

Graphical abstract
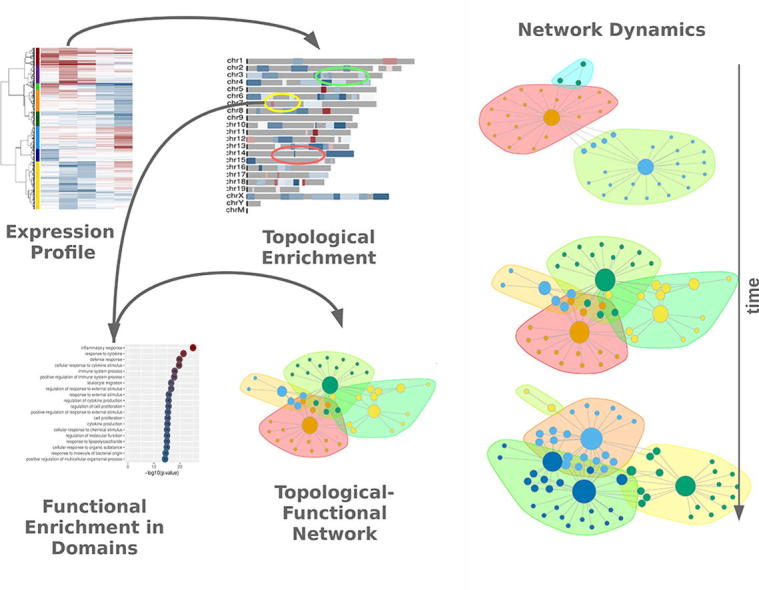
Keywords: Genome organization, Gene regulation, Biological networks, TNF, Cytokine response
Abstract
Genes in linear proximity often share regulatory inputs, expression and evolutionary patterns, even in complex eukaryote genomes with extensive intergenic sequences. Gene regulation, on the other hand, is effected through the co-ordinated activation (or suppression) of genes participating in common biological pathways, which are often transcribed from distant loci. Existing approaches for the study of gene expression focus on the functional aspect, taking positional constraints into account only marginally.
In this work we propose a novel concept for the study of gene expression, through the combination of topological and functional information into bipartite networks. Starting from genome-wide expression profiles, we define extended chromosomal regions with consistent patterns of differential gene expression and then associate these domains with enriched functional pathways. By analyzing the resulting networks in terms of size, connectivity and modularity we can draw conclusions on the way genome organization may underlie the gene regulation program.
Implementation of this approach in a detailed RNASeq profiling of sustained Tnf stimulation of mouse synovial fibroblasts, allowed us to identify unexpected regulatory changes taking place in the cells after 24 h of stimulation. Bipartite network analysis suggests that the cytokine response set by Tnf, progresses through two distinct transitions. An early generalization of the inflammatory response, that is followed by a late shutdown of immune-related functions and the redistribution of expression to developmental and cell adhesion pathways and distinct chromosomal regions.
We show that the incorporation of topological information may provide additional insights in the complex propagation of Tnf activation.
1. Introduction
From the analysis of the first genome-wide expression experiments, it became clear that gene expression levels were related to their relative localization in the linear dimension [1]. More recent studies have shown that transcriptional activation may spread in “waves” that affect nearby genes [2] and that a significant proportion of gene expression events may be attributed to the genomic position [3]. Such tendencies are reflected on evolutionary constraints, with genes in close proximity showing similar patterns of evolution [4], [5], [6]. These constraints inevitably lead to genes, involved in common functional pathways to be lying closer to each other in the linear genome [7], [8], but also extending this “structural proximity” to the genomic three-dimensional structure [9]. On the other hand, there exist regions with characteristic epigenetic modification patterns [10], [11], different combinations of which have been shown to delineate epigenetic “chromatin states”, that reflect different levels of regulatory and transcriptional activity [12], [13], [14]. The importance of genome compartmentalization and its underlying regulatory and transcriptional activity is evident in recent approaches on cis-regulatory domains [15] and in attempts to model gene expression levels on the basis of genomic position [3]. The biomedical importance of this organization is particularly important in various types of cancer, where extensive genomic translocations are the main cause for the aberrant regulation of genes and eventually for pathogenesis [16].
At the same time, the study of neighbour effects in the regulation of gene expression and of possible underlying mechanisms has been limited. Early works in the simple eukaryotic genome of S. cerevisiae have shown the existence of regions of gene expression correlation [17] and referred to their dynamics [18]. At the level of functional gene regulation, we have sufficient knowledge of how transcription programs are performed through the activation of specific pathways and gene regulatory networks [19]. We also have a variety of tools for assessing the importance of cellular processes and biological pathways through functional analyses of gene expression [20], [21]. Nevertheless, there is a growing need for methods that will incorporate spatial information and, until recently, the use of chromosomal linear distance as a predictive marker of gene expression has been limited to a few, large-scale projects [22].
In the past we have studied the positional footprint on gene deregulation in models of genome compartmentalization in yeast, where we have shown both transcriptional regulation [23] and nucleosomal structure [24] to be delineating distinct genomic domains. In this work we propose an approach that can act complementary to existing functional enrichment analyses [20], [25], [26] with an additional layer of information coming from the level of genome organization. By analyzing the spatial distribution of differential gene expression, we define regions with consistent gene deregulation profiles, that form extensive chromosomal domains (of the order of Mbp), where limited fluctuation of gene expression changes is suggestive of underlying organizing principles. Association of these regions with functional terms and pathways, through typical gene enrichment analyses, leads to the creation of topological-functional bipartite networks that may then be studied at multiple levels. While bipartite networks are not new to biology, their use has been limited to the mapping of associations between genes and respective ontologies (pathways, diseases etc). Here we implement a novel approach, in which genes (and their expression) are uses as proxies to deduce enrichments at the level of functional ontologies and genomic positions and then use these two as nodes in the creation of the network. We showcase this concept in the context of a well-studied gene regulatory program, the activation of cells by the prolonged exposure to Tnf.
Tnf is the archetype of major activating cytokines, which orchestrate the process of the inflammatory response. The succession of steps upon Tnf induction has been shown to involve dynamic RNA turnover [27], [28] but to be also accompanied by changes in the chromatin landscape [29] and the organization of transcription factories [30]. Sustained expression of Tnf can have devastating effects as is evident in transgenic animal models of inflammatory diseases [31], [32] but the prolonged activation of cells by Tnf has proved very difficult to study due to significant cross-talk that generates conflicting effects. While a number of activated signaling pathways coalesce, thus obscuring the direct response, the spatial aspect of gene expression may provide more robust signatures that can assist us in the decomposition of the multiple, ongoing signals. We thus employed a spatial-functional approach to show that fibroblasts, under prolonged Tnf exposure, undergo two major transitions that are reflected not only in the activated pathways but also in the clustering of deregulated genes in particular genomic domains. The two transition points mark both quantitative but, more importantly, qualitative differences and suggest a role for Tnf in the shaping of differentiation potential.
2. Methods
2.1. Gene expression profiling
Mouse synovial fibroblasts were isolated from C57BL/6 littermate mice. All animals were housed under specific pathogen–free conditions. Three biological replicates were isolated per experimental condition, and for each condition, a mixed-sex pool of 3 mice was used. Purity of all isolations was assessed by fluorescence-activated cell sorting, with the following acceptance criteria: 0.85% positive for CD90.2 and 2.5% positive for CD45. RNA was extracted from mouse synovial fibroblasts with the use of an Absolutely RNA Miniprep kit (Agilent Technologies). All library preparations, next-generation sequencing, and quality control steps were performed at the McGill University and Genome Quebec Innovation Centre (Montreal, Quebec, Canada). TruSeq RNA libraries were prepared and samples were run on an Illumina HiSeq2000 platform using a 100-bp paired-end setup.
2.2. Differential expression analysis
RNA-seq was performed with three replicates for 5 different timepoints at 1, 3, 6, 24 h and 7 days after Tnf exposure, alongside a 0 h control. Mapping was performed with TopHat2 [33] and differential expression was calculated against the 0 h control profile with Cufflinks/CuffDiff [34]. Differentially expressed genes were defined on the basis of standard thresholds for analysis with |log2(FC)| ≥ 1, p-value ≤ 0.05, after adjusting for multiple comparisons (Supplementary Table 1).
Functional analysis was performed with the use of gProfileR [26] through its R implementation. Enrichments were studied at the levels of Gene Ontology (GO), KEGG pathways, Transcription Factors (TF) and Human Phenotypes (HP).
Clustering was performed with agglomerative hierarchical clustering using Ward’s minimum variance criterion. The number of clusters was defined based on a simple elbow rule on the within sums of squares values of a k-means clustering approach. Profile similarity calculation was performed through the calculation of euclidean differences in mean cluster differential expression as described in [35].
2.3. Creation of domains of focal deregulation
We implemented a method based on unbiased recursive partitioning as described in [36]. Differential gene expression data (as log2FC) were used as values and their genomic coordinates as a discrete “time-like” variable. A custom R function was written with the used of the R function “breakpoints” from the Package “strucchange” [37], [38]. The function performs genome partitioning on the basis of an F-test (Chow Test), which tests the equality of the coefficients of two linear models and is, in this case, applied on consecutive linear models built on gene expression data read in a sliding window of 50 genes (see Supplementary Code and Data). Once the breakpoints are defined, the function creates a complete partitioning of the genome in discrete regions, each of which is described by a) the number of contained genes and b) their mean differential expression score. An arbitrary criterion of an absolute mean differential expression score ≥0.1 was used to call significant DFDs. These were used in the creation of bipartite networks.
Overlaps between DFDs and differentially expressed genes, gene clusters or other sets of genomic coordinates were reported as Jaccard Indexes and assessed statistically through a permutation test, performed as described in [39].
2.4. Topological-functional bipartite networks
These were created in the following way:
-
1.
Starting from a given expression profile, a list of differentially expressed genes is extracted and a set of significant DFDs is called (see above).
-
2.
For each DFD, the differentially expressed genes are being extracted and then passed to gProfileR for gene set enrichment analysis.
-
3.
Functional categories fulfilling significance criteria (Number of Genes in Category ≥30, adjusted p-value ≤0.05) are associated with the given DFD.
-
4.
The bipartite network is created as an edge list with one vertex being the DFD and the other being its enriched functions.
Networks were analyzed for modularity and visualized with the use of R’s igraph Package [40].
3. Results
3.1. Complex patterns of differentially expressed genes in the prolonged stimulation of fibroblasts by Tnf
Clustering of the 1595 differentially expressed genes in at least one of the analyzed timepoints revealed some very interesting aspects regarding the prolonged exposure of fibroblasts to Tnf. An initial small set (∼330) of deregulated genes (at 1 h) is replaced by a much larger (∼640) at 3 h of stimulation, with less than one third (97) of the genes being shared between the two timepoints. A similar pattern of expression is observed between 3 and 6 h of stimulation before another abrupt transition at 24 h, with only 55 genes being commonly deregulated between 6 h (378 DE genes) and 24 h (190 genes). A much longer period extending to 7 days for the last timepoint shows only mild further changes in the expression profile. Thus it seems that the state acquired by the cells at 24 h remains relatively stable. Overall, the expression data suggest two clear transition points between 1 h → 3 h (early) and 6 h → 24 h (late), which may be seen more clearly through the clustering of genes in 8 distinct clusters (Fig. 1A, top). It should be noted that most of the DE genes are timepoint-specific as shown in an analysis of set overlaps (Supplementary Fig. 1), where more than 450 genes are unique for 7 d and more than 350 unique for 3 h.
Fig. 1.

Gene expression clustering and functional analysis. A. (Top) Clustering of gene expression for 1595 genes that were differentially expressed (|log2FC| ≥ 1, p value ≤ 0.05) in at least one timepoint. Red corresponds to over- and blue to under-expression. The 8 clusters are shown in different colours in the left side of the heatmap. (Bottom) Summarization of the extended heatmap with mean differential expression value for each cluster. Clusters are shown in the same colour coding as above. Cluster names on the right correspond to a general description based on their expression patterns. B, C. Functional analysis of over- (B) and under- (C) expressed clusters. Names of clusters refer to (Fig. 1A, bottom), with the exception of Late Repression where both “Late Down” clusters from (A) are pooled together. Enriched terms were deduced from a gProfileR analysis. The top 20 enriched terms on the basis of p-values are reported for each cluster. (Functional analysis of the same type for the “EarlyUpLateDown” cluster provided as separate Supplementary Fig. 2). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
The aforementioned clustering helps us distinguish between genes that are initially over-expressed but gradually restored to normal (unstimulated) levels (Dark red cluster), or even reversed to under-expression (Light green cluster). More importantly, we find most of the clusters to be reflecting clear timepoint-specific expression tendencies. Thus there are clearly under-expressed clusters for early (1 h, Dark blue cluster), intermediate (3 h and 6 h, Yellow cluster) or late (24 h, 7 d, Orange and Dark Green Clusters) timepoints. Similarly the Dark red, Purple and Light Blue clusters are also reflecting time-specific for early-intermediate (1 h, 3 h), intermediate (3 h and 6 h) and late (24 h and primarily 7 d) over-expression respectively (Fig. 1A, bottom and Supplementary Fig. 2).
3.2. Functional enrichment analysis suggest two points of transition in the cytokine response
Clustering of gene expression profiles allows us to better dissect the dynamics of the implicated functions (Supplementary Table 2). We thus analyzed the functional enrichments of the genes in each of the 8 clusters. A clear progression of gene activation may be seen in the functional enrichments of the over-expressed gene clusters (Fig. 1B). Going from early (1 h) to intermediate (3 h, 6 h) to late (24 h, 7 d) timepoints, the functions are shifting from an initial, acute inflammatory response, to more generalized functions of the immune system, to finally become associated with functions related to developmental pathways, apoptosis and the extracellular matrix. It thus appears that fibroblasts, after initially sensing the cytokine cue of Tnf, undergo a slow process of switching on major developmental and apoptotic pathways. Given our limited time resolution we can position this transition sometime between 6 h and 24 h after the initial stimulation.
An interesting cluster (Light Green, Fig. 1A) perhaps better reflects this gradual transformation. It contains 64 genes with very high early over-expression which, in time, subsides and is eventually turned into strong under-expression at the late stages. Functional analysis of this particular cluster reveals a prominence of the Tnf, MAPK and IL-17 signaling pathways, all known downstream targets of Tnf, exerted mostly through the activation of key transcriptional regulators (Supplementary Fig. 2). This is a clear example of an initial, very focused activation, which in time, expands to incorporate more generalized pathways, but eventually switches off to become strongly repressed. Such a reversal of immediate Tnf targets has been only suggested up to now but is very clearly shown here. We will return to this important observation when we discuss the dynamics of the functional repertoire in later sections.
The shift to developmental functions is also apparent in the down-regulated gene clusters (Fig. 1C). Under-expression is strongly associated with transcriptional regulation in the early stage (1 h after stimulation). Clusters related to under-expression after 3 h are mostly associated with developmental functions, while those that are specific to the later stages are additionally related to cell–cell interactions such as adhesion and migration. This may suggest that an initial regulatory program gets underway since relatively early. It crystalizes, later on, into major transfoming functions that significantly alter the properties of the cells. Again, this long-term effect of Tnf sustained stimulation is shown here for the first time in such a detailed fashion and is described more explicitly in the following.
3.3. Domains of focal deregulation (DFD) reflect spatial preferences of gene expression
Functional enrichment analyses are important but they cannot provide insight into the mechanisms, with which the cells deploy their genome in effecting dynamic changes in the regulatory program. We thus set out to investigate the positional aspect of the gene expression process using a topological enrichment approach. Our goal was to identify chromosomal regions with consistent differential expression in our search for links between the genome architecture and the effected gene expression program.
Through a computational approach inspired by signal processing and applied to gene expression data (see Methods) we were able to create segmentation maps of the genome based on the underlying differential gene expression values. An average of ∼300 such regions were defined in each of the five conditions in our dataset, each of which was assigned with a mean differential expression score, directly calculated from the values of the genes it contained. Regions with increased negative or positive score corresponded to areas where gene deregulation was topologically consistent. We will from hereon refer to these regions as Domains of Focal Deregulation (DFD). The DFD chromosomal positions, size in bases as well as their mean expression scores are visualized in Fig. 2A, which reveals significant differences between the five timepoints. Few DFDs in the early timepoint (1 h) undergo two waves of expansion at the already discussed transition points of 3 h and 24 h. At the same time, this quantitative expansion in genome coverage is not coming from the same chromosomal regions, as a number of DFDs emerge and others are depleted between consecutive timepoints. This may be seen in Fig. 2B, where we plot the percentage of genome coverage at each timepoint, alongside the corresponding coverage percentages of the sets of overlapping DFDs between them.
Fig. 2.

Domains of Focal Deregulation. A. Mouse genome Domainograms showing significant Domains of Focal Deregulation (DFD) with a mean absolute score ≥ 0.1. Red is positive (over-expression), blue is negative (under-expression) color-coded for intensity. The expansion of the size and number of DFDs is evident, as is the predominance of negative domains, suggestive of an increased clustering of under-expressed genes. B. Genome coverage by DFD as a function of time. Values in the diagonal correspond to genome coverage for each timepoint separately, while the rest of the values correspond to the percentage of the genome covered by DFDs that are overlapping between timepoints. Notice how coverage is clustered in three groups (early: 1 h, intermediate: 3 h, 6 h and late: 24 h, 7 d). C. Size distributions of DFDs for different timepoints. Significant expansions in the size of the DFDs (p. value ≤ 0.05) are observed for the two transition points (1 h → 3 h and 6 h → 24 h). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
It is obvious from Fig. 2A and 2B that there are two points of expansion, with the percentage of genome covered almost doubling at 3 h and then again at 24 h. At the same time, the transitions we have underlined from the functional analyses are also reflected in the similarity between DFD distribution, as may be seen in the dendrogram of Fig. 2B. Almost 75% of the DFDs are shared between 24 h and 7 days and more than half are common between 3 h and 6 h, but less than one third of the early DFDs are overlapping with those of subsequent timepoints.
The expansion of DFDs, associated with the two transition points, is also evident in their size distribution as DFDs at 3 h and 24 h are significantly longer (Fig. 2C). This cannot be directly attributed to the extent of differential gene expression as the corresponding DEG numbers are quite different (643 and 190 for 3 h and 24 h respectively). Rather, it may be that it is a reflection of a general reorganization of the genome at topological level that occurs through a more broad distribution of genes with particular functional roles. It thus seems that in the transition from 6 h to 24 h the cells undergo a spatial “dispersion” in terms of gene deregulation, a fact that is linked to an extensive re-programming, with different genes being regulated in much wider genomic areas (see also Fig. 1A, Supplementary Fig. 2). In the following we provide evidence in support of this.
In all, we see that changes in gene regulation associated with the transition from 1 h to 3 h and from 6 h to 24 h are also reflected in the topological distribution of genes in linear chromosomal space. At both transition points we observe an expansion in the number and size of DFDs, which is a clear indication that extensive gene expression changes are not confined to the activation of functional pathways but are also associated with changes in the general chromosomal environment. In fact, a plausible hypothesis is that altered chromosomal accessibility may driving such generalized changes. In the following section we turn our attention to the study of this spatial enrichment of gene deregulation, coupled with its functional fingerprint.
3.4. DFDs reflect variable clustering of differentially expressed genes and are primarily associated with gene repression
Comparison of the extent and size of DFDs in the domainograms of Fig. 2A and the numbers of deregulated genes (see Supplementary Table 1) suggests that they are not directly related. The emergence of DFDs is due to a local clustering of differentially expressed genes rather than to their overall abundance. In order to test this we calculated the global and local enrichments of DFDs in differentially expressed genes in each timepoint as described in Methods (Fig. 3A). Even though, general enrichment of DEGs in DFDs is expected by definition, the observed enrichment patterns show great variability that reflects the progression of gene expression. We find a decreasing degree in the overall enrichment with time, suggesting that the general trend is one towards more s dispersed gene deregulation patterns.
Fig. 3.

Gene Expression in Domains of Focal Deregulation. A. Overlap enrichments of differentially expressed genes (DEGs) in DFDs for all, over- and under-expressed DEGs. Height of bars corresponds to enrichments calculated as observed/expected ratios of overlap. Significance levels shown on top of bars are: ***: <0.001, **:<0.01, *:<0.05. B. Distributions of DEG enrichment values on a per DFD basis. Significant changes (p ≤ 0.05) are observed between 1 h → 3 h, 6 h → 24 h and 24 h → 7 d. The change in the first transition is towards smaller enrichments (diffusion of DEGs) while the one in the second transition is towards greater enrichments (clustering of DEGs).
Early (1 h) DFDs were more enriched in differentially expressed genes but this effect is gradually diminished. More interestingly the largest part of DEG clustering occurs in the case of gene repression as may be seen when we plot enrichments separately for over- and under-expressed genes (Fig. 3A). Under-expressed genes are more than 3-fold enriched in the early timepoint, with their preference for DFDs dropping to almost half at the first transition point (3 h). In the opposite manner, clustering of over-expressed genes increases in DFDs after the first transition and then also stably returns to the levels of random expectancy. The tendency of under-expressed genes to cluster in DFDs may be also seen directly in the domainograms of Fig. 2A (where under-expression is shown in blue and over-expression in red), as well as in the distribution of differential expression scores (Supplementary Fig. 4), where the majority of DFDs show significantly low (negative) scores. These observations are indicative of connections with the three-dimensional genome structure as topologically-associated domains (TADs) are commonly associated with strongly repressive chromatin. It appears thus that spatial clustering of gene expression may partly reflect underlying genomic properties at the structural level and that, moreover, such properties come into play during the progression of cellular response to Tnf stimulation.
The overall enrichments shown above can be masked by effects that are related to the size distributions of DFDs, which as we saw earlier are also significantly variable between timepoints. In order to have a clearer view of DEG clustering, we calculated DEG enrichments at local level, that is for each DFD separately. Here we find a significant clustering at both the initial (1 h) and the 24 h timepoints (Fig. 3B). It thus appears that, while the initial timepoint may represent an early bookmarking of particular regions, a general redistribution of differential gene expression occurs at 24 h. Notice that in Fig. 3B both transitions (1 h → 3 h) and (6 h → 24 h) show highly significant changes in terms of enrichments. The first of the two is accompanied by a drop (1 h → 3 h) while the second by an increase, which are suggestive of different topological shifts between them. Hence, the transition from 1 h to 3 h appears to be more one of DFD expansion, with the subsequent “dilution” of DEG enrichment, while the one from 6 h → 24 h is more likely reflecting a general re-distribution of DFDs with the overall degree of DEG clustering in them increasing. This fits well with what we already knew for the early response, meaning a generalization of the initial immediate response that activates downstream inflammatory and immune-related pathways. On the other hand, it indicates something that was not expected for the second transition point, suggesting that around 24 h, there is a radical shift in gene deregulation at all levels (genes, pathways and genomic regions).
When we analyzed the enrichments of DFDs for genes belonging to the eight time-dependent gene expression clusters, we also found few but representative enrichments, in agreement with the overall tendency for under-expressed genes to cluster in DFDs (Supplementary Fig. 5). Early under-expressed genes were particularly enriched in DFDs (see also Fig. 3A), while over-expressed genes in both intermediate (3 h, 6 h) and late (24 h, 7 d) timepoints showed a general avoidance for DFD clustering. These findings are suggestive of different topological clustering tendencies for different functional categories, the study of which is the primary goal of this work. We next turned to the combined analysis of spatial/topological and functional enrichments in gene expression profiles through the introduction of bipartite topological-functional networks.
3.5. Topological-functional bipartite networks monitor gene regulation progression
Having already established a methodology to assess enrichments at both functional and positional level, we went on to combine the two aspects. We defined bipartite positional-functional gene enrichment networks as described in Methods. After constructing bipartite networks for each of the five timepoints, we performed an edge-betweeness modularity analysis [41] in order to identify network modules, which in this context, would represent genomic regions strongly connected with a particular set of functions. Inspection of the resulting networks (Fig. 4) leads to a number of interesting observations.
Fig. 4.

Bipartite Networks for early, intermediate and late stages of stimulation Bipartite networks for early, intermediate and late stages. Only names of DFD coordinates are shown, while functions are summarised in distinct modules. Module functions written in green are associated with over-expression, while those in red with under-expression. The expansion with time is evident as is the increase in modularity, which corresponds to increasing compartmentalization of functions. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Firstly, the number of DFDs and associated functions are, to some extent, reflected on the size of the networks. The early (1 h) consists of only four modules while the late (24 h and 7 d) contain 7 and 10 modules respectively. The modularity of the networks is also increasing with time, rising from 0.45 to 0.80 for 24 h, which is suggestive of stronger links between chromosomal regions and associated functions as time progresses.
More detailed examination of the networks reveals a number of key elements related to the way cells are affected by the prolonged stimulation by Tnf. Initial stimulation (1 h) affects a pair of core modules associated with cytokine and chemokine signaling. These are located in chromosomes 4 and 5 respectively but they are not both affected in the same way. The cytokine module (chr4) consists primarily of over-expressed genes, while the chemokine and chemotaxis is enriched in under-expressed genes.
This core pair of modules is also part of the first of the intermediate networks (3 h), in which it is expanded through the addition of an interferon/Tnf-related module (chr11). One major change in the 1 h → 3 h transition is the activation of the chemokine/chemotaxis module, which leads to the formation of a core of three activated modules (cytokine, chemokine and interferon) bridging parts of chromosomes 4, 5 and 11 respectively. A set of additional, smaller modules, associated with the immune response, development and apoptosis also arise at this stage. Progression to 6 h brings about two major changes. First, an expansion of the chemokine module with an additional module associated to chr9 and second, the switch of the cytokine module, which still forms part of the network core, to under-expression. This is an indication of the first signs of a shutdown of inflammatory functions, which will be generalised in later stages. A set of under-expressed modules related to cell adhesion, apoptosis and development complete the network.
The second major transition from 6 h to 24 h is accompanied by the complete disappearance of the cytokine-chemokine-interferon core. The highly modular 24 h network is the most fragmented one, comprising a set of isolated modules among which a number of functions such as cell adhesion, development and differentiation are associated with under-expression. These last three modules become over-expressed in the network of day 7, in which, we additionally observe the re-emergence of the cytokine-chemokine initial core, now strongly associated with under-expression. This module is strongly associated with the small cluster of genes (Light Green, Fig. 1A), which undergo the most intense deregulation from a strong initial over-expression to an equally strong late under-expression. Follow up experiments on the dynamics of the chromatin environment of these gene loci would be a first priority.
3.6. Functional dynamics of bipartite networks are consistent with two transition points at 3 h and 24 h
Having observed extensive changes in the bipartite networks we went on to assess the changes in a quantitative manner by examining the number and type of functional modules that are emerging, removed, expanded or contracted in the networks. A simple analysis of the number of times a function appears in each network is shown in Fig. 5A and is again representative of two major transition points in the system under study. Fig. 5A shows an initial, large increase in the number of functions as we move from 1 h to 3 h. The 3 h-acquired functions are related to chemotaxis and interferon signaling as suggested by the network modules. There are overall large similarities in the functional patterns of 3 h and 6 h, but these are followed by a radical depletion of the largest part of the functions in 24 h. Thus, this second transition point is marked by extensive re-organization of functions.
Fig. 5.

Bipartite network dynamics. A. DFDs numbers per function in bipartite networks. Each cell in the heatmap shows the number of DFDs associated with each function. Functions with 0 DFD (blue) are absent from the corresponding bipartite network. An expansion at 3 h and a subsequent decrease and redistribution at 24 h is evident. B. Percentage of DFDs belonging to each dynamic category for comparisons between consecutive timepoints. Emergence is prominent for the early transition, while deletion is quite significant for the late one. C. Number of gained/lost functions per DFD in the bipartite network comparisons between consecutive timepoints. 1 h → 3 h transition is marked by an acquisition of functions while 6 h → 24 h by a general loss. D. Functions clustered to DFDs behave differently between timepoints. Enriched functions were divided into those associated with a DFD in bipartite networks and those that were not. Mean enrichment of functions that are associated with DFDs is smaller than the one of functions that are not attached to a DFD for the early stage. The situation is inversed for the 24 h timepoint. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
Most of the inflammation and immune response-related functions are not present at 24 h. Instead they have been substituted by pathways associated with development, cell adhesion and cell motility, which are suggestive of major transformations occurring in fibroblasts under prolonged Tnf stimulation. A subsequent expansion of similar developmentally-related functions occurs in the latest timepoint (7 days). Most of the intermediate stage (3 h, 6 h) functions are absent but functional terms such as cell adhesion, cell differentiation and cell migration are prominent.
3.7. Dual dynamics of positional and functional enrichments suggest intermediate expansion followed by late re-organization
The changes in the functional footprint through time, discussed above, are also accompanied with changes in the domains of focal deregulation. As domain boundaries are more difficult to identify directly, we implemented a computational approach based on chromosomal coordinate overlap to assess the qualitative nature of the change. In this way, domains that are present in a given timepoint but not overlapping any corresponding domain in a subsequent timepoint are considered to be “depleted”, while the opposite are assigned as “emergent”. More complex cases of domains which overlap, were identified as either “contracted” or “expanded” depending on whether their boundaries were stretched or withdrawn between consecutive timepoints.
The results of this analysis are shown in Fig. 5B, where the percentage of each of the four categories of domain changes was calculated over the total extent of DFD coverage. The two transition points (1 h → 3 h) and (6 h → 24 h) show the greatest degree of domain changes but in different ways. The early transition point (1 h → 3 h) is marked by the emergence of a large number of new domains (>50% of the total), while the rest correspond to expansions. One the other hand, the 6 h → 24 h transition is the only such that contains contracted domains with a similar proportion of expansion, deletion and emergence signifying a general domain re-organization.
3.8. Prolonged Tnf stimulation goes through an initial expansion and a late-stage contraction of activated functions
Fig. 5A suggests two major shifts in the number of enriched functions between timepoints. Since these changes are tightly linked to DFD dynamics we wanted to test this further by looking into each DFD separately. We calculated the number of functions attributed to each DFD and compared them between consecutive timepoints, distinguishing between “gained” and “lost” functions. We then plotted the corresponding numbers for each DFD and grouping for the four transitions (1 h → 3 h, 3 h → 6 h, 6 h → 24 h and 24 h → 7d) in Fig. 5C. The plot shows two distinct trends in the shape of the bubbles (with horizontal and vertical orientations corresponding to predominant gain and loss of functions respectively). One can see that 1 h → 3 h and 24 h → 7 d transitions are associated with function gain and 3 h → 6 h and 6 h → 24 h with function loss, while it is also clear that the effects are stronger for the 1 h → 3 h expansion and the 6 h → 24 h contraction. Indeed, it is these two transitions that are also statistically significant in terms of number of acquired/lost functions per DFD (Supplementary Fig. 6).
An overview of the analyses of our bipartite network approach strongly suggests that the two major transition points in the prolonged stimulation of fibroblasts by Tnf are also qualitatively different, with an early (1 h → 3 h) expansion that is probably reflecting the generalization of the immune signaling response, followed by a major shutdown of immune-related pathways in 24 h.
3.9. Focal functions attract a greater number of differentially expressed genes
Our data suggest complex dynamics of enriched functions, being lost and gained from equally volatile DFDs. This dynamics is, however, confined to the subset of functions that are associated with DFDs. A large number of functional categories are also enriched in differentially expressed genes without being linked to focal deregulation. These are being enriched in genes that are distributed more broadly in genome space and may thus be subject to different expression biases. We tested these differences through a comparison of enrichment p-values for functional terms associated with DFDs (focal functions) against non-focal functions, that are enriched but not linked to particular chromosomal regions (Fig. 5D). Interestingly, we find significant differences between early (1 h) and late (24 h) stages. At the early stage, focal functions are significantly more enriched than non-focal ones, while the opposite holds for the 24 h timepoint. This result, also implied by the bipartite network modularity analysis, is suggestive of an increased fragmentation of differential gene expression into distinct regions and functions.
Together, the results from the bipartite network analyses provide a solid framework for the understanding of cellular response to a sustained cytokine cue. In the particular case of Tnf activation, an initial generalization of an immediate acute response is readily dampened and eventually reversed. This change is moreover, accompanied with a broad redistribution of gene deregulation in the genome.
4. Discussion
Our work constitutes one of the first attempts to incorporate positional information in the analyses of gene expression. The starting hypothesis is that differential gene expression may be clustered in confined regions of the linear genome due to regulatory, epigenetic and structural constraints. Indeed, elements of the three-dimensional genome structure such as TADs [42] have been shown to delineate genomic space with particular transcriptional tendencies, while we [23] and others [43] have demonstrated focally increased transcription in TAD boundaries. More recent works have focused on the regulatory potential of self-contained linear genomic regions, thus called cis-regulatory domains (CRDs) [15]. Through a relatively simple approach, we herein demonstrate that regions of consistent differential expression are common and may, moreover, provide insight in the way a gene expression program develops in time.
Implementation of this approach on the prolonged activation of epithelial cells by Tnf leads to the observation of significant enrichments for under-expressed compared to over-expressed genes. Even though we cannot exclude that it may be a particular characteristic of the system under study, it is worth noticing that repressive domains are readily formed in eukaryotic genomes mediated by Polycomp-Group (PcG) proteins [44] or through the association of genomic regions with the nuclear lamina [45]. Thus, it will not be surprising to find that there is a stronger overall clustering tendency for repressive genes in order to maintain transcriptional silencing.
Another interesting aspect that comes out of our time-dependent study is related to the progressive expansion of the genome space that is covered by DFDs. Since this is not correlated with the number of differentially expressed genes (the coverage by DFDs peaks at 24 h, where we have the smallest number of DEGs) we may assume that it reflects a propensity for increased genome compartmentalization. Indeed, we find that, with time, and independently of the number of differentially expressed genes, expression tends to become segmented into a greater number of regions and this is, moreover, accompanied by an increase of the bipartite network modularity. Again, while this may be a singular property of the cytokine response, it deserves to be studied in more detail and in different systems.
Besides providing insight on the functional modularity of gene expression profiles, the bipartite networks that we describe in this work may also assist in the formulation of hypotheses on genome organization. Interacting modules observed in the bipartite networks of the intermediate stages (3 h and 6 h) show strong functional interactions between regions from different chromosomes (chr4, chr5, chr9 and chr11 in particular). It would be really interesting to investigate whether such interactions are also reflected upon the three-dimensional organization of the genome. Even though inter-chromosomal interactions are inherently difficult to detect, new methodological approaches such as SPRITE [46] and GAM [47] would probably allow us to test similar hypotheses.
Another promising aspect of our work is related to the analysis of genes belonging to focal vs non-focal functional categories. Functional categories that tend to have their genes clustered in close proximity are more likely to be enriched depending on the stage of the process under study and this may be an indication of a more focal or more widespread expression program. One interesting question would be to examine genes, whose expression may be attributed to their relative position rather than their participation in a certain pathway. We would call these “by-stander genes” as, in essence, one could suggest that their mis-expression is driven by nearest neighbor effects. Modeling the likelihood for positional vs functional drive of such by-standers is a very interesting prospect especially in the context of stimulation by cytokines as they (and Tnf in particular) have been associated with the development of auto-immunity. The expansion of gene expression in broad areas of the genome that we describe herein, may accidentally trigger the activation of genes and pathways that are not directly related to the initial response, which could, under given circumstances, eventually lead to the development of pathological conditions.
Funding
This work was supported by the Operational Program “Human Resources Development, Education and Lifelong Learning” and is co-financed by the European Union (European Social Fund) and Greek National Funds. (Grant Number: 10038).
Acknowledgments
Acknowledgments
The authors are grateful to Prof. George Kollias for providing access to experimental facilities at BSRC “Alexander Fleming”, Greece. We would also like to thank Dr. Vangelis Ntougkos for assistance in the performance of experiments and Eleni Lianoudaki for proofreading this manuscript. We also acknowledge three anonymous reviewers for valuable comments and suggestions.
Conflicting interests
The authors declare no conflicting interests.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.01.001.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Common differentially expressed genes (DEG) across all time points. Horizontal coloured bars correspond to total DEG for each time point (ranked according to gene number), while vertical bars denote number of common DEG corresponding to set intersection shown in the grid below (ranked from higher to lower gene numbers).
Functional Enrichment analysis of the High-Low expression cluster (top 20 enriched terms). Functionally enriched terms almost entirely belonged to transcriptional regulatory factors affecting primary functions such as metabolism, in particular of macromolecules.
Distributions of differential expression values for each of the 8 defined clusters across timepoints.
Distributions of mean DFD differential expression scores across timepoints.
DFD enrichments (in log2 scale) for genes belonging in each of the 8 defined clusters. Few positive enrichments are all associated with down-regulated clusters. Negative enrichments, suggesting overall avoidance are associated with up-regulated clusters.
Distribution of the difference between Gained and Lost functions (Ngain-Nloss) for bipartite networks of consecutive timepoints. An initial gain for the 1h→3h transition is reversed to a predominance of lost functions for 6 h → 24 h.
Numbers of differentially expressed genes (over-expressed, under-expressed and general total) for each timepoint.
Complete list of enriched terms (GO and KEGG) for each of the 8 differentially expressed gene clusters.
Data Availability
Processed Gene Expression data, differential gene expression lists, details on the gene clusters, DFDs and bipartite networks as well as custom R scripts used in the analyses are deposited in Mendeley Data. Doi:https://doi.org/10.17632/fytpjj5ny5.4
References
- 1.Caron H., van Schaik B., van der Mee M., Baas F., Riggins G., van Sluis P. The human transcriptome map: clustering of highly expressed genes in chromosomal domains. Science. 2001;291:1289–1292. doi: 10.1126/science.1056794. [DOI] [PubMed] [Google Scholar]
- 2.Ebisuya M., Yamamoto T., Nakajima M., Nishida E. Ripples from neighbouring transcription. Nat Cell Biol. 2008;10:1106–1113. doi: 10.1038/ncb1771. [DOI] [PubMed] [Google Scholar]
- 3.Rennie S., Dalby M., van Duin L., Andersson R. Transcriptional decomposition reveals active chromatin architectures and cell specific regulatory interactions. Nat Commun. 2018;9(1):487. doi: 10.1038/s41467-017-02798-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ghanbarian A.T., Hurst L.D. Neighboring genes show correlated evolution in gene expression. Mol Biol Evol. 2015;32:1748–1766. doi: 10.1093/molbev/msv053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Batada N., Hurst L. Evolution of chromosome organization driven by selection for reduced gene expression noise. Nat Genet. 2007;39:945–949. doi: 10.1038/ng2071. [DOI] [PubMed] [Google Scholar]
- 6.Hurst L.D., Pál C., Lercher M.J. The evolutionary dynamics of eukaryotic gene order. Nat Rev Genet. 2004;5:299–310. doi: 10.1038/nrg1319. [DOI] [PubMed] [Google Scholar]
- 7.Lee J.M., Sonnhammer E.L.L. Genomic gene clustering analysis of pathways in eukaryotes. Genome Res. 2003;13:875–882. doi: 10.1101/gr.737703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tiirikka T., Siermala M., Vihinen M. Clustering of gene ontology terms in genomes. Gene. 2014;550:155–164. doi: 10.1016/j.gene.2014.06.060. [DOI] [PubMed] [Google Scholar]
- 9.Krefting J., Andrade-Navarro M.A., Ibn-Salem J. Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biol. 2018;16:87. doi: 10.1186/s12915-018-0556-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coolen M.W., Stirzaker C., Song J.Z., Statham A.L., Kassir Z., Moreno C.S. Consolidation of the cancer genome into domains of repressive chromatin by long-range epigenetic silencing (LRES) reduces transcriptional plasticity. Nat Cell Biol. 2010;12:235–246. doi: 10.1038/ncb2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanay A., Cavalli G. Chromosomal domains: epigenetic contexts and functional implications of genomic compartmentalization. Curr Opin Genet Dev. 2013;23:197–203. doi: 10.1016/j.gde.2012.12.009. [DOI] [PubMed] [Google Scholar]
- 12.Ernst J., Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28:817–825. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ernst J., Kellis M. Interplay between chromatin state, regulator binding, and regulatory motifs in six human cell types. Genome Res. 2013;23(7):1143–1154. doi: 10.1101/gr.144840.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kharchenko P.V., Alekseyenko A.A., Schwartz Y.B., Minoda A., Riddle N.C., Ernst J. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature. 2011;471:480–485. doi: 10.1038/nature09725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delaneau O., Zazhytska M., Borel C., Giannuzzi G., Rey G., Howald C. Chromatin three-dimensional interactions mediate genetic effects on gene expression. Science. 2019;364:eaat8266. doi: 10.1126/science.aat8266. [DOI] [PubMed] [Google Scholar]
- 16.Mitelman F., Johansson B., Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer. 2007;7:233–245. doi: 10.1038/nrc2091. [DOI] [PubMed] [Google Scholar]
- 17.Cohen B.A., Mitra R.D., Hughes J.D., Church G.M. A computational analysis of whole-genome expression data reveals chromosomal domains of gene expression. Nat Genet. 2000;26:183–186. doi: 10.1038/79896. [DOI] [PubMed] [Google Scholar]
- 18.Kosak S.T., Groudine M. Gene order and dynamic domains. Science. 2004;306:644–647. doi: 10.1126/science.1103864. [DOI] [PubMed] [Google Scholar]
- 19.Gerstein M.B., Kundaje A., Hariharan M., Landt S.G., Yan K.-K., Cheng C. Architecture of the human regulatory network derived from ENCODE data. Nature. 2012;489:91–100. doi: 10.1038/nature11245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang D.W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiaromonte F., Miller W., Bouhassira E.E. Gene length and proximity to neighbors affect genome-wide expression levels. Genome Res. 2003;13:2602–2608. doi: 10.1101/gr.1169203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsochatzidou M., Malliarou M., Papanikolaou N., Roca J., Nikolaou C. Genome urbanization: clusters of topologically co-regulated genes delineate functional compartments in the genome of Saccharomyces cerevisiae. Nucleic Acids Res. 2017;45:5818–5828. doi: 10.1093/nar/gkx198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nikolaou C. Invisible cities: segregated domains in the yeast genome with distinct structural and functional attributes. Curr Genet. 2018;64:247–258. doi: 10.1007/s00294-017-0731-6. [DOI] [PubMed] [Google Scholar]
- 25.Chen E.Y., Tan C.M., Kou Y., Duan Q., Wang Z., Meirelles G.V. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinf. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reimand J., Kull M., Peterson H., Hansen J., Vilo J. G:Profiler-a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007;35:W193–W200. doi: 10.1093/nar/gkm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hao S., Baltimore D. RNA splicing regulates the temporal order of Tnf-induced gene expression. Proc Natl Acad Sci USA. 2013;110(29):11934–11939. doi: 10.1073/pnas.1309990110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hao S., Baltimore D. The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol. 2009;10:281–288. doi: 10.1038/ni.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Diermeier S., Kolovos P., Heizinger L., Schwarz U., Georgomanolis T., Zirkel A. Tnfα signalling primes chromatin for NF-κB binding and induces rapid and widespread nucleosome repositioning. Genome Biol. 2014;15:536. doi: 10.1186/s13059-014-0536-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Papantonis A., Kohro T., Baboo S., Larkin J.D., Deng B., Short P. Tnfα signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J. 2012;44:1–11. doi: 10.1038/emboj.2012.288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keffer J., Probert L., Cazlaris H., Georgopoulos S., Kaslaris E., Kioussis D. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. EMBO J. 1991;10:4025–4031. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ntougkos E., Chouvardas P., Roumelioti F., Ospelt C., Frank-Bertoncelj M., Filer A. Genomic responses of mouse synovial fibroblasts during tumor necrosis factor-driven arthritogenesis greatly mimic those in human rheumatoid arthritis. Arthritis Rheumatol (Hoboken, N.J.) 2017;69:1588–1600. doi: 10.1002/art.40128. [DOI] [PubMed] [Google Scholar]
- 33.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapnell C., Williams B.A., Pertea G., Mortazavi A., Kwan G., van Baren M.J. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karagianni N., Kranidioti K., Fikas N., Tsochatzidou M., Chouvardas P., Denis M.C. An integrative transcriptome analysis framework for drug efficacy and similarity reveals drug-specific signatures of anti-Tnf treatment in a mouse model of inflammatory polyarthritis. PLoS Comput Biol. 2019;15 doi: 10.1371/journal.pcbi.1006933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hothorn T., Hornik K., Zeileis A. Unbiased recursive partitioning: a conditional inference framework. J. Comput. Graph. Stat. 2006;15:651–674. [Google Scholar]
- 37.Zeileis A., Leisch F., Hornik K., Kleiber C. Strucchange: an R package for testing for structural change in linear regression models. J Stat Softw. 2002;7(2):1–38. [Google Scholar]
- 38.Zeileis A., Kleiber C., Krämer W., Hornik K. Testing and dating of structural changes in practice. Comput Stat Data Anal. 2003;44(1–2):109–123. [Google Scholar]
- 39.Andreadis C., Nikolaou C., Fragiadakis G.S., Tsiliki G., Alexandraki D. Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Res. 2014;42(20):12650–12667. doi: 10.1093/nar/gku915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Csardi G., Nepusz T. The igraph software package for complex network research. InterJournal. 2006:1695. Complex Sy. [Google Scholar]
- 41.Barber M.J. Modularity and community detection in bipartite networks. Phys Rev E - Stat Nonlinear Soft Matter Phys. 2007;76 doi: 10.1103/PhysRevE.76.066102. [DOI] [PubMed] [Google Scholar]
- 42.Dixon J.R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ulianov S., Razin S., Shevelyov Y. Active chromatin and transcription play a key role in chromosome partitioning into TADs. Genome Res. 2015;26(1):70–84. doi: 10.1101/gr.196006.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kundu S., Ji F., Sunwoo H., Jain G., Lee J.T., Sadreyev R.I. Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol Cell. 2017;65:432–446.e5. doi: 10.1016/j.molcel.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Steensel B., Belmont A.S. Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell. 2017;169(5):780–791. doi: 10.1016/j.cell.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quinodoz S.A., Ollikainen N., Tabak B., Palla A., Schmidt J.M., Detmar E. Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell. 2018;174:744–757.e24. doi: 10.1016/j.cell.2018.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Beagrie R.A., Scialdone A., Schueler M., Kraemer D.C.A., Chotalia M., Xie S.Q. Complex multi-enhancer contacts captured by genome architecture mapping. Nature. 2017;543(7646):519–524. doi: 10.1038/nature21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Common differentially expressed genes (DEG) across all time points. Horizontal coloured bars correspond to total DEG for each time point (ranked according to gene number), while vertical bars denote number of common DEG corresponding to set intersection shown in the grid below (ranked from higher to lower gene numbers).
Functional Enrichment analysis of the High-Low expression cluster (top 20 enriched terms). Functionally enriched terms almost entirely belonged to transcriptional regulatory factors affecting primary functions such as metabolism, in particular of macromolecules.
Distributions of differential expression values for each of the 8 defined clusters across timepoints.
Distributions of mean DFD differential expression scores across timepoints.
DFD enrichments (in log2 scale) for genes belonging in each of the 8 defined clusters. Few positive enrichments are all associated with down-regulated clusters. Negative enrichments, suggesting overall avoidance are associated with up-regulated clusters.
Distribution of the difference between Gained and Lost functions (Ngain-Nloss) for bipartite networks of consecutive timepoints. An initial gain for the 1h→3h transition is reversed to a predominance of lost functions for 6 h → 24 h.
Numbers of differentially expressed genes (over-expressed, under-expressed and general total) for each timepoint.
Complete list of enriched terms (GO and KEGG) for each of the 8 differentially expressed gene clusters.
Data Availability Statement
Processed Gene Expression data, differential gene expression lists, details on the gene clusters, DFDs and bipartite networks as well as custom R scripts used in the analyses are deposited in Mendeley Data. Doi:https://doi.org/10.17632/fytpjj5ny5.4