

Abstract
Ribonucleotide reductases (RNRs) employ a complex radical-based mechanism during nucleotide reduction involving multiple active site cysteines that both activate the substrate and reduce it. Using an engineered allo-tRNA, we substituted two active site cysteines with distinct function in the class Ia RNR of Escherichia coli for selenocysteine (U) via amber codon suppression, with efficiency and selectivity enabling biochemical and biophysical studies. Examination of the C439U α2 mutant protein interactions with nucleotide substrates and the cognate β2 subunit demonstrates that the endogenous Y122• of β2 is reduced under turnover conditions, presumably through radical transfer >35 Å to form a transient U439• species. This putative U439• species is formed in a kinetically competent fashion, but is incapable of initiating nucleotide reduction via 3′-H abstraction. An analogous C225U α2 protein is also capable of radical transfer from Y122•, but the radical-based substrate chemistry partitions between turnover and stalled reduction akin to the reactivity of mechanism-based inhibitors of RNR. The results collectively demonstrate the essential role of cysteine redox chemistry in the class I RNRs and establish a new tool for investigating thiyl radical reactivity in biology.
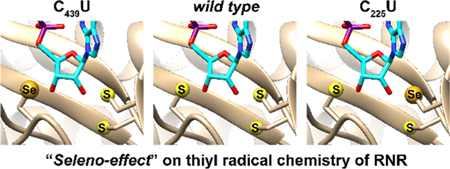
Graphical Abstract
Introduction
Redox active amino acids endow enzymes with intrinsic cofactor(s) and play critical roles in a plethora of enzymatic reactions.1 Among the redox active amino acids, cysteine (C) is unique in that the thiol sidechain can supply an electron (and a proton) individually, forming a thiyl radical, or in tandem with a second sterically accessible cysteine, forming a disulfide bond. Both thiol-thiyl radical and thiol-disulfide redox reactions involve proton-coupled electron transfer (PCET), and are exploited extensively in enzyme catalysis. Contrary to the thiol/disulfide couple, the role of thiyl radicals in enzymology remains poorly understood, yet thiyl radicals continue to be invoked in a broad range of enzymatic transformations. Identifying and defining the role of thiyl radicals is challenging for several distinctive reasons. First, the one electron reduction potential (E0) of the cysteine thiyl radical is the highest known among the physiologically relevant redox active amino acid radicals, which follow the general trend selenocysteine (U)2 < tyrosine (Y) < tryptophan (W) < glycine (G) < cysteine (C) (pH = 7).1 Second, thiyl radicals are quite reactive towards elements of all proteins including C=O and C-H bonds.3 Third, thiyl radicals are challenging to detect by conventional biophysical spectroscopic techniques including UV-vis absorption due to low extinction coefficients,4 and paramagnetic techniques due to broadening.5 Lastly, the controlled generation of thiyl radicals requires site-specific delivery of a potent oxidant, often through endothermic radical transfer (RT) from another protein, cofactor, or substrate based radical. These properties of the protein based thiyl radical present significant barriers to the study of their function in biology.
Both thiol-thiyl radical and thiol-disulfide redox reactions figure prominently in the function of ribonucleotide reductase (RNR), which catalyzes the reduction of nucleotides (di- or triphosphates) to deoxynucleotides, a committed step in de novo DNA biosynthesis and repair (Figure 1A).6,7 Early identification of conserved and essential cysteines of the enzyme,8,9 structural homology of the active site (Figure 1B),10 and reactivity studies11–13 suggest that a radical-based mechanism is employed by all RNRs involving a conserved cysteine on the “top face” that forms a thiyl radical and activates the substrate towards reduction by abstracting the 3′-H of the nucleotide. Two additional cysteines, or a cysteine, methionine, and formate, serve as radical substrate reductants located in the “bottom face.” The mechanism of cysteine oxidation on the top face has formed the basis of class differentiation: class I RNRs utilize a second subunit harboring a redox active (metallo-)cofactor for thiyl radical generation, class II utilize adenosylcob(II)alamin (AdoCbl), and class III utilize a radical-SAM activating enzyme that produces a glycyl radical.
Figure 1.

RNR mechanism of nucleotide reduction and active site structural homology. A General reaction catalyzed by RNR in all organisms. N = nucleoside base. B Structural alignment of class Ia (E. coli, green; PDB 1RLR), class II (L. leichmannii, orange; PDB 1L1L), and class III (Enterobacteria phage T4, light blue; PDB 1B8B) RNRs aligned based on the “top” and “bottom-face” cysteines. Top-face cysteines are on the top of the image, and bottom-face cysteines (disulfide for both class I and II, cysteine and methionine for class III) shown on the bottom. Some of the class III RNRs contain a methionine in place of one of the cysteines, and formate serves as the ultimate electron donor. Cysteine/methionine sulfurs shown in yellow spheres. C Putative mechanism of nucleotide reduction following the generation of the thiyl radical in 1. Cysteine numbering corresponds to the E. coli class Ia.
Thiyl radical-based catalysis on the top face has been most clearly demonstrated in the class II RNRs. The AdoCbl-dependent nature of the class II enzymes, and fortuitous reaction kinetics, proved essential in trapping a thiyl radical during turnover by rapid freeze-quench (RFQ) EPR spectroscopy.14 Analysis of the early reaction products resulting from mixing Lactobacillus leichmannii class II ribonucleotide triphosphate reductase (RTPR), substrate ATP, and AdoCbl yielded an exchange-coupled cob(II)alamin-thiyl radical (C408-S•, L. leichmannii numbering), generated in a kinetically competent fashion, and shown to be chemically competent for nucleotide reduction. 14,15 All available data thus far indicates that the thiyl radical abstracts the substrate 3′-H (Figure 1C, step i) in all RNRs, but this has yet to be explicitly demonstrated outside this specific class II enzyme.
The role of the bottom-face cysteines in substrate reduction has also been investigated, requiring perturbations to the enzyme active site or substrate to observe intermediates. Using several mechanism-based inhibitors (MBIs) it was shown that removal of the 2′-OH is assisted by proton transfer (PT) from one of the bottom-face cysteines in both class I and class II RNRs (Figure 1C, step ii).11 The resulting thiolate then reduces the 3′-keto-2′-deoxynucleotide radical (3) to form a disulfide radical anion (step iii), which then reduces the 3′-ketone regenerating the 3′-hydroxyl radical (5, step iv). Site-directed mutagenesis (SDM) affecting the proton inventory in the active site stabilizes the disulfide radical anion, which has been characterized in the Escherichia coli class Ia RNR (E441Qα2) by high-field pulsed EPR.16 Unfortunately, the disulfide radical anion, a key intermediate in the proposed mechanism, has only been observed by significantly altering protein or substrate.
We sought to develop methods to examine the PCET chemistry of cysteines in the active site with minimal protein or substrate perturbations. We now demonstrate the use of U as a cysteine surrogate with distinct electron and proton affinities, thus enabling the rational perturbation of the PCET chemistry at both the top and bottom face of the RNR active site. U is advantageous as it has a depressed U reduction potential relative to both C• and Y• 2,17. We targeted the α2 subunit cysteines of the paradigmatic class Ia RNR from E. coli, C439 (C439U, top face) and C225 (C225U, bottom face), for mutagenesis via a recently developed amber suppression protocol18 in good yield and purity. Mixing of the U-labeled α2 protein with the diferric-tyrosyl radical harboring β2 subunit, substrate, and effector, results in RT from the tyrosyl radical and effector, results in RT from the tyrosyl radical in β (Y122•) to the active site through a pathway of redox active amino acids (Y122• [β] • 19 and provide kinetic and chemical evidence for the involvement of thiyl radical chemistry on the top and bottom face. The results are consistent with reaction energetics rising from redox and pKa contributions, and the implication of this new method reported herein is discussed with regard to mechanistic consensus among all RNRs, and the broader implications of the method as a new tool to examine the multitude of thiyl radical enzymes.
Materials and Methods
Materials.
Adenosine, adenosine-5′-triphosphate (ATP), agarose, ampicillin, L-arabinose, 1 mg/L 99.99% arsenic in 2% HNO3 ICP-MS standard, bovine pancreas chymotrypsin (>40 U/mg), cytidine, cytosine, cytosine-5′-diphosphate (CDP), deoxycytidine, ethylenediaminetetraaceticacid (EDTA), glycerol (99%), guanidinium hydrochloride (GdHCl), (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES acid), kanamycin, LB medium, magnesium sulfate heptahydrate (MgSO4), methanol (HPLC grade), reduced nicotinamide adenine dinucleotide phosphate (NADPH), ruthenium (II) hexamine dichloride, 1 mg/L 99.99% selenium in 2% HNO3 ICP-MS standard, L-sele no cystine, sodium selenite, and bovine pancreas trypsin (>6,000 U/mg) were purchased from Sigma Aldrich at reagent grade purity or higher. [5-3H]-CDP was purchased from ViTrax and diluted with “cold” CDP to a final concentration of 32,100 cpm/nmol. Calf intestine alkaline phosphatase (AP, 1 U/μL) and 10x AP dephosphorylation buffer were purchased from Roche. Isopropyl β-D-1-thiogalactopyranoside (IPTG, 98%), dithiothreitol (DTT, 98%) were purchased from Promega. NdeI, XhoI, NcoI, Gibson Master Mix, GoTaq Green, and Phusion polymerase were purchased from New England Biolabs. Primers were purchased from Integrative DNA Technologies (IDT). Template pET-28a plasmids for wild (His)6-WT 0α2 were available from previous studies,20 and the pSec-EVOL2 plasmid was kindly provided by the Soll lab from previous studies.18 The pET-14b plasmid was purchased from EMD-Millipore. E. coli thioredoxin (Tr, 40 μmol/min/mg), thioredoxin reductase (TrR, 1800 μmol/min/mg), wt β2 (1.1 Y•/β2, 6000 nmol/min*mg), photoβ2, and 2,3,5-F3Y356 photoβ2 and 2,3,6-F3Y356 photoβ2 were available from previous studies.20−22 MilliQ(18 MΩ) was used for preparation of all solutions.
Construction of plasmids.
The previously developed pET-28a nrdA plasmid was sub-cloned into pET-14b by Gibson assembly.23 The nrdA fragment of the pET-28a construct was amplified by PCR with the primers:
Forward: 5′-ctttgttagcagccggatcctcgagCCGTAGGCCGGATAAG-3′ Reverse :3′-caatcgtcggcctaggagctcGGCATCCGGCCTATTCCGC-5′
where lower case bases indicate the pET-14b complementary multiple cloning site sequence, and upper-case bases indicate nrdA. The amplified nrdA fragment was extracted and purified by a Qiagen DNA gel purification kit. A pET-14b plasmid was doubly digested with XhoI and NcoI restriction enzymes as per the manufacturer’s instructions and purified similarly. Gibson assembly of the two DNA fragments was performed as per the manufacturer’s instructions. The assembly product was directly transformed into XL10 competent cells and streaked on fresh ampicillin plates. Colonies were screened for cloned nrdA pET-14b by PCR using the T7 promoter/terminator primers and GoTaq Green polymerase, performed as directed by the manufacturer, and 1% agarose gel electrophoresis of the colony PCR products identified several successfully cloned plasmids. Colonies containing the nrdA pET-14b were selected for further growth and DNA isolation by Qiagen miniprep. Sanger sequencing through Quintara Bio confirmed the nrdA pET-14b authenticity.
The wild type (wt) nrdA pET-14b was subjected to three individual SDM protocols to construct C439S, C439U and C225U nrdA pET-14b with the following primers:
C225U Forward: 5′-AGCTCCTAGGTACTGATC-3′ Reverse: 3′-AAGTCGAGGATCCATGAC-5′ C439U Forward: 5′-AACCTGTAGCTGGAGATA-3′ Reverse: 3′-AGATTGGACATCGACCTC-5′ C439S Forward: 5′-AACCTGTCCCTGGAGATA-3′ Reverse: 3′-AGATTGGACAGGGACCTC-5′
where bold bases represent sites of mutation. SDM by PCR was performed using Phusion polymerase as per the manufacturer’s instructions and the success of the reaction estimated by 1% agarose gel electrophoresis. Plasmids were amplified in XL10 cells, DNA isolated by Qiagen mini-prep kit, and their sequence was confirmed by Sanger sequencing by Quintara Bio.
Protein expression.
The C439U RNR was produced by co-transformation of ME6 cells18 (containing a scar-less quadruple deletion ΔselABC ΔfdhF from E. coli strain BW25113) with the pSec-EVOL2 and C439U pET-14b nrdA plasmids via heat shock by immersion of the samples in a 42 °C water bath for 30 s then placed on ice for 2 min. Following transformation, the cell suspension was diluted 1:1 v:v with SOC media and allowed to recover for 20 min at 37 °C prior to being plated onto 50 μg/mL kanamycin and 100 μg/mL ampicillin supplemented LB plates. Single colonies were selected for further growth. To test expression conditions, small scale growths (10 mL) were employed, where a single colony was inoculated into LB media containing 50 μg/mL kanamycin, 100 μg/mL ampicillin and various levels of Na2SeO3 (0–100 μM). Temperature, shaking, induction levels, and induction timing were optimized on this scale and evaluated by SDS-PAGE analysis of total protein expression and amber codon suppression. Shaking rates of 200 rpm and inoculation temperatures of 37 °C were found to be appropriate. Protein expression in 10 mL cultures did not scale to higher volumes, all other things being equal. We found that it was essential to inoculate single colonies directly into the larger cultures (>1 L) necessary for protein isolation and purification for biophysical methods. For large scale growth (4 × 2 L) single colonies were inoculated into 2 L of the above media in a 6 L Erlenmeyer flask at 37 °C shaking at 200 rpm. At OD600 = 0.1 (~6 h), 30 Na2SeO3 was added. At OD600 = 0.4, 0.1% L-arabinose was added to induce pSec-EVOL2, and the temperature was reduced to 30 °C, then the pET-14b nrdA plasmid was induced by addition of 0.25 mM IPTG at OD600 = 0.8, and the culture was maintained for an additional 20 h. Cells were harvested by centrifugation, collected, flash frozen in liquid N2, and stored until needed. An identical protocol was used for expression of all nrdA constructs for consistency.
Protein purification was performed identically as previously reported,21 with the exception of increased reductant in the final buffer exchange, yielding protein stored in 2 mM DTT, 50 mM HEPES, 5% glycerol pH = 7.6. The truncated proteins of both C439U and C225U expression were insoluble and precipitated in the cell pellet or in the DNA pellet during purification. All proteins were flash frozen in liquid N2 following purification and stored until used.
Selenium quantitation.
Selenium incorporation into α was quantitated by inductively coupled plasma mass spectrometry (ICP-MS) on an Agilent 7900. The Na2SeO3 used as the selenium source during protein expression was used as a selenium standard for calibration and 75As was used as an internal standard in all samples. Instrument response for all isotopes except 80Se yielded linear response (R = 0.99) between from 0.5 ppb to >> 100 ppb for all isotopes examined (74Se, 76Se, 77Se, 78Se, 82Se). Due to the overlap of the most abundant isotope (80Se) with the an Ar2+ product of the plasma 78Se was routinely used (23.7% relative abundance) for quantitation. Protein samples analyzed by ICP-MS were digested by trypsin and chymotrypsin to improve solubility in the 2% HNO3 preparation solution. Protein aliquots corresponding a theoretical final concentration of 100 ppb 78Se in 1.5 mL (typically 2–10 μL) were digested in 150 μL of 100 mM Tris, 10 mM CaCl2, and 2 M GdHCl at pH = 8.0 with 1 U of both trypsin and chymotrypsin for 24 h. The resulting digested peptide solution was diluted with 2% HNO3 to 1.5 mL supplemented with 50 ppb arsenic in 2% HNO3 and directly analyzed by ICP-MS. The ion integration time was 0.3 s, and ion counts, observed at 78Se unless otherwise stated, were converted to ppb via a standard curve.
Steady state assays.
Assays were performed as previously described in a coupled assay followed spectrophotometrically.21 Briefly, 0.2 μM α2, 2.5 μM β2, 1 mM CDP, 3 mM ATP, 60 μM Tr, 1 μM TrR and 0.2 mM NADPH were mixed in assay buffer (50 mM HEPES, 15 mM MgSO4, 1 mM EDTA, and 5% glycerol at pH 7.6), and NADPH consumption was monitored spectrophotometrically by a Cary 50 UV-vis spectrometer at 340 nm over 1 min. One unit of activity is defined as 1 nmol CDP reduced per minute per mg of protein. Assays were performed in triplicate and data is reported as the mean with error reported as one standard deviation.
Single turnover assays.
All α2 samples were buffer exchanged from storage buffer into assay buffer under an inert atmosphere in a glove box maintained at 4 °C to remove excess DTT unless otherwise noted. The buffer exchanged α2 was then added to a solution containing 10 μM β2, 0.2 mM 5-3H CDP (32,100 cpm/nmol), and 1 mM ATP to a final concentration of 10 μM α2 and a final volume of 60 μL. The reaction was quenched by addition of 10 μL of 2% HClO4 for 5–10 min then neutralized by addition of 5 μL of 0.5 M KOH. The precipitated protein was removed by centrifugation and 50 μL supernatant was dephosphorylated by either (A) dilution to 450 μL in 1× AP dephosphorylation buffer supplemented with 14 units AP, or (B) dilution to 60 μL in 10× AP dephosphorylation buffer and supplemented with 14 units AP. Solutions from both protocols were then reacted for 2 h at 37 °C.
Protocol A was employed for product isolation by the method of Steeper and Steward.24 Radioactivity was used to quantitate CDP reduction products and was measured by scintillation counting. Results are reported as the mean of three independent measurements and reported error reflects a single standard deviation.
Protocol B was employed for product isolation and characterization by HPLC on a Waters 717/515/2487 HPLC equipped with an Absorbosphere Nucleotide-Nucleoside column (250 mm × 4.6 mm). 45 μL of dephosphorylated reaction mixture (vida supra) was loaded onto the column equilibrated with 5 mM KPi pH = 6.8. Flow rate was maintained at 2 mL/min and a stepwise gradient was applied as follows; isocratic 0% MeOH from 0–3 min, linear gradient from 0–5% methanol from 3–15 min, and linear gradient from 5–20% MeOH from 15–20 min. Eluent was monitored by a photodiode array from 220–400 nm and HPLC traces are reported from absorption at 270 nm.
X-Band EPR spectroscopy.
Samples prepared for EPR spectroscopy were prepared by two protocols (C) and (D). In protocol C, wt β2 was mixed with CDP and ATP, and reaction was initiated with addition of C439(U/S) α2 to a final concentration of 50 μM α2β2, 1 mM CDP and 3 mM ATP in assay buffer in a total volume of 220 μL and then transferred to a Wilmad 4 mm precision quartz EPR tube, all under an inert atmosphere in a glove box at 4 °C. The reaction was quenched by hand following transfer (~15 s) into liquid N2 and stored cryogenically. In protocol D, the EPR tube was initially loaded with wt β2 (19 μL), and a premade solution of C439(U/S) α2, CDP and ATP in assay buffer supplemented with 20% glycerol (201 μL) was swiftly added in a single step in the EPR tube and rapidly frozen in liquid N2 cooled isopentane. This protocol reduced the quenching time to ~3 s. Samples were measured directly after freezing on a Bruker EMX X-band EPR spectrometer equipped with a recirculating helium cryostat cooled ER4199HS cavity. EPR parameters in general were as follows: microwave frequency, 9.3–9.8 GHz; modulation amplitude, 1.00 G; modulation frequency, 100 kHz; and time constant, 40.96 ms. The temperature and microwave power were varied over a wide range and individual spectra are reported with corresponding values.
Stopped Flow (SF) UV-vis spectroscopy.
SF measurements were performed on an Applied Photophysics DX 17MV instrument and data was recorded on a PC. The reaction was thermostated at 25 °C by a recirculating water bath. The contents of two syringes (A and B) were mixed 1:1 v:v to a total volume of 60 μL. Syringe A contained 10–20 μM α2 mutant of interest, 6 mM ATP in assay buffer, and syringe B contained 10–20 μM wt β2 and 2 mM CDP in assay buffer. Transients were monitored at 340 nm (εSe• ~ 1,100 M−1 cm−1, estimated by comparing to absorption at 460 nm), 460 nm (εSe• = 560 M−1 cm−1)2 and 412 nm (εy• = 3,600 M−1 cm−1) at 4 nm resolution. The data represent the average of at least eight transients and individual fitting parameters for mono-, bi- and triexponential fits to experimental data starting at 3 ms are reported.
Transient absorption spectroscopy.
Transient absorption (TA) lifetime and spectra measurements were performed on a home built nanosecond time resolved instrument described previously.25 Samples were prepared with 30 μM photoβ2, 50 μM α2, 1 mM CDP, 3 mM ATP and 10 mM Ru(NH3)6Cl3 in assay buffer. The sample volume (650 μL) was recirculated and filtered by an inline 0.22 μm PFE membrane filter (Millipore Sigma). The probe light was generated by a Xe-arc lamp. For TA lifetime measurements, the observation wavelength was selected by the spectrophotometer (λobs = 430 nm for 2,3,6-F3Y356 photo β2 at 0.45 nm resolution). Transient signals were collected at 2 ns digital resolution and linearly binned to 100 ns resolution to decrease noise. 1000 shots were averaged in 100 shot collection intervals to ensure samples did not degrade during the course of the measurement. TA spectra were collected on a thermoelectrically cooled CCD camera with 0.45 nm resolution (as determined by slits) and pixels were binned to a final resolution of 2 nm (4 pixels/nm) from 1–1.05 μs after excitation. Again, 1000 shots were averaged and spectra were collected in a −log[(pump on:probe on)/(pump off:probe off) − (pump off:probe on)/(pump off: probe off)] controlled by shutters and delay generators to eliminate signatures induced by the probe light.
Results
Protein preparation and characterization.
Two of the three catalytic cysteines within the 0α2 subunit of the E. coli class Ia RNR, C439 and C225, were chosen for substitution with U, representing redox active residues on both top and bottom face, respectively, of the active site of RNR. To accomplish U substitution for C, a newly developed strategy for heterologous selenoprotein expression was employed, wherein U is site specifically installed by amber codon suppression. We thus cloned the nrdA (β2) RNR gene into a pET-14b plasmid and performed site-directed mutagenesis to generate the (His)6-C439Z or (His)6-C22sZ α2, where Z indicates the amber codon mutation. A second plasmid was employed for selenocysteine biosynthesis and site-specific incorporation into the amber codon labeled α2 variant. This second plasmid controls expression of an engineered tRNA (allo-tRNAUTu)18 and proteins SelA (Aeromonas salmonicida subsp. pectinolytica 34mel), SelD (A. salmonicida), and Tr (Treponema denticola). The allo-tRNAUTu is crucial in that it has been engineered to interact with E. coll’s native elongation factor Tu (EF-Tu), circumventing the necessity for a selenocysteine insertion sequence (SECIS) structural recognition element in the gene of interest mRNA, thus eliminating the need for the bacterial SelB enzyme. Allo-tRNAUTu was also chosen for its inherent capability to be charged with serine by E. coli serine tRNA synthetase (serRS). Once charged with serine, S-allo-tRNAUTu is converted to U-allo-tRNAUTu by SelA, with the selenium source being selenophosphate derived from SelD and selenide. Tr serves to maintain U in the reduced state.
Evaluation of C439U expression and proper U incorporation was optimized on the 10 mL scale under a variety of conditions and analyzed by SDS-PAGE (Figure S1). From the gel analysis expression of both C439U α2 and selenocysteine/tRNA encoding plasmids are required for full length C439U α2 expression, and that expression of both plasmids results in a ~1:1 ratio of truncated to full length protein. Encouraged by this result, we attempted to scale up expression by sequential culture saturation (5 mL → 100 mL → 2 L) for biophysical characterization. Interestingly, expression was significantly lower at larger scales (>1 L) by this method resulting in very low yields (<0.5 mg/L) and a drop off of both total protein expression and amber codon suppression. We observed expressed protein yield could be recovered to some extent by inoculating a 2 L culture with a single colony from a freshly prepared plate (transformed, plated and grown for ~10 h) with total expression levels and amber codon suppression efficiency similar to small scale expression by SDS-PAGE. The truncated protein precipitated during cell lysis, and the resultant full-length protein, following Ni-affinity chromatography, was estimated to be >95% pure by SDS-PAGE (Figure S2).
Due to the nature of the selenocysteine biosynthesis machinery and incorporation method, it is possible to incorporate S rather than U at the stop codon site. This can occur if SelA/SelD activity cannot compete with S-allo-tRNAUTu charging by serRS and amber codon suppression. Fortunately, S is redox inert under the conditions employed here, rendering C439S α2 a mere loss in overall yield, minimizing complications in the subsequent analysis. The efficiency of U vs. S incorporation was evaluated by inductively coupled plasma mass spectrometry (ICP-MS) analysis of the purified protein to quantitate [Se] relative to [α2] (Table S1). Following digestion of the purified α2 by trypsin and chymotrypsin, ICP-MS analysis revealed that selenocysteine incorporation was dependent on α2 expression levels; induction with 1 mM IPTG exhibited 0.54 (0.06) Se/α, whereas with 0.25 mM IPTG, ratios of >0.9 Se/α were achieved. The lower IPTG concentration came at the cost of total protein isolated (1–2 mg/g cell paste), but improved the amber suppression efficiency. The pET-14b nrdA vector of the C439U α2 gene was also found to be somewhat “leaky,” expressing α2 in the absence of IPTG at an effective IPTG level of 100 μM. Under similar conditions, C225U was poorly expressed, but pure protein with 0.82 (0.07) Se/α was produced at 0.5 mg/g cell paste. The origin of this difference is still under investigation. It is also possible that U can be over-incorporated. While unlikely, we examined this possibility by expressing both wt and C439S α2 under identical conditions of U incorporation. Both proteins exhibited <0.05 Se/α, verifying that U is site-specifically incorporated at the amber codon. These proteins were employed as controls in further experiments. Care was taken to keep all selenoproteins microaerobic with excess (2 mM) DTT to ensure selenium remained in the selenol/selenide oxidation state.
Top-face reactivity.
To assess the effect of U incorporation at the top-face C439 on radical-based substrate activation we performed multi-turnover activity assays aerobically on C439U α2. No activity was found above background (20 (30) units) in these experiments, identical to activity observed for C439S (Table S2) and consistent with contaminating wt protein. By contrast, the activity measured for wild type α2 produced under identical conditions is 1,920 (30) U, nearly identical to the known activity of this subunit isolated under standard conditions (2,200 units).26 These data imply that the C439U mutation results in either an α2β2 complex that is incapable of RT, a RT event that is arrested at C439U (C439U•), or non-canonical radical chemistry with the substrate/enzyme that does not couple to bottom-face oxidation. It is also possible that, despite the presence of excess reductant, the selenocysteine has been oxidized by molecular oxygen to the corresponding selenic or selinic acid.
Radical-based reactions in enzyme active sites can often access off-pathway outcomes, either quenching the radical or generating new products that can inactivate the enzyme; indeed, this is the case with RNR To address this possibility in C439U α2, we performed single turnover assays in which the external reductant system (TR/TRR/NADPH) is omitted from the reaction, and only the endogenous reducing equivalents, C225/C462 in the bottom face and a second cysteine pair in the C-terminus, can drive nucleotide reduction. In these experiments, one α2 can therefore theoretically produce 4 dNDPs. In practice, values between 1.5–3.6 dCDP/α2 are obtained. Consistent with this product yield, wild type α2 produced under U incorporation conditions yielded 1.6 dCDP/α2. The C439U α2 gave no observable product when submitted to single turnover assay conditions in an inert atmosphere, either by the method of Steeper and Stuart (Table S2),24 or by HPLC analysis of the reaction products (Figure 2, Figure S3). Addition of DTT did not affect single turnover reactivity. These data suggest that either RT is impeded altogether by the C439U mutation or that RT is arrested at this site.
Figure 2.

HPLC chromatograms of single turnover reaction products for 10 μM wt (blue), C225U (green) and C439U (red) α2 with 10 μM β2 and an authentic standard (black). The standard contained 20 μM of cytosine (RT = 2.6 min), cytidine (RT = 4.6 min), and deoxycytidine (RT = 7.3 min). Peak splitting was observed under all flow rates and all concentrations suggesting column degradation (channeling, see Figure S3). Asterisk indicated the residual protein absorption profile that elutes in the void volume.
To evaluate the potential for C439U to act as a radical trap we turned to X-band EPR spectroscopy. Under similar conditions as those employed in single turnover experiments C439U and C439S α2 were mixed separately with β2, CDP and ATP and quenched by hand in liquid nitrogen after 10–15 s in an X-band EPR tube and submitted to X-band EPR analysis (Figure 3). Thiyl and selenyl radicals, while extremely rare in the literature, are generally observed to have strong g-anisotropy and relatively fast spin relaxation rates.4,27–30 Our EPR analysis reveals that the reaction of C439U α2 with wt β2 (1.1 Y•/β2), CDP, and ATP yields a net loss of ~60% of the Y122• signature as determined by double integration of the derivative spectrum, and no new resonance consistent with a selenyl radical was observed. Further experiments with 2-deoxy-2-azido cytidine diphosphate (N3CDP), a mechanism-based inhibitor that forms a distinct N-centered substrate radical following activation by 3′-H abstraction during the reaction with wt α2 and β231 yielded slightly lower levels of Y122• loss (~40%, Figure S4), but no new resonance was observed. Radical trapping experiments were performed under a variety of conditions: (i) with and without reductant as well as (ii) in a completely inert atmosphere. In either case, no new species were observed in the EPR spectrum. We also varied the quenching method to access faster quenching times of ~3 s to no avail.
Figure 3.

X-band EPR spectra of the Y122• of wt β2 in complex with C439S (black) and C439U (red) α2. Samples were prepared by protocol C resulting in 50 μM C439X (X = S or U) α2, 50 μM wt β2, 1 mM CDP, 3 mM ATP in assay buffer. Spectra recorded at 80 K with a modulation frequency of 100 kHz, a modulation amplitude of 1 G, and microwave power of 20 μW.
The kinetics of RT in the E. coli class Ia RNR have been characterized at each Y within the RT pathway by stopped-flow (SF) UV-vis; these experiments reveal that RT is gated by conformational changes that enable long range PCET at ~8 s−1.21,26,32 Here we applied SF UV-vis to define the kinetics of RT from Y122•[β] to C439U[α] by monitoring both the sharp absorption feature of the 33 and the broad selenyl radical absorption (300–500 nm, ε < 1,000 M−1 cm−1).2 Mixing of C439U α2 and ATP with β and CDP to a final concentration of 10 μM α2β2, 1 mM CDP, and 3 mM ATP resulted in a decay of the 412 nm feature with bimolecular kinetics characterized by kobs1 = 7.1 (0.1) s−1 and kobs2 = 0.40 (0.01) s−1 (Figure 4, red). A temporally concerted induced absorption signal was observed at both at 340 nm (Figure 4 blue, kobs1 = 6.5 (0.3) s−1 and kobs2 = 0.42 (0.03) s−1) and 460 nm (Figure S5), with a markedly lower intensity. This induced absorption feature at 460 nm appeared to be stable for 10 s and decayed with a rate of 0.02 s−1 (Figure S6). The induced absorbances at 340 and 460 nm, while consistent with a weak absorption profile of C439U•, also coincide with the broad absorption profile of the di-iron cluster. Control experiments with wt α2 probed at 340 and 460 nm yielded kinetically similar, yet markedly weaker signals, thus complicating interpretation (Figure S7). Fitting amplitudes and rates are reported in Table S3.
Figure 4.

Stopped-flow UV-vis kinetics for wt β2 and effector mixed with C439U α2 and substrate. Transients at 412 nm (left axis) shown in red and 340 nm (right axis) in blue, with associated biexponential fits in black. Proteins at 10 μM final concentration.
The rate determining conformational gating of RT and active site chemistry during the reaction of 0α2 with β2 precludes insight into the role of the C439U substitution in long range PCET.26 We have previously employed a photosensitized β2, termed photoβ2, to rapidly generate kinetically and chemically competent radicals on the RT pathway at the subunit interface in an intact α2β2 complex.34 This photoβ2 is constructed by the covalent attachment of a rhenium (I) 1,10-phenanthroline tricarbonyl bromomethylpyridine chromophore ([ReI]) adjacent to the pathway residue Y356 through a S355C substitution. Upon excitation of the [ReI] the excited state can directly oxidize Y356 followed by charge recombination, or the [Re1]* state can be oxidatively quenched, resulting in a [ReII] that can subsequently oxidize Y356 to form a long-lived Y356• for spectroscopic and kinetic characterization.20,21 To address the influence of the C439U mutation in 0α2 on RT kinetics in the absence of conformational gating we employed a 2,3,6-F3Y356-photoβ2. The 2,3,6-Y356 substitution enables specific spectroscopic observation of 2,3,6-F3Y356• among the potential Y• background due to the distinct absorption profile21 and also increases the one electron reduction potential of the (fluoro)phenoxy radical relative to tyrosine by +135 mV,35 biasing forward RT into α2 and the remainder of the RT pathway. By monitoring the kinetics of 2,3,6-F3Y356• in the presence of the radical trap C439U α2, and comparing this rate to that obtained in the presence of the RT pathway block, Y731F, we can deduce the rate of PCET across the subunit interface.21 Figure 5 shows the kinetics of 2,3,6-F3Y356• decay in the presence of C439U α2 at pH 8.2. The rate of decay was determined to be 9.9 (0.1) × 104 s−1 (kobs) as compared to the rate in the presence of Y731F previously determined to be 6.8 (0.3) × 104 s−1 (k0), yielding a radical injection rate of kPCET = 3.1 (0.5) × 104 s−1. This observation is consistent with the above data indicating that C439U is redox active and modulates RT analogous to 3-aminotyrosine (NH2Y).21
Figure 5.

Transient absorption kinetics of 2,3,6-F3Y356• decay (red) in the presence of C439U α2, substrate and effector in spectroscopy buffer and associated monoexponential fit. Data represents the average of three independent trials and fit is displayed for illustrative purposes.
Bottom-face reactivity.
The redox properties of the C225U α2 are distinct from the C439U, C225S, or wt counterparts. Single turnover and multi-turnover assays performed either aerobically or anaerobically rendered no NDP reduction activity, or very little in the case of C225U. Single turnover activity was observed under anaerobic conditions if the C225U α2 protein was directly used from DTT containing storage buffer, resulting in a final DTT concentration of 200 in the assay mixture. The residual DTT can serve as an external reductant, allowing the oxidized cysteines of the bottom face to be re-reduced for subsequent turnovers, yielding a maximal theoretical turnover of 24 dCDP/α2. Under these conditions, re-analysis of the single turnover activity of C225U revealed 7.9 (0.9) dCDP/α2, similar to the activity observed for wt α2 (11 dCDP/α2) as determined by the method of Steeper and Steuart (Table S2).24 This method of turnover quantitation does not discriminate between dCDP and cytosine production, a byproduct of substrate degradation due to incomplete reduction of the 3′-keto intermediate (Figure 1C, intermediate 4), resulting in release of pyrophosphate, cytosine (base) and furanone (Figure S8).36–38 Cytosine and dCDP formation by C225U α2 was evidenced by HPLC analysis of the reaction products under similar conditions (Figure 2, green). This reactivity is analogous to that of 2′-deoxy-2′-X cytidine diphosphate (X = F or Cl), mechanism-based inhibitors of RNR. Efforts to characterize intermediates during substrate activation and selenyl radical chemistry in the C225U α2 protein were unsuccessful by X-band EPR following the mixing of C225U α2 with wt β2, CDP and ATP (Figure S9) on the 3–15 s timescale.
Discussion
The thiyl radical form of cysteine is the active cofactor implicated in RNR and in glycyl radical enzymes (GREs), a growing class of enzymes important in anaerobic metabolism.39,40 In particular GREs are pivotal in host-microbiome interactions in the anaerobic environment of the gut, where they endow microorganisms with the ability to exploit host derived metabolites41 and xenobiotics42 for energy and biosynthesis. The reactivity of thiyl radicals represents a natural target for small molecule mechanism-based inhibitor design and are a target of current chemotherapeutic drugs.43,44 The rational design of inhibitors and the greater understanding of thiyl radicals in biology requires a detailed understanding of their reactivity which has proven experimentally challenging. In this work, we substitute the redox active cysteines of the paradigmatic thiyl radical enzyme RNR of E. coli for selenocysteine in an attempt to attenuate radical reactivity such that intermediates may be observed and reactivity trends revealed. Selenocysteine has been demonstrated to be quite distinct in reactivity relative to cysteine, particularly in its ability to scavenge reactive oxygen species reversibly and have diminished activity towards deleterious radical-based protein degradation.45 We have shown that the method of site-specific U incorporation developed by Söll et al.18 is robust in producing relatively high quantities of quantitatively substituted selenoproteins on the scale necessary for biochemical and biophysical characterization. While a strategy was ultimately developed for large scale expression, significant challenges were encountered en route. The observation of poor expression following sequential culture saturations as opposed to direct inoculation of the large volume media preparations suggests that either replication events or growth stagnation affect protein expression. This observation must be reconciled with the additional observation of no observable effect on growth rates in the presence of antibiotics that select for each plasmid. We do not observe any issues with growth or expression using just pET based vectors for nrdA expression, indicating that the likely culprit is the pSec-EVOL2 vector instability. The pSec-EVOL2 plasmid contains a high copy number origin of replication that may place some burden on the co-transformed cells. We hypothesize that horizontal gene transfer of the kanamycin resistance cassette during stationary phase conditions, and subsequent elimination of the pSec-EVOL2 plasmid during replication may rationalize the observed phenomena.
An interesting and unexpected consequence of the C439U mutation is that multiple methods of assessing RT provide strong kinetic evidence that radicals on pathway appear to funnel towards U439, but no U439• is directly observed, and no product deoxynucleotide is formed. SF UV-vis and transient absorption spectroscopies demonstrate that Y122• and Y356• respectively are highly susceptible to reduction by RT, and behave analogously to prior observations of radical trap NH2Y labeled α2 proteins.21,26 We have exhaustively sought to observe the putative C439U• intermediate by SF UV-vis, EPR, and transient absorption (TA) using a photoβ2 construct. SF UV-vis data indicates that a new optical feature is formed concomitant to Y122• reduction that resembles an authentic U•, but no evidence of this species was obtained by either EPR or TA. We hypothesize that either the radical exhibits an extreme level of broadening of the EPR signal rendering it unresolved in the current experiments, or that the transiently formed U• reacts with another element of the protein structure on a rapid timescale and is reduced (Figure 6). Thus, the reactivity of the corresponding putative U439• appears to be quite high, but distinct from C allowing insight to be gained regarding top-face thiyl radical catalysis. This is not unexpected as the C-H bond dissociation energy (BDE) of the 3-H is estimated to be ~390 kJ/mol for aliphatic alcohols,46 whereas the BDE of Se-H of U is ~310 kJ/mol and the Se•/Se- couple ~500 mV lower than the corresponding S•/SH.2 With regard to the latter, the reactivity of S• towards elements of protein structures is well documented and they can attack C-H bonds of the protein backbone, with known reactivity towards the relatively weak amino acid a-H (BDE ~ 355 kJ/mol).47,48 The reactivity is kinetically accelerated due to polar resonance effects of the SH/S•exchange.49,50 Furthermore, C acts as a nucleophile in the GRE pyruvate formate lyase, where it is proposed to attack the carbonyl carbon resulting in a transient substrate based oxyl radical.51,52 Analysis of the active site of the E. coli α2 near C439 does not immediately reveal a likely site of backbone or sidechain attack. Furthermore, selenyl radical attack of the protein backbone should produce a new radical, likely a carbon centered radical, which is not observed under any of the conditions applied herein. It is also possible that the protonation state of U439, anticipated to be a selenate, affects PCET during radical transfer, as C439 is both the proton and electron donor during radical transfer from Y730• → C439. In this scenario the Y730• → C439 would require protonation of the Y730-O•. The H-bonding environment of the Y730-O• has been examined by electron nuclear double resonance (ENDOR)53,54 and hyperfine sublevel correlation spectroscopy (HYSCORE), and contains H-bonds from both Y731 and C439 as well as a water molecule observed in several X-ray structures.55,56 Current evidence points to water as a proton donor during reduction of Y356,54 and thus this is a plausible alternative proton donor to Y730 during radical transfer to U439.
Figure 6.

Proposed reaction sequences for C439U (top) and C225U (bottom) α2.
When cysteine-based thiols are involved in enzymatic catalysis they often serve as electron donors or nucleophiles. In such instances, the substitution for selenocysteine may impact the reaction outcome, significantly affecting some reactions through the “seleno effect.”57 This effect, measured as ks/kse for the reaction of interest, can be significantly greater than or less than 1, depending on the reaction mechanism and role of the particular chalcogen. For the catalytic C/U at position 439 of 0α2 the seleno effect appears to be quite large , rendering the reaction mute in the seleno form. This is anticipated to be a conserved effect for all thiyl radical enzymes that involve C-H activation, including RNR and most GREs. Conversely, during substrate radical reduction (Figure 1C, steps ii-iv), the bottom-face cysteines serve to reduce the substrate. Given the higher nucleophilicity of U relative to C, and the lower reduction potential of the mixed selenosulfide relative to the canonical disulfide, it may be expected here that the seleno effect of the overall reaction for C225U would be low . Kinetic analysis of the seleno effect in this instance is impossible due to the conformational gating of the overall reaction,58 and the rapid active site chemistry,59 but we do observe substrate reduction at the earliest timepoints examined, suggesting that the ks/kse is not significantly raised.
The proton inventory during substrate radical reduction is essential to proper orchestration of radical chemistry at the active site. This has been examined in several ways including active site mutations that inhibit sidechain ionization (C225S, C462S, E441Q)9,60,61 and mechanism-based inhibitor substrates whose leaving groups are not protonated upon release (2′-deoxy-2′-X, X = Cl or F). In the latter case, the substrate radical can lose the 2′-X as X−, whereas the native substrate loses the 2′-OH as H2O, taking a proton of the active site from C225. Based on the pKa of selenocysteine (5.2–5.4), it is anticipated that the U225 sidechain is deprotonated, absent significant pKa perturbations at this site due to the protein environment. This would render U225 unable to protonate the 2′-OH and alter the proton inventory. Thus, with mechanism-based inhibitors, the proton inventory is too large, whereas in the C225U protein, it is too small. The selenate is a better reductant than the corresponding thiolate, and thus it may be expected to reduce the substrate radical, but can only reduce the substrate by ET rather than PCET. Product analysis of the reaction of C225U with CDP, ATP, and wt β2 shows that dCDP is indeed produced, but that the vast majority of substrate decomposes from the radical/3-keto state, producing cytosine. The small fraction of dCDP formed in the single turnover reaction of C225U could be due to a small fraction of selenol (seH) that can donate an H-atom during reduction of the 2′ radical, or it could be due to a small quantity of contaminating wt protein, and at this point we cannot disentangle these two possibilities. SDS-PAGE analysis of the resultant protein indicated no cleavage of the C225U protein following the reaction (data not shown), which is distinct behavior relative to the C225S protein, which is cleaved near the 225 site during the reaction with wt β2, substrate and effector.13 Furthermore, EPR analysis of the reaction mixture following single turnover preparations of C225U α2 showed little radical loss over the course of the reaction. Collectively, these data suggest that the substrate radical generated by H-atom abstraction by the C439 thiyl radical is not reduced by the U225 in the absence of a selenol, but rather by the top-face C439, resulting in a furanone that decomposes to release cytosine, 2-methylene-3(2H)-furanone, and pyrophosphate, leaving the protein based radical intact (Figure 6). Inhibition on the longer timescale is caused by alkylation of the active site or C-terminal cysteines. The analogous reaction products produced by wt α2 with mechanism-based inhibitors (excess H+) and the C225U 0α2 with native substrates (deficient H+) illustrate the essential role of proton coupling in PCET during radical-based active site catalysis.
Conclusions
The results presented herein have established a facile method for site specifically substituting redox active cysteines for selenocysteine. They also reveal for the first time the seleno effect on thiyl radical chemistry in a thiyl radical enzyme and render parallels between observations made on the thiyl radical reactivity of the class II RNRs with those of the class I enzymes, relevant to human health and common bacterial infections. The successful generation and analysis of these selenoproteins suggest that this technique may be broadly useful for interpreting reactivity trends in the diverse GREs, which can cleave C-C, C-N, C-O, C-S, and C-P bonds through diverse mechanisms that are challenging to examine by the conventional techniques of biochemistry and biophysics.
Supplementary Material
ACKNOWLEDGMENT
We gratefully acknowledge Dr. Ana Crnković and Prof. Dieter Söll for providing the pSec-EVOL2 plasmid, ME6 cells, and generous insight and advice. This work was supported by the NIH under grant numbers GM047274 (DGN) and GM029595 (JS).
ABBREVIATIONS
- RNR
Ribonucleotide reductase
- U
selenocysteine
- C
cysteine
- PCET
proton-coupled electron transfer
- Y
tyrosine
- W
tryptophan
- G
glycine
- RT
radical transfer
- AdoCbl
adenosylcob(II)alamin
- RFQ
rapid freeze quench
- EPR
electron paramagnetic resonance
- RTPR
ribonucleotide triphosphate reductase
- ATP
adenosine triphosphate
- MBI
mechanism-based inhibitor
- PT
proton transfer
- SDM
site-directed mutagenesis
- E. coli
Escherichia coli
- CDP
cytosine-5′-diphosphate
- EDTA
ethylenediaminetetraaceticacid
- ENDOR
electron nuclear double resonance
- GdHCl
guanidinium hydrochloride
- HEPES
(4-(2-hydroxy-ethyl)-1-piperazineethanesulfonic acid
- HYSCORE
hyperfine sublevel correlation
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- AP
calf intestine alkaline phosphatase
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- DTT
dithiothreitol
- Tr
E. coli thioredoxin
- TrR
E. coli thioredoxin reductase
- PCR
polymerase chain reaction
- SDS PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- ICP-MS
inductively coupled plasma mass spectrometry
- HPLC
high performance liquid chromatography
- SF
stopped-flow
- TA
transient absorption
- EF-Tu
elongation factor Tu
- SECIS
selenocysteine insertion sequence
- serRS
serine tRNA synthetase
- [ReI]
rhenium (I) 1,10-phenanthroline tricarbonyl bromomethylpyridine chromophore
- NH2Y
3-aminotyrosine
- GREs
glycyl radical enzymes
- BDE
bond dissociation energy
Footnotes
ASSOCIATED CONTENT
Supporting Information
SDS-PAGE analysis of selenoprotein expression and purification, selenium quantitation, activity measurements, HPLC standards, EPR spectrum of C439S/C439U α2 and wt β2 with N3CDP, SF UV-vis kinetics of C439U α2, SF UV-vis kinetics of wt α2, mechanism of RNR inhibition by 2’-F/Cl substituted nucleotides, EPR spectrum of C225U α2 and wt β2 with CDP, mechanism of RNR inhibition by 2′-N3 substituted nucleotides. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Stubbe J, and van der Donk WA (1998) Protein radicals in enzyme catalysis. Chem. Rev 98, 705–762. [DOI] [PubMed] [Google Scholar]
- (2).Nauser T, Dockheer S, Kissner R, and Koppenol WH (2006) Catalysis of Electron Transfer by Selenocysteine. Biochemistry 45, 6038–6043. [DOI] [PubMed] [Google Scholar]
- (3).Schöneich C (2011) Cysteine residues as catalysts for covalent peptide and protein modification: a role for thiyl radicals? Biochem. Soc. T 39, 1254–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Everett SA, Schöneich C, Stewart JH, and Asmus KD (1992) Perthiyl Radicals, Trisulfide Radical Ions, and Sulfate Formation. A Combined Photolysis and Radiolysis Study on Redox Processes with Organic Di- and Trisulfides. J. Phys. Chem 96, 306–314. [Google Scholar]
- (5).van Gastel M, Lubitz W, Lassmann G, and Neese F (2004) Electronic Structure of the Cysteine Thiyl Radical: A DFT and Correlated ab Initio Study. J. Am. Chem. Soc 126, 2237–2246. [DOI] [PubMed] [Google Scholar]
- (6).Reichard P (1993) From RNA to DNA, why so many ribonucleotide reductases? Science 260, 1773–1777. [DOI] [PubMed] [Google Scholar]
- (7).Stubbe JA, and van der Donk WA (1995) Ribonucleotide reductases: radical enzymes with suicidal tendencies. Chem. Biol 2, 793–801. [DOI] [PubMed] [Google Scholar]
- (8).Aberg A, Hahne S, Karlsson M, Larsson A, Ormö M, Ahgren A, and Sjöberg BM (1989) Evidence for two different classes of redox-active cysteines in ribonucleotide reductase of Escherichia coli. J. Biol. Chem 264, 12249–12252. [PubMed] [Google Scholar]
- (9).Mao SS, Holler TP, Yu GX, Bollinger JM Jr., Booker S, Johnston MI, and Stubbe J (1992) A Model for the Role of Multiple Cysteine Residues Involved in Ribonucleotide Reduction: Amazing and Still Confusing. Biochemistry 31, 9733–9743. [DOI] [PubMed] [Google Scholar]
- (10).Sintchak MD, Arjara G, Kellogg BA, Stubbe J, and Drennan CL (2002) The crystal structure of class II ribonucleotide reductase reveals how an allosterically regulated monomer mimics a dimer. Nat. Struct. Biol 9, 293–300. [DOI] [PubMed] [Google Scholar]
- (11).Harris G, Ator M, and Stubbe J (1984) Mechanism of Inactivation of Escherichia coli and Lactobacillus leichmannii Ribonucleotide Reductases by 2′-Chloro-2′-deoxynucleotides: Evidence for Generation of 2-Methylene-3(2H)-furanone. Biochemistry 23, 5214–5225. [DOI] [PubMed] [Google Scholar]
- (12).Thelander L, and Larsson B (1976) Active site of ribonucleoside diphosphate reductase from Escherichia coli. Inactivation of the enzyme by 2′-substituted ribonucleoside diphosphates. J. Biol. Chem 251, 1398–1405. [PubMed] [Google Scholar]
- (13).Mao SS, Holler TP, Bollinger MJ Jr., Yu GX, Johnston MI, and Stubbe J (1992) Interaction of C225SR1 Mutant Subunit of Ribonucleotide Reductase with R2 and Nucleoside Diphosphates: Tales of a Suicidal Enzyme. Biochemistry 31, 9744–9751. [DOI] [PubMed] [Google Scholar]
- (14).Licht S, Gerfen GJ, and Stubbe JA (1996) Thiyl Radicals in Ribonucleotide Reductases. Science 271, 477–481. [DOI] [PubMed] [Google Scholar]
- (15).Gerfen GJ, Licht S, Willems JP, Hoffman BM, and Stubbe J (1996) Electron Paramagnetic Resonance Investigations of a Kinetically Competent Intermediate Formed in Ribonucleotide Reduction: Evidence for a Thiyl Radical-Cob(II)alamin Interaction. J. Am. Chem. Soc 118, 8192–8197. [Google Scholar]
- (16).Lawrence CC, Bennati M, Obias HV, Bar G, Griffin RG, and Stubbe J (1999) High-field EPR detection of a disulfide radical anion in the reduction of cytidine 5′-diphosphate by the E441Q R1 mutant of Escherichia coli ribonucleotide reductase. Proc. Natl. Acad. Sci. U.S.A 96, 8979–8984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Steinmann D, Nauser T, and Koppenol WH (2006) Kinetics of tyrosyl radical repair by selenocysteine. Free Rad. Res 40, S136–S136. [Google Scholar]
- (18).Mukai T, Sevostyanova A, Suzuki T, Fu X, and Söll D (2018) A facile method for producing selenocysteine-containing proteins. Angew. Chem. Int. Ed 57, 7215–7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Minnihan EC, Nocera DG, and Stubbe J (2013) Reversible, Long-Range Radical Transfer in E. coli Class la Ribonucleotide Reductase. Acc. Chem. Res 46, 2524–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Greene BL, Stubbe J, and Nocera DG (2018) Photochemical Rescue of a Conformationally Inactivated Ribonucleotide Reductase. J. Am. Chem. Soc 140, 15744–15752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Greene BL, Taguchi AT, Stubbe J, and Nocera DG (2017) Conformationally Dynamic Radical Transfer within Ribonucleotide Reductase. J. Am. Chem. Soc 139, 16657–16665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Olshansky L, Stubbe J, and Nocera DG (2016) Charge-Transfer Dynamics at the α/β Subunit Interface of a Photochemical Ribonucleotide Reductase, J. Am. Chem. Soc 138, 1196–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, and Smith HO (2009) Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Meth 6, 343–345. [DOI] [PubMed] [Google Scholar]
- (24).Steeper JR, and Steuart CD (1970) A rapid assay for CDP reductase activity in mammalian cell extracts. Anal. Biochem 34, 123–130. [DOI] [PubMed] [Google Scholar]
- (25).Pizano AA, Olshansky L, Holder PG, Stubbe J, and Nocera DG (2013) Modulation of Y356 Photooxidation in E. coli Class la Ribonucleotide Reductase by Y731 Across the α2:β2 Interface. J. Am. Chem. Soc 135, 13250–13253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Minnihan EC, Seyedsayamdost MR, Uhlin U, and Stubbe J (2011) Kinetics of Radical Intermediate Formation and Deoxynucleotide Production in 3-Aminotyrosine-Substituted Escherichia coli Ribonucleotide Reductases. J. Am. Chem. Soc 133, 9430–9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Nelson DJ, Petersen RL, and Symons MCR (1977) Unstable Intermediates. Part 178. The Structure of Intermediates Formed in the Radiolysis of Thiols. J. Chem. Soc. Perk. Trans 2, 2005–2015. [Google Scholar]
- (28).Budzinski EE, and Box HC (1971) Oxidation and Reduction of Organic Compounds by Ionizing Radiation: L-Penicillamine Hydrochloride. J. Phys. Chem 75, 2564–2570. [DOI] [PubMed] [Google Scholar]
- (29).Kennedy MC, Antholine WE, and Beinert H (1997) An EPR Investigation of the Products of the Reaction of Cytosolic and Mitochondrial Aconitases with Nitric Oxide. J. Biol. Chem 272, 20340–20347. [DOI] [PubMed] [Google Scholar]
- (30).Kolberg M, Bleifuss G, Gräslund A, Sjöberg BM, Lubitz W, Lendzian F, and Lassmann G (2002) Protein thiyl radicals directly observed by EPR spectroscopy. Arch. Biochem. Biophys 403, 141–144. [DOI] [PubMed] [Google Scholar]
- (31).van der Donk WA, Stubbe J, Gerfen GJ, Bellew BF, and Griffin RG (1995) EPR Investigations of the Inactivation of Escherichia coli Ribonucleotide Reductase with 2′-Azido-2′-Deoxyuridine 5′-Diphosphate: Evidence for the Involvement of the Thiyl Radical of C225-R1. J. Am. Chem. Soc 117, 8908–8916. [Google Scholar]
- (32).Kasanmascheff M, Lee W, Nick TU, Stubbe J, and Bennati M (2016) Radical transfer in E. coli ribonucleotide reductase: a NH2Y731/R411A-α mutant unmasks a new conformation of the pathway residue 731. Chem. Sci 7, 2170–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bollinger JM Jr., Hangtong W, Ravi N, Huynh BH, Edmondson D, and Stubbe J (1995) Use of rapid kinetics methods to study the assembly of the diferric-tyrosyl radical cofactor of E. coli ribonucleotide reductase. Method. Enzymol 258, 278–303. [DOI] [PubMed] [Google Scholar]
- (34).Pizano AA, Lutterman DA, Holder PG, Teets TS, Stubbe J, and Nocera DG (2012) Photo-ribonucleotide reductase β2 by selective cysteine labeling with a radical phototrigger. Proc. Natl. Acad. Sci. U.S.A 109, 39–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Ravichandran KR, Zong AB, Taguchi AT, Nocera DG, Stubbe J and Tommos C (2017) Formal Reduction Potentials of Difluorotyrosine and Trifluorotyrosine Protein Residues: Defining the Thermodynamics of Multistep Radical Transfer. J. Am. Chem. Soc 139, 2994–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Thelander L, Larsson B, Hobbs J, and Eckstein F (1976) Active Site of Ribonucleoside Diphosphate Reductase from Escherichia coli. J. Biol. Chem 251, 1398–1405. [PubMed] [Google Scholar]
- (37).Stubbe J and Kozarich JW (1980) Inorganic Pyrophosphate Is Released from 2′-Chloro-2′-deoxyuridine 5′-Diphosphate by Ribonucleoside Diphosphate Reductase. J. Am. Chem. Soc 102, 2505–2507. [Google Scholar]
- (38).Stubbe J and Kozarich JW (1980) Fluoride, Pyrophosphate, and Base Release from Z′-Deoxy-Z′-fluoronucleoside 5′-Diphosphates by Ribonucleo-side-Diphosphate Reductase. J. Biol. Chem 255, 5511–5513. [PubMed] [Google Scholar]
- (39).Backman LRF, Funk MA, Dawson CD, and Drennan CL (2017) New tricks for the glycyl radical enzyme family. Crit. Rev. Biochem. Mol 52, 674–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Selmer T, Pierik AJ, and Heider J (2005) New glycyl radical enzymes catalysing key metabolic steps in anaerobic bacteria. Biol. Chem 386, 981–988. [DOI] [PubMed] [Google Scholar]
- (41).Levin BJ, Huang YY, Peck SC, Wei Y Martinez-del Campo A, Marks JA, Franzosa EA, Huttenhower C, and Balskus EP (2017) A prominent glycyl radical enzyme in human gut microbiomes metabolizes trans-4-hydroxy-L-proline. Science 355, eaai8386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Koppel N, Rekdal VM, and Balskus EP (2017) Chemical transformation of xenobiotics by the human gut microbiota. Science 356, eaag2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, Todd GC, and Grindey GB (1990) Evaluation of the antitumoractivity of gemcitabine (2′ ,2′-difluoro-2’-deoxycytidine). Cancer Res. 50, 4417–4422. [PubMed] [Google Scholar]
- (44).Wang J, Lohman GJS, and Stubbe J (2009) Mechanism of Inactivation of Human Ribonucleotide Reductase with p53R2 by Gemcitabine 5′-Diphosphate. Biochemistry 48, 11612–11621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Reich HJ and Hondal RJ (2016) Why Nature Chose Selenium. ACS Chem. Biol 11, 821–841. [DOI] [PubMed] [Google Scholar]
- (46).McMillen DF and Golden DM (1982) Hydrocarbon bond dissociation energies. Annu. Rev. Phys. Chem 33, 493–532. [Google Scholar]
- (47).Mozziconacci O, Kerwin BA, and Schöneich C (2010) Reversible intramolecular hydrogen transfer between protein cysteine thiyl radicals and αC-H bonds in insulin: control of selectivity by secondary structure. J. Phys. Chem. B 112, 15921–15932. [DOI] [PubMed] [Google Scholar]
- (48).Rauk A, Yu D, Taylor J, Shustov GV, Block DA, and Armstrong DA. (1999) Effects of structure on αC-H bond enthalpies of amino acid residues: relevance to H transfers in enzyme mechanisms and protein oxidation. Biochemistry 38, 9089–9096. [DOI] [PubMed] [Google Scholar]
- (49).Dang H-S, Roberts BP and Tocher DA (2001) Selective radical-chain epimerization at electron-rich chiral tertiary C-H centres using thiols as protic polarity reversal catalysts. J. Chem. Soc. Perkin Trans 1, 2452–2461. [Google Scholar]
- (50).Reid DL, Shustov GV, Armstrong DA., Rauk A, Schuchmann MN, Akhlaq S and von Sonntag C (2002) H-atom abstraction from thiols by C-centered radicals: a theoretical and experimental study of reaction rates. Phys. Chem. Chem. Phys 4, 2965–2974. [Google Scholar]
- (51).Becker A, Fritz-Wolf K, Kabsch W, Knappe J, Schultz S, and Volker Wagner AF (1999) Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat. Struct. Biol 6, 969–975. [DOI] [PubMed] [Google Scholar]
- (52).Becker A and Kabsch W (2002) X-ray structure of pyruvate formate-lyase in complex with pyruvate and CoA. How the enzyme uses the Cys-418 thiyl radical for pyruvate cleavage. J. Biol. Chem 277, 40036–40042. [DOI] [PubMed] [Google Scholar]
- (53).Argirevic T, Riplinger C, Stubbe J, Neese F, Bennati M (2012) ENDOR Spectroscopy and DFT Calculations: Evidence for the Hydrogen-Bond Network Within α2 in the PCET of E. coli Ribonucleotide Reductase. J. Am. Chem. Soc 134, 17661–17670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Nick TU, Lee W, Koßmann S, Neese F, Stubbe J, and Bennati M (2015) Hydrogen Bond Network between Amino Acid Radical Intermediates on the Proton-Coupled Electron Transfer Pathway of E. coli α2 Ribonucleotide Reductase. J. Am. Chem. Soc 137, 289–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Uhlin U and Ecklund H (1994) Structure of ribonucleotide reductase protein R1. Nature 370, 533–539. [DOI] [PubMed] [Google Scholar]
- (56).Yokoyama K, Uhlin U, and Stubbe J (2010) Site-Specific Incorporation of 3-Nitrotyrosine as a Probe of pKa Perturbation of Redox-Active Tyrosines in Ribonucleotide Reductase. J. Am. Chem. Soc 132, 8385–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).O’Keefe JP, Dustin CM, Barber D, Snider GW, and Holand RJ (2018) A “Seleno Effect” Differentiates the Roles of Redox Active Cysteine Residues in Plasmodium falciparum Thioredoxin Reductase. Biochemistry 57, 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ge J, Yu G, Ator MA, and Stubbe J (2003) Pre-Steady-State and Steady-State Kinetic Analysis of E. coli Class I Ribonucleotide Reductase. Biochemistry 42, 10071–10083. [DOI] [PubMed] [Google Scholar]
- (59).Licht S and Stubbe J (1999) Mechanistic Investigations of Ribonucleotide Reductase. Comp. Nat. Prod. Chem 5, 163–203. [Google Scholar]
- (60).van der Donk W, Yu G, Silva DJ, Stubbe J, McCarthy JR, Jarvi ET, Matthews DP, Resvick RJ, and Wagner E (1996) Inactivation of Ribonucleotide Reductase by (E)-2′-Fluoromethylene-2′-deoxycytidine 5’-Diphosphate: A Paradigm for Nucleotide Mechanism-Based Inhibitors. Biochemistry 35, 8381–8391. [DOI] [PubMed] [Google Scholar]
- (61).Persson AL, Eriksson M, Katterle B, Pötsch S, Sahlin M, and Sjöberg BM (1997) A New Mechanism-based Radical Intermediate in a Mutant R1 Protein Affecting the Catalytically Essential Glu441 in Escherichia coli Ribonucleotide Reductase. J. Biol. Chem 272, 31533–31541. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.