

Historically, venous thromboembolism (VTE) has been the main and very often the only vascular disease entity noted in cancer patients. Furthermore, the risk of VTE has been considered to be present in these patients even irrespective of cancer therapy. Thus, there has not been much concern for vascular toxicities as a consequence of cancer therapy in the past, with the exception of 5-FU and radiation therapy. The introduction of targeted therapies, especially those that inhibit the VEGF signaling pathway, however, has changed this view and has drawn more attention to the topic of vascular toxicities with cancer therapies.
As outlined in Figure 1, the vascular disease spectrum that can be seen in cancer patients is very broad. It can affect all vascular territories, can involve both, the venous and the arterial circulation, can be functional or structural in nature, and can be of lasting or only temporary duration. For this very reason, it is difficult to devise a uniform classification system of vascular toxicities of cancer therapies. For practical purposes, an approach by type of presentation might be preferred and will be the structure for this review. The focus will be on clinical aspects and less so on basic and translational science.
Figure 1.

Outline of the spectrum of common vascular toxicities and related cancer therapeutics.
Cerebrovascular events
Cerebrovascular events (CVAs) including transient ischemic attack (TIA) and stroke have been only rarely reported with classical chemotherapeutics in the past. This changed with the introduction of vascular endothelial growth factor (VEGF) inhibitors, including the monoclonal antibody bevacizumab and a number of tyrosine kinase inhibitors (TKIs) that target the VEGF receptor II (e.g. sunitnib, sorafenib). VEGF inhibitors increase the risk of ischemic and hemorrhagic strokes. The relative risk of either type of stroke is increased 3-fold in patients on bevacizumab with an absolute incidence of ischemic and hemorrhagic strokes of 0.5% and 0.3%, respectively.1 A higher relative risk of CVAs was noted in colorectal cancer patients (6.4-fold increased risk) and the highest absolute incidence of CVAs was reported in patients with mesothelioma (1.9%).1 The risk of any CVA doubled with doubling of the dose of bevacizumab, i.e. CVA incidence 2% and 4% with 2.5 and 5 mg/kg/week bevacizumab dose regimen, respectively.1 Intracranial hemorrhages tend to occur earlier (median time 2.6 months), often in the setting of tumor progression with a poor prognosis (owing to tumor progression and 50% mortality of intracranial bleeding events).2 On the contrary, ischemic strokes tend to be seen later on in the course of therapy (median 16.2 months) and do not associate with a rapidly fatal outcome in most cases. Key risk factors for intracranial bleeding are use of additional medications that increase the risk for bleeding and thrombocytopenia but, importantly, not CNS tumors or metastases.3,4 In regards to ischemic stroke, vascular risk factors apply as in the general population.
The most common type of stroke in cancer patients is thrombo-embolic in nature, and the rate of cryptogenic strokes is nearly twice as high (50% versus 30% in the general population). 5 The risk of arterial thromboembolic events emerges 5 months before and peaks within one month window before and after cancer diagnosis (Figure 2).5–7 The risk is highest in patients with GI tract, lung cancers, and non-Hodgkin lymphoma. It is also seen more commonly with advanced stages (Stage III and IV).5–7 These dynamics and clear similarities with VTE suggest that general thrombophilia in cancer patients is an important determining factor. 8 A nidus for thrombus in the heart can be the valves (marantic endocarditis), the left atrium/left atrial appendage (atrial fibrillation), and indwelling central venous catheters in the setting of a patent foramen oval. Other sources for emboli to the brain in cancer patients include septic emboli and tumor emboli. Platinum drugs, next to VEGF inhibitors, are the main cancer drugs that associate with thrombo-embolic events. 9
Figure 2.
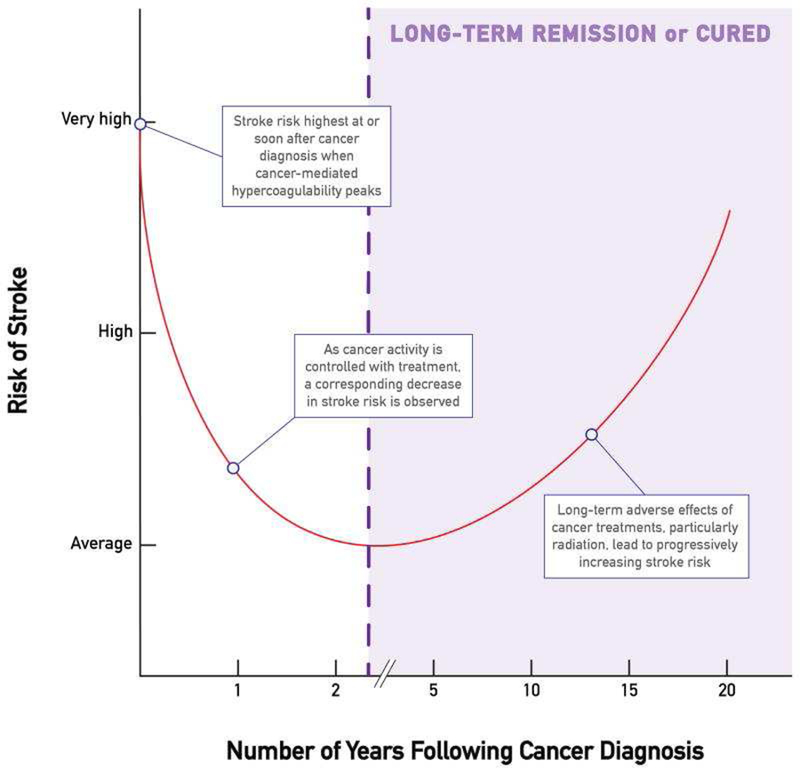
Illustration of the risk of stroke relative to the time of cancer diagnosis. From Navi BB, Iadecola C. Ischemic stroke in cancer patients: A review of an underappreciated pathology. Ann Neurol. 2018;83(5):873–883; with permission.
The second group to consider (after thromboembolism) in cancer patients with ischemic stroke are those with vasculopathy. This is mainly atherosclerosis of the carotid arteries though the intracranial vasculature can be affected as well. Again, against the premise that targeted therapies would be more specific and associated with less toxicity, progressive atherosclerosis has been reported with the Bcr-Abl inhibitors nilotinib and ponatinib.10–12 The drugs can cause acute CVAs with evidence of carotid artery disease or intracranial pathology similar to Moya Moya disease. 13,14 While these drugs cause injury to the endothelium, this does not seem to provide a full explanation.15 For instance, other drugs such as cisplatin also bestow endothelial toxicity but have been associated more with thrombotic events rather than atherosclerosis. 16,17 Other factors are therefore likely playing an important role in determining the outcome of endothelial injury with cancer therapeutics.
Abnormal vasoreactivity is less likely to be a prominent contributing factor for stroke in cancer patients even though stroke cases have been described in patients on 5-FU and capecitabine, which are known to impact vascular reactivity, most of the time without an identifiable cause. In fact the term “chemotherapy-induced stroke mimic” has been used 18 Another stroke mimic in cancer patients is posterior reversible encephalopathy syndrome (PRES), which can present as an acute cerebral event with headache, confusion, visual symptoms, and seizures. Posterior cerebral white matter edema on neuroimaging is the pathognomic sign, related to impaired autoregulation of the cerebral vasculature, often in the setting of severe hypertension. Numerous cases have been reported with a broad range of cancer therapeutics, especially those that can cause endothelial/vascular injury such as VEGF signaling pathway inhibitors but also immune checkpoint inhibitors. 19–21
Finally, stroke presentations in cancer patients can be the consequence of cerebral artery dissections or compression of vessels by tumors. 22 Both, venous compression/thrombosis and arterial compression are possibilities.
From a management standpoint, each patient should undergo a standard work-up within the guideline recommended time metrics for stroke evaluation. 23 This includes a timely head CT, which is key evaluation step in cancer patients, important to identify hemorrhage and/or CNS masses. If these are not seen, an ischemic etiology is to be assumed and further work-up should include evaluation for carotid artery disease and cardio-embolic causes as well as dissections and venous thrombosis.
Chest pain
Various chemotherapeutic agents have been associated with a broad spectrum of chest pain presentations including typical effort angina and atypical variant and microvascular angina.24 The classical example is 5-fluorouracil (5-FU) and its oral pro-drug capecitabine, which can cause coronary vasospasm and related symptoms. 25 Cardiac injury seen with the use of these drugs is considered to be a consequence of the ischemia induced by these drugs (though direct cardiotoxic effects have been discussed as well).26 Cardiac dysfunction can evolve to the point of cardiogenic shock. 27 Takotsubo’s is of differential diagnostic consideration even in these cases.28 The latter and various chest pain syndromes can also be encountered with paclitaxel and docetaxel, similarly related to the induction of abnormal vasoreactivity. 25 Finally, cisplatin, often in combination with bleomycin, can lead to presentations of chest pain, commonly in a manner that should raise concern for acute coronary syndrome (ACS).29–31 While these drugs lead to endothelial injury and can thus provoke signs and symptoms of endothelial dysfunction, especially platinum drugs are well known to induce thrombotic events. Abnormal vasoreactivity, arterial thrombosis, and progressive atherosclerosis have all been noted with VEGF inhibitor therapy.32,33 For the VGEF inhibitor sunitinib it has furthermore been shown that it alters coronary microcirculatory structure with a reduction in coronary flow reserve (CFR).34 This effect is due to inhibition of PDGF beta signaling with impairment of pericyte viability.34
The possibility of pre-existing coronary artery disease should not be forgotten in cancer patients and needs to be entertained in the evaluation of these patients. Of importance, patients with a history of ischemic heart disease and especially myocardial infarction have an 8-fold increased risk of developing “cardiotoxicity” with the use of 5-FU. Other etiologies of ischemic chest pain in cancer patients to consider include coronary artery compression by cardiac and noncardiac tumors.35–37 True invasion in the coronary arteries should raise suspicion for angiosarcoma. On the other hand, non-obstructive encasement of the right coronary artery in the right ventricular grove by diffuse large B-cell lymphoma or thymoma has become known as the “floating artery sign”. 38 Last but not least, other causes of chest pain need to be entertained in cancer patients just as they are in the general population including aortopathies, pericardial diseases, even aortic stenosis, costochondritis, and gastro-intestinal and pulmonary etiologies.
From a management perspective, most patients can be started empirically on vasodilator therapy with sublingual nitroglycerin, long-acting nitroglycerin or the long-acting calcium channel blockers amlodipine unless hypotension poses concerns. In patients with vasospastic disease, this intervention is curative (and thereby diagnostic). In individuals with endothelial dysfunction or microvascular disease, vasodilator therapy can ameliorate symptom burden. Other CCBs that might need to be used include nifedipine and felodipine, or diltiazem or verapamil when cardiac function is normal in view of their negative inotropic effect. Beta-blocker have traditionally been used to favorably manage patients below their ischemic threshold. For patients in VEGF inhibitors or other endothelial toxic agents such as cisplatin, various interventions can be considered to improve vascular health (Figure 3). Vasoreactivity testing can be used as a parameter of vascular health and can be assessed, for instance, with an Endo-PAT. Additional evaluation steps include stress testing, as in the general population. In those cases with microvascular dysfunction, expected reduced CFR, quantitative PET would be ideal, which measured regional myocardial blood flow. However, in view of the multiple possible etiologies, a number of them being structural and a key question being the need for structural intervention, an anatomic imaging approach, possibly combined with functional assessment, might be recommendable. Coronary computed tomography angiography (CCTA) with virtual fractional flow reserved and myocardial perfusion imaging provides such as tool. “Triple rule out” chest CTs address the concerns related to aortic and pulmonary disease processes, pulmonary embolism, as well as coronary artery disease. For those patients with coronary artery disease not controlled by medications or signs of severe extent (including any left main disease), an invasive coronary angiogram should be performed to define the next best step in terms of revascularization. Anemia and thrombocytopenia are important considerations in cancer patients and need to be carefully weighted into the decision making. Estimates of the duration of cytopenias are important as well as is the overall prognosis of the patient. However, a case can be made for percutaneous coronary intervention (PCI) as a palliative measure to improve quality of life not accomplished but medical therapy alone.
Figure 3.
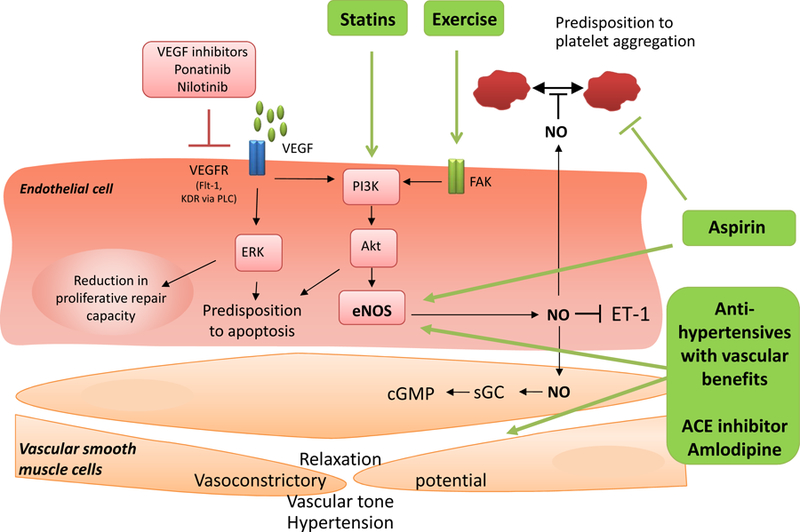
Therapeutic intervention to improve vascular health, especially in patients on cancer therapeutics with an inhibitory effect on the VEGF signaling pathway. From Touyz RM, Herrmann SMS, Herrmann J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J Am Soc Hypertens. 2018;12(6):409–425; with permission.
Acute coronary syndrome
All cancer therapeutics associated with chest pain can also cause ACS; in fact, unstable angina may be considered not infrequently in those with resting chest pain secondary to abnormal vasoreactivity of the epicardial and/or microvascular circulation. Furthermore profound coronary vasospasm as seen with 5-FU, particularly with continuous infusion (less so with bolus infusion of 5-FU and capecitabine), can lead to ST segment elevation and if prolonged to myocardial infarction, ventricular arrhythmias such as ventricular tachycardia and fibrillation, and even sudden cardiac death.39–41 Profound and prolonged vasospasm is considered to also underlie ACS presentations with paclitaxel, gemcitabine, rituximab, and sorafenib.42–47
The level of suspicion for an acute thrombotic event should be high in patients who present with chest pain while on cisplatin (and especially in those on concomitant therapy with additional endothelial toxic drugs such as vinca alkaloids, bleomycin, or gemcitabine). 48,49 Intravascular evaluations in patients presenting with such a constellation indicated plaque erosion as the underlying pathology. 50 Plaque hemorrhage can destabilize plaques in patients receiving treatment with vascular disrupting agents.51 Even if not acutely, plaque hemorrhage fosters the growth and vulnerability of atherosclerotic plaques. 52 VEGF inhibitors would be expected to elicit an anti-angiogenic response and thereby plaque stabilization rather than plaque destabilization. 53,54 Any increased risk in thromboembolic events may therefore be more due to impairment in the viability of surface-lining endothelial cells. VEGF inhibitors also suppress endothelial repair to injury, and this combined effect may ultimately be a key factor (similar to cisplatin in this regard). 55 Conceivably, immune checkpoint inhibitors may increase plaque inflammation and thereby predispose to classical plaque rupture. However, this has not yet been proven. Alternative etiologies of thrombotic coronary artery occlusions and related ACS that need to be entertained in cancer patients (similar to stroke) are embolic event via a patent foramen ovale, from the cardiac chambers, and/or even tumor embolization.56,57 Further to consider is spontaneous coronary artery dissection, possibly as a result of cancer therapy on vascular remodeling.58–60
As outlined in a recent study on ACS in patients with active hematological malignancies, type II myocardial infarctions were adjudicated in two thirds of MIs patients who underwent coronary angiography.61,62 Other than altered vasoreactivity, tachycardia, hypotension, hypoxia, and anemia may predispose to demand-supply mismatch in those with significant coronary artery disease, or potentially with patho-anatomical variants such as (severe) myocardial bridging. 62 Of note, severe coronary artery disease (three vessel and left main disease) was seen in half of the patients with active hematological malignancies and ACS who underwent coronary angiography.61
Another very important observation pertains to the impact of guideline-recommended therapies on mortality outcomes.61 Aspirin, beta-blocker, and ACE inhibitor/angiotensin receptor blocker reduced mortality whereas heparin use and an invasive approach did not. 61 The latter might be explained by the fact that type I MIs constituted only the minority and medical management sufficed in most patients. Other studies support these observations in broader cohorts of patients including those with solid tumors. The SCAI-based algorithm advises on an approach based on a modified TIMI risk score and takes the platelet count of the patient into consideration (Figure 4).63Revascularization strategies should follow practice guidelines (taking into account regional differences). 64,65,66,67 If stenting is performed, the ESC guidelines recommend drug-eluting stents (DES) regardless of (patient and lesion) presentation type. 67 This is based on the fact that improvements in DES design has reduced their thrombotic risk so much that it may be lower than with bare-metal stents (BMS). 68,69 In the LEADERS FREE trial in patients at high bleeding risk (including 15% with anemia, 15% with anticipated surgery in the next year, and 10% with malignancy), for instance, the combination with just one month of dual anti-platelet therapy (DAPT) a polymer-free biolimus A9-coated DES was superior to BMS in terms of safety (stent thrombosis, death, and MI) and efficacy (repeat revascularization). 70 It is important to point out that most reports on acute stent thrombosis in cancer patients have been in the setting of BMS, often shortly after discontinuation of DAPT. 71 This, however, does not exclude the possibility of this event after DES. In fact, malignancy has been listed as a key risk factor for early stent thrombosis and the most important patient-related risk factor for late stent thrombosis.72 For patients with ACS DAPT duration should be for a minimum of 1 year, regardless of coronary management strategy, i.e. even when medical therapy alone is chosen or surgery rather than stenting. This recommendation may even more so apply to cancer patients; however, the bleeding risk of these patients (especially with anticipated thrombocytopenia) needs to be taken in consideration. At this point it is not known if prediction score for thrombosis and bleeding risk such as the DAPT and PRECISE-DAPT server equally well in cancer patients (with a higher risk of both). 73,74
Figure 4.
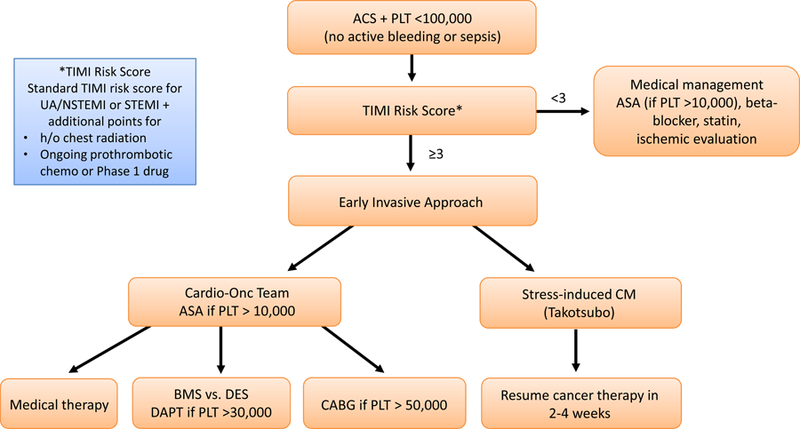
SCAI algorithm for the management of ACS in cancer patients. Adapted from Iliescu CA, Grines CL, Herrmann J, et al. SCAI Expert consensus statement: Evaluation, management, and special considerations of cardio-oncology patients in the cardiac catheterization laboratory (endorsed by the cardiological society of india, and sociedad Latino Americana de Cardiologia intervencionista). Catheter Cardiovasc Interv. 2016;87(5):E202–223; with permission.
Claudication/ acute limb ischemia
Other than in patients with pre-existing peripheral arterial disease, signs and symptoms of chronic limb ischemia were very rarely seen in cancer patients as a consequence of cancer therapy, other than, e.g. radiation therapy to the pelvis. This, however, changed with the introduction of TKIs targeting the Bcr-Abl oncogenic fusion gene product, namely nilotinib and ponatinib.12,75 These drugs were found to cause what has been termed progressive peripheral arterial occlusion disease (POAD), characterized by diffuse stenosis of the lower arterial circulation, vascular occlusions and formation of collateral circulation.76 The dynamics of these changes were perceived to be out of proportion to the risk factor profile. Indeed, in a carefully conducted matched-control study CML patients on nilotinib experienced a significantly higher rate of arterial occlusive events, 80% and 20% involving the peripheral and coronary circulation, respectively.15 Furthermore, atherosclerotic cardiovascular disease (ASCVD) risk factors models such as the EURO score were still predictive of risk.15 Collectively, these findings support the view that nilotinib plays a causal role and that the disease process is consistent with accelerated atherosclerosis rather than a different disease process. The same seems to hold true for ponatinib.12,25,77While the underlying pathophysiology is not fully defined, recent experimental studies indicate that nilotinib downregulates VEGF receptor II and thereby shares VEGF signaling pathway inhibition with ponatinib.15,78 Both drugs have a profoundly negative impact on endothelial cell viability, even in a therapeutic dose spectrum.79,80 In an animal model, nilotinib and ponatinib were found to shift the balance more towards vascular instability including both, the solid as well as the fluid phase.81 Plaque rupture with acute thrombosis is a conceivable mechanisms of acute events in patients undergoing therapy with these drugs but they have not necessarily been confirmed in all cases. Rather, it seems that at least in some patient events of acute ischemia are due to rapid progression to the point of occlusion with insufficient collateral formation. 82
In cancer patients at large, however, thromboembolism likely remains the more common mechanism of acute limb ischemia.83 Similar to acute thrombotic events in other vascular territories, the source of embolism can be anywhere proximal to the occlusion point in the peripheral arteries, aorta, the valves, the left ventricle, the left atrium/appendage or even on the venous side with paradoxical emboli. Patients with acute promyelocytic leukemia are particularly prone to acute arterial thrombosis in all vascular territories.84
Management of cancer patients with suspected peripheral arterial disease is as recommended by AHA/ACC guidelines. 85 Pulse status on physical exam is the first step followed by assessment of ankle-brachial index (ABI), which takes center stage. Patients with non-compressible vessels should have an assessment of toe-brachial index, whereas those with symptoms of claudication and normal or borderline normal ABU should have an exercise ABI. Those with any abnormal findings should be started on GDMT including aspirin or clopidogrel, statin, and optimal blood pressure control, considering an ACE inhibitor/ARB. If symptoms persist, anatomic assessment of the lower extremity vascular is to be pursued including Duplex ultrasound, CT angiography or MR angiography. For patients with suspected chronic limb ischemia, i.e. physical examination suggestive of PAD with rest pain, nonhealing wound, or gangrene, any abnormality on ABI testing is to be followed in by additional perfusion assessment including TBI with waveforms, TcPO2, and skin perfusion pressure, and if any of these are abnormal anatomic assessment as above or by invasive angiography. Patients presenting with acute limb ischemia need to be immediately recognized (acutely cold and painful leg) with motor and sensory assessment, ultrasound for arterial and venous Doppler signals. The differentiation then is between a viable limb, a marginally or immediately threatened limb, or a nonviable limb. Amputation is indicated for the latter scenario whereas all other should undergo emergent revascularization and anticoagulation, unless contraindicated.
At present it is unknown how to best assess patients to be started on nilotinib or ponanitib at baseline and during follow-up (Table 1).78,86 Various recommendations have been provided including alteration of dose and overall treatment regimens.11,12 In principle, for PAD and other atherosclerosis diseases with these drugs, the intensity for screening and preventive efforts should increase and the threshold for intervention and cessation of therapy should decrease with increasing risk category. As outlined above, the EURO score may be a unique tool to tailor screening efforts. The SCAI vascular surveillance algorithm takes into account duration of risk as well the impact of radiation therapy (Figure 5).
Table 1.
Recommendation for vascular assessment of patients on Bcr/Abl therapy, namle nilotinib and ponatinib
| Symptoms | Peripheral Pulse status | Risk Factors | Vascular tests | F/u |
|---|---|---|---|---|
| None | Normal | None | None | Every 12 months |
| None | Reduced +/− | None or present | ABI | If ABI ≥ 0.9: 12 mo. f/u |
| If ABI <0.9: additional tests* and 6 mo. f/u | ||||
| Present +/− | Reduced +/− | None or present | ABI | Vascular specialist |
| 3–6 mo. f/u | ||||
| If ABI <0.7: stop |
Additional tests include exercise ABI, Duplex U/S, and lower extremity CT angiography as well as evaluation of carotid IMT and coronary CTA or stress test
Data from Breccia M, Arboscello E, Bellodi A, et al. Proposal for a tailored stratification at baseline and monitoring of cardiovascular effects during follow-up in chronic phase chronic myeloid leukemia patients treated with nilotinib frontline. Crit Rev Oncol Hematol. 2016;107:190–198.
Figure 5.
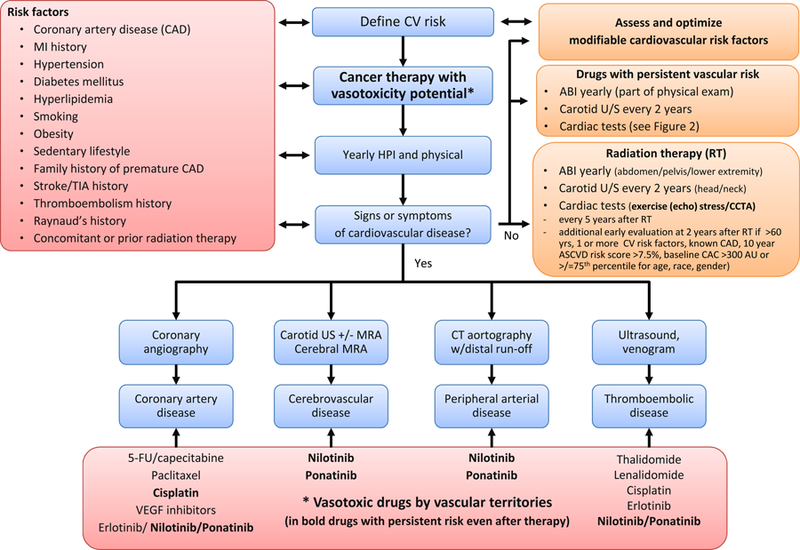
SCAI algorithm for the surveillance of vascular toxicities with cancer therapies. Adapted from Iliescu CA, Grines CL, Herrmann J, et al. SCAI Expert consensus statement: Evaluation, management, and special considerations of cardio-oncology patients in the cardiac catheterization laboratory (endorsed by the cardiological society of india, and sociedad Latino Americana de Cardiologia intervencionista). Catheter Cardiovasc Interv. 2016;87(5):E202–223; with permission.
Raynaud’s
Raynaud’s is a clinical diagnosis with a distinction between primary or secondary Raynaud’s. Abnormal vasoconstriction/ vasospasm of digital arteries and cutaneous arterioles is thought to be the underlying mechanism and in its most severe form can lead to ischemic fingertip necrosis. In most cancer patients, an underlying cause can be identified, especially medications. Raynaud’s has been reported with bleomycin, vinca alkaloids, cisplatin, carboplatin, gemcitabine, and IFN alpha, even as early as after the first dose.87–92 Endothelial injury has been a leading consideration; but evolving endothelial dysfunction may not suffice to explain this phenomenon. For IFN-alpha, for instance, immune-mediated vasculitis has been discussed in addition to thrombus formation and vasospasm.93 Raynaud’s can also occur as a paraneoplastic phenomenon, even before the diagnosis of malignancy is made.94
Usually no additional testing is performed and vasodilator therapy is the treatment of choice. Calcium channel blockers are the preferred agents whereas beta-blocker should be avoided (or switched to carvedilol if absolutely needed due to its additional alpha blocking properties which may ease peripheral vasoconstriction, but there is no guarantee that patients will benefit or tolerate this beta-blocker either). As with all vascular toxicities the risks and benefits of continuing versus discontinuing any culprit cancer therapeutic need to be carefully weighed.
Venous thromboembolic disease
As cancer patients face a higher risk of venous thromboembolism in general, it has been challenging to assign a high risk potential for venous thromboembolic events (VTEs) to specific cancer therapeutics.95 However, a few drugs do seem to increase the risk beyond what would be expected in the population. These include cisplatin, bevacizumab, TKIs targeting the VEGF signaling pathway as well as other, mTOR inhibitors, immunomodulatory agents including thalidomide and lenalidomide, possibly even immune checkpoint inhibitors, and anti-hormonal agents such as tamoxifen. The PROTECHT risk prediction score for VTE in cancer patients takes platinum-based and gemcitabine chemotherapy into consideration and may perform better than other risk prediction scores, including the extensively validated Khorana risk score.96 Despite the promise of such risk scores and promising results in clinical trials, generalized risk score-guided VTE prophylaxis is not recommended in cancer outpatients. In fact, primary prophylaxis is recommended only for patients with multiple myeloma, either aspirin if no additional risk factors, or LMWH/warfarin if additional risk factors are present including the use of IMDs with steroids.97
In cancer patients with VTE, guideline recommendations for treatment vary by society (Table 2). Importantly, the most recent 2019 NCCN guideline now lists rivaroxaban as a viable option for monotherapy aside from dalteparin as well as apixaban for patients who cannot or will not take LMWH (e.g. due to HIT). Furthermore, LMWH or unfractionated heparin for 5–10 days followed by edoxaban (or dabigatran) is another viable option. For many years LMWHs had been the preferred choice over warfarin given greater efficacy in preventing recurrent VTE. However, there are many obstacles to LMWH including administration and costs. DOACs have been found to be as effective as LMWH but with a higher bleeding risk potential, especially upper gastrointestinal bleeding (and possibly also genitorurinary leeding).98 The International Society of Thrombosis and Haemostasis Guidance Statement therefore suggests the use of specific DOACs (edoxaban and rivaroxaban) only for cancer patients with an acute diagnosis of VTE who have a low risk of bleeding and no drug–drug interactions with current systemic therapy.99 They continue to recommend LMWHs for cancer patients with an acute diagnosis of VTE and a high risk of bleeding, including patients with luminal gastrointestinal cancers with an intact primary, patients with cancers at risk of bleeding from the genitourinary tract, bladder, or nephrostomy tubes, or patients with active gastrointestinal mucosal abnormalities such as duodenal ulcers, gastritis, esophagitis, or colitis.
Table 2.
Treatment of cancer-related venous thrombosis
| 2016 ITAC-CME consensus recommendations | 2015 International Society of Thrombosis and Haemostasis Guidance Statements on Diagnosis and treatment of the incidental venous thrombosis in cancer patients | 2019 NCCN Guidelines on Cancer-Associated Venous Thromboembolic Disease | 2016 ACCP Guidelines |
|---|---|---|---|
|
Initial treatment of established venous thromboembolism (VTE): first 10 days of anticoagulation 1. Low-molecular-weight heparin (LMWH) is recommended for the initial treatment of established VTE in patients with cancer (grade 1B). 2. Fondaparinux and unfractionated heparin can also be used for the initial treatment of established VTE in patients with cancer (grade 2D). 3. Thrombolysis in patients with cancer with established VTE should only be considered on a case-by-case basis, with specific attention paid to contraindications, especially bleeding risk—eg, specifically if brain metastasis (guidance, based on evidence of very low quality and the high bleeding risk of thrombolytic therapy). 4. In the initial treatment of VTE, inferior vena cava filters can be considered in the case of contraindication for anticoagulant treatment or in the case of pulmonary embolism recurrence under optimal anticoagulation. Periodic reassessment of contraindications for anticoagulation is recommended, and anticoagulation should be resumed when safe. Early maintenance (10 days to 3 months) and long-term (beyond 3 months) 1. LMWHs are preferred over vitamin K antagonists (VKAs) for the treatment of VTE in patients with cancer (grade 1A). 2. LMWH should be used for a minimum of 3 months to treat established VTE in patients with cancer (grade 1A). 3. Direct oral anticoagulants can be considered for VTE treatment of patients with stable cancer not receiving systemic anticancer therapy, and in cases where VKA is an acceptable, but not an available, treatment choice (guidance). 4. After 3–6 months, termination or continuation of anticoagulation (LMWH, VKA, or direct oral 5. anticoagulants) should be based on individual assessment of the benefit-to-risk ratio, tolerability, drug availability, patient preference, and cancer activity (guidance, in the absence of data). Treatment of VTE recurrence in patients with cancer given anticoagulant treatment: 1. increase in LMWH dose (by 20–25%) in patients treated with LMWH 2. switch from VKA to LMWH in patients treated with VKA; and 3. inferior vena cava filter insertion—with continued anticoagulant therapy, unless contraindicated Treatment of established catheter-related thrombosis 1. For the treatment of symptomatic catheter-related thrombosis in patients with cancer, anticoagulant treatment is recommended for a minimum of 3 months; in this setting, LMWHs are suggested. Direct comparisons between LMWHs and VKAs have not been made in this setting. 2. The central venous catheter can be kept in place if it is functional, well positioned, and non-infected with good resolution of symptoms under close surveillance; irrespective of whether the central venous catheter is kept or removed, no standard approach in terms of duration of anticoagulation is established (guidance) |
• In cancer patients with a diagnosis of incidental VTE, we recommend a careful review of the history to exclude symptomatic VTE. • In patients with incidental PE involving the main, lobar, segmental or multiple subsegmental pulmonary arteries, we suggest that no further testing is required to confirm the diagnosis. • In patients with isolated SSPE, we recommend careful review of the images by radiologists, and suggest that compression ultrasonography of the lower limbs be performed to detect concomitant incidental DVT. • In patients with incidental ileofemoral DVT on CT of the abdomen and pelvis, we suggest confirming the diagnosis with Doppler ultrasonography of the pelvis and compression ultrasonography of the lower limbs. • In cancer patients with incidental VTE, we recommend standard anticoagulation with LMWH in those with symptoms compatible with VTE. • In patients with incidental proximal DVT, or PE of the main, lobar, segmental or multiple subsegmental pulmonary arteries, we recommend therapeutic anticoagulation for at least 6 months. • In patients with isolated SSPE with proximal DVT, we recommend therapeutic anticoagulation for at least 6 months. • In patients with isolated SSPE with distal DVT or without DVT, we suggest that the decision to provide anticoagulation be made on a case-by-case basis, considering the risk of bleeding, the presence of risk factors for recurrent thrombosis, the performance status of the patient, and patient preference. If the decision is not to anticoagulate, we suggest clinical monitoring and serial bilateral compression ultrasonography after 1 week in those with distal DVT, to detect thrombus extension. • In patients with incidental splanchnic vein thrombosis, we suggest anticoagulant therapy in patients with thrombosis that appears to be acute, or that shows progression or extension over time, and in those who are neither actively bleeding nor have a very high risk of bleeding. • In cancer patients with evidence of disease or ongoing systemic or locoregional therapy, we suggest periodic re-evaluation of the risks of bleeding and VTE recurrence, as well as patient preferences, to guide the decision of whether to extend LMWH beyond 6 months. |
Monotherapy • LWMH: Dalteparin 200 U/kg SC daily for 30 days, then 150 U/kg once daily for 2–6 months (category 1), enoxaparin (category 2A) • Rivaroxaban 15 mg BID for 21 days, then 20 mg daily (category 2A) • Fondaprarunux 5, 75., 10 mg [<50, 50–100, and >100 kg] (category 2A) • UFH (categpry 2B) • UFH IV then SC (category 2A) • UFH SC (category 2A) • For patients who refuse or have compelling reasons to avoid LMWH: Apixaban (category 2A) Combination therapy with edoxaban • LMWH (dalteparin 200 U/kg SC daily or enoxaparin 1 mg/kg SC BID) (category 1) or UFH IV or SC for 5–10 days, then edoxaban 60 mg daily (or 30 mg if CrCl 30–50 mL or <60 kg weight or concomitant p-glycoprotein inhibitors or inducers) for at least 6 months Combination therapy with warfarin • LMWH (as above), fondaparinux, or UFH IV or SC for 5–10 days, then warfarin with INR 2–3 for at least 6 months • Combination therapy with dabigatran • LMWH (dalteparin 200 U/kg SC daily or enoxaparin 1 mg/kg SC BID) (category 1) or UFH IV or SC for 5–10 days, then dabigatran 150 mg BID (as long as CrCl >30 mL/min) for at least 6 months Duration: • Minimum of 3 months • For non-catheter-associated DVT/PE indefinite while cancer is active, under treatment or risk factors for recurrence persist •For catheter-associated thrombosis, anticoagulation as long as catheter is in place, recommended at least 3 months |
• In patients with DVT of the leg or PE and cancer (“cancer-associated thrombosis”), as long-term (first 3 months) anticoagulant therapy, we suggest LMWH over VKA therapy (Grade 2B), dabigatran (Grad e 2C), rivaroxaban(Grade 2C), apixaban (Grade 2C), or edoxaban (Grade 2C). • In patients with DVT of the leg or PE who receive extended therapy, we suggest that there is no need to change the choice of anticoagulant after the first 3 months (Grade 2C). • In patients with DVT of the leg or PE and active cancer (“cancer-associated thrombosis”) and who (i) do not have a high bleeding risk, we recommend extended anticoagulant therapy (no scheduled stop date) over 3 months of therapy (Grade 1B), or (ii) have a high bleeding risk, we suggest extended anticoagulant therapy (no scheduled stop date) over 3 months of therapy (Grade 2B). • In patients with an unprovoked proximal DVT or PE who are stopping anticoagulant therapy and do not have a contraindication to aspirin, we suggest aspirin over no aspirin to prevent recurrent VTE (Grade 2B). • In patients with acute DVT or PE who are treated with anticoagulants, we recommend against the use of an inferior vena cava (IVC) filter (Grade 1B). • In patients with acute DVT of the leg, we suggest not using compression stockings routinely to prevent PTS (Grade 2B). |
It is recommended that the same anticoagulant is used for 3 months and that treatment should continue as long as cancer is active, under active treatment, or risk factors for recurrence persist. In case of recurrent VTE on anticoagulation, patients should be switched to LMWH if not on it already, otherwise, the dose of LMWH should be increased by 25% (anti-Xa levels and HIT should be considered). For patients with thrombocytopenia, the NCCN guidelines have listed enoxaparin as the only agent at full dose, half-dose, or no dose/combination with platelet transfusion in case of platelet counts of >50k, 25–50k, or <25k. Cost considerations are an important aspect, as different insurance plans may cover one but not either anticoagulant.
Pulmonary hypertension
Pulmonary hypertension is another example of a vascular toxicity that received a new level of attention with the introduction of targeted therapy. A clustering of nine patients on dasatinib therapy, a TKI of BCL-ABL, used in patients with Philadelphia Chromosome-positive (Ph+) leukemias, was noted in the French Pulmonary Hypertension Registry. Common to all patients, pulmonary capillary wedge pressure was normal and all but one patient had not response to vasodilator therapy. Mean pulmonary artery pressure was 46 mmHg and the average right ventricular systolic pressure was 65 mmHg. 100 In a larger follow-up study of 21 patients, pulmonary vascular resistance and arterial pressure dropped upon discontinuation of dasatinib but remained elevated in one third of the patients over an average follow-up period of 24 months. Whether one treatment strategy (e.g. endothelin receptor antagonists) is better than another is unknown at present. Universal screening has not been endorsed, but patients developing dyspnea or signs and symptoms of right heart failure on dasatinib therapy need to be evaluated by echocardiography and additional right heart catheterization if found to have pulmonary hypertension (Figure 6).101,102 As many as 1 in 10 patients on dasatinib may develop pulmonary.100,103 Pathomechanistically, dasatinib leads to endothelial injury and increases the susceptibility to experimental pulmonary hypertension, e.g. by chronic hypoxia, with structural alterations of the pulmonary arteries.104 In addition, immune mechanisms have been discussed to contribute to dasatinib-induced pulmonary hypertension based on the frequently concomitant exudative pleural and pericardial effusions with lymphocytic accumulations.105
Figure 6.
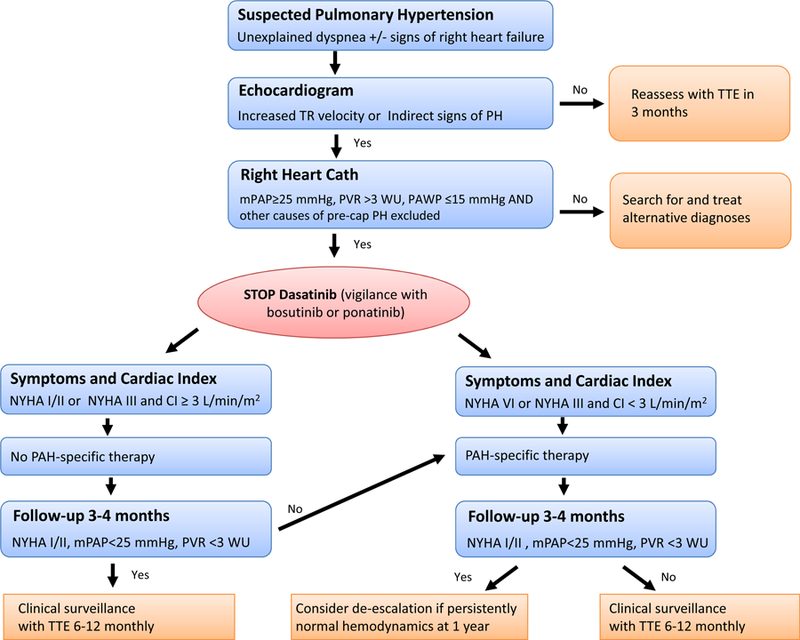
Suggested algorithm for the evaluation for pulmonary hypertension in patients on dasatinib. Adapted from Weatherald J, Chaumais MC, Savale L, et al. Long-term outcomes of dasatinib-induced pulmonary arterial hypertension: a population-based study. Eur Respir J. 2017;50(1); with permission.
The combination of a VEGF receptor 2 inhibitor with chronic hypoxia likewise results in reproducible pulmonary hypertension in experimental models.106 Furthermore, VEGF receptor 2 deficiency, even if confined to endothelial cells only, impairs vascularization and resolution of intra-pulmonary artery thrombi, which may contribute to chronic thromboembolic pulmonary hypertension.107 In addition to structural alteration, Rho kinase-mediated vasoconstriction is a contributing factor to severe occlusive pulmonary hypertension under the outlined conditions.108 Other newer agents that have implicated in pulmonary hypertension include nilotinib, ponatinib, carfilzomib, and ruxolitinib, but causality is not confirmed, especially in view of contradictory findings.109 Trastuzumab emtansine, rituximab, and bevacizumab have been implied in isolated case reports of pulmonary hypertension.109
Another chemotherapeutic that historically has been associated with pulmonary hypertension is bleomycin. Approximately 1 in 10 patients treated are affected and the risk emerges gradually over the course of therapy and even years later.110 The underlying pathology is pulmonary fibrosis as a consequence of the stimulation and transformation of fibroblasts into collagen-producing myofibroblasts by activated alveolar macrophages and epithelial cells in a response-to-injury pattern to inflammation.110 Statins have been shown to ameliorate bleomycin-induced lung injury as have Rho kinase inhibition, endothelin receptor antagonism, arginase inhibition, and provision of inhaled or even dietary NO, and sildenafil.111–118
Finally, interferon (IFN) alpha can induce pulmonary vasculitis and pulmonary hypertension for unknown reasons.91 Immune mechanisms are discussed among others, similar to the discussion on the effects of IFN alpha on the peripheral arterial vasculature.
Systemic hypertension
Increase in systemic blood pressure is a notorious characteristic of agents designed to target the VEGF signaling pathway. On average, systolic and diastolic blood pressure increase by 10 to 20 mmHg and 5 to 15 mmHg, respectively. Absolute numbers as well as the reported incidence rates of hypertension, however, are influenced by the monitoring techniques and definitions used. Ambulatory blood pressure monitoring has the advantage of detecting early and mild forms of hypertension. Chemotherapy-related systemic hypertension occurs over the course of the first few cycles of therapy, in fact, as early as within hours of therapy initiation, especially with TKIs.119 Reported incidences of hypertension also tend to be as much as two times higher with TKIs than with bevacizumab (up to 70% for all grade and up to nearly 20% high grade, life-threatening hypertensive crisis is uncommon still, <5%).25,120,121 Patients with pre-existing hypertension are at greater risk of developing worsening blood pressure control.122,123 Age ≥ 60 to 65 years, smoking, hypercholesterolemia, and obesity may further increase the risk; but these factors have not been universally confirmed as predictors. Ethnicity may play a role as does cancer type (higher in Asians and renal cell carcinoma patients).124
The mechanisms by which VEGF inhibitors increase blood pressure remain debated.125 Changes in systemic vascular resistance, secondary to endothelial dysfunction and capillary rarefication, are potential mechanisms of systemic hypertension with VEGF signaling pathway inhibitors.119 This is in keeping with the well documented effects of VEGF in angiogenesis and nitric oxide (NO) production by endothelial NO synthase (eNOS), with NO being crucial for normal endothelial function, vascular homeostasis and angiogenesis.125 In addition, inhibition of renal NO signaling leads to a rightward shift of the renal pressure-natriuresis curve with impaired sodium excretion, fluid retention and thus salt-dependent hypertension.126
The other class of chemotherapeutics that have been associated with hypertension are the mammalian target of rapamycin (mTOR) inhibitors. Everolimus carries a higher risk (up to 30%, hypertensive crisis 1%) than temsiolimus (overall <10%). The mechanisms of mTOR inhibitor-induced hypertension are not well defined. The same holds true for carfizomib; hypertension is seen in over 40% of patients treated with this proteasome inhibitor. Hypertensive crisis and emergency is rate but can occur.
In view of the potential worsening of systemic blood pressures to the point of life-threatening levels, it is generally recommended to control blood pressure prior to initiation of therapy and to follow close especially early in the course of therapy (Figures7 and 8). In view of the higher cardiovascular event risk in patients on VEGF inhibitor therapy, an argument for more intensive blood pressure targets can be made. 126 No anti-hypertensive has been shown to be superior per se, but some studies suggest more favorable survival outcomes with angiotensin converting enzyme inhibitors, though this has not been universally confirmed. 127–130 Non-dihydropyridines should be avoided as they inhibit cytochrome P450 3A4 and can result in higher levels of VEGF inhibitors.125 Fluid/volume management is an important factor in patients on carfilzomib. In severe, resistant hypertension, cancer therapy should be interrupted, which promptly and effectively decreases blood pressures.
Figure 7.
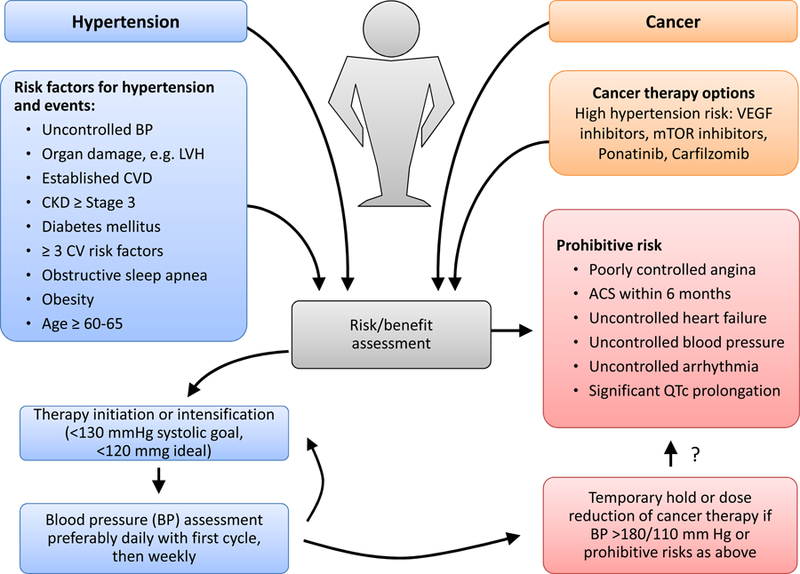
Evaluation proposal for cancer patients undergoing chemotherapy with hypertension risk such as those targeting the vascular endothelial growth factor (VEGF) pathway. Baseline evaluation should take into account risk factors for cardiovascular (CV) events, including uncontrolled blood pressure (BP), left ventricular hypertrophy (LVH), cardiovascular disease (CVD), chronic kidney disease (CKD), diabetes. Ideally patients should be optimized before starting chemotherapy and should be followed more closely early after starting therapy. In case of severe BP elevation or complications related or aggravated by it, cessation of therapy is to be considered. A blood pressure goal for patients on VEGF inhibitor therapy of <130 mmHg systolic (2017 hypertension guideline) and <120 mmHg systolic ideally (SPRINT trial target) is proposed. From Touyz RM, Herrmann SMS, Herrmann J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J Am Soc Hypertens. 2018;12(6):409–425; with permission.
Figure 8.
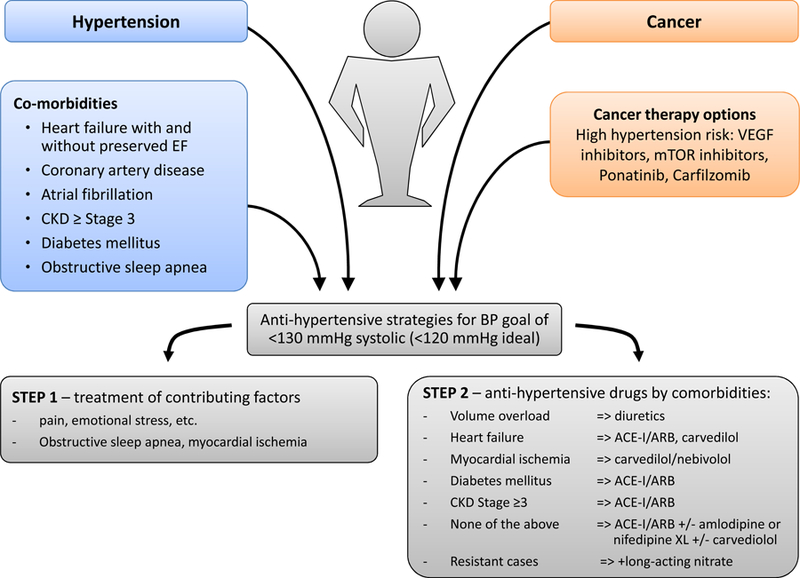
Management proposal for blood pressure (BP) control of cancer patients undergoing chemotherapy with hypertension risk. As outlined in the text, we propose that patients on VEGF inhibitor therapy should be treated toward a goal of <130 mmHg systolic (2017 hypertension guideline) and <120 mmHg systolic ideally (SPRINT trial target). Two steps toward reaching this goal are to be pursued: (1) treatment of contributing and aggravating factors and (2) antihypertensive therapy by comorbidity. From Touyz RM, Herrmann SMS, Herrmann J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J Am Soc Hypertens. 2018;12(6):409–425; with permission.
Synopsis.
The introduction of targeted agents into modern cancer therapy pursued the goal of molecularly more specific and thereby more effective and safer therapies. Paradoxically, however, a number of toxicities were brought to greater attention, among these not only cardiac but also vascular toxicities. The latter reach far beyond venous thromboembolism and include a broad spectrum of presentations based on vascular territories and pathomechanisms involved, including abnormal vascular reactivity, acute thrombosis or accelerated atherosclerosis. Herein we provide an overview of the most common presentations and their management strategies.
Key Points.
A broad spectrum of vascular toxicities has been recognized in the cancer patient and even more so since the introduction of targeted therapies.
Vascular toxicities of cancer therapies can involve all vascular territories, can be functional or structural in nature, and can be of lasting or only temporary duration.
The management of cancer therapy-related vascular toxicities is directed towards the underlying pathological mechanism: thrombosis, abnormal vasoreactivity, or structural alteration (remodeling).
Acknowledgments:
National Institute of Health (HL116952 and CA233610).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: None related to this content.
References
- 1.Zuo PY, Chen XL, Liu YW, Xiao CL, Liu CY. Increased risk of cerebrovascular events in patients with cancer treated with bevacizumab: a meta-analysis. PLoS One. 2014;9(7):e102484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Letarte N, Bressler LR, Villano JL. Bevacizumab and central nervous system (CNS) hemorrhage. Cancer chemotherapy and pharmacology. 2013;71(6):1561–1565. [DOI] [PubMed] [Google Scholar]
- 3.Sandler A, Hirsh V, Reck M, von Pawel J, Akerley W, Johnson DH. An evidence-based review of the incidence of CNS bleeding with anti-VEGF therapy in non-small cell lung cancer patients with brain metastases. Lung cancer. 2012;78(1):1–7. [DOI] [PubMed] [Google Scholar]
- 4.Khasraw M, Holodny A, Goldlust SA, DeAngelis LM. Intracranial hemorrhage in patients with cancer treated with bevacizumab: the Memorial Sloan-Kettering experience. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012;23(2):458–463. [DOI] [PubMed] [Google Scholar]
- 5.Navi BB, Iadecola C. Ischemic stroke in cancer patients: A review of an underappreciated pathology. Ann Neurol. 2018;83(5):873–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Navi BB, Reiner AS, Kamel H, et al. Risk of Arterial Thromboembolism in Patients With Cancer. J Am Coll Cardiol. 2017;70(8):926–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Navi BB, Reiner AS, Kamel H, et al. Arterial thromboembolic events preceding the diagnosis of cancer in older persons. Blood. 2019;133(8):781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oren O, Herrmann J. Arterial events in cancer patients-the case of acute coronary thrombosis. J Thorac Dis. 2018;10(Suppl 35):S4367–S4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.El Amrani M, Heinzlef O, Debroucker T, Roullet E, Bousser MG, Amarenco P. Brain infarction following 5-fluorouracil and cisplatin therapy. Neurology. 1998;51(3):899–901. [DOI] [PubMed] [Google Scholar]
- 10.Aichberger KJ, Herndlhofer S, Schernthaner GH, et al. Progressive peripheral arterial occlusive disease and other vascular events during nilotinib therapy in CML. Am J Hematol. 2011;86(7):533–539. [DOI] [PubMed] [Google Scholar]
- 11.Valent P, Hadzijusufovic E, Hoermann G, et al. Risk factors and mechanisms contributing to TKI-induced vascular events in patients with CML. Leuk Res. 2017;59:47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valent P, Hadzijusufovic E, Schernthaner GH, Wolf D, Rea D, le Coutre P. Vascular safety issues in CML patients treated with BCR/ABL1 kinase inhibitors. Blood. 2015;125(6):901–906. [DOI] [PubMed] [Google Scholar]
- 13.Coon EA, Zalewski NL, Hoffman EM, Tefferi A, Flemming KD. Nilotinib treatment-associated cerebrovascular disease and stroke. American journal of hematology. 2013;88(6):534–535. [DOI] [PubMed] [Google Scholar]
- 14.Mayer K, Gielen GH, Willinek W, Muller MC, Wolf D. Fatal progressive cerebral ischemia in CML under third-line treatment with ponatinib. Leukemia. 2014;28(4):976–977. [DOI] [PubMed] [Google Scholar]
- 15.Hadzijusufovic E, Albrecht-Schgoer K, Huber K, et al. Nilotinib-induced vasculopathy: identification of vascular endothelial cells as a primary target site. Leukemia. 2017;31(11):2388–2397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dieckmann KP, Struss WJ, Budde U. Evidence for acute vascular toxicity of cisplatin-based chemotherapy in patients with germ cell tumour. Anticancer Res. 2011;31(12):4501–4505. [PubMed] [Google Scholar]
- 17.Dursun B, He Z, Somerset H, Oh DJ, Faubel S, Edelstein CL. Caspases and calpain are independent mediators of cisplatin-induced endothelial cell necrosis. American journal of physiology Renal physiology. 2006;291(3):F578–587. [DOI] [PubMed] [Google Scholar]
- 18.Nguyen MT, Stoianovici R, Brunetti L. Chemotherapy induced stroke mimic: 5-Fluorouracil encephalopathy fulfilling criteria for tissue plasminogen activator therapy. Am J Emerg Med. 2017;35(9):1389–1390. [DOI] [PubMed] [Google Scholar]
- 19.Baytan B, Ozdemir O, Demirkaya M, Evim MS, Gunes AM. Reversible posterior leukoencephalopathy induced by cancer chemotherapy. Pediatr Neurol. 2010;43(3):197–201. [DOI] [PubMed] [Google Scholar]
- 20.Hottinger AF. Neurologic complications of immune checkpoint inhibitors. Curr Opin Neurol. 2016;29(6):806–812. [DOI] [PubMed] [Google Scholar]
- 21.How J, Blattner M, Fowler S, Wang-Gillam A, Schindler SE. Chemotherapy-associated Posterior Reversible Encephalopathy Syndrome: A Case Report and Review of the Literature. Neurologist. 2016;21(6):112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen T, DeAngelis LM. Stroke in cancer patients. Curr Neurol Neurosci Rep. 2006;6(3):187–192. [DOI] [PubMed] [Google Scholar]
- 23.Powers WJ, Rabinstein AA, Ackerson T, et al. 2018 Guidelines for the Early Management of Patients With Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke. 2018;49(3):e46–e110. [DOI] [PubMed] [Google Scholar]
- 24.Sestito A, Sgueglia GA, Pozzo C, et al. Coronary artery spasm induced by capecitabine. J Cardiovasc Med (Hagerstown). 2006;7(2):136–138. [DOI] [PubMed] [Google Scholar]
- 25.Herrmann J, Yang EH, Iliescu CA, et al. Vascular Toxicities of Cancer Therapies: The Old and the New--An Evolving Avenue. Circulation. 2016;133(13):1272–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sara JD, Kaur J, Khodadadi R, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. 2018;10:1758835918780140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi N, Hata N, Yokoyama S, Shinada T, Shirakabe A, Mizuno K. A case of Takotsubo cardiomyopathy during 5-fluorouracil treatment for rectal adenocarcinoma. Journal of Nippon Medical School = Nippon Ika Daigaku zasshi. 2009;76(1):27–33. [DOI] [PubMed] [Google Scholar]
- 28.S YH, Tornvall P, Tornerud M, Henareh L. Capecitabine caused cardiogenic shock through induction of global Takotsubo syndrome. Cardiovasc Revasc Med. 2013;14(1):57–61. [DOI] [PubMed] [Google Scholar]
- 29.Dixon A, Nakamura JM, Oishi N, Wachi DH, Fukuyama O. Angina pectoris and therapy with cisplatin, vincristine, and bleomycin. Annals of internal medicine. 1989;111(4):342–343. [DOI] [PubMed] [Google Scholar]
- 30.Rodriguez J, Collazos J, Gallardo M, Hernando G. Angina pectoris following cisplatin, etoposide, and bleomycin in a patient with advanced testicular cancer. The Annals of pharmacotherapy. 1995;29(2):138–139. [DOI] [PubMed] [Google Scholar]
- 31.Fukuda M, Oka M, Itoh N, et al. Vasospastic angina likely related to cisplatin-containing chemotherapy and thoracic irradiation for lung cancer. Internal medicine. 1999;38(5):436–438. [DOI] [PubMed] [Google Scholar]
- 32.Pantaleo MA, Mandrioli A, Saponara M, et al. Development of coronary artery stenosis in a patient with metastatic renal cell carcinoma treated with sorafenib. BMC Cancer. 2012;12:231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winnik S, Lohmann C, Siciliani G, et al. Systemic VEGF inhibition accelerates experimental atherosclerosis and disrupts endothelial homeostasis--implications for cardiovascular safety. Int J Cardiol. 2013;168(3):2453–2461. [DOI] [PubMed] [Google Scholar]
- 34.Chintalgattu V, Rees ML, Culver JC, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med. 2013;5(187):187ra169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinberg BA, Conces DJ Jr., Waller BF. Cardiac manifestations of noncardiac tumors. Part I: Direct effects. Clinical cardiology. 1989;12(5):289–296. [DOI] [PubMed] [Google Scholar]
- 36.Zeymer U, Hirschmann WD, Neuhaus KL. Left main coronary stenosis by a mediastinal lymphoma. The Clinical investigator. 1992;70(11):1024–1026. [DOI] [PubMed] [Google Scholar]
- 37.Orban M, Tousek P, Becker I, Augustin N, Firschke C. Cardiac malignant tumor as a rare cause of acute myocardial infarction. The international journal of cardiovascular imaging. 2004;20(1):47–51. [DOI] [PubMed] [Google Scholar]
- 38.Juan YH, Chatzizisis YS, Saboo SS, Rocha T, Steigner ML. Tumor encasement of the right coronary artery: role of anatomic and functional imaging in diagnosis and therapeutic management. Open Cardiovasc Med J. 2014;8:110–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lu JI, Carhart RL, Graziano SL, Gajra A. Acute coronary syndrome secondary to fluorouracil infusion. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(18):2959–2960. [DOI] [PubMed] [Google Scholar]
- 40.Cardinale D, Colombo A, Colombo N. Acute coronary syndrome induced by oral capecitabine. The Canadian journal of cardiology. 2006;22(3):251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frickhofen N, Beck FJ, Jung B, Fuhr HG, Andrasch H, Sigmund M. Capecitabine can induce acute coronary syndrome similar to 5-fluorouracil. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2002;13(5):797–801. [DOI] [PubMed] [Google Scholar]
- 42.Schrader C, Keussen C, Bewig B, von Freier A, Lins M. Symptoms and signs of an acute myocardial ischemia caused by chemotherapy with Paclitaxel (Taxol) in a patient with metastatic ovarian carcinoma. European journal of medical research. 2005;10(11):498–501. [PubMed] [Google Scholar]
- 43.Shah K, Gupta S, Ghosh J, Bajpai J, Maheshwari A. Acute non-ST elevation myocardial infarction following paclitaxel administration for ovarian carcinoma: a case report and review of literature. Journal of cancer research and therapeutics. 2012;8(3):442–444. [DOI] [PubMed] [Google Scholar]
- 44.Gemici G, Cincin A, Degertekin M, Oktay A. Paclitaxel-induced ST-segment elevations. Clin Cardiol. 2009;32(6):E94–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ozturk B, Tacoy G, Coskun U, et al. Gemcitabine-induced acute coronary syndrome: a case report. Med Princ Pract. 2009;18(1):76–80. [DOI] [PubMed] [Google Scholar]
- 46.Armitage JD, Montero C, Benner A, Armitage JO, Bociek G. Acute coronary syndromes complicating the first infusion of rituximab. Clin Lymphoma Myeloma. 2008;8(4):253–255. [DOI] [PubMed] [Google Scholar]
- 47.Arima Y, Oshima S, Noda K, et al. Sorafenib-induced acute myocardial infarction due to coronary artery spasm. J Cardiol. 2009;54(3):512–515. [DOI] [PubMed] [Google Scholar]
- 48.Jafri M, Protheroe A. Cisplatin-associated thrombosis. Anti-cancer drugs. 2008;19(9):927–929. [DOI] [PubMed] [Google Scholar]
- 49.Karabay KO, Yildiz O, Aytekin V. Multiple coronary thrombi with cisplatin. J Invasive Cardiol. 2014;26(2):E18–20. [PubMed] [Google Scholar]
- 50.Ito D, Shiraishi J, Nakamura T, et al. Primary percutaneous coronary intervention and intravascular ultrasound imaging for coronary thrombosis after cisplatin-based chemotherapy. Heart and vessels. 2012;27(6):634–638. [DOI] [PubMed] [Google Scholar]
- 51.Michel JB, Martin-Ventura JL, Nicoletti A, Ho-Tin-Noe B. Pathology of human plaque vulnerability: mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis. 2014;234(2):311–319. [DOI] [PubMed] [Google Scholar]
- 52.Michel JB, Virmani R, Arbustini E, Pasterkamp G. Intraplaque haemorrhages as the trigger of plaque vulnerability. Eur Heart J. 2011;32(16):1977–1985, 1985a, 1985b, 1985c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jain RK, Finn AV, Kolodgie FD, Gold HK, Virmani R. Antiangiogenic therapy for normalization of atherosclerotic plaque vasculature: a potential strategy for plaque stabilization. Nat Clin Pract Cardiovasc Med. 2007;4(9):491–502. [DOI] [PubMed] [Google Scholar]
- 54.Kolodgie FD, Narula J, Yuan C, Burke AP, Finn AV, Virmani R. Elimination of neoangiogenesis for plaque stabilization: is there a role for local drug therapy? J Am Coll Cardiol. 2007;49(21):2093–2101. [DOI] [PubMed] [Google Scholar]
- 55.Ramcharan KS, Lip GY, Stonelake PS, Blann AD. Effect of standard chemotherapy and antiangiogenic therapy on plasma markers and endothelial cells in colorectal cancer. Br J Cancer. 2014;111(9):1742–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kushiyama S, Ikura Y, Iwai Y. Acute myocardial infarction caused by coronary tumour embolism. European heart journal. 2013;34(48):3690. [DOI] [PubMed] [Google Scholar]
- 57.Diaz Castro O, Bueno H, Nebreda LA. Acute myocardial infarction caused by paradoxical tumorous embolism as a manifestation of hepatocarcinoma. Heart. 2004;90(5):e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mir MA, Patnaik MM, Herrmann J. Spontaneous coronary artery dissection during hematopoietic stem cell infusion. Blood. 2013;122(19):3388–3389. [DOI] [PubMed] [Google Scholar]
- 59.Ghosh N, Chow CM, Korley V, Chisholm R. An unusual case of chronic coronary artery dissection: did cisplatin play a role? The Canadian journal of cardiology. 2008;24(10):795–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Abbott JD, Curtis JP, Murad K, et al. Spontaneous coronary artery dissection in a woman receiving 5-fluorouracil--a case report. Angiology. 2003;54(6):721–724. [DOI] [PubMed] [Google Scholar]
- 61.Park JY, Guo W, Al-Hijji M, et al. Acute coronary syndromes in patients with active hematologic malignancies-Incidence, management, and outcomes. Int J Cardiol. 2018. [DOI] [PMC free article] [PubMed]
- 62.Thygesen K, Alpert JS, Jaffe AS, et al. Fourth Universal Definition of Myocardial Infarction (2018). J Am Coll Cardiol. 2018;72(18):2231–2264. [DOI] [PubMed] [Google Scholar]
- 63.Iliescu CA, Grines CL, Herrmann J, et al. SCAI Expert consensus statement: Evaluation, management, and special considerations of cardio-oncology patients in the cardiac catheterization laboratory (endorsed by the cardiological society of india, and sociedad Latino Americana de Cardiologia intervencionista). Catheter Cardiovasc Interv. 2016;87(5):E202–223. [DOI] [PubMed] [Google Scholar]
- 64.Rodriguez F, Mahaffey KW. Management of Patients With NSTE-ACS: A Comparison of the Recent AHA/ACC and ESC Guidelines. J Am Coll Cardiol. 2016;68(3):313–321. [DOI] [PubMed] [Google Scholar]
- 65.Amsterdam EA, Wenger NK, Brindis RG, et al. 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;130(25):e344–426. [DOI] [PubMed] [Google Scholar]
- 66.Roffi M, Patrono C, Collet JP, et al. 2015 ESC Guidelines for the management of acute coronary syndromes in patients presenting without persistent ST-segment elevation: Task Force for the Management of Acute Coronary Syndromes in Patients Presenting without Persistent ST-Segment Elevation of the European Society of Cardiology (ESC). Eur Heart J. 2016;37(3):267–315. [DOI] [PubMed] [Google Scholar]
- 67.Neumann FJ, Sousa-Uva M, Ahlsson A, et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur Heart J. 2018. [DOI] [PubMed]
- 68.Gogas BD, McDaniel M, Samady H, King SB 3rd. Novel drug-eluting stents for coronary revascularization. Trends Cardiovasc Med. 2014;24(7):305–313. [DOI] [PubMed] [Google Scholar]
- 69.Palmerini T, Biondi-Zoccai G, Della Riva D, et al. Stent thrombosis with drug-eluting and bare-metal stents: evidence from a comprehensive network meta-analysis. Lancet. 2012;379(9824):1393–1402. [DOI] [PubMed] [Google Scholar]
- 70.Urban P, Meredith IT, Abizaid A, et al. Polymer-free Drug-Coated Coronary Stents in Patients at High Bleeding Risk. N Engl J Med. 2015;373(21):2038–2047. [DOI] [PubMed] [Google Scholar]
- 71.Smith SC, Winters KJ, Lasala JM. Stent thrombosis in a patient receiving chemotherapy. Cathet Cardiovasc Diagn. 1997;40(4):383–386. [DOI] [PubMed] [Google Scholar]
- 72.Gori T, Polimeni A, Indolfi C, Raber L, Adriaenssens T, Munzel T. Predictors of stent thrombosis and their implications for clinical practice. Nat Rev Cardiol. 2019;16(4):243–256. [DOI] [PubMed] [Google Scholar]
- 73.Yeh RW, Secemsky EA, Kereiakes DJ, et al. Development and Validation of a Prediction Rule for Benefit and Harm of Dual Antiplatelet Therapy Beyond 1 Year After Percutaneous Coronary Intervention. JAMA. 2016;315(16):1735–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Costa F, van Klaveren D, James S, et al. Derivation and validation of the predicting bleeding complications in patients undergoing stent implantation and subsequent dual antiplatelet therapy (PRECISE-DAPT) score: a pooled analysis of individual-patient datasets from clinical trials. Lancet. 2017;389(10073):1025–1034. [DOI] [PubMed] [Google Scholar]
- 75.Herrmann J, Lerman A. An update on cardio-oncology. Trends Cardiovasc Med. 2014;24(7):285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aichberger KJ, Herndlhofer S, Schernthaner GH, et al. Progressive peripheral arterial occlusive disease and other vascular events during nilotinib therapy in CML. American journal of hematology. 2011;86(7):533–539. [DOI] [PubMed] [Google Scholar]
- 77.Herrmann J Tyrosine Kinase Inhibitors and Vascular Toxicity: Impetus for a Classification System? Curr Oncol Rep. 2016;18(6):33. [DOI] [PubMed] [Google Scholar]
- 78.Moslehi JJ, Deininger M. Tyrosine Kinase Inhibitor-Associated Cardiovascular Toxicity in Chronic Myeloid Leukemia. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(35):4210–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gover-Proaktor A, Granot G, Pasmanik-Chor M, et al. Bosutinib, dasatinib, imatinib, nilotinib, and ponatinib differentially affect the vascular molecular pathways and functionality of human endothelial cells. Leuk Lymphoma. 2018:1–11. [DOI] [PubMed]
- 80.Gover-Proaktor A, Granot G, Shapira S, et al. Ponatinib reduces viability, migration, and functionality of human endothelial cells. Leuk Lymphoma. 2017;58(6):1455–1467. [DOI] [PubMed] [Google Scholar]
- 81.Pouwer MG, Pieterman EJ, Verschuren L, et al. The BCR-ABL1 Inhibitors Imatinib and Ponatinib Decrease Plasma Cholesterol and Atherosclerosis, and Nilotinib and Ponatinib Activate Coagulation in a Translational Mouse Model. Front Cardiovasc Med. 2018;5:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Herrmann J, Bell MR, Warren RL, Lerman A, Fleming MD, Patnaik M. Complicated and Advanced Atherosclerosis in a Young Woman With Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia: Success and Challenges of BCR/ABL1-Targeted Cancer Therapy. Mayo Clin Proc. 2015;90(8):1167–1168. [DOI] [PubMed] [Google Scholar]
- 83.Tsang JS, Naughton PA, O’Donnell J, et al. Acute limb ischemia in cancer patients: should we surgically intervene? Annals of vascular surgery. 2011;25(7):954–960. [DOI] [PubMed] [Google Scholar]
- 84.Kalk E, Goede A, Rose P. Acute arterial thrombosis in acute promyelocytic leukaemia. Clin Lab Haematol. 2003;25(4):267–270. [DOI] [PubMed] [Google Scholar]
- 85.Gerhard-Herman MD, Gornik HL, Barrett C, et al. 2016 AHA/ACC Guideline on the Management of Patients With Lower Extremity Peripheral Artery Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2017;69(11):e71–e126. [DOI] [PubMed] [Google Scholar]
- 86.Breccia M, Arboscello E, Bellodi A, et al. Proposal for a tailored stratification at baseline and monitoring of cardiovascular effects during follow-up in chronic phase chronic myeloid leukemia patients treated with nilotinib frontline. Crit Rev Oncol Hematol. 2016;107:190–198. [DOI] [PubMed] [Google Scholar]
- 87.Staff S, Lagerstedt E, Seppanen J, Maenpaa J. Acute digital ischemia complicating gemcitabine and carboplatin combination chemotherapy for ovarian cancer. Acta obstetricia et gynecologica Scandinavica. 2011;90(11):1296–1297. [DOI] [PubMed] [Google Scholar]
- 88.Vogelzang NJ, Bosl GJ, Johnson K, Kennedy BJ. Raynaud’s phenomenon: a common toxicity after combination chemotherapy for testicular cancer. Annals of internal medicine. 1981;95(3):288–292. [DOI] [PubMed] [Google Scholar]
- 89.Kuhar CG, Mesti T, Zakotnik B. Digital ischemic events related to gemcitabine: Report of two cases and a systematic review. Radiol Oncol. 2010;44(4):257–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zeidman A, Dicker D, Mittelman M. Interferon-induced vasospasm in chronic myeloid leukaemia. Acta haematologica. 1998;100(2):94–96. [DOI] [PubMed] [Google Scholar]
- 91.Al-Zahrani H, Gupta V, Minden MD, Messner HA, Lipton JH. Vascular events associated with alpha interferon therapy. Leukemia & lymphoma. 2003;44(3):471–475. [DOI] [PubMed] [Google Scholar]
- 92.McGrath SE, Webb A, Walker-Bone K. Bleomycin-induced Raynaud’s phenomenon after single-dose exposure: risk factors and treatment with intravenous iloprost infusion. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(4):e51–52. [DOI] [PubMed] [Google Scholar]
- 93.Raanani P, Ben-Bassat I. Immune-mediated complications during interferon therapy in hematological patients. Acta haematologica. 2002;107(3):133–144. [DOI] [PubMed] [Google Scholar]
- 94.Madabhavi I, Revannasiddaiah S, Rastogi M, Gupta MK. Paraneoplastic Raynaud’s phenomenon manifesting before the diagnosis of lung cancer. BMJ case reports. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122(10):1712–1723. [DOI] [PubMed] [Google Scholar]
- 96.van Es N, Di Nisio M, Cesarman G, et al. Comparison of risk prediction scores for venous thromboembolism in cancer patients: a prospective cohort study. Haematologica. 2017;102(9):1494–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lyman GH, Bohlke K, Khorana AA, et al. Venous thromboembolism prophylaxis and treatment in patients with cancer: american society of clinical oncology clinical practice guideline update 2014. J Clin Oncol. 2015;33(6):654–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Raskob GE, van Es N, Verhamme P, et al. Edoxaban for the Treatment of Cancer-Associated Venous Thromboembolism. N Engl J Med. 2018;378(7):615–624. [DOI] [PubMed] [Google Scholar]
- 99.Khorana AA, Noble S, Lee AYY, et al. Role of direct oral anticoagulants in the treatment of cancer-associated venous thromboembolism: guidance from the SSC of the ISTH. J Thromb Haemost. 2018;16(9):1891–1894. [DOI] [PubMed] [Google Scholar]
- 100.Montani D, Bergot E, Gunther S, et al. Pulmonary arterial hypertension in patients treated by dasatinib. Circulation. 2012;125(17):2128–2137. [DOI] [PubMed] [Google Scholar]
- 101.Weatherald J, Chaumais MC, Montani D. Pulmonary arterial hypertension induced by tyrosine kinase inhibitors. Curr Opin Pulm Med. 2017;23(5):392–397. [DOI] [PubMed] [Google Scholar]
- 102.Weatherald J, Chaumais MC, Savale L, et al. Long-term outcomes of dasatinib-induced pulmonary arterial hypertension: a population-based study. Eur Respir J. 2017;50(1). [DOI] [PubMed] [Google Scholar]
- 103.Jeon Y-W, Lee S-E, Kim S-H, et al. Six-Year Follow-Up Of Dasatinib-Related Pulmonary Arterial Hypertension (PAH) For Chronic Myeloid Leukemia In Single Center. Blood 2013;122(21):4017. [Google Scholar]
- 104.Guignabert C, Phan C, Seferian A, et al. Dasatinib induces lung vascular toxicity and predisposes to pulmonary hypertension. J Clin Invest. 2016;126(9):3207–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bergeron A, Rea D, Levy V, et al. Lung abnormalities after dasatinib treatment for chronic myeloid leukemia: a case series. American journal of respiratory and critical care medicine. 2007;176(8):814–818. [DOI] [PubMed] [Google Scholar]
- 106.Sakao S, Tatsumi K. The effects of antiangiogenic compound SU5416 in a rat model of pulmonary arterial hypertension. Respiration; international review of thoracic diseases. 2011;81(3):253–261. [DOI] [PubMed] [Google Scholar]
- 107.Alias S, Redwan B, Panzenbock A, et al. Defective angiogenesis delays thrombus resolution: a potential pathogenetic mechanism underlying chronic thromboembolic pulmonary hypertension. Arteriosclerosis, thrombosis, and vascular biology. 2014;34(4):810–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Oka M, Homma N, Taraseviciene-Stewart L, et al. Rho kinase-mediated vasoconstriction is important in severe occlusive pulmonary arterial hypertension in rats. Circulation research. 2007;100(6):923–929. [DOI] [PubMed] [Google Scholar]
- 109.McGee M, Whitehead N, Martin J, Collins N. Drug-associated pulmonary arterial hypertension. Clin Toxicol (Phila). 2018;56(9):801–809. [DOI] [PubMed] [Google Scholar]
- 110.Reinert T, #xe1, Baldotto CSdR, Nunes FAP, Scheliga AAdS. Bleomycin-Induced Lung Injury. Journal of Cancer Research. 2013;2013:9. [Google Scholar]
- 111.Lee AH, Dhaliwal R, Kantores C, et al. Rho-kinase inhibitor prevents bleomycin-induced injury in neonatal rats independent of effects on lung inflammation. American journal of respiratory cell and molecular biology. 2014;50(1):61–73. [DOI] [PubMed] [Google Scholar]
- 112.Bei Y, Hua-Huy T, Duong-Quy S, et al. Long-term treatment with fasudil improves bleomycin-induced pulmonary fibrosis and pulmonary hypertension via inhibition of Smad2/3 phosphorylation. Pulm Pharmacol Ther. 2013;26(6):635–643. [DOI] [PubMed] [Google Scholar]
- 113.Schroll S, Lange TJ, Arzt M, et al. Effects of simvastatin on pulmonary fibrosis, pulmonary hypertension and exercise capacity in bleomycin-treated rats. Acta physiologica. 2013;208(2):191–201. [DOI] [PubMed] [Google Scholar]
- 114.Baliga RS, Milsom AB, Ghosh SM, et al. Dietary nitrate ameliorates pulmonary hypertension: cytoprotective role for endothelial nitric oxide synthase and xanthine oxidoreductase. Circulation. 2012;125(23):2922–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Van Rheen Z, Fattman C, Domarski S, et al. Lung extracellular superoxide dismutase overexpression lessens bleomycin-induced pulmonary hypertension and vascular remodeling. American journal of respiratory cell and molecular biology. 2011;44(4):500–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schroll S, Arzt M, Sebah D, Nuchterlein M, Blumberg F, Pfeifer M. Improvement of bleomycin-induced pulmonary hypertension and pulmonary fibrosis by the endothelin receptor antagonist Bosentan. Respir Physiol Neurobiol. 2010;170(1):32–36. [DOI] [PubMed] [Google Scholar]
- 117.Hemnes AR, Zaiman A, Champion HC. PDE5A inhibition attenuates bleomycin-induced pulmonary fibrosis and pulmonary hypertension through inhibition of ROS generation and RhoA/Rho kinase activation. American journal of physiology Lung cellular and molecular physiology. 2008;294(1):L24–33. [DOI] [PubMed] [Google Scholar]
- 118.Grasemann H, Dhaliwal R, Ivanovska J, et al. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. American journal of physiology Lung cellular and molecular physiology. 2015;308(6):L503–510. [DOI] [PubMed] [Google Scholar]
- 119.Izzedine H, Ederhy S, Goldwasser F, et al. Management of hypertension in angiogenesis inhibitor-treated patients. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2009;20(5):807–815. [DOI] [PubMed] [Google Scholar]
- 120.Qi WX, He AN, Shen Z, Yao Y. Incidence and risk of hypertension with a novel multi-targeted kinase inhibitor axitinib in cancer patients: a systematic review and meta-analysis. British journal of clinical pharmacology. 2013;76(3):348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Qi WX, Lin F, Sun YJ, et al. Incidence and risk of hypertension with pazopanib in patients with cancer: a meta-analysis. Cancer chemotherapy and pharmacology. 2013;71(2):431–439. [DOI] [PubMed] [Google Scholar]
- 122.Wicki A, Hermann F, Pretre V, et al. Pre-existing antihypertensive treatment predicts early increase in blood pressure during bevacizumab therapy: the prospective AVALUE cohort study. Oncology research and treatment. 2014;37(5):230–236. [DOI] [PubMed] [Google Scholar]
- 123.Hamnvik OP, Choueiri TK, Turchin A, et al. Clinical risk factors for the development of hypertension in patients treated with inhibitors of the VEGF signaling pathway. Cancer. 2015;121(2):311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tomita Y, Uemura H, Fujimoto H, et al. Key predictive factors of axitinib (AG-013736)-induced proteinuria and efficacy: a phase II study in Japanese patients with cytokine-refractory metastatic renal cell Carcinoma. European journal of cancer. 2011;47(17):2592–2602. [DOI] [PubMed] [Google Scholar]
- 125.Small HY, Montezano AC, Rios FJ, Savoia C, Touyz RM. Hypertension due to antiangiogenic cancer therapy with vascular endothelial growth factor inhibitors: understanding and managing a new syndrome. Can J Cardiol. 2014;30(5):534–543. [DOI] [PubMed] [Google Scholar]
- 126.Touyz RM, Herrmann SMS, Herrmann J. Vascular toxicities with VEGF inhibitor therapies-focus on hypertension and arterial thrombotic events. J Am Soc Hypertens. 2018;12(6):409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Keizman D, Huang P, Eisenberger MA, et al. Angiotensin system inhibitors and outcome of sunitinib treatment in patients with metastatic renal cell carcinoma: a retrospective examination. Eur J Cancer. 2011;47(13):1955–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mc Menamin UC, Murray LJ, Cantwell MM, Hughes CM. Angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in cancer progression and survival: a systematic review. Cancer Causes Control. 2012;23(2):221–230. [DOI] [PubMed] [Google Scholar]
- 129.Izzedine H, Derosa L, Le Teuff G, Albiges L, Escudier B. Hypertension and angiotensin system inhibitors: impact on outcome in sunitinib-treated patients for metastatic renal cell carcinoma. Ann Oncol. 2015;26(6):1128–1133. [DOI] [PubMed] [Google Scholar]
- 130.Sorich MJ, Kichenadasse G, Rowland A, Woodman RJ, Mangoni AA. Angiotensin system inhibitors and survival in patients with metastatic renal cell carcinoma treated with VEGF-targeted therapy: A pooled secondary analysis of clinical trials. Int J Cancer. 2016;138(9):2293–2299. [DOI] [PubMed] [Google Scholar]