

SUMMARY
Immune checkpoint inhibitors have improved patient survival in melanoma, but the innate resistance of many patients necessitates the investigation of alternative immune targets. Many immune checkpoint proteins lack proper characterization, including V-domain Ig suppressor of T cell activation (VISTA). VISTA expression on immune cells can suppress T cell activity; however, few studies have investigated its expression and regulation in cancer cells. In this study, we observe that VISTA is expressed in melanoma patient samples and cell lines. Tumor cell-specific expression of VISTA promotes tumor onset in vivo, associated with increased intratumoral T regulatory cells, and enhanced PDL-1 expression on tumor-infiltrating macrophages. VISTA transcript levels are regulated by the stemness factor Forkhead box D3 (FOXD3). BRAF inhibition upregulates FOXD3 and reduces VISTA expression. Overall, this study demonstrates melanoma cell expression of VISTA and its regulation by FOXD3, contributing to the rationale for therapeutic strategies that combine targeted inhibitors with immune checkpoint blockade.
In Brief
VISTA is an understudied immune checkpoint protein. Through the analysis of patient samples and studies in mouse models, Rosenbaum et al. investigate the functional consequences of VISTA expression on melanoma cells. Furthermore, they demonstrate that the BRAF-regulated transcription factor FOXD3 negatively regulates VISTA expression.
Graphical Abstract
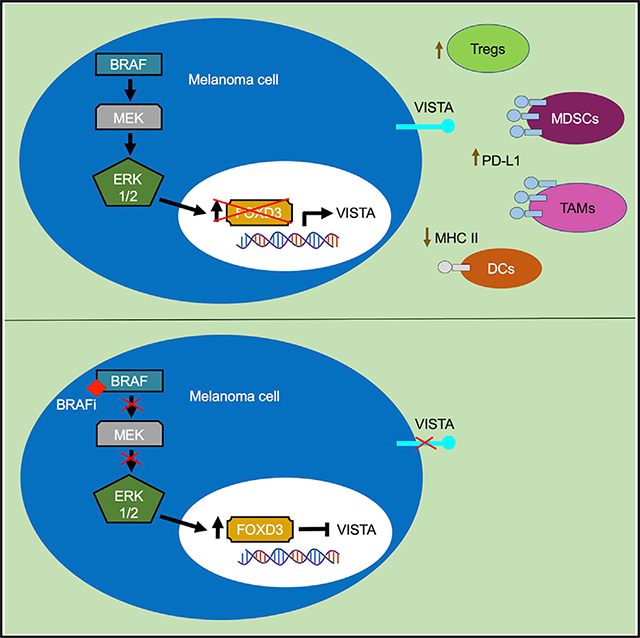
INTRODUCTION
Inhibitory immune checkpoint proteins promote cancer growth by suppressing T cell responses; thus, releasing these checkpoints enhances the ability of the immune system to kill cancer cells. US Food and Drug Administration (FDA) approval of checkpoint inhibitors targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and programmed cell death 1 (PD-1), such as ipilimumab, pembrolizumab, and nivolumab, has improved patient survival and increased response durability in melanoma. However, objective response rates range from ~19% to 58% of patients (Hodi et al., 2018). Mechanisms of acquired and adaptive resistance to immune checkpoint blockade include T cell dysfunction, melanoma cell dedifferentiation, and upregulation of compensatory immune checkpoint proteins (Jenkins et al., 2018; Mehta et al., 2018). As such, additional immune modulatory proteins such as LAG-3, TIM-3, and TIGIT are under investigation in clinical trials for their potential as therapeutic targets in melanoma (clinical trials: , , and ). Targeting multiple immune checkpoints is synergistic, suggesting that checkpoint proteins may engage in distinct, non-redundant activities (Nirschl and Drake, 2013). Hence, the specific role of each immune checkpoint protein in modulating the anti-tumor immune response requires further study.
V-domain immunoglobulin (Ig) suppressor of T cell activation (VISTA) has been identified as a putative immune checkpoint protein that inhibits T cell activation and proliferation (Le Mercier et al., 2014; Wang et al., 2011). VISTA is primarily expressed on hematopoietic cells and only a few studies have evaluated its expression on cancer cells (Villarroel-Espindola et al., 2018; Wang et al., 2011). The role of VISTA is controversial. Some studies propose that VISTA acts as a ligand expressed on antigen-presenting cells to inhibit T cells (Le Mercier et al., 2014; Wang et al., 2011), while other studies suggest that it behaves as an inhibitory receptor on T cells (Flies et al., 2014). Moreover, VISTA has been linked to the phagocytic clearance of cancer cells (Yoon et al., 2015) and the regulation of MT1-MMP for cancer cell invasion (Sakr et al., 2010). Thus, VISTA modulation of the tumor immune microenvironment remains unclear, and its precise role in cancer requires further characterization. In melanoma, VISTA expression on antigen-presenting cells promotes tumor growth independently of PD-1/PD-L1 (Liu et al., 2015), and it is upregulated on tumor-infiltrating macrophages after ipilimumab treatment (Gao et al., 2017). These studies suggest that VISTA can behave in a compensatory mechanism when another immune checkpoint is blocked.
VISTA has also been implicated in embryogenesis, playing a critical role in bone morphogenetic protein 4 (BMP4)-dependent differentiation of mouse embryonic stem cells and the temporal regulation of stemness markers (Aloia et al., 2010). These wide-ranging studies highlight a potential, unusual connection between immune modulatory and stemness pathways. In melanoma, stemness and dedifferentiation are associated with resistance to targeted therapy and immune checkpoint blockade (Mehta et al., 2018; Tsoi et al., 2018). Targeting agents against VISTA have entered clinical trials (e.g., ); however, a thorough understanding of the role of VISTA in cancer and, specifically, in melanoma is lacking.
Forkhead box D3 (FOXD3) is a transcription factor known for its role in neural crest cell differentiation, with the potential to act as both an initiator and a repressor of transcription (Krishnakumar et al., 2016). Our lab previously demonstrated that FOXD3 is upregulated in response to BRAF inhibitors and mediates adaptive resistance to therapy in melanoma (Abel et al., 2013). Targeted inhibition of BRAF can enhance immune cell infiltration into melanoma tumors (Wilmott et al., 2012). The extent to which FOXD3 regulates targets that may alter the anti-tumor immune response remains unknown. In the present study, we evaluated the tumor-specific expression of VISTA in melanoma. We observed that VISTA promotes tumor onset in immunocompetent mouse melanoma models and that the mutant BRAF-regulated transcription factor, FOXD3, suppresses VISTA expression at the transcript level. Overall, we provided insight into the role of tumor-specific VISTA expression and identified a regulatory connection between FOXD3 and VISTA.
RESULTS
VISTA Expression Is Associated with T Cell Dysfunction in Melanoma
Several studies suggest that VISTA behaves as an inhibitory immune checkpoint protein; however, its precise role requires further characterization. In melanoma, we evaluated VISTA association with T cell dysfunction using a workflow that was previously generated for the determination of anti-tumor immune response regulators (Jiang et al., 2018). We performed survival analysis on The Cancer Genome Atlas (TCGA) cutaneous melanoma dataset of non-recurrent stage III patients with a regional lymph, cutaneous, or subcutaneous tumor sample (n = 186) (Broad Institute TCGA Genome Data Analysis Center, 2016). These 186 samples were stratified by low versus high VISTA RNA sequencing (RNA-seq) expression (The Cancer Genome Atlas Network, 2015). Since tumor-infiltrating lymphocytes (TILs) and cytolytic activity are associated with improved survival in melanoma (The Cancer Genome Atlas Network, 2015), samples were further classified by cytotoxic lymphocyte (CTL) level (sum of CD8A, CD8B, GZMA, GZMB, and PRF1 expression) and survival time from date of biospecimen accession was plotted for each expression group. In patients who expressed low levels of VISTA, there was a statistically significant decrease in survival in the low CTL versus the high CTL cohort (~4.07 years versus ~8.75 years for 10-year restricted mean survival time [RMST]; Figure 1A). Conversely, when patients exhibited high levels of VISTA, the difference in survival between the low and high CTL cohorts was no longer evident (~5.40 years versus ~5.02 years RMST; Figure 1B). VISTA expression was also associated with a positive Z score of interaction with T cell dysfunction when analyzed within the tumor immune dysfunction and exclusion (TIDE) Cox proportional hazards model (Figure 1C) (Jiang et al., 2018). These data suggest that high VISTA expression is associated with decreased CTL function and that in melanoma patients with low VISTA expression, high CTL is associated with improved survival.
Figure 1. VISTA Is Expressed in Patient Samples and Correlates with T Cell Dysfunction.

(A and B) Survival analysis was performed on TCGA’s cutaneous melanoma dataset using non-recurrent stage III patients with a regional lymph, cutaneous, or subcutaneous tumor sample (n = 186). Patients were stratified by VISTA RNA-seq expression (high = Z score > 1) and by expression-based estimation of cytotoxic lymphocyte (CTL) level (combined expression of CD8A, CD8B, GZMA, GZMB, and PRF1). Shown are Kaplan-Meier curves using overall survival times from date of biospecimen accession for patients with low versus high CTL, concurrent with (A) low or (B) high VISTA levels. The 10-year restricted mean survival times (RMSTs) were calculated for each cohort.
(C) The T cell dysfunction score for VISTA was calculated in melanoma using the tumor immune dysfunction and exclusion (TIDE) computational framework. VISTA was found to be significantly synergistic with T cell dysfunction.
(D) Whole pieces of human tumor samples were homogenized, lysed, and probed for VISTA expression by western blot. B, BRAF mutant; N, NRAS mutant; WT, BRAF/NRAS wild type. ACTIN was used as a loading control.
(E) Patient tumor samples were stained with fluorescent antibodies for flow cytometry analysis. Cells were gated for CD45 positivity to select for immune cells, and the VISTA expression level was determined and analyzed in this subset compared to negative controls and expression in other cell subsets in the same sample.
(F) Melanoma IHC samples from TCGA were previously scored for lymphocyte density and lymphocyte distribution. These two scores were summed to give an Lscore (no samples had an Lscore of 1). Samples were categorized by Lscore, and VISTA expression was analyzed in each category. Lscores > 3 demonstrate a statistically significant enhancement of VISTA expression; however, VISTA expression is still easily detectable in samples determined to have no or few immune infiltrates (Lscore = 0, 2). *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we analyzed VISTA expression in melanoma patient samples obtained from surgeries (Table S1). By western blotting of whole-tumor samples, we observed varying levels of VISTA expression (Figure 1D). Since these samples contained multiple cell types, we analyzed the specific expression of VISTA on immune cells by flow cytometry. Tumor-associated immune cells, defined by CD45 positivity, displayed a range of VISTA positivity (39.5%–75.7%) (Figures 1E, S1A, and S1B). We analyzed RNA-seq datasets from the melanoma TCGA to further assess immune cell-specific expression of VISTA. Each patient sample in the melanoma TCGA is annotated with an Lscore, the sum of lymphocyte distribution and lymphocyte density determined by immunohistochemistry. We observed that VISTA expression was significantly elevated in samples with high Lscores (Figure 1F). We still observed high levels of VISTA expression in samples that showed no evidence of lymphocyte infiltration (Lscore = 0) or low levels of lymphocyte infiltration (Lscore = 2) (Figure 1F). These findings indicate that VISTA is expressed in patient samples but that its expression may not be exclusive to immune cells.
VISTA Is Expressed on Melanoma Cells
To determine whether VISTA is specifically expressed on melanoma cells, we analyzed patient samples by flow cytometry. VISTA expression was observed with a range of 14.9%–57% on CD45−S100+/Melan-A+/GP100+ melanoma cells (Figures 2A, S1A, and S1B). VISTA expression was further analyzed by immunohistochemistry (IHC), and tumor-specific staining was determined by a pathologist. High tumor-specific positivity was observed in 4 of 13 samples, moderate/low positivity was observed in 8 of 13 samples, and 1 of 13 samples was negative (Figure 2B; Table S1). A tumor microarray of stage III and stage IV melanoma patient samples was analyzed for VISTA expression by IHC and tumor cell-specific expression of VISTA was again detected. When scored by a pathologist in a blinded manner and normalized to the number of nuclei in each sample, the levels of tumor cells expressing VISTA ranged from 0%–5.9% (Figure 2C). No significant difference was observed in tumor-specific VISTA expression between tumor types, mutation statuses, stages, or sexes (Figures S1C–S1F). We also observed VISTA expression in monocultures of melanoma cell lines analyzed by western blot (Figure 2D). The reagents used to detect VISTA by western blot were validated by small interfering RNA (siRNA) knockdown of VISTA (Figure 2E). VISTA showed a complex banding pattern and inhibition of N-linked glycosylation led to a dramatic band shift (Figures S2A and S2B), indicating that VISTA is posttranslationally modified by glycosylation. VISTA expression was analyzed in other cell types and found to be low or undetectable on melanocytes, but highly expressed on keratinocytes, fibroblasts, and some cancer-associated fibroblast (CAF)-like cells (Capparelli et al., 2015, 2018) (Figure S1G). These data demonstrate that VISTA is expressed on multiple cell types in the tumor microenvironment.
Figure 2. VISTA Is Expressed in Melanoma Patient Samples and Cell Lines.

(A) Patient tumor samples were stained with fluorescent antibodies for flow cytometry analysis. Cells were gated for CD45 negativity, then gated for S100/MELANA/GP100 positivity to select for melanoma cells. The VISTA expression level in this subset was determined by comparing fluorescence levels to negative controls and to other cell subsets in the same sample.
(B) IHC staining for VISTA was performed on melanoma patient samples. Tumor cell-specific membranous VISTA staining was judged by a pathologist, and the expression level was defined as high, 15%–65% VISTA+ tumor cells; moderate/low, 1%–14%; negative, 0%. Representative samples were imaged at 40× magnification; scale bars, 50 μm.
(C) IHC staining for VISTA was performed on a tissue microarray (TMA) of melanoma patient samples. Tumor cell-specific membranous VISTA staining was scored by a pathologist, and the number of large nuclei was counted using Visiopharm software. The number of tumor-positive VISTA cells was divided by total nuclei for each sample, and the number of patients showing a range of tumor-specific VISTA expression was plotted. Representative samples were imaged at 40× magnification; scale bars, 50 μm.
(D) A panel of lysates from melanoma cell lines was probed for VISTA expression by western blot. An empty lane was eliminated, denoted by the solid black line.
(E) Cells were transfected with a non-targeting control siRNA or siRNA against VISTA for 72 h. Cells were lysed and protein expression was probed by western blot.
See also Figures S1 and S2 and Table S1.
Melanoma-Specific VISTA Expression Promotes Tumor Onset
We tested the role of VISTA expression on melanoma cells in an immunocompetent setting. The mouse melanoma cell line, D4M, was mutagenized by UVB irradiation to create UV2 cells with increased neoantigen expression. The endogenous expression of VISTA was undetectable to low in vitro and in vivo in D4M UV2 cells; thus, we engineered cells to overexpress VISTA (Figures 3A, 3B, and S3A). VISTA overexpression did not alter cell growth in in vitro IncuCyte assays (Figure 3C). Furthermore, VISTA knockdown in human melanoma cells had little effect on in vitro cell proliferation, 2-dimensional (2D) wound healing, or 3-dimensional (3D) invasion (Figures S2C–S2G).
Figure 3. Tumor-Specific Expression of VISTA Promotes Tumor Onset.

(A) The mouse melanoma cell line, D4M UV2, was engineered to express a V5-tagged VISTA, and expression was verified by western blot.
(B) As for (A), except that expression was verified by flow cytometry.
(C) In vitro cell growth of D4M UV2 cells expressing VISTA was evaluated using the IncuCyte live cell imager. No significant difference in cell growth was found. Data are representative of 3 independent experiments.
(D) Cells were injected into C57BL/6 mice, and tumors were measured by caliper every 2–3 days. Tumors were considered fully formed when they reached ~50mm3, at which point it was considered the time of tumor onset. Data were collected from a total of 18 mice per group from 2 independent experiments. *p < 0.05.
(E) Cells were injected into NSG mice and time-to-tumor onset was tracked, as in (D). Data were collected from a total of 5 mice per group.
(F) YUMM1.7 cells were engineered and injected as in (A). Tumors were considered fully formed when they reached ~50 mm3. Data were collected from a total of 6 mice per group from 2 independent experiments. *p < 0.05.
(G) Cells were injected into NSG mice and time-to-tumor onset was tracked, as in (F). Data were collected from a total of 5 mice per group.
See also Figures S2 and S3.
VISTA may exert tumor-extrinsic effects on the immune microenvironment. To determine VISTA effects in vivo, D4M UV2 cells were injected intradermally into C57BL/6 mice and tumor growth was monitored. We observed that tumor-specific VISTA expression in D4M UV2 led to faster time-to-tumor onset, defined as reaching a tumor volume of ~50 mm3 (Figures 3D and S3B). Similar effects were observed in another UV-mutagenized mouse melanoma cell line, YUMM1.7, engineered to express VISTA (Figures 3F and S3C–S3E) (Wang et al., 2017). In contrast, tumor-specific VISTA expression had no effect on tumor onset in immunodeficient NSG mice (Figures 3E and 3G). Furthermore, tumor-specific expression of VISTA in the poorly immunogenic parental cell lines D4M and YUMM1.7 had no effect on time-to-tumor onset (Figures S3F–S3H). Although we observed significant effects on tumor growth at early time points, tumor cell-specific VISTA had little effect on long-term tumor growth and survival (Figures S3I and S3J). These data suggest that VISTA expression in melanoma cells promotes tumor onset in immunocompetent models.
VISTA-Expressing Tumors Have Alterations in TIL Populations and Respond to Immune Checkpoint Blockade
To assess the effects of melanoma-specific VISTA expression on TILs in vivo, we harvested D4M UV2 and D4M UV2 VISTA tumors after 7 days and analyzed TILs by flow cytometry. In VISTA-expressing tumors, we observed an increase in the percentage of immunosuppressive T regulatory cells (Tregs; FOXP3+CD4+CD3+CD45+) compared to tumors from parental cells (Figure 4A). No differences were detected in the infiltration or activation potential of CD8+ or CD4+ T cells (Figures S4A–S4F). Furthermore, VISTA expression on mouse melanoma cells did not alter T cell killing in in vitro cytotoxicity assays (Figures S4J and S4K).
Figure 4. VISTA Expression Promotes an Immunosuppressive Microenvironment, but Does Not Alter Response to PD-1.

(A) Tumors were analyzed for tumor-infiltrating lymphocytes 7 days after injection. The presence of FOXP3+CD4+CD3+ T regulatory cells was determined by flow cytometry as a percentage of cells gated as Live and CD45+. Data were collected from 9 mice per group, combined from 2 independent experiments. *p < 0.05.
(B) As in (A), dendritic cells (gated as Live F4/80−CD11c+MHCNhiCD3−CD45+) were analyzed for MHC II levels by flow cytometry, and mean fluorescence intensity (MFI) was quantified. *p < 0.05.
(C) As in (A), tumor-associated macrophages (TAMs) (gated as Live CD11b+F4/80+CD3−CD45+) were analyzed for PD-L1 positivity. MFI of PD-L1+ cells was quantified in the TAM immune cell population. **p < 0.01.
(D) As in (A), myeloid-derived suppressor cells (MDSCs) (gated as Live CD11b+GR-1+CD3−CD45+) were analyzed for PD-L1 positivity. MFI of PD-L1+ cells was quantified in the MDSC immune cell population. *p < 0.05.
(E) D4M UV2 VISTA cells were injected into C57BL/6 mice. When tumors reached ~50 mm3, animals were treated with either anti-PD-1 antibody or the corresponding isotype control (rat IgG2a) every 2–3 days. Data were collected from 5 mice treated with isotype and 6 mice treated with anti-PD-1. One animal in the anti-PD-1 treatment group with zero tumor volume was excluded from the statistical analyses of tumor growth curves. For the remaining animals, the fitted group average tumor growth curves are depicted by a bold line. Anti-PD-1 treatment delayed tumor growth on average by 12.2 days (95% CI 6.5–17.8, p = 0.001) as compared to the control group.
(F) D4M UV2 VISTA cells were injected into C57BL/6 mice. When tumors reached ~50 mm3, animals were treated with either anti-VISTA antibody or the corresponding isotype control (hamster polyclonal IgG) every 2–3 days. Data were collected from 7 mice treated with isotype and 8 mice treated with anti-VISTA.
(G) The time to doubling was defined as the first observation day when the tumor volume exceeded twice the volume at day 0. Animals with tumor volumes that did not double at any time were censored on the last day of observation. The time to doubling was analyzed using the Kaplan-Meier estimator of the survival curves and Cox proportional hazards model.
See also Figures S4 and S5.
Next, we evaluated effects on innate immune cell populations and observed a decrease in major histocompatibility complex (MHC) class II levels, quantified by mean fluorescence intensity, on dendritic cells (DCs; F4/80−CD11c+MHCIIhiCD3−CD45+), suggesting that these antigen-presenting cells are less activated (Figures 4B and S4I). Comparable numbers of tumor-associated macrophages (TAMs; CD11b+F4/80+CD3−CD45+) and myeloid-derived suppressor cells (MDSCs; CD11b+GR-1+CD3−CD45+) were isolated from D4M UV2 tumors and D4M UV2 VISTA tumors (Figures S4G and S4H); however, TAMs and MDSCs from D4M UV2 VISTA tumors expressed increased cell surface levels of PD-L1 (Figures 4C and 4D). Since PD-L1-expressing macrophages are important targets of PD-1/PD-L1 blockade (Lin et al., 2018), we tested whether animals bearing D4M UV2 VISTA tumors could benefit from therapeutic targeting of the PD-1/PD-L1 immune checkpoint axis. Treatment with a PD-1 blocking antibody (RMP1–14) led to a growth delay of 12.2 days on average compared with isotype control (95% confidence interval [CI]: 6.5–17.8; p = 0.001) and to improved survival (Figures 4E and S5E). Compared to mice with parental tumors, mice with VISTA-expressing tumors experienced a trend toward decreased survival under anti-PD-1 treatment, although this effect did not reach statistical significance (Figures S5A–S5D).
Next, we tested the efficacy of a VISTA-blocking antibody (clone 13F3) in D4M UV2 VISTA-expressing tumors. Within the control treatment group, 6 of 7 mice progressed rapidly; however, in the anti-VISTA antibody treatment group, we observed a highly heterogeneous response (Figure 4F). Only 2 of 8 mice treated with anti-VISTA antibody showed rapid tumor progression, while the remaining 6 mice treated with anti-VISTA antibody showed a delayed response, and 2 tumors ultimately progressed (Figure 4F). The heterogeneity of response precluded statistical analysis using standard tumor growth curve models; however, anti-VISTA treatment increased median tumor volume doubling time from 7 to 29 days, resulting in a hazard ratio of 2.84 (90% CI: 1.02–7.95, p = 0.095) (Figure 4G). In addition, there was a trend toward increased survival with anti-VISTA treatment, but this was not statistically significant (Figure S5F). We conclude that VISTA expression in tumors promotes an increase in several immunosuppressive cell populations and phenotypes. Furthermore, these tumors still respond to anti-PD-1 therapy, which is consistent with VISTA being independent of the PD-1/PD-L1 immune checkpoint axis (Liu et al., 2015).
The Transcription Factor FOXD3 Regulates VISTA Levels in Melanoma Cells
VISTA plays roles in both the immune system and embryogenesis as a critical component of BMP4-mediated signaling (Aloia et al., 2010). In melanoma cells, VISTA knockdown did not alter BMP4 downstream signaling, as measured by levels of phospho-SMAD1/5 and expression of ID2 (Figures S6A and S6B). Next, we asked whether VISTA is regulated by embryonic pathways in melanoma. FOXD3 regulates the melanocytic switch during neural crest cell differentiation and is upregulated in response to BRAF inhibition (Abel et al., 2013; Weiss et al.,2014). In the pan-cancer TCGA dataset, FOXD3 was highly expressed in melanoma compared to other cancers (Figure 5A) (Cerami et al., 2012; Gao et al., 2013). We reevaluated a microarray dataset from A375 cells induced to express either FOXD3 or LacZ (control) (Abel et al., 2013). FOXD3 selectively downregulated the expression of VISTA, but did not alter the expression of other immune modulatory proteins (Figure 5B). Furthermore, expression of FOXD3, but not LacZ, was sufficient to reduce the total cellular expression and cell surface levels of VISTA (Figures 5C–5E, and S6D). Reagents used to detect VISTA by flow cytometry were validated by siRNA knockdown (Figure S6D).
Figure 5. FOXD3 Is Highly Expressed in Melanoma and Represses VISTA Expression.

(A) RNA-seq data from TCGA was analyzed for FOXD3 expression across all of the cancer studies.
(B) Microarray data were reevaluated for FOXD3 regulation of immune checkpoint proteins.
(C) Expression of a V5-tagged FOXD3 was induced by treatment with doxycycline (100 ng/mL). After 48 h, doxycycline was refreshed and IFNγ (100 ng/mL) was added to select wells for an additional 48 h. Cells were lysed and protein expression was probed by western blot. Data are representative of 3 independent experiments.
(D) Expression of a V5-tagged LacZ control was induced by treatment with doxycycline for 96 h. Cells were lysed and protein expression was probed by western blot. Data are representative of 3 independent experiments.
(E) FOXD3 expression was induced by treatment with doxycycline for 96 h, and the cell surface levels of VISTA were analyzed by flow cytometry. Shown is the MFI. Data are representative of 3 independent experiments. *p < 0.05.
(F) A panel of lysates from various melanoma cell lines was probed for the expression of endogenous levels of VISTA and FOXD3 by western blot.
See also Figure S6.
PD-L1 and indoleamine 2,3-dioxygenase 1 (IDO1) were undetectable at basal levels, but they were induced following interferon γ (IFNγ) treatment. These immunomodulatory proteins were minimally downregulated or unaltered by FOXD3 expression (Figures 5C and S6C). Analysis of the TCGA RNA-seq data showed a significant positive correlation between VISTA and PD-L1 (Figure S6E). Furthermore, using flow cytometry, we detected the co-expression of VISTA and PD-L1 on melanoma cells and the co-expression of VISTA and PD-1 on immune cells in a patient tumor sample (Figure S6F). While PD-L1 is an IFNγ-stimulated protein, VISTA levels were unaltered by treatment with IFNγ, as well as by treatment with other cytokines and growth factors (Figures 5C, S6G, and S6H). Analysis across a panel of mutant BRAF melanoma cell lines showed that VISTA levels are inversely correlated with basal levels of FOXD3 (Figure 5F). These data identify an unusual connection between the stemness factor FOXD3 and VISTA in melanoma cells.
FOXD3 Regulates VISTA Transcript Levels
To determine how FOXD3 regulates VISTA expression, we introduced two point mutations, N187A and H191A, that are known to ablate the DNA-binding capabilities of FOXD3 (Yaklichkin et al., 2007). While the expression of wild-type FOXD3 reduced VISTA levels, the DNA-binding mutant FOXD3 N187/H191A had no effect on VISTA expression (Figures 6A and 6B). Alternatively, mutations that disrupt FOXD3 interaction with Groucho proteins for transcriptional repression (F378E) (Yaklichkin et al., 2007) or prevent the phosphorylation of FOXD3 at serine site 46 (S46A) (Figure S7A) did not interfere with FOXD3-mediated repression of VISTA (Figure S7B). These data demonstrated that FOXD3 regulation of VISTA is dependent on its DNA-binding domain. Furthermore, FOXD3 expression reduced VISTA transcript levels by qRT-PCR (Figures 6C and S7D). Reagents to detect VISTA expression by qRT-PCR were validated using siRNA knockdown (Figure S7C). VISTA protein was not altered by proteasome or autophagy inhibitors (Figures S7E and S7F), indicating that protein stability and autophagy are not predominant regulators of VISTA protein levels. These findings indicate that FOXD3 regulates VISTA transcript levels.
Figure 6. FOXD3 Regulates Transcription of VISTA mRNA.

(A) A375 cells were treated with doxycycline for 96 h to induce the overexpression of a DNA-binding mutant FOXD3. VISTA and FOXD3 levels were detected by western blot. Data are representative of 3 independent experiments.
(B) A375 cells were treated with doxycycline for various lengths of time to induce the expression of either WT or DNA-binding mutant FOXD3.
(C) Cells were treated with doxycycline for varying lengths of time to induce FOXD3 expression. RNA was isolated and qPCR was performed using 2 different Taqman assay probes for VISTA mRNA. Data are representative of 3 independent experiments. *p < 0.05, **p < 0.01.
(D) ChIP-seq data of V5-FOXD3 were mined for FOXD3 binding peaks. Shown is a map of the locus for the VISTA gene VSIR and aligned reads. Data were visualized in the Integrated Genomics Viewer.
(E) FOXD3 binding at identified peaks was verified by ChIP. A375TR/V5-FOXD3 cells were treated with doxycycline for 48 h to induce V5-FOXD3 expression. V5 antibody was used to pull down FOXD3 protein, and specific primers (Table S2) were used to probe binding at each peak (labeled in 5D) by qRT-PCR. Data are representative of 3 independent experiments. *p < 0.05, **p < 0.01.
Next, we reevaluated a chromatin immunoprecipitation sequencing (ChIP-seq) dataset of FOXD3 expression (Abel et al., 2013). We observed three putative FOXD3 binding peaks associated with intron 1 of the VSIR gene encoding VISTA (Figure 6D). Next, we performed ChIP assays in A375 cells inducibly expressing V5-FOXD3 to verify FOXD3 binding at these peaks. Using primers designed to be specific to each peak, we observed V5-FOXD3 binding at peaks 1 and 2 (Figure 6E; Table S2). ChIP specificity in these samples was confirmed by FOXD3 binding to a known target, ERBB3 (Abel et al., 2013) (Figure S7G). These findings show that FOXD3 reduces VISTA transcript levels and directly binds intron 1 of the VSIR gene.
BRAF Inhibition Reduces VISTA Protein and Transcript Levels
FOXD3 is upregulated in response to BRAF inhibitors (Abel et al., 2013). Since FOXD3 suppresses VISTA expression in melanoma cells, we predicted that pharmacological inhibition of mutant BRAF would also affect VISTA levels. PLX4720 is the non-clinical tool compound of the FDA-approved BRAF inhibitor vemurafenib, and the effects of these compounds are indistinguishable (Bollag et al., 2010; Joseph et al., 2010; Tsai et al., 2008). Mutant BRAF melanoma cells were treated with PLX4720 to inhibit BRAF signaling, as evidenced by reduced levels of phospho-MEK and phospho-ERK1/2 (Figure 7A). PLX4720 treatment reduced VISTA protein and transcript levels over time, as detected by western blot and qRT-PCR, respectively (Figures 7A and 7C). The combination of BRAF and MEK inhibitors is FDA approved for the treatment of BRAF V600E/K melanoma (Larkin et al., 2014). We treated mutant BRAF melanoma cells with a combination of PLX4720 and the MEK inhibitor PD0325901 and detected a reduction in VISTA expression over time (Figure 7B). BRAF knockdown by siRNA also reduced VISTA transcript levels (Figure 7D). These data demonstrated that BRAF inhibition reduces the expression of VISTA at the transcript level. We treated a short-term culture of the human BRAF V600E mutant melanoma patient sample TJUMEL54 with the combination of PLX4720 and PD0325901. BRAF and MEK inhibition led to the upregulation of FOXD3 and the downregulation of VISTA (Figure 7E). Western blot detection of VISTA in TJUMEL54 was verified by siRNA knockdown of VISTA in a short-term culture (Figure 7F). These data demonstrate that BRAF inhibition upregulates FOXD3 and suppresses the expression of VISTA.
Figure 7. BRAF Inhibition Suppresses VISTA Expression.

(A) Cells were treated with the BRAF inhibitor PLX4720 (1 μM) for the indicated lengths of time. Cell lysates were analyzed by western blot. Shown is a representative set of blots from 3 independent experiments.
(B) Cells were treated with a combination of the BRAF inhibitor PLX4720 (1 μM) and the MEK inhibitor PD0325901 (35 nM) for the indicated lengths of time. Cell lysates were analyzed by western blot. Shown is a representative set of blots from 3 independent experiments.
(C) Cells were treated as in (A) and RNA was isolated. qPCR was performed using 2 independent Taqman probes for VISTA mRNA. Data are representative of 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
(D) Cells were transfected with siRNAs for 72 h, and then RNA was isolated. qPCR was performed using 2 independent Taqman probes for VISTA mRNA. Data are representative of 3 independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001.
(E) A short-term culture of a human melanoma patient sample was treated with a combination of 1 μM PLX4720 and 35 nM of the MEK inhibitor PD0325901 for varying lengths of time. Lysates were probed by western blot.
(F) A short-term culture, as in (D), was transfected with siRNA against VISTA or a non-targeting control for 72 h. Lysates were probed by western blot.
DISCUSSION
The role of VISTA in cancer requires further characterization. Furthermore, the melanoma cell-specific expression and regulation of VISTA have not been investigated. In this study, we observed that VISTA was expressed on melanoma cells in patient samples and cell lines, and its tumor-specific expression promoted tumor onset in immunocompetent mouse models. VISTA expression was regulated by FOXD3, and the targeted inhibition of BRAF signaling reduced VISTA transcript levels. Thus, we show melanoma cell expression of this immune checkpoint protein and identify mechanisms regulating its expression.
In the melanoma TCGA dataset, patients with immune-rich tumors showed longer survival in the low VISTA subset, and this effect was lost in high VISTA expressers. Survival time is reported from the date of diagnosis. Since many samples were procured long after diagnosis, analyses of molecular data on survival should be interpreted with caution (Liu et al., 2018). Nonetheless, an overall trend showed that survival times were altered by CTL and VISTA expression levels. A novel aspect of our work is the identification of VISTA expression on melanoma cells. We observed a range of VISTA expression in patient samples and noted that even low levels of positivity can be meaningful. By comparison, PD-L1 positivity is defined by a cutoff of 1%–5% tumor cell staining (Weber et al., 2013). Immune checkpoint expression on diverse cell populations within the tumor can have distinct influences on the anti-tumor immune response. In head and neck cancer and adult T cell leukemia/lymphoma, high PD-L1 expression in immune cells and stromal cells, respectively, is a favorable prognostic factor, while high PD-L1 expression in tumor cells trends toward a negative prognostic factor (Kim et al., 2016; Miyoshi et al., 2016). Thus, tumor-specific VISTA expression can have important implications for its role in melanoma.
VISTA has been detected on non-melanoma cancer cells (Mulati et al., 2019; Villarroel-Espindola et al., 2018), and its expression in tumor cells may have significant implications for its role in cancer. For example, VISTA expression on ovarian and endometrial cancer cells can suppress T cell proliferation, infiltration, and cytokine production (Mulati et al., 2019). We observed that VISTA expression on melanoma cells did not inhibit T cell killing in vitro. In light of a recent study that shows that VISTA effects on T cells may be pH dependent, the examination of additional experimental conditions may be required to see the full range of VISTA effects (Johnston et al., 2019). In the present study, we demonstrated that melanoma cell-specific expression of VISTA promoted tumor onset in vivo. VISTA-expressing tumors showed a decrease in DC activation potential and an increase in FOXP3+ Tregs, which is consistent with studies that show that VISTA promotes the induction and maintenance of Treg pools (Le Mercier et al., 2014). Furthermore, TAMs and MDSCs in VISTA-expressing tumors showed increased PD-L1 expression, which could contribute to a more immunosuppressive tumor microenvironment (Gibbons Johnson and Dong, 2017). The interaction between various immune cell types within the tumor microenvironment is highly complex. Macrophages can promote intratumoral Tregs and Tregs can suppress DC-mediated anti-tumor immunity (Curiel et al., 2004; Jang et al., 2017; Sun et al., 2017); thus, the mechanism of VISTA effects on tumor onset may involve effects on multiple cell types. We observed that tumor-specific expression of VISTA had little effect on long-term tumor growth. This observation may be explained by its ability to promote tumor cell phagocytosis (Yoon et al., 2015) and further highlights the potential complexity of the role of VISTA on cancer cells and the need to target multiple immune checkpoints for disease control (Nirschl and Drake, 2013).
VISTA-expressing tumors still respond to PD-1 blockade, indicating that the immune inhibitory effects of VISTA are not strong enough to outweigh the influence of PD-1/PD-L1 in naive tumors. Furthermore, since VISTA and PD-L1 are differentially regulated by IFNγ, VISTA expression or targeting is unlikely to be affected by defects in IFNγ signaling associated with resistance to immune checkpoint blockade (Gao et al., 2016). Our observation that the response to VISTA inhibition is heterogeneous, alongside the work of others showing that VISTA and PD-1 act independently (Liu et al., 2015), further suggests that the combined targeting of VISTA and PD-1 may show enhanced efficacy compared to single agent alone.
In the present study, we identified VISTA as a FOXD3 gene target in melanoma cells. We previously discovered that ERBB3 and TWIST1 are direct targets of FOXD3 in melanoma, and the FOXD3-binding pattern at the VSIR locus was consistent with FOXD3 binding at intron 1 of the genes encoding ERBB3 and TWIST1 (Abel et al., 2013; Weiss et al., 2014). During embryogenesis, FOXD3 represses melanomagenesis in neural crest cells during a critical lineage switch to neural/glial precursors (Weiss et al., 2014). Studies in mouse embryonic stem cells and epiblast cells identified a FOXD3 binding peak near the VSIR gene (Krishnakumar et al., 2016). Whether FOXD3 also regulates VISTA expression during embryonic development requires further investigation. Various mechanisms are used to protect developing cells from the maternal immune system, including the altered expression of MHC antigen-presenting proteins and the upregulation of immune-inhibitory proteins such as IDO1 and PD-L1 (D’Addio et al., 2011; Trowsdale and Betz, 2006). Our data provide additional connections between transcription factors involved in embryonic development and immunomodulatory proteins. Dedifferentiation is a common mechanism of resistance to both targeted therapy and immunotherapy in melanoma (Mehta et al., 2018; Tsoi et al., 2018). This phenomenon may be explained in part due to the intersection between stemness transcription factors and modulation of immune privilege during embryonic development.
We observed that mutant BRAF inhibition led to the upregulation of FOXD3 and downregulation of VISTA transcript and protein levels. BRAF inhibitors enhance immune cell infiltration into the tumor (Wilmott et al., 2012); however, the effect of FOXD3 upregulation on the tumor immune microenvironment has not yet been studied. Our observations that BRAF inhibition and subsequent FOXD3 upregulation decrease VISTA levels at the transcript level suggest that therapeutic targeting of the BRAF pathway will reduce tumor expression of VISTA. It is not uncommon for oncogenic signaling pathways to regulate the expression of immune checkpoints; for example, the expression of PD-L1 can be regulated by signaling downstream of RAS, epidermal growth factor receptor (EGFR), cyclin dependent kinase 5 (CDK5), and rac family small GTPase 1 (RAC1) (Coelho et al., 2017; Dorand et al., 2016; Li et al., 2018; Vu et al., 2015). Studies in melanoma and other cancers show a similar modulation of the immune checkpoint protein PD-L1 following treatment with therapeutic agents, including inhibitors of vascular endothelial growth factor receptor 2 (VEGFR2), BRD/BET proteins, MEK, and phosphatidylinositol 3-kinase (PI3K) (Hogg et al., 2017; Jiang et al., 2013; Zheng et al., 2018). These data contribute to the rationale for therapeutic strategies combining targeted therapies with immune checkpoint blockade (Vanneman and Dranoff, 2012). Targeting agents against VISTA have entered clinical trials, and our findings further define the cell types that may be affected by its therapeutic targeting.
STAR⋆METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. All requests should be directed to and will be fulfilled by the Lead Contact, Andrew Aplin (Andrew.Aplin@jefferson.edu).
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell lines
A375 cells (purchased from ATCC in 2005) were cultured in DMEM with 10% FBS. WM35 and 1205Lu cells (donated by Dr. Meenhard Herlyn, Wistar Institute, Philadelphia, PA in 2005) were cultured in MCDB153 with 2% FBS, 20% Leibowitz L-15 medium, and 5 μg/mL insulin. D4M3.A cells (donated by Dr. Constance Brinckerhoff and Dr. David Mullins, Geisel School of Medicine at Dartmouth, Hanover, NH; 2016) (Jenkins et al., 2014), and D4M UV2 (generated at Mass General Hospital) were cultured in Advanced DMEM with 10% FBS and 1% L-glut. YUMM1.7 and YUMMER1.7 (donated by Dr. Marcus Bosenberg, Yale School of Medicine; 2014 and 2017, respectively) were cultured in DMEM/F12 with 10% FBS and 1% non-essential amino acids. All cells were maintained at 37°C in 5% CO2. Human cell lines were authenticated by sequencing at NRAS and BRAF loci and by STR analysis.
Animals
6–8 week old male C57BL/6 mice were purchased from Jackson Laboratories. Only male C57BL/6 mice were used because the melanoma cell lines in this study were originally derived from male mice, which could generate an immune reaction when injected into immunocompetent female mice. Male and female NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice and C57BL/6-Tg(TcraTcrb) 1100Mjb/J (OT-1) mice were originally purchased from Jackson Laboratories and bred at Thomas Jefferson University. Animals were randomly assigned to experimental groups, or evenly distributed between sexes for NSG experiments. All animal experiments were approved by the IACUC (protocol #1052) and performed in a facility at Thomas Jefferson University accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care (AAALAC).
Patient samples
20 tumor samples from 17 patients were collected from surgeries performed by Dr. Adam Berger at Thomas Jefferson University under an Institutional Review Board approved protocol that included written informed consent and was in accordance with recognized ethical guidelines. 8 patients were male, and 9 patients were female. Patients ranged in age from 36 to 84 years old. 8 patients harbored BRAF mutations, 6 patients harbored NRAS mutations, and 3 patients were wild-type for BRAF and wild-type for NRAS. Detailed information on these samples is provided in Table S1. Tissue microarrays (TMA) of Stage III and Stage IV patients were generated at The University of Texas MD Anderson Cancer Center containing 676 tumor samples from 317 patients; samples were collected from 4 patients at both stage III and stage IV and were considered separately. Of the samples collected, 250 samples were from 95 stage III patients and 426 samples were from 226 stage IV patients. 95 patients were identified as female, while 226 patients were identified as male. Patients ranged in age from 25 to 110 years old. 116 patients harbored BRAF mutations, 34 patients harbored NRAS mutations, 129 patients were wild-type BRAF/ wild-type NRAS, and 42 patients had no reported mutation data. Tumor types included samples of acral melanoma from 19 patients, cutaneous melanoma from 99 patients, lentigo melanoma from 5 patients, mucosal melanoma from 4 patients, and nodular melanoma from 29 patients.
METHOD DETAILS
In vivo studies
Cells were injected intradermally onto the backs of C57BL/6 or NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice. Cell numbers were based on previous publications and past experiments (D4M UV2 = 1×106 cells; D4M = 3×105 cells; YUMMER1.7 = 5×105 cells; YUMM1.7 = 3×105 cells) (Jenkins et al., 2014; Wang et al., 2017). Tumors were considered fully formed when they reached ~50mm3. For immune checkpoint blockade experiments, animals began treatment when tumors reached ~50mm3 and received 250 μg anti-PD-1 antibody (RMP1–14) or the corresponding isotype control (Rat IgG2a clone 2A3), or 300 μg anti-VISTA antibody (13F3) or the corresponding isotype control (polyclonal Armenian hamster IgG) (BioXCell; West Lebanon, NH) administered by intraperitoneal injection every 2–3 days. Treatments were determined based on previous publications (Le Mercier et al., 2014; Wang et al., 2017). Animals were sacrificed when tumors exceeded 750mm3.
Inhibitors, growth factors, and reagents
Recombinant IFNγ and BMP4 were purchased from R&D Systems (Minneapolis, MN). PLX4720 and PD0325901 were purchased from Selleck Chemicals (Houston, TX).
Immunohistochemical analysis
TJUMEL samples were fixed in formalin, paraffin-embedded, and stained manually. After deparaffinization with xylene, rehydration with ethanol, and antigen retrieval with Citra Plus 1x, samples were stained with 1:200 anti-VISTA antibody from Cell Signaling Technology (D1L2G). The intensity of staining and percentage of positive cells were evaluated by Dr. Peter McCue in a blinded manner. Expression level was defined as: high, > 15% VISTA+ tumor cells; moderate/low, 1%–14%; negative, 0%. Entire areas of staining were analyzed. The Ventana Ultraview Benchmark was used for automated staining of the tumor microarray, with 1:100 anti-VISTA antibody from Cell Signaling Technology (D1L2G). The number of positive tumor cells was determined by Dr. Peter McCue and medical resident Dr. Mehri Mollaee in a blinded manner. The number of large, rounded cell nuclei in each sample was quantified by Dr. Zhijiu Zhong utilizing Visiopharm software, and VISTA-positive tumor cells were normalized to the number of nuclei in each sample. The normalized value calculated for each sample was averaged across all samples from the same patient.
Lentiviral construction and transduction
A375TR and 1205LuTR FOXD3 and LacZ cells were previously engineered (Abel and Aplin, 2010). Transgene expression was induced with doxycycline (100ng/mL). Mouse VISTA was amplified from a Vsir (NM_028732) expression plasmid (OriGene; Rockville, MD), cloned into pENTR/D-TOPO (Invitrogen; Carlsbad, CA), and LR recombined into pLenti-4/TO/V5-DEST. Expression constructs and packaging plasmids pLP1, pLP2, pLP/VSVG were cotransfected into HEK293FT cells to generate viral particles. Cells were transduced with particles for 48 hours and then selected with zeocin, as previously described (Abel and Aplin, 2010).
Western blot analysis
Protein lysates were prepared in Laemmli sample buffer, separated by SDS-PAGE, and proteins transferred to PVDF membranes. Immunoreactivity was detected using horseradish peroxidase-conjugated secondary antibodies (CalBioTech; Spring Valley, CA) and chemiluminescence substrate (ThermoScientific; Waltham, MA) on a Versadoc Imaging System (Bio-Rad; Hercules, CA). For detection of human VISTA (#D1L2G), p-ERK1/2 T202/T204 (#9101), HSP90 (#C45G5), PD-L1 (#E1L3N), PD-L2 (#D7U8C), IDO1 (#D5J4E), and V5 (#D3H8Q) antibodies were purchased from Cell Signaling Technology (Danver, MA). BRAF V600E (VE1) antibody was purchased from Spring Bioscience (Pleasanton, CA). ACTIN (A2066) antibody was purchased from Sigma (St. Louis, MO). FOXD3 antibody (#631702) was purchased from BioLegend (San Diego, CA). Mouse VISTA antibody (#Ab214933) was purchased from Abcam (Cambridge, United Kingdom).
siRNA transfection
Cells were transfected for 4 hours with chemically synthesized siRNA at a final concentration of 25nmol/L using Lipofectamine RNAi-MAX (Invitrogen) transfection reagent. Cells were harvested after 72 hours of knockdown. Target sequences used were as follows: Non-targeting control (UGGUUUACAUGUCGACUAA), VISTA #1 (GAUGUGACCUUCUACAAGA), VISTA #2 (GUGCCUGCAUCG UAGGAAU), BRAF #1 (ACAGAGACCUCAAGAGUAAUU), BRAF #2 (CCGAGACAGUCUAAAGAAAUU) siRNAs were purchased from Dharmacon (Lafayette, CO).
Flow cytometry
Cells were treated with doxycycline for 96 hours to induce FOXD3 expression, fixed in formalin/BSA/PBS, and then stained with 1 mg/mL of either VISTA antibody (Cell Signaling Technology #D1L2G) or rabbit IgG (Calbiochem) for 1 hour. Next, cells were stained with Alexa Fluor 647 anti-rabbit secondary antibody (Molecular Probes; Eugene, OR) at 1:750 dilution for 30 minutes. For human patient samples and mouse tumors, tumor pieces were minced with the gentleMACS™ Octo Dissociator using C Tubes (Miltenyi Biotec; Bergisch Gladback, Germany) in digestion media (1x HBSS, 0.1mg/ml Collagenase IA, 60 U/ml DNase I) and incubated at 37°C for 30 minutes with continuous rotation. Cells were washed with medium (RPMI 1640 with L-glutamine, 10% FBS, 1% Pen-Strep, and 5×10−5 β-mercaptoethanol), filtered through a 70μm nylon filter, fixed in formalin/BSA/PBS, and then incubated with Zombie Fixable Viability Dye (BioLegend) for 10 mins. For human samples, cells were then stained for 1 hour with fluorochrome-conjugated antibodies. To identify melanoma cells, live CD45-cells were evaluated for expression of a cocktail of S100, GP100, and MART-1 antibodies (Cho et al., 2015; Weinstein et al., 2014). Antibodies used for these analyses included: CD45 (BioLegend #2D1), VISTA (Cell signaling Technology #D1L2G), PD-1 (BioLegend #NAT105), PD-L1 (BioLegend #29E.2A3), S100 (Abcam #ab76749), GP100 (Abcam #ab137078), MART-1 (Abcam #ab51061). FITC was conjugated to GP100 and MART-1 antibodies using the FITC fast conjugation kit (Abcam). Cells were analyzed on an LSR II, BD Celesta, or Fortessa flow cytometer (BD Biosciences; Franklin Lakes, NJ) using Flowjo software (TreeStar, Ashaland, OR). For TIL analysis of mouse tumors, after live/dead stain, cells were stained for 30 mins with fluorochrome-conjugated antibodies. For Treg staining, cells were fixed and nuclear permeabilized using the eBioscience™ FOXP3/Transcription factor buffer staining set and an antibody specific for FOXP3 (clone FJK-16 s) following company instructions. ForT cell re-stimulation assays, cytokine production byT cells was assessed after ex vivo stimulation with the eBioscience™ cell stimulation cocktail 500X. Cells were incubated in TCM for 5 hours at 37°C in 5% CO2, with 1x stimulation cocktail and 1 μg/ml brefeldin A (GolgiPlug; BD Biosciences). Cells were then washed with ice-cold FACS, stained with Zombie UV, then antibodies specific for surface proteins, and then antibodies specific for intracellular cytokines. Mouse tumors were stained with a cocktail of antibodies against CD45.2 (Biolegend, clone 104), FOXP3 (Thermo, clone FJK-16 s), CD4 (Biolegend, clone RM4.4), CD3 (Biolegend, clone 17A2), PD-L1 (Biolegend, clone 10F.9G2), CD11b (Biolegend, clone M1/70), F4/80 (Biolegend, clone BM8), GR-1 (Biolegend, clone RB6–8C5), IFNγ (Biolegend, clone XMG1.2), TNFα (Biolegend clone MP6-XT22), CD8α (Biolegend, clone 53.6.7), CD11c (Biolegend, clone N418), I-A/I-E (Biolegend, clone M5/114.15.1).
Chromatin immunoprecipitation
A375TR/V5-FOXD3 cells were treated with doxycycline to induce FOXD3 expression for 48 hours. Chromatin immunoprecipitation was performed, as previously described (Abel et al., 2013). V5 (Invitrogen #R960–25) and IgG (Cell Signaling #G3A1) antibodies were used at a concentration of 5 μg/mL for immunoprecipitation. Purified DNA was analyzed by qPCR using iQ SYBR Green Supermix (Bio-Rad), 0.8 μM oligonucleotide primers, and 5 μL ChIP product. Primer sequences are detailed in Table S2. Primers were designed using the Integrated Genome Viewer (IGV 2.3) program to visualize sequences located under peaks of interest, and designed to amplify 206–212bp within the sequence located under each peak. Primer specificity was confirmed by melt curve analysis. Reaction conditions were as follows: denaturation at 94°C for 30 s, annealing at 50°C for 30s, and elongation at 72°C for 30 s, with 50 cycles in total. PCR was performed on an iCycler with MyiQ version 1.0 software (Bio-Rad).
Quantitative RT-PCR
RNA was extracted using the RNeasy Plant Mini Kit (QIAGEN; Hilden, Germany). Conversion to cDNA was achieved through the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative RT-PCR was carried out using Taqman probes (ThermoFisherScientific), according to the manufacturer’s instructions. Relative fold change was calculated after normalization to GAPDH or 18S rRNA using the comparative Ct method. Taqman probes used were VISTA#1-AssayID#Hs00735289_m1; VISTA#2- AssayID#Hs00735287_m1; 18S rRNA-AssayID#Hs99999901_s1; and GAPDH- AssayID#Hs02786624_g1.
IncuCyte® live cell analysis
Cells were trypsinized and plated onto a 6-well or 96-well plate. For scratch wound assays, the IncuCyte® WoundMaker™ was utilized to create a uniform scratch across all wells. Photomicrographs were taken every 2 hours using an Incucyte® Live cell imager (Essen Biosciences; Ann Arbor, MI). Plate confluence and wound closure were measured using IncuCyte® software and presented as percentages.
Spheroid Invasion Assay
siRNA transfection was utilized to knockdown VISTA. Twenty-four hours post transfection, cells were plated onto agarose to form spheroids. Cells were allowed to form spheroids for 4 days, then placed into 3D collagen matrix and incubated for an additional 2 days. Cells were incubated with calcein to mark live cells, and images were taken. The area of the invasive front was quantified using ImageJ and normalized to the area of the spheroid.
LC-MS/MS
FOXD3 was purified from melanoma cells, and TiO2-based immobilized metal ion affinity chromatography (IMAC) was used to enrich phosphopeptides. Peptides were run through LC-MS/MS and the doubly charged precursor of m/z = 862.36 was identified as a phosphorylated DSDAGCDSpPAGPPELR. The phosphorylation site at serine-46 was identified by a neutral loss of H3PO4 from y9 fragment after β-elimination.
T cell cytotoxicity assay
CD8+ T cells were expanded from C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-1) transgenic mouse spleens (CD8+ T cells are specific for OVA peptide), and activated in vitro with 1 μg/mL OVA peptide (257–264) (Genemed Synthesis Inc; San Antonio, TX) and 30U/mL IL-2 (BioLegend). IL-2 and media were refreshed after 48 hours. D4MTR/VISTA cells were treated with or without doxycycline (100ng/mL) for 48 hours prior to the assay to induce VISTA expression. 7 days post T cell expansion, cancer cells were pulsed with or without 1 μM OVA peptide (257–264) for 1 hour, and then labeled with high (15 μM) or low (5 μM) concentrations of carboxyfluorescein succinimidyl ester (CSFE) for 20 minutes to distinguish pulsed versus non-pulsed cells. Pulsed and non-pulsed cancer cells were plated at a 1:1 ratio. T cell expansion was verified by CD8+ flow cytometry staining, and T cells were incubated with cancer cells at varying ratios for 24 hours. After 24 hours, cells were analyzed by flow cytometry. Target cancer cells were gated by FSC-A/SSC-A, then the ratio of CSFEhi to CSFElow cells was evaluated and specific lysis was calculated (% Specific Lysis = [1-(Non-transferred control ratio/Experimental ratio)] x 100).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
For TMA analyses, significance was tested using a non-parametric Mann-Whitney U Test. All other statistical analyses were performed using a two-tailed Student t test. In vitro studies were conducted three independent times; values were averaged and representative images are shown. For in vivo studies, survival curves and curves showing % tumor-free mice were analyzed using a Logrank (Mantel-Cox) test. For growth delay analysis of the anti-PD-1 experiment, tumor volumes in animals that had actual tumor growth were analyzed using a non-linear mixed effects model with the random effects of animal in the parameters of the tumor growth curves. The animal with zero tumor volume at the end of experiment (1 in anti-PD-1 group) was excluded from analyses of the tumor growth curves. The tumor volumes were modeled as exponential functions of Day. For tumor volume doubling analysis of the anti-VISTA experiment, the time to doubling was defined as the first observation day when the tumor volume exceeded twice the volume at day zero. Animals with tumor volumes not exceeding twice the volume at day zero at any time were censored at the last day of observation. The time to doubling was analyzed using the Kaplan-Meier estimator of the survival curves and Cox proportional hazard model. A p value of < 0.05 was considered statistically significant. Significance is denoted by *p < 0.05, **p < 0.01, ***p < 0.001.
TCGA analyses
The Cancer Genome Atlas (TCGA) SKCM RNA sequencing (RNA-seq) V2 RSEM normalized counts data were retrieved from the latest Broad GDAC Firehose data run (doi:10.7908/C11G0KM9) (Broad Institute TCGA Genome Data Analysis Center, 2016). Cutaneous melanoma survival outcome data and tumor biospecimen data were collected from the TCGA Pan-Cancer Clinical Data Resource (TCGA-CDR) repository (Liu et al., 2018) and the TCGA Pan-Cancer Atlas publication page (https://gdc.cancer.gov/about-data/publications/pancanatlas), respectively. Overall survival times were calculated from date of biospecimen accession. Patient sample data were refined to only include non-recurrent stage III patients with a regional lymph, cutaneous, or subcutaneous tumor sample (n = 186). Subsequent analyses on this sample set were based on the TIDE workflow previously generated for determination of anti-tumor immune response regulators (Jiang et al., 2018). TCGA patients were stratified by VISTA RNA-seq expression (High = z-score >1), and further classified by cytotoxic lymphocyte (CTL) level, which was calculated by taking the sum of expression values for CD8A, CD8B, GZMA, GZMB, and PRF1. Kaplan-Meier survival plots were generated using the survminer package (v0.4.3 https://CRAN.R-project.org/packages/survminer/index). Data analyses were performed in R (v3.5.1 http://www.R-project.org/). T cell dysfunction score was calculated using the Tumor Immune Dysfunction and Exclusion (TIDE) Cox proportional hazards model (Jiang et al., 2018). For analysis of lymphocyte levels in TCGA samples, each sample was classified by Lscore. Lscore is equal to the sum of lymphocyte distribution and lymphocyte density scores determined by sample histology (The Cancer Genome Atlas Network, 2015).
DATA AND CODE AVAILABILITY
This study did not generate any datasets or code. TCGA data are publicly available from cbioportal.org. The microarray dataset of A375 cells induced to express either FOXD3 or LacZ (Abel et al., 2013) was previously deposited in the GEO database (GSE43962).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| VISTA | Cell Signaling Technology | Cat# 64953; RRID:AB_2799671 |
| p-ERK1/2 T202/T204 | Cell Signaling Technology | Cat# 9101; RRID:AB_331646 |
| HSP90 | Cell Signaling Technology | Cat# 4877; RRID:AB_2233307 |
| PD-L1 | Cell Signaling Technology | Cat# 13684; RRID:AB_2687655 |
| PD-L2 | Cell Signaling Technology | Cat# 82723; RRID:AB_2799999 |
| IDO1 | Cell Signaling Technology | Cat# 86630; RRID:AB_2636818 |
| V5 | Cell Signaling Technology | Cat# 13202; RRID:AB_2687461 |
| BRAF V600E | Spring Bioscience | Cat# E19290; RRID:AB_11203850 |
| ACTIN | Sigma | Cat# A5316; RRID:AB_476743 |
| FOXD3 | BioLegend | Cat# 631702; RRID:AB_2105424 |
| VISTA | Abcam | Cat# Ab214933; RRID:AB_2313773 |
| PD-1 | BioLegend | Cat# 329736; RRID:AB_2629582 |
| S100 | Abcam | Cat# ab76749; RRID:AB_1566703 |
| GP100 | Abcam | Cat# Ab137078; RRID:AB_2732921 |
| MART-1 | Abcam | Cat# ab51061; RRID:AB_880693 |
| CD45.2 | BioLegend | Cat# 109832; RRID:AB_2565511 |
| FOXP3 | Thermo | Cat# 14-5773-82; RRID:AB_467576 |
| CD4 | BioLegend | Cat# 116004; AB_313689 |
| CD3 | BioLegend | Cat# 100216; RRID:AB_493697 |
| PD-L1 | BioLegend | Cat# 124321; RRID:AB_2563635 |
| CD11b | BioLegend | Cat# 101259; RRID:AB_2566568 |
| F4/80 | BioLegend | Cat# 123116; RRID:AB_893481 |
| GR-1 | BioLegend | Cat# 108406; RRID:AB_313371 |
| IFNγ | BioLegend | Cat# 505810; RRID:AB_315404 |
| TNFα | BioLegend | Cat# 506333; RRID:AB_2562450 |
| CD8α | BioLegend | Cat# 100742; RRID:AB_2563056 |
| CD11c | BioLegend | Cat# 117308; RRID:AB_313777 |
| I-A/I-E | BioLegend | Cat# 107630; RRID:AB_2069376 |
| Anti-mouse PD-1 | BioXcell | Cat# BE0146; RRID:AB_10949053 |
| Rat IgG2a isotype control | BioXcell | Cat# BE0089; RRID:AB_1107769 |
| Anti-mouse VISTA | BioXcell | Cat# BE0310; RRID:AB_2736990 |
| Armenian Hamster IgG isotype control | BioXcell | Cat# BE0091; AB_1107773 |
| Biological Samples | ||
| Patient tumor samples | Thomas Jefferson University Surgery Department | TJUMELXXX |
| Patient Short Term Culture | Thomas Jefferson University Surgery Department | TJUMEL54 |
| Tumor microarray | MD Anderson Surgery Department | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PLX4720, BRAF inhibitor | Selleck Chemicals | PLX4720 |
| PD0325901, MEK inhibitor | Selleck Chemicals | PD0325901 |
| DharmaFECT Duo Transfection Reagent | Dharmacon Inc. | #T-2010-01 |
| CellTrace CSFE | ThermoFisher | C34554 |
| 500x Cell Stimulation Cocktail | eBioscience | #00-4970-03 |
| BD Cytofix/cytoperm kit | BE Biosciences | #554714 |
| FoxP3/Transcription factor buffer staining set | ebioscience | #00-5523-00 |
| BMP4 | R&D Systems | 314-BP-010 |
| IFNγ | R&D Systems | 285-IF-100 |
| IL-2 | BioLegend | 714604 |
| OVA peptide (257–264) | Genemed Synthesis Inc. | Custom order |
| 18S rRNA Taqman Probe | ThermoFisher | Assay ID Hs99999901_s1 |
| GAPDH Taqman Probe | ThermoFisher | Assay ID Hs02786624_g1 |
| VISTA Taqman Probe #1 | ThermoFisher | Assay ID Hs00735289_m1 |
| VISTA Taqman Probe #2 | ThermoFisher | Assay ID Hs00735287_m1 |
| iQ SYBR Green Supermix | Bio-Rad | N/A |
| Critical Commercial Assays | ||
| FITC fast conjugation kit | Abcam | N/A |
| RNeasy Plant Mini Kit | QIAGEN | N/A |
| iScript cDNA Synthesis Kit | Bio-Rad | N/A |
| Experimental Models: Cell Lines | ||
| Human: A375 BRAF V600E Melanoma Cell line | ATCC | A375 |
| Human: 1205Lu Melanoma Cell Line | The Wistar Institute | 1205Lu |
| Human: WM35 Melanoma Cell Line | The Wistar Institute | WM35 |
| Mouse: D4M3.A BRAF V600E Melanoma Cell Line | Dartmouth University | D4M3.A |
| Mouse: YUMM1.7 BRAF V600E Melanoma Cell Line | Yale University | YUMM1.7 |
| Mouse: D4M UV2 BRAF V600E Melanoma Cell Line-mutagenized | Mass General Hospital, Harvard Medical School | D4M UV2 |
| Mouse: YUMMER1.7 BRAF V600E Melanoma Cell Line-mutagenized | Yale University | YUMMER1.7 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 Mice | Jackson Labs | C57BL/6 |
| Mouse: NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ | Jackson Labs then bred at Thomas Jefferson University | NSG |
| Mouse: C57BL/6-Tg(TcraTcrb)1100Mjb/J | Jackson Labs then bred at Thomas Jefferson University | OT-1 |
| Oligonucleotides | ||
| VISTA siRNA #1 | Dharmacon Inc. | D-032651-02-0005 |
| VISTA siRNA #2 | Dharmacon Inc. | D-032651-04-0005 |
| Non-targeting CTL siRNA | Dharmacon Inc. | D-001810-01 |
| Primers for ChIP, See Table S1 | This paper | N/A |
| Recombinant DNA | ||
| pLenti4/TO/V5-mVISTA | Generated by S. Rosenbaum | N/A |
| pLenti4/TO/V5-FOXD3 | Generated by E. Abel | N/A |
| pLenti4/TO/EGFP-FOXD3 | Generated by E. Abel | N/A |
| pLenti4/TO/EGFP-FOXD3 N187A/H191A | Generated by E. Abel | N/A |
| pLenti4/TO/EGFP-FOXD3 S46A | Generated by E. Abel | N/A |
| pLenti4/TO/EGFP-FOXD3 F378E | Generated by E. Abel | N/A |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | N/A |
| Quantity One | BioRad, Hercules, CA | N/A |
| Graphpad Prism | GraphPad | N/A |
| Integrated Genome Viewer (IGV 2.3) program | Broad Institute | N/A |
| MyiQ version 1.0 software | Bio-Rad | N/A |
| R project | N/A | http://www.R-project.org |
Highlights.
VISTA is heterogeneously expressed on melanoma cells
VISTA expression promotes tumor onset and alters the immune microenvironment
FOXD3 suppresses the expression of VISTA
BRAF/MEK inhibitor treatment reduces levels of VISTA
ACKNOWLEDGMENTS
This work is supported by grants from National Institutes of Health (NIH) R01 CA196278 and R01 CA160495 to A.E.A.; R01 AR043369-22, R01 CA222871-02, R01 AR072304-02, and 5P01 CA163222-05 to D.E.F.; and in part by 1F99CA245552-01 and T32 GM 100836-5 to S.R.R. In addition, the study was supported by Dr. Miriam and Sheldon G. Adelson Medical Research Foundation awards to A.E.A., M.A.D., and D.E.F. The Sidney Kimmel Cancer Center Flow Cytometry, Translational Pathology, and Meta-Omics core facilities are supported by NIH/National Cancer Institute Support Grant P30 CA056036. We are grateful to Dr. Meenhard Herlyn (Wistar Institute), Dr. Marcus Bosenberg (Yale School of Medicine), and Dr. Constance E. Brinckerhoff and Dr. David Mullins (both at Geisel School of Medicine at Dartmouth) for generously providing cell lines; Christina Nickerson and Christine Sheehan at the Albany Medical Research Histology Core for histology staining; and Conroy Field for optimizing conditions in the animal studies.
DECLARATION OF INTERESTS
A.E.A. reports receiving a commercial research grant from Pfizer Inc. (2013–2017), has ownership interest in patent number 9880150, and has consulted for SpringWorks Therapeutics and Fortress Biotech within the last 3 years. D.E.F. has a financial interest in Soltego Inc., a company developing SIK inhibitors for topical skin darkening treatments that may be used for a broad set of human applications. D.E.F.’s interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. The other authors declare no competing interests.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.036.
REFERENCES
- Abel EV, and Aplin AE (2010). FOXD3 is a mutant B-RAF-regulated inhibitor of G(1)-S progression in melanoma cells. Cancer Res. 70, 2891–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel EV, Basile KJ, Kugel CH 3rd, Witkiewicz AK, Le K, Amaravadi RK, Karakousis GC, Xu X, Xu W, Schuchter LM, et al. (2013). Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J. Clin. Invest 123, 2155–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aloia L, Parisi S, Fusco L, Pastore L, and Russo T (2010). Differentiation of embryonic stem cells 1 (Dies1) is a component of bone morphogenetic protein 4 (BMP4) signaling pathway required for proper differentiation of mouse embryonic stem cells. J. Biol. Chem 285, 7776–7783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, et al. (2010). Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broad Institute TCGA Genome Data Analysis Center (2016). Broad GDAC Firehose 2016_01_28 run (Broad Institute of MIT and Harvard; ). 10.7908/C11G0KM9. [DOI] [Google Scholar]
- Capparelli C, Rosenbaum S, Berger AC, and Aplin AE (2015). Fibroblast-derived neuregulin 1 promotes compensatory ErbB3 receptor signaling in mutant BRAF melanoma. J. Biol. Chem 290, 24267–24277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capparelli C, Purwin TJ, Heilman SA, Chervoneva I, McCue PA, Berger AC, Davies MA, Gershenwald JE, Krepler C, and Aplin AE (2018). ErbB3 Targeting Enhances the Effects of MEK Inhibitor in Wild-Type BRAF/NRAS Melanoma. Cancer Res. 78, 5680–5693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JH, Robinson JP, Arave RA, Burnett WJ, Kircher DA, Chen G, Davies MA, Grossmann AH, VanBrocklin MW, McMahon M, and Holmen SL (2015). AKT1 activation promotes the development of melanoma metastases. Cell Rep. 13, 898–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelho MA, de Carné Trécesson S, Rana S, Zecchin D, Moore C, Molina-Arcas M, East P, Spencer-Dene B, Nye E, Barnouin K, et al. (2017). Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 47, 1083–1099.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, et al. (2004). Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med 10, 942–949. [DOI] [PubMed] [Google Scholar]
- D’Addio F, Riella LV, Mfarrej BG, Chabtini L, Adams LT, Yeung M, Yagita H, Azuma M, Sayegh MH, and Guleria I (2011). The link between the PDL1 costimulatory pathway and Th17 in fetomaternal tolerance. J. Immunol 187, 4530–4541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorand RD, Nthale J, Myers JT, Barkauskas DS, Avril S, Chirieleison SM, Pareek TK, Abbott DW, Stearns DS, Letterio JJ, et al. (2016). Cdk5 disruption attenuates tumor PD-L1 expression and promotes antitumor immunity. Science 353, 399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, and Chen L (2014). Coinhibitory receptor PD-1H preferentially suppresses CD4+ T cell-mediated immunity. J. Clin. Invest 124, 1966–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Ward JF, Pettaway CA, Shi LZ, Subudhi SK, Vence LM, Zhao H, Chen J, Chen H, Efstathiou E, et al. (2017). VISTA is an inhibitory immune checkpoint that is increased after ipilimumab therapy in patients with prostate cancer. Nat. Med 23, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons Johnson RM, and Dong H (2017). Functional Expression of Programmed Death-Ligand 1 (B7-H1) by Immune Cells and Tumor Cells. Front. Immunol 8, 961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob J-J, Rutkowski P, Cowey CL, Lao CD, Schadendorf D, Wagstaff J, Dummer R, et al. (2018). Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 19, 1480–1492. [DOI] [PubMed] [Google Scholar]
- Hogg SJ, Vervoort SJ, Deswal S, Ott CJ, Li J, Cluse LA, Beavis PA, Darcy PK, Martin BP, Spencer A, et al. (2017). BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep. 18, 2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang J-E, Hajdu CH, Liot C, Miller G, Dustin ML, and Bar-Sagi D (2017). Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep. 20, 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, Mullins DW, and Brinckerhoff CE (2014). Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res. 27, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Barbie DA, and Flaherty KT (2018). Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Zhou J, Giobbie-Hurder A, Wargo J, and Hodi FS (2013). The activation of MAPK in melanoma cells resistant to BRAF inhibition promotes PD-L1 expression that is reversible by MEK and PI3K inhibition. Clin. Cancer Res. 19, 598–609. [DOI] [PubMed] [Google Scholar]
- Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, Li Z, Traugh N, Bu X, Li B, et al. (2018). Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med 24, 1550–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, Corbett M, Rankin AL, Dibella R, Campbell L, et al. (2019). VISTA is an acidic pH-selective ligand for PSGL-1. Nature 574, 565–570. [DOI] [PubMed] [Google Scholar]
- Joseph EW, Pratilas CA, Poulikakos PI, Tadi M, Wang W, Taylor BS, Halilovic E, Persaud Y, Xing F, Viale A, et al. (2010). The RAF inhibitor PLX4032 inhibits ERK signaling and tumor cell proliferation in a V600E BRAF-selective manner. Proc. Natl. Acad. Sci. USA 107, 14903–14908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Ha S-J, Hong MH, Heo SJ, Koh YW, Choi EC, Kim EK, Pyo KH, Jung I, Seo D, et al. (2016). PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for head and neck cancer patients. Sci. Rep 6, 36956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnakumar R, Chen AF, Pantovich MG, Danial M, Parchem RJ, Labosky PA, and Blelloch R (2016). FOXD3 Regulates Pluripotent Stem Cell Potential by Simultaneously Initiating and Repressing Enhancer Activity. Cell Stem Cell 18, 104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, et al. (2014). Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med 371, 1867–1876. [DOI] [PubMed] [Google Scholar]
- Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, Noelle RJ, and Wang L (2014). VISTA Regulates the Development of Protective Antitumor Immunity. Cancer Res. 74, 1933–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C-W, Lim S-O, Chung EM, Kim Y-S, Park AH, Yao J, Cha J-H, Xia W, Chan L-C, Kim T, et al. (2018). Eradication of Triple-Negative Breast Cancer Cells by Targeting Glycosylated PD-L1. Cancer Cell 33, 187–201.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, Szeliga W, Herbst R, Harms PW, Fecher LA, et al. (2018). Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest 128, 805–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD, Miller HE, Guleria I, Barth RJ, Huang YH, and Wang L (2015). Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc. Natl. Acad. Sci. USA 112, 6682–6687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, et al. ; Cancer Genome Atlas Research Network (2018). An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 173, 400–416.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Kim YJ, Robert L, Tsoi J, Comin-Anduix B, Berent-Maoz B, Cochran AJ, Economou JS, Tumeh PC, Puig-Saus C, and Ribas A (2018). Immunotherapy Resistance by Inflammation-Induced Dedifferentiation. Cancer Discov. 8, 935–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi H, Kiyasu J, Kato T, Yoshida N, Shimono J, Yokoyama S, Taniguchi H, Sasaki Y, Kurita D, Kawamoto K, et al. (2016). PD-L1 expression on neoplastic or stromal cells is respectively a poor or good prognostic factor for adult T-cell leukemia/lymphoma. Blood 128, 1374–1381. [DOI] [PubMed] [Google Scholar]
- Mulati K, Hamanishi J, Matsumura N, Chamoto K, Mise N, Abiko K, Baba T, Yamaguchi K, Horikawa N, Murakami R, et al. (2019). VISTA expressed in tumour cells regulates T cell function. Br. J. Cancer 120, 115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirschl CJ, and Drake CG (2013). Molecular pathways: coexpression of immune checkpoint molecules: signaling pathways and implications for cancer immunotherapy. Clin. Cancer Res 19, 4917–4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakr MA, Takino T, Domoto T, Nakano H, Wong RW, Sasaki M, Nakanuma Y, and Sato H (2010). GI24 enhances tumor invasiveness by regulating cell surface membrane-type 1 matrix metalloproteinase. Cancer Sci. 101, 2368–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Wei F-Q, Li W-J, Wei J-W, Zhong H, Wen Y-H, Lei W-B, Chen L, Li H, Lin H-Q, et al. (2017). A positive-feedback loop between tumour infiltrating activated Treg cells and type 2-skewed macrophages is essential for progression of laryngeal squamous cell carcinoma. Br. J. Cancer 117, 1631–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Network (2015). Genomic Classification of Cutaneous Melanoma. Cell 161, 1681–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trowsdale J, and Betz AG (2006). Mother’s little helpers: mechanisms of maternal-fetal tolerance. Nat. Immunol 7, 241–246. [DOI] [PubMed] [Google Scholar]
- Tsai J, Lee JT,Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, et al. (2008). Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc. Natl. Acad. Sci. USA 105, 3041–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoi J, Robert L, Paraiso K, Galvan C, Sheu KM, Lay J, Wong DJL, Atefi M, Shirazi R, Wang X, et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanneman M, and Dranoff G (2012). Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 12, 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villarroel-Espindola F, Yu X, Datar I, Mani N, Sanmamed M, Velcheti V, Syrigos K, Toki M, Zhao H, Chen L, et al. (2018). Spatially Resolved and Quantitative Analysis of VISTA/PD-1H as a Novel Immunotherapy Target in Human Non-Small Cell Lung Cancer. Clin. Cancer Res 24, 1562–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu HL, Rosenbaum S, Purwin TJ, Davies MA, and Aplin AE (2015). RAC1 P29S regulates PD-L1 expression in melanoma. Pigment Cell Melanoma Res. 28, 590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y, Lu L-F, Gondek D, Wang Y, Fava RA, et al. (2011). VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med 208, 577–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Perry CJ, Meeth K, Thakral D, Damsky W, Micevic G, Kaech S, Blenman K, and Bosenberg M (2017). UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment Cell Melanoma Res. 30, 428–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, Zhao X, Martinez AJ, Wang W, Gibney G, et al. (2013). Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol 31, 4311–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein D, Leininger J, Hamby C, and Safai B (2014). Diagnostic and prognostic biomarkers in melanoma. J. Clin. Aesthet. Dermatol 7, 13–24. [PMC free article] [PubMed] [Google Scholar]
- Weiss MB, Abel EV, Dadpey N, and Aplin AE (2014). FOXD3 modulates migration through direct transcriptional repression of TWIST1 in melanoma. Mol. Cancer Res 12, 1314–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, Kefford RF, Hersey P, and Scolyer RA (2012). Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin. Cancer Res 18, 1386–1394. [DOI] [PubMed] [Google Scholar]
- Yaklichkin S, Steiner AB, Lu Q, and Kessler DS (2007). FoxD3 and Grg4 physically interact to repress transcription and induce mesoderm in Xenopus. J. Biol. Chem 282, 2548–2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon KW, Byun S, Kwon E, Hwang S-Y, Chu K, Hiraki M, Jo S-H, Weins A, Hakroush S, Cebulla A, et al. (2015). Control of signaling-mediated clearance of apoptotic cells by the tumor suppressor p53. Science 349, 1261669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B, Ren T, Huang Y, and Guo W (2018). Apatinib inhibits migration and invasion as well as PD-L1 expression in osteosarcoma by targeting STAT3. Biochem. Biophys. Res. Commun 495, 1695–1701. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any datasets or code. TCGA data are publicly available from cbioportal.org. The microarray dataset of A375 cells induced to express either FOXD3 or LacZ (Abel et al., 2013) was previously deposited in the GEO database (GSE43962).