

Abstract
In systemic sclerosis (SSc), previously healthy adults develop an inflammatory prodrome with subsequent progressive fibrosis of the skin and viscera. SSc has a weak signature for genetic contribution, and there are few pathogenic insights or targeted treatments for this condition. Here, chromatin accessibility and transcriptome profiling coupled with targeted epigenetic editing revealed constitutive activation of a previously unannotated transforming growth factor β2 (TGFB2) enhancer maintained through epigenetic memory in SSc. The resulting autocrine TGFβ2 signaling enforced a pro-fibrotic synthetic state in ex vivo fibroblasts from patients with SSc. Inhibition of nuclear factor kappa-B (NF-kB) or bromodomain-containing protein 4 (BRD4) achieved sustained inhibition of TGFB2 enhancer activity, mitigated pro-fibrotic gene expression, and reversed dermal fibrosis in patient skin explants. These findings suggest an epigenetic mechanism of fibrosis in SSc and inform a regulatory mechanism of TGFB2, a major pro-fibrotic cytokine.
One Sentence Summary:
NF-kB and BRD4 epigenetically activate a TGFB2 enhancer to lock sclerotic patient fibroblasts into a pro-fibrotic synthetic state.
Introduction
Systemic sclerosis (SSc) is a rare and complex disorder in which previously healthy young adults show intractable fibrosis of the skin and internal organs (1, 2). This complex disease associates with an overt inflammatory prodrome, but it is unclear whether this is a marker or driver of disease. Although SSc is among the most devastating rheumatic diseases with a 10-year mortality rate approximating 30% (3, 4), its pathogenic mechanism remains unknown, and treatments are largely symptomatic and often ineffective.
The TGFβ family of cytokines is a dominant regulator of both physiologic and pathologic collagen synthesis and deposition in the extracellular matrix (5, 6). TGFβ has been directly implicated in the differentiation of invasive and synthetic myofibroblasts from a variety of progenitor cell types, and for the initiation and maintenance of transcriptional programs intrinsic to fibrosis. Lesional skin from patients with SSc shows a signature for high TGFβ signaling that is sustained in isolated dermal fibroblasts maintained in culture (7–10). The mechanism of enforced TGFβ signaling in SSc however remains unknown. Obstacles to progress include the lack of a major defined genetic contribution to the condition, resulting in a lack of animal models that faithfully recapitulate the predisposition for and pathogenesis of human disease. Familial recurrence of SSc is exceedingly rare, and although the susceptibility loci identified by genome-wide association studies are broadly indicative of a general autoinflammatory tendency, they are not specific for SSc (11). Moreover, twin studies have shown minimal heritability for SSc (12). In the absence of a major genetic determinant, it is unclear how SSc fibroblasts sustain elevated TGFβ signaling. A possible explanation for the maintenance of a fibrotic synthetic repertoire in cultured SSc fibroblasts might be the integration of more subtle genetic and environmental influences into a stable predisposing epigenetic state (12). To investigate a potential epigenetic mechanism of SSc, we applied a variety of complementary discovery-based approaches to profile and functionally interrogate primary tissue and cells from patients with this condition.
Results
Autocrine TGFβ2 signaling “locks” ex vivo SSc fibroblasts into a pro-fibrotic state
RNA-sequencing (RNA-seq) analysis revealed that primary dermal fibroblasts derived from lesional skin of patients with diffuse SSc (hereafter called SSc fibroblasts; n=6) showed concordant and stable expression differences when compared to control fibroblasts isolated from healthy donors (n=5). SSc fibroblasts maintained high expression of genes associated with extracellular matrix (ECM) organization and TGFβ signaling (Fig. 1A, fig. S1). Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) confirmed the elevated expression of pro-fibrotic genes, such as type 1 collagen (COL1A1) and the collagen-specific chaperone SERPINH1, in patient-derived primary fibroblast lines (Fig. 1B). SSc fibroblasts also showed elevated mRNA and protein expression of TGFβ2 (but not β1 or β3) (Fig. 1B, 1C). Inhibition of RNA transcription by actinomycin D normalized TGFB2 mRNA expression in SSc fibroblasts, indicating that the heightened amount of TGFB2 mRNA in SSc fibroblasts related to increased transcription (fig. S2). The equivalent expression of the myofibroblast differentiation marker smooth muscle actin (ACTA2) in control and SSc fibroblasts indicated that gene expression changes in SSc fibroblasts reflected differences in the performance of comparable cell types, rather than a transition to myofibroblasts in SSc (Fig. 1B). Of note, we observed no differences in gene expression between control fibroblasts and fibroblasts derived from non-lesional skin of patients with SSc (fig. S3). We detected specific upregulation of TGFB2 mRNA expression in vivo by RT-qPCR of homogenized lesional skin biopsies (Fig. 1D) and in situ mRNA hybridization of sectioned lesional biopsies from patients with diffuse SSc (Fig. 1E, fig. S4, fig. S5). Taken together, these data document that patient fibroblasts from lesional skin actively maintain pro-fibrotic gene expression and transcriptional upregulation of TGFB2 ex vivo.
Fig. 1. Patient-derived SSc fibroblasts maintain a fibrotic synthetic repertoire ex vivo.
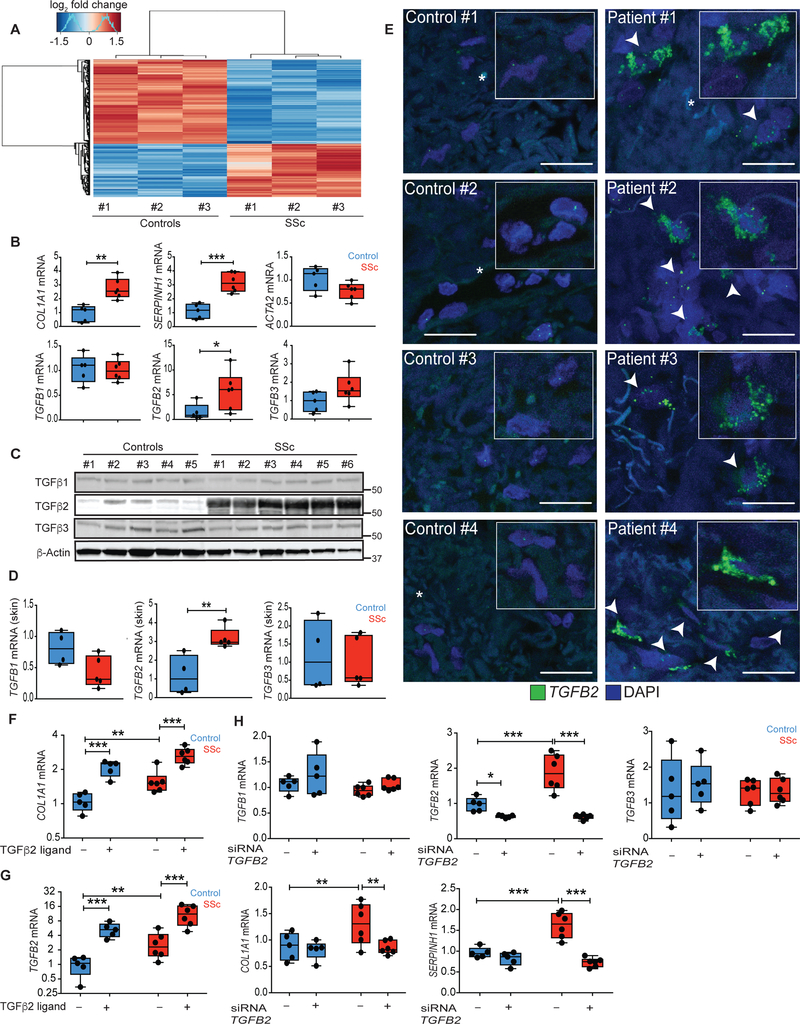
(A) Heatmap of genes differentially expressed (rows) between healthy control and lesional SSc fibroblasts by RNA-seq (n = 3 biological replicates, FDR < 0.05). (B and C) mRNA and protein expression in control and SSc fibroblast lines. (D) mRNA expression in homogenized biopsies taken from diffuse SSc lesional skin (n = 5) versus healthy control skin (n = 4). (E) TGFB2 mRNA expression (green) measured by mRNA in situ hybridization of healthy control skin and lesional SSc skin sections (n = 4). Arrowheads indicate cells with high TGFB2 expression. Scale bars, 20 um. (F and G) mRNA expression upon stimulation of fibroblasts with exogenous TGFβ2 ligand. (H) TGFB ligand and TGFβ target gene expression upon siRNA knockdown of TGFB2 in primary fibroblasts. *P < 0.05; **P < 0.01; ***P < 0.001. ncontrol = 5 and nSSc = 6. Two-way Student t-test for single comparisons, or one-factor ANOVA with FDR correction for all experiments, except panel A and D-TGFB2 mRNA: Mann-Whitney. Black points represent individual biological replicates. Y-axes are varied across similar plots to aid visualization.
Stimulation with exogenous TGFβ2 ligand further amplified the already heightened expression of COL1A1 and TGFB2 in SSc fibroblasts (Fig. 1F, 1G). This result suggested that autocrine TGFβ2 signaling maintains pro-fibrotic gene expression in SSc fibroblasts. To test this hypothesis, we used TGFB2-specific siRNA to knock down expression of TGFβ2 (but not β1 or β3). We observed normalization of COL1A1 and SERPINH1 expression in patient fibroblasts (Fig. 1H), attesting to the pathogenic importance of TGFβ2.
Epigenetic modulation of a discrete TGFβ2 enhancer mitigates pro-fibrotic gene expression in SSc fibroblasts
Given the absence of a strong genetic signature of SSc, we hypothesized that epigenetic events initiate and maintain a pro-fibrotic state in SSc fibroblasts. To test this, we performed Assay for Transposase Accessible Chromatin with high-throughput sequencing (ATAC-seq) to monitor chromatin accessibility in parallel with RNA-seq in control and patient fibroblasts from lesional skin (13). Although there was no global signature for enhanced chromatin accessibility at transcription start site (TSS) regions in SSc fibroblasts compared to controls, the TGFβ signaling pathway was enriched among those genes that showed increased chromatin accessibility in SSc fibroblasts (fig. S6A, S6B).
We hypothesized that SSc fibroblasts exhibited increased chromatin accessibility at the TGFB2 locus. We compared ATAC-seq signals in control and SSc fibroblasts with reference to the proximal promoter or enhancer sequences predicted by ENCODE. We observed no differences in chromatin accessibility at the proximal promoter of TGFB2 between control and SSc fibroblasts (Fig. 2A). However, the TGFB2 locus contained numerous predicted enhancers.
Fig. 2. Epigenetic activation of the TGFB2 enhancer promotes pro-fibrotic gene expression in SSc fibroblasts.

(A) ATAC-seq signals between healthy control and SSc fibroblasts at the TGFB2 locus were compared to ENCODE predictions for proximal promoter (orange) or putative enhancers (green), with the candidate enhancer of interest highlighted on the right. Genome-wide significant peaks at the TGFB2 enhancer are indicated by asterisks. (B) TGFB2 mRNA expression was regressed on estimated chromatin accessibility of the putative enhancer in control and patient fibroblasts. R2, co-efficient for determination of linear regression. (C) Chromatin immunoprecipitation followed by qPCR (ChIP-qPCR) demonstrated H3K27ac or EP300 occupancy at the putative TGFB2 enhancer in fibroblasts. Values are represented as percent input normalized by IgG control. (D) mRNA expression after targeted histone acetylation of the TGFB2 enhancer by co-expression of TGFB2 enhancer-specific guide RNA (sgRNA) and dCas9-EP300 (histone acetyltransferase, dCas9-EP300core) in control and SSc fibroblasts. dCas9-EP300Δ (dCas9-EP300D1399Y) contained a nonfunctional residue substitution at the acetyltransferase domain and was used as a negative control. (E) Targeting of dCas9-KRAB (histone methyltransferase) to the TGFB2 enhancer using TGFB2 enhancer-specific guide RNAs in control and SSc fibroblasts. *P < 0.05; **P < 0.01; ***P < 0.001. ncontrol = 5 and nSSc = 6. Two-way Student’s t-test or one-factor ANOVA with FDR correction were used for all experiments, except for panel C-H3K27ac: Mann-Whitney; panel D-TGFB1 mRNA, panel E-COL1A1 mRNA and panel E-SERPINH1 mRNA: Kruskall-Wallis with FDR correction. Black points represent individual biological replicates. Y-axes are varied across similar plots to aid visualization.
To prioritize the predicted enhancers found near TGFB2, we compared ENCODE predicted enhancer sequences in cell lines that did or did not express the cytokine. We identified a distal cluster of putative enhancers that showed a signature of open chromatin specifically among TGFB2-expressing cell lines (fig. S7). Within this cluster, patients with SSc exhibited higher chromatin accessibility at one of these putative enhancers (Fig. 2A). Three of three patients with SSc contained chromatin accessibility peaks at this putative enhancer with Bonferroni-corrected genome-wide significance (P = 1E-30, P = 1E-24, P = 1E-27), a signal only observed in one of three controls (P = 1E-18). Furthermore, we found a correlation between the extent of chromatin accessibility at the enhancer with TGFB2 mRNA expression in both control and patient cells (Fig. 2B, R2 = 0.78, P = 0.03), a finding not observed for any of the other putative enhancers near the TGFB2 locus (fig. S8).
Consistent with our hypothesis, the putative enhancer in SSc fibroblasts exhibited epigenetic marks of enhancer activity, including elevated acetylation of H3K27 (H3K27ac) and occupancy by the histone acetyltransferase EP300 (Fig. 2C) (14, 15). These marks were not observed in patient fibroblasts derived from non-lesional skin (fig. S9). We observed no difference in tri-methylation of H3K27, a repressive epigenetic mark, at this enhancer between control and lesional SSc fibroblasts (fig. S10). Taken together, these results suggest that the predicted enhancer has functional potential and remains operative in patient fibroblasts.
To assess the function of the putative enhancer, we targeted histone acetyltransferase activity using TGFB2 enhancer-specific guide RNAs with deactivated Cas9 tethered to the histone acetyltransferase domain of EP300 (dCas9-EP300core) (16). Targeted histone acetylation of the putative enhancer specifically induced TGFB2 (but not TGFB1 or TGFB3) expression in both control and patient fibroblasts (Fig. 2D). This occurred in association with elevated COL1A1 and SERPINH1 expression. These patterns were not observed when an inactive form of EP300 (dCas9-EP300D1399Y) was used as a negative control (Fig. 2D). These results indicated that histone acetylation of the putative enhancer was sufficient to induce TGFβ2- and TGFβ-target gene expression. Furthermore, targeting of histone methyltransferase activity to the putative enhancer using deactivated Cas9 tethered to a Krueppel-associated box domain (dCas9-KRAB) was sufficient to normalize TGFB2 mRNA and pro-fibrotic gene expression in patient fibroblasts (Fig. 2E) (17). These results suggest that epigenetic modifications that activate the TGFB2 enhancer contribute to the maintenance of a pro-fibrotic state in SSc fibroblasts. In keeping with this hypothesis, Sanger sequencing did not reveal any patient-specific genetic variation in the TGFB2 enhancer sequence (data file S1).
TGFB2 enhancer activity rebounds after histone acetyltransferase inhibition
We next asked if EP300 inhibition might decommission the TGFB2 enhancer. Treatment of lesional SSc fibroblasts with a small molecule competitive inhibitor of EP300 (SGC-CBP30) decreased TGFB2 expression, EP300 occupancy and H3K27ac at the TGFB2 enhancer (Fig. 3A, 3B) (18). Unexpectedly, however, TGFB2 expression rebounded quickly upon removal of the drug, recapitulating steady-state amounts of TGFB2 expression, EP300 occupancy, and H3K27ac within 5 days after drug removal. These results suggested a mechanism of epigenetic memory that maintains heightened activity of the TGFB2 enhancer in SSc fibroblasts.
Fig. 3. BRD4 and NF-kB maintain epigenetic activation of the TGFB2 enhancer in SSc.

(A and B) TGFB2 expression and epigenetic modification of the TGFB2 enhancer were assayed immediately after treatment with EP300 inhibitor (SGC-CBP30) or after 5 days in fresh media sans SGC-CBP30. (C) ChIP-qPCR for BRD4 occupancy at the TGFB2 enhancer in baseline conditions (left) and in response to SGC-CBP30 treatment (right) in fibroblasts ex vivo. (D) ChIP-qPCR for acetylated p65 (activated NF-kB) occupancy at the TGFB2 enhancer in baseline conditions ex vivo. (E) Chem-seq data revealed specific binding of JQ1, BRD4 and RNA polymerase II at the TGFB2 enhancer highlighted on the right (21). (F) mRNA expression and epigenetic modifications of the TGFB2 enhancer upon siRNA knockdown of BRD4 mRNA in fibroblasts ex vivo. (G and H) Pulse chase analysis of TGFB2 enhancer activity in response to JQ1 or BAY 11–7085 treatment, up to 5 days after drug removal. (I) TGFβ2 protein expression in vehicle or JQ1-treated fibroblasts ex vivo. (J) TGFβ2 target gene expression in cultured control or SSc fibroblasts upon BRD4 inhibition by siRNA knockdown compared to scrambled siRNA control. (K) Collagen and TGFB2 expression upon co-treatment of JQ1 and TGFβ2 ligand in SSc fibroblasts ex vivo. (L) Heatmap of differentially expressed genes between vehicle or JQ1-treated control or SSc fibroblasts by RNA-seq. n = 3, FDR < 0.05. JQ1-treated SSc fibroblasts clustered together with control fibroblasts in principle component analysis (PC1 and PC2 account for 65% and 14% of total variance, respectively). *P < 0.05; **P < 0.01; ***P < 0.001. ncontrol = 5 and nSSc = 6, except for ChIP studies where ncontrol = 4 and nssc = 4. Two-way Student’s t-test for single comparisons or one-factor ANOVA with FDR correction were used for all experiments, except for panel A, panel F-H3K27ac and BRD4 occupancy: Kruskall-Wallis with corrected FDR, and panel D: Mann-Whitney. Black points represent individual biological replicates.
BRD4 and NF-kB enforce epigenetic memory in SSc
BRD4 is a bromodomain and extra-terminal domain (BET) family member that imposes epigenetic memory during mitosis, and activates inflammation-associated enhancers through the pro-inflammatory transcription factor NF-kB (19, 20). In support of the hypothesis that NF-kB and BRD4 activate the TGFB2 enhancer in SSc fibroblasts, ChIP-qPCR revealed higher occupancy of BRD4 and activated NF-kB (acetylated p65; Fig. 3C, 3D) at the TGFB2 enhancer in SSc fibroblasts derived from lesional skin, but not in fibroblasts derived from non-lesional skin of patients compared to controls (fig. S9). We also observed increased NF-kB signaling in SSc fibroblasts, as assessed by co-culturing of control or SSc fibroblasts with a cell line harboring a NF-kB reporter allele (fig. S11). In keeping with a contribution to epigenetic memory, BRD4 occupancy remained higher in lesional SSc fibroblasts than controls throughout SGC-CBP30 treatment (Fig. 3C).
We independently validated these results using Chem-seq to identify genomic regions enriched upon pull-down of DNA-protein complexes that interact with the drug JQ1, a competitive inhibitor that blocks BRD4 and (with lower affinity) other BET family members from binding to acetylated lysine residues (21, 22). Strikingly, JQ1 was specifically bound to the TGFB2 enhancer under consideration in this study, with direct overlap of ChlP-seq signals for BRD4 and RNA polymerase II observed (Fig. 3E).
We hypothesized that if NF-kB partners with BRD4 to activate the TGFB2 enhancer, NF-kB signaling would induce TGFB2 expression. We also reasoned that inhibition of NF-kB or BRD4 would decommission the TGFB2 enhancer in SSc fibroblasts. Indeed, stimulation with tumor necrosis factor α (TNFα, an activator of NF-kB) potently induced TGFB2 expression in control and SSc fibroblasts (fig. S12). Pre-treatment with either an NF-kB inhibitor (BAY 11–7082) or JQ1 suppressed the effects of TNFα and abrogated TGFB2 expression. siRNA knockdown of BRD4 also suppressed TGFB2 mRNA expression in SSc fibroblasts, in addition to H3K27ac and BRD4 occupancy at the TGFB2 enhancer (Fig. 3F, fig. S13).
BRD4 and NF-kB inhibition results in durable TGFB2 enhancer decommission
We also found that JQ1 treatment was sufficient to suppress TGFB2 expression in SSc fibroblasts, an effect that was sustained after drug removal (Fig. 3G). These effects occurred in association with sustained normalization of H3K27ac and BRD4 occupancy at the TGFB2 enhancer. Similar findings were observed upon BAY 11–7082 treatment (Fig. 3H), suggesting that BRD4 and NF-kB stabilize epigenetic activation of the TGFB2 enhancer in SSc fibroblasts.
We next hypothesized that BRD4 inhibition with consequent suppression of TGFβ2 expression would normalize the fibrotic synthetic repertoire of lesional SSc fibroblasts. Indeed, BRD4 inhibition abrogated TGFβ2 protein expression, in association with normalized type 1 collagen expression in SSc fibroblasts (Fig. 3I, 3J). Treatment with TGFβ2 ligand in the presence of JQ1 potently induced collagen expression in SSc fibroblasts, supporting the concept that collagen suppression by JQ1 is secondary to TGFβ2 inhibition (Fig. 3K). TGFβ2 ligand stimulation in the presence of JQ1 (and hence BRD4 inhibition) failed to induce TGFB2 expression. Furthermore, RNA-seq and principle component analyses confirmed that JQ1 treatment mitigated gene expression differences between control and SSc lesional fibroblasts (Fig. 3L).
JQ1 treatment represses collagen synthesis and promotes collagen clearance in SSc skin explants
To test if JQ1 treatment was sufficient to normalize pro-fibrotic gene expression in a more physiologic context, control or SSc lesional skin biopsies were maintained in organ culture in the presence of vehicle or JQ1 for ten days. JQ1 treatment normalized TGFB2 and COL1A1 expression in the dermis of SSc skin explants (Fig. 4). Picrosirius red and Masson’s Trichrome staining revealed reduction of collagen in the superficial dermis of JQ1-treated patient skin (Fig. 5A, fig. S14). These results led us to hypothesize that BRD4 inhibition results in elevated collagenase expression in SSc skin. Notably, TGFβ signaling is known to suppress the expression of matrix metallopeptidase 1 (MMP1), the major type 1 collagenase in the skin (23, 24). We observed potent induction of MMP1 expression in JQ1-treated patient skin (Fig. 5B). In SSc fibroblasts, JQ1 treatment induced MMP1 expression, an effect abrogated by co-treatment with TGFβ2 ligand (fig. S15A). In keeping with these results, siRNA knockdown of TGFB2 increased MMP1 expression in SSc fibroblasts (fig. S15B). Taken together, these data suggest that the potential therapeutic efficacy of BRD4 inhibition in SSc encompasses both suppression of pro-fibrotic gene expression and the clearance of excessive extracellular matrix.
Fig. 4. Organ culture with JQ1 mitigates pro-fibrotic gene expression in SSc skin.
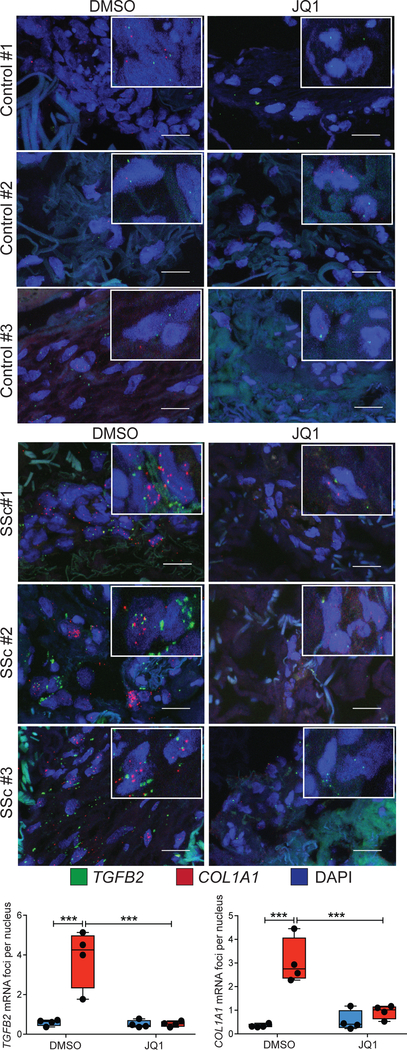
mRNA in situ hybridization for TGFB2 (green foci) and COL1A1 (red foci) in the dermis of healthy control skin and SSc lesional skin biopsies maintained in organ-culture with DMSO or JQ1 for 10 days. Scale bars, 20 um. Total number of fluorescent foci normalized to the number of nuclei was quantified and averaged from three different sections per biological replicate. ***P < 0.001. ncontrol = 4, nssc = 4. One-factor ANOVA with FDR correction was used for all experiments. Black points represent individual biological replicates.
Fig. 5. Organ culture with JQ1 promotes collagen clearance in SSc skin.
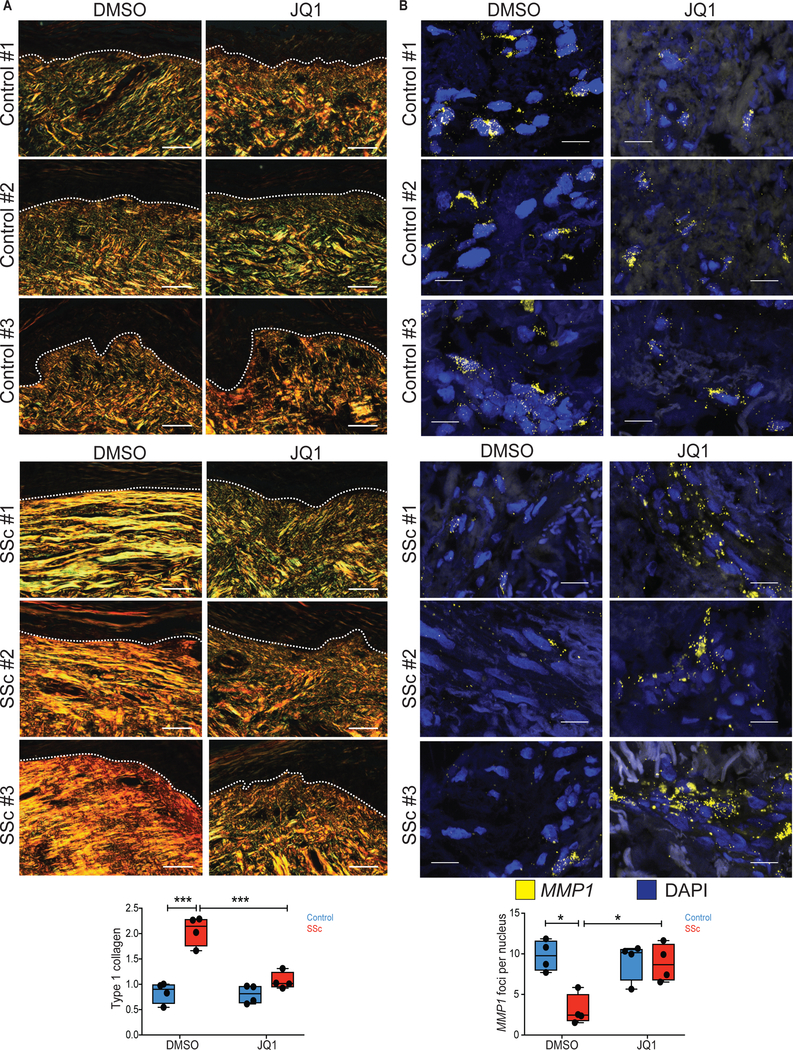
(A) Picrosirius red staining in the dermis of healthy control skin and SSc lesional skin biopsies that were maintained in organ-culture with DMSO or JQ1 for 10 days. Type 1 collagen (yellow/orange fibers) was visualized and quantified based on light birefringence under polarized light. Dashed lines indicate the epidermal-dermal junction. Scale bars, 50 um. (B) mRNA in situ hybridization for MMP1 (yellow foci) in the dermis of control and SSc skin biopsies maintained in organ-culture with DMSO or JQ1 for 10 days. Scale bars, 20 um. Total number of fluorescent foci were normalized by the total number of nuclei and averaged from two different sections per biological replicate. *P < 0.05; **P < 0.01; ***P < 0.001. ncontrol = 4, nssc = 4. One-factor ANOVA with FDR correction was used for all experiments except for panel B: Kruskall-Wallis with FDR correction. Black points represent individual biological replicates.
Discussion
SSc is an etiologically mysterious disease in which previously healthy adults acquire an inflammatory prodrome that progresses to include a strong and unrelenting predisposition for fibrosis of the skin and viscera. Here we show that constitutive epigenetic activation of a newly identified TGFB2 enhancer maintains a pro-fibrotic state in lesional SSc fibroblasts in a mechanism dependent on BRD4 and NF-kB. We found that excessive TGFβ2 production by SSc fibroblasts resulted in heightened collagen deposition not only by inducing COL1A1 expression, but also by stimulating the expression of SERPINH1 (a collagen-specific chaperone) and suppressing the expression of MMP1 (a dominant collagenase in human skin). TGFβ2 production by cells with a permissive epigenetic landscape could further amplify transcription at the TGFB2 locus, plausibly constituting a feed-forward mechanism relevant to the persistence of disease once established.
Analogous to the regulation of inflammatory enhancers initiated by inflammatory mediators like NF-kB and enforced by BRD4 (25, 26), we determined that NF-kB and BRD4 are necessary to maintain activity at the critical TGFB2 enhancer and the fibrotic synthetic repertoire in SSc. NF-kB is a signaling intermediate that is integrated into the inflammatory response to various environmental stimuli previously implicated in SSc such as lipopolysaccharides, toll-like receptor agonists and TNFα (27–29). This supports the hypothesis that the effects of various proposed triggers of inflammation in SSc (for example, microangiopathy, tissue injury, and infectious disease) converge, at least in part, through the activation of NF-kB (30, 31). Thus, this study both elucidates mechanism and suggests a vulnerability for fibrosis in SSc.
The question regarding how SSc patients acquire epigenetic activation of the TGFB2 enhancer remains unanswered. Our data suggest a pathogenic sequence for SSc that initiates with an aberrant inflammatory response and is maintained through epigenetic memory at a specific enhancer for TGFB2. We hypothesize that environmental factors and the genetic pro-inflammatory susceptibility of SSc patients combine to activate TGFB2 enhancer activity (11). It also remains unclear how various autoimmune disorders such as SSc, Grave’s disease, rheumatoid arthritis, systemic lupus, primary biliary cirrhosis and psoriasis are pre-determined in patients who share common susceptibility alleles. The possibility of disease-specific perturbations of epigenetic regulation deserves attention, but remains speculative at this time.
Limitations of this study include the potentially limited pathophysiologic relevance of dermal fibroblasts studied ex vivo. Although we corroborated gene expression patterns observed in SSc fibroblasts in primary tissue, the cellular response to pharmacological inhibitors of BRD4, NF-kB or P300 may differ in vivo, and we had limited ability to explore the chronicity of therapeutic effects in cell or organ culture systems. These experimental systems are also highly simplified and incapable of fully recapitulating paracrine effects, complex microenvironments or the influence of immunologic mediators of disease. Lastly, we documented epigenetic dysregulation of TGFβ2 expression in cell lines or skin explants from diffuse SSc patients. Additional work will be needed to determine if this mechanism extends to other organs systems, subgroups of SSc patients, or presentations of scleroderma.
In light of this study, treatment strategies for SSc that may warrant additional testing include selective TGFβ2, NF-kB and/or BRD4 antagonists. We predict that TGFβ ligand targeting strategies that prioritize the potency of TGFβ2 neutralization will show the greatest efficacy. Additionally, these data may help to predict the contexts within which inhibition of alpha(v) integrin-mediated activation of TGFβ holds potential for the treatment of fibrosis. Notably, whereas both TGFβ1 and TGFβ3 can be activated by integrins, TGFβ2 is refractory due to lack of an RGD sequence in its latency associated peptide (10). Lastly, these results reveal a basic biological mechanism for regulating TGFB2 enhancer activity, and may inform pathogenic mechanisms for other TGFβ2-driven diseases, such as chronic obstructive pulmonary disease (COPD), glioblastoma, and glaucoma (32–34).
Materials and Methods
Study Design.
Our objective was to identify and target potential epigenetic mechanisms regulating pro-fibrotic gene expression in primary dermal fibroblasts of patients with SSc. We hypothesized that SSc fibroblasts maintain transcriptomic differences (particularly of genes related to TGFβ and extracellular matrix synthesis) through an epigenetic mechanism. To test this, we studied skin explants or primary dermal fibroblast lines established from skin biopsies of healthy volunteers or patients with diffuse SSc in vitro. We further characterized SSc fibroblasts using dedicated functional studies (siRNA, dCas9-EP300core, dCas9-KRAB) to support mechanistic conclusions. We tested potential therapeutic applications using fibroblasts or skin explants with the pharmacologic inhibitors for NF-kB, EP300 or BRD4. Power analysis was not used to calculate sample sizes. Once a protocol had been optimized, all experiments were included in the analysis if both control and experimental groups were performed in parallel, and if internal controls were met. No outliers were excluded. All experiments were performed at least twice (fully independent experiments). Technical replicates for each patient were averaged, and each average served as a data point for comparison.
Participants
Healthy control donors and patients with diffuse SSc were recruited from the Johns Hopkins Scleroderma Center (table S1). 4 mm skin biopsies were taken from healthy control volunteers or from the lesional skin of patients with active diffuse SSc. Non-lesional skin biopsies were taken from four patients with diffuse SSc. Biopsies were taken from the forearm and cultured as described previously (10). Autoantibody assays were performed as part of routine clinical care and/or using a commercially available immunoblot platform (EuroImmune, Systemic Sclerosis [Nucleoli] profile). Patients were considered positive for a given autoantibody if they were positive by either method. All skin biopsies and research protocols were performed in compliance with the Johns Hopkins School of Medicine Institutional Review Board after informed consent.
Cell culture
Control and patient fibroblasts were maintained in complete media (DMEM + 10% FCS + Anti/Anti + GlutaMAX).
SGC-CBP30, BAY11–7082, JQ1 pulse-chase analysis.
Control and patient fibroblasts were treated with SGC-CBP30 (2.5 uM, Sigma), BAY 11–7082 (20 nM, Sigma) or JQ1 (0.25 uM, ApexBio) for 72 hours in complete media, followed by incubation in fresh complete media sans inhibitor for 5 days.
TNFα or TGFβ stimulation.
Control and patient fibroblasts were pre-conditioned with BAY 11–7082 or JQ1 for 24 hours prior to TNFα or TGFβ stimulation (10 ng/mL, 12 hours, R&D Systems) in complete media.
RNA isolation, quantitative PCR and RNA-sequencing
RT-qPCR:
Total RNA was isolated using Trizol (Invitrogen) according to manufacturer’s protocols. RNA was reverse transcribed to cDNA using a cDNA synthesis kit (Applied Biosystems). Relative transcript abundances for target genes were quantified using Taqman probes (Applied Biosystems) per manufacturer’s protocol. Relative quantification of each target was normalized to GAPDH transcript abundance using the expression 2-CT / 2-CT(GAPDH).
RNA-sequencing (RNA-seq):
To profile baseline transcriptomic differences, total RNA was isolated from three control fibroblast lines and three SSc fibroblast lines (derived from lesional skin) using Trizol and RNeasy isolation columns (Qiagen) per manufacturer’s protocol. DNA was digested with on-column DNase (Qiagen) treatment. The quality of the RNA used for sequencing was determined using an Agilent 2100 Bioanalyzer; all samples had RNA integrin numbers (RIN) of at least 9.60. mRNA was enriched by poly-A selection, prepped using an Illumina TruSeq mRNA sample preparation kit, and sequenced by Illumina HiSeq 2000. Adapter sequences from one-hundred base-pair paired-end FASTQ reads were trimmed using Trimgalore (https://github.com/FelixKrueger/TrimGalore). Trimmed sequence reads were aligned to hg19 using RSEM (35). Differential gene expression analysis was performed using DESeq2 package (36) with the gene count output from RSEM. The top differentially expressed genes (false discovery rate [FDR] < 0.05) were used to generate heatmaps and for subsequent gene ontology (GO) analyses. To assess the therapeutic efficacy of JQ1, total RNA was isolated from three control fibroblast lines and three SSc fibroblast lines using Trizol after 48 hours treatment with DMSO or JQ1. Samples were processed for RNA-seq as detailed above.
Western blotting
Cultured cells were washed with PBS (Gibco), and lysate was collected in MPER (Thermo Fisher Scientific) with phosphatase inhibitor and protease inhibitor (Roche). Lysate concentration was quantified using a BSA quantification kit (Thermo Fisher Scientific), and 8 ug of protein were loaded per sample onto 10% BisTris gels (Criterion). Western blotting was performed using the Bio-Rad and LiCor Odyssey detection systems. Antibodies used included TGFβ1 (Abcam), TGFβ2 (Abcam), TGFβ3 (Abcam), and β-Actin (Abcam).
siRNA transfection
Cultured cells were transfected with siRNA against TGFB2 (Dharmacon) or BRD4 (Dharmacon) using Dharmafect 1 transfection reagent, followed by mRNA isolation and RT-qPCR at 5 day post-transfection.
ATAC-seq
Nuclei were isolated from three control and three SSc fibroblast lines using ATAC lysis buffer, transposed with Tn5 transposase (Illumina) for 30 minutes, and ligated with barcoded adapter sequences as previously described (13). Library quantitation and a quality check for proper nucleosomal laddering was performed using an Agilent 2100 Bioanalyzer prior to sequencing as previously described (13), and 50 base-pair paired-end FASTQ reads were generated by Illumina HiSeq 2000. Adapter sequences were trimmed from high quality FASTQ reads using TrimGalore (https://github.com/FelixKrueger/TrimGalore) and aligned to hg19 using Bowtie2 using the parameter -X2000 allowing fragments of up to 2 kb to be aligned (37). Duplicate sequence reads were identified and removed using PICARD tools (http://picard.sourceforge.net) with default settings as previously described (13). Remaining reads were filtered for alignment quality greater than Q30. Reads that mapped to mitochondria, unmapped contigs, or the Y chromosome were removed. Sequence reads were subsequently filtered for nucleosome-free reads based on read fragment size limits (0 to 100 bp) (13), followed by quantile normalization. We used MACS2 to call peaks with the parameters (--nomodel --nolambda --keep-dup all --call-summits) (38), a subset of which were removed using the consensus excludable ENCODE blacklist (http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeMapability/). Peaks were mapped to nearest coding genes using bedtools (39),and the height of each peak was used to identify genes with differential chromatin accessibility analysis between control and SSc fibroblasts using the EBSeq package (40).
Genome annotation
Proximal promoter and enhancer predictions:
Browser extensible data (BED) tracks containing predictions for proximal promoters (DNase hypersensitive regions, H3K4me1+ regions) or distal regulatory elements (DNAse hypersensitive regions, H3K27ac+ regions) for ten cell lines (astrocyte, endothelial cell, fibroblast, keratinocyte, epithelial cells, gastric cells, hepatic stellate cell, placental cells, GM12878, HepG2, K562) were downloaded from ENCODE (http://promoter.bx.psu.edu/ENCODE/download.html). Transcript per million (TPM) values for these ten cell lines were also queried using the ENCODE database (http://promoter.bx.psu.edu/ENCODE/search_human.php), and were used to group cell lines as expressors (TGFB2 TPM equal to or greater than 1) or non-expressors of TGFB2 (TGFB2 TPM less than 1).
Chem-seq:
Chem-seq tracks for JQ1 and ChIP-seq tracks for BRD4 and RNApol II were downloaded from GEO accessions GSE44098 and GSE43743, respectively (21), and aligned using Integrative Genomics Viewer (41) to qualitatively identify genomic regions that exhibited signals for JQ1, BRD4 and RNApol II occupancies.
Transfection with dCas9-EP300 and dCas9-KRAB constructs
Cultured cells were transfected with expression constructs encoding dCas9-EP300core, dCas9-EP300D1399Y, or dCas9-KRAB (gifts from Charles Gersbach’s lab, Addgene) with or without guide RNA specifically designed to target the TGFB2 enhancer (designed using Crispr Design, Addgene). Constructs were transfected using Lipofectamine (Invitrogen) in OptiMEM (Gibco) per manufacturer’s protocols. Total RNA was isolated from EP300- and KRAB-transfected cells three and five days after transfection, respectively.
Chromatin immunoprecipitation
Cultured cells were washed with PBS and crosslinked with 1% formaldehyde, followed by cell lysis using SDS buffer. Lysate was sonicated with Bioruptor UCD-200 (Diagenode), followed by incubation with Dynabeads (Invitrogen) conjugated with rabbit-derived antibodies: BRD4 (Bethyl Laboratories), EP300 (Bethyl Laboratories), acetylated p65 (NF-kB), H3K27ac (Abcam) H3K27me3 (Millipore), or isotype control (Abcam). Protein:DNA:bead complexes were washed with RIPA buffer, RIPA+NaCl, LiCl, and TE buffer. protein:DNA complexes were eluted with elution buffer. Reverse crosslinking was performed overnight at 65 degrees Celsius, followed by DNA purification and qPCR. ChIP signal was normalized to total chromatin input (percent input), which was was calculated as 100*2(CTinput-CTtarget). Primers used for TGFB2 enhancer ChIP were AGCCAGTTGAGGAGTTTCACA and AAGCATTTGGTAGTGAGTCATCC (forward and reverse, respectively).
Organ culture
4 mm biopsies taken from healthy controls or the lesional skin of patients with diffuse SSc were cut into two 2 mm pieces. One piece was cultured in DMEM + 10% FCS + Anti/Anti with DMSO, while the other was cultured in DMEM + 10% FCS + Anti/Anti with JQ1 (2.5 uM). After 10 days of treatment, samples were formalin-fixed and paraffin-embedded (FFPE) for histological assessment and in situ hybridization.
mRNA in situ hybridization
FFPE sections of control and SSc skin were de-paraffinized and prepared for in situ hybridization as per manufacturer’s protocol (Advanced Cell Diagnostics). Probes used included TGFB2-C1, COL1A1-C2, and MMP1-C1 (Advanced Cell Diagnostics). Fluorescent images were taken by confocal microscopy. Total numbers of fluorescent foci were quantified by the Spots function of Imaris microscopy imaging software.
NF-kB luciferase reporter cell line co-culture
Control or SSc dermal fibroblast lines were co-cultured with NF-kB luciferase reporter NIH 3T3 stable cell lines (Signosis) in DMEM + 10% FCS for three days. Cells were trypsinized, lysed, and given Firefly substrate (Signosis). Luciferase activity was quantified using BioTek luminometer.
Sanger sequencing
Genomic DNA from three healthy control or three SSc fibroblast lines were isolated using DNeasy Blood and Tissue Kit (Qiagen), and PCR was performed using three primer pairs, which amplified three overlapping amplicons spanning the TGFB2 enhancer. The PCR products were purified using a Gel Extraction Kit (Qiagen) and sequenced using the same primer pairs and a Big Dye terminator kit v3.1 (Applied Biosystems). Linear amplification products were separated in an automated capillary sequencer (Applied Biosystems). Three overlapping amplicons were stitched together using Sequencher software 4.8 to form a continuous DNA sequence for the TGFB2 enhancer. Primer sequences were as follows:
Enhancer part 1 F : TACAAAAGCAGGCAATGAGC
Enhancer part 1 R: CGTCAGAAACCTGGACAACA
Enhancer part 2 F: TTGCTAGCATTGTGTCAGCAC
Enhancer part 2 R: GAGGGGGATATAATGGGAACA
Enhancer part 3 F: AGACCCGGTAAAAGCCAGTT
Enhancer part 3 R: GCCAGGCACAACACAGAATA
Statistical methods
Data are shown as standard box-and-whisker plots with individual data points produced using Prism (Graphpad) software and R. Lower and upper margins of each bar (blue bar: control, red bar: SSc) indicate 25th and 75th percentiles, respectively; the internal line indicates the mean, and whiskers indicate the range. Black points represent individual biological replicates. Heatmaps were generated using R, Homer and Java TreeView. All independent technical replicates for each patient were averaged into a single data point. A two-tailed t-test was used for single comparisons between two groups, and one-factor ANOVA with Benjamini, Krieger and Yeuktieli’s two-stage method for controlling for multiple hypotheses was used for multiple comparisons of normally distributed datasets (confirmed by Shapiro-Wilk normality test, data file S2). For non-normal data, Mann-Whitney tests were used for single comparisons between two groups, and the Kruskal-Wallis test followed by Benjamini, Krieger and Yeuktieli’s two-stage method for controlling for multiple hypotheses was used for multiple comparisons. P-values for ATAC-seq peaks generated by MACS2 statistical package were Bonferroni-corrected for genome-wide significance (p < 6.6 × 10−9). Statistical analyses were calculated using Prism software, DESeq2, EBSeq, MACS2 and R. P < 0.05 and FDR < 0.05 were used as thresholds for statistical significance. All experiments except for RNA-seq and ATAC-seq were repeated at least twice.
Supplementary Material
Acknowledgments:
We thank A. Rosen, H. Bjornsson, J.C. Shin for consultation on experimental design, and S. Cooke and R. Bagirzadeh for technical support.
Funding: These studies were funded by the Scleroderma Research Foundation (H.C.D.), the Howard Hughes Medical Institute (H.C.D.); NIH R01 (H.C.D., D.W., R01AR068379), Staurulakis Family Discovery Fund (A.A.S.), NIH U01 (M.A.B., U01HG009380), and NIH R01 (M.A.B., R01HG007348). The Experimental and Computational Genomics Core at the Johns Hopkins Sidney Kimmel Comprehensive Cancer Center was supported by NIH P30 CA006973.
Footnotes
Competing interests: H.C.D is a consultant for GlaxoSmithKline, and founder and consultant for Blade Therapeutics. A provisional patent titled “Targeted epigenetic therapy against distal regulatory element of TGFB2 expression” has been filed (U.S. Provisional Patent Application No. 62/624,024).
Data and materials availability: The plasmids used for dCas9-mediated epigenome targeting were provided through a material transfer agreement with Addgene. The high-throughput sequencing datasets generated during the current study (RNA-seq, ATAC-seq) have been deposited to the GEO repository (GSE130313).
References and Notes:
- 1.Shah AA, Wigley FM, My approach to the treatment of scleroderma. Mayo Clin Proc 88, 377–393 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denton CP, Khanna D, Systemic sclerosis. The Lancet 390, 1685–1699 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Toledano E et al. , A meta-analysis of mortality in rheumatic diseases. Reumatol Clin 8, 334–341 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Ferri C et al. , Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine (Baltimore) 81, 139–153 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Sonnylal S et al. , Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum 56, 334–344 (2007). [DOI] [PubMed] [Google Scholar]
- 6.Massague J, TGFbeta signalling in context. Nat Rev Mol Cell Biol 13, 616–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ihn H, Yamane K, Kubo M, Tamaki K, Blockade of endogenous transforming growth factor beta signaling prevents up-regulated collagen synthesis in scleroderma fibroblasts: association with increased expression of transforming growth factor beta receptors. Arthritis Rheum 44, 474–480 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Sfikakis PP et al. , Immunohistological demonstration of transforming growth factorbeta isoforms in the skin of patients with systemic sclerosis. Clin Immunol Immunopathol 69, 199–204 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Querfeld C, Eckes B, Huerkamp C, Krieg T, Sollberg S, Expression of TGF-beta 1, - beta 2 and -beta 3 in localized and systemic scleroderma. J Dermatol Sci 21, 13–22 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Gerber EE et al. , Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature 503, 126–130 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farh KK et al. , Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feghali-Bostwick C, Medsger TA Jr., Wright TM, Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum 48, 1956–1963 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Visel A et al. , ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calo E, Wysocka J, Modification of enhancer chromatin: what, how, and why? Mol Cell 49, 825–837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilton IB et al. , Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33, 510–517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thakore PI et al. , Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12, 1143–1149 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conery AR et al. , Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang B, Yang XD, Zhou MM, Ozato K, Chen LF, Brd4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol 29, 1375–1387 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dey A, Nishiyama A, Karpova T, McNally J, Ozato K, Brd4 marks select genes on mitotic chromatin and directs postmitotic transcription. Mol Biol Cell 20, 4899–4909 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anders L et al. , Genome-wide localization of small molecules. Nat Biotechnol 32, 92–96 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Filippakopoulos P et al. , Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brennan M et al. , Matrix metalloproteinase-1 is the major collagenolytic enzyme responsible for collagen damage in UV-irradiated human skin. Photochem Photobiol 78, 43–48 (2003). [DOI] [PubMed] [Google Scholar]
- 24.Bujor AM et al. , Akt blockade downregulates collagen and upregulates MMP1 in human dermal fibroblasts. J Invest Dermatol 128, 1906–1914 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Brown JD et al. , NF-kappaB directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell 56, 219–231 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xiao X et al. , Guidance of super-enhancers in regulation of IL-9 induction and airway inflammation. J Exp Med 215, 559–574 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farina GA et al. , Poly(I:C) drives type I IFN- and TGFbeta-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol 130, 2583–2593 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lawrence T, The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol 1, a001651 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oeckinghaus A, Ghosh S, The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol 1, a000034 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moritz F et al. , Tie2 as a novel key factor of microangiopathy in systemic sclerosis. Arthritis Res Ther 19, 105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson ME et al. , Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PLoS One 10, e0114017 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wotton D, A CREB1-TGFbeta2 self-sustaining loop in glioblastoma. Cancer Discov 4, 1123–1125 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Inatani M et al. , Transforming growth factor-beta 2 levels in aqueous humor of glaucomatous eyes. Graefes Arch Clin Exp Ophthalmol 239, 109–113 (2001). [DOI] [PubMed] [Google Scholar]
- 34.Cho MH et al. , Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir Med 2, 214–225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li B, Dewey CN, RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y et al. , Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Quinlan AR, Hall IM, BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leng N et al. , EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29, 1035–1043 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Robinson JT et al. , Integrative genomics viewer. Nat Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.