

Somatic mutation levels in human liver increase with age and are higher in differentiated hepatocytes than in residential LSCs.
Abstract
Accumulating somatic mutations have been implicated in age-related cellular degeneration and death. Because of their random nature and low abundance, somatic mutations are difficult to detect except in single cells or clonal cell lineages. Here, we show that in single hepatocytes from human liver, an organ exposed to high levels of genotoxic stress, somatic mutation frequencies are high and increase substantially with age. Considerably lower mutation frequencies were observed in liver stem cells (LSCs) and organoids derived from them. Mutational spectra in hepatocytes showed signatures of oxidative stress that were different in old age and in LSCs. A considerable number of mutations were found in functional parts of the liver genome, suggesting that somatic mutagenesis could causally contribute to the age-related functional decline and increased incidence of disease of human liver. These results underscore the importance of stem cells in maintaining genome sequence integrity in aging somatic tissues.
INTRODUCTION
Genome integrity is critically important for cellular function. Evidence has accumulated that loss of genome integrity and the increasingly frequent appearance of various forms of genome instability, from chromosomal aneuploidy to base substitution mutations, are hallmarks of aging (1, 2). However, thus far, of all mutation types, only chromosomal alterations could readily be studied directly during in vivo aging using cytogenetic methods (3). Because of their small size, random nature, and low abundance, most somatic mutations are difficult to detect, except in single cells or in clonal lineages (4). In the past, using transgenic reporters, mutations have been found to accumulate with age in a tissue-specific manner (5). However, this approach does not allow a genome-wide, direct analysis of somatic mutations in human primary cells. More recently, using single-cell whole-genome sequencing (WGS), somatic mutations were found to accumulate with age in human neurons (6) and B lymphocytes (7). Others also reported increased somatic mutations in human primary cells isolated from intestine, colon, and liver, albeit in clones propagated from human tissue-specific stem cells (8), which may not be representative of the differentiated cells that ultimately provide tissue function. Nevertheless, together, these studies confirmed that mutations in different somatic cell types of humans accumulate with age.
Here, we present single-cell genome-wide somatic mutation profiles of differentiated human liver hepatocytes as compared with adult liver stem cells (LSCs). Human liver is of particular interest for studying genome instability because of its high metabolic activity and its role in detoxification of xenobiotics, which makes this organ the most important target for genotoxicity in the body. In humans, accumulation of de novo mutations could contribute to the observed age-related loss of liver function, most notably a severe reduction in metabolic capacity, and multiple pathologies, including fatty liver disease, cirrhosis, hepatitis, infections, and cancer (9, 10). Our results indicate high spontaneous mutation frequencies in differentiated hepatocytes that significantly increase with age. By contrast, mutation frequencies in adult LSCs, defined as the cells that give rise to clonal outgrowths, were fairly low. In differentiated hepatocytes, a considerable number of mutations were found in functional parts of the genome. These results indicate that the human liver is subject to a high burden of genotoxicity and that adult stem cells are a critical component in maintaining overall genome integrity within a tissue.
RESULTS
Age-related accumulation of somatic mutations in differentiated human hepatocytes
The quantitative detection of de novo somatic mutations in single cells after whole-genome amplification (WGA) and WGS remains a challenge because of the high chance of errors. Here, we used a well-validated, highly accurate method, single-cell multiple displacement amplification (single-cell MDA, or SCMDA) (11), to analyze somatic mutations in single primary hepatocytes from human donors varying in age between 5 months and 77 years. These cells were isolated shortly after death through perfusion of whole livers from healthy human individuals after informed consent by the donor’s family (Lonza Walkersville Inc.). Cell viability was higher than 80% and, after Hoechst staining, individual, diploid hepatocytes were isolated via fluorescence-activated cell sorting (FACS) into individual polymerase chain reaction (PCR) tubes (fig. S1A). In total, we sequenced four single hepatocytes and bulk genomic liver DNA for each of 12 human donors (table S1). Each cell was subjected to our recently developed procedure for WGA and WGS (11, 12). Somatic single-nucleotide variants (SNVs) in single cells were identified relative to bulk genomic DNAs at a depth of ≥20× using VarScan2, MuTect2, and HaplotypeCaller with certain modifications (Materials and Methods and table S2). Overlapping mutations from this tricaller procedure were exclusively considered for further analysis. The results were essentially confirmed by using two alternative variant callers: SCcaller (11) and LiRA (Linked Read Analysis) (13).
After adjusting for genomic coverage, the number of SNVs per cell for 48 hepatocytes from 12 donors was found to vary between 357 and 5206 with four extreme outliers of 20,557 to 37,897 SNVs per cell excluded from the statistical model (Fig. 1A and table S2). The number of mutations per cell was found to increase significantly with the age of the donor (P = 1.22 × 10−9), with median values of 1222 ± 855 SNVs per cell in the young group (≤36 years, n = 21 cells), and 4054 ± 1168 SNVs per cell in the aged group (≥46 years, n = 23 cells), excluding the four outliers (Fig. 1A). The median number of mutations per cell in hepatocytes from the youngest donor was in the same range as what we recently reported for primary human fibroblasts from young donors, i.e., 1027 and 926 SNVs per cell from the 5-month-old and 6-year-old donors, respectively (11, 12). However, during aging, mutation levels increased over the same age range up to 2.5 times higher than in our previously analyzed human B lymphocytes (7) or human neurons analyzed by others (6) (fig. S2A).
Fig. 1. SNV levels in normal human liver cells.
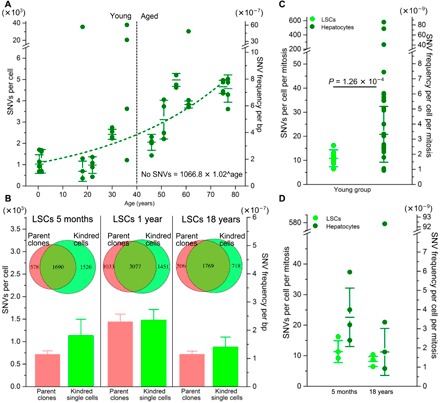
(A) SNV levels in individual differentiated hepatocytes. The y axis on the left indicates the number of mutations per cell, and the y axis on the right indicates mutation frequency per base pair. The median values with SDs among four cells of each subject are indicated. Data indicate an exponential increase in mutation frequency with donor age (R = 0.892, P = 1.16 × 10−6). bp, base pair. (B) SNV levels in LSC-derived parent clones (red) and their kindred cells (light green) from three young donors. The Venn diagrams indicate the fraction of SNVs detected in the parent clones (collectively for each individual; n = 3) that were also detected in the kindred LSCs. The bars indicate the median mutation frequencies in clones (red) and kindred single cells (light green). (C) Comparison of SNV levels in differentiated hepatocytes (dark green dots; n = 24 from six donors) and LSCs (light green; n = 10 from three donors), all within the young donor group ≤36 years. Mutation frequencies were corrected for the estimated number of cell divisions. (D) SNV levels in LSCs and differentiated hepatocytes from the same participants, corrected for the estimated number of cell divisions.
At this stage, we were interested in the possible cause of the high mutation frequencies in the four outlier cells. Three of the four outliers with the highest SNV levels revealed multiple mutations in genes involved in DNA repair (table S3) (14), which could conceivably underlie the observed accelerated mutation accumulation in these cells. Of note, individual outlier cells with high mutation levels have been detected in other tissues (6, 7).
Together, these findings indicate that the liver is prone to high levels of de novo somatic mutations, which could possibly be related to its major role in the metabolization and detoxification of xenobiotics.
Validation of liver single-cell sequencing data
The mutation frequencies observed in human hepatocytes from older subjects were higher than those previously found in human neurons and B lymphocytes (6, 7). They were also higher than the mutation frequencies reported for stem cell–derived liver organoids (fig. S2B) (8). It is critically important to validate the results obtained with single-cell mutation analysis to rule out possible amplification artifacts. In our previous studies on human primary fibroblasts, we validated single-cell data by also analyzing unamplified DNA from clones derived from cells in the same population (11). Here, we generated liver-specific clones from young donors by plating the prepurified hepatocyte cell suspensions in selective medium for LSC expansion (Materials and Methods). Under these conditions, the differentiated hepatocytes died within 5 to 7 days, while the residential LSCs could be propagated without differentiation. The latter was confirmed using biomarker analysis (Materials and Methods and fig. S1B) (15, 16). In addition, we obtained from a commercial source one sample of human postnatal LSCs from a 1-year-old donor at passage 9 (approximately 27 population doublings), which were expanded and also grown into clones in the same way.
LSC clones could be established only from young individuals, i.e., hepatocyte samples from the 1-year-old, 5-month-old, and 18-year-old participants. This is in keeping with observations that resident stem cell properties change with age, with a general reduction in proliferative capacity and increased cellular senescence (17).
Both LSC clones and kindred single cells derived from the young individuals were processed and subjected to WGS, as described above for differentiated hepatocytes. We then tested for the fraction of mutations called in the clones that were also found in the single cells derived from them. As shown in the Venn diagrams (Fig. 1B and fig. S3, A and B), most of these mutations were indeed confirmed in the single cells. This is very similar to what we previously reported for human single fibroblasts and clones derived from the same population of cells (11), which underscores the validity of our single-cell mutation detection method, also in liver cells. Of note, most of the mutations found in the single cells, but not in their parental clones, are likely to be also real. These are likely either mutations missed during variant calling in the clone or de novo mutations arising in the individual cells during clone culture and expansion.
Reduced somatic mutation levels in adult LSCs compared with differentiated hepatocytes
Once we confirmed the validity of our single-cell data, we directly compared mutation frequencies between the single cells defined as LSCs and differentiated hepatocytes, both from the young donor group. Previous studies have provided evidence for lower spontaneous mutation frequencies in stem as compared with differentiated cells (18, 19). For this comparison to be valid, we compared mutation frequencies per cell division in both cell types. This was necessary because the number of cell divisions is a major factor in causing base substitution mutations through replication errors. We first estimated the number of cell divisions that had occurred in human somatic cells of the young age group since the zygote, as described previously (20) (Materials and Methods). We then added, only to the LSCs, the estimated additional numbers of cell divisions during culture (Materials and Methods). The results show that, on a per cell division basis, somatic mutation frequencies were indeed lower in the LSCs than in the differentiated hepatocytes (about twofold), i.e., 11 SNVs versus 21 SNVs per cell per mitosis, respectively (P = 1.26 × 10−4, two-tailed Student’s t test) (Fig. 1C and table S2). A reduced mutation rate in LSCs could explain the fairly modest age-related increase reported previously for stem cell–derived organoids (figs. S2B and S3C) (8). The tendency of differentiated hepatocytes to accumulate mutations to a much higher level than stem cells is further confirmed by the significantly higher cell-to-cell variation among the former (P = 1.42 × 10−3, Levene’s test; Fig. 1, C and D). These observations are in keeping with the idea that stem cells are superior to differentiated cells in preserving their genome integrity, possibly through an enhanced capability to prevent or repair DNA damage (21, 22).
Differences in somatic mutation spectra between adult LSCs and differentiated hepatocytes
Next, we analyzed the mutational spectra in LSCs and differentiated hepatocytes. In differentiated hepatocytes, the most common mutation types were GC-to-AT transitions and GC-to-TA transversions (Fig. 2A and fig. S4, A and B). These mutations are known to be induced by oxidative damage (23), which itself has often been considered as a main driver of aging and age-related diseases (24). However, the most rapidly increased mutation type with age was the AT-to-GC transition (P = 2.16 × 10−10, two-tailed Student’s t test; table S4 for Pearson’s χ2 test). This mutation can be caused by mispairing of hydroxymethyluracil (5-hmU), another common oxidative DNA lesion. Alternatively, AT-to-GC mutations are induced by mutagenic alkyl-DNA adducts formed as a result of thymine residue alkylation (25, 26). Notably, certain minor alkyl-pyrimidine derivatives can escape repair, accumulate during aging, and lead to mutations much later (26, 27).
Fig. 2. Mutational spectra in normal human liver cells.
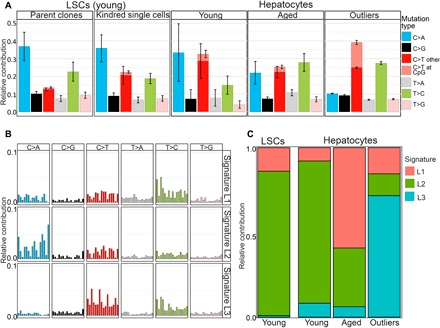
(A) Relative contribution of the indicated six mutation types to the point mutation spectrum for the five indicated liver sample groups. Data are represented as the mean relative contribution of each mutation type in sample groups of young and aged differentiated hepatocytes (21 cells from six donors ≤36 years, and 23 cells from six donors ≥46 years), adult LSC-derived parent clones and their kindred single cells separately, and a group of outlier cells (n = 4). (B) Three mutational signatures (L1, L2, and L3) were de novo identified by non-negative matrix factorization analysis from the somatic mutations in the different groups in (A). (C) Contributions of signatures L1, L2, and L3 to all SNVs in young and aged hepatocytes, young LSCs, and outlier cells.
Mutation spectra of the LSCs and LSC clones revealed a lower fraction of GC-to-AT transitions as compared with differentiated hepatocytes from the young group (Fig. 2A and figs. S3D and S4, A and B). This could be due to the virgin state of these cells, not participating in metabolizing xenobiotics, which is associated with oxidative DNA damage. However, we cannot rule out that, instead, the altered spectrum is related to in vitro culturing, which may alter the ratio of GC-to-AT transitions and GC-to-TA transversions. In the human LSCs derived from clones, the relative frequency of the GC-to-AT transition mutations is slightly, albeit significantly, increased as compared with the parent clones themselves (P = 7.43 × 10−4, two-tailed Student’s t test; table S4 for Pearson’s χ2 test; Fig. 2A and fig. S4A). Kindred single LSCs, which were derived from parent LSC clones, representing the original LSCs, have undergone multiple rounds of cell division with ample opportunity for replication errors, for example, as a consequence of ambient oxygen to which these cells have been inevitably exposed during subculture. Hence, this would suggest that cell culture has the opposite effect of what we observed from the stem cell versus differentiated cell difference, i.e., increasing rather than decreasing the fraction of GC-to-AT transitions.
To analyze mutation spectra more precisely, we performed non-negative matrix factorization (Materials and Methods) to extract three de novo mutation signatures, signatures L1, L2, and L3, from the mutation spectra of the four groups of human liver cells analyzed, i.e., combined LSCs and clones collectively, differentiated hepatocytes from young participants, differentiated hepatocytes from aged participants, and the four combined outlier cells. We compared these signatures to the COSMIC (Catalogue Of Somatic Mutations in Cancer) signatures described for various human tumors (Fig. 2B and table S5). Signature L1 substantially increased in differentiated hepatocytes from the aged group as compared with hepatocytes and LSCs from young individuals (Fig. 2C). This signature highly correlated with the liver-specific and age-associated mutation signature A dominant in human organoids of liver-specific origin in the aforementioned organoid study (8), as well as with COSMIC signature SBS5, strongly associated with aging (fig. S4C and table S5) (28, 29). Signature L2, with its increased level of oxidative GC > TA transversions, dominated the mutation spectrum of both LSCs and differentiated hepatocytes from young donors (Fig. 2C) and was significantly reduced in cells from the aged donors. Signature L2 highly correlated with COSMIC signatures SBS18 and SBS36, known to be associated not only with oxidative stress (fig. S4C and table S5) but also with proliferation signature C (table S5), found in all in vitro propagated cell types in the aforementioned organoid study (8). Since this signature was dominant in the LSCs, it possibly reflects the stem/progenitor-like origin of hepatocytes and remains dominant in differentiated hepatocytes of the young individuals (Fig. 2C). Signature 3, dominant in the outlier cells, highly correlated with COSMIC signature SBS5, the aging signature, but also correlated with SBS6 and SBS1, signatures associated with DNA mismatch repair deficiency (29).
The above analysis was confirmed when we, instead of extracting de novo signatures from our four groups of liver cell mutation spectra, tested which of the reference COSMIC signatures could be found in these groups (fig. S4C).
Relative protection of the functional genome against mutations in LSCs as compared with differentiated hepatocytes
Next, we analyzed the distribution of the somatic mutations in human liver cells across the genome. After pooling all mutations of the 21 differentiated cells from the young and the 23 differentiated cells from the old individuals, excluding the four outliers, the large majority of mutations distributed randomly across the genome in both groups (Fig. 3A). We then tested the possibility that during aging, mutations in functionally relevant sequences were selected against, as we previously observed for age-related mutation accumulation in B lymphocytes (7). Here, the functional liver genome was defined as the transcribed liver exome, using available data on gene expression levels in 175 previously described total liver samples [Genotype-Tissue Expression (GTEx) Consortium] (30), and its regulatory regions, identified as promoters of active genes or open chromatin regions, e.g., transcription factor binding regions, identified by ATAC (Assay for Transposase-Accessible Chromatin) sequencing in total liver tissue (ENCODE) (31). Of note, since the databases used were from whole liver, these definitions would not necessarily apply to LSCs or other subpopulations. However, it is reasonable to assume that whole liver is a good surrogate even for those fairly rare liver-specific cells.
Fig. 3. SNV distributions across total and functional genome in human liver.
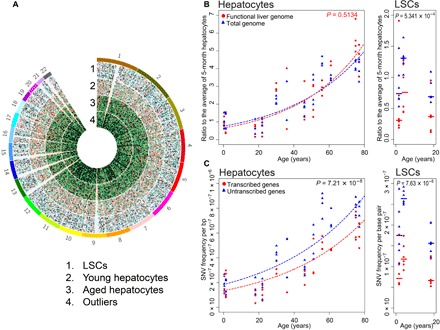
(A) Circos diagram of genomic SNV distribution in four groups: pooled LSCs, young and aged hepatocytes, and outlier cells. (B) SNV levels in the functional genome and genome overall in differentiated hepatocytes (left) and in LSCs (right) as a function of age. Each data point represents the ratio of the number of mutations per cell to the median number of mutations of the four cells from the 5-month-old subject. Mutations in the functional genome are shown in red and those in the genome overall in blue. (C) Mutation frequency per base pair in the transcribed part of the liver genome (red) and the nontranscribed part (blue) in differentiated hepatocytes (left) and LSCs (right) as a function of age.
The ratio of total to functional SNVs in differentiated hepatocytes was found to remain about 1 across the different age levels (P = 0.5134, Wilcoxon signed-rank test, two tailed) (Fig. 3B), indicating no selection against deleterious somatic mutations in low-proliferating hepatocyte populations during aging. By contrast, the same ratio in pooled adult LSCs was about 2 and significantly different from that in differentiated hepatocytes (P = 5.34 × 10−4, Wilcoxon signed-rank test, two tailed). This suggests selection against deleterious mutations during the cell proliferation cycles that gave rise to these stem cells. It also suggests that LSCs may have an increased capacity to protect their genome simply by remaining quiescent. We also compared mutation frequencies in transcribed versus untranscribed liver cell genes. Transcribed liver genes were defined as genes with expression values ≥1 transcripts per kilobase per million (TPM), while nontranscribed genome included all sequences with expression values <1 TPM in liver tissue (GTEx) (30). The results indicated a significantly lower number of SNVs affecting transcribed liver genes than nontranscribed genes across all donor ages (P = 7.21 × 10−8, Wilcoxon signed-rank test, two tailed) as well as in the LSCs and clones (P = 7.63 × 10−6, Wilcoxon signed-rank test, two tailed) (Fig. 3B), suggesting active transcription-coupled repair in normal human liver (32).
DISCUSSION
Somatic mutations have long been implicated as a cause of aging (33, 34). However, thus far, it has not been possible to test this hypothesis directly because of a lack of advanced methods to analyze random somatic mutagenesis in vivo, which requires high-throughput sequencing of single cells. Using our advanced single-cell sequencing method, we show that the number of somatic base substitution mutations in normal human liver significantly increases with age, reaching as much as 3.3 times more mutations per cell in aged humans than in young individuals. Of note, the numbers of mutations in aged liver are significantly higher than what has previously been reported for aged human liver organoids (fig. S2B) (8) and also higher than recent results reported for aged human neurons (fig. S2A) and B cells (7). Since we essentially ruled out that many of these mutations are artifacts of the amplification system, the most likely cause of this high mutagenic activity in the human liver is the high metabolic and detoxification activity in this organ, which is known to be associated with genotoxicity (35).
Out of 48 hepatocyte cells analyzed, 4 cells revealed extremely elevated mutation loads, over 10 times exceeding SNV levels in age-matched normal hepatocytes even from the same subject. These outliers have also been observed in the only two studies of somatic mutations in human tissues in vivo using a single-cell WGS approach (6, 7). Of the four outliers observed in this present study, multiple de novo SNVs were found to reside in DNA repair genes, strongly suggesting that these mutations were responsible for mutator phenotypes similar to what has been shown for cancers (36). While we cannot know when the mutations that gave rise to rapid mutation accumulation in these cells occurred, this may have been fairly recently, with imminent death of the cells likely. On average, almost 60 nonsynonymous mutations in the functional exome of these cells were found, suggesting a likely functional effect (table S6). However, since we could not longitudinally follow mutation loads in the same single cells, our data do not allow any conclusions on the cause and effect of the observed mutations.
Somatic mutation frequencies in normal differentiated hepatocytes were found to be much higher than in residential LSCs. This means that in vitro clonal surrogates for cells do not always accurately represent the mutation loads of in vivo differentiated cells, which makes predictions of a functional impact of somatic mutations from these clonal data difficult. While we do not know the mechanism(s) of reduced spontaneous mutation loads in stem as compared with differentiated cells, such evidence has also been reported by others (18, 19), and it is possible that stem cells have superior genome maintenance systems as compared with their differentiated counterparts. However, a caveat in this respect is that the LSCs that we enriched for may not in fact be the LSCs giving rise to most of the differentiated hepatocytes. Hence, we cannot be sure that a direct comparison between a stem cell and differentiated cells derived from this stem cell was in fact made.
Another important question is the possible functional impact of random somatic mutagenesis on the aging phenotype. While from our current data we cannot conclude direct cause-and-effect relationships, our observation that the functional part of the genome accumulated numerous mutations suggests that aging-related cellular degeneration and death could at least, in part, be due to somatic mutations. While the occurrence of no more than 11 nonsynonymous mutations in the transcribed exome of liver hepatocytes from humans in their 70s suggests a minor contribution of changes in the protein-coding part of the genome, the well over 100 de novo mutations in gene regulatory sequences may point toward an important role for stochastic gene expression changes in age-related loss of organ function and increased disease incidence. These mutations could possibly increase transcriptional noise, a molecular phenotype that appears characteristic for cells from aged individuals (37–39).
Last, while in our current work only base substitution mutations were analyzed, other types of mutations are likely to occur as well. The frequency of most of these mutations, e.g., small insertions and deletions, copy number variation, and genome structural variation, is likely to be much lower than the frequencies of base substitutions observed to rise to thousands of mutations per cell. However, their effects are possibly much larger since they affect a larger part of the genome and, when in exomes, almost always lead to loss of function. It is conceivable that, taken together, de novo mutations could have serious effects on the function of human somatic cells in vivo above and beyond their causal relevance in liver cancer.
MATERIALS AND METHODS
Human specimens
Frozen human hepatocyte samples were purchased from Lonza Walkersville Inc. Whole livers for hepatocyte isolation were obtained with the informed consent of families of registered organ donors. The obtained liver organs were rejected for transplant due to either lack of a donor match or morphological alterations (e.g., tearing and hematoma). All 12 selected hepatocyte donors were healthy participants of various age, gender, and ethnicity (table S1) without any liver cancer or other liver pathology history. These cells had been isolated using a gold standard, two-step liver/liver lobe perfusion procedure. Cells were suspended in 2 to 5 ml of media and counted with Trypan blue to estimate viability (higher than 80%), and frozen in dimethyl sulfoxide/liquid nitrogen (www.lonza.com). One specimen of frozen human neonatal LSCs from a 1-year-old donor was purchased from Kerafast Inc. (www.kerafast.com). These cells had been derived by the Sherley laboratory (Boston, MA, USA) and characterized to confirm their stem cell identity (40–42).
Single hepatocyte collection
After thawing, hepatocyte suspensions were used to collect single hepatocytes into individual 0.2-ml PCR tubes with 2.5 μl of phosphate-buffered saline (PBS) by means of FACS (FACSAria, Becton Dickenson). Selection of the target hepatocyte population was based on the large cell size of hepatocytes (forward-scatter/side-scatter parameters) along with the additional fluorescence staining for DNA content and cell viability. Briefly, bulk hepatocyte suspension samples were prior stained according to the manufacturer’s protocol with the viable DNA-binding dye Hoechst 33342 (Life Technologies) to discriminate cells with a standard diploid chromosome set and LIVE/DEAD Cell Vitality Assay Kit C12 Resazurin/SYTOX Green (Thermo Fisher Scientific) to select viable healthy cells. Typical FACS layout is shown in fig. S1A. Upon sorting, tubes with single cells were frozen on dry ice and kept at −80°C until use.
Adult LSC polarization and culture
Neonatal LSCs of passage 9 (one passage corresponds to approximately three cell population doublings for these cells according to the manufacturer’s protocol) from the 1-year-old donor were purchased from Kerafast Inc. The commercial LSCs were cultured in polarization media [Dulbecco’s modified Eagle’s medium, 10% dialyzed fetal bovine serum (Invitrogen), 1.5 mM xanthosine (Sigma), 1× penicillin/streptomycin, epidermal growth factor human (20 ng/ml; Invitrogen), transforming growth factor–β human recombinant (0.5 ng/ml Sigma)] according to the manufacturer’s protocol (Kerafast Inc.) (40–42). These cells served as controls to characterize de novo isolated and polarized LSCs.
Additional LSC cultures were isolated and polarized and characterized from the bulk commercial hepatocyte suspensions (Lonza Walkersville Inc.) from young donors using previously described protocols with specific modifications (15, 16) combined with the aforementioned Kerafast protocol for neonatal LSCs. Briefly, bulk suspension hepatocytes (0.5 × 106 to 1 × 106 of cells) were transferred to polarization media as described for the neonatal LSCs and cultured on cell-adhesive 12-well plates for 5 to 7 days. Then, all nonattached hepatocytes were removed, and fresh media were added to the small remaining population of attached progenitor cells. After 1 to 1.5 weeks of culture and media changes, attached cells symmetrically divided, growing to mixed clonal populations of polarized adult LSCs. These cultures were frozen at early passage (p = 3 to 5) until further use. Only LSCs from donors of younger age (≤22 years) could be isolated in this way.
Phenotypes of the polarized cells were analyzed for the presence of specific surface stem cell and epithelial progenitor cell epitopes, e.g. EpCAM (epithelial cell adhesion molecule), Lgr5, CD90, CD29, CD105, and CD73, upon staining with antibodies by means of multicolor flow cytometry analysis (LSRII, Becton Dickinson) as recommended previously (15, 16, 43, 44). Characteristic FACS profiles and specific phenotypes for commercial LSCs (control) and two manually isolated and polarized LSC lineages are shown in fig. S1B.
Adult LSC clones and single-cell establishment
Single-cell derived parent clones and their kindred single cells were prepared and collected using CellRaft arrays (Cell Microsystems) as described previously (11). Briefly, an LSC suspension was plated on a CellRaft array consisting of 12,000 individual portable rafts for single cells at the required density of 5000 cells per array. After 4 to 8 hours, individual LSCs were elongated and attached to the array surface locating on individual rafts. After attachment, the medium with floating cells was replaced, and single-cell positions were marked and tracked during the following 7 to 10 days to detect dividing cells and growing individual single-cell derived clones. Once the colony/clone reached confluence on the raft (8 to 10 cells per raft), it was dislocated from the array with a positioned automatic needle and transferred with a magnetic wand to a 96-well plate. Upon reaching confluence, single-cell derived clones were trypsinized and subsequently transferred to 24-well plates, then 12-well plates, 6-well plates, and, lastly, 10-cm plates to reach a total amount of 1.5 × 106 to 3 × 106 cells per parent clone. Together, the process of establishing a clone from a single cell took about 25 to 30 days.
Individual single cells from the parent clones were collected, also using CellRafts, and transferred to a 0.2-ml PCR tube containing 2.5 μl of PBS. The presence of a single raft was observed under a magnifying glass. Upon single-cell collection, tubes were fast frozen on dry ice and kept on −80°C until further use.
Single-cell WGA
Single hepatocytes from each subject were subjected to WGA using our modified procedure of low-temperature cell lysis and DNA denaturation followed by MDA as described (11). As positive and negative controls for WGA, we used 1 ng of human genomic DNA and DNA-free PBS solution, respectively. Resultant MDA products were purified using AMPureXP beads (Beckman Coulter), and the amplified DNA concentration was measured with the Qubit High Sensitivity dsDNA kit (Invitrogen Life Sciences). To verify sufficient and uniformly amplified single-cell MDA products, we performed the eight-target locus-dropout test as described previously (11). Selected confirmed samples (four single-cell MDA products per subject) were further subjected to library preparation and WGS.
Genomic DNA and clone-derived DNA extraction
Human bulk genomic DNA was collected from total cell suspensions using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s protocol. LSC clone–derived DNA was extracted from clones of at least 1.5 × 106 to 2.5 × 106 cells in a similar way. DNA concentration was quantified with the Qubit High Sensitivity dsDNA kit (Invitrogen Life Sciences), and DNA quality was evaluated by 1% agarose gel electrophoresis.
Library preparation and WGS
The libraries for Illumina next-generation WGS were generated from 0.2 to 0.4 μg of genomic DNA, clone-derived bulk DNA, and single-cell MDA DNA human samples using the NEBNext Ultra II FS DNA Library Prep Kit for Illumina (New England BioLabs). The libraries were sequenced with 2 × 350–base pair paired-end reads on an Illumina HiSeq X Ten sequencing platform by Novogene Inc.
Next-generation WGS at a minimal depth of 20X base coverage was performed on four individual mature hepatocytes per human subject (12 human subjects, 48 single cells in total) (table S2). Bulk DNA from two or three LSC-derived clones and MDA products from three to four corresponding kindred single cells per donor (three donors, eight parent clones, and 10 kindred single LSCs) were sequenced similarly.
Alignment for WGS
For all samples, adapter and low-quality reads were trimmed by Trim Galore (version 0.3.7). Quality checks were performed before and after read trimming by FastQC (version 0.11.4). The trimmed reads were aligned to the human reference genome (GRCh37 with decoy) by BWA mem (version 0.7.10) (45). Duplications were removed using samtools (version 0.1.19) (46). The known indels and single-nucleotide polymorphism (SNPs) were collected from the 1000 Genomes Project (phase 1) and Single Nucleotide Polymorphism Database (dbSNP) (build 144). Then, the reads around known indels were locally realigned, and their base quality scores were recalibrated on the basis of known indels and SNVs, both via the Genome Analysis Toolkit (GATK, version 3.5.0) (47).
Calling somatic SNVs
Somatic mutations between each single cell and the corresponding bulk and between each clone and corresponding bulk were identified using three different variant callers: VarScan2 (48), MuTect2 (49), and HaplotypeCaller (47). To obtain high-quality mutation calls and avoid high false-positive rates in individual callers, we applied a comprehensive procedure in filtering. First, we only considered mutations on autosomes. Then, we considered mutations with a GATK phred-scaled quality score of at least 30 and excluded mutations overlapping with known SNPs from dbSNP. Furthermore, we required a minimum base depth of 20X and filtered mutations with variant-supporting reads in bulk. Moreover, mutations present in at least two cells in each individual were also removed to further exclude potential germline mutations. The mutations present in all three variant callers were considered as true de novo mutations. Last, considering that amplification errors and/or nonuniform coverage could induce false-positive mutations in no more than one-eighth of the reads, we used a binomial distribution to filter these potential false-positive mutations, which excluded most mutations present in 25% of the reads or less. To further check the power of the used pipeline in filtering amplification errors, we also called the somatic mutations using our alternative, the SCcaller tool (11) and the LiRA pipeline (13) (figs. S2A and S3B).
Estimating mutation frequencies
The frequency of somatic SNVs per cell was estimated after normalizing genomic coverage
As the reads were aligned to the haploid reference genome, the frequency of somatic SNVs per base pair was calculated by dividing the frequency of somatic SNVs per cell by genome size and ploidy of the genome (ploidy = 2)
The surveyed genome per single cell/clone was calculated as the number of nucleotides with read mapping quality ≥20 and position coverage ≥20X.
The outliers of the hepatocytes were defined using Tukey’s range test: Four cells were defined as extreme outliers as their frequencies were higher than Q3 + 3 * IQR, where Q3 is the third quartile of the frequencies and IQR is the interquartile range. The outlier cells were excluded from the statistical model.
For the LSC-differentiated hepatocyte comparison, the absolute de novo mutation frequencies were corrected for the number of cell divisions undergone since the zygote (table S2). We used 45.1 as the number of developmental mitoses (20) and assumed a subsequent turnover rate of one cell division per year, based on empirical evidence from rodents (50, 51). In total, 45.5, 46.3, and 61.6 cell divisions were estimated for both LSCs and differentiated hepatocytes from 5-month-old, 1-year-old, and 18-year-old individuals, respectively. For LSCs from 5-month-old, 1-year-old, and 18-year-old individuals, we then added, respectively, an estimated 33, 41.7, and 33 cell divisions during the enrichment process of stem cells, and 21.9, 24.5, and 21.9 cell divisions associated with clonal outgrowth of the single LSCs.
Detecting overlap between clone and kindred cell
To determine the overlap between SNVs called in the clones and the single cells derived from them, genome coverage in the clone was normalized to that in its kindred single cell. Mutations found in a single cell and appearing in at least 1 read in the parent clone were considered as overlapping. When there were no variant-supporting reads in the clone, the mutation was determined as kindred cell specific. This assignment left some mutations with an unknown status more likely to be de novo mutations arising in the individual cells during clone culture and expansion.
Identifying mutation signatures
The identified mutations in all individuals were pooled into four groups: LSC cells/clones from young donors, hepatocytes from young and aged donors, and outlier hepatocytes. The integrated spectra of six mutation types in each group were plotted using the R package “MutationalPatterns” (52). Using non-negative matrix factorization (NMF) decomposition in the same package, we revealed group-specific mutational signatures as well as de novo identified three signatures in normal human liver cells. To identify the potential origin of the mutational spectra, the group mutational signatures and newly revealed signatures to the published signatures associated with liver-specific organoids and various cancer tissues. Three tissue-specific organoid signatures were obtained from a recent study (8); 67 cancer mutation signatures were downloaded from the latest version 3 of the COSMIC database (https://cancer.sanger.ac.uk/cosmic/signatures/SBS/) (28, 29). The cosine similarity between newly identified and published signatures was calculated for comparisons (table S5).
Annotation of functional genomes
All reported mutations were annotated based on the gene definitions of GRCh37.87. Mutations were further extracted from the functional genome, including transcribed genes, promoters, and open chromatin regions. The nonsynonymous and synonymous mutations were identified by analysis of variance (ANOVA) (53), while damaging and tolerated mutations were checked by SIFT (54) and PROVEAN (55). When damaging (Sorting Intolearnt From Tolerant, SIFT) or deleterious (Protein Variation Effect Analyzer, PROVEAN), the mutation was marked as damaging, and when tolerated (SIFT) and neutral (PROVEAN), a tolerated mutation.
The open chromatin regions were identified by ENCODE transcription factor binding regions in whole genome and ATAC sequencing data in the functional genome in liver tissue samples. Raw ATAC sequencing data were downloaded from ENCODE (experiment name: ENCSR373TDL) (31). The adapter and low-quality ATAC sequencing reads were filtered using Trim Galore (version 0.3.7). Clean reads were aligned to the human reference genome (GRCh37) with Bowtie2 (version 2.2.3; option: -X 2000). Duplicated reads were removed with the Picard tool (version 1.119). Open chromatin regions were determined by MACS2 (version 2.1.1; option: callpeak -g hs --nomodel --shift −100 --extsize 200) (56).
Gene expression levels for total human liver tissue were obtained from GTEx (https://gtexportal.org/) (30). We defined the transcribed genes as those with expression level ≥1 TPM in all samples. Also, we separated the transcribed and nontranscribed genome by TPM ≥1 and < 1 in all samples, respectively.
Supplementary Material
Acknowledgments
We thank X. Hao (Vijg Lab) for assisting with data analysis and the Flow Cytometry Core at the Albert Einstein College of Medicine for assistance in single-cell sorting and collection. Funding: This study was supported by NIH grant P01 AG017242 (J.V.) and Liver Research Center NIH/NIDDK5 grant P30 DK041296. Author contributions: J.V., A.Y.M., and K.B. conceived this study and designed the experiments. O.A., M.K., and A.W.W. provided field-specific study expertise and logistics. K.B. performed the experiments. S.S. and X.D. analyzed the data. K.B., S.S., and J.V. wrote the manuscript. Competing interests: A.Y.M., X.D., and J.V. are cofounders of SingulOmics Corp. related to this work. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. WGS data (accession number lphs001956) can be accessed at https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001190.v1.p1. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaax2659/DC1
Fig. S1. Phenotypic characterization of human hepatocytes and adult LSCs by means of flow cytometry.
Fig. S2. Somatic mutation levels in human liver and brain.
Fig. S3. Mutational landscape in LSC clones/single cells from young donors and liver organoids from adult/aged individuals (8).
Fig. S4. Mutational spectra in human liver.
Table S1. Human liver donor information list.
Table S2. Final WGS data and mutation calling results on human liver cells.
Table S3. List and description of the mutations in DNA repair–related genes identified in outlier cells with high mutation levels.
Table S4. Statistical analysis of mutation type contributions for spectra of different liver cells and groups: Pearson’s χ2 test and two-tailed Student’s t test.
Table S5. Correlation of spectral liver group patterns and de novo signatures identified in human liver cells (L1, L2, and L3 for hepatocytes versus LSCs versus outliers and LSC1 and LSC2 for LSC cells/clones versus liver organoids) with cancer-related signatures (COSMIC) and organoid-specific signatures (8).
Table S6. Average number of SNVs per cell in indicated groups of pooled human liver cells distributed across total and functional liver genome within specific genome sequences.
REFERENCES AND NOTES
- 1.Maslov A. Y., Vijg J., Genome instability, cancer and aging. Biochim. Biophys. Acta 1790, 963–969 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vijg J., Somatic mutations, genome mosaicism, cancer and aging. Curr. Opin. Genet. Dev. 26, 141–149 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Iourov I. Y., Vorsanova S. G., Yurov Y. B., Chromosomal mosaicism goes global. Mol. Cytogenet. 1, 26 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L., Vijg J., Somatic mutagenesis in mammals and its implications for human disease and aging. Annu. Rev. Genet. 52, 397–419 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dollé M. E., Snyder W. K., Dunson D. B., Vijg J., Mutational fingerprints of aging. Nucleic Acids Res. 30, 545–549 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lodato M. A., Rodin R. E., Bohrson C. L., Coulter M. E., Barton A. R., Kwon M., Sherman M. A., Vitzthum C. M., Luquette L. J., Yandava C. N., Yang P., Chittenden T. W., Hatem N. E., Ryu S. C., Woodworth M. B., Park P. J., Walsh C. A., Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L., Dong X., Lee M., Maslov A. Y., Wang T., Vijg J., Single-cell whole-genome sequencing reveals the functional landscape of somatic mutations in B lymphocytes across the human lifespan. Proc. Natl. Acad. Sci. U.S.A. 116, 9014–9019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blokzijl F., de Ligt J., Jager M., Sasselli V., Roerink S., Sasaki N., Huch M., Boymans S., Kuijk E., Prins P., Nijman I. J., Martincorena I., Mokry M., Wiegerinck C. L., Middendorp S., Sato T., Schwank G., Nieuwenhuis E. E., Verstegen M. M., van der Laan L. J., de Jonge J., JN I. J., Vries R. G., van de Wetering M., Stratton M. R., Clevers H., Cuppen E., van Boxtel R., Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538, 260–264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.A. J. Sanyal, T. D. Boyer, N. A. Terrault, K. D. Lindor, Zakim and Boyer’s Hepathology: A Testbook Of Liver Disease (Elsevier, ed. 7, 2017). [Google Scholar]
- 10.Cieslak K. P., Baur O., Verheij J., Bennink R. J., van Gulik T. M., Liver function declines with increased age. HPB 18, 691–696 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong X., Zhang L., Milholland B., Lee M., Maslov A. Y., Wang T., Vijg J., Accurate identification of single-nucleotide variants in whole-genome-amplified single cells. Nat. Methods 14, 491–493 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gundry M., Li W., Maqbool S. B., Vijg J., Direct, genome-wide assessment of DNA mutations in single cells. Nucleic Acids Res. 40, 2032–2040 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.C. L. Bohrson, A. R. Barton, M. A. Lodato, R. E. Rodin, V. Viswanadham, D. Gulhan, I. Cortes, M. A. Sherman, L. J. Luquette, M. Kwon, M. E. Coulter, C. A. Walsh, P. J. Park, Linked-read analysis identifies mutations in single-cell DNA sequencing data. bioRxiv:10.1101/211169 (29 October 2017). [DOI] [PMC free article] [PubMed]
- 14.Wood R. D., Mitchell M., Lindahl T., Human DNA repair genes, 2005. Mutat. Res. 577, 275–283 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Herrera M. B., Bruno S., Buttiglieri S., Tetta C., Gatti S., Deregibus M. C., Bussolati B., Camussi G., Isolation and characterization of a stem cell population from adult human liver. Stem Cells 24, 2840–2850 (2006). [DOI] [PubMed] [Google Scholar]
- 16.Herrera Sanchez M. B., Previdi S., Bruno S., Fonsato V., Deregibus M. C., Kholia S., Petrillo S., Tolosano E., Critelli R., Spada M., Romagnoli R., Salizzoni M., Tetta C., Camussi G., Extracellular vesicles from human liver stem cells restore argininosuccinate synthase deficiency. Stem Cell Res. Ther. 8, 176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alt E. U., Senst C., Murthy S. N., Slakey D. P., Dupin C. L., Chaffin A. E., Kadowitz P. J., Izadpanah R., Aging alters tissue resident mesenchymal stem cell properties. Stem Cell Res. 8, 215–225 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Cervantes R. B., Stringer J. R., Shao C., Tischfield J. A., Stambrook P. J., Embryonic stem cells and somatic cells differ in mutation frequency and type. Proc. Natl. Acad. Sci. U.S.A. 99, 3586–3590 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rouhani F. J., Nik-Zainal S., Wuster A., Li Y., Conte N., Koike-Yusa H., Kumasaka N., Vallier L., Yusa K., Bradley A., Mutational history of a human cell lineage from somatic to induced pluripotent stem cells. PLOS Genet. 12, e1005932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milholland B., Dong X., Zhang L., Hao X., Suh Y., Vijg J., Differences between germline and somatic mutation rates in humans and mice. Nat. Commun. 8, 15183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mohyeldin A., Garzón-Muvdi T., Quiñones-Hinojosa A., Oxygen in stem cell biology: A critical component of the stem cell niche. Cell Stem Cell 7, 150–161 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Beerman I., Accumulation of DNA damage in the aged hematopoietic stem cell compartment. Semin. Hematol. 54, 12–18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang D., Kreutzer D. A., Essigmann J. M., Mutagenicity and repair of oxidative DNA damage: Insights from studies using defined lesions. Mutat. Res. 400, 99–115 (1998). [DOI] [PubMed] [Google Scholar]
- 24.Pomatto L. C. D., Davies K. J. A., Adaptive homeostasis and the free radical theory of ageing. Free Radic. Biol. Med. 124, 420–430 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pauly G. T., Moschel R. C., Mutagenesis by O6-methyl-, O6-ethyl-, and O6-benzylguanine and O4-methylthymine in human cells: Effects of O6-alkylguanine-DNA alkyltransferase and mismatch repair. Chem. Res. Toxicol. 14, 894–900 (2001). [DOI] [PubMed] [Google Scholar]
- 26.De Bont R., van Larebeke N., Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 19, 169–185 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Lindahl T., Instability and decay of the primary structure of DNA. Nature 362, 709–715 (1993). [DOI] [PubMed] [Google Scholar]
- 28.Bamford S., Dawson E., Forbes S., Clements J., Pettett R., Dogan A., Flanagan A., Teague J., Futreal P. A., Stratton M. R., Wooster R., The COSMIC (Catalogue Of Somatic Mutations in Cancer) database and website. Br. J. Cancer 91, 355–358 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.L. B. Alexandrov, J. Kim, N. J. Haradhvala, M. N. Huang, A. W. Ng, A. Boot, K. R. Covington, D. A. Gordenin, E. Bergstrom, N. Lopez-Bigas, L. J. Klimczak, J. R. McPherson, S. Morganella, R. Sabarinathan, D. A. Wheeler, V. Mustonen; PCAWG Mutational Signatures Working Group, G. Getz, S. G. Rozen, M. R. Stratton, The repertoire of mutational signatures in human cancer. bioRxiv: 10.1101/322859 [Preprint] 3 July 2019. 10.1101/322859. [DOI]
- 30.GTEx Consortium , The genotype-tissue expression (GTEx) project. Nat. Genet. 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerstein M. B., Kundaje A., Hariharan M., Landt S. G., Yan K.-K., Cheng C., Mu X. J., Khurana E., Rozowsky J., Alexander R., Min R., Alves P., Abyzov A., Addleman N., Bhardwaj N., Boyle A. P., Cayting P., Charos A., Chen D. Z., Cheng Y., Clarke D., Eastman C., Euskirchen G., Frietze S., Fu Y., Gertz J., Grubert F., Harmanci A., Jain P., Kasowski M., Lacroute P., Leng J. J., Lian J., Monahan H., O’Geen H., Ouyang Z., Partridge E. C., Patacsil D., Pauli F., Raha D., Ramirez L., Reddy T. E., Reed B., Shi M., Slifer T., Wang J., Wu L., Yang X., Yip K. Y., Zilberman-Schapira G., Batzoglou S., Sidow A., Farnham P. J., Myers R. M., Weissman S. M., Snyder M., Architecture of the human regulatory network derived from ENCODE data. Nature 489, 91–100 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jinks-Robertson S., Bhagwat A. S., Transcription-associated mutagenesis. Annu. Rev. Genet. 48, 341–359 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Szilard L., On the nature of the aging process. Proc. Natl. Acad. Sci. U.S.A. 45, 30–45 (1959). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Failla G., The aging process and cancerogenesis. Ann. N. Y. Acad. Sci. 71, 1124–1140 (1958). [DOI] [PubMed] [Google Scholar]
- 35.Lebel M., de Souza-Pinto N. C., Bohr V. A., Metabolism, genomics, and DNA repair in the mouse aging liver. Curr. Gerontol. Geriatr. Res. 2011, 859415 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Loeb K. R., Loeb L. A., Significance of multiple mutations in cancer. Carcinogenesis 21, 379–385 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Enge M., Arda H. E., Mignardi M., Beausang J., Bottino R., Kim S. K., Quake S. R., Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell 171, 321–330.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Halpern K. B., Shenhav R., Matcovitch-Natan O., Tóth B., Lemze D., Golan M., Massasa E. E., Baydatch S., Landen S., Moor A. E., Brandis A., Giladi A., Avihail A. S., David E., Amit I., Itzkovitz S., Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 542, 352–356 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martinez-Jimenez C. P., Eling N., Chen H.-C., Vallejos C. A., Kolodziejczyk A. A., Connor F., Stojic L., Rayner T. F., Stubbington M. J. T., Teichmann S. A., de la Roche M., Marioni J. C., Odom D. T., Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 355, 1433–1436 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee H.-S., Crane G. G., Merok J. R., Tunstead J. R., Hatch N. L., Panchalingam K., Powers M. J., Griffith L. G., Sherley J. L., Clonal expansion of adult rat hepatic stem cell lines by suppression of asymmetric cell kinetics (SACK). Biotechnol. Bioeng. 83, 760–771 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Paré J.-F., Sherley J. L., Biological principles for ex vivo adult stem cell expansion. Curr. Top. Dev. Biol. 73, 141–171 (2006). [DOI] [PubMed] [Google Scholar]
- 42.Sherley J. L., Asymmetric cell kinetics genes: The key to expansion of adult stem cells in culture. Stem Cells 20, 561–572 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Huch M., Dorrell C., Boj S. F., van Es J. H., Li V. S., van de Wetering M., Sato T., Hamer K., Sasaki N., Finegold M. J., Haft A., Vries R. G., Grompe M., Clevers H., In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 494, 247–250 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huch M., Gehart H., van Boxtel R., Hamer K., Blokzijl F., Verstegen M. M., Ellis E., van Wenum M., Fuchs S. A., de Ligt J., van de Wetering M., Sasaki N., Boers S. J., Kemperman H., de Jonge J., Ijzermans J. N., Nieuwenhuis E. E., Hoekstra R., Strom S., Vries R. R., van der Laan L. J., Cuppen E., Clevers H., Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160, 299–312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H., Durbin R., Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; 1000 Genome Project Data Processing Subgroup , The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koboldt D. C., Zhang Q., Larson D. E., Shen D., McLellan M. D., Lin L., Miller C. A., Mardis E. R., Ding L., Wilson R. K., VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cibulskis K., Lawrence M. S., Carter S. L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E. S., Getz G., Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alison M. R., Lin W.-R., Hepatocyte turnover and regeneration: Virtually a virtuoso performance. Hepatology 53, 1393–1396 (2011). [DOI] [PubMed] [Google Scholar]
- 51.Duncan A. W., Dorrell C., Grompe M., Stem cells and liver regeneration. Gastroenterology 137, 466–481 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Blokzijl F., Janssen R., van Boxtel R., Cuppen E., MutationalPatterns: Comprehensive genome-wide analysis of mutational processes. Genome Med. 10, 33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang K., Li M., Hakonarson H., ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ng P. C., Henikoff S., SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31, 3812–3814 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Choi Y., Chan A. P., PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 31, 2745–2747 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Feng J., Liu T., Qin B., Zhang Y., Liu X. S., Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/5/eaax2659/DC1
Fig. S1. Phenotypic characterization of human hepatocytes and adult LSCs by means of flow cytometry.
Fig. S2. Somatic mutation levels in human liver and brain.
Fig. S3. Mutational landscape in LSC clones/single cells from young donors and liver organoids from adult/aged individuals (8).
Fig. S4. Mutational spectra in human liver.
Table S1. Human liver donor information list.
Table S2. Final WGS data and mutation calling results on human liver cells.
Table S3. List and description of the mutations in DNA repair–related genes identified in outlier cells with high mutation levels.
Table S4. Statistical analysis of mutation type contributions for spectra of different liver cells and groups: Pearson’s χ2 test and two-tailed Student’s t test.
Table S5. Correlation of spectral liver group patterns and de novo signatures identified in human liver cells (L1, L2, and L3 for hepatocytes versus LSCs versus outliers and LSC1 and LSC2 for LSC cells/clones versus liver organoids) with cancer-related signatures (COSMIC) and organoid-specific signatures (8).
Table S6. Average number of SNVs per cell in indicated groups of pooled human liver cells distributed across total and functional liver genome within specific genome sequences.