplasmic reticulum (ER) [12, 26, 27]. Expressed alone, neither protein exits the ER due to an –RKR- tripeptide cytoplasmic ER retention/retrieval motif [27]. These motifs are likely concealed upon complete channel assembly to allow ER exit, ensuring only properly assembled channels traffic to the cell surface [27]. A member of the inward rectifier potassium channel family, Kir6.2 has two transmembrane helices and intracellular N- and C-termini [28]. SUR1 is classified as a member of the ABCC subfamily in the ATP-binding cassette (ABC) transporter superfamily [29]. In addition to a characteristic ABC core structure comprising two transmembrane domains (TMD1 & 2) and two cytoplasmic nucleotide binding domains (NBD1 & 2), SUR1 has an N-terminal transmembrane domain (TMD0) which is connected to the ABC core by a long cytoplasmic loop called L0 [30–32] (Fig. 1A). The co-dependence of SUR1 and Kir6.2 on each other for surface expression sets them apart from other ABC transporters and Kir channels and raises interest in the molecular interactions involved in their co-evolution into a functional unit. Several channel protein domains had been implicated in channel assembly, including TMD0 of SUR1 [26, 33, 34], and the N-terminus and the first transmembrane helix of Kir6.2 [26]. However, detailed mechanisms underlying subunit-subunit interactions remained unresolved until the recent development of high resolution 3D structures, discussed later.
Figure 1. KATP channel architecture.

(A) Topology of SUR1 and Kir6.2 with domains labeled and colored to match the 3D structures shown below. Locations of the two N-linked glycosylation sites (branched sticks) in SUR1 and the RKR ER retention signal (black circle) in SUR1 and Kir6.2 are marked. (B) Structural model (side view and top view) of the KATP channel with glibenclamide bound to SUR1 shown as red spheres and ATP bound to Kir6.2 in green spheres. Model (PDB: 6BAA) is built using a 3.7Å cryoEM map (EMD-7073).
Although not the focus of this review, functional regulation of KATP channels is briefly summarized as it pertains to the mechanisms of pharmacological chaperones all of which are also channel inhibitors, as well as channel structures solved in the presence of various gating molecules. KATP channel function is governed mainly by intracellular ATP and ADP (reviewed in [7, 9, 35], the concentrations of which are determined by glucose levels. In a simplified scheme, ATP binds to Kir6.2 in an Mg2+-independent manner to close the channel, while MgATP and MgADP bind to the NBDs of SUR1, causing dimerization of the NBDs, which counteracts the inhibitory action of ATP on Kir6.2 and thereby stimulate channel activity. When blood glucose levels rise, the intracellular ATP to ADP ratio increases as a result of glycolysis such that ATP inhibition dominates to suppress KATP channel activity. Conversely, when blood glucose levels fall, the ATP to ADP ratio decreases such that MgATP/MgADP stimulation prevails to increase channel activity. In this way, KATP channels link glucose metabolism to β-cell excitability, which in turn regulates voltage-gated calcium channels, Ca2+ influx, and insulin secretion (reviewed in [36, 37]).
KATP channel trafficking mutations and pharmacological chaperones
A mutation in human SUR1, ΔF1388, was the first CHI-associated genetic defect found to prevent KATP channel trafficking to the β-cell plasma membrane [38]. The mutant protein was retained in the ER, unable to reach its mature complex-glycosylated state. Subsequent studies of other mutations carried by CHI patients identified additional mutations that greatly reduce or abolish channel expression at the cell surface [34, 39–44]. Most were shown to be retained intracellularly, suggesting they are misfolded and/or unable to assemble with Kir6.2. These mutations are collectively referred to as trafficking mutations. To date, more than forty mutations have been reported to disrupt normal channel expression at the cell surface, mostly missense mutations in SUR1. These mutations typically manifest as recessive phenotypes [45]. Affected CHI patients with such mutations do not respond to drugs that stimulate channel function due to lack of surface expression and often undergo pancreatectomy in infancy or childhood to prevent severe hypoglycemia [22]. Understanding how mutations impair channel surface expression and how such defects may be corrected would likely improve treatments for the disease.
Pharmacological chaperones are small molecules whose binding to cognate proteins facilitate protein folding and assembly, thus chaperoning proteins to their correct cellular destinations. They have been exploited to correct folding and trafficking defects of a number of genetically mutated proteins or to boost surface expression of WT proteins, such as CFTR, vasopressin type 2 receptors, gonadotropin releasing hormone receptors, and d-opioid receptors [46, 47], Yan et al. first explored the concept of pharmacological chaperones in KATP trafficking mutants by testing drugs known to target KATP channels, sulfonylureas and diazoxide, on two SUR1 trafficking mutations, A116P and V187D located in TMD0 [40]. The β-cell KATP channel is inhibited by sulfonylurea drugs, which are used clinically to improve insulin secretion in type 2 diabetes mellitus. Yan et al. found that a high affinity sulfonylurea, glibenclamide, and a lower affinity sulfonylurea tolbutamide both significantly improved surface expression of the two mutant channels. In contrast, the channel opening drug diazoxide was without effect. Metabolic pulse-chase and co-immunoprecipitation experiments showed that SUR1 proteins with these mutations are degraded faster [40] and have reduced association with Kir6.2 [34], suggesting the mutations disrupt SUR1 folding and assembly with Kir6.2 to render increased endoplasmic reticulum associated degradation (ERAD) of mutant SUR1. In addition to rescuing mutant channels to the cell surface, sulfonylureas also improved the biogenesis efficiency and thereby surface expression of wild-type (WT) channels [40], suggesting that these drugs also facilitate WT channel protein folding and/or assembly. Importantly, the study by Yan et al. showed that upon washout of the low affinity tolbutamide, both A116P and V187D mutant channels that had been chaperoned to the cell surface were then as normally responsive to ATP inhibition and MgATP/MgADP stimulation as WT channels [40] (see Table 1). This demonstrates that trafficking mutants can be rescued to recover functions and thus the potential of pharmacological chaperones in treating those CHI patients harboring trafficking mutations.
Table 1:
KATP trafficking mutations1
| SUR1 mutation | Rescue by pharmacological chaperones | Rescued channel function2 | Reference |
|---|---|---|---|
| C6G | Barely | N. D.3 | [39] |
| G7R | Yes | Functional (efflux) | [41] |
| V21D | Yes | Functional (efflux, patching) | [39] |
| N24K | Yes | Functional (efflux) | [41] |
| F27S | Yes | Functional (efflux, patching) | [41, 58] |
| D29G | Yes | Functional (efflux, patching) | [39] |
| A30T | Yes | Functional (efflux, patching) | [39] |
| L31P | Barely | N.D. | [39] |
| L40R | Barely | N.D. | [39] |
| G70E | Yes | Functional (efflux, patching) | [39] |
| R74W | Yes | Decreased ATP sensitivity4 | [41, 92] |
| M80R | Yes | Functional (efflux, patching) | [39] |
| G92D | Yes | Functional (efflux, patching) | [39] |
| G111R | Yes | Functional (efflux, patching) | [39] |
| A113V | Yes | Functional (efflux, patching) | [39] |
| A116P | Yes | Functional (efflux, patching) | [40] |
| E128K | Yes | Decreased ATP sensitivity (patching)4 | [41, 92] |
| R168C | Yes | Functional (efflux, patching) | [39] |
| G173R | Yes | Functional (efflux, patching) | [39] |
| V187D | Yes | Functional (efflux, patching) | [40] |
| R495Q | No | N.D. | [41] |
| E501K | No | N.D. | [41] |
| L503P | No | N.D. | [41] |
| F686S | No | N.D. | [41] |
| G716V | No | N.D. | [41] |
| L1350Q | No | N.D. | [41] |
| ΔF1388 | No | No MgADP response5 | [38, 40] |
| D1472H | No | N.D. | [41] |
| L1544P | No | No MgADP response5 | [40, 44] |
Only missense SUR1 mutations our group has tested for response to pharmacological chaperones (glibenclamide, tolbutamide, repaglinide, and/or carbamazepine) are included.
Functional properties were assessed by 86Rb+ efflux assay in response to metabolic inhibition (efflux) or inside-out patch-clamp recordings (patching) which measure channel sensitivity to ATP inhibition and MgADP stimulation, as described in [39–41]. Note functional studies were performed after washout of pharmacological chaperones to remove the inhibitory effects of the chaperones.
N.D.: Not determined due to lack of surface expression and lack of response to pharmacological chaperones.
In addition to causing trafficking defects these two mutations also render the channels less sensitive to ATP inhibition.
Functions were determined of channels escaped to the cell surface by mutating the RKR ER retention signal to AAA.
Following the initial report, many more disease mutations were subjected to expression studies to test their impact on channel expression at the cell surface and response to pharmacological chaperones (Table 1). A striking finding that emerged from these studies is that although SUR1 trafficking mutations occur throughout the protein, only those within TMD0 were rescued by sulfonylureas [39, 41, 48]. In addition, the rescue effect of sulfonylureas is dependent on Kir6.2 [49, 50]. Without Kir6.2 co-expression, pharmacological chaperones are unable to rescue mutant SUR1 to the cell surface even when the RKR ER retention signal was mutated to AAA, which allows wild-type SUR1 to traffic to the cell surface independent of Kir6.2. This observation argues against a simple pharmacochaperoning mechanism wherein sulfonylureas correct folding defects of mutant SUR1 protein, and rather suggest that sulfonylureas act further on mutant SUR1-Kir6.2 interactions. A third interesting aspect about KATP pharmacochaperones is that they are all channel inhibitors. Sulfonylureas have been used to treat type 2 diabetes for more than half a century as they close KATP channels to stimulate insulin secretion [20]. A distinct class of antidiabetic compounds developed more recently known as glinides, such as repaglinide, also acts by inhibiting KATP channels to stimulate insulin secretion [51]. Like the sulfonylurea glibenclamide, repaglinide was found to be highly effective in restoring surface expression of KATP trafficking mutants with SUR1 TMD0 mutations [50, 52]. By contrast, as with diazoxide, additional KATP channel openers including NN414 and VU0071063 failed to show any effect [39, 40]. Curiously, these channels openers seem to actually reduce biogenesis efficiency of WT channels [39]. Thus, inhibitors generally help to stabilize the channel complex while openers have the opposite effect.
Discovery of carbamazepine as a novel KATP channel pharmacological chaperone and inhibitor
Since sulfonylureas and glinides only rescue SUR1 with trafficking mutations in TMD0, a question arises as to whether there are other compounds that can correct SUR1 trafficking defects caused by mutations in its ABC core domain. The ABC core of SUR1 is similar to other ABCC proteins such as CFTR [31]. CFTR has been a subject of intense research in the field of pharmacological chaperones because the cellular mechanism for the most prevalent cystic fibrosis-causing mutation, ΔF508 located in the NBD1, is CFTR misfolding and failure to traffic to the plasma membrane [53]. Numerous drug screening studies have been conducted in search of compounds that correct the trafficking defect of ΔF508 CFTR. It was hypothesized that these CFTR correctors may also be effective in facilitating the folding of SUR1 harboring mutations in the ABC core that shares structural homology with CFTR. Studies testing CFTR correctors on KATP trafficking mutants did show a number of promising compounds with some salutary effects [54]. In this initial study, the compounds were tested against the TMD0 mutations A116P and V187D, and it was therefore unclear whether the small rescue effect applies to other trafficking mutations. A subsequent study tested one of the compounds, carbamazepine, against 19 SUR1 trafficking mutations distributed throughout the protein [48]. Surprisingly, contrary to the expectation that a CFTR ΔF508 corrector might also rescue SUR1 ABC core trafficking mutations, the study showed that carbamazepine only rescued TMD0 trafficking mutations, just like sulfonylureas and glinides.
Carbamazepine is best known as an anticonvulsant that inhibits voltage-gated Na+ channels [55, 56]. The finding that carbamazepine is a KATP channel chaperone raised questions about the nature of its interaction with the channel. To test whether carbamazepine binds directly to the channel, Devaraneni et al. conducted competition binding assays between carbamazepine and the sulfonylurea glibenclamide [49]. Glibenclamide is a high affinity ligand for KATP channels with an estimated KD of ~1–5 nM [57]. Carbamazepine was found to compete with glibenclamide for binding to the channel with an estimated KD of ~25nM [49]. Moreover, like glibenclamide, carbamazepine inhibits channel activity and abolishes channel stimulation by MgATP/MgADP [58]. Finally, mutating SUR1 residues previously implicated in glibenclamide binding diminished the ability of both glibenclamide and carbamazepine to act as a channel inhibitor and pharmacological chaperone [49]. These results provide strong evidence that carbamazepine and glibenclamide exert their effects on the channel via similar mechanisms and related sites.
The chemical structures of sulfonylureas, glinides and carbamazepine are quite distinct. How do these diverse compounds inhibit KATP channels and chaperone the same set of trafficking mutants? Although the studies summarized above suggest these drugs likely interact with the channel complex similarly to affect channel assembly and function, fundamental aspects of the underlying mechanisms remained a mystery in the absence of a high resolution 3D channel structure.
CryoEM structures of KATP channels
Early biochemical and biophysical studies have suggested that the KATP channel complex is a hetero-octamer of four pore-forming Kir6.2 subunits and four regulatory SUR1 subunits [59–61]. The large size of the complex estimated to be ~ 950 kDa made solving the structure of the channel by X-ray crystallography a formidable task. In 2005, using an engineered construct that fuses the C-terminus of SUR1 with the N-terminus of Kir6.2, Mikhailov et al. reported an initial three-dimensional KATP structure obtained by single-particle negative stain cryo-EM [62]. The structure shows a central tetrameric Kir6.2 core embraced by four SUR1 proteins. Although the resolution was too low to resolve molecular details, the study set the stage for subsequent efforts that eventually led to several near atomic resolution structures of the channel.
With the advances in single-particle cryo-EM techniques, three groups have published seven studies in the past two years reporting KATP channel structures at subnanometer resolutions using different construct designs and in different states [52, 63–68] (Table 2). Our group solved multiple structures of channels formed by co-expression of SUR1 and Kir6.2 proteins, including the apo state (no ligand), as well as in the presence of ATP alone, or the presence of ATP and glibenclamide, repaglinide, or carbamazepine [52, 66, 67]. The group led by Lei Chen published an initial structure of channels formed by separate SUR1 and Kir6.2 proteins in the presence of GBC [65], and several subsequent structures using a fusion protein with a 39-amino acids linker between the C-terminus of SUR1 and the N-terminus of Kir6.2 (SUR1–39a.a.-Kir6.2) in the presence of various ligands including ATPγS, MgADP, glibenclamide, repaglinide, and the channel opener NN414 [63, 68]. A third group led by Jue Chen and Roderick MacKinnon reported KATP structures in the presence of MgATP, using a fusion of human SUR1 C-terminus to Kir6.2 N-terminus via a 6-aa linker [64]. All structures confirmed a central core formed by the Kir6.2 tetramer surrounded by four SUR1 (Fig. 1B). In all structures, Kir6.2 is closed. In the apo state, or in the presence of pharmacological chaperones, the SUR1 shows an inward-facing conformation with the two NBDs separate. In the presence of MgATP, MgADP, or MgADP plus NN414, the SUR1 NBDs are dimerized. These structures have offered unprecedented views of the channel and resolved important ligand binding sites. Detailed comparison of structures with SUR1 NBDs in different states has been reviewed recently by others [69]. Below we will focus on structural information that is most relevant to understanding how KATP pharmacological chaperones work.
Table 2:
KATP channel cryoEM structures
| Constructs | Ligands (PDB/ EMDB ID; resolution) | Structure highlights | Reference |
|---|---|---|---|
| hamster SUR1 (Q608K)+mouse Kir6.2 | Glibenclamide (5WUA/EMD-6689; 5.6Å) | Kir6.2 closed and SUR1 inward-facing Glibenclamide and PIP2 density tentatively assigned; however, glibenclamide density assignment was later corrected in Wu et al. |
[65] |
| hamsterSUR1+ratKir6.2 | Glibenclamide+ATP (5TWV/5.8Å) | Resolved ATP cryoEM density in Kir6.2 | [67] |
| hamsterSUR1+ratKir6.2 | Glibenclamide+ATP (6BAA/EMD-7073; 3.7Å) | Resolved glibenclamide cryoEM density in SUR1 | [66] |
| humanSUR1-(6aa linker)-humanKir6.2 fusion1 | MgATP+PIP2 Class 1: quatrefoil (6C3O/EMD-7339; 3.9Å) Class 2: propeller (6C3P/EMD-7338; 5.6Å) | SUR1’s NBDs dimerized, with MgATP bound at NBD1 and MgADP at NBD2; Kir6.2 closed, bound to ATP Two conformations were resolved: quatrefoil and propeller, with the quatrefoil form being the dominant class |
[64] |
| hamsterSUR1-(39aa linker)-mouseKir6.2 fusion1 | ATPγS (5YW8/EM D-6848; 4.4Å) Glibenclamide/ATPγS (5YKE/EMD-6831; 4.1Å) MgADP/NN414/PIP2 (5YWC/EMD-6852; 4.3Å) |
Glibenclamide density assigned Only propeller-like conformation was observed in MgADP bound, NBDs-dimerized structure Tentative assignment of NN414 density |
[68] |
| hamsterSUR1-(39aa linker)-mouseKir6.2 fusion | Repaglinide+ATPγS (6JB1/EMD-9787; 3.3Å) | Repaglinide density assigned | [63] |
| hamsterSUR1+ratKir6.2 | SUR1 bound to: Glibenclamide+ATP | SUR1 NBD1 bound to ATP in all structures except the apo state structure | [52] |
| (6PZA/EM D-20530; 3.7Å) Repaglinide+ATP (6PZ9/EMD-20528; 3.7Å) Carbamazepine+ATP (6PZC/EMD-20530; 4.3Å) ATP only (6PZI/EMD-20535; 4.5Å) Apo (6PZB/EMD-20533; 4.6Å) |
Glibenclamide, repaglinide, and carbamazepine cryoEM density found in the same binding pocket, and no density observed in the pocket in ATP only or apo state structures Kir6.2 N-term density assigned in the drug-bound structures |
Only the dominant class or the best resolution structure for each liganded state is included.
Channel assembly domains
Biochemical and biophysical studies prior to cryoEM structures implicated several domains in SUR1 and Kir6.2 as critical for channel assembly. Using chimeras of Kir6.2 with Kir2.1, which is known to not associate with SUR1, the first transmembrane helix and the cytoplasmic N-terminus of Kir6.2 were found to be important for specific assembly with SUR1 [26]. Using chimeric SUR1 proteins containing TMD0 from MRP1, an ABC transporter that cannot interact with Kir6.2, it was shown that the TMD0 in SUR1 confers specific interaction with Kir6.2 [26]. Further evidence for TMD0 in mediating interactions with Kir6.2 came from studies showing that SUR1-TMD0 alone can assemble with Kir6.2 and modulate the trafficking as well as gating of Kir6.2 [34, 70].
In the cryoEM structures, TMD0 of each SUR1 subunit makes direct contact with a Kir6.2, serving as the primary anchor between SUR1 and Kir6.2 (Fig. 2A). In particular, the first transmembrane helix in TMD0 of SUR1 (SUR1-TM1) runs parallel to the first transmembrane helix (M1) of Kir6.2 (Fig. 2B). In addition, the cytoplasmic loops in TMD0 and the proximal end of the long cytoplasmic loop L0, which connects TMD0 to the SUR1-ABC core domain, form extensive interactions with the cytoplasmic domain of Kir6.2 near the lipid-cytoplasm interface. The 3D structure explains why so many TMD0 mutations disrupt channel assembly and trafficking (Fig. 2C). Interestingly, mutation of Kir6.2 Q52 to E or D has been shown to suppress assembly and trafficking defects caused by certain SUR1-TMD0 mutations, including F27S and A116P (Fig. 2D) [71], highlighting that in addition to transmembrane interactions, the cytoplasmic loops of SUR1 and Kir6.2 also serve to “glue” the two subunits together during channel assembly.
Figure 2. Primary assembly domains of the KATP channel.

(A) A side view of the channel structure showing the close contact between SUR1-TMD0 and Kir6.2. (B) A close-up view showing the interactions between the TM1 in TMD0 of SUR1 and M1 of Kir6.2, and the cytoplasmic loops of Kir6.2, TMD0, and the proximal part of L0. (C) Trafficking mutations mapped on the cryoEM structure of KATP channels. The red spheres mark the following mutations: C6G, G7R, V21D, N24K, F27S, D29G, A30T, L31P, L40R, G70E, R74W/Q. M80R, G92D, G111R, A113V, A116P, W128K, R168C, G173R, and V187D, which are all responsive to pharmacological chaperones. The green spheres mark the following mutations: R495Q, E501K, L503P, F686S, G716V, E1324K, L1350Q, R1353P, DF1388, M1395R, D1472H, R1494W, and L1544P, which are not responsive to pharmacological chaperone rescue. (D) Two SUR1 TMD0 trafficking mutations, F27S and A116P, were rescued by a second site mutation in Kir6.2 Q52E. The side chains of the mutated residues are shown in the structural model.
Pharmacological chaperone binding pocket
For decades, sulfonylureas have been used to treat T2DM and there has been vast interest in understanding how these drugs inhibit KATP channel mechanistically. Early sequence comparison and chimeric studies between SUR1 and SUR2, which exhibit differential sensitivities to the low affinity sulfonylurea tolbutamide and the high affinity sulfonylurea glibenclamide led to identification of a key residue critical for tolbutamide and glibenclamide binding, SUR1 S1238 [72]. Subsequent biochemical and biophysical studies pointed to additional structural components, including the L0 linker of SUR1 and the N-terminus of Kir6.2, that are needed for high affinity glibenclamide binding [73, 74]. Specifically, a mutation Y230A in L0 of SUR1 diminished the ability of glibenclamide to interact with and inhibit the channel [50, 73], and deletion of the distal N-terminus of Kir6.2 also reduced the binding of glibenclamide as well as channel inhibition by sulfonylureas [75–77]. Despite the abundance of structure-function correlation data, how the drugs and the various structural elements implicated are related in 3D space was left for imagination and the precise drug binding site(s) remained obscure, until the cryoEM structures became available.
In the first two cryoEM structure papers by Li et al. and Martin et al. glibenclamide was present in the sample. However, due to the moderate 5–6Å resolutions and uncertainty in modeling the L0 linker which had no existing homologous structures in the protein databank, the glibenclamide density was only tentatively assigned to a location in between the L0 region where Y230 is located and SUR1 TMD bundle above NBD1 where S1238 is located [65], or not assigned [67]. A later improved cryoEM map at 3.7Å by Martin et al. showed unambiguous glibenclamide density located within the TM bundle above NBD1 [66]. Molecular modeling revealed a binding pocket formed by residues from both TMD1 and TMD2 including S1238 (Fig. 3A). Mutation of residues lining the pocket diminished glibenclamide inhibition of the channel, providing functional validation of the binding site model [66]. Interestingly, the L0 loop residue Y230 previously implicated in binding is not directly in contact with the drug but interacts structurally with residues forming the binding pocket, and thus is playing an indirect structural role in glibenclamide binding (Fig. 3A). The glibenclamide binding site identified by Martin et al. is also confirmed by another cryoEM structure from Wu et al. using the SUR1–39a.a.-Kir6.2 fusion protein [68]. In the glibenclamide bound structure, the two NBDs are separate, a conformation referred to as “inward-facing” in the ABC transporter literature [31]. Interestingly, in KATP structures where SUR1 NBDs are bound to MgATP/MgADP and dimerized, the space in the TM bundle above NBD1 becomes too small to accommodate glibenclamide [64, 68]. Glibenclamide is known to inhibit channel activity in part by abolishing the ability of MgATP/MgADP to stimulate channels [58, 78]. The structures suggest that by occupying the space in the TM bundle above NBD1 glibenclamide interferes with conformational change associated with NBD dimerization thus preventing MgATP/MgADP stimulation.
Figure 3. Pharmacological chaperone binding pocket.
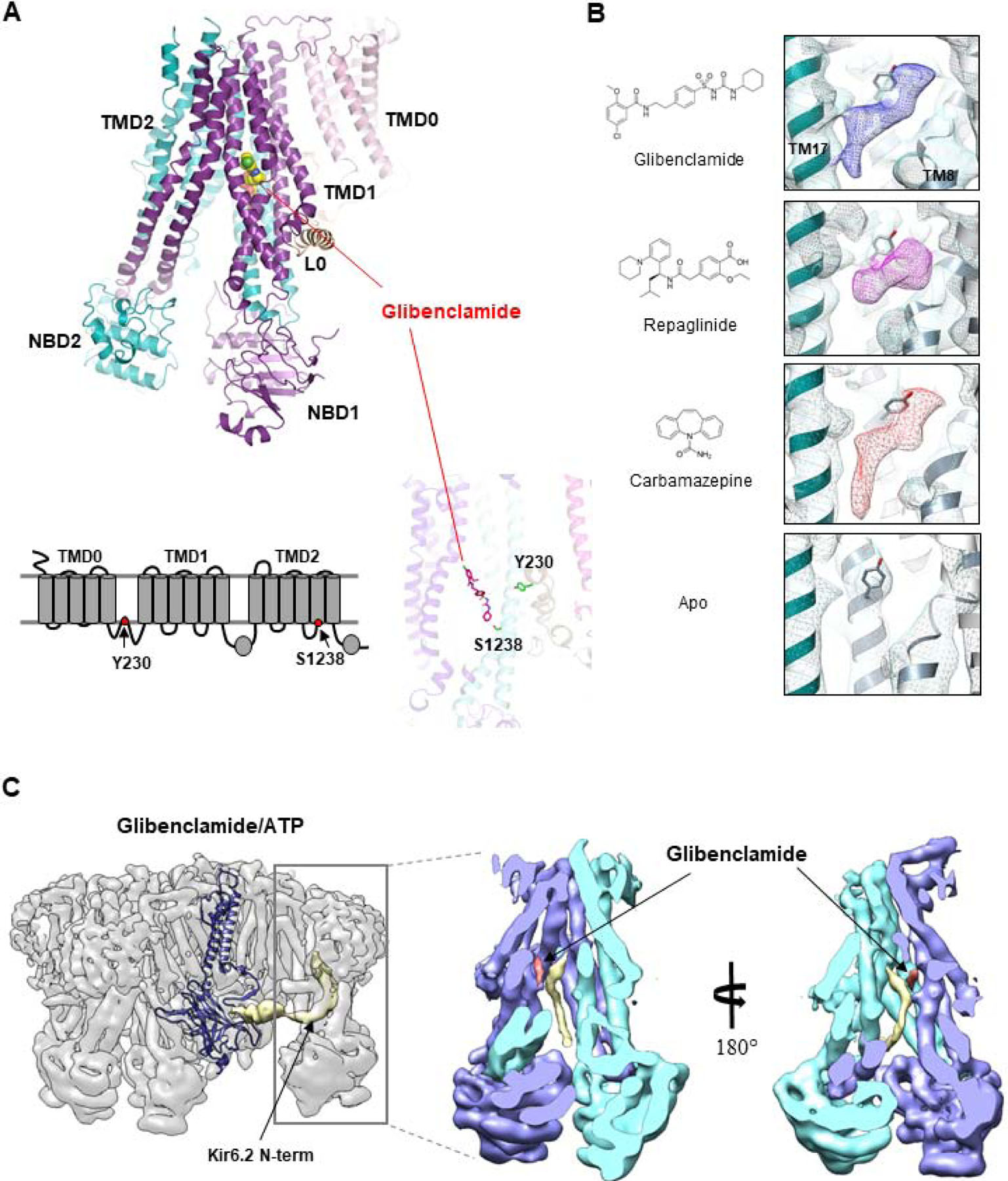
(A) Top: Structure of the SUR1 subunit showing the location of the bound glibenclamide (space-filling model). Bottom: 2D topology model (left) and 3D structure of SUR1 showing the location of the two residues previously implicated in glibenclamide (shown in stick model). (B) Chemical structures of pharmacological chaperones and their corresponding cryoEM densities in the SUR1 binding pocket. The empty binding pocket in the apo state is shown for comparison. (C) CryoEM map of the channel in complex with glibenclamide and ATP showing the density of the Kir6.2 N-term (gold) extending into the SUR1 ABC core central cavity. The structure of a Kir6.2 subunit is shown to illustrate the connectivity between the low resolution Kir6.2 N-terminus (filtered to 6Å) density with the modeled Kir6.2 structure. Right: Two slice views of the SUR1 ABC core showing the spatial relationship between glibenclamide and the Kir6.2 N-terminus.
Having identified the glibenclamide binding site, our group further determined the binding site for repaglinide and carbamazepine to test the hypothesis that these drugs bind to the same site. CryoEM structures of channels determined in the presence of ATP and repaglinide or carbamazepine showed the same conformation as channels bound to ATP and glibenclamide [52]. CryoEM densities corresponding to repaglinide and carbamazepine were found in the glibenclamide binding pocket (Fig. 3B). Intriguingly, although the cryoEM density of repaglinide is well fitted by a single molecule of the drug, that of carbamazepine is much larger, about twice the expected size of a single carbamazepine, suggesting the molecule might bind as a dimer or can adopt different binding positions in the pocket to generate an averaged density bigger than a single, rigidly bound carbamazepine [52]. Although all three pharmacological chaperones are located in the same pocket, differences in contacting SUR1 residues are noted. In particular, S1238, which is adjacent to glibenclamide and carbamazepine, is far away from repaglinide. This is consistent with previous studies showing that S1238 is not important for rapaglinide’s action, in contrast to glibenclamide or carbamazepine [49, 50, 79]. The binding site models for the drugs were further supported by additional studies showing that mutation of residues surrounding the densities of the various drugs also attenuated their ability to chaperone KATP trafficking mutants [52]. Concurrent with our study, Lei Chen’s group also determined the repaglinide binding site using the SUR1–39a.a.-Kir6.2 fusion protein and reported similar findings [63]. These studies answer a long standing question in the KATP channel pharmacological field and help explain how chemically diverse compounds share a common binding pocket to influence channel behavior.
The Kir6.2 N-terminus
The distal N-terminus of Kir6.2 of ~30 amino acids (referred to as Kir6.2 N-term) is a disordered region with few predicted secondary structures. Interestingly, as discussed above, the Kir6.2 N-term has been shown to be important for channel assembly, gating, and drug interactions. Deletion of the Kir6.2 N-term increases the open probability of the channel [77, 80, 81], decreases the binding affinity of glibenclamide and repaglinide to the channel complex [76], and decreases channel sensitivity to inhibition by sulfonylureas and glinides [75, 77]. Studies looking into its role in channel assembly have shown that it is also critical for efficient assembly and trafficking of the channel and for pharmacological chaperones to rescue SUR1-TMD0 trafficking mutants [49]. How this stretch of amino acids has such a profound role in channel regulation, and whether the effects of its deletion on the myriad of channel properties are related to one another have been a major puzzle.
In the initial published cryoEM structures, the Kir6.2 N-terminus is unresolved, suggesting it is dynamic. Using site-directed incorporation of a photoactivatable unnatural amino acid p-azidophenylalaine at the N-terminus of Kir6.2 it has been shown that pharmacochaperones such as glibenclamide and carbamazepine enhanced crosslinking between Kir6.2 N-term and SUR1 upon photoactivation [49], indicating these drugs promote association between Kir6.2 N-term and SUR1. Further crosslinking studies were conducted using lysine targeted crosslinkers such as DSP and CBDPS (Cyanurbiotindimercaptopropionylsuccinimide) on purified channels bound to glibenclamide, followed by trypsin digestion and mass spectrometry, to identify possible interactions. These studies identified a SUR1-Kir6.2 crosslink that connects K5 of Kir6.2 N-term to K602 located in TMD1 of SUR1 lining the central cavity [52]. The finding suggests that the Kir6.2 N-term may reside in the central cavity of SUR1’s ABC core near the drug binding site, which would be consistent with previous studies implicating a role of Kir6.2 N-term in drug binding. Wu et al. first proposed that a low resolution density in the central cavity seen in glibenclamide bound channel structures published by Li et al. and Martin et al was likely the Kir6.2 N-term [68]. However, in these structures the proposed cryoEM density of Kir6.2 N-term and the structured region of the Kir6.2 (starting from residue 32) are not contiguous, leaving uncertainty about the assignment. Interestingly, this density was absent in the structure of channels formed by the SUR1–39a.a.-Kir6.2 fusion protein in the presence of glibenclamide and it was reasoned that this was due to the long linker which could not be accommodated by the space of the cavity [68]. Our group compared multiple structures of channels formed by SUR1 and Kir6.2 co-expression in the presence or absence of drugs (glibenclamide, repaglinide, or ATP). Aided by improved focus refinement of the SUR1 ABC-core, we were able to observe a density in the central cavity that is contiguous with the first structured residue of Kir6.2 and is right next to the drug binding site (Fig. 3C). This density is clear in all drug-bound structures particularly those bound to the high affinity drugs glibenclamide and repaglinide. By contrast, the density is very weak and barely detectable in the apo state structure. Together these structural data indicate that the Kir6.2 N-term reaches into the central cavity of the SUR1-ABC core and this interaction is stabilized by pharmacological chaperones.
Insight into KATP channel assembly and gating regulation by pharmacological chaperones
From the cryoEM structures, which reveal close physical interactions between SUR1-TMD0 and Kir6.2, the pharmacological chaperone binding pocket, and the location of the Kir6.2 N-terminus in drug-bound states, and the wealth of structure-function correlation data in the literature discussed above, a mechanistic model emerges accounting for the ability of pharmacological chaperones to overcome trafficking defects caused by TMD0 mutations (Fig. 4A). During channel biogenesis, SUR1 and Kir6.2 must form initial contacts. Extension of the Kir6.2 N-term into the central cavity of the SUR1 ABC core likely plays a critical role at this initial stage by forming a transient handhold to guide the docking of TMD0 onto the Kir6.2 outer helix (M1) and allow additional cytoplasmic contacts to take place. This interaction, however, appears labile and transient as the Kir6.2 N-term is not visible in the apo state structure but is stabilized by binding of pharmacological chaperones in the TM bundle above the NBD1, as evidenced by the clear Kir6.2 N-term density in the drug-bound states. This model explains why deletion of the Kir6.2 N-term dramatically reduces channel biogenesis and surface expression [49], even though Kir6.2 outer helix is still available for SUR1-TMD0 interaction. For SUR1 containing TMD0 trafficking mutations, likely conformational defects prevent efficient and stable docking of TMD0 onto Kir6.2 such that the transient interaction between the Kir6.2 N-term is unable to sustain stable assembly, resulting in reduced channel biogenesis and surface expression. In the presence of pharmacological chaperones, Kir6.2 N-term is stabilized in SUR1’s ABC core central cavity, providing a firm handle to prolong interactions between mutant TMD0 and Kir6.2 for channel assembly and trafficking to the cell surface. Trafficking mutations outside TMD0 have not been found to respond to known KATP pharmacological chaperones. A possible explanation is that these mutations cause severe misfolding of the ABC core structure sufficient to engage ER quality control such that the mutant proteins are triaged by ERAD [82].
Figure 4. Model of KATP channel assembly and gating regulation by pharmacological chaperones.

(A) Cartoon showing in the absence of pharmacological chaperones (PC), mutant TMD0 is unable to form stable interactions with Kir6.2, leading to ER-associated degradation (ERAD) of the mutant SUR1. Binding of PCs stabilizes Kir6.2 N-terminus (~30 amino acids shown as dotted blue line) in the ABC core central cavity of SUR1 to allow assembly of mutant SUR1 with Kir6.2 and trafficking to the plasma membrane. (B) PCs inhibit channel activity by stabilizing SUR1 in an inward-facing conformation unable to be stimulated by Mg-nucleotides, as well as by trapping the Kir6.2 N-terminus in the SUR1 ABC core central cavity to prevent opening of the Kir6.2 channel. The grey rectangle represents the lipid bilayer of the ER membrane in (A) and plasma membrane in (B).
In addition to facilitating channel assembly, all KATP pharmacological chaperones are also channel inhibitors. The structures show that by lodging into the space in the TM bundle formed by transmembrane helices from both TMD1 and TMD2, these compounds stabilize the ABC core in an inward-facing conformation (Fig. 4B). This would prevent dimerization of the NBDs in the presence of MgATP/MgADP, and thus abolish channel stimulation by Mg-nucleotides, as well documented in the literature. In addition, in the drug-bound structures, the Kir6.2 N-term is seen in the ABC core cavity adjacent the drug binding site and perhaps even contributes to drug binding (Fig. 3B). The Kir6.2 N-term is known to modulate channel open probability independent of adenine nucleotides and its deletion results in increased channel activity. Crosslinking of an engineered cysteine at L2 position of Kir6.2 (L2C) with an endogenous cysteine in SUR1 (C1142) lining the central cavity reduced channel activity in a redox-sensitive manner, indicating residence of the Kir6.2 N-term in the SUR1 central cavity is associated with channel closure [52]. Thus, pharmacological chaperones also reduce channel activity by trapping the Kir6.2 N-term in the SUR1 ABC core (Fig. 4B). As noted earlier, potassium channel openers have been shown to be ineffective in rescuing KATP trafficking mutants, and in fact reduce biogenesis efficiency of WT channels. These openers are thought to stimulate channel activity by stabilizing SUR1 in a NBDs-dimerized conformation. In this conformation the SUR1 ABC-core central cavity would not be able to accommodate the Kir6.2 N-term needed for efficient channel assembly. Currently, it is unknown where the potassium channel openers bind. Wu et al. reported a structure bound to MgADP and NN414 and tentatively assigned the NN414 cryoEM densities [68]. However, more work is needed to validate the assignment.
Conclusions and Perspectives
Pharmacological chaperones are promising therapeutic tools for diseases resulting from defective protein folding and/or trafficking. Many of these chaperones are cognate physiological or pharmacological ligands of target proteins. Novel correctors are also being discovered through chemical library screens. In the case of membrane proteins, which are particularly difficult to crystallize for structural determinations, the structural mechanisms through which a drug’s binding affects a chaperone correction have until recently remained elusive. The rapid advances in single-particle cryoEM have now made it possible to obtain structures of challenging protein targets [83], presenting new opportunities to address mechanistic questions. The KATP channel complex exemplifies how detailed structural information is crucial for understanding how chemically diverse compounds may exert similar effects on channel assembly and function. This knowledge provides a foundation for structure-based development of new or improved chaperones.
A major challenge in clinical application of current KATP pharmacological chaperones is that the inhibitory chaperones need to be removed to recover channel functions. Moreover, while most mutants rescued to the cell surface exhibit normal function upon removal of the chaperones, some have been found to also cause reduced ATP sensitivity (Table 1). How to improve surface expression as well as function is a major goal for the field. In this regard, further therapeutic opportunities will derive from a fuller understanding of the channel biogenesis pathway that identifies molecular partners during folding, assembly, trafficking, and even degradation. In this regard, many questions remain unanswered. How are transcription and nascent polypeptide translation coupled with folding and assembly? What cellular chaperones are involved in monitoring and facilitating folding and assembly other than Hsp70 and Hsp90 we have reported previously [84]? Also, inhibition of ERAD has been shown to boost WT channel surface expression and at least one ERAD machinery protein derlin-1 is known to be involved [85]. Can ERAD protein interactions with the channel be manipulated to overcome channel trafficking defects? These are all issues for future research emphasis.
Finally, the KATP channel pharmacological chaperone studies may have implications for other diseases caused by folding/trafficking mutations in ABC transporters or Kir channels. In searching for correctors of KATP channels with mutations in the SUR1 ABC core domain, we had exploited compounds identified from CFTRΔF508 corrector screens. While the studies on carbarmazepine led to unexpected findings that helped illuminate KATP assembly and gating mechanisms, a lingering question remains as to how this drug corrects the trafficking defect of CFTRΔF508. Several CFTR cryoEM structures have recently been published but none in complex with correctors [86–91]. One would predict carbamazepine to bind to a similar pocket in CFTR as in SUR1. Since CFTR does not have an assembly partner like Kir6.2, the mechanism of surface expression rescue might be different. Perhaps carbamazepine binding stabilizes the mutant CFTR to prevent it from rapid degradation and thus allow escape to the cell surface. There are also additional CFTR correctors that should be studied in detail for their potential effects on SUR1 trafficking mutations in the ABC core domain. Future comparative studies among ABC transporters will undoubtedly cross fertilize to resolve the remaining questions in the field.
Acknowledgements
This work was supported by National Institutes of Health grant DK057699 and DK066485 (to Show-Ling Shyng). We thank Dr. Bruce Patton for comments on the manuscript.
Abbreviations:
- CFTR
cystic fibrosis transmembrane conductance regulator
- CHI
congenital hyperinsulinism
- KATP
ATP-sensitive potassium channel
- Kir6.2
inwardly rectifying potassium channel 6.2
- NBD
nucleotide binding domain
- SUR1
sulfonylurea receptor 1
- TMD
transmembrane domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES
- 1.Noma A, ATP-regulated K+ channels in cardiac muscle. Nature, 1983. 305(5930): p. 147–8. [DOI] [PubMed] [Google Scholar]
- 2.Ashcroft FM, Harrison DE, and Ashcroft SJ, Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature, 1984. 312(5993): p. 446–8. [DOI] [PubMed] [Google Scholar]
- 3.Cook DL and Hales CN, Intracellular ATP directly blocks K+ channels in pancreatic B-cells. Nature, 1984. 311(5983): p. 271–3. [DOI] [PubMed] [Google Scholar]
- 4.Murphy KP and Greenfield SA, ATP-sensitive potassium channels counteract anoxia in neurones of the substantia nigra. Exp Brain Res, 1991. 84(2): p. 355–8. [DOI] [PubMed] [Google Scholar]
- 5.Weik R and Neumcke B, ATP-sensitive potassium channels in adult mouse skeletal muscle: characterization of the ATP-binding site. J Membr Biol, 1989. 110(3): p. 217–26. [DOI] [PubMed] [Google Scholar]
- 6.Kovacs RJ and Nelson MT, ATP-sensitive K+ channels from aortic smooth muscle incorporated into planar lipid bilayers. Am J Physiol, 1991. 261(2 Pt 2): p. H604–9. [DOI] [PubMed] [Google Scholar]
- 7.Aguilar-Bryan L and Bryan J, Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr Rev, 1999. 20(2): p. 101–35. [DOI] [PubMed] [Google Scholar]
- 8.Ashcroft FM, Adenosine 5’-triphosphate-sensitive potassium channels. Annu Rev Neurosci, 1988. 11: p. 97–118. [DOI] [PubMed] [Google Scholar]
- 9.Nichols CG, KATP channels as molecular sensors of cellular metabolism. Nature, 2006. 440(7083): p. 470–6. [DOI] [PubMed] [Google Scholar]
- 10.Flagg TP, et al. , Muscle KATP channels: recent insights to energy sensing and myoprotection. Physiol Rev, 2010. 90(3): p. 799–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seino S, ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu Rev Physiol, 1999. 61: p. 337–62. [DOI] [PubMed] [Google Scholar]
- 12.Inagaki N, et al. , Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science, 1995. 270(5239): p. 1166–70. [DOI] [PubMed] [Google Scholar]
- 13.Chutkow WA, et al. , Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channels. Diabetes, 1996. 45(10): p. 1439–45. [DOI] [PubMed] [Google Scholar]
- 14.Inagaki N, et al. , A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron, 1996. 16(5): p. 1011–7. [DOI] [PubMed] [Google Scholar]
- 15.Yamada M, et al. , Sulphonylurea receptor 2B and Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. J Physiol, 1997. 499 (Pt 3): p. 715–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Remedi MS and Koster JC, K(ATP) channelopathies in the pancreas. Pflugers Arch, 2010. 460(2): p. 307–20. [DOI] [PubMed] [Google Scholar]
- 17.Ashcroft FM, ATP-sensitive potassium channelopathies: focus on insulin secretion. J Clin Invest, 2005. 115(8): p. 2047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nichols CG, Adenosine Triphosphate-Sensitive Potassium Currents in Heart Disease and Cardioprotection. Card Electrophysiol Clin, 2016. 8(2): p. 323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smeland MF, et al. , ABCC9-related Intellectual disability Myopathy Syndrome is a KATP channelopathy with loss-of-function mutations in ABCC9. Nat Commun, 2019. 10(1): p. 4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Costello RA and Shivkumar A, Sulfonylureas, in StatPearls. 2019: Treasure Island (FL). [Google Scholar]
- 21.Flanagan SE, et al. , Update of mutations in the genes encoding the pancreatic beta-cell K(ATP) channel subunits Kir6.2 (KCNJ11) and sulfonylurea receptor 1 (ABCC8) in diabetes mellitus and hyperinsulinism. Hum Mutat, 2009. 30(2): p. 170–80. [DOI] [PubMed] [Google Scholar]
- 22.Snider KE, et al. , Genotype and phenotype correlations in 417 children with congenital hyperinsulinism. J Clin Endocrinol Metab, 2013. 98(2): p. E355–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin GM, et al. , Pharmacological rescue of trafficking-impaired ATP-sensitive potassium channels. Front Physiol, 2013. 4: p. 386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee I, et al. , Therapies and outcomes of congenital hyperinsulinism-induced hypoglycaemia. Diabet Med, 2019. 36(1): p. 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vajravelu ME and De Leon DD, Genetic characteristics of patients with congenital hyperinsulinism. Curr Opin Pediatr, 2018. 30(4): p. 568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwappach B, et al. , Molecular basis for K(ATP) assembly: transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron, 2000. 26(1): p. 155–67. [DOI] [PubMed] [Google Scholar]
- 27.Zerangue N, et al. , A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron, 1999. 22(3): p. 537–48. [DOI] [PubMed] [Google Scholar]
- 28.Hibino H, et al. , Inwardly rectifying potassium channels: their structure, function, and physiological roles. Physiol Rev, 2010. 90(1): p. 291–366. [DOI] [PubMed] [Google Scholar]
- 29.Dassa E, Natural history of ABC systems: not only transporters. Essays Biochem, 2011. 50(1): p. 19–42. [DOI] [PubMed] [Google Scholar]
- 30.Aguilar-Bryan L, et al. , Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science, 1995. 268(5209): p. 423–6. [DOI] [PubMed] [Google Scholar]
- 31.Thomas C and Tampe R, Multifaceted structures and mechanisms of ABC transport systems in health and disease. Curr Opin Struct Biol, 2018. 51: p. 116–128. [DOI] [PubMed] [Google Scholar]
- 32.Tusnady GE, et al. , Membrane topology distinguishes a subfamily of the ATP-binding cassette (ABC) transporters. FEBS Lett, 1997. 402(1): p. 1–3. [DOI] [PubMed] [Google Scholar]
- 33.Babenko AP and Bryan J, Sur domains that associate with and gate KATP pores define a novel gatekeeper. J Biol Chem, 2003. 278(43): p. 41577–80. [DOI] [PubMed] [Google Scholar]
- 34.Chan KW, Zhang H, and Logothetis DE, N-terminal transmembrane domain of the SUR controls trafficking and gating of Kir6 channel subunits. EMBO J, 2003. 22(15): p. 3833–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vedovato N, Ashcroft FM, and Puljung MC, The Nucleotide-Binding Sites of SUR1: A Mechanistic Model. Biophys J, 2015. 109(12): p. 2452–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jacobson D and Shyng SL, Ion Channels of the Islets in Type 2 Diabetes. J Mol Biol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rorsman P and Ashcroft FM, Pancreatic beta-Cell Electrical Activity and Insulin Secretion: Of Mice and Men. Physiol Rev, 2018. 98(1): p. 117–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cartier EA, et al. , Defective trafficking and function of KATP channels caused by a sulfonylurea receptor 1 mutation associated with persistent hyperinsulinemic hypoglycemia of infancy. Proc Natl Acad Sci U S A, 2001. 98(5): p. 2882–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin GM, et al. , Pharmacological Correction of Trafficking Defects in ATP-Sensitive Potassium Channels Caused by Sulfonylurea Receptor 1 Mutations. J Biol Chem, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yan F, et al. , Sulfonylureas correct trafficking defects of ATP-sensitive potassium channels caused by mutations in the sulfonylurea receptor. J Biol Chem, 2004. 279(12): p. 11096–105. [DOI] [PubMed] [Google Scholar]
- 41.Yan FF, et al. , Congenital hyperinsulinism associated ABCC8 mutations that cause defective trafficking of ATP-sensitive K+ channels: identification and rescue. Diabetes, 2007. 56(9): p. 2339–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Crane A and Aguilar-Bryan L, Assembly, maturation, and turnover of K(ATP) channel subunits. J Biol Chem, 2004. 279(10): p. 9080–90. [DOI] [PubMed] [Google Scholar]
- 43.Tornovsky S, et al. , Hyperinsulinism of infancy: novel ABCC8 and KCNJ11 mutations and evidence for additional locus heterogeneity. J Clin Endocrinol Metab, 2004. 89(12): p. 6224–34. [DOI] [PubMed] [Google Scholar]
- 44.Taschenberger G, et al. , Identification of a familial hyperinsulinism-causing mutation in the sulfonylurea receptor 1 that prevents normal trafficking and function of KATP channels. J Biol Chem, 2002. 277(19): p. 17139–46. [DOI] [PubMed] [Google Scholar]
- 45.Boodhansingh KE, et al. , Novel dominant KATP channel mutations in infants with congenital hyperinsulinism: Validation by in vitro expression studies and in vivo carrier phenotyping. Am J Med Genet A, 2019. 179(11): p. 2214–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Convertino M, Das J, and Dokholyan NV, Pharmacological Chaperones: Design and Development of New Therapeutic Strategies for the Treatment of Conformational Diseases. ACS Chem Biol, 2016. 11(6): p. 1471–89. [DOI] [PubMed] [Google Scholar]
- 47.Leidenheimer NJ and Ryder KG, Pharmacological chaperoning: a primer on mechanism and pharmacology. Pharmacol Res, 2014. 83: p. 10–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen PC, et al. , Carbamazepine as a novel small molecule corrector of trafficking-impaired ATP-sensitive potassium channels identified in congenital hyperinsulinism. J Biol Chem, 2013. 288(29): p. 20942–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Devaraneni PK, et al. , Structurally distinct ligands rescue biogenesis defects of the KATP channel complex via a converging mechanism. J Biol Chem, 2015. 290(12): p. 7980–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yan FF, Casey J, and Shyng SL, Sulfonylureas correct trafficking defects of disease-causing ATP-sensitive potassium channels by binding to the channel complex. J Biol Chem, 2006. 281(44): p. 33403–13. [DOI] [PubMed] [Google Scholar]
- 51.Seino S, et al. , beta-Cell signalling and insulin secretagogues: A path for improved diabetes therapy. Diabetes Obes Metab, 2017. 19 Suppl 1: p. 22–29. [DOI] [PubMed] [Google Scholar]
- 52.Martin GM, et al. , Mechanism of pharmacochaperoning in a mammalian KATP channel revealed by cryo-EM. Elife, 2019. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mijnders M, Kleizen B, and Braakman I, Correcting CFTR folding defects by small-molecule correctors to cure cystic fibrosis. Curr Opin Pharmacol, 2017. 34: p. 83–90. [DOI] [PubMed] [Google Scholar]
- 54.Sampson HM, et al. , Compounds that correct F508del-CFTR trafficking can also correct other protein trafficking diseases: an in vitro study using cell lines. Orphanet J Rare Dis, 2013. 8: p. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang YC and Kuo CC, An inactivation stabilizer of the Na+ channel acts as an opportunistic pore blocker modulated by external Na+. J Gen Physiol, 2005. 125(5): p. 465–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ambrosio AF, et al. , Mechanisms of action of carbamazepine and its derivatives, oxcarbazepine, BIA 2–093, and BIA 2–024. Neurochem Res, 2002. 27(1–2): p. 121–30. [DOI] [PubMed] [Google Scholar]
- 57.Hu S, et al. , Pancreatic beta-cell K(ATP) channel activity and membrane-binding studies with nateglinide: A comparison with sulfonylureas and repaglinide. J Pharmacol Exp Ther, 2000. 293(2): p. 444–52. [PubMed] [Google Scholar]
- 58.Zhou Q, et al. , Carbamazepine inhibits ATP-sensitive potassium channel activity by disrupting channel response to MgADP. Channels (Austin), 2014. 8(4): p. 376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shyng S and Nichols CG, Octameric stoichiometry of the KATP channel complex. J Gen Physiol, 1997. 110(6): p. 655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Inagaki N, Gonoi T, and Seino S, Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett, 1997. 409(2): p. 232–6. [DOI] [PubMed] [Google Scholar]
- 61.Clement J.P.t., et al. , Association and stoichiometry of K(ATP) channel subunits. Neuron, 1997. 18(5): p. 827–38. [DOI] [PubMed] [Google Scholar]
- 62.Mikhailov MV, et al. , 3-D structural and functional characterization of the purified KATP channel complex Kir6.2-SUR1. EMBO J, 2005. 24(23): p. 4166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding D, et al. , The Structural Basis for the Binding of Repaglinide to the Pancreatic KATP Channel. Cell Rep, 2019. 27(6): p. 1848–1857 e4. [DOI] [PubMed] [Google Scholar]
- 64.Lee KPK, Chen J, and MacKinnon R, Molecular structure of human KATP in complex with ATP and ADP. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li N, et al. , Structure of a Pancreatic ATP-Sensitive Potassium Channel. Cell, 2017. 168(1–2): p. 101–110 e10. [DOI] [PubMed] [Google Scholar]
- 66.Martin GM, et al. , Anti-diabetic drug binding site in a mammalian KATP channel revealed by Cryo-EM. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin GM, et al. , Cryo-EM structure of the ATP-sensitive potassium channel illuminates mechanisms of assembly and gating. Elife, 2017. 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu JX, et al. , Ligand binding and conformational changes of SUR1 subunit in pancreatic ATP-sensitive potassium channels. Protein Cell, 2018. 9(6): p. 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Puljung MC, Cryo-electron microscopy structures and progress toward a dynamic understanding of KATP channels. J Gen Physiol, 2018. 150(5): p. 653–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Babenko AP and Bryan J, SUR domains that associate with and gate KATP pores define a novel gatekeeper. J Biol Chem, 2003. 26: p. 26. [DOI] [PubMed] [Google Scholar]
- 71.Zhou Q, Pratt EB, and Shyng SL, Engineered Kir6.2 mutations that correct the trafficking defect of K(ATP) channels caused by specific SUR1 mutations. Channels (Austin), 2013. 7(4): p. 313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ashfield R, et al. , Identification of the high-affinity tolbutamide site on the SUR1 subunit of the K(ATP) channel. Diabetes, 1999. 48(6): p. 1341–7. [DOI] [PubMed] [Google Scholar]
- 73.Bryan J, et al. , Toward linking structure with function in ATP-sensitive K+ channels. Diabetes, 2004. 53 Suppl 3: p. S104–12. [DOI] [PubMed] [Google Scholar]
- 74.Winkler M, et al. , Testing the bipartite model of the sulfonylurea receptor binding site: binding of A-, B-, and A + B-site ligands. J Pharmacol Exp Ther, 2007. 322(2): p. 701–8. [DOI] [PubMed] [Google Scholar]
- 75.Koster JC, Sha Q, and Nichols CG, Sulfonylurea and K(+)-channel opener sensitivity of K(ATP) channels. Functional coupling of Kir6.2 and SUR1 subunits. J Gen Physiol, 1999. 114(2): p. 203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kuhner P, et al. , Importance of the Kir6.2 N-terminus for the interaction of glibenclamide and repaglinide with the pancreatic K(ATP) channel. Naunyn Schmiedebergs Arch Pharmacol, 2012. 385(3): p. 299–311. [DOI] [PubMed] [Google Scholar]
- 77.Reimann F, et al. , Involvement of the n-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J Physiol, 1999. 518 (Pt 2): p. 325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gribble FM, Tucker SJ, and Ashcroft FM, The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J Physiol, 1997. 504 (Pt 1): p. 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hansen AM, et al. , Differential interactions of nateglinide and repaglinide on the human beta-cell sulphonylurea receptor 1. Diabetes, 2002. 51(9): p. 2789–95. [DOI] [PubMed] [Google Scholar]
- 80.Babenko AP, Gonzalez G, and Bryan J, The N-terminus of KIR6.2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels. Biochem Biophys Res Commun, 1999. 255(2): p. 231–8. [DOI] [PubMed] [Google Scholar]
- 81.Koster JC, et al. , ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. J Physiol, 1999. 515 (Pt 1): p. 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yan FF, et al. , Role of ubiquitin-proteasome degradation pathway in biogenesis efficiency of {beta}-cell ATP-sensitive potassium channels. Am J Physiol Cell Physiol, 2005. 289(5): p. C1351–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cheng Y, et al. , A primer to single-particle cryo-electron microscopy. Cell, 2015. 161(3): p. 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yan FF, et al. , Role of Hsp90 in biogenesis of the beta-cell ATP-sensitive potassium channel complex. Mol Biol Cell, 2010. 21(12): p. 1945–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang F, Olson EM, and Shyng SL, Role of Derlin-1 protein in proteostasis regulation of ATP-sensitive potassium channels. J Biol Chem, 2012. 287(13): p. 10482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fay JF, et al. , Cryo-EM Visualization of an Active High Open Probability CFTR Anion Channel. Biochemistry, 2018. 57(43): p. 6234–6246. [DOI] [PubMed] [Google Scholar]
- 87.Liu F, et al. , Molecular Structure of the Human CFTR Ion Channel. Cell, 2017. 169(1): p. 85–95 e8. [DOI] [PubMed] [Google Scholar]
- 88.Liu F, et al. , Structural identification of a hotspot on CFTR for potentiation. Science, 2019. 364(6446): p. 1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang Z and Chen J, Atomic Structure of the Cystic Fibrosis Transmembrane Conductance Regulator. Cell, 2016. 167(6): p. 1586–1597 e9. [DOI] [PubMed] [Google Scholar]
- 90.Zhang Z, Liu F, and Chen J, Conformational Changes of CFTR upon Phosphorylation and ATP Binding. Cell, 2017. 170(3): p. 483–491 e8. [DOI] [PubMed] [Google Scholar]
- 91.Zhang Z, Liu F, and Chen J, Molecular structure of the ATP-bound, phosphorylated human CFTR. Proc Natl Acad Sci U S A, 2018. 115(50): p. 12757–12762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pratt EB, et al. , Sulfonylurea receptor 1 mutations that cause opposite insulin secretion defects with chemical chaperone exposure. J Biol Chem, 2009. 284(12): p. 7951–9. [DOI] [PMC free article] [PubMed] [Google Scholar]