

Abstract
Background
Chloroplast (cp) genome information would facilitate the development and utilization of Taxodium resources. However, cp genome characteristics of Taxodium were poorly understood.
Results
We determined the complete cp genome sequences of T. distichum, T. mucronatum, and T. ascendens. The cp genomes are 131,947 bp to 132,613 bp in length, encode 120 genes with the same order, and lack typical inverted repeat (IR) regions. The longest small IR, a 282 bp trnQ-containing IR, were involved in the formation of isomers. Comparative analysis of the 3 cp genomes showed that 91.57% of the indels resulted in the periodic variation of tandem repeat (TR) motifs and 72.46% single nucleotide polymorphisms (SNPs) located closely to TRs, suggesting a relationship between TRs and mutational dynamics. Eleven hypervariable regions were identified as candidates for DNA barcode development. Hypothetical cp open reading frame 1(Ycf1) was the only one gene that has an indel in coding DNA sequence, and the indel is composed of a long TR. When extended to cupressophytes, ycf1 genes have undergone a universal insertion of TRs accompanied by extreme length expansion. Meanwhile, ycf1 also located in rearrangement endpoints of cupressophyte cp genomes. All these characteristics highlight the important role of repeats in the evolution of cp genomes.
Conclusions
This study added new evidence for the role of repeats in the dynamics mechanism of cp genome mutation and rearrangement. Moreover, the information of TRs and hypervariable regions would provide reliable molecular resources for future research focusing on the infrageneric taxa identification, phylogenetic resolution, population structure and biodiversity for the genus Taxodium and Cupressophytes.
Keywords: Taxodium, chloroplast genome, repeat, indel, single nucleotide polymorphisms, arrangement
Background
Taxodium belongs to the family Cupressaceae, is native to North America and Mexico, and contains three tax: bald cypress, pond cypress and montezuma cypress. However, there have been continuous debates concerning the taxonomy of these three taxa as one, two, or three species from the nineteenth century to the present [1–3]. In this study, we temporarily consider treating them as three species. Taxodium have strong resistance to biotic and abiotic stresses, and their life span can be as long as thousands of years [4]. Since 1973, Institute of Botany, Jiangsu Province and Chinese Academy of Sciences has been vigorously engaged in interspecific hybridization breeding of Taxodium. A batch of new varieties named ‘Zhongshanshan’ have been selected from the hybrids with the advantages of high ornamental value, rapid growth and strong stress resistance. They have been popularized and applied in 18 provinces and municipalities of China, bringing better ecological and social benefits in the urban landscaping, ecological civilization construction, sponge city construction, and ecological restoration of the Yangtze River economic zone [4]. ‘Zhongshanshan’ has become an important tree species with huge market demand in China. Although it has great economic value for the development and utilization of Taxodium resources, the research basis of phylogenetics, species/variety identification and genetic diversity of this genus is weak at present.
As an semi-autonomous replication organelles, the chloroplast genome has some unique advantages compared with nuclear and mitochondrion genome [5]. Cp genome is much smaller than the nuclear genome, and it’s easy to obtain the complete cp genome sequence. The gene density of cp genome is larger and the evolution rate is moderate, and segments with different evolution rates can be selected for different research purposes. The cp genomes of higher plants are highly conserved in organization, gene order and content, which can ensure homology among distant evolutionary groups. Besides, genes of cp genomes are single-copy, which ensures the direct homology of genes among species, and there is almost no interference of side-line homologous genes. Therefore, cp genome has unique value in phylogenetics, species identification and population genetics of higher plants.
Typically, the circular genomes of cp are organized into large and small single-copy regions, separated by an inverted repeat (IR). IR region is a pair of sequences with the same sequence and opposite direction, named IRA and IRB respectively. The sequence between IRA and IRB can produce triggered flip-flop recombination, which can stabilize the single-copy regions. Although the gene content and arrangement of cp genomes are relatively conservative, a series of changes have taken place from point to surface, including RNA editing (base insertion/deletion and substitution/transition), gene transfer and loss, inversion events, etc. The typical feature of the cp genomes of conifers were the loss of IR region [6]. Some small inverted repeats (sIRs) were found in cp genomes facilitating the stabilization. Wu et al. analyzed the plastomes of 24 representative genera in all of the five cupressophyte families, and found that every cupressophyte family has evolved its own specific and novel sIR systems, for example the rpoC2-IR in Sciadopitysis [7], the trnN-IR in Podocarpaceae [8], and the rrn5-IR in Araucariaceae [8]. TrnQ-UUG containing sIRs were found in almost all families of Cupressaceae and Taxaceae, except Callitris [8], and were found mediating HR [6, 9, 10]. Some other sIRs can also mediate HR in Cupressaceae and Taxaceae. For example, a 335 bp trnN-GUU containing sIR and a 211 bp sIR in the IGS of Torreya fargesii [10]. Due to the loss of IR region, conifer cps are also characterized by the extensive genomic rearrangements compared with most angiosperms [11]. The mechanisms underlying indel (insertion/deletion), Single nucleotide polymorphism (SNP) and rearrangement of cp genomes have attracted the attentions of many researchers [12–14].
Wu et al. [8] published the cp genome of Taxodium distichum, which is the only published cp genome of Taxodium. Nevertheless, the aim of its development is to systematically study the high variation in cp size and organization of Conifers II (cupressophytes) [8]. Changes in the gene and structure of cp genomes in the genus has not been referred. Due to the controversial taxonomy of the genus Taxodium, it is impossible to determine which taxa the published chloroplast genome belongs to. Therefore, in addition to development of cpDNA of T. ascendens and T. mucronatum, the cp genome of T. distichum was also re-sequenced in this study. We analyzed the structure characteristics of Taxodium cp genomes, conducted comparative analysis between the 3 cp genomes, and look insights into the dynamics of cp genome mutation and rearrangement of Taxodium. The results would advance our current understanding of the complexity, dynamics, and evolution of cp in conifers.
Results
Sequencing of Taxodium plastid genomes
Illumina 150-bp paired-end sequencing of long-range PCR-amplified plastid DNA generated 5045–6946 Mb clean reads for the three sampled Taxodium species (Table 1). Using the combination of de novo and reference-guided assembly, we obtained complete plastid nucleotide sequences for all three species. The nucleotide sequences of the four plastid genomes range from 131,947 bp in T. distichum to 132,613 bp in T. ascendens (Table 2). Like the cp genomes of other cupressophyte species, they lack the IR region and have no distinct quadruple structure. The gene map of the T. ascendens plastid genome is presented in Fig. 1 as a representative. The three genomes encode an identical set of 120 genes (Additional file 1), and the arrangements of these 120 genes are totally collinear (Additional file 2). The 120 unique genes include 83 protein-coding genes (Table 2), 33 transfer RNA (tRNA) genes, and 4 ribosomal RNA (rRNA)genes. They also have similar GC contents of 35.22–35.26%. This is similar to other gymnosperm plastid genomes.
Table 1.
Sequencing and assembly results of three chloroplast genomes of Taxodium
| Species | Raw data (Mb) | Clean data (Mb) | Clean data GC(%) | Clean data Q20(%) | Clean data Q30(%) | GC Content(%) | N rate (%) |
|---|---|---|---|---|---|---|---|
| T. ascendens | 5329 | 4895 | 37.85 | 98.02 | 93.94 | 35.22% | 0% |
| T. distichum | 5045 | 4695 | 35.74 | 98.11 | 94.2 | 35.26% | 0% |
| T. mucronatum | 6946 | 6595 | 34.19 | 98.38 | 94.91 | 35.25% | 0% |
Note: Read length: read length of valid data; Clean data GC: average GC content of valid data; Clean data Q20: Q20 value of valid data; Clean data Q30: Q30 value of valid data. Total Length (bp): The total length of the sample assembly result; GC Content (%): GC content of the sample assembly sequence; N rate (%): the content of unknown base N in the sample assembly sequence
Table 2.
Gene information statistics
| Species | Accession No. | Genome size (bp) | Coding Gene number (#) | CDS total length (bp) | CDS average length (bp) | CDS length / Genome (%) |
|---|---|---|---|---|---|---|
| T. ascendens | MN535012 | 132,613 | 83 | 74,469 | 897 | 56.16 |
| T. distichum | MN535013 | 131,947 | 83 | 74,217 | 894 | 56.25 |
| T. mucronatum | MN535011 | 132,037 | 83 | 74,217 | 894 | 56.21 |
Accession No.: Accession number of the complete chloroplast genome in genebank database. CDS: coding sequence
Fig. 1.

Circular gene map of the chloroplast genome of T. ascendens. Genes drawn within the circle are transcribed clockwise, while those drawn outside are transcribed counterclockwise. Genes are color-coded according to their functional groups. Inner circle represents GC content
Tandem repeats analysis
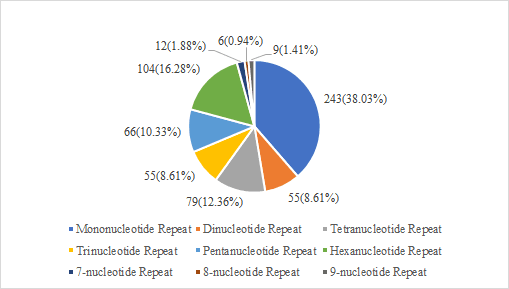
A TR is a repetitive sequence of adjacent specific nucleic acid sequence patterns repeated twice or more, including simple sequence repeats (SSR), whose repeat motif is 1–6 nucleotides, and long sequence repeats, whose repeat motif is ≥7 nucleotides. A total of 639 TRs were detected in the T. ascendens cp genome using Phobos (Additional file 3); the total length of repeats was 8462 bp, and TRs were widely distributed in the coding and non-coding regions of the cp genome (Fig. 2, Circle 3). Repeated motifs ranged from mononucleotide to 95-nucleotide. Among these, 601 were SSRs and 38 were long sequence repeats. Mononucleotide repeats were the most abundant SSRs, accounting for 38.03% (243) of the total, of which 238 repeat units were A/T and only five were G/C (Additional file 4). Of the 55 (8.61%) dinucleotide repeats, 34 were AT/TA type, 17 were AG/TC type, four were AC/TG type, and none were GC/CG type. For trinucleotide repeats, except for four AGC/TCG and two AGG/TCC types, the rest were all repeat types with A/T ratio higher than GC. Among the 79 (12.36%) tetranucleotide repeats, 64 had a higher A/T ratio than GC. Because of the high A/T content of repeat motifs, an increase in repeats will lead to a low GC content of chloroplast genes. The total length of repeats ranged from 7 bp to 453 bp, of which 325 (50.86%) were short and less than 10 bp, 286 (44.76%) were medium-sized and TRs ranging from 10 bp to 20 bp, and 28 (4.38%) were long and TRs ranging from 20 bp to 50 bp. Long TRs were mostly distributed in non-coding regions (Fig. 2, Circle 3, Green dots). The total length of seven long TRs was more than 50 bp (Table 3). Their total lengths were 98 bp (ycf2), 110 bp (psbJ-clpP), 116 bp (rps18), 145 bp (trnI-ycf2), 152 bp (clpP-accD), 333 bp (ycf1) and 453 bp (clpP-accD) (Additional file 3).
Fig. 2.

Distribution of conserved gene blocks, TRs, indels, and SNPs in the plastomes of T. ascendens. Circle 1: plastome map of T. ascendens with coding genes labeled in blue, tRNAs labeled in red, and rRNAs in purple. Circle 2: conserved blocks of genes relative to the cp genome of Cycas. Circle 3: location of 639 TRs reported by phobos software. TRs of different length are marked with different colors, with green representing repeats ≥20 bp, rose red representing 10~19 bp, and orange representing < 10 bp. The relative height of the dot position in the figure represents the relative number of polymorphic loci within non-overlapping bins of 200 bp. Repeats in the three different colors were treated separately in statistical analysis. Dots with a high relative position represent more loci belonging to TRs within the 200 bp window. Circle 4: counts of indels (blue). Circle 5: counts of SNPs (red). Dots with a high relative position, represent more polymorphic loci in the 200 bp window. The red rectangle (HR01-HR11) showed the locations of the 11 selected hypervariable regions. The number or relative position of dots of indel and/or SNP inner rectangles were higher that outside
Table 3.
Basic information for long tandem repeats > 50 bp
| Repeat Class | Minimum | Maximum | Length | Location | Normalised Repeat Length | Percentage Perfection |
|---|---|---|---|---|---|---|
| 95-nucleotide Repeat | 116,478 | 116,930 | 453 | clpP-accD | 453 | 99.78% |
| 63-nucleotide Repeat | 101,809 | 102,141 | 333 | ycf1 | 333 | 100% |
| 38-nucleotide Repeat | 115,180 | 115,289 | 110 | psbJ-clpP | 110 | 98.18% |
| 32-nucleotide Repeat | 75,866 | 75,963 | 98 | ycf2 | 96 | 94.79% |
| 24-nucleotide Repeat | 110,842 | 110,957 | 116 | rps18 | 116 | 96.55% |
| 22-nucleotide Repeat | 73,789 | 73,933 | 145 | trnI-ycf2 | 145 | 98.62% |
| 19-nucleotide Repeat | 116,921 | 117,072 | 152 | clpP-accD | 152 | 100% |
Researches have shown that there are many TRs on the coding DNA sequence (CDS) of accD gene and its surrounding regions of gymnosperms [12]. In order to study the general features of hypothetical cp open reading frame 1(ycf1) genes in cupressophytes, we analyzed the ycf1 gene sequences in 44 species (Fig. 3). The length of ycf1 gene in Pinaceae is similar to that of Ginkgo biloba and Cycas, which were about 5000 bp. However, the ycf1 gene length in cupressophytes experienced an extraordinary expansion, ranging from 6666 bp to 8931 bp, with Taxus baccata and Sciadopitys verticillata has the shortest and longest CDS, respectivelly. There were no TRs in Ginkgo biloba and Cycas cp gemones, but, except for four species, TRs were detected in most conifers. In the Taxodium, TRs were only detected in T. ascenden. The same situation happened in Cupressus, with TRs are detected in Cu. chengiana and Cu. gigantea but not in Cu. jiangeensis. It can be seen from Fig. 3 that the insertion positions of TRs on the ycf1 CDS were family specific. For Cupressaceae, there are two major insertion positions, one located in the middle of the CDS, the other one is near the C-terminal region.
Fig. 3.

The tandem repeats of ycf1 gene in conifers. The phylogenetic tree was constructed based on the sequence alignments of ycf1 genes using Mafft. The right side of the phylogenetic tree showed the position of tandem repeats on the ycf1 gene. The length of the horizontal line was drawn according to the length of multiple sequence alignment, which included the length of gaps. Therefore, the position of repeats in different species can be mapped to each other. The rightmost column listed the actual length of ycf1 genes (excluding gaps)
Dispersed repeats
Fifty dispersed repeats were detected in the T. distichum, T. mucronatum, and T. ascenden cp genomes, respectively, using REPuter (Additional file 5). In the T. distichum cp genome, there were 24 forward repeats, 21 palindromic repeats, three complement repeats, and two reverse repeats. In the T. mucronatum cp genome, there were 26 forward repeats, 18 palindromic repeats, one complement repeat, and five reverse repeats. In the T. ascendens cp genome, only 36 forward repeats and 14 palindromic repeats were detected.
In cupressophyte cp genomes, the highly reduced IRs are replaced by short repeats that have the potential to mediate homologous recombination [6, 9, 10]. Three sIRs >100 bp were detected in T. ascendens (Table 4), and their sequences were identical in all three Taxodium cp genomes. Among them, sIR1 and sIR3 contained complete trnQ-UUG and trnI-CAU genes, respectively. For sIR2, one copy was located in the intergenic region (IGS) region of petA-ccsA and the other in the psbJ-clpP IGS.
Table 4.
SIRs in the cpDNA of Taxodium ascendens
| Name | Copy | Location | Length | Mismatch | Contained gene |
|---|---|---|---|---|---|
| sIR1 | a | 7412–7693 | 282 | 3 | trnQ-UUG |
| b | 45,569–45,850 | trnQ-UUG | |||
| sIR2 | a | 99,641–99,759 | 119 | 2 | Null (PetA-ccsA) |
| b | 115,292–115,410 | Null (psbJ-clpP) | |||
| sIR3 | a | 73,129–73,241 | 111 | 3 | trnI-CAU |
| b | 132,385–132,497 | trnI-CAU |
The trnQ-UUG gene was the only one in a sIR longer than 200 bp. If the 282-bp IR is able to mediate homologous recombination (HR), we would expect the presence of two type isomers. Semi-quantitative PCR with a variable number of cycles was conducted to verify the presence of the two isomers. The isomers illustrated in Fig. 1 is designated as the type I, and the other is the type II. All four reactions generated products, which verified the presence of both the I and II forms. It was also apparent that there were minor differences in amplification efficiency between the four PCR reactions. With 30 PCR cycles, the electrophoresis bands of type 1(rps4/chlB products) are very bright, while those of type 2(psbK/trnL products) are much weaker (Fig. 4). These results suggest that the type I is predominant in T. ascendens, in agreement with our assembly results.
Fig. 4.

Co-existence of two isomeric chloroplasts in T. distichum. The corresponding PCR amplicons are shown, and the numbers above each lane of gel photos denote the PCR cycles conducted
To quantify the relative frequency of the two isomeric genomic forms, Illumina paired-end reads were mapped to the genome and isomer frequencies were calculated using the method of Guo [9]. There were 297 read pairs that spanned the trnQ-containing IR copies, of which 293 pairs (98.65%) supported the type I isomer while four pairs (1.35%) supported the type II isomer.
Phylogenetic and rearrangements analysis
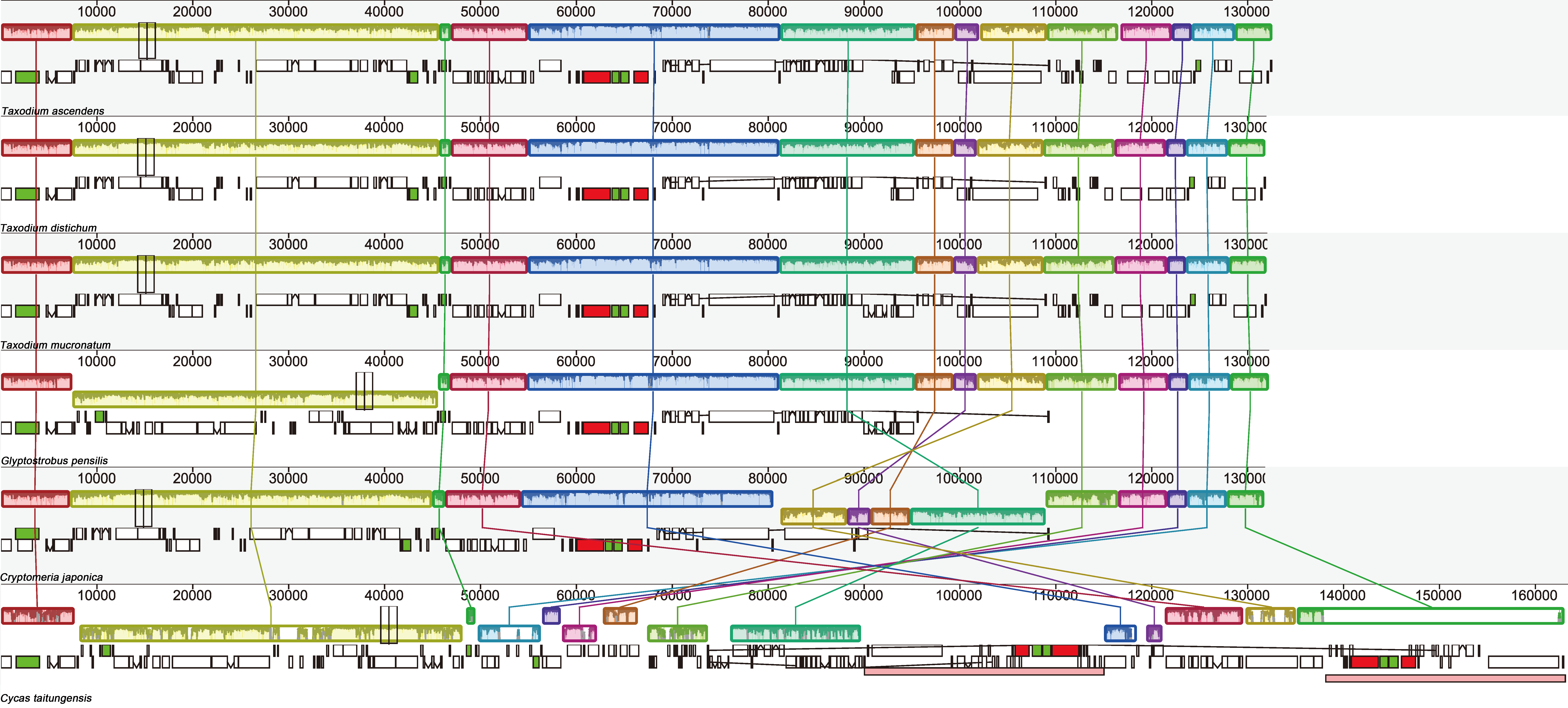
Phylogenetic tree based on the 51 single copy coding genes of 44 species were constructed (Fig. 5). Among the genus Taxodium, T. mucronatum and T. distichum were clustered together. The genus Taxodium has the closest relationship with Glyptostrobus, and then forms a group with Cryptomeria. Unanimously, mauve alignment showed that no rearrangements occurred between Taxodium and Glyptostrobus pensilis, while at least four rearrangements occurred between Taxodium and Cryptomeria japonica (Additional file 6).
Fig. 5.

Phylogenetic analysis of 44 chloroplast genomes. Numbers below each node are bootstrap support values
Previous studies have shown that cycads possess the oldest sequence of genes in seed plants. We conducted mauve alignment between Taxodium and Cycad taitungensis cp genomes. Compared with Cycas, there were 13 conserved gene clusters in Taxodium cp genomes, which were labeled S01 to S13 (Table 5) (Fig. 2, Circle 2). Therefore, there were at least 13 rearrangements in the process of transformation from cycad chloroplast genome structure to T. ascendens genome structure. The size of the conversed gene blocks ranged from 1236 bp to 40489 bp. Five of the 13 inversion endpoints occurred near tRNAs, including trnI, trnT, trnQ, trnF, and trnM. Interesting, there is a sequence between S07 and S08 that can't find homologous sequence on Cycad, and its position (101807-102061) overlaped with the 63-nucleotide repeat TR493 (101809-102141) on ycf1(Additional file 3). Two other inversion endpoints were also in TRs. The inversion endpoint (99473) between conversed gene blocks S07 and S08 was in the mononucleotide repeat TR484 (99466-99478) located in petA-ccsA. The inversion endpoint (116761) between conserved gene blocks S10 and S11 was in the 95-nucleotide repeat TP571 (116478-116930) located on clpP-accD.The accD gene or its adjacent region is a hot rearrangement area of cupressophyte cp genomes, Li et al. found that there are five types of gene order in cupressophytes, and speculated that many inversion events have occurred here during the evolution of cupressophyte cp genomes [12]. We also analysis the sequence variability of the genes adjacent to the ycf1 gene in Taxodium. The gene order around ycf1 of the 44 analyzed species could be classified into twelve types (Fig. 6). Cycas, Ginkgo, and Taxaceae have the same gene order: ndhH-rps15-ycf1-chlN-chlL gene order (type I). As we can see from Fig. 6, at least one side (right side/left side) of ycf1 gene in type II (Pinaceae), type III (Podocarpuceae), type IV (Podocarpuceae) and type V (Podocarpuceae) maintains the same gene order of type I. However, in Cupressaceae (type VI to XII), gene orders of both sides (right side and left side) of ycf1 gene were totally different from type I. Therefore, the arrangement frequency around ycf1 gene in Cupressaceae was much higher. In the Cupressaceae family, Glyptostrobus, Taxodium,Metasequoia, Taiwannia and Cunninghamia have a conversed trnL (UAG)- trnP (GGG)-ycf1-rpl20 –rps18(type VI) gene order. And in Juniperus, Cupressus and Hesperocyparis, the gene order is mainly: trnL (UAG)-ccsA-ycf1-trnL (CAA)-ycf2(type XI). In view of the diversity of gene organization around ycf1 gene, it is speculated that ycf1 gene may be frequently involved in the rearrangement events of cupressophytes cp genomes.
Table 5.
Information of the 13 conserved gene blocks of Taxodium cp genomes compared with Cycas taitungensis
| Name | Start | End | Length (bp) |
|---|---|---|---|
| S01 | 1(trnI-psbA) | 7831(trnT) | 7831 |
| S02 | 7832(trnT) | 45,722(psbK-trnQ) | 37,891 |
| S03 | 45,723(psbK-trnQ) | 46,958(trnF-ndhD) | 1236 |
| S04 | 46,959(trnF-ndhD) | 54,967(rps15-rpl32) | 8009 |
| S05 | 54,968(rps15-rpl32) | 95,456(psbB-psaI) | 40,489 |
| S06 | 95,457(psbB-psaI) | 99,472(petA-ccsA) | 4016 |
| S07 | 99,473(petA-ccsA) | 101,806(ycf1) | 2334 |
| S08 | 102,062(ycf1) | 108,715(ycf1-rpl20) | 6654 |
| S09 | 108,715(ycf1-rpl20) | 116,760(clpP-accD) | 8045 |
| S10 | 116,761(clpP-accD) | 122,206(rbcL-atpE) | 5446 |
| S11 | 122,207(rbcL-atpE) | 124,233(atpB-trnM) | 2027 |
| S12 | 124,234(atpB-trnM) | 128,797(ndhJ-chlN) | 4564 |
| S13 | 128,798(ndhJ-chlN) | 132,613(trnI- psbA) | 3815 |
Fig. 6.

Gene organization around the ycf1 gene in gymnosperms. The direction of the arrow of genes was from the N-terminal to the C-terminal. The roman numbers I–XII denotes 12 types of gene organization around the ycf1
Comparative analysis of genomic structure
Compared with T. ascendens, 83 indels, including 43 deletions and 40 insertions of different origins were detected in T. distichum and T. mucronatum (Fig. 2, Circle 4). Among them, 82 indels occurred in IGS regions, and only one 252 bp indel occurred in the CDS region of ycf1. Therefore, the total CDS length of T. ascendens is 252 bp longer than of T. distichum and T. mucronatum. The indel did not caused frame shifts or stop codons. Among the 83 indels, only seven (8.43%) were located outside repeat regions, and the remaining 76 (91.57%) were located within 51 TRs (Additional file 7). Among these, 64 indel sequences were integer multiples of repeat motifs, that is, the generation of indel sequences created differences in the number of complete repeat motifs. Twelve indel sequences were non-integer multiples of repeat motifs, i.e., the indel sequences contained partial incomplete repeat motif sequences. Of the 51 TRs containing indels, 47 (92.16%) were SSR indels of 1–4 nucleotides. Among these, 30 belonged to mononucleotide repeat type A/T, 14 belonged to dinucleotide repeat type AT/TA, two belonged to trinucleotide repeat type AAT/ATT, and one belonged to tetranucleotide repeat type AAAT/ATTT. All SSR indels contained only A/T. The remaining four large indels were 19-, 22-, 28-, and 95-nucleotide repeats.
A total of 45 SNPs were detected in T. distichum compared with T. ascendens, representing 31 in IGS regions and 14 in CDS regions (Fig. 2, Circle 5), with six synonymous mutations and eight non-synonymous mutations (Table 6). A total of 50 SNPs were detected in T. mucronatum compared with T. ascendens, representing 35 in IGS regions and 15 in CDS regions, with six synonymous mutations and nine non-synonymous mutations. No mutations appeared on start/stop codon or caused the triplet codon of the site mutates into a termination codon. We merged SNPs occurring at the same site into one for statistical analysis, so a total of 69 SNPs of diverse origin were found. Among these, 45 (65.22%) SNPs were found in non-coding regions and 24 (34.78%) SNPs were found in 16 coding regions, including rps16, psbC, rpoB, rpoC2, ndhA, rps12, rpl2, rps3, cemA, ycf1, clpP, accD, rbcL, atpB, ndhK, and chlN. Of the 69 SNPs, 18 (26.09%) were located within the TR sequences, 32 (46.38%) were located within 100 bp windows adjacent to the repeats, and only 19 (27.54%) were located outside the 100 bp windows adjacent to repeats (Fig. 7).
Table 6.
Statistics of SNP annotation results
| CDS | Intergenic | Total | |||
|---|---|---|---|---|---|
| Synonymous | Nonsynonymous | Total | |||
| T. distichum | 6 | 8 | 14 | 31 | 45 |
| T. mucronatum | 6 | 9 | 15 | 35 | 50 |
Note: Synonymous: Synonymous mutation in the gene region; Nonsynonymous: Nonsynonymous mutation in the gene region; Intergenic: SNP in the intergenic region
Fig. 7.

Number of SNPs within 0–300 bp windows adjacent to the tandem repeat regions. The SNPs within 0 bp windows represent SNPs located inside the repeat regions
Analysis of chloroplast genome hypervariable regions of Taxodium
Regions enriched with indels and SNPs can be considered as hypervariable regions in the complete cp genome. Since most indels show periodic variation of repeat motifs, the number of polymorphic sites contained in an indel is affected not only by the degree of periodic variation but also by the size of the repeat motif. When two TRs have the same number of sites, compared with repeats with longer repeat motifs, the repeat sequence with shorter repeat motif has more repeat cycles, indicating a higher polymorphic potential. Therefore, when we select hypervariable regions, these should not only be based on the number of polymorphic sites, but also regions with more indel/SNPs of different origins to ensure that the region has higher polymorphic potential. Based on this principle, the most variable regions of the Taxodium cp genome are clpP-accD, trnV-rps12, ndhF-trnN, trnV-ndhC, trnI-trnL, ycf1-rpl20, atpI-atpH, rpl36-rps11, ycf1, psbJ-clpP, and trnI-rrn16(Fig. 2, HR01-HR11). These hypervariable regions contained at least four SNPs and/or indels of diverse origin in this study (Additional file 8). These regions can be considered interspecies mutational hotspots in Taxodium and could be potentially high-resolution DNA barcodes in the study of population genetics.
Discussion
Phylogenetic and rearrangements analysis
Godfrey [15] considered T. ascendens (pond cypress) as a varied form of bald cypress due to temporal differences in phylogeny. Farjon [16] considered that Taxodium has two species, T. distichum and T. mucronatum, but T. distichum has two varieties, var. distichum and var. imbricatum [17]. Both of these views support a closer relationship between baldcypress and pondcypress. However, The phylogenetic trees constructed by chloroplast whole genome sequence and ycf1 gene sequence here all supported a closer genetic relationship between T. distichum and T. mucronatum. The closer genetic relationship between Taxodium and Glyptostrobus is consistent with their similar growth habits. The two genera are both pioneer waterlogging-tolerant tree species, and can develop unique cypress knees acting as pneumatophores thought to help in oxygenation to the roots [11].
SIR> 200 bp are thought to be effective substrates for HR [18]. However, there is a distinct difference between Pinaceae and cupressophyte species, in that the former has more of these sIRs than the latter, and species of the subclade Cupressaceae all have relatively shorter sIRs compared to other two subclades within the cupressophyte clade [11]. Unanimously, cp genome rearrangements are much more frequent in cupressophytes, especially in the subclade Cupressaceae, than in Pinaceae [11]. In our research, only one short sIR greater than 200 bp (282 bp) was found in T. ascendens, and we identified at least 13 rearrangements in the process of transformation from Cycad to T. ascendens, which was consistent with the features of Cupressaceae described above. Suggested mechanisms responsible for cp rearrangements are diverse [8]. Among the 13 inversion endpoints, five located near tRNAs; the same was found in Trachelium caeruleum [19]; thus, the rearrangement breakpoints may selectively constrain some regions of the cp genome [20, 21]. Besides, three rearrangement breakpoints were in TRs, suggesting potential association between the rearrangement and repeats.
Chloroplast genome isomers
The trnQ-containing sIRs of Cupressaceae and Taxaceae have been widely proved to be capable of mediating HR, such as the 544 bp sIRs in Cephalotaxus oliveri [6], the ~ 250 bp sIR in Juniperus [9], and the 298 bp sIRs in Torreya fargesii [10]. Although, the trnQ-UUG is duplicated, this gene is not retained from the IR region. It is proposed that the trnQ-IR originated in the common ancestor of Cupressaceae and Taxaceae after they split from Sciadopitysis and was transformed from the trnQ-UUG forward TRs to reverse repeats located in different regions through cp genome rearrangement [7, 12]. Not surprisingly, there is also a 283 bp trnQ-containing sIR in Taxodium that can mediate HR. There is also a 111 bp trnI-CAU containing sIR in Taxodium, which was also detected in Torreya fargesii [10], Cephalotaxaceae [6], and Sciadopitys verticillata [7], but none of them were detected to mediate HR. The TrnI-CAU containing sIR was thought to be retained from the ancient IR region [22].
Since, the trnQ-IR can mediate HR in Cupressaceae and Taxaceae, sequences around trnQ showed two types of arrangement [9]. In type I, the gene orders were chlB- trnQ (UUG)- trnT (UGU) & psbK- trnQ (UUG)-trnL (UAA). In type II, gene orders were chlB- trnQ (UUG)-psbK & trnT (UGU) - trnQ (UUG)-trnL (UAA). Due to the HR mediating by trnQ-IR, the two isomers co-exist in individual plant, but the proportions are very different [6, 9, 10]. In some conifers, type I isomer was predominant, for example, J. scopulorum has 95% of type I isomer [9]. However, in Juniperus virginiana, Cephalotaxus oliver and Ce. wilsoniana type II isomer was predominant. Just like J. scopulorum and Cryptomeria japonica, type I isomer is predominant in Taxodium, and accounting for 98.65% in T. ascendens. The cause of the different contents between the two isomers in individuals is unclear, and the length of sIR may be one factor [10].
Mutational dynamics in Taxodium cp genomes
The relationship between indels and repeats has been widely concerned by many researchers, but they only found that the locations of indels were highly correlated with the location of repeats [6, 14], and they did not study in detail whether indels appeared in the internal, boundary or external region of repeats. This study analyzed the relative location of indels and repeats in detail and found that 83.53% of indels resulted in/caused by periodic variation of repeat motifs, and two indels were accompanied by point mutations of repeat motifs. There are many hypotheses about the molecular mechanism of TR sequence formation, including the unequal exchange hypothesis, the reorganization hypothesis, and the DNA sliding hypothesis. At present, the more accepted view is the sliding hypothesis; that is, TR mutation is the result of the interaction of DNA slippage and DNA replication repair systems. The hypothesis holds that the repeat sequence is formed by the increase in length of the original repeat motif by copy slip, while the original microsatellite sequence may come from a random point mutation. Local de-chaining and re-pairing sometimes occur between the new chains and template chains in the process of replication. When the new strand and template strand are mismatched occasionally, DNA polymerase synthesis on the mismatched DNA strand will cause the length of the new strand to change. If this mismatch is not correctly repaired by the DNA replication repair system in vivo, the next round of replication can produce double-stranded DNA with mutated sequence length [23]. There were 16.47% of indels in Taxodium that did not show differences in repetition period. This finding may be explained by the existence of other non-repeat polymerase-stalling sequence motifs; another possible explanation is that repeat sequences were destroyed by mutation, while the indel remained [24].
We identified 72.46% of SNPs around repeat sequences, including 18 locations within repeat sequences and 32 locations within 100 bp adjacent to the TR regions. McDonald et al. proposed that repeat-sequence-induced recurrent repair is the mechanism inducing SNPs [24]. Repeat sequence is one of the main cause of DNA replication fork arrest [25], as a result, DNA polymerases, including high-fidelity DNA polymerase and error-prone repair polymerases, are widely recruited to restart replication [26–28]. Persistent recruitment of error-prone repair polymerases will increase the chance of DNA replication being restarted by an error-prone polymerase and the DNA surrounding the region being synthesized with a higher rate of error [26, 27, 29], leading to an increased likelihood of mutations. A few SNPs located closely to indels, and indel effect may also be an inducement of mutation, which can be explained by the mutagenic-when-heterozygous hypothesis and will vanish over evolutionary time-scales [24].
Highly significant correlations between cp genome polymorphisms (indels and SNPs) and repeats have been reported in some studies [6, 14], but not in others [30]. In our study, although 639 TRs were found to be widely spread on cp genome sequences of T. ascendens, only 83 indels and 69 SNPs of diverse origin were detected, leaving no polymorphic sites around most repeats. Not all repeat sequences are surrounded by indels or SNPs, and this may result from the mutation rate of repeat sequences themselves being affected by many other factors, such as repeat sequence length, chromosome location, flanking sequence characteristics, age, etc. [23, 31–33]. Thus, it can be concluded that repeats play a pivotal role in the generation of indel and SNP mutations, but they do not necessarily lead to polymorphism. And we cannot predict mutational hotspot regions based solely on the distribution of repeat sequences.
Analysis of chloroplast genome hypervariable regions of Taxodium
Eleven hypervariable regions of the Taxodium cp genome were identified in this study. Among them, clpP- accD IGS and ycf1 CDS were the most unique regions. Containing the two largest TRs, they were both the hypervariable regions and arrangement endpoints of Taxodium. AccD gene and its adjacent sequences were found to extremely expanded due to the insertion of TRs, and they were the hotspots of mutation and rearrangement of cupressophytes [6, 12]. Here, we also analyzed the ycf1 gene characteristics throughout cupressophytes, and got similar results. The length of the ycf1 genes showed an extraordinary expansion, and there were universal insertion of TRs. They also located in potential rearrangement endpoints. Besides, in Cupressaceae, the insertion position of TR on ycf1 and the arrangement of its surrounding genes were quite different from those of other conifers. Thus, similar to accD gene and its surrounding regions, ycf1 gene may play an important role in the cp genome structural evolution of cupressophytes.
Conclusion
The cp gnomes of Taxodium were characterized by several unusual features, such as the loss of the typical IRA copy, the wide spread of TRs, extensive genomic inversions, the presence of isomeric plastomes, and the big variation of ycf1 genes among genus. All these characteristics highlight the potentially important role of repeats in the dynamics of cp genome mutation and rearrangement. Moreover, the information of TPs and hypervariable regions would provide reliable molecular resources for future research focusing on the infrageneric taxa identification, phylogenetic resolution, population structure and biodiversity for the genus Taxodium and Cupressophytes. The comparative chloroplast genomics of the genus Taxodium advances our understanding of the dynamics, complexity, and evolution of cp genomes in Cupressophytes.
Methods
DNA extraction and sequencing
Fresh buds were harvested from adult trees of T. distichum, T. mucronatum, and T. distichum planted at the Institute of Botany, Jiangsu Province & Chinese Academy of Sciences, Nanjing, China. Total DNA was isolated using an improved extraction method [34]. After DNA isolation, 1 μg of purified DNA was fragmented and used to construct short-insert libraries (insert size 430 bp) according to the manufacturer’s instructions (Illumina), then sequenced on an Illumina Hiseq 4000 [35].
Genome assembly and annotation
High-quality reads were mapped to the reference cp genome of T. distichum (NC_034941) using Bowtie 2 [36] with default parameters. Three coding gene sequence with the highest coverage was used as a seed sequence for de novo assembly of the chloroplast genome by NOVOPlasty v.2.6.2 [37] with the reference genome as a template. Then CAP3 [38] was useds for contigs merging and de-redundancy. Cp cyclization and initiation site determination is done by manual processing.
Cp genes were annotated using an online Dual Organellar GenoMe Annotator tool [39], using default parameters to predict protein-coding genes, tRNA genes, and rRNA genes. The annotation results were further checked and adjusted manually. A whole chloroplast genome Blast [40] search (E-value ≤1e-5, minimal alignment length percentage ≥ 40%) was performed against five databases: Kyoto Encyclopedia of Genes and Genomes (https://www.kegg.jp/), Clusters of Orthologous Groups (http://clovr.org/docs/clusters-of-orthologous-groups-cogs/), Non-Redundant Protein Database, Swiss-Prot (https://web.expasy.org/docs/swiss-prot_guideline.html) and GO (http://geneontology.org/). The circular chloroplast genome map was drawn using OrganellarGenomeDRAW v1.2 [41].
Estimate of repeats and plastomic inversions
Phobos-v3.3.12 software [42] was used to detect TRs in the cp genomes. The repeat motif length range was set to 1–100 nt (repeat unit length) and the consistency (perfection) was set to be more than 85%. The repeat length of ycf1 genes TRs with length ≥ 25 bp were detected in ycf1 genes, and repeat location structure figure were drawn based on the repeat location information. Dispersed repeat sequences were detected by REPuter [22] software, with a minimum size of 30 bp and a maximum of three nucleotides mismatch between the two repeat copies. The repeat sequences were searched using the following four ways: forward, reverse, completion, palindromic.
We downloaded the whole cp genome sequences of 44 species online and from this study, including three species of Pinaceae (Conifers I), 39 cupressophytes (Conifers II), as well as Ginkgo biloba and Cycas taitungensis as outgroups The 39 cupressophytes covered Cupressaceae, Taxaceae, Sciadopityaceae and Podocarpaceae. Fifty-one single copy coding genes of the 44 species were used for multiple sequence alignment. Mafft version 7 (https://mafft.cbrc.jp/alignment/software/) was used to carry out multiple sequence alignment, and then fasttree 2.1 (http://www.microbesonline.org/fasttree/)was used to build Maximum-likelihood phylogenetic trees. The gene content of related species was visually detected and compared by Mauve [43] with default settings. According to the genome annotation information of each species, sequences information of two genes located before and after ycf1 genes respectively were extracted, and gene organization containing the five adjacent genes were drawn.
Detection of isomers
The primers used by Guo et al. [9] were used to test whether the trnQ-containing sIR could mediate homologous recombination. The primer sequences were as follows: rps4 (5′-CCTGGTAAAGTTTTGABACG-3′), psbK (5′-CAAATGAAAAGCGGCATCG-3′), chlB (5′-GTTCCAATATGAGCAGGACCAG-3′), and trnL-UAA (5′-GTTTCCATACCAAGGCTC-3′). PCR was performed using the following primer combinations (rps4 + chlB, rps4 + trnL-UAA, psbK+chlB, psbK+trnL-UAA). Each reaction was 50 ul in volume and included 100 ng DNA. PCR reaction: 94 °C for 3 min → (94 °C for 15 s, 55 °C for 15 s, 72 °C for 1 min) × 35 cycles →72 °C for 3 min → 4 °C for storage. To quantify the relative frequency of the two isomeric genomic forms, Illumina paired-end reads were mapped to the genome using Bowtie 2 [36] with default parameters. The custom Perl script of Guo et al. [9] was used to count repeat-spanning read pairs, enabling us to quantify the frequency of the repeat in each possible genomic arrangement.
Comparative analysis of genomic structure
InDel refers to the insertion and deletion of sequences in the genome. Samples and reference sequences were compared using LASTZ [44]. The best comparison results were then selected through the processing of axt_correction, axtSort, and axtBest programs, and InDel results were preliminarily obtained. InDel loci were then compared with the sequencing Reads of the samples 150 bp upstream and downstream of the reference sequence using BWA [(http://bio-bwa.sourceforge.net/) software and SAMtools [45] (http://samtools.sourceforge.net/). After filtering, reliable inDels were obtained.
By using MUMmer [46] software, each sample was compared with the reference sequence globally. Sites with differences between the sample sequence and the reference sequence were identified and preliminarily filtered, and potential SNP sites were detected. The sequence of 100 bp on each side of the reference sequence SNP locus was extracted, and then the extracted sequence was compared with the assembly result using BLAST [40] to verify the SNP locus. If the aligned length was less than 101 bp, the unreliable SNP was removed; if the extracted sequence and the assembly result were aligned several times, the SNP considered to be a duplicate region was also removed, and finally a reliable SNP was obtained.
Supplementary information
Additional file 1 Basic information of the 120 genes in Taxodium distichum, Taxodium mucronatum, and Taxodium ascenden chloroplast genomes.
{kind=link}
Additional file 2 Dot plot analysis of Taxodium chloroplast genomes.(A) Taxodium ascenden &Taxodium distichum, (B) Taxodium ascenden &Taxodium mucronatum.
Additional file 3 Tandem repeats detected in the Taxodium ascendens chloroplast genome using Phobos software.
{kind=link}
Additional file 4 Distribution of 1–9 nucleotide repeat motifs with different numbers in Taxodium ascendens.
Additional file 5 Dispersed repeats detected in the Taxodium distichum, Taxodium mucronatum, and Taxodium ascenden chloroplast genomes.
{kind=link}
Additional file 6 Mauve alignment of T. ascendens, T. distichum, T. mucronatum, Glyptostrobus pensilis, Cryptomeria japonica, and Cycad taitungensis. Locally collinear blocks are denoted by different color boxes. Histograms within each block represent the degree of sequence similarity.
Additional file 7 Information of indels and SNPs in Taxodium distichum and/or Taxodium mucronatum chloroplast genomes with Taxodium ascenden chloroplast genome as reference.
Additional file 8 List of the 8 highly variable regions in Taxodium chloroplast genomes.
Acknowledgements
We sincerely thank Shanghai BIOZERON Biotechnology Co., Ltd. for performing the high throughput sequencing.
Abbreviations
- CDS
Coding DNA sequence
- Cp
Chloroplast
- HR
Homologous recombination
- IR
Inverted repeat
- rRNA
Ribosomal RNA
- SIR
small inverted repeat
- SNP
Single nucleotide polymorphism
- SSR
Simple sequence repeat
- TR
Tandem repeat
- tRNA
Transfer RNA
- Ycf1
Hypothetical chloroplast open reading frame 1
Authors’ contributions
HD performed the experiments, analyzed the data, prepared figures and/or tables,authored drafts of the paper. JG prepared samples. LX prepared figures and Tables. ZW and YL Y collected and identified field materials. ML analyzed the data. YY conceived and designed the experiments, reviewed and revised the drafts of the paper. All the authors read and approved the final manuscript.
Funding
This research was supported by the National Natural Science Foundation of China (31700588), the Jiangsu Key Laboratory for the Research and Utilization of Plant Resources (JSPKLB201842), the Natural Science Foundation of Jiangsu (BK20160601) and the Biological Resources Service Network (kfj-brsn-2018-6-003).
Availability of data and materials
Sequence information of the 3 cp genomes is available in the NCBI database under the accession number MN535011- MN535013. The reference cp genome of T. distichum and the whole cp genome sequences of 44 species analysed in this study were all downloaded from the NCBI database with their accession numbers listed in Fig. 3 and Fig. 5.Other datasets supporting the conclusions of this article are included within the article and its additional files.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
Mingzhi Li is employed by Biodata Biotechnology Co. Ltd.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-020-6532-1.
References
- 1.Denny GC, Arnold MA. Taxonomy and nomenclature of Baldcypress, Pondcypress, and Montezuma Cypress: one, two, or three species? Horttechnology. 2007;17(1):125–127. doi: 10.21273/HORTTECH.17.1.125. [DOI] [Google Scholar]
- 2.Tiwari SP, Yadav D, Kumar P, Chauhan DK. Comparative palynology and wood anatomy of Taxodium distichum (L.) rich. and Taxodium mucronatum ten. Plant Syst Evol. 2012;298(4):723–730. doi: 10.1007/s00606-011-0582-4. [DOI] [Google Scholar]
- 3.Lickey EB, Walker GL. Population genetic structure of baldcpress (Taxodium distichum [L.] RICH. var. distichum) and pondcpress (T. distichum var. imbricarium [NUTTALL] croom): biogeographic and taxonomic implications. Southeast Nat. 2002;1(2):131–148. doi: 10.1656/1528-7092(2002)001[0131:PGSOBT]2.0.CO;2. [DOI] [Google Scholar]
- 4.Creech D, Eguiluz-Piedra T. Can taxodium be improved? Piedra. 2011;69(2):11–20. [Google Scholar]
- 5.Xue S, Shi T, Luo W, Ni X, Iqbal S, Ni Z, Huang X, Yao D, Shen Z, Gao Z. Comparative analysis of the complete chloroplast genome among Prunus mume, P. armeniaca, and P. salicina. Hortic Res. 2019;6(1):89. doi: 10.1038/s41438-019-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yi X, Gao L, Wang B, Su Y, Wang T. The complete chloroplast genome sequence of Cephalotaxus oliveri (Cephalotaxaceae): evolutionary comparison of cephalotaxus chloroplast DNAs and insights into the loss of inverted repeat copies in gymnosperms. Genome Biol Evol. 2013;5(4):688–698. doi: 10.1093/gbe/evt042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsu C, Wu C, Chaw S. Birth of four chimeric plastid gene clusters in Japanese umbrella pine. Genome Biol Evol. 2016;8(6):1776–1784. doi: 10.1093/gbe/evw109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu C, Chaw S. Large-scale comparative analysis reveals the mechanisms driving plastomic compaction, reduction, and inversions in Conifers II (Cupressophytes) Genome Biol Evol. 2016;8:w278. doi: 10.1093/gbe/evw278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guo W, Grewe F, Cobo-Clark A, Fan W, Duan Z, Adams RP, Schwarzbach AE, Mower JP. Predominant and substoichiometric isomers of the plastid genome coexist within juniperus plants and have shifted multiple times during cupressophyte evolution. Genome Biol Evol. 2014;6(3):580–590. doi: 10.1093/gbe/evu046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao K. Chloroplast genomic research of Torreya fargesii: insights into the structural variation of conifer chloroplast genomes. Huairou: University of Chinese Academy of Sciences; 2015. [Google Scholar]
- 11.Hao Z, Cheng T, Zheng R, Xu H, Zhou Y, Li M, Lu F, Dong Y, Liu X, Chen J. The complete chloroplast genome sequence of a relict conifer Glyptostrobus pensilis: comparative analysis and insights into dynamics of chloroplast genome rearrangement in cupressophytes and pinaceae. PLoS One. 2016;11(8):e161809. doi: 10.1371/journal.pone.0161809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J. Molecular evolution of accD in gymnosperm and the evolutionary chloroplast genomics of cupressophyte. 2015. [Google Scholar]
- 13.Weng M, Blazier JC, Govindu M, Jansen RK. Reconstruction of the ancestral plastid genome in Geraniaceae reveals a correlation between genome rearrangements, repeats, and nucleotide substitution rates. Mol Biol Evol. 2014;31(3):645–659. doi: 10.1093/molbev/mst257. [DOI] [PubMed] [Google Scholar]
- 14.Ahmed I, Biggs PJ, Matthews PJ, Collins LJ, Hendy MD, Lockhart PJ. Mutational dynamics of aroid chloroplast genomes. Genome Biol Evol. 2012;4(12):1316. doi: 10.1093/gbe/evs110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Godfrey R. Trees, shrubs and woody vines of northern Florida and adjacent Georgia and Alabama. Athens: University of Georgia Press; 1988. [Google Scholar]
- 16.Farjon A. World checklist and bibliography of conifers. 2. Kew: The Royal Botanic Garden; 2001. [Google Scholar]
- 17.Croom H. A catalogue of the plants native or naturalized in the vicinity of New Bern, North Carolina. 2. New York: Scott and Co; 1837. [Google Scholar]
- 18.Day A, Madesis P. DNA replication, recombination, and repair in plastids. 2007. [Google Scholar]
- 19.Haberle RC, Fourcade HM, Boore JL, Jansen RK. Extensive rearrangements in the chloroplast genome of trachelium caeruleum are associated with repeats and tRNA genes. J Mol Evol. 2008;66(4):350–361. doi: 10.1007/s00239-008-9086-4. [DOI] [PubMed] [Google Scholar]
- 20.Green BR. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011;66(1):34–44. doi: 10.1111/j.1365-313X.2011.04541.x. [DOI] [PubMed] [Google Scholar]
- 21.Wicke S, Schneeweiss GM, Depamphilis CW, Kai FM, Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol Biol. 2011;76(3–5):273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurtz S, Choudhuri JV, Ohlebusch E, Schleiermacher C, Stoye J, Giegerich R. REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001;29(22):4633–4642. doi: 10.1093/nar/29.22.4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heale SM, Petes TD. The stabilization of repetitive tracts of DNA by variant repeats requires a functional DNA mismatch repair system. Cell. 1995;83(4):539. doi: 10.1016/0092-8674(95)90093-4. [DOI] [PubMed] [Google Scholar]
- 24.McDonald MJ, Wang W, Huang H, Leu J. Clusters of nucleotide substitutions and insertion/deletion mutations are associated with repeat sequences. PLoS Biol. 2011;9(6):e1000622. doi: 10.1371/journal.pbio.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mirkin EV, Mirkin SM. Replication fork stalling at natural impediments. Microbiol Mol Biol Rev. 2007;71(1):13–35. doi: 10.1128/MMBR.00030-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Napolitano R, Janel-Bintz R, Wagner J, Fuchs RP. All three SOS-inducible DNA polymerases (Pol II, Pol IV and Pol V) are involved in induced mutagenesis. EMBO J. 2014;19(22):6259–6265. doi: 10.1093/emboj/19.22.6259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wei Y, Roger W. What a difference a decade makes: insights into translesion DNA synthesis. Proc Natl Acad Sci U S A. 2007;104(40):15591–15598. doi: 10.1073/pnas.0704219104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ponder RG, Fonville NC, Rosenberg SM. A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell. 2005;19(6):791–804. doi: 10.1016/j.molcel.2005.07.025. [DOI] [PubMed] [Google Scholar]
- 29.Tang M, Pham P, Shen X, Taylor JS, O'Donnell M, Woodgate R, Goodman MF. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404(6781):1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 30.Hsu CY, Wu CS, Chaw SM. Ancient nuclear plastid DNA in the yew family (taxaceae) Genome Biol Evol. 2014;6(8):2111–2121. doi: 10.1093/gbe/evu165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunkel TA. Slippery DNA and diseases. Nature. 1993;365(6443):207–208. doi: 10.1038/365207a0. [DOI] [PubMed] [Google Scholar]
- 32.Ellegren H. Microsatellite mutations in the germline: : implications for evolutionary inference. Trends Genet. 2000;16(12):551–558. doi: 10.1016/S0168-9525(00)02139-9. [DOI] [PubMed] [Google Scholar]
- 33.Wierdl M, Dominska M, Petes TD. Microsatellite instability in yeast: dependence on the length of the microsatellite. Genetics. 1997;146(3):769. doi: 10.1093/genetics/146.3.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McIntosh E, Edwards MA, Henry RJ, Rymer Capturing chloroplast variation for molecular ecology studies: a simple;next generation sequencing approach applied to a rainforest tree. BMC Ecol. 2013;13(1):8. doi: 10.1186/1472-6785-13-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borgström E, Lundin S, Lundeberg J. Large scale library generation for high throughput sequencing. PLoS One. 2011;4(6):e19119. doi: 10.1371/journal.pone.0019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;4(9):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dierckxsens N, Mardulyn P, Smits G. NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 2017;45(4):w955. doi: 10.1093/nar/gkw955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res. 1999;9(9):868. doi: 10.1101/gr.9.9.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wyman SK, Jansen RK, Boore JL. Automatic annotation of organellar genomes with DOGMA. Bioinformatics. 2004;20(17):3252–3255. doi: 10.1093/bioinformatics/bth352. [DOI] [PubMed] [Google Scholar]
- 40.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 41.Lohse M, Drechsel O, Bock R. OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr Genet. 2007;52(5–6):267–274. doi: 10.1007/s00294-007-0161-y. [DOI] [PubMed] [Google Scholar]
- 42.Mayer C, Leese F, Tollrian R. Genome-wide analysis of tandem repeats in Daphnia pulex - a comparative approach. BMC Genomics. 2010;11(1):277. doi: 10.1186/1471-2164-11-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14(7):1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chiaromonte F, Yap VB, Miller W. Scoring pairwise genomic sequence alignments. Pac Symp Biocomput. 2002;7(12):115–126. doi: 10.1142/9789812799623_0012. [DOI] [PubMed] [Google Scholar]
- 45.Heng L. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27(21):2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1 Basic information of the 120 genes in Taxodium distichum, Taxodium mucronatum, and Taxodium ascenden chloroplast genomes.
Additional file 2 Dot plot analysis of Taxodium chloroplast genomes.(A) Taxodium ascenden &Taxodium distichum, (B) Taxodium ascenden &Taxodium mucronatum.
Additional file 3 Tandem repeats detected in the Taxodium ascendens chloroplast genome using Phobos software.
Additional file 4 Distribution of 1–9 nucleotide repeat motifs with different numbers in Taxodium ascendens.
Additional file 5 Dispersed repeats detected in the Taxodium distichum, Taxodium mucronatum, and Taxodium ascenden chloroplast genomes.
Additional file 6 Mauve alignment of T. ascendens, T. distichum, T. mucronatum, Glyptostrobus pensilis, Cryptomeria japonica, and Cycad taitungensis. Locally collinear blocks are denoted by different color boxes. Histograms within each block represent the degree of sequence similarity.
Additional file 7 Information of indels and SNPs in Taxodium distichum and/or Taxodium mucronatum chloroplast genomes with Taxodium ascenden chloroplast genome as reference.
Additional file 8 List of the 8 highly variable regions in Taxodium chloroplast genomes.
Data Availability Statement
Sequence information of the 3 cp genomes is available in the NCBI database under the accession number MN535011- MN535013. The reference cp genome of T. distichum and the whole cp genome sequences of 44 species analysed in this study were all downloaded from the NCBI database with their accession numbers listed in Fig. 3 and Fig. 5.Other datasets supporting the conclusions of this article are included within the article and its additional files.