

Abstract
New drugs are desperately needed to combat methicillin-resistant Staphylococcus aureus (MRSA) infections. Here we report screening commercial kinase inhibitors for anti-bacterial activity and found the anti-cancer drug sorafenib as major hit effectively killing MRSA strains. Varying the key structural features led to the identification of a potent analogue, PK150, that showed anti-bacterial activity against several pathogenic strains at sub-micromolar concentrations. Furthermore, this antibiotic eliminated challenging persisters as well as established biofilms. PK150 holds promising therapeutic potential as it did not induce in vitro resistance, shows oral bioavailability and in vivo efficacy. Analysis of the mode of action using chemical proteomics revealed several targets, including interference with menaquinone biosynthesis by inhibiting demethylmenaquinone methyltransferase and stimulation of protein secretion by altering the activity of signal peptidase IB. Reduced endogenous menaquinone levels along with enhanced levels of extracellular proteins of PK150-treated bacteria support this target hypothesis. The associated antibiotic effects, especially the lack of resistance development, likely stem from the compound’s polypharmacology.
Current treatment of bacterial infections is challenged by a dramatic increase of multidrug-resistant strains.1 In addition to infections caused by Gram-negative multidrug-resistant (MDR) pathogens, Gram-positive pathogens, such as Staphylococcus aureus, remain among the top causes of healthcare-associated infections with high priority in drug development.2 Especially methicillin-resistant Staphylococcus aureus (MRSA) has become difficult to treat and causes severe infections.3 Adding to the challenge is a phenomenon termed persistence, by which a sub-population of bacteria switch into a dormant state and thereby become tolerant towards antibiotics.4 These persister cells can reinitiate growth after termination of antibiotic therapy, causing a relapse of the infection and failure of treatment.4 Moreover, many antibiotics lack the ability to eradicate established biofilms, which are multicellular surface-bound bacterial communities largely composed of bacteria in a dormant persister state.
Although there have been some recent discoveries based on natural products, e.g. teixobactin,5 acyldepsipeptides6 and arylomycins,7 the scope of targets for antibiotics is still extremely narrow. Almost all marketed antibiotics target cell-wall biosynthesis, membrane integrity, DNA synthesis or protein-biosynthesis, all of which have already been counteracted via numerous resistance mechanisms. Considering the intricate cellular processes regulating virulence and viability, a large number of bacterial proteins awaits exploitation as potential antibacterial targets. Research into drugs directed against essential bacterial kinases is still in its infancy8 and a wealth of eukaryotic kinase inhibitors, originally developed to fight cancer, awaits bacterial testing and putative drug repurposing.9 Recently, sorafenib, a marketed anticancer drug (Nexavar), and a few derivatives thereof were shown to be effective against MRSA.10,11 However, at the time, it was uncertain if the bacterial target of sorafenib was indeed a kinase. While sorafenib’s core N,N’-diarylurea motif is also present in other antibacterial compounds, including triclocarban (TCC) - a N,N’-diarylurea that was commonly used as an antimicrobial agent in personal care products - the corresponding modes of action are largely unresolved.12,13
Herein, we present our findings based on mining small molecule kinase inhibitors for activity against S. aureus and confirm sorafenib as one of the most potent hits. Chemical dissection of the sorafenib scaffold by organic synthesis of 72 analogues resulted in a compound (PK150) with 10-fold enhanced anti-MRSA activity, lack of resistance development under laboratory conditions, killing of persisters, elimination of established biofilms, and in vivo efficacy in a mouse model. Chemical proteomic studies did not reveal a known kinase as target, but interference with menaquinone biosynthesis and dysregulation of protein secretion as putative target mechanisms.
Results
Antibacterial screen of kinase inhibitors
To access new antibiotic targets with essential functions in S. aureus physiology we screened a library of 232 commercial kinase inhibitors (Supplementary Data 1) for antibacterial activity against the methicillin-sensitive (MSSA) strain NCTC 8325. Two structurally related compounds, sorafenib (SFN) and regorafenib, exhibited the lowest minimal inhibitory concentrations (MIC) of 3 μM (1.4 μg/mL). The antibiotic scope of SFN was subsequently evaluated against a panel of Gram-positive and Gram-negative bacteria (Fig. 1a, Supplementary Data 2). Importantly, SFN showed activity against antibiotic-sensitive and multidrug-resistant S. aureus reference strains as well as ten clinical MRSA isolates, suggesting that the compound acts via a mechanism unrelated to established resistance pathways. While no activity was detected against Gram-negative bacteria, inhibition of Mycobacterium tuberculosis growth was observed with an MIC of 25 μM (11.6 μg/mL).
Figure 1. Antibacterial properties of SFN and PK150.

(a) Activity spectra of SFN and PK150 against pathogenic bacteria. See also Supplementary Data 2 for details on the specific strains tested.
(b) Time-dependent killing of exponentially growing S. aureus NCTC 8325 by different concentrations of SFN and PK150. Dashed line represents 99.9% of killed bacteria. Data represent average values ± SD (n = 3 per group).
(c) Persister cell assay. S. aureus ATCC 29213 bacteria were incubated with ciprofloxacin (78 μM, 100-fold MIC) for 24 hours to isolate persister cells. Bacterial cells were then washed with PBS, resuspended in PBS + 1% (v/v) MH2 medium, and treated with rifampicin (0.1 μM, 10-fold MIC), PK150 (2.4 μM, 8-fold MIC) or DMSO for 196 h. Data represent average values ± SD (n = 3 per group); data points marked with an asterisk fall below the limit of detection (33 CFU/mL, dashed line): no viable bacteria were detected in these cases.
(d) Eradication of S. aureus ATCC 29213 biofilm after treatment (24 h) with various concentrations of PK150. Data are normalized to DMSO (100% biofilm) and represent average values ± SD (n = 6 per group).
(e) Resistance development during serial passaging in the presence of sub-MIC concentrations of antimicrobials. Ofloxacin (OFL) served as positive control. For SFN, a 20-fold MIC was the highest concentration tested (solubility limit). The figure is representative for three independent experiments.
Chemical dissection of sorafenib results in a compound with enhanced antibiotic potency
To gain a better understanding of the role of key structural features of SFN for its antibiotic properties, we chemically dissected the SFN scaffold in an in-depth structure-activity relationship (SAR) study. In total, 72 analogues were synthesized with 40 compounds bearing systematic modifications in all three parts of SFN, namely the lateral 4-chloro-3-(trifluoromethyl) phenyl, the central urea, and the terminal aryl heteroaryl ether moieties (Table 1). The synthesis of all analogues followed a modular blueprint as depicted in Supplementary Fig. 1 and their structures are summarized in Supplementary Data 1. Analogues in which the lateral 4-chloro-3-(trifluoromethyl) phenyl moiety was replaced by diverse aliphatic or aromatic groups resulted in inactive molecules. Even minor alterations, such as the removal of substituents (1-134, 1-163, SFN-C), abolished antibiotic activities. In addition, modifications of the central urea motif were restricted (3-001). In contrast, the aryl heteroaryl ether motif could be identified as an area for high optimization potential (2-013, 3-004, 3-005, 3-006). Importantly, a 2,2-difluoro-1,3-benzodioxole analogue (PK150) exhibited an MIC of 0.3 μM (118 ng/mL) and thus revealed the highest antibacterial potency among all compounds. This structural motif appears to be crucial as removal of the fluorine substituents (1-159) or opening of the acetal resulting in a dimethoxy analogue (1-160) was associated with a significant drop in activity. Changing the regiochemistry by placement of the acetal moiety more proximal to the urea motif (1-164) led to a slight decrease in activity. Again, removal of substituents of the 4-chloro-3-(trifluoromethyl) phenyl moiety (PK150-C) abolished the antibiotic effect highlighting the central role of this structural motif.
Table 1.
Representative derivatives of SFN with their corresponding antibacterial activities (MIC against S. aureus NCTC 8325), shift of activity in the presence of menaquinone-4 (MK-4; 100 μg/mL), and SpsB activation potential. Data are derived from three biologically independent experiments performed in triplicates and SpsB activity is given as mean value ± SD. A full list of compounds and corresponding activities can be found in Supplementary Data 1 and MIC shifts of PK150, SFN and 1-164 at different concentrations of MK-4 are listed in Supplementary Data 2.
| MIC (μM) |
MIC at 100 μg/mL MK-4 (μM) |
SpsB activity (normalized) at 50 μM |
||
|---|---|---|---|---|
| SFN | 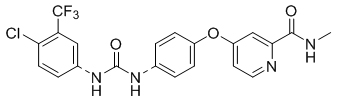 |
3 | 20 | 1.64 ± 0.12 |
| 1-134 | 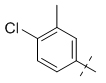 |
> 100 | n.a. | 1.12 ± 0.08 |
| 1-163 | 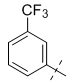 |
> 100 | n.a. | 1.33 ± 0.20 |
| SFN-C | 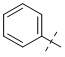 |
> 100 | n.a. | 1.18 ± 0.16 |
| 3-001 | 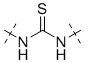 |
10 | 30 | 1.58 ± 0.28 |
| 2-013 | 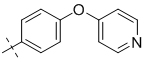 |
3 | 7 | 1.91 ± 0.25 |
| 3-004 | 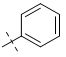 |
3 | >100 | 1.49 ± 0.16 |
| 3-005 | 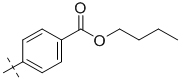 |
0.6 | 3-5 | 2.12 ± 0.25 |
| 3-006 | 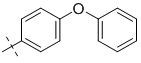 |
0.5 | 5 | 2.10 ± 0.14 |
| PK150 | 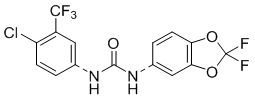 |
0.3 | 1-3 | 2.54 ± 0.39 |
| PK150-C | 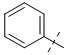 |
> 100 | n.a. | 1.15 ± 0.01 |
| 1-159 | 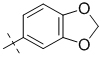 |
10 | >100 | 1.40 ± 0.01 |
| 1-160 | 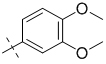 |
30 | 50 | 1.64 ± 0.16 |
| 1-164 | 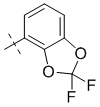 |
0.5 | 3 | 1.42 ± 0.08 |
PK150 was more potent against S. aureus NCTC 8325 compared to vancomycin (MIC 1 μM; 1.4 μg/mL) and linezolid (MIC 3 μM; 1.0 μg/mL) (Supplementary Data 2). In addition, activity against the panel of MRSA strains was retained and, in contrast to SFN, also killing of vancomycin-resistant enterococci (VRE) was observed (MIC 3 μM; 1.0 μg/mL; Fig. 1a, Supplementary Data 2). The compound was active against mycobacteria, including M. tuberculosis with an MIC of 2 μM (0.93 μg/mL), but inactive against all tested Gram-negative bacteria (Fig. 1a, Supplementary Data 2).
PK150 rapidly kills exponentially growing S. aureus, largely reduces persisters and established biofilms, and does not induce in vitro resistance
In-depth evaluation of biological properties revealed a bactericidal effect of PK150 and SFN against exponentially growing MSSA cells (Fig. 1b, Supplementary Fig. 2). In line with this observation, particularly PK150 disintegrated cells in both a concentration- and time-dependent manner as shown by enhanced membrane permeability (Supplementary Fig. 3). Moreover, the amount of persister cells generated from stationary S. aureus cultures by ciprofloxacin treatment was effectively reduced in the presence of PK150. Unlike rifampicin,14 PK150 fully eradicated persister cells (Fig. 1c). Persister killing was confirmed in an independent assay format, by which cells at different optical densities were treated with PK150, SFN, inactive controls and ciprofloxacin or with a combination of these compounds with oxacillin.15 Again, only PK150 and SFN significantly reduced CFUs of bacterial cultures by several orders of magnitude throughout all experiments (Supplementary Fig. 4). In addition, staphylococcal biofilms, largely composed of persisters,16 were effectively eradicated upon PK150-treatment (80% reduction in biofilm after 24 h treatment), whereas the antibiotic vancomycin did not significantly affect biofilm integrity (Fig. 1d, Supplementary Fig. 5).
Given these properties, we next tested resistance development against SFN and PK150. S. aureus cells were repeatedly passaged in the presence of different compound concentrations (0.25-4-fold MIC) for 27 days. While control antibiotic ofloxacin and SFN exhibited a rapid drop in antibacterial activity after a few passages, PK150 remained active over the complete course of the study (Fig. 1e, Supplementary Fig. 6). Even addition of the mutagen ethyl methanesulfonate17 did not trigger resistance development in an agar-plate-based assay format.
Chemical proteomics reveal cellular targets with putative roles in the antibiotic mechanism
Next, we performed target identification studies to understand the mode of action of SFN and PK150, respectively, by chemical proteomics. The core scaffolds of SFN and PK150 were synthetically equipped with a diazirine photocrosslinker and an alkyne tag (Supplementary Fig. 7) required for affinity-based protein profiling (AfBPP) (Fig. 2a).18,19 Satisfyingly, the resulting photoprobes SFN-P and 3-005-P (due to the restricted SAR of PK150 derived from the closest analogue 3-005) exhibited only a slight increase in MIC of about 3-fold compared to SFN and PK150, respectively (Supplementary Data 1). SFN-P was used to establish optimal labelling conditions: Intact S. aureus cells were incubated with the photoprobe, then irradiated with UV-light to form a covalent linkage with the target protein, lysed and labelled proteins clicked to rhodamine-biotin azide. Gel-based analysis revealed an optimal concentration of 10-50 μM in both the soluble and insoluble fraction (Fig. 2b, Supplementary Fig. 8). To unravel the identity of targeted proteins, we performed a quantitative, gel-free AfBPP analysis by conjugation to rhodamine-biotin azide, enrichment on avidin beads, and mass-spectrometric (LC-MS/MS) analysis (Fig. 2c). To exclude unspecific binding events, three control experiments were performed: i) incubation with DMSO to account for unspecific avidin binding, ii) competitive labelling in the presence of an excess of SFN to confirm that SFN-P and SFN address the same binding site, and iii) comparison to a small portfolio of minimal photocrosslinker-probes (DA-1, DA-2 or DA-3)20 to determine background binding. Tryptic peptides of each experiment were labelled by stable dimethyl isotopes21 and isotope ratios of detected peptides were statistically evaluated (student’s t-test) for enrichment by the photoprobe, competition with parent SFN, and background photocrosslinker binding (Figs. 2c, 2d, 2g, Supplementary Figs. 9 and 10, Supplementary Data 3).
Figure 2. Target identification by chemical proteomic profiling in S. aureus.

(a) Schematic experimental workflow for target identification by affinity-based protein profiling (AfBPP). Intact cells are treated with probe or DMSO (as control), UV-irradiated, lysed and labelled proteins clicked to rhodamine-biotin-azide (Note that the probe is capable of binding extracellular as well as intracellular targets). Following enrichment on avidin beads, proteins are enzymatically digested and finally appended to either light (L) or heavy (H) isotopes via dimethyl labelling. Ratios of H/L peptides are determined via subsequent LC-MS/MS measurements.
(b) Fluorescence SDS-PAGE showing labelling of S. aureus NCTC 8325 cells with SFN-P (50 μM) and competition with 10-fold excess of SFN (0.5 mM) before enrichment on avidin beads. The gel is representative for three biologically independent experiments.
(c and d) AfBPP experiments using SFN-P. Volcano plots show log2 fold enrichment of proteins in the soluble fraction after treatment of S. aureus NCTC 8325 cells with SFN-P (50 μM) compared to DMSO (c) or to pretreatment with the competitor SFN (500 μM; d). Dashed lines represent a log2 enrichment ratio of 3 and a –log10 p-value of 2 (two-sided one sample t-test over normalized protein ratios), respectively. Benjamini-Hochberg significance threshold is at a –log10 p-value of 1.7 for labelling and 3.3 for competition studies, respectively (FDR of 0.05). The red dot represents the essential protein SpsB. Blue dots represent proteins for which respective transposon mutants were tested for MIC shifts of SFN and PK150 in comparison to the MIC in the wild-type; no MIC shifts were observed. The grey dot represents lytic regulatory protein (LrP) whose transposon mutant has shown a shift in MIC to 10 μM (SFN) and 0.3-1 μM (PK150); for transposon mutants tested, see Supplementary Data 2. Data represent average values; n = 3 biologically independent experiments performed in triplicates.
(e) Fluorescence SDS-PAGE showing labelling of S. aureus NCTC 8325 cells with 3-005-P (5 μM) after enrichment on avidin beads. The gel is representative for three biologically independent experiments.
(f) AfBPP experiment using 3-005-P. Volcano plot shows log2 fold enrichment of proteins after treatment of S. aureus NCTC 8325 cells with 3-005-P (5 μM) compared to DMSO in a label-free proteomic profiling experiment. Soluble and insoluble proteins were not separated and measured together. Dashed lines represent a log2 enrichment ratio of 3 and a –log10 p-value of 2 (two-sided two sample t-test over LFQ intensities), respectively. Blue dots represent proteins whose respective transposon mutants were tested and did not exhibit an MIC shift against SFN and PK150. The grey, red, and purple dot denote LrP, SpsB, and demethylmenaquinone methyltransferase MenG, respectively. Data represent average values from three biologically independent experiments performed in triplicates.
(g) Table allocating the six major protein hits from AfBPP experiments with both photoprobes SFN-P and 3-005-P to the volcano plots in panels (c), (d), (f) and Supplementary Fig. 9.
Only one protein, signal peptidase IB (SpsB, Q2FZT7), an essential membrane serine-endopeptidase involved in the S. aureus protein secretion (Sec) pathway,22 consistently exhibited high enrichment ratios (log2-fold > 3) and confidences (p < 0.01) in probe labelling and competition studies and at least 8-fold less enrichment in background photocrosslinking (Figs. 2c and 2d, Supplementary Figs. 9 and 10, red dot).
We continued target deconvolution with PK150 derived 3-005-P via quantitative, gel- and label-free AfBPP analysis23 (Figs. 2e and 2f). These studies revealed only minor alterations in the specificity profile compared to labelling with SFN-P. Most importantly, demethylmenaquinone methyltransferase (MenG) pertained as one of the strongest hits that was less enriched with SFN-P (Figs. 2f and 2g, purple dot). MenG catalyses the final step in the biosynthesis of menaquinone, a vitamin crucial for electron transport during bacterial respiration and energy metabolism.24,25
For proteins showing high enrichment ratios in AfBPP experiments with SFN-P (Figs. 2c, 2d, 2g, Supplementary Fig. 9, dots in blue and grey) and/or with 3-005-P (Figs. 2f and 2g), MIC-shifts of the corresponding transposon mutants (Nebraska transposon mutant library)26 were determined upon treatment with SFN and PK150, respectively (Supplementary Data 2). Only a mutant in the gene encoding lytic regulatory protein (LrP, Q2FWA8), a transmembrane protein of unknown function, revealed a slightly decreased susceptibility towards SFN and PK150 (3.3-fold), indicating a contribution to the overall antibiotic mechanism (Supplementary Data 2). No transposons were available for essential SpsB and MenG. In case of SpsB, a direct knockout is not available due to its essential function. However, a deletion strain bearing a resistance-induced ABC transporter that bypasses peptidase-mediated secretion is viable, albeit with impaired fitness.27 In this strain no MIC-shift occurred upon SFN and PK150 treatment, respectively, reinforcing that other target(s) may additionally contribute to the overall mechanism.
PK150 lowers menaquinone levels and stimulates SpsB activity
To evaluate whether SFN and PK150 interfere with menaquinone biosynthesis, endogenous levels were determined in viable S. aureus cells treated with the compounds. Lysis, menaquinone extraction and quantification by LC-MS indeed revealed a reduction upon treatment (Fig. 3a). Moreover, addition of exogenous menaquinone rescued compound-treated S. aureus cells, rendering them less susceptible to SFN and PK150 (Table 1). The MIC shift was dependent on menaquinone concentration and also observed with a variety of bioactive compounds from our diphenyl urea compound library (Table 1, Supplementary Data 2). As we cannot exclude the contribution of other interfering effects, such as aggregation between menaquinone and the compounds, we independently validated these results by measuring inhibition of MenG directly. A radioactive enzymatic assay that allows a sensitive readout of the methylation process catalysed by MenG28 was used for monitoring the cellular fate of the radiolabelled methyl group of cofactor S-adenosyl-l-methionine ([3H]SAM). Cellular lysate of S. aureus NCTC 8325 overexpressing MenG was shown to incorporate significantly less radioactive tritium from [3H]SAM into menaquinone in the presence of PK150 compared to a DMSO-treated sample (Fig. 3b). In addition, methylation of non-naturally occurring demethylmenaquinone-2 by lysates of MenG-overexpressing and wild type S. aureus NCTC 8325 cells was analysed by quantitative LC-MS measurements. Menaquinone-2 synthesis was enhanced in MenG-overexpressing cells and again significantly inhibited by PK150 (Figs. 3c and 3d). Overall, similar results were also observed with 1-164, a regioisomer of PK150 (Figs. 3a, 3b and 3d), whereas SFN caused a weaker inhibition in these assays similar to that observed with structurally related, but antibiotically inactive SFN-C and PK150-C (Fig. 3d). Furthermore, as menaquinone deficiency is likely to influence membrane charge distribution, we measured the membrane potential of S. aureus cells treated with PK150 or with SFN and found both compounds to reduce the potential (see Supplementary Fig. 11 and the Supplementary Discussion for details).
Figure 3. Validation of cellular targets with putative roles in the antibiotic mechanism.

(a) Metabolic profiling of endogenous menaquinone levels in S. aureus NCTC 8325 cells upon compound treatment. Bacteria were treated with sub-inhibitory concentrations (0.1-fold to 0.5-fold the respective MIC) of PK150, 1-164, SFN or vancomycin (VANCO). Menaquinone-8 (MK-8) was extracted by solid phase extraction and quantified by LC-MS, using single ion monitoring and menaquinone-9 as internal standard. MK-8 levels are normalized to DMSO-treated samples. Each colour represents an individual independent extraction experiment, where differently shaped symbols (circles, triangles, diamonds) indicate independent biological samples. Error bars denote mean values ± SD (n = 7 for PK150, SFN; n = 4 for 1-164; n = 6 for VANCO).
(b) Radioactive enzymatic assay monitoring the transfer of the radiolabelled methyl group of S-adenosyl-l-methionine ([3H]SAM) to demethylmenaquinone-2 (DMK-2). Cellular lysate of S. aureus pRMC2-MenG (20 mg/mL total protein concentration) was incubated with [3H]SAM, DMK-2, and different concentrations of PK150, 1-164 or SFN. After 80 min incubation time, menaquinone was extracted with hexane/2-propanol 3:2 and the radioactivity in the organic phase measured by liquid scintillation counting. “No substrate” indicates the background activity in the absence of DMK-2. Values represent mean ± SD of independent replicates (n = 5 for DMSO, 10 μM conditions and no substrate; n = 3 for 2 μM and 20 μM conditions) and are normalized to the DMSO-treated control.
(c and d) Enzymatic assay monitoring the methylation of demethylmenaquinone-2 (DMK-2) by LC-MS. Cellular lysate of either S. aureus pRMC2-MenG or S. aureus NCTC 8325 (20 mg/mL total protein concentration) was treated with S-adenosyl-l-methionine, DMK-2 and different concentrations of PK150, 1-164, SFN or respective controls (SFN-C, PK150-C). After 80 min incubation time, menaquinones were extracted by solid phase extraction and production of menaquinone-2 (MK-2) was quantified by LC-MS, using single ion monitoring (SIM) with menaquinone-4 (MK-4) as internal standard. MenG-overexpressing strain pRMC2-MenG (MenG(+)) produces more MK-2 compared to the wild type NCTC 8325 (WT), as indicated by the total area under the curve (AUC) ± SD, n = 3, of the SIM peak of MK-2 (c). MK-2 quantities were normalized to the respective DMSO-treated samples and represent the averaged values ± SD from three independent experiments (d).
(e) Labelling of recombinant SpsB. Fluorescence gel shows labelling patterns after pretreatment of E. coli BL21(DE3)pLysS cells harbouring pET-55-DEST-SpsB (either induced (+) or not induced (-) for expression of SpsB (MW 24951 Da)) with DMSO (control), SFN or PK150 (500 μM; competition) and subsequent incubation with SFN-P (50 μM). Note that two lanes representing purified SpsB were omitted for clarity and are depicted in Supplementary Fig. 12 together with the corresponding Coomassie stain. The gel is representative for two independent experiments.
(f) Schematic representation of the fluorescence resonance energy transfer (FRET)-assay for measuring the activity of membrane-bound SpsB via the cleavage of a FRET-peptide substrate. The FRET-peptide substrate is based on the signal peptide sequence of Staphylococcus epidermidis SceD preprotein modified with the fluorescent donor 5-((2-aminoethyl)amino)-1-naphthalenesulfonic acid (EDANS) and the quenching acceptor 4-((4-(dimethylamino)phenyl)azo)benzoic acid (DABCYL).
(g) SFN- and PK150-induced concentration-dependent cleavage of the FRET-substrate by membrane-bound SpsB (50 μg/mL total membrane protein concentration). Substrate cleavage rates are normalized to DMSO-treated samples from the induced membranes. Membranes were extracted from E. coli BL21(DE3)pLysS cells harbouring pET-55-DEST-SpsB (either induced (+) or not induced (-) for expression of SpsB). Bars highlight SpsB activity at 10 μM compound concentration relative to DMSO. All data represent mean values ± SD of averaged triplicates of n = 3 biologically independent experiments per group; n.s., not significant, p = 0.2023; *, p = 0.0249; ***, p = 0.00045; ****, p = 1.76 ⋅ 10-5 (two-sided unpaired parametric t-test) for compound- vs. DMSO-treated groups.
We next tested if SFN and PK150 bind to SpsB and if this binding affects peptidase activity. SpsB was cloned, overexpressed in Escherichia coli and labelled by SFN-P in situ (Fig. 3e, Supplementary Fig. 12). Pre-incubation with SFN and PK150 significantly reduced the signal, validating target binding of the parent compound. No competition was observed with PK150-C. SFN-C still reduced the signal, indicating residual binding affinity (Supplementary Fig. 12).
Utilizing a FRET-based assay (Fig. 3f),29,30 E. coli membranes induced for SpsB expression (Fig. 3g) and S. aureus membranes containing native SpsB (Supplementary Fig. 13) were incubated with a fluorogenic SpsB substrate. Substrate turnover was low in E. coli membranes lacking SpsB expression. Surprisingly, addition of SFN resulted in significant stimulation of enzymatic substrate hydrolysis by 27% at 10 μM and up to 32% at 50-100 μM compared to the DMSO control in E. coli membranes (Fig. 3g). Similar results were obtained with S. aureus membranes containing endogenous SpsB (Supplementary Fig. 13). Interestingly, PK150 enhanced substrate turnover even stronger with a 1.7-fold elevated activity compared to SFN in induced E. coli and native S. aureus membranes (corresponding to a maximum of a 2.3-2.9-fold increase over base activity; Fig. 3g, Supplementary Fig. 13). Very limited or no activation was achieved with the structurally related, but antibiotically inactive analogues SFN-C and PK150-C. Activation of SpsB was seen with several antibiotically active members of the library (Table 1, Fig. 4a, Supplementary Fig. 14, Supplementary Data 1). Elevated turnover comparable to the membrane assays was also observed with the enzyme purified from the cytosolic fraction (Supplementary Fig. 13). Further results on the validation of this assay and the activation of SpsB can be found in the Supplementary Discussion and in Supplementary Figs. 15-18.
Figure 4. Structure activity relationship study of SFN and mode of action analysis by chemical proteomics.

(a) Structure activity relationship study with 40 analogues of SFN highlighting the role of various structural key elements for the activation of SpsB (boxplot) as well as antibacterial activity (colour code). Antibacterial activity was tested against S. aureus NCTC 8325; MIC values are represented according to the introduced colour code. SpsB activity was determined by a FRET-based activity assay using S. aureus NCTC 8325 membranes containing endogenous SpsB (0.2 mg/mL total membrane protein concentration; 50 μM compound concentration). Substrate cleavage rates are normalized to DMSO-treated samples. A full list of compounds, structures and activities can be found in Supplementary Data 1.
(b) Secretome analysis. Volcano plots show log2 fold change of protein levels in the secretome after treatment of S. aureus NCTC 8325 cells with PK150 (0.15 μM, 0.5-fold MIC; left panel) or SFN (1.5 μM, 0.5-fold MIC; right panel) compared to DMSO treatment. Dark red dots represent proteins whose secretion has been found to be inhibited by Arylomycin C16 (experimentally proposed SpsB substrates).35 Light red dots represent proteins that are predicted to have a SpsB signal peptide motif (according to UniProtKB as annotated by SignalP).61 Purple circles represent peptidoglycan hydrolase domain-containing proteins (Pfam annotations).60 Data represent average values and p-values were calculated using a two-sided two sample t-test; n = 4 independent experiments per group.
(c) Surfaceome analysis. Volcano plots show log2 fold change of protein levels in the surfaceome after treatment of S. aureus NCTC 8325 cells with PK150 or SFN (0.5-fold MIC) compared to DMSO. Colour coding is equivalent to panel (b). Data represent average values and p-values were calculated using a two-sided two sample t-test; n = 4 independent experiments per group.
We next investigated the molecular basis for SpsB activation by docking together with molecular dynamics (MD) simulations.31 The refined docking pose with the best binding free energy suggested PK150 to be located adjacent to the active site with an average distance of 12 Å between its centre and the Cα atom of the active site residues S36 and K77 (Supplementary Fig. 19). Here, the CF3 alkyl unit of PK150 is predicted to interact predominantly with four non-polar amino acids, namely L41, V47, V64 and V170, which form a hydrophobic surface patch yielding strong hydrophobic interactions (Supplementary Fig. 19). In addition, the urea group of PK150 forms crucial hydrogen bonds with D147 (Supplementary Fig. 19). Supported by SAR data (Fig. 4a), these two invariant structural moieties seem to be hallmarks of PK150 binding into the SpsB groove.
MD simulations were applied to elucidate the origin of activation. Interestingly, PK150 binding initiates changes in the secondary structure including both destabilization as well as stabilization (Supplementary Fig. 19), thereby rigidifying peptide substrate binding areas, the active site Ser containing loop region, as well as residues in direct proximity of the active site dyad (brown, grey and pink boxes, Supplementary Fig. 19).
SFN and PK150 dysregulate protein secretion and induce autolysis
While inhibition of SpsB with arylomycin, which is believed to cause cell death by accumulation of unprocessed proteins, has been studied by proteomic analysis of the bacterial secretome,32–34 little is known about the underlying stimulating effects. In order to understand how activation of SpsB affects the secretome, we analysed extracellular proteins in the presence of SFN and PK150 as well as in the presence of inactive SFN-C and PK150-C (Table 2, Fig. 4b, Supplementary Fig. 20). Secretomes of treated cells were precipitated, digested with trypsin and quantified by label-free MS.23 Statistical analysis of antibiotic-treated cells revealed elevated levels of SpsB-processed proteins in the secretome compared to DMSO- and inactive control compound-treated cells (Fig. 4b, Supplementary Fig. 20, Supplementary Data 4).35
Table 2.
Enrichment analysis of peptidoglycan hydrolase domain-containing proteins (Pfam annotations:60 CHAP domain, LysM domain, amidase, transglycolase, glucosaminidase and peptidase M23 domain) using a two-sided Fisher‘s exact test among proteins that have shown a log2-fold enrichment of > 0.5 in the secretome (full measured secretome of n = 806 total proteins was used as background). Further details in Supplementary Table 1.
| Category | PK150/DMSO | SFN/DMSO | Secretome |
|---|---|---|---|
| Proteins with log2(protein ratio) > 0.5 | |||
| Total proteins | 83 | 121 | 806 |
| „PGH domain AND Signal Predicted/AC16-responder“ proteins | 10 | 9 | 12 |
| Not „PGH domain AND Signal Predicted/AC16-responder“ proteins | 73 | 112 | 794 |
| Fisher Exact p-value (Secretome as background) | 4.47 ⋅ 10-9 | 4.38 ⋅ 10-6 | |
Proteins bound to the membrane surface of SFN- and PK150-treated bacteria were shaved by tryptic digest and analysed via LC-MS/MS. In line with the proposed mechanism of activation, the abundance of membrane-residing SpsB substrate proteins was reduced in SFN- and PK150-treated cells compared to treatment with DMSO or inactive SFN-C and PK150-C, respectively (Fig. 4c, Supplementary Fig. 20).
Bioinformatic analysis of proteins with enhanced secretion upon SpsB stimulation revealed that peptidoglycan hydrolase (PGH) domain-containing proteins (autolysins; Supplementary Data 4) were significantly overrepresented in the secretomes of PK150- and SFN-treated cells (unlike in the inactive control samples; Table 2, Supplementary Table 1). These autolysins break down peptidoglycan (PGN) and facilitate cell division, which is controlled by PGN-bound teichoic acid molecules. In fact, previous studies with inducible autolysin expression vectors revealed that minor imbalances of secreted autolysins result in dysregulation of cell wall biosynthesis and consequently induce rapid cell death.36,37 The extracellular accumulation of PGH domain containing proteins upon antibiotic treatment as seen here (Supplementary Data 4) could likely provoke such imbalances resulting in the induction of autolysis. Interestingly, analysis of autolysin activity via zymography of S. aureus cells treated with SFN and PK150 revealed a characteristic induction of several hydrolytic enzymes in the higher MW range (Supplementary Fig. 21). These bands were also observed with penicillin G. In contrast, no such induction occurred upon treatment with inactive SFN-C, PK150-C and DMSO. Taken together, these results indicate that SFN and PK150 likely kill S. aureus by altering protein secretion, including disbalances in secreted autolysin levels.
To obtain additional insights into the antibiotic mechanism, we performed field emission scanning (FESEM) as well as transmission (TEM) electron microscopy examination. While FESEM micrographs of bacteria treated with PK150-C (Fig. 5a) or DMSO (Supplementary Fig. 22) showed no cellular abnormalities, PK150 incubation induced extracellular vesicle (EV) formation at the septum of dividing cells (Fig. 5b, Supplementary Fig. 22). Moreover, lysis and full degradation of cells were observed only in bacteria treated with PK150 (Figs. 5c and 5d, Supplementary Fig. 22). A closer inspection of membranes in ultrathin sections of PK150-treated S. aureus via TEM showed EV formation to be initiated by protrusions of cytoplasmic membrane (CM) through gaps in the cell wall (CW), which was absent in both controls (Figs. 5e and 5f, Supplementary Fig. 22). Two different cytoplasmic cargos consisting of intracellular material (Fig. 5e) or DNA (Supplementary Fig. 22) were detected in EV. Moreover, cellular disintegration by PK150 could be visualized at various stages (Supplementary Fig. 22). The lack of peptidoglycan at the sites of vesicle formation (Fig. 5f) suggests active CW degradation and burst of the extruded CM as mode of action. Given the known location of autolysins at the septum of dividing cells,38 their dysregulation at these sites supports the antibiotic mechanism of PK150.
Figure 5. Field emission scanning and transmission electron micrographs of S. aureus NCTC 8325.

(a-d) Field emission scanning electron micrographs (FESEM) of S. aureus NCTC 8325 treated with the inactive control compound PK150-C (2.4 μM, a) or PK150 (1.2 μM, b; 2.4 μM, c and d). Round and smooth shape indicates healthy cells for control- (a) as well as DMSO-treated (Supplementary Fig. 22) cells, whereas PK150-treated cells show deviations in cell appearance (b-d). Arrow heads point to damaged S. aureus (c). (b) and (d) depict two modes of action of PK150, namely extracellular vesicle formation with cytoplasmic content, predominantly in the division zone of bacteria (b, arrows) and extrusion of DNA containing material (d). Images are representative for 75 recordings from two independent preparations.
(e and f) Transmission electron micrographs (TEM) of S. aureus NCTC 8325 treated with PK150 (2.4 μM). The two modes of action of PK150 can also be observed in TEM images – formation of extracellular vesicles (e, black arrow points towards cytoplasmic membrane of vesicle), which probably burst and release the entire cytoplasmic content leaving cell wall relicts (f). DNA, deoxyribonucleic acid; CY, cytoplasm; CM, cytoplasmic membrane; CW, cell wall. Images are representative for 210 recordings from three independent preparations.
Autolysin secretion and SpsB activity are decreased in SFN-resistant strains
To further elucidate the overall mechanism, we selected SFN-resistant S. aureus isolates from our resistance development assays (Fig. 1e, Supplementary Fig. 6) and subjected them to whole genome sequencing. Aside from few mutations found only in part of the sequenced isolates, all clones carried frameshift mutations in SAOUHSC_01359 (mprF) encoding phosphatidylglycerol lysyltransferase fmtC (Q2G2M2) (for details, including a list of all mutations, see Supplementary Table 2 and Supplementary Data 5). MIC shift assays with the corresponding transposon mutants of all affected genes did not result in altered sensitivity towards SFN compared to the wild type (Supplementary Data 2). We thus assumed a more general mechanism, which indirectly influences target activity. In order to investigate this hypothesis in more detail, we first tested whether resistance provoked changes on the proteome level. No corresponding peptides of fmtC (mprF; Q2G2M2) were found in our MS measurements (Fig. 6a, Supplementary Data 4), which corresponds well to the results from whole-genome sequencing. Moreover, downregulation of several peptidoglycan hydrolase (PGH) domain-containing proteins was observed in the proteome of SFN-resistant isolates (Fig. 6a, Supplementary Data 4). This downregulation likely counteracts their elevated extracellular secretion upon antibiotic treatment. Interestingly, the transcriptional regulator ArcR (HTH-type transcriptional regulator ArcR; Q2FUY1) and several enzymes of the corresponding arginine deiminase pathway (Fig. 6a, Supplementary Data 4), responsible for ATP production via arginine catabolism under oxygen limiting conditions,39 were found to be upregulated, suggesting a switch to an alternative energy metabolism of resistant cells.
Figure 6. In-depth analysis of SFN-resistant S. aureus isolates and accompanying consequences for compound-induced SpsB stimulation.

(a) Proteome analysis of SFN-resistant isolates. Volcano plots show log2 fold change of protein levels in the full proteome of SFN-resistant S. aureus isolates compared to wild-type S. aureus NCTC 8325. SFN-resistant isolates were obtained in three independent experiments by sequential passaging in sub-MIC concentration of SFN (Fig. 1e, Supplementary Fig. 6). Dark red dots represent proteins whose secretion has been found to be inhibited by Arylomycin C16 (experimentally proposed SpsB substrates).35 Light red dots represent proteins that are predicted to have a SpsB signal peptide motif (according to UniProtKB as annotated by SignalP).61 Purple circles represent peptidoglycan hydrolase domain-containing proteins (Pfam annotations).60 Proteins highlighted include fmtC (mprF, phosphatidylglycerol lysyltransferase; Q2G2M2), emp (extracellular matrix protein-binding protein emp; Q2G012), sbi (immunoglobulin-binding protein sbi; Q2FVK5), sle1 (N-acetylmuramoyl-L-alanine amidase sle1; Q2G0U9), spa (immunoglobulin G-binding protein A; P02976), ssaA (staphylococcal secretory antigen SsaA; Q2FV55), ssaA2 (staphylococcal secretory antigen ssaA2; Q2G2J2), arcR (HTH-type transcriptional regulator ArcR; Q2FUY1), arcA (arginine deiminase; Q2FUX7), arcC2 (carbamate kinase 2; Q7X2S2), and arcF (ornithine carbamoyltransferase; Q2FUX8). Data represent average values and p-values were calculated using a two-sided two sample t-test; n = 4 independent experiments per group.
(b) Resistance towards SFN provokes changes in compound-induced SpsB stimulation. SpsB activity was determined by a FRET-based peptidase activity assay using membranes of SFN-resistant S. aureus or transposon mutant NE1360 (insertion into mprF) as the source of endogenous SpsB (0.2 mg/mL total membrane protein concentration). Substrate cleavage rates are normalized to DMSO-treated samples. Data represent mean values ± SD of averaged triplicates of n = 4 (50 μM conditions) or n = 3 (rest) biologically independent experiments (individual membrane preparations) for wild type and SFN-resistant isolates, and n = 5 independent experiments (individual measurements on different days, derived from two different membrane preparations) for NE1360.
(c) Secretome analysis of SFN-resistant isolates. Volcano plots show log2 fold change of protein levels in the secretome after treatment of SFN-resistant S. aureus NCTC 8325 cells with SFN (1.5 μM, 0.5-fold MIC) compared to DMSO treatment. Colour coding is equivalent to panel (a). Data represent average values and p-values were calculated using a two-sided two sample t-test; n = 4 independent experiments per group.
FmtC is involved in the modification of membrane lipid phosphatidylglycerol (PG) with positively charged amino-groups (lysine).40 It regulates biophysical properties of the membrane and thereby might influence the activity of key enzymes of the cell wall.41,42 In line with this function, SpsB stimulation by SFN in the resistant isolates was diminished as shown in FRET-based peptidase assays using membrane fractions from SFN-resistant isolates as source for endogenous SpsB (Fig. 6b). Interestingly, membranes derived from the corresponding transposon mutant of fmtC were still activated by both SFN and PK150, albeit at a lower level than in the wildtype (Fig. 6b). In-depth secretome analysis further revealed no increase in Sps-dependent protein secretion upon SFN-treatment in resistant bacteria (Fig. 6c). In particular, levels of peptidoglycan hydrolase (PGH) domain-containing proteins (Supplementary Data 4) seem to be tightly controlled even after antibiotic treatment. For further findings related to the SFN-resistant isolates, please refer to the Supplementary Discussion, Supplementary Data 4, Supplementary Table 3, and Supplementary Fig. 23.
PK150 is orally bioavailable and effective in treatment of in vivo S. aureus infections
Based on its potency and lack of resistance development, we further evaluated the pharmacological profile of PK150. First, we utilized the kinobeads technology43 to evaluate if PK150 still exhibited kinase affinity, which could contribute to toxic side effects. Lysates of four human cancer cell lines (K-562, Colo 205, SK-N-BE(2) and MV-4-11) were incubated with kinobeads, capturing a vast variety of more than 250 human kinases, in presence or absence of SFN and PK150. MS analysis of these competitive affinity pull-downs revealed that in contrast to SFN, which was bound to 8 human kinases, PK150 exhibited no affinity to any kinase (Supplementary Data 6, Supplementary Fig. 24).
In vitro toxicity of PK150 was determined by MTT assays against a panel of human and murine cell lines revealing IC50 values ranging from 7 to 15 μM (Supplementary Table 4). Corresponding selectivity ratios of cytotoxicity to antibiotic activity (ratios between 23 and 52) suggest a therapeutic window for treatment of bacterial infections in vivo. PK150 does not cause haemolysis of red blood cells and, similarly to SFN, exhibits excellent plasma stability with no observable degradation after 6 h incubation (Supplementary Figs. 25 and 26).
With this promising in vitro pre-evaluation, PK150 was subject to pharmacokinetic studies (PK) in mice (Figs. 7a and 7b). No obvious signs of toxicity were observed for 10 and 20 mg/kg p.o. and for 10 mg/kg i.v. Higher i.v. dosing of 20 mg/kg resulted in severe toxic effects and was thus avoided for subsequent therapeutic models. Oral bioavailability was approximately 63% and the mean residence time (MRT) was slightly enhanced via this administration route.
Figure 7. Pharmacokinetic and pharmacodynamic parameters of PK150 and in vivo efficacy.

(a) PK150 pharmacokinetic parameters in murine plasma following oral (p.o.) or intravenous (i.v.) administration. Data represent means ± SD (n = 3 per group).
(b) Pharmacokinetic analysis of PK150 in murine plasma following oral (p.o.) or intravenous (i.v.) administration. Time dependent plasma concentrations after the administration of 10 mg/kg p.o. (diamonds), 20 mg/kg (squares) p.o. or 10 mg/kg i.v. (circles), respectively, are shown. Compound levels in plasma were determined by LC-MS/MS analysis. Data represent means ± SD (n = 3 per group).
(c) Efficacy of PK150 and levofloxacin (LVX) against S. aureus ATCC 33591 (MRSA) in the neutropenic murine thigh model. PK150 (20 mg/kg p.o.) and the corresponding vehicle were administered p.o. 30 min, 4 h and 8 h after bacterial inoculation, whereas LVX (5 mg/kg) and the corresponding vehicle were administered intraperitoneally 2 h, 6 h and 10 h after bacterial inoculation. Data are expressed as mean values ± SD; n = 5 for PK150 and vehicle p.o.; n = 6 for LVX and vehicle i.p.; ***, p = 0.00093; **, p = 0.0028 (two-sided Student’s t-test).
(d and e) Efficacy of PK150 against S. aureus SH1000 in a murine bloodstream infection model. Bacterial loads in the liver (d) and heart (e) of S. aureus-infected mice treated with PK150 (20 mg/kg p.o.; squares) or vehicle alone (circles) via the p.o. administration route or with levofloxacin (LVX; triangles) via the i.p. administration route. Each symbol represents an individual mouse. Compilation data from three independent experiments are presented. Horizontal lines represent the mean values ± SD: (d) n = 14 for vehicle and LVX; n = 13 for PK150. **, p = 0.0040; *, p = 0.014 (two-sided Student’s t-test). (e) n = 16 for for vehicle; n = 14 for PK150; n = 13 for LVX. **, p = 0.0016 (PK150) and p = 0.0032 (LVX, two-sided Student’s t-test).
A neutropenic mouse thigh model was used to determine the pharmacodynamic efficacy of repeated PK150 dosing (20 mg/kg p.o.) against MRSA strain ATCC 33591 (Fig. 7c). A 10-fold reduction in CFU/g in thighs was observed in PK150-treated mice in comparison with vehicle-treated mice. The same range of reduction was determined for mice treated with the positive control levofloxacin upon i.p. administration (5 mg/kg).
In vivo efficacy of PK150 against MSSA (strain SH1000) was further demonstrated in a murine bloodstream infection model. While no effect was observed in the kidneys (Supplementary Fig. 27), bacterial loads in liver and heart were both significantly reduced by approximately 100-fold (Figs. 7d and 7e). In comparison to the marketed antibiotic levofloxacin (LVX) as benchmark control, PK150 exhibited not only equal potency but also convenient dosing intervals of up to two days and good bioavailability.44,45
Discussion
The repurposing of drugs has become an attractive strategy in the search for innovative medicine without going through enormous efforts of de novo pharmacological optimization procedures.46 Given the drought in the discovery of new antibiotics on the one hand and the large number of about 300 bacterial proteins with predicted essential function47 on the other, mining bacterial proteomes with known drugs for untargeted pathways represents an attractive strategy to unravel new and resistance-free drug candidates. In our phenotypic screen of small molecule kinase inhibitors for anti-staphylococcal activity, SFN stood out as the best antibiotic hit molecule. However, chemical proteomic profiling with its corresponding probe revealed not a single known kinase as a potential bacterial target. Moreover, in-depth SAR revealed the pyridine moiety, a hallmark of kinase inhibition,48,49 to be dispensable for antibiotic activity. Instead several other putative bacterial protein targets, foremost two essential proteins, MenG and SpsB, were identified. Of note, we cannot exclude that additional non-proteinogenous targets may exist and contribute to the overall mode of action.
MenG catalyses the final step in menaquinone biosynthesis and disruption of MenG activity by small molecules led to antibiotic activity in other bacterial strains.28,50 We identified MenG as one of the strongest hits in our AfBPP experiment with the PK150-derived photoprobe 3-005-P. In accordance with this finding, PK150 reduced menaquinone levels in living bacteria, inhibited MenG in cellular lysates and exhibited reduced antibiotic activity in the presence of exogenous menaquinone, whereas with SFN these effects were less pronounced. Of note, although SFN does not exhibit a strong effect on menaquinone biosynthesis, up-regulation of the alternative arginine pathway in whole proteomes of SFN-resistant cells provides an independent link to alterations in the cellular energy metabolism.
Inhibition of SpsB is a challenging task as the enzyme is insensitive to classical serine protease inhibitors.51 Besides synthetic molecules, natural products of the arylomycin family represent the most potent inhibitors.22,32 Contrary to the well-explored inhibitory path, we discovered the first activation mechanism of SpsB. How can SpsB activation contribute to killing of bacterial cells? Analysis of bacteria treated with PK150 or SFN at sub-MIC concentrations confirmed that the secretion of SpsB-dependent proteins was significantly elevated. SpsB controls a key enzymatic step facilitating mature protein release after translocation in the membrane. Although little is known about the rate-determining step in protein secretion, studies of surface and extracellular proteomes revealed an accumulation of Sec- and SpsB-dependent proteins in the surfaceome.52–54 Indeed, we observed a reduction of cell surface proteins prone to triggered SpsB processing upon compound addition. In principle, a depot of SpsB substrates could be available on the cell surface under normal conditions and readily mobilized upon enzyme stimulation to respond to external challenges. It is thus likely that, in contrast to SpsB inhibition, where proteins are trapped in the membrane and thereby propagate cell lysis, the general dysregulation of secretion induces a similar stress to bacterial cell integrity. This is supported by the observed induction of lysis, rapid bactericidal killing of exponentially growing bacterial cells, as well as by lesions visible in electron microscopy examination. In light of the upregulation of several autolysins in the secretome, it is likely that, in line with previous literature studies, a slight imbalance of these degradative and tightly controlled enzymes could trigger autolysis.36,37 Interestingly, we observed down-regulation of several autolysins in proteomes of SFN-resistant strains.
PK150´s descent from a marketed drug turned out to be a beneficial property as the molecule already exhibited excellent stability and pharmacokinetic properties including oral bioavailability. Optimization from SFN to PK150 resulted in a loss of human kinase affinity (see also Supplementary Discussion). While in vitro toxicity only slightly changed, the greater antibacterial potency provided a suitable therapeutic window. PK150 exhibits optimal stability and oral bioavailability as well as suitable in vivo efficacy. Ongoing medicinal chemistry studies are performed to further optimize solubility, toxicity and spectrum of bacteria addressed to further exploit the antibiotic potential of this promising drug candidate in pre-clinical disease models.
Methods
See Supporting Information for detailed methods and protocols.
Activity-based protein profiling in S. aureus NCTC 8325
S. aureus NCTC 8325 overnight cultures were diluted 1:10 in B medium and incubated at 37°C, 200 rpm. After 7 h, bacterial cells were harvested, washed and resuspended in PBS to give a final theoretical OD600 of 40, then aliquoted and photoprobe (SFN-P, 50 μM, or 3-005-P, 5 μM) or DMSO (1% final total concentration) were added, followed by incubation at 25°C, 700 rpm for 45 min. For competition experiments, cultures were first incubated with competitor SFN (0.5 mM) or DMSO only for 45 min before addition of the photoprobe. After compound treatment, samples were diluted with PBS (5 mL final volume) and irradiated for 30 min with UV light (360 nm). Subsequently, Bacteria were harvested, washed and resuspended in PBS (supplemented with EDTA-free protease inhibitors), and lysed mechanically (Precellys Lysing Kit and Homogenizer). The lysate was treated with 8 μg/mL lysostaphin (37°C, 20 min). Separation of soluble and insoluble fraction was performed by centrifugation (21,000 x g, 4°C, 1 h). The insoluble fraction was washed twice with PBS. Note that no separation of soluble and insoluble fractions was done for labelling with 3-005-P. Protein concentration was measured by BCA assay and samples were adjusted to equal protein amounts. For click chemistry (CuACC), click reagents were added to each sample at the indicated final concentrations: trifunctional linker (TFL, 60 μM),55 TCEP (1 mM), TBTA ligand (0.1 mM) and CuSO4 (1 mM). Samples were incubated at RT for 1 h. Following CuACC, proteins were precipitated with ice-cold acetone (4 volumes) at -20°C overnight, washed twice with ice-cold methanol, and resuspended in 0.4% (w/v) SDS in PBS. Upon avidin-bead enrichment, samples were reduced with 5 mM TCEP (1 h at 37°C), alkylated using 10 mM iodoacetamide (30 min at 25°C) and quenched with 10 mM dithiothreitol (30 min at 25°C) on-bead. Proteins were pre-digested using LysC (2 h at 25°C) and then digested with trypsin (16 h at 37°C). Samples were desalted using SepPak C18 cartridges (50 mg, Waters) prior to MS measurement. For SFN-P, this was accompanied by stable isotope dimethyl labelling on-column, whereas for 3-005-P, label-free quantification was used. LC-MS/MS analysis was performed with an Ultimate3000 Nano-HPLC system (Thermo Fisher Scientific) coupled to an Orbitrap Fusion (for SFN-P, Thermo Fisher Scientific) or to a Q Exactive Plus (for 3-005-P, Thermo Fisher Scientific). Peptide and protein identification was performed using MaxQuant56 with Andromeda57 as search engine and the Uniprot database for S. aureus NCTC 8325. Statistical analysis was performed in Perseus.58 Three biological replicates consisting of three technical replicates each were analysed. Putative contaminants, reverse hits and proteins, identified by side only, were removed. For SFN-P, normalized protein ratios were log2(x) transformed and filtered to contain at least one valid value within technical replicates. Ratios were z-score normalized within replicates and average values of technical replicates were calculated. Mean differences in enrichment and respective p-values were obtained by a two sided one sample t-test over the three biological replicates. For 3-005-P, LFQ intensities were grouped into DMSO- and probe-treated condition and filtered to contain at least six valid values within at least one group. LFQ intensities were then averaged between technical replicates and log2(x) transformed. Missing values were imputated from normal distribution and p-values were obtained by a two-sided two sample t-test over the three biological replicates.
See supporting information for a detailed description of the proteomic workflow.
Supplementary Material
Acknowledgements
We thank the Network on Antimicrobial Resistance in Staphylococcus aureus (NARSA) for the supply of the Nebraska Transposon Mutant Library (NTML). Furthermore, we thank Prof. Stephanie Grond for providing arylomycin and Prof. Floyd Romesberg for providing S. aureus N315 ARC0001ΔSpsB. We also like to thank Shoko Miami and Prof. Eric Rubin for determining antimicrobial activities against M. tuberculosis. We thank Annett Klaschwitz, Florian Kortmann, Stefan Hifinger and Dr. Christoph Lierse v. Gostomski for scintillation measurement of radioactively labelled menaquinone. S.A.S. was funded by the Center for Integrated Protein Science Munich (CIPSM), Deutsche Forschungsgemeinschaft SFB1035 and European Research Council (ERC) and the European Union’s Horizon 2020 research and innovation programme (grant agreement No 725085, CHEMMINE, ERC consolidator grant). E.K. was supported by the doctoral fellowship of the Fonds der Chemischen Industrie. R.M. was supported by a doctoral fellowship of the Boehringer Ingelheim Fonds. K.R. was supported by the German Centre for Infection Research (DZIF) (TTU 09.710). I.A. acknowledges funding by Deutsche Forschungsgemeinschaft SFB1035. W.M.W. was funded by National Science Foundation (CHE-1454116) and the National Institute of General Medical Sciences (R35 GM119426). M.C.J. acknowledges a National Science Foundation predoctoral grant (DGE-1144462). S.M.H. acknowledges financial support by a Liebig fellowship of the Fonds der Chemischen Industrie. M.W.H., C.F. and F.A.M.M. were funded by the Federal Ministry for Education and Research (BMBF) under the framework programme ‘VIP+’ – project ‘aBacter’. We like to thank Dietrich Mostert for excellent experimental support, Mona Wolff, Katja Bäuml, Katja Gliesche, Linda Nguyen and Janine Schreiber for excellent technical support, and Matthias Stahl for critical comments on the manuscript.
Footnotes
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE59 partner repository with the dataset identifier PXD012946.
Whole Genome Sequencing data and metadata are available on the SRA repository under the Bioproject number PRJNA525411.
Bacterial strains and plasmids used in this work are readily available from the authors, or can be purchased commercially as stated in the Supplementary Information.
Code availability
All computer code used is either publicly available software, described in prior publications31, or available from the authors upon request. For details, also on the versions and parameters used, please refer to the respective section in the Supplementary Information.
Animal studies
The animal studies were conducted in accordance with the recommendations of the European Community (Directive 86/609/EEC, 24 November 1986). All animal procedures were performed in strict accordance with the German regulations of the Society for Laboratory Animal Science (GV- SOLAS) and the European Health Law of the Federation of Laboratory Animal Science Associations (FELASA). Animals were excluded from further analysis if sacrifice was necessary according to the humane endpoints established by the ethical board. All experiments were approved by the ethical board of the Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit, Oldenburg, Germany (LAVES; permit No. 33.9-42502-04-13/1195 and 33.19-42502-04-15/1857).
Author contributions
P.L., E.K., R.M. and S.A.S. designed experiments, interpreted results and wrote the manuscript with input from all authors. P.L. synthesized library compounds and probes and performed structure-activity relationship studies. V.S.K. assisted in the chemical synthesis of AfBPP probes. E.K. and P.L. performed gel- and MS-based labelling, analysis of the MS data as well as SpsB target deconvolution and validation experiments. E.K. analysed mass spectrometry-based data and conducted bioinformatics analyses. R.M. performed target identification, MS data analysis, and validation experiments in the context of the menaquinone biosynthesis pathway and assisted in further validation experiments. E.K., P.L. and S.M.H. performed bacterial resistance development studies. E.K. carried out persister assays and time-kill assays. M.W.H., C.F. and F.A.M.M. performed time-kill assays as well as biofilm and persister studies. M.C.J. and W.M.W. helped in biofilm studies. J.L. performed microbiological studies in Mycobacteria. D.C.-M. and D.H.P. conducted whole-genome sequencing of resistant bacterial isolates and analysed related data. I.U. and I.A. performed molecular docking and dynamic studies and interpreted related data. K.R. and M.Ro. performed electron microscopy studies and analysed related data. M.Re. and B.K. performed kinobead pulldown experiments and analysed related data. K.R. and E.M. performed animal studies and analysed related data.
Competing financial interests
P.L., E.K. and S.A.S. are co-inventors on a European patent (EP 16 171 906.7) covering the structure of PK150. All other authors declare no competing financial interests.
References
- [1].Cassini A, et al. Attributable deaths and disability-adjusted life-years caused by infections with antibiotic-resistant bacteria in the EU and the European Economic Area in 2015: a population-level modelling analysis. Lancet Infect Dis. 2019;19:56–66. doi: 10.1016/S1473-3099(18)30605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tacconelli E, et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect Dis. 2018;18:318–327. doi: 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- [3].Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Harms A, Maisonneuve E, Gerdes K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science. 2016;354 doi: 10.1126/science.aaf4268. aaf4268. [DOI] [PubMed] [Google Scholar]
- [5].Ling LL, et al. A new antibiotic kills pathogens without detectable resistance. Nature. 2015;517:455–459. doi: 10.1038/nature14098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sass P, et al. Antibiotic acyldepsipeptides activate ClpP peptidase to degrade the cell division protein FtsZ. Proc Natl Acad Sci. 2011;108:17474–17479. doi: 10.1073/pnas.1110385108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Smith PA, et al. Optimized arylomycins are a new class of Gram-negative antibiotics. Nature. 2018;561:189–194. doi: 10.1038/s41586-018-0483-6. [DOI] [PubMed] [Google Scholar]
- [8].Kurosu M, Begari E. Bacterial protein kinase inhibitors. Drug Dev Res. 2010;71:168–187. [Google Scholar]
- [9].Miller JR, et al. A class of selective antibacterials derived from a protein kinase inhibitor pharmacophore. Proc Natl Acad Sci. 2009;106:1737–1742. doi: 10.1073/pnas.0811275106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chang H-C, et al. In vitro and in vivo activity of a novel sorafenib derivative SC5005 against MRSA. J Antimicrob Chemother. 2016;71:449–459. doi: 10.1093/jac/dkv367. [DOI] [PubMed] [Google Scholar]
- [11].Roberts JL, et al. GRP78/Dna K is a target for Nexavar/Stivarga/Votrient in the treatment of human malignancies, viral infections and bacterial diseases. J Cell Physiol. 2015;230:2552–2578. doi: 10.1002/jcp.25014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Pujol E, et al. Pentafluorosulfanyl-containing triclocarban analogues with potent antimicrobial activity. Molecules. 2018;23 doi: 10.3390/molecules23112853. 2853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Walsh SE, et al. Activity and mechanisms of action of selected biocidal agents on Gram-positive and -negative bacteria. J Appl Microbiol. 2003;94:240–247. doi: 10.1046/j.1365-2672.2003.01825.x. [DOI] [PubMed] [Google Scholar]
- [14].Conlon BP, et al. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature. 2013;503:365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Springer MT, Singh VK, Cheung AL, Donegan NP, Chamberlain NR. Effect of clpP and clpC deletion on persister cell number in Staphylococcus aureus. J Med Microbiol. 2016;65:848–857. doi: 10.1099/jmm.0.000304. [DOI] [PubMed] [Google Scholar]
- [16].Waters EM, Rowe SE, O’Gara JP, Conlon BP. Convergence of Staphylococcus aureus persister and biofilm research: Can biofilms be defined as communities of adherent persister cells? PLOS Pathog. 2016;12:e1006012. doi: 10.1371/journal.ppat.1006012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hamamoto H, et al. Lysocin E is a new antibiotic that targets menaquinone in the bacterial membrane. Nat Chem Biol. 2015;11:127–133. doi: 10.1038/nchembio.1710. [DOI] [PubMed] [Google Scholar]
- [18].Evans MJ, Cravatt BF. Mechanism-based profiling of enzyme families. Chem Rev. 2006;106:3279–3301. doi: 10.1021/cr050288g. [DOI] [PubMed] [Google Scholar]
- [19].Fonović M, Bogyo M. Activity-based probes as a tool for functional proteomic analysis of proteases. Expert Rev Proteomics. 2008;5:721–730. doi: 10.1586/14789450.5.5.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kleiner P, Heydenreuter W, Stahl M, Korotkov VS, Sieber SA. A whole proteome inventory of background photocrosslinker binding. Angew Chem Int Ed. 2017;56:1396–1401. doi: 10.1002/anie.201605993. [DOI] [PubMed] [Google Scholar]
- [21].Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJR. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc. 2009;4:484–494. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- [22].Rao CVS, Waelheyns ED, Economou A, Anné J. Antibiotic targeting of the bacterial secretory pathway. Biochim Biophys Acta. 2014;1843:1762–1783. doi: 10.1016/j.bbamcr.2014.02.004. [DOI] [PubMed] [Google Scholar]
- [23].Cox J, et al. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Boersch M, Rudrawar S, Grant G, Zunk M. Menaquinone biosynthesis inhibition: a review of advancements toward a new antibiotic mechanism. RSC Adv. 2018;8:5099–5105. doi: 10.1039/c7ra12950e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kurosu M, Begari E. Vitamin K2 in electron transport system: are enzymes involved in vitamin K2 biosynthesis promising drug targets? Molecules. 2010;15:1531–1553. doi: 10.3390/molecules15031531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Fey PD, et al. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio. 2013;4:e00537–12. doi: 10.1128/mBio.00537-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Craney A, Dix MM, Adhikary R, Cravatt BF, Romesberg FE. An alternative terminal step of the general secretory pathway in Staphylococcus aureus. mBio. 2015;6:e01178–15. doi: 10.1128/mBio.01178-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Benkovic SJ, et al. Identification of borinic esters as inhibitors of bacterial cell growth and bacterial methyltransferases, CcrM and MenH. J Med Chem. 2005;48:7468–7476. doi: 10.1021/jm050676a. [DOI] [PubMed] [Google Scholar]
- [29].Rao CVS, et al. Enzymatic investigation of the Staphylococcus aureus type I signal peptidase SpsB – implications for the search for novel antibiotics. FEBS J. 2009;276:3222–3234. doi: 10.1111/j.1742-4658.2009.07037.x. [DOI] [PubMed] [Google Scholar]
- [30].Therien AG, et al. Broadening the spectrum of β-lactam antibiotics through inhibition of signal peptidase type I. Antimicrob Agents Chemother. 2012;56:4662–4670. doi: 10.1128/AAC.00726-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Antes I. DynaDock: A new molecular dynamics-based algorithm for protein-peptide docking including receptor flexibility. Proteins. 2010;78:1084–1104. doi: 10.1002/prot.22629. [DOI] [PubMed] [Google Scholar]
- [32].Craney A, Romesberg FE. The inhibition of type I bacterial signal peptidase: Biological consequences and therapeutic potential. Bioorg Med Chem Lett. 2015;25:4761–4766. doi: 10.1016/j.bmcl.2015.07.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Smith PA, Romesberg FE. Mechanism of action of the arylomycin antibiotics and effects of signal peptidase I inhibition. Antimicrob Agents Chemother. 2012;56:5054–5060. doi: 10.1128/AAC.00785-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Walsh SI, Craney A, Romesberg FE. Not just an antibiotic target: Exploring the role of type I signal peptidase in bacterial virulence. Bioorg Med Chem. 2016;24:6370–6378. doi: 10.1016/j.bmc.2016.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schallenberger MA, Niessen S, Shao C, Fowler BJ, Romesberg FE. Type I signal peptidase and protein secretion in Staphylococcus aureus. J Bacteriol. 2012;194:2677–2686. doi: 10.1128/JB.00064-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chao MC, et al. Protein complexes and proteolytic activation of the cell wall hydrolase RipA regulate septal resolution in mycobacteria. PLOS Pathog. 2013;9:e1003197. doi: 10.1371/journal.ppat.1003197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Frankel MB, Hendrickx APA, Missiakas DM, Schneewind O. LytN, a murein hydrolase in the cross-wall compartment of Staphylococcus aureus, is involved in proper bacterial growth and envelope assembly. J Biol Chem. 2011;286:32593–32605. doi: 10.1074/jbc.M111.258863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Pinho MG, Kjos M, Veening J-W. How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat Rev Microbiol. 2013;11:601–614. doi: 10.1038/nrmicro3088. [DOI] [PubMed] [Google Scholar]
- [39].Makhlin J, et al. Staphylococcus aureus ArcR controls expression of the arginine deiminase operon. J Bacteriol. 2007;189:5976–5986. doi: 10.1128/JB.00592-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ernst CM, Peschel A. Broad-spectrum antimicrobial peptide resistance by MprF-mediated aminoacylation and flipping of phospholipids. Mol Microbiol. 2011;80:290–299. doi: 10.1111/j.1365-2958.2011.07576.x. [DOI] [PubMed] [Google Scholar]
- [41].Jones T, et al. Failures in clinical treatment of Staphylococcus aureus infection with daptomycin are associated with alterations in surface charge, membrane phospholipid asymmetry, and drug binding. Antimicrob Agents Chemotherapy. 2008;52:269–278. doi: 10.1128/AAC.00719-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Roy H. Tuning the properties of the bacterial membrane with aminoacylated phosphatidylglycerol. IUBMB Life. 2009;61:940–953. doi: 10.1002/iub.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Médard G, et al. Optimized chemical proteomics assay for kinase inhibitor profiling. J Proteome Res. 2015;14:1574–1586. doi: 10.1021/pr5012608. [DOI] [PubMed] [Google Scholar]
- [44].Fish DN, Chow AT. The clinical pharmacokinetics of levofloxacin. Clin Pharmacokinet. 1997;32:101–119. doi: 10.2165/00003088-199732020-00002. [DOI] [PubMed] [Google Scholar]
- [45].Scaglione F, Mouton JW, Mattina R, Fraschini F. Pharmacodynamics of levofloxacin and ciprofloxacin in a murine pneumonia model: Peak concentration/MIC versus area under the curve/MIC ratios. Antimicrob Agents Chemother. 2003;47:2749–2755. doi: 10.1128/AAC.47.9.2749-2755.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Nosengo N. Can you teach old drugs new tricks? Nature. 2016;534:314–316. doi: 10.1038/534314a. [DOI] [PubMed] [Google Scholar]
- [47].Xu HH, et al. Staphylococcus aureus TargetArray: Comprehensive differential essential gene expression as a mechanistic tool to profile antibacterials. Antimicrob Agents Chemother. 2010;54:3659–3670. doi: 10.1128/AAC.00308-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wan PTC, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- [49].Wu P, Nielsen TE, Clausen MH. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol Sci. 2015;36:422–439. doi: 10.1016/j.tips.2015.04.005. [DOI] [PubMed] [Google Scholar]
- [50].Sukheja P, et al. A novel small-molecule inhibitor of the Mycobacterium tuberculosis demethylmenaquinone methyltransferase MenG is bactericidal to both growing and nutritionally deprived persister cells. mBio. 2017;8:e02022–16. doi: 10.1128/mBio.02022-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Paetzel M, Dalbey RE, Strynadka NCJ. Crystal structure of a bacterial signal peptidase in complex with a β-lactam inhibitor. Nature. 1998;396:186–190. doi: 10.1038/24196. [DOI] [PubMed] [Google Scholar]
- [52].Dreisbach A, van Dijl JM, Buist G. The cell surface proteome of Staphylococcus aureus. Proteomics. 2011;11:3154–3168. doi: 10.1002/pmic.201000823. [DOI] [PubMed] [Google Scholar]
- [53].Gatlin CL, et al. Proteomic profiling of cell envelope-associated proteins from Staphylococcus aureus. Proteomics. 2006;6:1530–1549. doi: 10.1002/pmic.200500253. [DOI] [PubMed] [Google Scholar]
- [54].Hempel K, et al. Quantitative cell surface proteome profiling for SigB-dependent protein expression in the human pathogen Staphylococcus aureus via biotinylation approach. J Proteome Res. 2010;9:1579–1590. doi: 10.1021/pr901143a. [DOI] [PubMed] [Google Scholar]
- [55].Eirich J, et al. Pretubulysin derived probes as novel tools for monitoring the microtubule network via activity-based protein profiling and fluorescence microscopy. Mol BioSyst. 2012;8:2067–2075. doi: 10.1039/c2mb25144b. [DOI] [PubMed] [Google Scholar]
- [56].Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–1372. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- [57].Cox J, et al. Andromeda: A peptide search engine integrated into the MaxQuant environment. J Proteome Res. 2011;10:1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- [58].Tyanova S, et al. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13:731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- [59].Vizcaíno JA, et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016;44:11033–11033. doi: 10.1093/nar/gkw880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Finn RD, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016;44:D279–D285. doi: 10.1093/nar/gkv1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Nielsen H. Predicting secretory proteins with SignalP. Methods Mol Biol. 2017;1611:59–73. doi: 10.1007/978-1-4939-7015-5_6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.