

Abstract
A complete inventory of the forces governing protein folding is critical for productive protein modeling, including structure prediction and de novo design, as well as understanding protein misfolding diseases of clinical significance. The dominant contributors to protein folding include the hydrophobic effect and conventional hydrogen bonding, along with Coulombic interactions and van der Waals interactions. Over the past few decades, important additional contributors have been identified, including C–H···O hydrogen bonding, n→π* interactions, C5 hydrogen bonding, chalcogen bonding, and interactions involving aromatic rings (cation–π, X–H···π, π–π, anion–π, and sulfur–arene). These secondary contributions fall into two general classes: (1) weak but abundant interactions of the protein main chain, and (2) strong but less frequent interactions involving protein side chains. Though interactions with high individual energies play important roles in specifying nonlocal molecular contacts and ligand binding, we estimate that weak but abundant interactions are likely to make greater overall contributions to protein folding, particularly at the level of secondary structure. Further research is likely to illuminate additional roles of these noncanonical interactions and could also reveal contributions yet unknown.
Graphical Abstract
Proteins are the principal molecular machines of the cell, capable of myriad activities that enable life. Each individual protein derives its function from the unique, three-dimensional arrangement of its chemical components.1 Seminal experiments by Anfinsen2 demonstrated that amino-acid sequences can contain all of the chemical information necessary to specify a particular stable structure.3 Decoding the chemical information present within the polypeptide chain should therefore allow one to predict the structure from its sequence alone. Given the number of putative protein sequences generated from DNA sequencing data,4 such technology would prove invaluable for addressing countless issues in biology. Moreover, the same insight could eventually allow physicians to predict the effects of particular mutations, empowering personalized medicine; this prospect could be especially important for the treatment of diseases caused by protein misfolding.5 Finally, a complete understanding of the factors governing protein structure could be leveraged toward the design of new proteins with emergent functions,6,7 the limits of which are hard to conceive. Understanding the molecular basis for protein structure has thus become one of the central scientific challenges of our age.
CANONICAL FORCES IN PROTEIN FOLDING
Under physiological conditions, the free energy of the folded state of a typical globular protein is 10–15 kcal/mol less than that of the unfolded state.8,9 The enthalpic and entropic energy differences between the two states are, however, several fold larger.10,11 The ensuing dichotomy in the Gibbs equation—∆G° is a small difference between large values of ∆H° and T∆S°—underlies the need for a comprehensive inventory of the forces that govern protein folding.
The interactions that stabilize protein structure also guide a polypeptide chain in attaining that structure.12 The folding of polypeptide chains is well known to be encouraged by a handful of noncovalent interactions: the hydrophobic effect, conventional hydrogen bonding, Coulombic interactions, and van der Waals interactions.8,13 A detailed understanding of these canonical forces has led to the development of many important technologies, including force fields for molecular dynamics simulations14–16 and automated methods for protein design,17,18 which in turn have yielded exciting results.19,20 To assess the limitations in these methods, the biophysics community has engaged in a recurring systematic evaluation called the Critical Assessment of Structure Prediction (CASP).21 The premise of this biannual competition is simple: given only the target sequence, computational biophysicists attempt to predict the three-dimensional structures of proteins that have been determined recently by experimental structural biology. The contest has been held twelve times since 1994, and has led to many insights.21 Though significant improvements were achieved during the early years of the CASP competition, progress of late has been slow.22 In particular, the accuracy of models for sequences bearing low identity to proteins of known structure remains poor.23 In the absence of homologous proteins or domains on which to base initial models, structure prediction relies increasingly on molecular mechanics approaches, which have been problematic.24 Similar limitations have been noted in the prediction of organic crystal structures,25,26 a problem that is conceptually similar to that of protein structure prediction in requiring an accurate inventory of relevant forces. (In addition, efficient packing is essential for both protein folding27 and crystal growth,28 as the interior of proteins has long been known to be closer to a solid than a liquid.29)
The ongoing challenges in protein structure prediction and design is suggestive of an incomplete understanding of the forces that govern protein folding, structure, and stability.24 Apparently, an understanding of the canonical forces alone is insufficient for properly describing protein biophysics. To address this problem, researchers have identified a suite of additional interactions that also contribute to the enthalpy of protein folding. We review the manifestations and contributions of those secondary interactions herein.
SECONDARY INTERACTIONS OF THE MAIN CHAIN
The most ubiquitous secondary interactions are those that occur between backbone atoms (Figure 1). These interactions enlist the lone pairs of the main-chain oxygen. In analogy to the hydrogen bonds donated by the main-chain nitrogen,30 nearly all of the lone pairs of the main-chain oxygen are engaged in intramolecular interactions.31
Figure 1.

Secondary interactions involving the main chain. (A) Structural model of an idealized β-sheet, showing conventional main-chain hydrogen bonds (black dashes), C–H···O hydrogen bonds (green dashes), and C5 hydrogen bonds (blue dashes). (B) Structural model of an idealized α-helix, showing main-chain hydrogen bonds (black dashes) and n→π* interactions (blue arrows). (C,D) Orbital overlap that underlies formation of n→π* interactions (C) and C5 hydrogen bonds (D).
C–H···O Hydrogen Bonds.
The first secondary interactions identified were noncanonical hydrogen bonds involving carbon-based donors.32 Though proteins typically feature weaker carbon acids than do some other organic molecules, there are some protons that are sufficiently acidic to engage in hydrogen bonding. For example, there is substantial evidence that histidine side chains can donate hydrogen bonds from Cε.33 By far, though, the most common C–H···O hydrogen bond donors are the Cα–H protons of the main chain.
C–H···O hydrogen bonds have been observed widely in crystal structures of small molecules, and were proposed to contribute to protein stability in the 1960s.34,35 Despite early debate, C–H···O hydrogen bonds are now well-accepted.36,37 They share many properties with canonical hydrogen bonds, such as directionality and cooperativity,38 though they notably induce blue shifts in vibrational spectra.39,40 Like canonical hydrogen bonds, they are predominantly electrostatic interactions, with smaller contributions from van der Waals attraction and charge transfer.38 Experimental characterizations of their energy within peptides or proteins remain scarce,41,42 owing to their small energies and the experimental challenge of probing the backbone. Calculations generally point to energies of 1–2 kcal/mol,43,44 which is approximately half that of canonical hydrogen bonds.45 Nevertheless, detailed analysis of the geometry of intermolecular contacts in proteins has shown a substantial propensity for carbon-based acids to engage with hydrogen-bond acceptors. Like other hydrogen bonds,46 these interactions are identified by a short donor–acceptor distance (typically, <2.5 Å), and relative linearity between the donor, acceptor, and their antecedents.
The most prevalent example of C–H∙∙∙O hydrogen bonds in proteins are the interstrand Cα–H∙∙∙O=C hydrogen bond in β-sheets (Figure 1A).47,48 These C–H∙∙∙O contacts occur at distances that are significantly shorter than expected for repulsive van der Waals interactions, and correspond closely to those observed in small-molecule crystal structures with validated interactions. Moreover, the approach of the donor to the carbonyl acceptor occurs largely within the plane of the peptide bond, where the carbonyl electron density is maximal. These interactions could affect some 35% of residues in β-sheets.31
Other, albeit less frequent, examples of C–H∙∙∙O hydrogen bonds occur in proteins. Contacts with carbonyl oxygens in the backbone of the α-helix have been noted,49 though these interactions usually involve less acidic β-protons, so the energy contributed by such contacts is likely modest. Additionally, α-helices might benefit from C–H∙∙∙O hydrogen bonds involving donation of a proline α-proton to carbonyl acceptors,50 which has the potential to attenuate the strong helix-breaking tendencies of proline residues. Backbone C–H···O hydrogen bonds are also a feature of the collagen triple helix,51 and might contribute to binding energy and discrimination at protein–protein interfaces.52 One notable example of the latter occurs between transmembrane helices. Transmembrane helices, whether within individual proteins or at interfaces within complexes of multiple proteins, often contact one another along ridges of small amino acids, typified by the GXXXG motif.53 These contacts are mediated by multiple C–H···O hydrogen bonds between helices54 and lead to a characteristic interaction geometry, which has been termed the GAS-right motif.55
n→π* Interactions.
A distinct interaction within the backbone has been posited to contribute to protein folding: the n→π* interaction.56,57 These weak interactions occur between adjacent carbonyl groups in the backbone due to donation of lone pair (n) electron density from a carbonyl oxygen into the π* orbital of another carbonyl group (Figures 1B and 1C). Originally invoked to explain the correlation of pyrrolidine ring pucker with the cis–trans conformation of prolyl peptide bonds in collagen,58 this interaction has now been recognized in a variety of systems.59–61 A signature of an n→π* interaction is a sub-van der Waals contact of the donor oxygen on the acceptor carbon along the Bürgi–Dunitz trajectory.62,63 Though these interactions were posited to be a particular example of dipolar interaction,64–66 extensive evidence67–69 indicates that these interactions harbor distinct, charge-transfer character, a view that has gained acceptance.70 The ensuing electronic donation can pyramidalize the acceptor carbonyl group, as has been observed in high-resolution protein crystal structures.71,72 Computational69 and experimental56 studies on small molecules have estimated the energy of a typical n→π* interaction to be between 0.3 and 0.7 kcal/mol; experimental measurements of the energy of an n→π* interaction have yet to be achieved in a folded protein. Nevertheless, prototypical n→π* interactions are strong enough to compete with canonical hydrogen bonds.73 Moreover, polarizing a acceptor carbonyl group with a hydrogen-bond acceptor increases the interaction energy of an n→π* interaction.74,75 Despite their modest energy, n→π* interactions are predicted to contribute significantly to protein stability because of their frequency: a third of all residues in folded proteins are poised to engage in n→π* interactions.57 Moreover, they contribute differentially to secondary structure formation: >70% of residues in α-helices are predicted to engage in an n→π* interaction, but <10% of β-sheet residues are predicted to do so.57 This interaction is implicated in stabilizing not only α-helices,76 but also other helical conformations such as 31057 and PPII geometries.77,78 Additional interactions are possible in amino acid side chains.79–81
C5 Hydrogen Bonds.
An analogous interaction has been identified in β-sheets.82,83 Specifically, amide protons in β-strands can donate an intraresidue hydrogen bond to its own carbonyl oxygen, forming a C5 hydrogen bond (Figures 1A and 1D). These interactions become significant at donor–acceptor distances below 2.5 Å. Despite their nonlinear geometry, they bear the hallmarks of traditional hydrogen bonding, and their perturbation also causes predictable changes in the stability of β-sheets. Though calculations suggest that these interactions are significantly weaker than traditional hydrogen bonds, often affording only around 0.25 kcal/mol, nearly 5% of residues in folded proteins are affected by such interactions, making the C5 hydrogen bond a key contributor to protein structure and stability. Moreover, bioinformatic83 and crystallographic84 analyses indicate that C5 hydrogen bonds likely contribute to amyloid formation, which is implicated in many neurodegenerative diseases.5
SECONDARY INTERACTIONS INVOLVING SIDE CHAINS
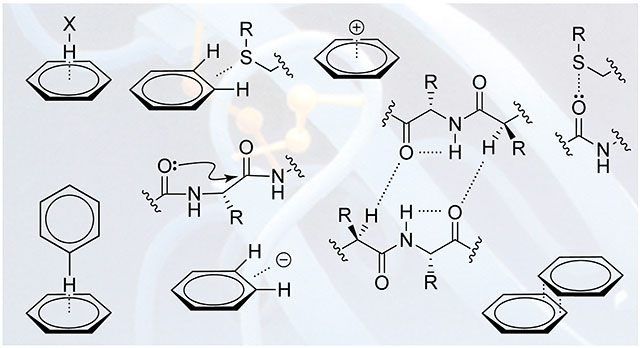
Many secondary interactions engage side-chain atoms (Figure 2). Among such secondary interactions, those involving the aromatic rings of phenylalanine, tyrosine, and tryptophan residues are most important.85 The unique electron-distribution in these side chains enables a number of possible interactions. The facial π cloud bears significant partial negative charge and is nucleophilic, whereas the ring edge bears a partial positive charge and is electrophilic. This charge deposition also creates a permanent electric quadrupole that forms strong electrostatic interactions with both cations and anions. Indeed, perhaps the most important single example of noncanonical forces in protein folding is the cation–π interaction.
Figure 2.

Secondary interactions involving side chains.
Cation–π Interactions.
The significance of the cation–π interaction derives not only from its specific roles, but also from its energy, which is distinctly larger than that of other secondary interactions in proteins; individual cation–π interactions can contribute 2–5 kcal/mol to the binding of ligands to their receptors.86 Originally articulated in the supramolecular chemistry of organic cations,87 these interactions are largely electrostatic attractions between electric monopoles and the electronically negative surfaces of aromatic rings, along with their corresponding quadrupole moments.88 Other contributions to the cation–π interaction, such as dispersion and charge transfer, exist and can be important, but predictions of binding affinity based on electrostatics alone are usually quite successful.89,90 Importantly, this energy is often sufficient to overcome the desolvation penalty for the binding of ions to protein pockets or cavities.91 Indeed, binding sites for a variety of organic cations feature an abundance of aromatic residues, making the cation–π interaction key for the recognition of small-molecule ligands86 or post-translational modifications on proteins such as histones.92
In addition to these critical functional roles, the abundance of aromatic and cationic residues in protein side chains presents an important opportunity for their contribution to protein folding. The cation–π interaction has been observed to perturb the pKa values of functional groups in proteins,93 demonstrating their influence unambiguously. Even under stringent criteria for identification, cation–π interactions affect approximately 1 in every 77 amino acid residues; using similar criteria, canonical salt bridges are twofold more common.94 As interactions between side chains, both cation–π interactions and salt bridges are much less abundant than are secondary interactions of the main chain. For example, whereas ~26% of tryptophan residues are engaged in a cation–π interaction,94 the frequency of tryptophan in eukaryotic proteins is only (1.2 ± 0.2)%,95 diminishing overall impact.
Arginine forms cation–π interactions more often than does lysine.94 This preference is not due to intrinsic interaction energies, but likely arises from the ability of arginine to participate in additional intermolecular interactions while proximal to an aromatic ring. Aromatic residues in proteins form cation–π interactions with the relative frequency: tryptophan more often than tyrosine more often than phenylalanine, which parallels the intrinsic interaction energies of the side chains. Analysis of cation–π distributions across structural motifs is less advanced, but studies of designed peptides indicate that cation–π interactions can make significant contributions to secondary structure.96,97 In addition, cation–π interactions are a common feature of protein–protein interfaces.98 Though the contributions of individual cation–π interactions to ligand binding can lead to several kcal/mol of stabilization,86 experimental measurements of contributions to protein stability provide somewhat lower values, suggesting that the energy of single cation–π interactions in peptides and proteins are generally in the range of 0.5–1.0 kcal/mol.85,99 Nonetheless, this energy has been sufficient for the design of model proteins stabilized by cation–π interactions.100
X–H···π Interactions.
Unsurprisingly, given their affinity for cations, aromatic rings are additionally capable of accepting hydrogen bonds101,102 even from weaker, carbon-based acids.103–105 Like the cation–π interaction, these X–H···π interactions appear to be especially relevant for ligand binding, where they contribute especially to carbohydrate recognition;106–108 the binding sites of lectins are often enriched in aromatic residues, particularly tryptophan,109 which can direct the binding mode of the carbohydrate with exquisite specificity via C–H···π interactions.107,108 Within protein structure, hydrogen bonds to aromatic acceptors are identified by short contact of the donor heavy atom with the center of the aromatic ring at a steep angle of elevation to the plane of the ring.102 By these criteria, they appear sufficiently common to contribute to protein folding; surveys estimate that approximate 10% of aromatic residues accept hydrogen bonds from nitrogen, oxygen, or sulfur donors.102 In addition, interactions of aromatic rings with backbone donors, although relatively infrequent,102 could demarcate changes in the secondary structure pattern and stabilize structural termini. Nonetheless, these interactions are uniformly weaker than cation–π interactions, commensurate with the reduction in electrostatic attraction. Gas-phase studies indicate that energies of these interactions are approximately 5–10 times weaker than for analogous cations; for example, the interaction energy for ammonia and benzene in the gas phase is 1.8 kcal/mol,85 whereas that for ammonium and benzene is 18 kcal/mol.88 Nevertheless, tuning the strength of an individual C–H···π interaction is sufficient to modulate the thermostability of designed miniproteins.110
π–π Interactions.
The unique electron-distribution in aromatic rings also allows them to interact favorably with one another. Inspired by the high aromatic contact of protein interiors, an early survey of aromatic–aromatic interactions in proteins found not only that such pairs were more common than expected by chance, but also that particular short contact distances were favored strongly, suggesting an attraction.111 Specific attraction between aromatic rings was first suggested by the dominance of enthalpy in the interaction, ruling out the previously hypothesized solvophobic nature.85 Extensive characterization has shown that aromatic rings interact primarily in two geometries: T-shaped (or edge-to-face) and displaced-stacked (or offset-stacked).85,111,112 Both arrangements are well described by a balance between contributions from dispersion85 and electrostatics.113 Geometries observed in crystalline or gas-phase arenes are recapitulated in proteins, though the exact preference of aromatic residues for different geometries differs between estimates,114 possibly due to the expanding number of known protein structures.115 Regardless, these contacts can clearly offer stability to proteins. Interestingly, thermophilic proteins have significantly more aromatic–aromatic contacts than do homologues from mesophilic organisms, consistent with a contribution to thermostability.116 Experimental characterizations of individual aromatic–aromatic interactions in peptide and protein model systems have estimated the energy of a single interaction to be approximately 0.5–1.5 kcal/mol, with smaller values being observed for solvent-exposed residues in peptides117,118 and larger values for residues in proteins.119 These experimental energies generally agree well with those from calculations.120,121 Importantly, over half of aromatic residues in proteins have been predicted to engage in attractive interactions, based on the simple criterion of an inter-centroid distance of less than 7 Å.111
Anion–π Interactions.
Aromatic rings can also interact with anions at their edges, which bear partial positive charge.122 Indeed, initial analyses revealed that carboxylates contact aromatic rings in proteins more frequently than would be expected by chance, and approach is predominantly edge-to-edge.123 Energy deconvolution indicates that these interactions are likely dominated by electrostatics rather than van der Waals interactions. Experimental measurements of the energy of a single anion–π interaction suggest that such interactions contribute approximately 0.5 kcal/mol.124 Detection of anion–π interactions in proteins is complicated by the number of degrees of freedom between the interacting partners. Indeed, both energy calculations and Boltzmann statistics find a differential potential of ring atoms to engage in anion–π interactions.125 Using an energy-based criterion for identification, anion–π interactions involving phenyalanine have been observed in approximately 70% of proteins.126 Using geometric identification criteria, tryptophan was found to have a higher propensity than phenylalanine or tyrosine to engage in anion–π interactions, possibly due to its size or dipole moment. Most anion–π interactions are distant in sequence, though local contacts in both α-helices and β-sheets are known.126 Preliminary analyses have also catalogued the coincidence and cooperativity of anion–π interactions with hydrogen bonds,125 π–π interactions,127 and cation–π interactions.127 Anion–π interactions might contribute to the formation of protein–protein interfaces125 and are likely to be especially important in protein–DNA interactions127 because DNA features both additional anionic phosphoryl groups and electron-deficient π-systems that encourage the approach of anionic amino acid residues. An especially strong anion–π interaction has been reported to stabilize a β-hairpin in the WW domain.128
Sulfur–Arene Interactions.
Aromatic rings are additionally capable of interacting with lone pairs, though these contacts generally involve electron-deficient rings, unlike those in proteins.129 Nevertheless, reports have documented an enrichment of sulfur atoms near aromatic rings in proteins130,131 and protein–protein interfaces,131 leading to postulation of a so-called sulfur–arene interaction.85 Early reports estimate that half of sulfur-containing residues form short contacts with aromatic rings.130 As in small-molecule crystal structures,132 sulfur atoms in proteins approach aromatic rings along the ring edge, though detailed geometries have not been catalogued for each type of residue (cysteine, cystine, and methionine). Experimental perturbations of this interaction in peptides have found stabilizing energies on the order of 0.5 kcal/mol.133,134 Although results from peptide studies suggest that these interactions are dominated by the hydrophobic effect rather than by a specific attraction,134 experiments in proteins suggest that sulfur–arene interactions cannot be replaced by purely hydrophobic interactions.131 Further research is needed to clarify the thermodynamic contributions of sulfur–arene interactions to protein folding.
Chalcogen Bonding.
Sulfur atoms can also participate in stereoelectronic interactions with electron-pair donors in a paradigm termed chalcogen bonding.135 In these interactions, electron density from a donor, often a carbonyl oxygen, is transferred into the σ* orbital of one of the bonds to the sulfur atom.136 These contacts were originally observed in surveys of small-molecule crystal structures,137 which first indicated the characteristic interaction geometry. The interaction has since been implicated in many examples of small-molecule structure and reactivity.138 Only in the early 2000s, however, was chalcogen bonding implicated in protein structure.139,140 Interaction geometries of methionine and disulfide sulfur atoms are broadly consistent with those observed in small-molecule crystal structures, and occur most frequently with main-chain oxygens in α-helices.140 Calculations on a prototypical interaction indicated an energy of 0.64 kcal/mol due to charge transfer;140 interactions with cystine disulfides are modestly stronger than those with methionine thioethers. Surveys of protein structures find that 13% of cystine residues and 7% of methionine residues engage in sub-van der Waals contacts with oxygen atoms, which could allow for energetically significant interactions. Chalcogen bonding has also been implicated in ligand binding,141 especially for heterocyclic ligands.142
RELATIVE CONTRIBUTIONS OF SECONDARY FORCES
This complex suite of interactions can be divided largely into two groups. The first is the set of strong interactions that are relatively uncommon in proteins, either because of geometric constraints or amino acid frequency, typified by the cation–π interaction. These interactions can contribute significant energy to the overall energy of folding, but more importantly, they direct the formation of specific contacts, particularly at positions remote in sequence. Moreover, as these interactions pertain largely to side-chain functionalities, their appreciation is likely to improve methods for predicting protein structure from sequence.
Contrast these interactions with the weaker, yet more abundant interactions, such as C–H···O hydrogen bonds or the n→π* interaction. There, individual interactions are likely to be of little importance, given that their energies fall below that of thermal energy at ambient temperatures; however, their cumulative effects over a large number of residues can make a substantial contribution to protein stability. Most are highly local interactions, occurring within a single residue or between adjacent residues, and could thereby guide the earliest events in the protein folding process. In addition, invoking the specific geometric preferences of these interactions might improve model accuracy and refinement. Finally, even crude estimates of the total contributions of pervasive, weak interactions suggest that they play critical roles in stabilizing the overall fold of proteins (Table 1 and Figure 3), perhaps comparable to some canonical interactions.
Table 1.
Estimated Frequency and Energy of Secondary Forces in Protein Folding
| Interaction | Approximate Frequency per 100 Residues | Approximate Energy (kcal/mol)a |
|---|---|---|
| n→π* Interactions | 3357 | 0.2569 |
| C–H···O Hydrogen bonds | 1031 | 0–141,42 |
| π–π Interactionsb | 5111 | 0.5–1.5117–119 |
| C5 Hydrogen bonds | 583 | 0.25–1.583 |
| Cation–π interactions | 1–294 | 0.5–285 |
| Sulfur–arene interactionsb | 2–3130 | 0.3–0.5133,134 |
| Anion–π interactions | 1–2125 | 0.5124 |
| Chalcogen bonds | <1140 | 0.64140 |
| X–H∙∙∙π Interactions | 1102 | 0.35143 |
Preference is given to experimental measurements in proteins and peptides. Computational values are used in the absence of experimental data.
Frequency per 100 residues was estimated by multiplying the frequency of relevant residues95 by the fraction of those residues that engage in the interaction.
Figure 3.

Bar graph of the estimated enthalpic contribution of secondary interactions to the conformational stability of globular proteins. Black bars, interactions of the main chain (Figure 1); gray bars, interactions involving side chains (Figure 2). Data are from Table 1. The sum of the energies is ~27 kcal/mol per 100 residues.
OUTLOOK
A comprehensive understanding of secondary contributions to protein structure should benefit computational force fields. Given the intimacy of these interactions, secondary interactions might encourage the dense packing commonly observed in folded proteins. In addition, because some of these interactions, such as the n→π* interaction or C5 hydrogen bonds, correlate with secondary structure, including these parameters could improve secondary structure prediction or refinement. Importantly, partitioning energetic contributions between individual interactions would allow them to be scrutinized independently. As is, force fields subsume a variety of interactions into relatively few terms. Consider, for example, the hydrogen bonds in an α-helix. There is significant evidence that the α-helix is stabilized by both hydrogen bonds and n→π* interactions; however, force fields account only for the hydrogen bonds. Hence, in order to achieve agreement with experimental results, hydrogen-bonding potentials have, in effect, absorbed the computational energy that should be attributed to the n→π* interaction. This approach might be sufficient for modeling an α-helix, but it distorts hydrogen-bonding energies in other regions. Likewise, the empirical optimization of electrostatic parameters might be distorted by absorbing contributions from cation–π interactions. Such canopies reside at the core of computational models, and success might be enhanced by dissecting contributions from secondary interactions and treating those contributions independently.
Significant progress has been made in inventorying the noncovalent interactions available to proteins. Still, additional interactions undoubtedly lack recognition, much less curation. In addition, many known interactions (Table 1) remain poorly characterized in terms of their nature, their precise energetic contributions, or their frequency. Probing the contributions of backbone interactions is particularly challenging given the lack of genetic approaches to perturbation. Data on the distribution of secondary interactions across secondary and tertiary structural motifs are limited, as are data on the interplay of these interactions with canonical hydrogen-bonding or Coulombic interactions (as well as with one another). In addition, relatively little work has been done to characterize secondary interactions involving post-translational modifications.143,144 For example, whereas the strong electrostatic consequences of phosphorylation are well described, the effects of acylation or oxidation remain largely opaque. Moreover, the nature, strength, and roles of many of these interactions in cellular environments remain poorly characterized, as most of the relevant studies to date have been performed in vitro—an important consideration given that many secondary interactions are sensitive to solvent and other environmental conditions. Finally, as protein design and engineering efforts advance, consideration of interactions not possible in natural, proteinogenic amino acids (such as halogen bonding145–147) could also warrant attention.
ACKNOWLEDGMENTS
Work on protein chemistry in the Raines Laboratory is supported by Grant R01 GM044783 (NIH).
KEYWORDS
- Protein folding
process by which a linear polypeptide adopts its three-dimensional conformation
- C–H···O Hydrogen bond
hydrogen bond between a carbon-based acid and an oxygen lone pair
- n→π* Interaction
stereoelectronic interaction between a lone pair and π antibonding orbital, especially that between two carbonyl groups
- C5 Hydrogen bond
intraresidue hydrogen bond between backbone N–H and C=O groups in a β-strand
- Cation–π interaction
interaction of a positive charge with the face of an aromatic ring
- X–H···π Interaction
hydrogen bond donated to the face of an aromatic ring
- π–π Interaction
interaction between two aromatic rings in either an edge-to-face or offset-stacked geometry
- Anion–π interaction
interaction of a negative charge with the edge of an aromatic ring
- Sulfur–arene interaction
short contact between sulfur atoms and aromatic rings
- Chalcogen bonding
stereoelectronic interaction between a lone pair and σ antibonding orbital of a C–S or S–S bond
Footnotes
The authors declare no competing financial interests.
REFERENCES
- (1).Dill KA; MacCallum JL The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [DOI] [PubMed] [Google Scholar]
- (2).Anfinsen CB; Haber E; Sela M; White FH Jr. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc. Natl. Acad. Sci. U. S. A 1961, 47, 1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Anfinsen CB Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [DOI] [PubMed] [Google Scholar]
- (4).Venter JC; Adams MD; Myers EW; Li PW; Mural RJ; Sutton GG; Smith HO; Yandell M; Evans CA; Holt RA; Gocayne JD; Amanatides P; Ballew RM; Huson DH; Wortman JR; Zhang Q; Kodira CD; Zheng XH; Chen L; Skupski M; Subramanian G; Thomas PD; Zhang J; Gabor Miklos GL; Nelson C; Broder S; Clark AG; Nadeau J; McKusick VA; Zinder N; Levine AJ; Roberts RJ; Simon M; Slayman C; Hunkapiller M; Bolanos R; Delcher A; Dew I; Fasulo D; Flanigan M; Florea L; Halpern A; Hannenhalli S; Kravitz S; Levy S; Mobarry C; Reinert K; Remington K; Abu-Threideh J; Beasley E; Biddick K; Bonazzi V; Brandon R; Cargill M; Chandramouliswaran I; Charlab R; Chaturvedi K; Deng Z; Francesco VD; Dunn P; Eilbeck K; Evangelista C; Gabrielian AE; Gan W; Ge W; Gong F; Gu Z; Guan P; Heiman TJ; Higgins ME; Ji R-R; Ke Z; Ketchum KA; Lai Z; Lei Y; Li Z; Li J; Liang Y; Lin X; Lu F; Merkulov GV; Milshina N; Moore HM; Naik AK; Narayan VA; Neelam B; Nusskern D; Rusch DB; Salzberg S; Shao W; Shue B; Sun J; Wang ZY; Wang A; Wang X; Wang J; Wei M-H; Wides R; Xiao C; Yan C; Yao A; Ye J; Zhan M; Zhang W; Zhang H; Zhao Q; Zheng L; Zhong F; Zhong W; Zhu SC; Zhao S; Gilbert D; Baumhueter S; Spier G; Carter C; Cravchik A; Woodage T; Ali F; An H; Awe A; Baldwin D; Baden H; Barnstead M; Barrow I; Beeson K; Busam D; Carver A; Center A; Cheng ML; Curry L; Danaher S; Davenport L; Desilets R; Dietz S; Dodson K; Doup L; Ferriera S; Garg N; Gluecksmann A; Hart B; Haynes J; Haynes C; Heiner C; Hladun S; Hostin D; Houck J; Howland T; Ibegwam C; Johnson J; Kalush F; Kline L; Koduru S; Love A; Mann F; May D; McCawley S; McIntosh T; McMullen I; Moy M; Moy L; Murphy B; Nelson K; Pfannkoch C; Pratts E; Puri V; Qureshi H; Reardon M; Rodriguez R; Rogers Y-H; Romblad D; Ruhfel B; Scott R; Sitter C; Smallwood M; Stewart E; Strong R; Suh E; Thomas R; Tint NN; Tse S; Vech C; Wang G; Wetter J; Williams S; Williams M; Windsor S; Winn-Deen E; Wolfe K; Zaveri J; Zaveri K; Abril JF; Guigó R; Campbell MJ; Sjolander KV; Karlak B; Kejariwal A; Mi H; Lazareva B; Hatton T; Narechania A; Diemer K; Muruganujan A; Guo N; Sato S; Bafna V; Istrail S; Lippert R; Schwartz R; Walenz B; Yooseph S; Allen D; Basu A; Baxendale J; Blick L; Caminha M; Carnes-Stine J; Caulk P; Chiang Y-H; Coyne M; Dahlke C; Mays AD; Dombroski M; Donnelly M; Ely D; Esparham S; Fosler C; Gire H; Glanowski S; Glasser K; Glodek A; Gorokhov M; Graham K; Gropman B; Harris M; Heil J; Henderson S; Hoover J; Jennings D; Jordan C; Jordan J; Kasha J; Kagan L; Kraft C; Levitsky A; Lewis M; Liu X; Lopez J; Ma D; Majoros W; McDaniel J; Murphy S; Newman M; Nguyen T; Nguyen N; Nodell M; Pan S; Peck J; Peterson M; Rowe W; Sanders R; Scott J; Simpson M; Smith T; Sprague A; Stockwell T; Turner R; Venter E; Wang M; Wen M; Wu D; Wu M; Xia A; Zandieh A; Zhu X The sequence of the human genome. Science 2001, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- (5).Chiti F; Dobson CM Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem 2006, 75, 333–366. [DOI] [PubMed] [Google Scholar]
- (6).Bryson JW; Betz SF; Lu HS; Suich DJ; Zhou HX; O’Neil KT; DeGrado WF Protein design: A hierarchic approach. Science 1995, 270, 935–941. [DOI] [PubMed] [Google Scholar]
- (7).Huang P-S; Boyken SE; Baker D The coming of age of de novo protein design. Nature 2016, 537, 320–327. [DOI] [PubMed] [Google Scholar]
- (8).Dill KA Dominant forces in protein folding. Biochemistry 1990, 29, 7133–7155. [DOI] [PubMed] [Google Scholar]
- (9).Richards FM Protein stability: Still an unsolved problem. Cell. Mol. Life Sci 1997, 53, 790–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Privalov PL; Gill SJ Stability of protein structure and hydrophobic interaction. Adv. Protein Chem 1988, 39, 191–234. [DOI] [PubMed] [Google Scholar]
- (11).Makhatadze GI; Privalov PI Energetics of protein structure. Adv. Protein Chem 1995, 47, 307–425. [DOI] [PubMed] [Google Scholar]
- (12).Weissman JS; Kim PS Reexamination of the folding of BPTI: Predominance of native intermediates. Science 1991, 253, 1386–1393. [DOI] [PubMed] [Google Scholar]
- (13).Pace CN; Scholtz JM; Grimsley GR Forces stabilizing proteins. FEBS Lett. 2014, 588, 2177–2184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem 1982, 4, 187–217. [Google Scholar]
- (15).Mayo SL; Olafson BD; Goddard III WA DREIDING: A generic force field for molecular simulations. J. Phys. Chem 1990, 94, 8897–8909. [Google Scholar]
- (16).Cornell WD; Cieplak P; Bayly CI; Gould IR; Kenneth M Merz J; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- (17).Dahiyat BI; Mayo SL De novo protein design: Fully automated sequence selection. Science 1997, 278, 82–87. [DOI] [PubMed] [Google Scholar]
- (18).Das R; Baker D Macromolecular modeling with Rosetta. Annu. Rev. Biochem 2008, 77, 363–382. [DOI] [PubMed] [Google Scholar]
- (19).Kuhlman B; Dantas G; Ireton GC; Varani G; Stoddard BL; Baker D Design of a novel globular protein fold with atomic-level accuracy. Science 2003, 302, 1364–1368. [DOI] [PubMed] [Google Scholar]
- (20).Jiang L; Althoff EA; Clemente FR; Doyle L; Röthlisberger D; Zanghellini A; Gallaher JL; Betker JL; Tanaka F; Barbas III CF; Hilvert D; Houk KN; Stoddard BL; Baker D De novo computational design of retro-aldol enzymes. Science 2008, 319, 1387–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Moult J A decade of CASP: Progress, bottlenecks and prognosis in protein structure prediction. Curr. Opin. Struct. Biol 2005, 15, 285–289. [DOI] [PubMed] [Google Scholar]
- (22).Kryshtafovych A; Fidelis K; Moult J CASP9 results compared to those of previous CASP experiments. Proteins 2011, 79, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Moult J; Fidelis K; Kryshtafovych A; Schwede T; Tramontano A Critical assessment of methods of protein structure prediction (CASP)—round X. Proteins 2014, 82, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Koehl P; Levitt M A brighter future for protein structure prediction. Nat. Struct. Biol 1999, 6, 108–111. [DOI] [PubMed] [Google Scholar]
- (25).Lommerse JPM; Motherwell WDS; Ammon HL; Dunitz JD; Gavezzotti A; Hofmann DWM; Leusen FJJ; Mooji WTM; Price SL; Schweizer B; Schmidt MU; van Eijck BP; Verwer P; Williams DE A test of crystal structure prediction of small organic molecules. Acta Crystallogr. 2000, B56, 697–714. [DOI] [PubMed] [Google Scholar]
- (26).Bardwell DA; Adjiman CS; Arnautova YA; Bartashevich E; Boerrigter SX; Braun DE; Cruz-Cabeza AJ; Day GM; Della Valle RG; Desiraju GR; van Eijck BP; Facelli JC; Ferraro MB; Grillo D; Habgood M; Hofmann DWM; Hofmann F; Jose KV; Karamertzanis PG; Kazantsev AV; Kendrick J; Kuleshova LN; Leusen FJJ; Maleev AV; Misquitta AJ; Mohamed S; Needs RJ; Neumann MA; Nikylov D; Orendt AM; Pal R; Pantelides CC; Pickard CJ; Price LS; Price SL; Scheraga HA; van de Streek J; Thakur TS; Tiwari S; Venuti E; Zhitkov IK Towards crystal structure prediction of complex organic compounds—a report on the fifth blind test. Acta Crystallogr. 2011, B67, 535–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Eriksson AE; Baase WA; Zhang X-J; Heinz DW; Blaber M; Baldwin EP; Matthews BW Response of a protein structure to cavity-creating mutations and its relation to the hydrophobic effect. Science 1992, 255, 178–183. [DOI] [PubMed] [Google Scholar]
- (28).Brock CP; Dunitz JD Towards a grammar of crystal packing. Chem. Mater 1994, 6, 1118–1127. [Google Scholar]
- (29).Klapper MH On the nature of the protein interior. Biochim. Biophys. Acta 1970, 229, 557–566. [DOI] [PubMed] [Google Scholar]
- (30).Fleming PJ; Rose GD Do all backbone polar groups in proteins form hydrogen bonds? Protein Sci. 2005, 14, 1911–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bartlett GJ; Woolfson DN On the satisfaction of backbone-carbonyl lone pairs of electrons in protein structures. Protein Sci. 2016, 25, 887–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Horowitz S; Trievel RC Carbon–oxygen hydrogen bonding in biological structure and function. J. Biol. Chem 2012, 287, 41576–41582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Derewenda ZS; Derewenda U; Kobos PM (His)Cε–H···O=C< hydrogen bond in the active sites of serine hydrolases. J. Mol. Biol 1994, 241, 83–93. [DOI] [PubMed] [Google Scholar]
- (34).Ramachandran GN; Sasisekharan V Refinement of the structure of collagen. Biochim. Biophy. Acta 1965, 109, 314–316. [DOI] [PubMed] [Google Scholar]
- (35).Ramachandran GN; Venkatachalam CM The stability of the two-bonded collagen triple helix. Biochim. Biophy. Acta 1966, 120, 457–458. [DOI] [PubMed] [Google Scholar]
- (36).Taylor R; Kennard O Crystallographic evidence for the existence of C–H···O, C–H···N, and C–H···Cl hydrogen bonds. J. Am. Chem. Soc 1982, 104, 5063–5070. [Google Scholar]
- (37).Gu Y; Kar T; Scheiner S Fundamental properties of the CH···O Interaction: Is it a true hydrogen bond? J. Am. Chem. Soc 1999, 121, 9411–6422. [Google Scholar]
- (38).Steiner T Unrolling the hydrogen bond properties of C–H···O interactions. Chem. Commun 1997, 8, 727–723. [Google Scholar]
- (39).Hobza P; Havlas Z Blue-shifting hydrogen bonds. Chem. Rev 2000, 100, 4253–4264. [DOI] [PubMed] [Google Scholar]
- (40).Qian W; Krimm S Vibrational spectroscopy of hydrogen bonding: Origin of the different behavoir of the C–H···O hydrogen bond. J. Phys. Chem. A 2002, 106, 6628–6636. [Google Scholar]
- (41).Yohannan S; Faham S; Yang D; Grosfeld D; Chamberlain AK; Bowie JU A Cα–H···O hydrogen bond in a membrane protein is not stabilizing. J. Am. Chem. Soc 2004, 126, 2284–2285. [DOI] [PubMed] [Google Scholar]
- (42).Arbely E; Arkin IT Experimental measurement of the strength of a Cα–H···O bond in a lipid bilayer. J. Am. Chem. Soc 2004, 126, 5362–5363. [DOI] [PubMed] [Google Scholar]
- (43).Scheiner S; Kar T; Gu Y Strength of the CαH···O hydrogen bond of amino acid residues. J. Biol. Chem 2001, 276, 9832–9837. [DOI] [PubMed] [Google Scholar]
- (44).Scheiner S Relative strengths of NH···O and CH···O hydrogen bonds between polypeptide chain segments. J. Phys. Chem. B 2005, 109, 16132–16141. [DOI] [PubMed] [Google Scholar]
- (45).Vargas R; Garza J; Dixon DA; Hay BP How strong is the Cα–H···O=C hydrogen bond? J. Am. Chem. Soc 2000, 122, 4750–4755. [Google Scholar]
- (46).McDonald IK; Thornton JM Satisfying hydrogen bonding potential in proteins. J. Mol. Biol 1994, 238, 777–793. [DOI] [PubMed] [Google Scholar]
- (47).Derewenda ZS; Lee L; Derewenda U The occurrence of C–H···O hydrogen bonds in proteins. J. Mol. Biol 1995, 252, 248–262. [DOI] [PubMed] [Google Scholar]
- (48).Scheiner S Contributions of NH···O and CH···O hydrogen bonds to the stability of β-sheets in proteins. J. Phys. Chem. B 2006, 110, 18670–18679. [DOI] [PubMed] [Google Scholar]
- (49).Manikandan K; Ramakumar S The occurrence of C–H···O hydrogen bonds in α-helices and helix termini in globular proteins. Proteins 2004, 56, 768–781. [DOI] [PubMed] [Google Scholar]
- (50).Chakrabarti P; Chakrabarti S C–H···O Hydrogen bond involving proline residues in α-helices. J. Mol. Biol 1998, 284, 867–873. [DOI] [PubMed] [Google Scholar]
- (51).Bella J; Berman HM Crystallographic evidence for Cα–H···O=C hydrogen bonds in a collagen triple helix. J. Mol. Biol 1996, 264, 734–742. [DOI] [PubMed] [Google Scholar]
- (52).Jiang L; Lai L CH···O hydrogen bonds at protein–protein interfaces. J. Biol. Chem 2002, 277, 37732–37740. [DOI] [PubMed] [Google Scholar]
- (53).Senes A; Gerstein M; Engelman DM Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with β-branched residues at neighboring positions. J. Mol. Biol 2000, 296, 921–936. [DOI] [PubMed] [Google Scholar]
- (54).Senes A; Ubarretxena-Belandia I; Engelman DM The Cα–H···O hydrogen bond: A determinant of stability and specificity in transmembrane helix interactions. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 9056–9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Mueller BK; Subramaniam S; Senes A A frequent, GxxxG-mediated, transmembrane association motif is optimized for the formation of interhelical Cα–H hydrogen bonds. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E888–E895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hinderaker MP; Raines RT An electronic effect on protein structure. Protein Sci. 2003, 12, 1188–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bartlett GJ; Choudhary A; Raines RT; Woolfson DN n→π* Interactions in proteins. Nat. Chem. Biol 2010, 6, 615–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Bretscher LE; Jenkins CL; Taylor KM; DeRider ML; Raines RT Conformational stability of collagen relies on a stereoelectronic effect. J. Am. Chem. Soc 2001, 123, 777–778. [DOI] [PubMed] [Google Scholar]
- (59).Singh SK; Das A The n→π* interaction: A rapidly emerging non-covalent interaction. Phys. Chem. Chem. Phys 2015, 17, 9596–9612. [DOI] [PubMed] [Google Scholar]
- (60).Newberry RW; Raines RT The n→π* interaction. Acc. Chem. Res 2017, 50, 1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Sahariah B; Sarma BK Relative orientation of the carbonyl groups determines the nature of orbital interactions in carbonyl–carbonyl short contacts. Chem. Sci 2019, 10, 909–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Bürgi HB; Dunitz JD; Shefter E Geometric reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc 1973, 95, 5065–5067. [Google Scholar]
- (63).Bürgi HB; Dunitz JD; Shefter E Chemical reaction paths. IV. Aspects of O···C=O interactions in crystals. Acta Crystallogr. 1974, B30, 1517–1527. [Google Scholar]
- (64).Fischer FR; Wood PA; Allen FH; Diederich F Orthogonal dipolar interactions between amide carbonyl groups. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 17290–17294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Fäh C; Hardegger LA; Ebert MO; Schweizer WB; Diederich F Self-association based on orthogonal C=O···C=O interactions in the solid and liquid state. Chem. Commun 2010, 46, 67–69. [DOI] [PubMed] [Google Scholar]
- (66).Worley B; Richard G; Harbinson GS; Powers R 13C NMR reveals no evidence of n→π* interactions in proteins. PLoS One 2012, 7, e42075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Choudhary A; Gandla D; Krow GR; Raines RT Nature of amide carbonyl–carbonyl interactions in proteins. J. Am. Chem. Soc 2009, 131, 7244–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Kamer KJ; Choudhary A; Raines RT Intimate interactions with carbonyl groups: Dipole–dipole or n→π*? J. Org. Chem 2013, 78, 2099–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Newberry RW; VanVeller B; Guzei IA; Raines RT n→π* Interactions of amides and thioamides: Implications for protein stability. J. Am. Chem. Soc 2013, 135, 7843–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Persch E; Dumele O; Diederich F Molecular recognition in chemical and biological systems. Angew. Chem., Int. Ed 2015, 54, 3290–3327. [DOI] [PubMed] [Google Scholar]
- (71).Choudhary A; Raines RT Signature of n→π* interactions in α-helices. Protein Sci. 2011, 20, 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Newberry RW; Bartlett GJ; Vanveller B; Woolfson DN; Raines RT Signatures of n→π* interactions in proteins. Protein Sci. 2014, 23, 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Newberry RW; Orke SJ; Raines RT n→π* Interactions are competitive with hydrogen bonds. Org. Lett 2016, 18, 3614–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Kuemin M; Nagel YA; Schweizer S; Monnard FW; Ochsenfeld C; Wennemers H Tuning the cis/trans conformer ratio of Xaa–Pro amide bonds by intramolecular hydrogen bonds: The effect on PPII helix stability. Angew. Chem., Int. Ed 2010, 49, 6324–6327. [DOI] [PubMed] [Google Scholar]
- (75).Shoulders MD; Kotch FW; Choudhary A; Guzei IA; Raines RT The aberrance of the 4S diastereomer of 4-hydroxyproline. J. Am. Chem. Soc 2010, 132, 10857–10865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Wenzell NA; Ganguly HK; Pandey AK; Bhatt MR; Yap GPA; Zondlo NJ Electronic and steric control of n→π* interactions: Stabilization of the α‐helix conformation without a hydrogen bond. ChemBioChem 2019, 20, 963–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Horng JC; Raines RT Stereoelectronic effects on polyproline conformation. Protein Sci. 2006, 15, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Wilhelm P; Lewandowski B; Trapp N; Wennemers H A crystal structure of an oligoproline PPII-helix, at last. J. Am. Chem. Soc 2014, 136, 15829–15832. [DOI] [PubMed] [Google Scholar]
- (79).Pal TK; Sankararamakrishnan R Quantum chemical investigations on intraresidue carbonyl–carbonyl contacts in aspartates of high-resolution protein structures. J. Phys. Chem. B 2010, 114, 1038–1049. [DOI] [PubMed] [Google Scholar]
- (80).Bartlett GJ; Newberry RW; Vanveller B; Raines RT; Woolfson DN Interplay of hydrogen bonds and n→π* interactions in proteins. J. Am. Chem. Soc 2013, 135, 18682–18688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Kilgore HR; Raines RT n→π* Interactions modulate the properties of cysteine residues and disulfide bonds in proteins. J. Am. Chem. Soc 2018, 140, 17606–17611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Toniolo C Intramolecularly hydrogen-bonded peptide conformations. CRC Crit. Rev. Biochem 1980, 9, 1–44. [DOI] [PubMed] [Google Scholar]
- (83).Newberry RW; Raines RT A prevalent intraresidue hydrogen bond stabilizes proteins. Nat. Chem. Biol 2016, 12, 1084–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Gallagher-Jones M; Glynn C; Boyer DR; Martynowycz MW; Hernandez E; Miao J; Zee CT; Novikova IV; Goldschmidt L; McFarlane HT; Helguera GF; Evans JE; Sawaya MR; Cascio D; Eisenberg DS; Gonen T; Rodriguez JA Sub-ångström cryo-EM structure of a prion protofibril reveals a polar clasp. Nat. Struct. Mol. Biol 2018, 25, 131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Meyer EA; Castellano RK; Diederich F Interactions with aromatic rings in chemical and biological recognition. Angew. Chem., Int. Ed 2003, 42, 1210–1250. [DOI] [PubMed] [Google Scholar]
- (86).Dougherty DA Cation–π interactions in chemistry and biology: A new view of benzene, Phe, Tyr, and Trp. Science 1996, 271, 163–168. [DOI] [PubMed] [Google Scholar]
- (87).Dougherty DA The cation–π interaction. Acc. Chem. Res 2013, 46, 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Ma JC; Dougherty DA The cation–π interaction. Chem. Rev 1997, 97, 1303–1324. [DOI] [PubMed] [Google Scholar]
- (89).Mecozzi S; West AP Jr.; Dougherty DA Cation–π interactions in simple aromatics: Electrostatics provide a predictive tool. J. Am. Chem. Soc 1996, 118, 2307–2308. [Google Scholar]
- (90).Mecozzi S; West AP,J; Dougherty DA Cation–π interactions in aromatics of biological and medicinal interest: Electrostatic potential surfaces as a useful qualitative guide. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 10566–10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Kumpf RA; Dougherty DA A mechanism for ion selectivity in potassium channels: Computational studies of cation–π interactions. Science 1993, 261, 1708–1710. [DOI] [PubMed] [Google Scholar]
- (92).Taverna SD; Li H; Ruthenburg AJ; Allis CD; Patel DJ How chromatin-binding modules interpret histone modifications: Lessons from professional pocket pickers. Nat. Struct. Mol. Biol 2007, 14, 1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Loewenthal R; Sancho J; Fersht AR Histidine–aromatic interactions in barnase: Elevation of histidine pKa and contribution to protein stability. J. Mol. Biol 1992, 224, 759–770. [DOI] [PubMed] [Google Scholar]
- (94).Gallivan JP; Dougherty DA Cation–π interactions in structural biology. Proc. Natl. Acad. Sci. U. S. A 1999, 98, 9459–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Echols N; Harrison P; Balasubramanian S; Luscombe NM; Bertone P; Zhang Z; Gerstein M Comprehensive analysis of amino acid and nucleotide composition in eukaryotic genomes, comparing genes and pseudogenes. Nucleic Acids Res. 2002, 30, 2515–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Tsou LK; Tatko CD; Waters ML Simple cation–π interaction between a phenyl ring and a protonated amine stabilizes an α-helix in water. J. Am. Chem. Soc 2002, 124, 14917–14921. [DOI] [PubMed] [Google Scholar]
- (97).Waters ML Aromatic interactions in peptides: Impact on structure and function. Biopolymers 2004, 76, 435–445. [DOI] [PubMed] [Google Scholar]
- (98).Crowley PB; Golovin A Cation–π interactions in protein–protein interfaces. Proteins 2005, 59, 231–239. [DOI] [PubMed] [Google Scholar]
- (99).Salonen LM; Ellermann M; Diederich F Aromatic rings in chemical and biological recognition: Energetics and structures. Angew. Chem., Int. Ed 2011, 50, 4808–4842. [DOI] [PubMed] [Google Scholar]
- (100).Craven TW; Cho MK; Traaseth NJ; Bonneau R; Kirshenbaum K A miniature protein stabilized by a cation–π interaction network. J. Am. Chem. Soc 2016, 138, 1543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Levitt M; Perutz MF Aromatic rings act as hydrogen bond acceptors. J. Mol. Biol 1988, 201, 751–754. [DOI] [PubMed] [Google Scholar]
- (102).Steiner T; Koellner G Hydrogen bonds with π-acceptors in proteins: Frequencies and role in stabilizing local 3D structures. J. Mol. Biol 2001, 305, 535–557. [DOI] [PubMed] [Google Scholar]
- (103).Brandl M; Weiss MS; Jabs A; Sühnel J; Hilgenfeld R CH···π-Interactions in proteins. J. Mol. Biol 2001, 307, 357–377. [DOI] [PubMed] [Google Scholar]
- (104).Plevin MJ; Bryce DL; Boisbouvier J Direct detection of CH/π interactions in proteins. Nat. Chem 2010, 2, 466–471. [DOI] [PubMed] [Google Scholar]
- (105).Perras FA; Marion D; Boisbouvier J; Bryce DL; Plevin MJ Observation of CH···π interactions between methyl and carbonyl groups in proteins. Angew. Chem., Int. Ed 2017, 56, 7564–7567. [DOI] [PubMed] [Google Scholar]
- (106).Chen W; Enck S; Price JL; Powers DL; Powers ET; Wong CH; Dyson HJ; Kelly JW Structural and energetic basis of carbohydrate–aromatic packing interactions in proteins. J. Am. Chem. Soc 2013, 135, 9877–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Hudson KL; Bartlett GJ; Diehl RC; Agirre J; Gallagher T; Kiessling LL; Woolfson DN Carbohydrate–aromatic interactions in proteins. J. Am. Chem. Soc 2015, 137, 15152–15160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Kiessling LL Chemistry-driven glycoscience. Bioorg. Med. Chem 2018, 26, 5229–5238. [DOI] [PubMed] [Google Scholar]
- (109).Vyas NK Atomistic features of protein–carbohydrate interactions. Curr. Opin. Struct. Biol 1991, 1, 732–740. [Google Scholar]
- (110).Baker EG; Williams C; Hudson KL; Bartlett GJ; Heal JW; Porter Goff KL; Sessions RB; Crump MP; Woolfson DN Engineering protein stability with atomic precision in a monomeric miniprotein. Nat. Chem. Biol 2017, 13, 764–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Burley SK; Petsko GA Aromatic–aromatic interaction: A mechanism of protein structure stabilization. Science 1985, 229, 23–28. [DOI] [PubMed] [Google Scholar]
- (112).Blundell T; Singh J; Thornton J; Burley SK; Petsko GA Aromatic interactions. Science 1986, 234, 1005.3775369 [Google Scholar]
- (113).Hunter CA; Sanders JKM The nature of π–π interactions. J. Am. Chem. Soc 1990, 112, 5525–5534. [Google Scholar]
- (114).Hunter CA; Singh J; Thornton JM π–π Interactions: The geometry and energetics of phenylalanine–phenylalanine interactions in proteins. J. Mol. Biol 1991, 218, 837–846. [DOI] [PubMed] [Google Scholar]
- (115).McGaughey GB; Gagne M; Rappe AK π-Stacking interactions: Alive and well in proteins. J. Biol. Chem 1988, 273, 15458–15463. [DOI] [PubMed] [Google Scholar]
- (116).Kannan N; Wishveshwara S Aromatic clusters: A determinant of thermal stability of thermophilic proteins. Protein Eng. 2000, 13, 753–761. [DOI] [PubMed] [Google Scholar]
- (117).Tatko CD; Waters ML Selective aromatic interactions in β-hairpin peptides. J. Am. Chem. Soc 2002, 124, 9372–9373. [DOI] [PubMed] [Google Scholar]
- (118).Butterfield SM; Patel PR; Waters ML Contribution of aromatic interactions to α-helix stability. J. Am. Chem. Soc 2002, 124, 9751–9755. [DOI] [PubMed] [Google Scholar]
- (119).Serrano L; Bycroft M; Fersht AR Aromatic–aromatic interactions and protein stability: Investigation by double-mutant cycles. J. Mol. Biol 1991, 218, 465–475. [DOI] [PubMed] [Google Scholar]
- (120).Burley SK; Petsko GA Dimerization energetics of benzene and aromatic amino acid side chains. J. Am. Chem. Soc 1986, 108, 7995–8001. [Google Scholar]
- (121).Chelli R; Gervasio FL; Procacci P; Schettino V Stacking and T-shape competition in aromatic–aromatic amino acid interactions. J. Am. Chem. Soc 2001, 124, 6133–6143. [DOI] [PubMed] [Google Scholar]
- (122).Frontera A; Gamez P; Mascal M; Mooibroek TJ; Reedijk J Putting anion–π interactions into perspective. Angew. Chem., Int. Ed 2011, 50, 9564–9583. [DOI] [PubMed] [Google Scholar]
- (123).Jackson MR; Beahm R; Duvvuru S; Narasimhan C; Wu J; Wang H-N; Philip VM; Hinde RJ; Howell EE A preference for edgewise interactions between aromatic rings and carboxylate anions: The biological relevance of anion–quadrupole interactions. J. Phys. Chem. B 2007, 111, 8242–8249. [DOI] [PubMed] [Google Scholar]
- (124).Shi Z; Olson CA; Bell AJ Jr.; Kallenbach NR Non-classical helix-stabilizing interactions: C–H···O H-bonding between Phe and Glu side chains in α-helical peptides. Biophys. Chem 2002, 101, 267–279. [DOI] [PubMed] [Google Scholar]
- (125).Chakravarty S; Ung AR; Moore B; Shore J; Alshamrani M A comprehensive analysis of anion–quadrupole interactions in protein structures. Biochemistry 2018, 57, 1852–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Philip V; Harris J; Adams R; Nguyen D; Spiers J; Baudry J; Howell EE; Hinde RJ A survey of aspartate–phenylalanine and glutamate–phenylalanine interactions in the protein data bank: Searching for anion–π pairs. Biochemistry 2011, 50, 2939–2950. [DOI] [PubMed] [Google Scholar]
- (127).Lucas X; Bauzá A; Frontera A; Quiñonero D A thorough anion–π interaction study in biomolecules: On the importance of cooperativity effects. Chem. Sci 2016, 7, 1038–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Smith MS; Lawrence EEK; Billings WM; Larsen KS; Bécar NA; Price JL An anion–π interaction strongly stabilizes the β-sheet protein WW. ACS Chem. Biol 2017, 12, 2535–2537. [DOI] [PubMed] [Google Scholar]
- (129).Egli M; Sarkhel S Lone pair–aromatic interactions: To stabilize or not to stabilize. Acc. Chem. Res 2007, 40, 197–205. [DOI] [PubMed] [Google Scholar]
- (130).Reid KSC; Lindley PF; Thornton JM Sulphur–aromatic interactions in proteins. FEBS J. 1985, 190, 209–213. [Google Scholar]
- (131).Valley CC; Cembran A; Perlmutter JD; Lewis AK; Labello NP; Gao J; Sachs JN The methionine–aromatic motif plays a unique role in stabilizing protein structure. J. Biol. Chem 2012, 287, 34979–34991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Zauhar RJ; Colbert CL; Morgan RS; Welsh WJ Evidence for a strong sulfur–aromatic interactions derived from crystallographic data. Biopolymers 2000, 53, 233–248. [DOI] [PubMed] [Google Scholar]
- (133).Viguera AR; Serrano L Side-chain interactions between sulfur-containing amino acids and phenylalanine in α-helices. Biochemistry 1995, 34, 8771–8779. [DOI] [PubMed] [Google Scholar]
- (134).Tatko CD; Waters ML Investigation of the nature of the methionine–π interaction in β-hairpin peptide model systems. Protein Sci. 2004, 13, 2515–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Iwaoka M Chalcogen bonds in protein architecture In Noncovalent Forces, Scheiner S, Ed. Springer: Cham, Switzerland, 2015; Vol. 19, pp 265–290. [Google Scholar]
- (136).Pascoe DJ; Ling KB; Cockroft SL The origin of chalcogen-bonding interactions. J. Am. Chem. Soc 2017, 139, 15160–15167. [DOI] [PubMed] [Google Scholar]
- (137).Rosenfield RE Jr.; Parthasarathy R; Dunitz JD Directional preferences of nonbonded atomic contacts with divalent sulfur. 1. Electrophiles and nucleophiles. J. Am. Chem. Soc 1977, 99, 4860–4862. [Google Scholar]
- (138).Vogel L; Wonner P; Huber SM Chalcogen bonding: An overview. Angew. Chem., Int. Ed 2019, 58, 1880–1891. [DOI] [PubMed] [Google Scholar]
- (139).Pal D; Chakrabarti P Non-hydrogen bond interactions involving the methionine sulfur atom. J. Biomol. Struct. Dyn 2001, 19, 115–128. [DOI] [PubMed] [Google Scholar]
- (140).Iwaoka M; Takemoto S; Okada M; Tomoda S Weak nonbonded S···X (X = O, N, and S) interactions in proteins. Statistical and theoretical studies. Bull. Chem. Soc. Japan 2002, 75, 1611–1625. [Google Scholar]
- (141).Kříž K; Fanfrlík J; Lepšík M Chalcogen bonding in protein–ligand complexes: PDB survey and quantum mechanical calculations. ChemPhysChem 2018, 19, 2540–2548. [DOI] [PubMed] [Google Scholar]
- (142).Mitchell MO Discovering protein–ligand chalcogen bonding in the protein data bank using endocyclic sulfur-containing heterocycles as ligand search subsets. J. Mol. Model 2017, 23, 287. [DOI] [PubMed] [Google Scholar]
- (143).Hughes RM; Waters ML Effects of lysine acetylation in a β-hairpin peptide: Comparison of an amide–π and a cation–π interaction. J. Am. Chem. Soc 2006, 128, 13586–13591. [DOI] [PubMed] [Google Scholar]
- (144).Beaver JE; Waters ML Molecular recognition of Lys and Arg methylation. ACS Chem. Biol 2016, 11, 643–653. [DOI] [PubMed] [Google Scholar]
- (145).Auffinger P; Hays FA; Westhof E; Ho PS Halogen bonds in biological molecules. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 16789–16794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Metrangolo P; Neukirch H; Pilati T; Resnati G Halogen bonding based recognition processes: A world parallel to hydrogen bonding. Acc. Chem. Res 2005, 38, 386–395. [DOI] [PubMed] [Google Scholar]
- (147).Voth AR; Khuu P; Oishi K; Ho PS Halogen bonds as orthogonal molecular interactions to hydrogen bonds. Nat. Chem 2009, 1, 74–79. [DOI] [PubMed] [Google Scholar]