

Significance
Necroptosis is a form of cell death implicated in many human diseases. A series of requirements has to be fulfilled before the cell makes the ultimate decision to die. One such essential event in necroptosis is the phosphorylation of RIPK3. It has previously been suggested that RIPK3 might phosphorylate itself. Here we discover that CK1 family kinases directly phosphorylate RIPK3. In cells without CK1 proteins, RIPK3 is not phosphorylated and the cells remain alive when exposed to necroptosis-inducing signals. Additionally, when cells express a form of RIPK3 which cannot be recognized by CK1, the cells also remain alive. This work defines a step in the necroptosis pathway and identifies potential targets for intervening necroptosis-associated diseases.
Keywords: necrosome, casein kinase 1, RIPK3 phosphorylation, necroptosis, MLKL
Abstract
Necroptosis is a regulated necrotic cell death pathway, mediated by a supermolecular complex called the necrosome, which contains receptor-interacting protein kinase 1 and 3 (RIPK1, RIPK3) and mixed-lineage kinase domain-like protein (MLKL). Phosphorylation of human RIPK3 at serine 227 (S227) has been shown to be required for downstream MLKL binding and necroptosis progression. Tandem immunoprecipitation of RIPK3 reveals that casein kinase 1 (CK1) family proteins associate with the necrosome upon necroptosis induction, and this interaction depends on the kinase activity of RIPK3. In addition, CK1 proteins colocalize with RIPK3 puncta during necroptosis. Importantly, CK1 proteins directly phosphorylate RIPK3 at S227 in vitro and in vivo. Loss of CK1 proteins abolishes S227 phosphorylation and blocks necroptosis. Furthermore, a RIPK3 mutant with mutations in the CK1 recognition motif fails to be phosphorylated at S227, does not bind or phosphorylate MLKL, and is unable to activate necroptosis. These results strongly suggest that CK1 proteins are necrosome components which are responsible for RIPK3-S227 phosphorylation.
Necroptosis is a form of regulated immunogenic cell death which is implicated in many pathological human conditions, such as infection, inflammation, neurodegeneration, and cancer (1–6). One of the best-studied necroptosis pathways is induced by tumor necrosis factor (TNF) (T), with simultaneous treatment of small molecules Smac-mimetic (S) to inhibit the NF-κB survival pathway and Z-VAD-FMK (Z) to inhibit apoptosis (7). The combination of TNF, Smac-mimetic, and Z-VAD-FMK (TSZ) treatment triggers a signaling cascade that ultimately results in membrane permeabilization. Specifically, TSZ treatment leads to the phosphorylation and activation of receptor interacting protein kinase 1 (RIPK1), which then recruits receptor interacting protein kinase 3 (RIPK3) (7–11). RIPK3 subsequently recruits mixed-lineage kinase domain-like protein (MLKL) to form a supermolecular complex called necrosome (12, 13), which is essential for cell death execution.
RIPK1 and RIPK3 each contain an N-terminal kinase domain followed by a RIP homotypic interaction motif (RHIM) (14). The RHIM domain mediates homo- or heterointeraction, which is the driving force for the oligomerization of RHIM-containing proteins to activate necroptotic death. For example, the RHIM domain of RIPK1 can interact with the RHIM domain of another RIPK1 molecule or the RHIM domain of RIPK3. In addition, many pathogens activate necroptosis through RHIM-containing proteins, such as TIR domain-containing adapter-inducing IFN-β (TRIF), or Z-nucleic acid binding protein 1 (ZBP1/DAI), which interact with RHIM domain of RIPK3 to promote RIPK3 oligomerization (15–18). During necroptosis, RIPK1 and RIPK3 interaction results in the formation of amyloid fibrils through the RHIM domain. The core of RHIM amyloids consists of stacked β-sheets, with many side chains extending out from the core, which potentially could bring in other signaling proteins (19–21). The assembly of this highly oligomeric signal platform is essential for necroptosis (22). RHIM domain mutations of RIPK1 or RIPK3 that abolish RHIM–RHIM interaction also abolish the ability of RIPK1 or RIPK3 to activate necroptosis (7, 23, 24). For instance, the RIPK3-RHIM mutant fails to interact with RIPK1 or form homooligomers and does not induce necroptosis.
This signaling pathway is tightly regulated by phosphorylation. RIPK1, RIPK3, and MLKL are all phosphorylated at multiple positions (7–13, 25–32). In particular, the phosphorylation of human RIPK3 on serine 227 (S227) has been shown to be a critical step, without which RIPK3 will not recruit MLKL and the cells do not die (12). Furthermore, phosphorylation of human MLKL at threonine 357 and serine 358 by RIPK3 is also essential (29). Importantly, the kinase activity of both RIPK1 and RIPK3 is required for necroptosis (7–13). Inhibitors of RIPK1 or RIPK3 block cell death, and the kinase-dead RIPK1 or RIPK3 cannot activate necroptosis (9, 33–35). Specifically, the kinase-dead form of RIPK3 still binds RIPK1, but is not phosphorylated at S227 (12, 31). Thus it is believed that S227 is not phosphorylated by RIPK1; rather it is autophosphorylated. However, one cannot exclude the possibility that unidentified kinases may be involved in the phosphorylation of S227 other than RIPK1 or RIPK3.
Casein kinase 1 (CK1) is a family of serine/threonine kinases, which have been shown to phosphorylate many substrates that are involved in multiple cellular processes, including circadian rhythm, Wnt signaling, membrane transportation, p53 regulation, and cell division (36–38). The canonical CK1 substrate is pS/pT-X1–2-S/T, with a priming phosphorylation of serine or threonine at the −3 or −2 position to the target site. Alternatively, the priming phosphorylation can be substituted with multiple acidic residues (D/E) upstream of the target site (36–38).
In this report, we demonstrated that CK1α, CK1δ, and CK1ε associated with RIPK3 and the necrosome upon necroptosis induction. CK1 proteins directly phosphorylated RIPK3 at S227 in vitro and in vivo. Loss of CK1 proteins abolished S227 phosphorylation and blocked necroptosis. Furthermore, RIPK3-E221/225A, a mutant which could not be phosphorylated by CK1, failed to bind and phosphorylate MLKL, and was unable to activate necroptosis. These results strongly suggest that CK1 proteins are responsible for RIPK3 S227 phosphorylation.
Results
Casein Kinase 1 Family Proteins CK1α, CK1δ, and CK1ε Associate with the Necrosome upon Necroptosis Induction.
In order to identify additional proteins which might associate with the necrosome, a previously established HeLa cell line stably expressing HA–3xFlag-tagged RIPK3 was used for tandem immunoprecipitation (31). These cells readily undergo necroptosis with combination treatment of TNF, Smac-mimetic (to block the NF-κB pathway), and ZVAD-FMK (to block apoptosis) (TSZ). Necrosulfonamide (NSA) is an MLKL inhibitor, which allows the necrosome to form, but blocks cell death (12) (Fig. 1A). Tandem immunoprecipitation of RIPK3 and silver staining revealed several unique bands present in the TSZ/NSA treated sample (Fig. 1B). Mass spectrometry (MS) analysis identified RIPK3 and another known necrosome component RIPK1, and the bands between 43 kDa and 55 kDa were found to contain both casein kinase 1 family proteins CK1δ and CK1ε. These two proteins share 84% sequence identity and many identified peptides were assigned to both proteins.
Fig. 1.

CK1α, CK1δ, and CK1ε associate with the necrosome upon necroptosis induction. (A) Current understanding of necrosome formation. Treatment with TNFα (T), Smac-mimetic (S), and Z-VAD-FMK (Z) leads to RIPK1 phosphorylation and recruitment of RIPK3. Autophosphorylation of serine 227 of human RIPK3 is required for MLKL binding and necrosome formation. Subsequent phosphorylation of MLKL by RIPK3 eventually results in cell death. Compound NSA blocks necroptosis downstream of necrosome formation. (B) Identification of CK1δ and CK1ε from tandem immunoprecipitation. HeLa cells expressing HA-3xFlag-RIPK3 were treated with DMSO or TSZ plus NSA. Cell lysates were subjected to tandem immunoprecipitation with anti-Flag and anti-HA antibodies and then silver staining. Arrow pointed bands were cut out for MS analysis and identified as RIPK1, RIPK3, and CK1δ/CK1ε, respectively. (C) HeLa cells stably expressing 3xFlag-RIPK3 (WT) or 3xFlag-RIPK3-K50A (K50A) were treated with or without TSZ/NSA. Cell lysates were subjected to immunoprecipitation with anti-Flag antibody and Western blotting was performed with indicated antibodies. (D) HeLa cells (2 × 106) were transfected with 1 μg DNA encoding 3xFlag-RIPK3 or 3xFlag-RIPK3-K50A and treated with DMSO or TSZ/NSA. Cell lysates were subjected to immunoprecipitation with anti-Flag antibody and Western blotting was performed with indicated antibodies. (E) HeLa:GFP-RIPK3 cells were transfected with a construct encoding RFP-CK1ε. Live cell imaging was recorded with TSZ addition at time 0. Images were shown at 20 min, 3 h 50 min, and 10 h 40 min. The last column shows the three-dimensional construction of images at 10 h 40 min. (Scale bar, 10 μm.)
To confirm the interaction, we used a pair of previously established cell lines, stably expressing 3xFlag-RIPK3 (wild type [WT]) or 3xFlag-RIPK3-K50A (K50A) which is kinase dead (12, 31). K50A cells do not die upon TSZ treatment, confirming that RIPK3 kinase activity is required for necroptosis (12, 31). As previously reported, RIPK1 interacted with RIPK3 upon TSZ/NSA treatment (Fig. 1C, lane 6), and that interaction did not need RIPK3 kinase activity (lane 8). However, signal-induced MLKL interaction with RIPK3 was dependent on RIPK3 kinase activity (Fig. 1C, compare lanes 6 and 8). Interestingly, CK1ε interacted with RIPK3 in the same way as MLKL, suggesting that it might be a necrosome component.
The interaction was also confirmed with transient transfection experiments. HeLa cells, which do not express endogenous RIPK3, were transfected with a low amount of DNA (1 μg/2 × 106 cells) encoding 3xFlag-RIPK3 or 3xFlag-RIPK3-K50A. At this DNA amount, ectopically expressed RIPK3 did not cause cell death by itself and the cells only underwent necroptosis with TSZ treatment. Again, CK1δ and CK1ε only interacted with WT-RIPK3 under TSZ/NSA treatment (Fig. 1D, lane 6), but not with kinase-dead K50A mutant (lane 8). In addition, another CK1 family protein CK1α which shares about 70% sequence identity with CK1δ in the kinase domain, bound RIPK3 in the same manner.
RIPK3-CK1 interaction was further confirmed by fluorescent microscopy. HeLa cells stably expressing GFP-RIPK3 were transfected with a construct encoding RFP-CK1ε. GFP-RIPK3 was initially diffuse in the cytosol and TSZ treatment resulted in the formation of GFP-RIPK3 puncta, which have been shown to be aggregates of necrosome (12). The number and size of GFP-RIPK3 puncta increased with time. RFP-CK1ε also formed puncta in a similar fashion and the majority of RFP-CK1ε puncta colocalized with GFP-RIPK3 (Fig. 1E), suggesting that CK1ε associates with the supermolecular complex necrosome.
RIPK3 Directly Interacts with CK1 and the Interaction Requires an Intact RHIM Domain.
The core of the necrosome is formed by RIPK1 and RIPK3, which share common domains (Fig. 2A). Both proteins have an N-terminal kinase domain and a C-terminal RHIM domain which mediates both homo-and heterooligomerization (14). Additionally, RIPK1 contains a C-terminal death domain (Fig. 2A). In order to identify which protein in the necrosome directly interacts with CK1, constructs encoding myc-tagged RIPK1 or RIPK3 were transfected into HeLa cells at 5 μg/2 × 106 cells. With this amount of DNA, overexpressed RIPK1 and RIPK3 induced cell death, bypassing signal requirement. Immunoprecipitation with anti-myc antibody revealed that RIPK3 bound endogenous CK1, while RIPK1 did not (Fig. 2B).
Fig. 2.

RIPK3 directly interacts with CK1 and the interaction requires an intact RHIM domain. (A) Diagrams of RIPK1, RIPK3, and RIPK3 truncations. RHIM, RIP homotypic interaction motif. In (324 to 518)-RHIMm, amino acids QVGD at 459 to 462 were all mutated to alanines, which abolishes RHIM-mediated interactions. (B) HeLa cells (2 × 106) were transfected with 5 μg DNA encoding myc-tagged RIPK3 or RIPK1. Cell lysates were harvested after 36 h and subjected to immunoprecipitation with anti-Myc antibody. Western blotting was performed with indicated antibodies. (C) HeLa cells (2 × 106) were transfected with 5 μg DNA encoding 3xFlag-tagged RIPK3 truncations. Cell lysates were harvested after 36 h and subjected to immunoprecipitation with anti-Flag antibody. Western blotting was performed with indicated antibodies. The 14-3-3 served as a negative control. (D) Summary of the interaction results. RIPK1 recruits RIPK3 through hetero-RHIM domain interaction. RIPK3 then forms homooligomers through its RHIM domain. Oligomerized RIPK3 recruits CK1 through the RHIM-containing region and recruits MLKL through its kinase domain. (E) HeLa cells (2 × 106) were transfected with 0.5 μg of each indicated DNA construct. Cell lysates were harvested after 36 h and subjected to immunoprecipitation with anti-Flag antibody. Western blotting was performed with indicated antibodies. The 14-3-3 served as a negative control.
To determine which region of RIPK3 interacts with CK1, Flag-tagged RIPK3 truncations (Fig. 2A) were transiently overexpressed in HeLa cells. Overexpression presumably bypasses the requirement of upstream signal for interaction. As previously reported (12, 39), amino acids (1 to 323) of RIPK3, which contains the kinase domain, interacted with MLKL (Fig. 2C, lanes 5 and 6). Interestingly, full-length RIPK3 and the C-terminal region (324 to 518) containing RHIM domain interacted with CK1, even though the expression level of the C-terminal region (324 to 518) was the lowest (Fig. 2C, lanes 3 and 7). However, the (324 to 518)-RHIMm mutant which abolishes RIPK3 hetero- and homooligomerization did not interact with CK1 (Fig. 2C, lane 8). These results suggest that RIPK3 interacts with MLKL through its kinase domain, and interacts with CK1 through the intact oligomerizable RHIM domain (Fig. 2D).
CK1 proteins failed to interact with RIPK3-K50A, which has an intact RHIM domain (Fig. 1 C and D). One possible explanation is that the RHIM domain of K50A cannot form homooligomers. To test this hypothesis, 3xFlag- and 3xmyc- tagged RIPK3 or RIPK3-K50A were coexpressed in HeLa cells at low level. As expected, 3xFlag-RIPK3 interacted with 3xmyc-RIPK3 upon TSZ/NSA treatment (Fig. 2E, lane 6). However, 3xFlag-RIPK3-K50A failed to interact with 3xmyc-RIPK3-K50A, and as shown before, it did not interact with CK1 and MLKL (Fig. 2E, lane 8). These results suggest that RIPK3 kinase activity is required for RIPK3 homooligomerization, which is essential for subsequent CK1 and MLKL recruitment and necrosome formation.
A Short β-Sheet–Containing Region in the Kinase Domain of CK1 Interacts with RIPK3.
Next we examined which region of CK1 interacted with RIPK3. CK1 proteins contain an N-terminal kinase domain and a C-terminal regulation domain, which might autoinhibit the kinase domain (Fig. 3A) (36–38). CK1 structures reveal that roughly the first 100 amino acids contain five β-sheets and the rest of the kinase domain is mainly α-helixes and loops (40, 41). It has been demonstrated that the RHIM domain of RIPK1 and RIPK3 forms amyloid fibrils consisting of stacked β-sheets upon necroptosis induction (19, 20). Intermolecular β-sheet interactions are widely observed for protein–protein interaction and amyloid aggregation (22). Since CK1 interacts with the oligomerized RHIM domain (Fig. 2), we examined whether the β-sheets in CK1 mediated the interaction. Indeed, overexpressed full-length CK1ε or CK1ε-(1 to 100), which contains the β-sheets, bound RIPK3 (Fig. 3B, lanes 4 and 5), but CK1ε-(101 to 417) did not (Fig. 3B, lane 6).
Fig. 3.

A short β-sheet–containing region in the kinase domain of CK1 interacts with RIPK3. (A) Diagrams of CK1α, CK1δ, CK1ε, and CK1ε truncations. Amino acids 1 to 100 of CK1ε contain five β-sheets. (B and C) HeLa cells stably expressing 3xFlag-RIPK3 (2 × 106) were transfected with 5 μg DNA encoding indicated tagged CK1ε truncations. Cell lysates were harvested after 36 h and subjected to immunoprecipitation with anti-Flag antibody. Western blotting was performed with indicated antibodies. (D) HeLa:GFP-RIPK3 cells were transfected with constructs encoding RFP-CK1ε, CK1ε-(1 to 50)-RFP or CK1ε-(51 to 100)-RFP and treated with TSZ/NSA before imaging. (Scale bar, 10 μm.) (E) Sequence alignment of the homologous regions of amino acids 50 to 100 of CK1ε. Secondary structures are labeled on top. (F) HeLa:3xFlag-RIPK3 cells were treated with DMSO, TSZ, or TSZ/NSA. Cell lysates were incubated with GST-fused CK1δ peptides for GST-pulldown experiment. Western blotting was performed with anti-Flag antibody (Top) and the protein input was stained with Coomassie blue (Bottom).
To further narrow down the region, amino acids 1 to 100 of CK1ε were divided in half and fused with RFP to stabilize the proteins. Only (51 to 100)-RFP fusion protein bound RIPK3, the (1 to 50)-RFP fusion protein did not (Fig. 3C). This result was confirmed by fluorescent microscopy. Upon TSZ/NSA treatment, GFP-RIPK3 formed puncta. Full-length CK1ε-RFP and (51 to 100)-RFP also formed puncta which colocalized with GFP-RIPK3, while (1 to 50)-RFP did not form puncta and remained diffuse in the cytoplasm (Fig. 3D).
The region of 51 to 100 of CK1ε is highly conserved among the three CK1 proteins (Fig. 3E) and contains two β-sheets. To identify the minimal region required for CK1/RIPK3 interaction, recombinant CK1δ peptides fused with GST were bound to glutathione beads and incubated with lysates from HeLa:3xFlag-RIPK3 cells under different treatments. Amino acids 60 to 88 of CK1δ bound RIPK3, but amino acids 68 to 83 barely did (Fig. 3F), suggesting that both β-sheets and some surrounding sequences are required for the interaction. Interestingly, this interaction was not signal dependent, as RIPK3 from lysates which had no necroptosis induction (Fig. 3F, lane 8) bound the peptide just as robustly as RIPK3 from cells treated with TSZ or TSZ/NSA (Fig. 3F, lanes 9 and 10). It is possible that the high concentration of GST-CK1δ peptide bypasses signal dependence. Furthermore, RIPK3 bound to GST-CK1δ (60 to 88) migrated much slower, suggesting that heavily phosphorylated RIPK3 preferentially interacted with CK1 peptide. Another possibility is that RIPK3 enriched on the GST beads could phosphorylate itself.
CK1α, CK1δ, and CK1ε Directly Phosphorylate RIPK3 on Serine 227.
Since CK1 proteins are kinases, next we wanted to determine if CK1 directly phosphorylated RIPK3. The Group-Based Prediction System (http://gps.biocuckoo.cn) predicted that one of the top CK1 phosphorylation sites in RIPK3 is serine 227, within the sequence ELPTEPSLVYEAV, containing two acidic residues before S227 (Fig. 4A). Previously S227 has been suggested to be an autophosphorylation site. This is supported by the observation that S227 is not phosphorylated in cells expressing catalytically inactive RIPK3, even after TSZ treatment (12). However, catalytically inactive RIPK3 is also unable to recruit CK1 family members (Fig. 1 C and D). This raises an alternative explanation that one or more CK1 family members might contribute to S227 phosphorylation. To test this hypothesis, recombinant RIPK3 (151 to 254) was incubated with constitutively active CK1δ in an in vitro kinase assay. RIPK3 (151 to 254) contained a portion of the kinase domain, but did not display any kinase activity on its own (Fig. 4B, lane 1). Recombinant CK1 was able to phosphorylate RIPK3 at S227, as shown by the use of a phospho-S227 specific antibody (Fig. 4B, lane 2), and this phosphorylation was erased by λppase treatment (Fig. 4B, lane 3).
Fig. 4.
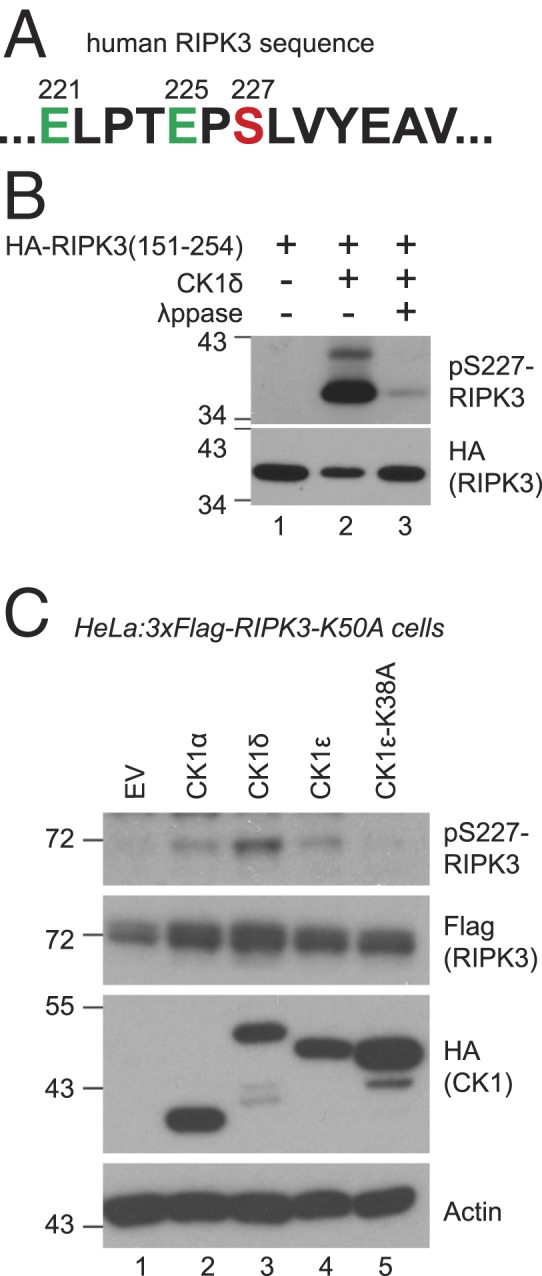
CK1 proteins directly phosphorylate RIPK3 at serine 227. (A) RIPK3 sequence around serine 227. (B) In vitro kinase assay was performed with recombinant CK1δ and recombinant HA-tagged RIPK3 (151 to 254). For lane 3, the sample from lane 2 was treated with λppase. Western blotting was performed with indicated antibodies. Antibody against phosphorylated S227 of RIPK3 is marked as pS227-RIPK3. (C) HeLa:3xFlag-RIPK3-K50A cells were transfected with constructs encoding indicated HA-tagged CK1 proteins. Cell lysates were subjected to Western blotting with indicated antibodies. EV, empty vector. CK1ε-K38A is kinase dead.
To investigate the ability of CK1 to phosphorylate RIPK3 in vivo, HA-tagged CK1α, CK1δ, CK1ε, or a catalytically inactive CK1ε mutant (CK1ε-K38A) were transiently overexpressed in HeLa:3xFlag-RIPK3-K50A cells. Because RIPK3-K50A lacks kinase activity, any RIPK3 phosphorylation cannot be the result of autophosphorylation. With overexpression, CK1α, CK1δ, and CK1ε could phosphorylate RIPK3 on S227 even without TSZ/NSA treatment (Fig. 4C). However, cells expressing catalytically inactive CK1ε did not show RIPK3-S227 phosphorylation (Fig. 4C, lane 5). Along with data from Fig. 4B, this suggests that CK1 may be directly responsible for RIPK3-S227 phosphorylation. Because RIPK3 is catalytically inactive in these cells, it is not possible to assess other downstream markers of necroptosis such as MLKL phosphorylation or cell survival after TSZ treatment.
Loss of CK1 Abolishes RIPK3-S227 Phosphorylation and Blocks Cell Death.
All three CK1 members bind RIPK3 upon necroptosis induction (Figs. 1 and 2) and are able to phosphorylate RIPK3 at S227 when overexpressed (Fig. 4), suggesting redundant function. To assess how loss of CK1 family members might impact necroptosis, single CK1α, CK1δ, and CK1ε knockout (KO) cell lines were generated using CRISPR-Cas9 (Fig. 5A), beginning with parental cells expressing 3xFlag-RIPK3. These single KO cells all exhibited more cell survival upon necroptosis induction compared to parental cells as measured by CellTiter-Glo (Fig. 5B). An attempt was made to further generate a CK1α/CK1δ/CK1ε triple knockout cell line. Unfortunately, a triple knockout cell line was never obtained. Due to the many functions of CK1 in the cells, loss of CK1α, CK1δ, and CK1ε is likely lethal. However, a CK1α/CK1ε double knockout cell line (1α1ε-DKO) was generated. These cells grew slower than parental cells, presumably because of the defects in many CK1-regulated processes including cell division. When compared to the parental cells, the 1α1ε-DKO cells exhibited much lower levels of RIPK3-S227 phosphorylation upon necroptosis induction (Fig. 5C, lanes 2 and 4). Upon longer exposure, a low level of phosphorylation was still observed in DKO cells. The decreased S227 phosphorylation corresponded to an increase in cell survival after TSZ treatment compared to parental cells (Fig. 5D). In addition, when the 1α1ε-DKO cells were passaged in culture, over time they displayed increased levels of CK1δ protein, which suggests that the cells may be compensating for the loss of CK1α and CK1ε.
Fig. 5.

Loss of CK1 abolishes RIPK3-S227 phosphorylation and blocks cell death. (A) Cell lysates from parental cells (HeLa:3xFlag-RIPK3) and KO cells were subjected to Western blotting with indicated antibodies. (B) Parental cells and KO cells were treated with DMSO or TSZ and cell viability was measured by the CellTiter-Glo assay. Viable cells are expressed as a percentage of DMSO-treated cells. Data are represented as mean ± SD of technical triplicates. (C) Parental cells or the CK1α/CK1ε double knockout (1α/1ε DKO) cells were treated with or without TSZ/NSA. Cell lysates were subjected to Western blotting with indicated antibodies. (D) Parental cells or the 1α/1ε DKO cells were treated with DMSO or TSZ and cell viability was measured by the CellTiter-Glo assay. (E) 1α/1ε DKO cells were transfected with siRNA against CK1δ or luciferase control for 72 h. The cells were then treated with TSZ/NSA for 4 h or 8 h and cell lysates were subjected to Western blotting. Antibody against phosphorylated MLKL-Ser358 is marked as p-MLKL. (F) The 1α/1ε DKO cells were transfected with siRNA against CK1δ or control for 72 h. The cells were then treated with DMSO or TSZ and cell viability was measured by the CellTiter-Glo assay. (G) HT-29 cells were transfected with two rounds of siRNA against control or a combination of siRNAs against CK1α, CK1δ, and CK1ε. The cells were then treated with or without TSZ/NSA and cell lysates were subjected to Western blotting with indicated antibodies. Antibody against phosphorylated S166 of RIPK1 is marked as p-RIPK1. (H) HT-29 cells were transfected with two rounds of siRNA as in G. The cells were then treated with DMSO or TSZ and cell viability was measured by the CellTiter-Glo assay.
To test whether the low level of S227 phosphorylation and small amount of cell death in the DKO cells was due to CK1δ, the remaining family member, CK1δ was knocked down in the 1α1ε-DKO cells using small interfering RNA (siRNA). These cells exhibited a further decrease in S227 phosphorylation (Fig. 5E) and increase in cell survival after TSZ treatment (Fig. 5F) compared to cells treated with control siRNA. Additionally, these cells did not exhibit MLKL phosphorylation, a critical step in necroptosis. These results strongly suggest that CK1 proteins are required for S227 phosphorylation and necroptosis progression.
Next we examined whether CK1 regulated RIPK3 phosphorylation in human colon cancer HT-29 cells. Transfection of siRNAs against CK1α, CK1δ, and CK1ε resulted in decreased expression of all three proteins (Fig. 5G, lanes 3 and 4). Compared to cells transfected with control siRNA, these cells showed markedly decreased phosphorylation of RIPK3-S227 and MLKL, but not RIPK1, upon TSZ/NSA treatment (Fig. 5G, lanes 2 and 4). Importantly, these cells exhibited much increased survival upon TSZ treatment (Fig. 5H). These results confirm the role of CK1 in RIPK3-S227 phosphorylation and necroptosis.
E221 and E225 in the CK1 Recognition Motif Are Essential for RIPK3-S227 Phosphorylation and Necrosome Formation.
To exclude the possibility that CK1 might phosphorylate other important residues in RIPK3 which might indirectly promote S227 phosphorylation, the acidic residues E221 and E225 upstream of S227 were mutated to alanine. Because these glutamate residues are located in the antigen used for preparing the phospho-specific antibody (42), there is a possibility that mutating them could disrupt the recognition by the antibody. A dot blot comparing nonphosphorylated control, phosphorylated WT, and phosphorylated E221/225A peptides revealed that mutation of glutamate at positions 221 and 225 to alanines did not impact recognition by the phospho-specific antibody (Fig. 6A). The lack of signal in the nonphosphorylated control peptide confirmed the specificity of the antibody.
Fig. 6.

Acidic residues E221 and E225 in the CK1 recognition motif are essential for RIPK3-S227 phosphorylation and necroptosis. (A) Different amounts of indicated peptides were spotted on a nitrocellulose membrane. Western blotting was performed with the pS227-RIPK3 antibody. pS, phosphoserine. ELPTEP(pS)LVYEAV was reported as the antigen used for developing the antibody (42). (B) In vitro kinase assay was performed with recombinant CK1δ and recombinant HA-tagged RIPK3 (151 to 254) mutants. Western blotting was performed with indicated antibodies. (C) HeLa cells stably expressing 3xFlag-RIPK3 (WT) or 3xFlag-RIPK3-E221/225A mutant were treated with or without TSZ/NSA. Cell lysates were subjected to Western blotting with indicated antibodies. (D) WT or E221/225A mutant cells were treated with or without TSZ/NSA. Cell lysates were subjected to immunoprecipitation with an anti-Flag antibody, and Western blotting was performed with indicated antibodies. (E) WT or E221/225A mutant cells were treated with DMSO or TSZ and stained with DAPI and PI. (Scale bar, 50 μm.) (F) WT or E221/225A mutant cells were treated with DMSO or TSZ, and cell viability was measured by the CellTiter-Glo assay. (G) Proposed working model. TSZ treatment leads to RIPK1 phosphorylation and recruitment of RIPK3. RIPK3 autophosphorylation favors RIPK3 homooligomerization over the RIPK1-RIPK3 heterooligomerization. CK1α/1δ/1ε interact with RIPK3 oligomers and phosphorylate RIPK3 at serine 227. Phosphorylated RIPK3 subsequently recruits MLKL to form the necrosome.
To investigate the importance of E221/E225 for S227 to be phosphorylated by CK1, recombinant CK1δ was incubated with recombinant HA-tagged RIPK3 (151 to 254) with different mutations in an in vitro kinase assay. Decreased levels of S227 phosphorylation were observed in mutants where one, or both glutamates, was mutated to alanine (Fig. 6B), suggesting that E221 and 225 are important for RIPK3-S227 to be recognized and phosphorylated by CK1.
To assess whether these mutations would have a functional impact, a stable cell line expressing 3xFlag-RIPK3-E221/225A was established. Compared to cells expressing WT-RIPK3, these cells exhibited similar levels of RIPK1 phosphorylation, but almost undetectable RIPK3-S227 phosphorylation and MLKL phosphorylation after treatment with TSZ/NSA (Fig. 6C), suggesting that E221 and E225 are essential for RIPK3-S227 phosphorylation and necroptosis progression in vivo.
Next we examined the necrosome formation in the mutant cells. Immunoprecipitation of RIPK3-E221/225A confirmed that it still interacted with CK1 (Fig. 6D, lane 8). However, this mutant did not interact with MLKL, confirming a previous report that RIPK3 must be phosphorylated at S227 to recruit MLKL to the necrosome (12).
Lastly, the ability of these cells to undergo TNF-induced necroptosis was examined. Cells expressing WT or E221/225A RIPK3 were treated with dimethyl sulfoxide (DMSO) or TSZ and stained with propidium iodide (PI), a cell impermeable DNA dye. Cells expressing WT RIPK3 exhibited massive cell death as expected, while the cells expressing RIPK3-E221/225A showed little cell death (Fig. 6E). This was further confirmed by CellTiter-Glo assay. After treatment with TSZ, cells expressing E221/225A mutant showed much higher levels of survival than cells expressing WT-RIPK3 (Fig. 6F). These results strongly suggest that phosphorylation of RIPK3-S227 by CK1 is essential for necrosome formation and necroptosis progression (Fig. 6G).
Discussion
The necrosome is a million-dalton complex, containing minimal components of RIPK1, RIPK3, and MLKL (12). Our results suggest that CK1α, CK1δ, and CK1ε are also part of the necrosome. CK1 proteins interact with the necrosome only after necroptosis induction (Fig. 1 B–D). In addition, under fluorescence microscopy, CK1 colocalizes with GFP-RIPK3 puncta (Figs. 1D and 3D), which have been shown to be the aggregations of necrosome (12). Specifically, CK1 proteins interact with the C-terminal region of oligomerized RIPK3 containing stacked β-sheets of the RHIM amyloid core through their own β-sheets within the N-terminal of the kinase domain (Figs. 2 and 3). Furthermore, loss of CK1 proteins abolish necrosome function and MLKL phosphorylation (Fig. 5C). Lastly, the RIPK3-E221/225A mutant which cannot be phosphorylated by CK1 fails to form the necrosome (Fig. 6D). These results suggest that CK1 proteins are essential components of the necrosome.
CK1 interaction with RIPK3 is dependent on RIPK3 kinase activity (Fig. 1 C and D), and an intact RIPK3 RHIM domain (Fig. 2C). Our results in Fig. 2E indicate that RIPK3 kinase activity is required for a conformational change of RIPK3 that allows the RIPK3 RHIM domain to form a homooligomer, which is then responsible for CK1 recruitment. Structural analysis of the RHIM amyloid suggests that RIPK1-RIPK3 heterooligomers are preferred over the RIPK3 homooligomers (19–21). Indeed, when RIPK1-RHIM peptides and RIPK3-RHIM peptides are coexpressed in bacteria, the predominant species formed are RIPK1-RIPK3 heteroamyloids (19, 20). However, an elegant study has demonstrated that RIPK3 homooligomers, not RIPK1-RIPK3 heterooligomers, are responsible for necroptosis progression (23). In particular, induced RIPK3 homodimerization in RIPK1 knockout cells is sufficient to induce necroptosis. In fact, under some circumstance, RIPK1 can inhibit necroptosis by sequestering RIPK3 through the hetero-RHIM interaction (24). Therefore, it is possible that RIPK3 kinase activity is required to phosphorylate itself to change RIPK3 conformation to favor RIPK3 homooligomers rather than RIPK1-RIPK3 heterooligomers, which is then able to recruit CK1 and subsequently MLKL for necroptosis to proceed (Fig. 6G).
Phosphorylation of human S227 has been proposed to be an autophosphorylation event, mainly because the kinase-dead form of RIPK3 fails to undergo S227 phosphorylation upon necroptosis induction. This kinase-dead RIPK3 still interacts with RIPK1 (12, 31), suggesting that RIPK1 is not responsible for S227 phosphorylation. However, the kinase-dead form of RIPK3 does not interact with CK1 proteins (Fig. 1 C and D), suggesting an alternative explanation that CK1 proteins might be responsible for S227 phosphorylation. This hypothesis is supported by the following evidence. First, constitutively active CK1δ phosphorylates RIPK3-S227 in vitro (Fig. 4B) and overexpression of CK1α, CK1δ, and CK1ε induces phosphorylation of RIPK3-S227 in vivo (Fig. 4C). Second, cells deficient in CK1 proteins fail to undergo S227 phosphorylation and are resistant to necroptosis (Fig. 5). Lastly, the RIPK3-E221/225A mutant with mutations in the CK1 recognition motif fails to be phosphorylated at S227 in vivo and cannot bind MLKL or activate necroptosis (Fig. 6). These results strongly suggest that CK1 proteins are responsible for RIPK3-S227 phosphorylation. However, we cannot exclude the possibility that CK1 proteins might phosphorylate other sites on RIPK3 which is required for RIPK3 autophosphorylation of S227. The E221/225A mutant result argues against this possibility, unless E221 and E225 are also important for RIPK3 autophosphorylation.
CK1α, CK1δ, and CK1ε all interact with the necrosome upon necroptosis induction (Figs. 1 and 2), and all three could phosphorylate S227 to a similar extent (Fig. 4C), suggesting redundant function. At the same time, CK1 proteins are involved in many other important cellular processes, some of which may be essential for cell survival (36–38). This is manifested in the fact that triple knockout cells were never obtained and CK1α/CK1ε double knockout cells grow much slower in the culture. This makes it difficult to use CK1 inhibitors to block necroptosis for therapeutic purposes. An alternative strategy is to inhibit CK1 binding to RIPK3. A minimal region of 29 amino acids (60 to 88 of CK1δ) are sufficient to bind RIPK3 (Fig. 3F). It will be interesting to examine whether these 29 amino acids fused to a cell-permeable tag will be able to block necroptosis. In theory, this peptide will block RIPK3-CK1 interaction, but may not interfere with other CK1 functions, because CK1 kinase activity is not affected.
In summary, we propose the following model for how necrosome formation is regulated. Upstream signal leads to the activation and phosphorylation of RIPK1, which recruits RIPK3 through hetero-RHIM domain interaction. This interaction activates RIPK3 autophosphorylation to favor RIPK3 homooligomerization. RIPK3 homooligomers in turn recruit CK1 family proteins through the C-terminal region containing the RHIM-amyloid core. Phosphorylation of RIPK3 at S227 by CK1 enable RIPK3 to recruit MLKL through its N-terminal kinase domain to form the necrosome (Fig. 6G).
Materials and Methods
General Reagents.
The following reagents and antibodies were used: ZVAD-FMK (ApexBio), NSA (Millipore), anti-Flag M2 antibody and affinity gel (Sigma), anti-HA and affinity gel (Sigma), anti-myc and affinity gel (Sigma), anti-phospho-S227 of RIPK3 (Abcam, ab209384), anti-human MLKL (GeneTex, GTX107538), anti-phospho-S358 of MLKL (Abcam, ab187091), anti-RIPK1 (BD, 551042), anti-phospho-S166 of RIPK1 (Cell Signaling, 65746), anti-CK1α (Santa Cruz, SC-6477), anti-CK1δ (Bethyl Laboratories, A302-136A), anti-CK1ε (Santa Cruz, SC-6471), and anti–14-3-3 (Santa Cruz, SC-629). TSZ treatment includes TNF (20 ng/mL), Smac-mimetic (100 nM), and ZVAD-FMK (20 µM). NSA was used at 5 µM.
Cell Culture and Stable Cell Lines.
HeLa cells were cultured in Dulbecco’s modified eagle media (DMEM) (high glucose) supplemented with 10% fetal bovine serum (FBS). The HeLa:HA-3xFlag-RIPK3 cell line has been reported before (12, 31). HeLa:3xFlag-RIPK3, HeLa:3xFlag-RIPK3-K50A, and HeLa:3xFlag-RIPK3-E221/225A stable lines were generated using the same method. They were all established in the background of HeLa-TetR cells that expressed the Tet repressor (TetR) and the transgene expression was induced with 50 ng/mL doxycycline (Dox) for 24 h.
Cell Lysates and Immunoprecipitation.
Cells were treated with DMSO or TSZ/NSA for 16 h, except when otherwise stated. Cells were then scraped and washed with phosphate-buffered saline (PBS) twice followed by lysing with five volumes of lysis buffer (50 mM Tris, pH 7.4, 137 mM NaCl, 1 mM ethylenediaminetetraacetic acid [EDTA], 1% Triton X-100, and 10% glycerol, supplemented with protease inhibitors). After 30 min incubation on ice, the cells were centrifuged at 20,000 × g for 12 min and supernatant was collected. Lysates (1 mg) were incubated with 20 µL anti-Flag or anti-myc agarose beads at 4 °C overnight. Beads were washed five times with lysis buffer and eluted with 60 µL elution buffer (0.2 M glycine, pH 2.8) and immediately neutralized with 6 µL of 1 M Tris, pH 7.4. All procedures were done at 4 °C.
Tandem Immunoprecipitation.
Cell lysates (10 mg) were incubated with 200 µL anti-Flag agarose beads at 4 °C overnight. Beads were washed five times with lysis buffer and eluted twice with 1 mL lysis buffer containing 0.1 mg/mL 3xFlag peptide at 4 °C for 4 h. Combined eluate was incubated with 40 µL anti-HA agarose beads at 4 °C overnight. Beads were washed five times with lysis buffer and eluted twice with 120 µL lysis buffer containing 0.1 mg/mL HA peptide at 4 °C for 4 h. The eluate was separated on a 4 to 12% NuPAGE gel (Thermo, NP0321) and stained with SilverQuest silver staining kit (Thermo, LC6070).
Constructs.
All cDNAs were PCR cloned from reverse transcription products from HT-29 cells. The primers were designed according to the reference sequences as follows: human RIPK3 (NM_006871.3), human RIPK1 (NM_003804.3), human CK1α (NM_001025105.3), human CK1δ (NM_001893.6), and human CK1ε (NM_152221.3). All point mutations were generated by site-directed mutagenesis and verified by sequencing.
Live Cell Imaging and Fluorescence Microscopy.
Live cell imaging was recorded with an Ultraview Spinning disk confocal microscope (Perkin-Elmer) and analyzed with the Imaris X64 7.6.0 software. Fluorescence images were taken with a LSM 700 confocal microscope (Carl Zeiss) and analyzed with the Zeiss LSM Image Browser.
GST Pulldown.
The cDNAs encoding CK1δ (68 to 83) or (60 to 88) were cloned into the pGEX vector. GST-fusion proteins were purified with glutathione beads (Thermo, 17-0756-01) from BL21 Escherichia coli as described before (43). GST-fusion proteins on the glutathione beads (5 µg) were incubated with 1 mg of cell lysates at 4 °C for 4 h, and washed with lysis buffer five times before being directly boiled in 1xSDS loading buffer.
Recombinant Protein Purification.
The cDNAs encoding RIPK3 (151 to 254) with a HA-tag and its mutants were cloned into the pET21b vector. His-fusion proteins were purified from BL21(DE3) E. coli as described before (44). Purified recombinant proteins were dialyzed against PBS buffer.
Kinase Assay.
Constitutively active CK1δ was purchased from Abcam (ab103955). HA-tagged RIPK3 peptides (1 µg) were incubated with 0.1 µg of CK1δ in the kinase buffer (50 mM Tris, pH 7.4, 10 mM MgCl2, 0.02% bovine serum albumin, 1 mM DL-dithiothreitol [DTT] and 0.1 mM adenosine 5′-triphosphate [ATP]) at 30 °C for 1 h. For λppase treatment, kinase assay reaction was denatured at 55 °C for 15 min, precipitated with acetone, and then incubated with 5 units of λppase (NEB, P0753S) in lambda phosphatase buffer at 30 °C for 1 h.
CRISPR-Cas9 Knockout Cell Lines.
All of the CK1 knockout lines were generated in the HeLa:3xFlag-RIPK3 background according to the protocol described in ref. 45. Briefly, oligos targeting different CK1 were cloned into the gRNA vectors harboring different resistant genes. Each gRNA vector was cotransfected with a Cas9-expressing vector into parental cells, and single clones were selected. Gene knockout was confirmed by Western blotting and sequencing. The following targeting sequences were used: CK1α, GAGGGAAATATAAACTGGTACGG; CK1δ, CAAAATCTACAAGATGATGCAGG; and CK1ε, GGAACAAGTACCGCCTGGGACGG.
siRNA Transfection.
For siRNA transfection, cells were plated at 2,000 cells per well in 96-well plates or 100,000 cells per well in 6-well plates, 24 h prior to transfection. Transfection was carried out as per GenMute (SignaGen) protocol with 5 nM siRNA for 96-well plates and 50 nM for 6-well plates. Cells were incubated in standard culture conditions for 72 h prior to treatment. The following siRNAs were used: si-ctrl, CGUACGCGGAAUACUUCGA; si-CK1δ, GGACAUUGCUGCAGGAGAA; si-CK1α, CCAGAUGAUCAGUAGAAUU; and si- CK1ε, CCACCAAGCGCCAGAAGUA. For HT-29 cells, two rounds of siRNA transfection were performed to increase knockdown efficiency. Cells were first transfected in a 6-well plate. After 48 h, these cells were split into a 96-well and a new 6-well plate and transfected again before treatment.
Cell Survival Assay.
Cell survival was measured using the CellTiter-Glo luminescent cell viability assay according to the manufacturer’s protocol (Promega). Cells were seeded at 2,000 cells per well in 96-well plates, 24 h prior to treatment. Luminescence was measured using a BioTek Synergy 2 plate reader.
Cell Staining.
Cells were seeded at 1 × 105 cells/well in a 12-well plate. PI and 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI) were added directly to culture media at 1 µM and incubated for 10 min prior to imaging with a BioTek Cytation 3 plate reader.
Dot Blot.
Nonphosphorylated or phosphorylated peptides (purchased from Biomatik) were serially diluted in PBS buffer to desired concentrations, and 1 µL of each was spotted on a nitrocellulose membrane and air dried for 15 min followed by Western blotting.
MS-Liquid Chromatography.
MS-liquid chromatography was performed as previously described (27). Briefly, the protein band of interest was excised, destained, and reduced followed by in-gel trypsin digestion. The peptides were extracted and analyzed by using a QSTAR XL mass spectrometer (AB SCIEX).
Statistical Analysis.
Statistical analyses was performed with Excel. Student’s t test was used to determine significance in cell death. P < 0.05 was used to determine significant differences in cell death. Data are presented as mean ± SD.
Data Availability.
All data are included in the manuscript.
Acknowledgments
We thank Hong Yu for excellent technical assistance and Dr. Zhirong Shen for reagents. This work is supported by grants from the Welch Foundation (I1827) and National Institute of General Medical Sciences (R01, RGM120502A) to Z.W., and fellowships to S.H.-A. (TL1TR1104). Z.W. is the Virginia Murchison Linthicum Scholar in Medical Research and Cancer Prevention and Research Institute of Texas (CPRIT) Scholar (R1222).
Footnotes
The authors declare no competing interest.
This article is a PNAS Direct Submission.
References
- 1.Galluzzi L., et al. , Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on cell death 2018. Cell Death Differ. 25, 486–541 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sun L., Wang X., A new kind of cell suicide: Mechanisms and functions of programmed necrosis. Trends Biochem. Sci. 39, 587–593 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Shan B., Pan H., Najafov A., Yuan J., Necroptosis in development and diseases. Genes Dev. 32, 327–340 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vandenabeele P., Galluzzi L., Vanden Berghe T., Kroemer G., Molecular mechanisms of necroptosis: An ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Weinlich R., Oberst A., Beere H. M., Green D. R., Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 18, 127–136 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Mocarski E. S., Guo H., Kaiser W. J., Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology 479–480, 160–166 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He S., et al. , Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137, 1100–1111 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Holler N., et al. , Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Degterev A., et al. , Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 4, 313–321 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cho Y. S., et al. , Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 137, 1112–1123 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang D. W., et al. , RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325, 332–336 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Sun L., et al. , Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Zhao J., et al. , Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. U.S.A. 109, 5322–5327 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun X., Yin J., Starovasnik M. A., Fairbrother W. J., Dixit V. M., Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J. Biol. Chem. 277, 9505–9511 (2002). [DOI] [PubMed] [Google Scholar]
- 15.He S., Liang Y., Shao F., Wang X., Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc. Natl. Acad. Sci. U.S.A. 108, 20054–20059 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Upton J. W., Kaiser W. J., Mocarski E. S., DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11, 290–297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Upton J. W., Kaiser W. J., Mocarski E. S., Cytomegalovirus M45 cell death suppression requires receptor-interacting protein (RIP) homotypic interaction motif (RHIM)-dependent interaction with RIP1. J. Biol. Chem. 283, 16966–16970 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X., et al. , Direct activation of RIP3/MLKL-dependent necrosis by herpes simplex virus 1 (HSV-1) protein ICP6 triggers host antiviral defense. Proc. Natl. Acad. Sci. U.S.A. 111, 15438–15443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li J., et al. , The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150, 339–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mompean M., et al. , The structure of the necrosome RIPK1-RIPK3 core, a human hetero-amyloid signaling complex. Cell 173, 1244–1253.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kajava A. V., Klopffleisch K., Chen S., Hofmann K., Evolutionary link between metazoan RHIM motif and prion-forming domain of fungal heterokaryon incompatibility factor HET-s/HET-s. Sci. Rep. 4, 7436 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu H., Fuxreiter M., The structure and dynamics of higher-order assemblies: Amyloids, signalosomes, and granules. Cell 165, 1055–1066 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu X. N., et al. , Distinct roles of RIP1-RIP3 hetero- and RIP3-RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 21, 1709–1720 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Orozco S., et al. , RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 21, 1511–1521 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McQuade T., Cho Y., Chan F. K., Positive and negative phosphorylation regulates RIP1- and RIP3-induced programmed necrosis. Biochem. J. 456, 409–415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhong C. Q., et al. , Quantitative phosphoproteomic analysis of RIP3-dependent protein phosphorylation in the course of TNF-induced necroptosis. Proteomics 14, 713–724 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Newton K., et al. , Activity of protein kinase RIPK3 determines whether cells die by necroptosis or apoptosis. Science 343, 1357–1360 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Vanlangenakker N., Bertrand M. J., Bogaert P., Vandenabeele P., Vanden Berghe T., TNF-induced necroptosis in L929 cells is tightly regulated by multiple TNFR1 complex I and II members. Cell Death Dis. 2, e230 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H., et al. , Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54, 133–146 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez D. A., et al. , Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23, 76–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z., Jiang H., Chen S., Du F., Wang X., The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Mandal P., et al. , RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 56, 481–495 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Najjar M., et al. , Structure guided design of potent and selective ponatinib-based hybrid inhibitors for RIPK1. Cell Rep. 10, 1850–1860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J. X., et al. , The B-Raf(V600E) inhibitor dabrafenib selectively inhibits RIP3 and alleviates acetaminophen-induced liver injury. Cell Death Dis. 5, e1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hildebrand J. M., et al. , Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. U.S.A. 111, 15072–15077 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Knippschild U., et al. , The casein kinase 1 family: Participation in multiple cellular processes in eukaryotes. Cell. Signal. 17, 675–689 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Venerando A., Ruzzene M., Pinna L. A., Casein kinase: The triple meaning of a misnomer. Biochem. J. 460, 141–156 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Xu P., et al. , Structure, regulation, and (patho-)physiological functions of the stress-induced protein kinase CK1 delta (CSNK1D). Gene 715, 144005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie T., et al. , Structural insights into RIP3-mediated necroptotic signaling. Cell Rep. 5, 70–78 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Xu R. M., Carmel G., Sweet R. M., Kuret J., Cheng X., Crystal structure of casein kinase-1, a phosphate-directed protein kinase. EMBO J. 14, 1015–1023 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longenecker K. L., Roach P. J., Hurley T. D., Three-dimensional structure of mammalian casein kinase I: Molecular basis for phosphate recognition. J. Mol. Biol. 257, 618–631 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Li D., et al. , A cytosolic heat shock protein 90 and cochaperone CDC37 complex is required for RIP3 activation during necroptosis. Proc. Natl. Acad. Sci. U.S.A. 112, 5017–5022 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu S., et al. , MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. U.S.A. 114, E7450–E7459 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Reynoso E., et al. , Thioredoxin-1 actively maintains the pseudokinase MLKL in a reduced state to suppress disulfide bond-dependent MLKL polymer formation and necroptosis. J. Biol. Chem. 292, 17514–17524 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cong L., et al. , Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data are included in the manuscript.