

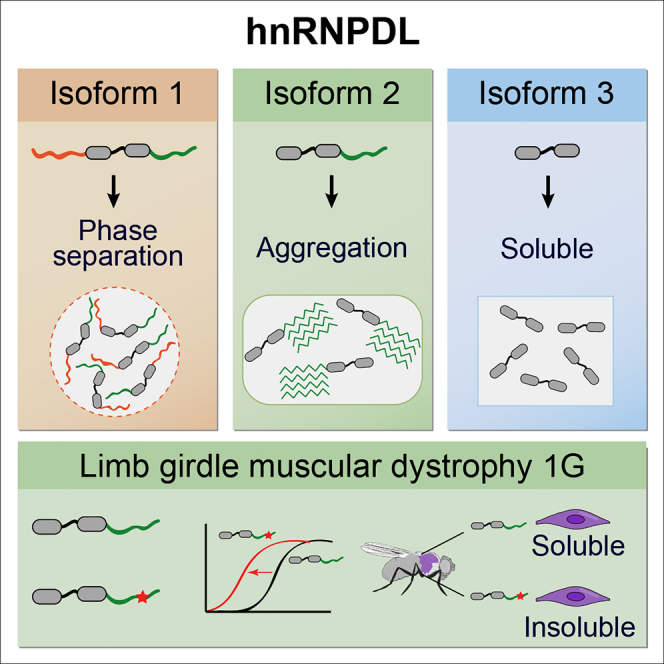
Summary
Prion-like proteins form multivalent assemblies and phase separate into membraneless organelles. Heterogeneous ribonucleoprotein D-like (hnRNPDL) is a RNA-processing prion-like protein with three alternative splicing (AS) isoforms, which lack none, one, or both of its two disordered domains. It has been suggested that AS might regulate the assembly properties of RNA-processing proteins by controlling the incorporation of multivalent disordered regions in the isoforms. This, in turn, would modulate their activity in the downstream splicing program. Here, we demonstrate that AS controls the phase separation of hnRNPDL, as well as the size and dynamics of its nuclear complexes, its nucleus-cytoplasm shuttling, and amyloidogenicity. Mutation of the highly conserved D378 in the disordered C-terminal prion-like domain of hnRNPDL causes limb-girdle muscular dystrophy 1G. We show that D378H/N disease mutations impact hnRNPDL assembly properties, accelerating aggregation and dramatically reducing the protein solubility in the muscle of Drosophila, suggesting a genetic loss-of-function mechanism for this muscular disorder.
Keywords: hnRNPDL, alternative splicing, isoforms, phase separation, aggregation, amyloid, prion-like, disease, mutation, LGMD1G
Graphical Abstract
Highlights
-
•
hnRNPDL requires both N- and C-terminal IDRs to phase separate
-
•
The absence of N-terminal IDR facilitates aggregation
-
•
The unique combination of IDRs in each isoform determines its cellular behavior
-
•
D378N/H mutations accelerate hnRNPDL aggregation and compromise its solubility
Batlle et al. show that alternative splicing controls heterogeneous ribonucleoprotein D-like (hnRNPDL) phase separation, aggregation, and solubility. Mutations that cause LGMD1G accelerate hnRNPDL aggregation and promote insolubility in Drosophila.
Introduction
Eukaryotic cells contain a variety of compartments or organelles with specialized functions. There are membrane-bound organelles like the nucleus or mitochondria, and membraneless organelles (MLOs) such as stress granules or P-bodies (Boeynaems et al., 2018). MLOs are enriched in a peculiar type of polypeptides known as prion-like proteins (March et al., 2016). These polypeptides consist of one or more globular domains with adjacent long intrinsically disordered regions (IDRs) of low complexity. These IDRs are enriched in specific amino acids, such as glutamine, asparagine, serine, glycine, and tyrosine, being similar in composition to the disordered domains of yeast prions, and thus referred to as prion-like domains (PrLDs) (King et al., 2012). Interestingly, prion-like proteins often have the ability to phase separate into liquid droplets and this may contribute to the formation of MLOs in the nucleus or cytoplasm (Boeynaems et al., 2018). MLOs are dynamic structures and their formation is usually reversible, but these assemblies may become irreversible when proteins aggregate within MLOs due to mutations, prolonged stress, or changes in protein concentration. Protein aggregation is linked to the onset of a growing list of human disorders (Harrison and Shorter, 2017). Not surprisingly, increasing evidences indicate a connection between pathological states and MLOs proteins malfunction (Ito et al., 2017). ATX2 (Kato et al., 2019), EWSR1 (Maharana et al., 2018), FUS (Patel et al., 2015), hnRNPA1 (Kim et al., 2013, Molliex et al., 2015), hnRNPA2 (Kim et al., 2013, Ryan et al., 2018), HTT (Peskett et al., 2018), TAF15 (Maharana et al., 2018), Tau (Wegmann et al., 2018), TDP43 (Babinchak et al., 2019), and TIA1 (Mackenzie et al., 2017) are well-characterized proteins involved in the formation of MLOs; their mutation being associated with age-related disorders such as amyotrophic lateral sclerosis (ALS), frontotemporal dementia (FTD), or inclusion-body myopathy (IBM) (Harrison and Shorter, 2017, Ito et al., 2017, Nedelsky and Taylor, 2019). Despite the increasing interest in MLOs, the molecular mechanisms that govern the transition between their functional and pathologic states are still not well understood.
Alternative splicing (AS) is an important mechanism underlying evolution complexity (Baralle and Giudice, 2017). Many MLOs proteins have AS isoforms with unknown functions (Gueroussov et al., 2017). Indeed, AS events are frequent in prion-like proteins, especially at their PrLDs, affecting their ability to establish multivalent interactions and to form higher-order complexes (Gueroussov et al., 2017). AS also alters the phase separation properties of prion-like proteins’ isoforms as observed for FUS protein with or without exon 8 inclusion (Gueroussov et al., 2017).
We focus this study on the heterogeneous ribonucleoprotein D-like (hnRNPDL), an RNA-binding protein displaying AS isoforms, linked to disease and with motifs similar to those associated with liquid-liquid phase separation (LLPS) in well-characterized human prion-like proteins.
hnRNPDL is a highly conserved nuclear RNA binding protein involved in mRNA biogenesis located in the genomic position 4q21 (Kamei et al., 1999). The HNRNPDL gene contains nine exons and eight introns, and three isoforms are produced by AS, named here as hnRNPDL isoform 1 (DL1), hnRNPDL isoform 2 (DL2), and hnRNPDL isoform 3 (DL3) (Figure 1A). DL2 was the first isoform discovered as a JKT41 binding protein 1 (JKTBP1) (Tsuchiya et al., 1998) and it is the predominant isoform in all mouse and human tissues (Akagi et al., 2000). It is 301 amino acids long, constituted by two contiguous canonical RNA recognition motifs (RRMs) and one predicted PrLD at the C terminus, enriched in Gly and Tyr residues. DL1 is a longer isoform of 420 amino acids comprising an additional predicted to be disordered and Arg-enriched domain at the N terminus (Kamei et al., 1999). DL1 expression levels are 4-fold lower than those of DL2 and the transcript is mainly present in brain and testis (Akagi et al., 2000). DL3 is the shorter and minor isoform, with only 244 amino acids missing both N and C terminus disordered regions (Kawamura et al., 2002).
Figure 1.

LLPS Propensity of hnRNDPL Isoforms
(A) Schematic diagram of hnRNPDL isoforms as SUMO fusion constructs. RNA recognition motifs (RRMs; blue) according to Pfam (El-Gebali et al., 2019), Arg-rich (orange), and Tyr-rich (green) IDR spliced regions according to Uniprot (Bateman et al., 2015) and their respective amino acid splicing positions are shown. hnRNPDL nuclear localization signal (NLS) sequence is as described in Kawamura et al. (2002).
(B) SUMO-hnRNPDL isoform 1 (DL1) LLPS at different protein concentrations in 50 mM HEPES pH 7.5 and 150 mM NaCl.
(C) 50-μM DL1 LLPS at different salt concentrations.
(D) 50-μM SUMO-DL2 and SUMO-DL3 LLPS in 150 mM salt with or without the presence of 10% Ficoll, and 50 mM salt.
(E) LLPS diagram of hnRNPDL isoforms in the absence of crowding agent. Green circles indicate positive and red diamonds indicate negative for the appearance of droplets at the indicated NaCl/protein concentration combinations.
hnRNPDL bears a 25-residue C-terminal PY nuclear localization signal (NLS) and its transport is mediated by the M9-transportin-1 (TNPO1) pathway (Kawamura et al., 2002). Interestingly, only DL2 and DL3 are able to shuttle between the cytoplasm and the nucleus, whereas DL1 remains strictly nuclear. hnRNPDL isoforms share the same shuttling sequence, except for DL3, which misses four residues that are not required for TNPO1 interaction (Figure 1A; Kawamura et al., 2002). Therefore, the basis of this differential translocation is unknown, but it may indicate hnRNPDL isoforms playing different roles in cells.
HNRNPDL constitutes one of the four genes present in the smallest deletion of the 4q21 microdeletion syndrome, being associated with growth retardation and hypotonia (Hu et al., 2017). Moreover, hnRNPDL expression is upregulated in different types of cancers, such as prostate cancer, chronic myeloid leukemia, colon cancer, and hepatocellular carcinoma (Zhou et al., 2014, Liu et al., 2007, Wu et al., 2008, Zhang et al., 2018). Finally, genome sequencing of Brazilian, Chinese, Uruguayan, and Argentinian families affected by limb-girdle muscular dystrophy 1G (LGMD1G, or LGMDD3 in the new nomenclature; Straub et al., 2018) detected D378N and D378H point substitutions in HNRNPDL, indicating a mutation hotspot for this disease (Berardo et al., 2019, Sun et al., 2019, Vieira et al., 2014). LGMD1G is an autosomal dominant inherited subtype of LGMD, the fourth most common muscular dystrophy, characterized by progressive weakness of hip- or shoulder-girdle muscles (Liewluck and Milone, 2018, Nigro and Savarese, 2014).
Interestingly, hnRNPDL D378N/H point mutations reside in the PrLD of hnRNPDL (Navarro et al., 2015). Disease-causing mutations in PrLDs have been discovered in other prion-like proteins, for example in hnRNPA1 and hnRNPA2 proteins (Kim et al., 2013), in both cases involving the replacement of a single Asp residue, as in hnRNPDL. These mutations have been reported to increase protein aggregation and result in the formation of cytoplasmic inclusions in patients (Harrison and Shorter, 2017, Kim et al., 2013, Mackenzie et al., 2017). Immunohistochemical analysis of LGMD1G patients identified nuclear condensates of hnRNPDL co-localizing with TNPO1 (Vieira et al., 2014). Congophilic deposits have however not been detected in LGMD1G patients and it remains unknown whether hnRNPDL mutations impact the protein aggregation propensity.
In this study, we show how AS results in hnRNPDL isoforms with dramatically different self-assembling properties in vitro and in vivo. We also demonstrate how Arg and Tyr residues, segregated in two distant IDRs in hnRNPDL, act as crucial determinants for both LLPS and aggregation. This spatial segregation of multivalent interacting residues explains how AS controls the propensity to form high-order intranuclear assemblies in mammalian cells, likely accounting for the different shuttling properties of the hnRNPDL isoforms. Finally, we confirm that, as in other prion-like proteins, hnRNPDL disease-causing mutations accelerate protein aggregation, resulting in completely insoluble variants when expressed in Drosophila muscle.
Results
hnRNPDL Alternative Splicing Isoforms Display Different Phase Separation Behavior
hnRNPDL has been shown to undergo LLPS in vitro (Wang et al., 2018). To evaluate the molecular determinants that govern this process, we took advantage of the different domain architectures of hnRNPDL isoforms and we tested their propensity to undergo phase transition. We expressed and purified hnRNPDL isoforms as fusions with solubility-enhancing His-SUMO tags (Figure 1A), hereinafter referred to as DL1, DL2, and DL3. As expected, the three hnRNPDL isoforms bear distinct LLPS propensity (Figures 1B–1E).
The DL1 isoform, containing Arg- and Tyr-enriched IDRs at the N- and C terminus, respectively, displays the strongest tendency to phase separate (Figure 1E). Upon salt dilution, the solution becomes turbid spontaneously, demixing from an aqueous phase to form liquid-like protein droplets under physiological ionic strength (Figure 1B). DL1 undergoes LLPS in a protein concentration-dependent manner in the absence of any crowding agent even at low protein concentrations (2 μM). For a given protein concentration, phase separation is enhanced with decreasing ionic strength (Figure 1C). DL2, missing the Arg-enriched disordered domain at its N terminus, also can undergo phase separation, but its propensity is much lower than that of DL1 (Figure 1E). At physiological ionic strength, DL2 requires the presence of a crowding agent (10% Ficoll) to phase separate (Figure 1D). When transferred to low ionic strength (50 mM NaCl), DL2 rapidly coalesces into micron-sized spherical structures without any crowding agent, but this process requires higher protein concentrations than for DL1 (Figures 1D and 1E). The fact that, in the absence of a crowding agent, DL2 only forms droplets at low ionic strength suggests that its LLPS depends on electrostatic interactions. Indeed, the C-terminal IDR is predicted to behave as a weak polyampholyte, a kind of molecule that displays reduced LLPS as the salt concentration increases (Das and Pappu, 2013). DL3, the isoform devoid of any IDRs, does not phase separate in any tested condition, neither at 50 mM NaCl nor after crowding agent addition (Figures 1D and 1E).
The radii of hydration of hnRNPDL isoforms were analyzed by dynamic light scattering (DLS) at physiological salt concentration without any crowding agent (Figure S1). The results correlated with the propensity to undergo LLPS (Figure 1E), with DL1 forming assemblies with a radius of hydration > 1,000 nm, which could not be observed in DL2 and DL3 proteins, that displayed average radii of hydration of 4.5 and 3.5 nm respectively, compatible with a monomeric state.
These data indicate that the absence of the N-terminus Arg-enriched IDR of hnRNPDL reduces LLPS propensity and the absence of both the N- and C-terminus IDRs completely abolishes LLPS, suggesting that these domains are required for hnRNPDL self-assembly. The results provide experimental support for the hypothesis that AS controls LLPS in hnRNPDL (Feng et al., 2019).
Interactions between Arg and Tyr Residues of DL1 IDRs Promote Its Phase Separation
Recent studies have reported the importance of Arg-rich domains in providing intermolecular interactions that contribute to LLPS (Boeynaems et al., 2017). Moreover, Arg residues have been shown to establish interactions with Tyr residues in FUS (Bogaert et al., 2018, Vernon et al., 2018, Wang et al., 2018). In the previous section, we have shown how the absence of the Arg- and Tyr-rich IDRs in hnRNPDL protein affects LLPS behavior. Therefore, we hypothesized that these residues might be interacting and promoting hnRNPDL phase separation. To confirm this hypothesis, we generated three hnRNPDL variants: (1) all N-terminus Arg mutated to Lys (R/K), (2) all C-terminus Tyr mutated to Phe (Y/F), and (3) both Arg to Lys and Tyr to Phe mutations (R/K+Y/F) (Figure 2A). These mutations were designed to maintain the aromatic and basic character of Tyr and Arg, respectively, while preserving the ability to establish π-cation contacts. The identity of the basic and positive residues determines the interaction strength, with Lys-Tyr, Arg-Phe, and Lys-Phe contacts all being weaker than Arg-Tyr (Wang et al., 2018). Therefore, these protein variants would allow us to evaluate the role of Arg and Tyr interaction strength in hnRNPDL LLPS.
Figure 2.

DL1 Mutants LLPS Behavior
(A) Schematic diagram of DL1 mutants as SUMO fusion constructs: (1) all N-terminus Arg mutated to Lys (R/K); (2) all C-terminus Tyr mutated to Phe (Y/F); (3) both Arg to Lys and Tyr to Phe mutations (R/K+Y/F); and (4) N-terminus (Nt) IDR alone.
(B) LLPS of DL1 and mutants at 50 μM in 50 mM HEPES pH 7.5 and 150 mM NaCl.
(C) LLPS of DL2 green labeled mixed with hnRNPDL-Nt red labeled, in a 1:1 ratio, at different protein concentrations and 150 mM salt.
The three hnRNPDL mutants showed a clear reduction of LLPS compared to DL1, being unable to phase separate at physiological conditions without crowding agents (Figures 2B and S2A). This supports the idea that hnRNPDL phase separation relies on the complementarity of Tyr and Arg contacts (Brady et al., 2017) and not on generic π-cation interactions, as observed also for FUS protein (Bogaert et al., 2018, Wang et al., 2018).
The reduced LLPS propensity of the R/K mutant (Figure S2A) is similar to that of the DL2 isoform (Figure 1E). The C terminus in DL2 is sufficient to promote LLPS, likely through Tyr-Tyr interactions (Burke et al., 2015, Murthy et al., 2019, Wang et al., 2018), but its ability to undergo LLPS under physiological ionic strength is weaker than that of DL1 (Figure 1E). To further confirm that the C-terminus Tyr-rich domain and the N-terminal Arg-rich domain can indeed form interactions responsible for the high LLPS propensity of DL1, we designed a construct consisting only of the Arg-rich domain of DL1 (hnRNPDL-Nt) (Figure 2A). hnRNPDL-Nt was unable to phase separate by itself (Figures 2B and S2A). However, when hnRNPDL-Nt was mixed with DL2 at a 1:1 ratio, we completely recover LLPS under physiological conditions and DL2 phase separated at concentrations as low as 6.25 μM in the absence of any crowding agent (Figure 2C).
The above results indicate that DL1 LLPS is likely governed by intermolecular interactions between the Arg residues at the N-terminal domain and Tyr residues at the C terminus.
DL1 Forms High-Order Complexes in the Nucleus of Mammalian Cells
In the previous sections, we observed how hnRNPDL isoforms bear different self-assembly propensities in vitro. hnRNPDL is located in the cell nucleus; consequently, we addressed their properties in the nuclear context. To this aim, we examined the isoforms localization in a HeLa hnRNPDL knockout (KO) cell line (HeLaDL-KO) (Figure S3A). Cells were transfected with EGFP-tagged fusion constructs or EGFP alone, as control, and immunostained with anti-G3BP antibody as a cytoplasmic marker (Figure 3A).
Figure 3.

Cellular Localization and Mobility of hnRNPDL Isoforms in Mammalian Cells
(A) Cellular localization by immunofluorescence of EGFP-DL1, EGFP-DL2, EGFP-DL3, and unfused EGFP after expression in HeLaDL-KO. Cells were stained with G3BP antibody (red) as cytoplasmic marker and DAPI (blue) as nuclear marker.
(B and C) Graph of normalized fluorescence intensity (B) and rate of recovery average (C) after FRAP in HeLaDL-KO cells expressing EGFP-DL1, EGFP-DL2, and EGFP-DL3 and unfused EGFP (∗∗∗p value < 0.0001).
(D) Example of a size exclusion chromatography elution pattern after individual EGFP-hnRNPDL isoforms expression in HeLaDL-KO cells. Elution volumes of EGFP-DL1 (74 kDa), EGFP-DL2 (61 kDa), and EGFP-DL3 (55 kDa) are indicated by arrows. Letters along the x axis indicate the elution volumes upon column calibration: A, column void volume; B, ferritin (440 kDa); C, aldolase (158 kDa); D, conalbumin (75 kDa); and E, ovalbumin (44 kDa).
All hnRNPDL isoforms are nuclear (Figure 3A). Both DL1 and DL2 are distributed throughout the nucleoplasm, but excluded from the nucleolus (Figure S4A). In contrast, DL3 was completely diffuse in the nucleus, suggesting that the hnRNPDL C-terminus IDR, the only region present in DL1 and DL2 and absent in DL3, might determine the intranuclear compartmentalization of the isoforms. DL1 and DL2 undergo LLPS in vitro, but they do not show nuclear puncta indicative of MLO formation. This behavior might be caused by the high RNA concentration in the nucleus, since high RNA/protein ratios have been shown to diminish LLPS in other prion-like RNA binding proteins, such as TDP43 and FUS (Maharana et al., 2018). In order to verify this idea, we added increasing concentrations of total RNA to DL1 in vitro and we observed a clear decrease in its LLPS propensity (Figures S2B and S2C). RNA completely dissolved DL1 droplets at 100 ng/μL (Figure S2B). This suggests that the nucleic acid titrates DL1 from the droplets. Accordingly, the droplets reappeared after RNase addition (Figure S2B).
We conducted fluorescent recovery after photobleaching (FRAP) analysis of a small region of the nucleus of HeLaDL-KO cells for all EGFP-tagged hnRNPDL isoforms. FRAP analysis showed that the three isoforms associate and dissociate within the nucleus on different timescales of seconds (Figure 3B). The fluorescence recovery half-times after photobleaching were 0.25, 0.84, and >7.0 s for DL3, DL2, and DL1, respectively (Figure 3C). The low mobility of DL1, relative to DL2 and DL3, suggests that it might be involved in the formation of larger or more stable complexes within the nucleoplasm. The DL2 and DL3 mobilities are also significantly different, indicating that these isoforms are also associated with nuclear complexes that differ in identity or stability, although the assemblies that they form are likely smaller and more dynamic than the ones formed by DL1.
To confirm that DL1, DL2, and DL3 could be involved in the formation of different complexes, we performed size exclusion chromatography (SEC) of individual HeLaDL-KO cellular extracts after transfection with each of the different constructs (Figures 3D and S5). The DL1 isoform elution pattern differed significantly from that of DL2 and DL3, the protein being eluted in the void volume of the column corresponding to molecular size complexes larger than 5 million Da. Instead, DL2 and DL3 were eluted in volumes consistent with them being in their monomeric form. In order to prove that the EGFP-tag does not alter the elution pattern of the isoforms, the retention of endogenous hnRNPDL in a HeLa wild-type (WT) cell line was analyzed (Figure S5). The elution profile of endogenous DL2, the predominant isoform, is similar to that of EGFP-DL2. The levels of endogenous DL1 and DL3 are too low to be detected in this experiment.
These results correlate well with the observed hnRNPDL isoforms in vitro self-assembly behavior. The presence of both hnRNPDL IDRs confers an increase in multivalency, enhancing protein-protein or protein-nucleic acids interactions and, consequently, the formation of larger or more stable complexes in the nucleus as reported also for other proteins (Gueroussov et al., 2017, Ying et al., 2017).
Transcription Inhibition Affects hnRNPDL Nuclear Localization
hnRNPs are predominantly located in the cell nucleus. Some hnRNPs, for example hnRNPA1, are known to shuttle between the nucleus and cytoplasm in a transcription-dependent manner, but others do not, for example hnRNPC (Dreyfuss et al., 2002, Piñol-Roma and Dreyfuss, 1991, Piñol-Roma and Dreyfuss, 1992). Actinomycin D (ActD) is an anti-tumor chemical that inhibits transcription by intercalating into transcriptionally active regions, resulting in a significant reduction in RNA synthesis in the nucleus (Su et al., 2013). This treatment is usually performed to determine whether protein localization is dependent on active transcription, its inhibition resulting in protein translocation to the cytoplasm (Bounedjah et al., 2014, Piñol-Roma and Dreyfuss, 1991, Piñol-Roma and Dreyfuss, 1992, Zhang et al., 2005). Interestingly, in contrast to untreated cells (Figure 3A), after 5 μg/ml ActD treatment for 3 h, hnRNPDL shows different transport depending on the considered isoform. Only the DL2 and DL3 isoforms were translocated to the cytoplasm indicating nuclear import that is dependent upon active transcription; meanwhile, the DL1 isoform remained nuclear (Figure 4), consistent with previous observations (Kamei and Yamada, 2002, Kawamura et al., 2002). The three isoforms bear the same shuttling sequence (Figure 1A) and, thus, in principle, they could be transported in the same manner. In the previous section, we showed that DL1 forms higher- or more-stable-order complexes (Figures 3B–3D), larger than 5 million Da in the nucleus, suggesting extensive protein-protein or protein-nucleic acid interaction networks. This observation could provide an explanation for DL1 nuclear retention, which ultimately would depend on the presence of both IDRs.
Figure 4.

Transcription Inhibition Effects on hnRNPDL Localization
EGFP-DL1, EGFP-DL2, and EGFP-DL3 and unfused EGFP localization after their expression in HeLaDL-KO cells and 5 μg/ml Actinomycin D (ActD) treatment for 3 h. Cells were stained with G3BP antibody (red) as cytoplasmic marker and DAPI (blue) as nuclear marker.
ActD treatment disrupts the nucleolus (Figure S4B) and relocates DL1 and DL2 isoforms within the nucleus, now exhibiting a speckled pattern (Figure 4). Interestingly, DNA staining with DAPI also showed nuclear puncta, but they do not colocalize with DL1 and DL2 foci. Indeed, the DL1 and DL2 foci tend to coincide with nuclear areas exhibiting poor DAPI staining, suggesting that high local DNA concentrations might prevent DL1 and DL2 foci formation. In contrast to DL1 and DL2, DL3 remains diffusely distributed in the nucleus. Therefore, we can ascribe the observed DL1 and DL2 localization patterns in the absence of ActD to the presence of the C-terminal Tyr-rich IDR. Similar observations have been reported for FUS and TAF15 proteins (Marko et al., 2012, Zinszner et al., 1997).
The data show that only DL2 and DL3 isoforms can shuttle between nucleus and cytoplasm after transcription inhibition, whereas only DL1 and DL2 form nuclear foci. This indicates that the unique combination of IDRs in each particular variant is an important determinant of its localization.
hnRNPDL Alternative Splicing Isoforms Have Different Aggregation Propensity
Prion-like proteins are well known for their aggregation propensity, usually mediated by their PrLDs (March et al., 2016). hnRNPDL isoforms were highly insoluble in bacteria, which is why we studied them as His-SUMO fusions. Once purified, we proceeded to analyze their in vitro aggregation properties (Figure 5). We used the amyloid-specific dye Thioflavin-T (ThT) (Biancalana and Koide, 2010) to follow the kinetics of hnRNPDL isoform aggregation at 50-μM protein concentration, 150 mM NaCl, and 37°C with agitation. Interestingly, after 2 days DL2 already exhibited a ThT signal in the plateau phase, DL1 started to bind ThT only after 3 days, and DL3 did not show any ThT binding after 4 days (Figure 5A). We also used the alternative amyloid-specific dye Congo Red (CR) to confirm the presence of amyloid-like assemblies (Wu et al., 2012). The absorbance spectra of CR shifts in the presence of amyloid aggregates, and in agreement with the ThT results, only the DL2 isoform promoted this red shift in CR spectrum after 4 days (Figure 5B). These results suggest that, from the three hnRNPDL isoforms, only DL2 displays significant amyloid aggregation propensity. In fact, the DL2 aggregates also bind the amyloid dyes Thioflavin-S (ThS) and Proteostat, and exhibit CR birefringence under polarized light (Figure S6A). Finally, we observed by transmission electron microscopy (TEM) the morphological features of the three hnRNPDL isoforms after 4 days. Negative staining indicated that DL2 formed typical amyloid fibrillary structures without any significant accumulation of amorphous material, DL1 forms amorphous aggregates, and DL3 formed only small aggregates (Figure 5C).
Figure 5.

hnRNPDL Isoform Aggregation Propensity
Evaluation of Thioflavin T (ThT) binding over time (A), Congo Red (CR) binding (B), and transmission electron microscopy (TEM) (C) at final aggregation time point of 50-μM SUMO-hnRNPDL isoforms in 50 mM HEPES pH 7.5 and 150 mM NaCl. Aggregation was conducted at 37°C and 600 rpm. Scale bars of TEM images in (C) represent 400 nm.
These results suggest that the Tyr-rich IDR, including a predicted PrLD (Figure 6A), is responsible for hnRNPDL self-assembly into ordered amyloid-like structures, but also that this reaction only occurs in the absence of the positively charged N-terminal IDR, which would act as a kind of intramolecular chaperone. The data correlate with the relative solubility of the endogenous or transfected isoforms in HeLa WT or hnRNPDL KO cells, respectively, DL2 being the more insoluble of the three variants (Figure S3).
Figure 6.

DL2 Disease-Causing Mutation Aggregation Propensity
(A) Schematic diagram of DL2, as a SUMO fusion construct. RRMs (blue) according to Pfam (El-Gebali et al., 2019) and PLAAC (Lancaster et al., 2014) predicted PrLD (light green) are indicated. The Tyr-rich IDR (dark green) and the disease-causing mutations (red) within the PrLD are shown with their respective amino acid positions.
(B–E) LLPS behavior (B), Thioflavin T (ThT) binding over time (C), transmission electron microscopy (TEM) at 24 h and 4 days (D), and Congo Red (CR) binding (E) at final aggregation time point of 50-μM DL2 and the disease-causing mutations D259N and D259H (DL2N and DL2H) in 50 mM HEPES pH 7.5 and 150 mM NaCl. Aggregation was conducted at 37°C and 600 rpm. Scale bars of TEM images in (D) represent 400 nm.
Disease-Causing Mutations Accelerate hnRNPDL Aggregation
Disease-causing mutations in PrLDs are common in prion-like proteins and they have been linked with an acceleration of the aggregation kinetics (Harrison and Shorter, 2017). Mutation of a specific Asp residue in the PrLDs of hnRNPA1 and hnRNPA2 proteins mapping to evolutionarily conserved regions of these IDRs are linked to ALS or MSP (Kim et al., 2013). The conserved Asp is involved in destabilizing electrostatic interactions and the removal of repulsion by mutation seems to be responsible for the increased propensity of the mutated PrLDs to self-associate and aggregate. Mutation of Asp378 of hnRNPDL to either Asn or His has been associated with LGMD1G (Berardo et al., 2019, Sun et al., 2019, Vieira et al., 2014). As in the case of hnRNPA1 and hnRNPA2, this Asp maps at the PrLD of hnRNPDL (Figure 6A; Navarro et al., 2015) and it is strictly conserved in vertebrates (Figure S7). Therefore, we examined how these mutations affect LLPS and the aggregation of hnRNPDL protein.
DL2 is reported to be the predominant isoform in tissues (Akagi et al., 2000), as observed by western blot of HeLa WT cells (Figure S3). Moreover, in the previous section we showed that DL2 is also the isoform with higher amyloid potential (Figure 5). Consequently, we focused the study on Asn and His mutants of the DL2 isoform (DL2N and DL2H) located at position D259; this position corresponds to D378 in DL1 because DL2 lacks the first 119 amino acids (Figure 6A).
We first checked how DL2 mutations affect LLPS behavior under physiological salt conditions (Figure 6B). In the absence of Ficoll, DL2H did not show detectable structures by light microscopy, whereas in DL2N small irregularly shaped and clustered structures were observed. In the presence of Ficoll, DL2H showed aggregated particles, whereas DL2N shows liquid droplets morphology similar to DL2 WT. These results indicate that the mutations in DL2 PrLD affect its LLPS propensity.
Comparison of DL2 mutants’ in vitro aggregation propensity over time with that of the WT isoform confirmed the impact of the Asp mutation on aggregation. Both DL2 mutants exhibited faster aggregation kinetics, as monitored by ThT, displaying a shorter lag phase and reaching the plateau phase significantly before than the WT form (Figure 6C). Accordingly, TEM analysis indicates that DL2H and DL2N proteins have already assembled into amyloid fibrils after 24 h, whereas the DL2 WT remains protofibrillar (Figure 6D). This is in agreement with the predictions of ZipperDB, a structure-based threading algorithm that scores six-amino acid-sequence stretches according to their propensity to form “steric zippers” in the spine of amyloid fibrils (Goldschmidt et al., 2010). ZipperDB predicted higher steric zipper propensity for DL2H and DL2N, relative to DL2 WT (Table S1). The AMYCO algorithm, which evaluates the impact of mutations on the aggregation of prion-like proteins (Iglesias et al., 2019), also predicts increases in aggregation propensities of 7% and 10% for the DL2H and DL2N variants, respectively.
The acceleration of the aggregation reaction in the mutant variants resulted in smaller and less-ordered amyloid fibrils at the end of the reaction (4 days), as observed by TEM (Figure 6D), which displayed a lower, but still significant, binding to CR than DL2 WT fibrils (Figure 6E). The amyloid-like nature of the DL2H and DL2N aggregates was further confirmed by staining with ThS, Proteostat, and by CR birefringence (Figure S6A).
The secondary structure content of the aggregates was assessed using attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR) in the amide I region of the spectrum (1,700–1,600 cm−1). Deconvolution of the FTIR absorbance spectra of DL2 WT, DL2H, and DL2N samples indicated the presence of a major band at 1,628 cm−1, which can be assigned to the presence of an intermolecular β sheet (Figure S6B). This component accounts for 25%–26% of the spectral area in the three cases (Table S2), which sharply coincides with the proportion of residues mapping at the PrLD in the respective DL2 constructs (25.8%). No major secondary structure differences were observed between the three amyloid assemblies, indicating that, in vitro, the impact of the mutations is mostly kinetic.
Overall, these results, provide evidences that mutation of Asp378 in hnRNPDL PrLD (position 259 in DL2) increases its aggregation propensity, as previously described for hnRNPA1 and hnRNPA2.
Disease-Causing Mutations Are Located in the Insoluble Fraction of Drosophila Muscle
To evaluate the effect of disease-causing mutations in hnRNPDL in vivo, we generated transgenic Drosophila expressing WT or mutant forms of human DL2 by using the UAS/GAL4 system. Multiple transgenic lines expressing a single copy of DL2 WT or mutant (DL2N or DL2H) were generated by site-specific ϕC31integrase-mediated transgenesis into Drosophila chromosome 3. This approach permits equal levels of expression between independent lines, as demonstrated empirically for the five independent lines expressing WT DL2 (Figure 7A). However, we observed that fly lines expressing mutant forms of DL2 (DL2N or DL2H) consistently exhibited lower levels of DL2 protein compared to flies expressing WT DL2 (Figure 7A).
Figure 7.

hnRNPDL Isoform 2 Disease-Causing Mutation Effects in Drosophila
(A) Expression levels of DL2 and disease-causing mutations (DL2N and DL2H) in transgenic flies. Thoraxes of adult flies were processed for western blot analysis with an antibody against hnRNPDL. Actin was blotted as a loading control.
(B) Adult flies were dissected to expose the dorsal longitudinal indirect fly muscle (DLM) and stained with Alexa Fluor 647-phalloidin (purple), hnRNPDL (red), and DAPI (blue).
(C) Thoraxes of adult flies were dissected. Sequential extractions were performed to examine the solubility profile of hnRNPDL.
Expression of either WT or mutant (DL2N or DL2H) forms of DL2 in Drosophila indirect flight muscle led to mild degeneration, showing disorganized muscle fibers as demonstrated by phalloidin staining (Figure 7B). Immunohistochemical analysis showed that both WT and mutant forms of DL2 localized predominantly to myonuclei (Figure 7B). Although we did not observe clear differences between WT and mutant proteins in their localization patterns, we did observe that the solubility of DL2 protein was strongly impacted by disease-causing mutations. Specifically, mutant DL2 (D259H or D259N) proteins were largely recovered from the detergent-insoluble fraction, whereas the WT DL2 protein is found both in the detergent-soluble and detergent-insoluble fractions (Figure 7C). This result is consistent with in vitro data that indicate enhanced aggregation propensity by disease-causing mutations (Figure 6C). The lower levels of DL2 mutant proteins relative to the WT protein in fly muscle might respond to an attempt of the proteostatic machinery to degrade misassembled insoluble species.
Discussion
In this work, we first characterized the molecular properties that govern the self-assembly of hnRNPDL isoforms in vitro. The three AS isoforms exhibit different LLPS propensities according to their IDRs composition: DL1, with both Arg-rich and Tyr-rich IDRs, displays a strong LLPS behavior; DL2, with only the Tyr-rich IDR, has a mild LLPS propensity, requiring a crowding agent to phase separate at physiological ionic strength; and DL3, with none of the IDRs, does not phase separate. Protein self-assembly driving forces can be mediated by π-cation, π-π, hydrophobic, electrostatic, dipole-dipole, or hydrogen bonding interactions (Boeynaems et al., 2018). Recently, it was described that FUS LLPS was governed by interactions between Tyr residues of the PrLD and Arg residues of the RNA binding domain (Wang et al., 2018). We hypothesized that hnRNPDL LLPS would follow the same mechanism than FUS, where Tyr residues from the C terminus and Arg residues from the N terminus would mediate interactions leading to phase separation. We experimentally validated this hypothesis generating hnRNPDL variants that maintain the aromatic and cationic residue content but are predicted to establish weaker interactions. These variants could not undergo LLPS under physiological conditions, indicating that, in hnRNPDL, Arg and Tyr residue interactions are required for phase separation. The absence of the N-terminus Arg-rich IDR significantly reduces the DL2 LLPS propensity. However, DL2 can still undergo LLPS, most probably by Tyr-Tyr interactions of the C-terminal PrLD, as it occurs with purified PrLDs of proteins such as FUS (Wang et al., 2018).
Different self-assembly properties of the hnRNPDL isoforms suggest that they might participate in distinct biological functions. We experimentally validated that hnRNPDL isoforms exhibit a significantly different behavior in mammalian cells, the data indicating that they might be involved in the formation of distinct nuclear complexes. DL1 and DL2 are excluded from the nucleolus, while DL3 is diffusely distributed. This exclusion from the nucleolus has been also reported for other prion-like proteins like FUS (Yang et al., 2014), TAF15 (Marko et al., 2012), or EWSR1 (Tannukit et al., 2008). The only common region in DL1 and DL2 and absent in DL3 is the C-terminus Tyr-rich PrLD. Therefore, hnRNPDL PrLD seems to determine the intranuclear compartmentalization of the isoforms. This is consistent with the observation that the deletion of the N-terminal PrLD of FUS resulted in a protein variant evenly distributed in the entire nucleus (Yang et al., 2014), as it occurs with the natural DL3 isoform. hnRNPDLs have LLPS propensity, but in the nucleus it is diffusely distributed, without the detection of nuclear puncta corresponding to MLOs. It has been shown that high RNA/protein ratios, as occur in the nucleus, prevent prion-like proteins from LLPS (Maharana et al., 2018). This could be the underlying reason of the diffuse distribution of the DL1 and DL2 variants in the nucleus. Accordingly, we experimentally validated that DL1 LLPS is strongly dependent on RNA levels. We were also interested in assessing whether hnRNPDL isoforms exhibit different mobilities in cells due to their differential self-assembly properties. In excellent agreement with in vitro behavior, DL2 and DL3 form dynamic and small complexes in the nucleus, whereas DL1 forms more rigid and bigger complexes. This last behavior can be univocally ascribed to the DL1 Arg-rich N terminus IDR, which increases DL1 multivalency thus favoring protein-protein and protein-nucleic acids interactions and, consequently, the formation of the observed large complexes.
We further validated that DL2 and DL3 are the only two isoforms able to shuttle between the nucleus and cytoplasm, their nuclear import being dependent on ongoing transcription, as previously reported (Kamei and Yamada, 2002, Kawamura et al., 2002). Similar differences in isoform shuttling have been reported for hnRNPD, a member of the same heterogeneous ribonucleoprotein (hnRNP) family, where p37 and p40 isoforms shuttle between nucleus and cytoplasm but p45 and p42 isoforms remain in the nucleus (Arao et al., 2000). hnRNPD p37 and p40 result from AS and lack exon 7, which encodes for a region of 50 residues corresponding to the only Tyr-rich IDR region (28% Tyr) in its PrLD. The p45 and p42 isoforms’ nuclear retention is suggested to be mediated through interaction with the nuclear SAF-B protein, p37 and p40 not interacting with it. SAF-B contains a large Arg-rich IDR region (26% Arg) and therefore it is plausible that π-cation contacts could contribute to the interactions with the p45 and p42 isoforms. DL1 possesses both types of IDRs and therefore can establish a potentially larger number of interactions. The fact that the protein is retained in the nucleus when RNA levels are decreased by ActD treatment suggests that these interactions are of proteic nature.
DL1 and DL2 form bright nuclear foci after RNA synthesis inhibition, while DL3 does not. This necessarily involves the Tyr-rich PrLD in the process. Interestingly, similar observations were described for hnRNPD, TAF15 and FUS after ActD treatment (Arao et al., 2000, Jobert et al., 2009, Marko et al., 2012, Zinszner et al., 1997). For the three proteins the formation of nuclear foci was depended on their PrLD (Marko et al., 2012, Zinszner et al., 1997). Nuclear foci after ActD may represent the retention of the protein in transient subnuclear compartments by interaction with proteins recruited to this site (Zinszner et al., 1997) thanks to the characteristic compositional bias of PrLDs.
Prion-like proteins are well known for their aggregation behavior and involvement in disease. Therefore, we evaluated the aggregation propensity of hnRNPDL isoforms. Not surprising, DL2, the predominant isoform in humans, hence the most probable isoform being involved in disease, is the isoform with higher amyloidogenic propensity. The data indicate that the amyloidogenic potential concentrates in the PrLD, since DL3, containing the two RRM domains, remains soluble. A reasonable explanation for the amyloidogenicity of DL2 is that it lacks the N-terminus IDR Arg-rich domain, which may act as a mechanism of protection in DL1, precluding rapid aggregation through electrostatic repulsion between positively charged regions. Therefore, this domain plays a different role in LLPS and amyloid formation, promoting the first reaction and inhibiting the second one. Importantly, this suggests that, for hnRNPDL, the process of phase separation plays a protective role against fibril formation.
As observed for other prion-like proteins, disease-causing mutations in the PrLD enhance the aggregation propensity of hnRNPDL. We experimentally demonstrated a significant acceleration of DL2 aggregation after introduction of the D378N and D378H LGMD1G-associated mutations (DL1 nomenclature). In Drosophila muscle, hnRNPDL mutants locate in the nucleus but show a clear reduction in solubility compared to the WT protein. Most pathogenic mutations in prion-like proteins are reported to promote the formation of cytoplasmic inclusions in patient tissues, a phenotype that can be recapitulated in cell-based assays and in animal models of the disease (Kim et al., 2013). This is not the case for hnRNPDL. Analysis of muscle tissues from LGMD1G patients bearing the hnRNPDL genetic mutations we studied here did not revealed the existence of congophilic deposits or any type of inclusions (Sun et al., 2019, Vieira et al., 2014), nor did we observe them in Drosophila. The mechanism by which hnRNPDL mutations elicits the LGMD1G phenotype is still unknown. Here, we uncover that DL2 is inherently aggregation prone and that the disease-causing mutations exacerbate this propensity, resulting in a protein that despite being diffusely distributed in the nucleus of Drosophila myocytes is totally insoluble. This evidence makes us think that LGMD1G is caused by a loss-of-function mechanism, in which DL2 function is lost because the protein aggregates or it is degraded by the cellular protein quality control machinery, in line with the lower levels of mutant proteins detected in Drosophila. Consistent with this view, blocking the translation of hnRNPDL in zebrafish to reduce the endogenous protein levels results in restricted and uncoordinated animal movements and disorganized myofibers (Vieira et al., 2014). Thus, it is feasible that LGMD1G patients would exhibit lower levels of soluble DL2 isoform in their muscular tissue, although this extent should be further confirmed. Importantly, like in the case of hnRNPA2 and hnRNPA1, aggregation in hnRNPDL is triggered by the mutation of a well-conserved Asp residue in the PrLD. This recurrent mutational change within three different hnRNP proteins indicates that these residues play a protective role against aggregation, likely by promoting electrostatic repulsion among side chains of neighboring monomers (Murray et al., 2018).
hnRNPDL protein is an RNA-binding protein involved in transcription and AS regulation (Li et al., 2019). The presence of the exon corresponding to its PrLD is responsible for the incorporation of hnRNPDL into multivalent hnRNP assemblies that, in turn, control the AS of other genes (Gueroussov et al., 2017). A recent study indicates that SRSF1 protein regulates AS of hnRNPDL, generating the isoforms containing the PrLD, and therefore the execution of the downstream splicing program (Feng et al., 2019). It was suggested that SRSF1 may serve as an upstream regulator of phase separation. We demonstrate here that this is the case. Indeed, we speculate that by regulating hnRNPDL AS, SRSF1 might also determine the size and dynamics of the complexes this protein forms in the nucleus, its nucleus-cytoplasm shuttling ability, and its amyloidogenic potential.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal to hnRNPDL | Abcam | Cat#ab183136 |
| Mouse monoclonal anti G3BP | BD Biosciences | Cat#611127; RRID: AB_398438 |
| Alexa Fluor 555 | Molecular Probes | Cat#A31572; RRID: AB_162543 |
| Mouse monoclonal anti GAPDH | Santa Cruz | Cat#sc47724; RRID: AB_627678 |
| Sheep polyclonal anti tubulin | Cytoskeleton Inc | Cat#ATN02; RRID:AB_10709401 |
| Mouse monoclonal anti C23 | Santa Cruz | Cat#sc-8031; RRID: AB_670271 |
| Bacterial and Virus Strains | ||
| One Shot TOP10 chemically competent E. coli | Life Technologies | Cat#C404003 |
| HI-Control CL21(DE3) chemically competent cells (SOLOS) | Lucigen Corporation | Cat#60435-1 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1x TrypLE Express | Thermo Fisher Scientific | Cat #12604-013 |
| Lipofectamine2000 | Thermo Fisher Scientific | Cat# 11668027 |
| His-SUMO-hnRNPDL isoform 1 | This study | N/A |
| His-SUMO-hnRNPDL isoform 2 | This study | N/A |
| His-SUMO-hnRNPDL isoform 3 | This study | N/A |
| His-SUMO-hnRNPDL isoform 2 D259N | This study | N/A |
| His-SUMO-hnRNPDL isoform 2 D259H | This study | N/A |
| Oregon Green Protein Labeling Kit | Molecular Probes | Cat #O10241 |
| Texas Red Protein Labeling Kit | Molecular Probes | Cat#T10244 |
| Q5 site directed mutagenesis kit | New England Biolabs | Cat#E0554S |
| Expresso T7 SUMO cloning and expression system | Lucigen Corporation | Cat#49003-2 |
| NEBuilder HiFi DNA Assembly Master Mix | New England Biolabs | Cat#E2621S |
| Ficoll400 | Sigma-Aldrich | Cat#F2637 |
| Secure seal imaging spacers 1.9x0.12 mm | Grace Biolabs | Cat#654002 |
| Proteostat® aggresome detection kit | Enzo life sciences | Cat#ENZ-51035-K100 |
| Experimental Models: Cell Lines | ||
| HeLa cells | ATCC | Cat #CCL-2; RRID: CVCL_0030 |
| HeLa hnRNPDL knock out cells | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| Drosophila melanogaster: cell line w1118 | Bloomington Drosophila Stock Center | Cat#3605 |
| Drosophila melanogaster: cell line Mhc-Gal4 | Bloomington Drosophila Stock Center | Cat#55133 |
| Drosophila melanogaster: cell line UAS-hnRNPDL isoform 2 | Bestgene Inc. | N/A |
| Drosophila melanogaster: cell line UAS-hnRNPDL isoform 2 D259N | Bestgene Inc. | N/A |
| Drosophila melanogaster: cell line UAS-hnRNPDL isoform 2 D259H | Bestgene Inc. | N/A |
| Oligonucleotides | ||
| See Table S3 for the primer list | ||
| Recombinant DNA | ||
| pET28a-hnRNPDL isoform 1 | GenScript | N/A |
| pETite-His-SUMO Kan vector | Lucigen Corporation | Cat#49003-1 |
| pETite-His-SUMO-hnRNPDL isoform 1 | This study | N/A |
| pETite-His-SUMO-hnRNPDL isoform 2 | This study | N/A |
| pETite-His-SUMO-hnRNPDL isoform 3 | This study | N/A |
| pETite-His-SUMO-hnRNPDL isoform 2 D259N | This study | N/A |
| pETite-His-SUMO-hnRNPDL isoform 2 D259H | This study | N/A |
| pEGFP-C3 vector | Clontech | Cat#6082-1 |
| pEGFP-C3-hnRNPDL isoform 1 | This study | N/A |
| pEGFP-C3-hnRNPDL isoform 2 | This study | N/A |
| pEGFP-C3-hnRNPDL isoform 3 | This study | N/A |
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Addgene | Cat# 62988; RRID:Addgene_62988 |
| pUASTattB | Drosophila Genomics Resource Center | Cat#1419 |
| Software and Algorithms | ||
| SigmaPlot 10.0 | Systal Software | http://www.sigmaplot.co.uk |
| GaphPad Prism 5 | GraphPad | https://www.graphpad.com/ |
| ImageJ 1.51J Software | NIH | https://imagej.nih.gov/ij/download.html |
| Igor Pro 7.0 | Wavemetrics | https://www.wavemetrics.com/ |
Lead Contact and Materials Availability
Further information and requests for resources and reagent should be directed to and will be fulfilled by the Lead Contact, S. Ventura (Salvador.ventura@uab.es).
All unique/stable reagents generated in this study will be made available on request but we may require a payment and/or a completed Materials Transfer Agreement if there is potential for commercial application.
Experimental Model and Subject Details
Bacterial Cell Culture
cDNA clones were transformed into One Shot TOP10 and BL21(DE3) SOLOS chemically competent E. coli cells (Thermo Fisher Scientific). Single colonies were grown overnight at 37°C and 220 rpm in LB media containing kanamycin selection antibiotic at a concentration of 50 mg/ml. All competent bacterial cells were stored at −80°C in 15% glycerol.
Mammalian cell Culture
HeLa cells (of female origin) were grown at 37°C and 5% CO2, and maintained in DMEM High Glucose (Hyclone SH30022.01) medium supplemented with 10% fetal bovine serum. Cells were passaged and plated using 1X TrypLE Express (Thermo Fisher Scientific). Cells were authenticated by short tandem repeat (STR) profiling.
Drosophila Culture
All Drosophila stocks were maintained on standard diet in a 25°C incubator with a 12 h day/night cycle.
Method Details
Isoforms sequence
hnRNPDL isoform 1, 2 and 3 (DL1, DL2 and DL3) sequences were obtained from Uniprot (Bateman et al., 2015):
MEVPPRLSHVPPPLFPSAPATLASRSLSHWRPRPPRQLAPLLPSLAPSSARQGARRAQRHVTAQQPSRLAGGAAIKGGRRRRPDLFRRHFKSSSIQRSAAAAAATRTARQHPPADSSVTMEDMNEYSNIEEFAEGSKINASKNQQDDGKMFIGGLSWDTSKKDLTEYLSRFGEVVDCTIKTDPVTGRSRGFGFVLFKDAASVDKVLELKEHKLDGKLIDPKRAKALKGKEPPKKVFVGGLSPDTSEEQIKEYFGAFGEIENIELPMDTKTNERRGFCFITYTDEEPVKKLLESRYHQIGSGKCEIKVAQPKEVYRQQQQQQKGGRGAAAGGRGGTRGRGRGQgqnwnqgfnnyydqgygnynsayggdqnysgyggydytgynygnygygqgyadysgqQSTYGKASRGGGNHQNNYQPY
Bold corresponds to the N terminus Arg-rich domain present in DL1; lowercase italic corresponds to the C terminus Tyr-rich domain present in DL1 and DL2.
Bacterial molecular cloning
pET28a-hnRNPDL vector was purchased from GenScript. This plasmid has assembled the synthetic human gene hnRNPDL isoform 1 (DL1) with optimized codon for bacterial expression. DL1 sequence was amplified by polymerase chain reaction (PCR) using plasmid pET28a-hnRNPDL as a template. The PCR fragment was cloned into pETite-His-SUMO kan vector using the Expresso T7 SUMO cloning and expression system (Lucigen Corporation) obtaining the pETite-SUMO-DL1 vector. The final vector was transformed into One Shot TOP10 chemically competent E. coli cells (Thermo Fisher Scientific).
pETite-SUMO-DL1 was used as a template for isoform 2 construct (DL2), missing the first 119 amino acids, using the Q5 site directed mutagenesis kit (New England Biolabs).
hnRNPDL isoform 2 point mutations D259N/H (DL2N and DL2H) and isoform 3 construct (DL3), missing the first 119 amino acids and residues at position 343-399, were obtained using pETite-SUMO-DL2 plasmid as a template and the Q5 site directed mutagenesis kit (New England Biolabs).
DNA fragments of R/K and Y/F mutated regions were purchased from GenScript in order to obtain pETite-SUMO-DL-R/K, Y/F and R/K+Y/F plasmids. pETite-SUMO-DL1 was used as a template for these constructs using the Q5 site directed mutagenesis kit (New England Biolabs) and DNA fragments were inserted using the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs).
hnRNPDL-Nt was obtained using pETite-SUMO-DL1 plasmid as a template and the Q5 site directed mutagenesis kit (New England Biolabs).
Once confirmed the sequences of the final vectors, they were transformed into chemically competent E. coli BL21 DE3 cells from Expresso T7 SUMO cloning and expression system (Lucigen Corporation) for protein expression.
Protein expression and purification
100 mL of Luria Broth (LB) with 50 mg/ml kanamycin (kan) were inoculated by single colony of BL21 cells with pETite-SUMO-DL1/2/2N/2H/3 and incubated overnight at 37°C and 220 rpm (New Brunswick Innova 44R shaker). 25 mL of saturated overnight culture was transferred into 1 L LB-kan and incubated at 37°C and 220 rpm. The protein expression was induced at OD600 = 0.5 by addition of isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.5 mM. The induced culture was incubated for additional 3 h at 37°C and 220 rpm. Cells were then harvested by centrifugation for 15 min at 4,000 g and 4°C (Sorvall LYNX 6000. Thermo Scientific). The cell pellet was frosted at −80°C.
Pellets from 2 L cell culture were resuspended in 40 mL binding buffer (50 mM HEPES pH 7.5, 1 M NaCl, 5% glycerol, 20 mM imidazole, 1 mM dithiothreitol (DTT)) supplemented with, 0.2 mg/ml lysozyme, 20 μg/ml DNase and 1 tablet of protease inhibitor cocktail EDTA free (Roche). The suspension was incubated for 20 min at 4°C with slow agitation and then lysed by passing through a LM10 microfluidizer (Microfluidics) at 18.000 psi. Lysate cells were centrifuged for 30 min at 30,000 g and 4°C. The supernatant was filtered through a 0.45 μm PVDF membrane and loaded onto a HisTrap FF Ni-column (GE Healthcare) at a flow rate of 2 ml/min. Protein was eluted by an imidazole gradient over 10 column volumes starting from 0 to 100% of elution buffer (50 mM HEPES pH 7.5, 1 M NaCl, 5% glycerol, 500 mM imidazole, 1 mM DTT). His purified protein was treated with 0.2 mg/ml RNase (Thermo Scientific) for 15 min at 37°C and 1 mM PMSF was added to avoid protein degradation. Then, protein was filtered using 0.22 μm PVDF membrane, concentrated using 10 K Amicon (Millipore) to 5 ml, filtered again and subjected to size exclusion chromatography on a Superdex 200 16/600 column (GE Healthcare) equilibrated in 50 mM HEPES pH 7.5, 1 M NaCl, 5% glycerol and 1 mM DTT. The fractions were analyzed by SDS-PAGE, pooled, concentrated, filtered and stored in small volume aliquots at −80°C. Absence of RNA was confirmed by 260/280 absorbance ratio using Nanodrop 8000 (Thermo Scientific). Protein concentration was determined by OD280 absorbance using Nanodrop 8000 (Thermo Scientific).
For SUMO-hnRNPDL-Nt purification, with a calculated isoelectric point of 10.24, an ion exchange chromatography with a HiTrap SP column (GE Healthcare) using a salt gradient from 50 mM to 1 M NaCl was performed before the size exclusion chromatography.
Phase separation
Purified proteins were diluted to desired protein and salt concentrations in 50 mM HEPES pH 7,5. Dilution of salt from 1 M NaCl (storage buffer) induced phase separation. A secure seal imaging spacer (Grace Biolabs) was used between slide and coverslip to visualize protein droplets in a Leica TCS SP8 microscope. Ficoll400 (Sigma) was used in addition to induce phase separation for DL2.
For DL2:hnRNPDL-Nt colocalization experiments, DL2 was green labeled with Oregon Green and hnRNPDL-Nt was red label with Texas Red using molecular probes protein labeling kits (Invitrogen) as described in the commercial protocol. A stock at 100 μM in 150 mM NaCl was prepared for each protein and then proteins were mixed at 1:1 ratio making posterior serial dilutions from the 50 μM stock mixture.
For RNA experiments, DL1 was used at 6.25 μM final concentration to obtain droplets of 2-6 μm and ensure that the surface was not fully covered with droplets to facilitate imaging. Total RNA was obtained from HeLa cells following the TRIzol Reagent user guide (Invitrogen). RNA diluted in RNase-free water was added at the indicated concentrations and posterior addition of 5 ng/μl of RNase (Thermo Scientific) was used to induce again LLPS.
Dynamic light scattering (DLS)
hnRNPDL isoforms protein size was determined using a DynaPro NanoStar (Wyatt technologies) in 50 mM HEPES pH 7.5 and 150 mM NaCl at different protein concentrations per duplicate. Protein radius was extracted from mass data.
Protein aggregation
Right before each experiment, the stock solutions were diluted to 50 μM in 50 mM HEPES buffer pH 7.4 and 150 mM NaCl. For aggregation assays the samples were incubated for 4 days at 37°C and 600 rpm.
Transmission Electron Microscopy (TEM)
The morphology of the aggregated proteins was evaluated by negative staining and using a JEOL TEM-1400Plus Transmission Electron Microscope, 80 KV. 50 μM aggregated protein solution was sonicated for 10 min in an ultrasonic bath (Fisher Scientific FB15052) and then diluted to 1 μM final concentration in H2O. 5 μl of the diluted solution was placed on carbon-coated copper grids and incubated for 5 min. The grids were then washed and stained with 5 μl of 2% w/v uranyl acetate for 2 min. Then, grids were washed again before analysis.
Congo Red (CR) binding
CR binding to aggregated proteins was analyzed using a Specord® 200 Plus spectrophotometer (Analyticjena). The absorbance spectra were recorded from 400 to 650 nm. 10 μL of 50 μM aggregated protein was added to 90 μl of 20 μM CR in 50 mM HEPES buffer pH 7,4 and 150 mM NaCl and was incubated at room temperature for 5 min before the measurement. The same buffer with 20 μM CR and without protein was employed as a control.
Thioflavin-T (Th-T) aggregation kinetics assay
The fluorescence spectra of Th-T were recorded using a Perkin Elmer EnSpire Multimode plate reader. Reactions were carried out at 50 μM protein concentration in a solution containing 20 μM of Th-T in 50 mM HEPES buffer pH 7,4 and 150 mM NaCl at 37°C. The aggregation kinetics were followed monitoring the changes in Th-T fluorescence intensity at 495 nm every 5 min with prior shaking at 100 rpm for 15 s. The same buffer with 20 μM ThT and without protein was employed as a control.
Thioflavin-S (ThS) and proteostat® staining
100 μL of aggregates protein was incubated for 1 h in the presence of 150 μM ThS (Sigma) or 1/2000 Proteostat® dilutuion (Enzo Life Sciences) in 50 mM HEPES buffer pH 7,4 and 150 mM NaCl. Then, the samples were washed two times with the same buffer. Finally, the precipitated fraction was resuspended in a final volume of 10 μL and placed on a microscope slide and sealed. Images were obtained at 20x magnification in an Eclipse 90i fluorescence microscope.
Congo Red (CR) birefringence
Aggregated protein was first sonicated for 10 min in an ultrasonic bath (Fisher Scientific FB15052). 10 μL sonicated aggregated protein was incubated for 1 h in the presence of 100 μM CR (Sigma). 5 μL sample was placed on a microscope slide and viewed at 10x magnification with a Leica stereoscopic microscope MZFLIII.
Fourier transform infrared (FT-IR) spectroscopy
100 μL aggregated protein was centrifuged and washed two times with H2O to remove the presence of salts. The final pellet was resupended in 10 μL H2O. FTIR experiments were performed using a Bruker Tensor 27 FT-IR spectrometer (Bruker Optics Inc) with a Golden Gate MKII ATR accessory. Each spectrum consists of 32 independent scans, measured at a spectral resolution of 4 cm-1 within 1800-1500 cm-1 range. All spectral data were acquired and normalized using the OPUS MIR Tensor 27 software. Data was afterward deconvoluted using the Peak Fit 4.12 program.
Mammalian molecular cloning
hnRNPDL isoform 1 sequence was amplified by PCR from cDNA extract of U2OS cells and assembled to pEGFP-C3 HindIII and BamHI digested plasmid using the NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs). Similar to bacterial molecular cloning, pEGFP-C3-DL1 plasmid was used as a template for isoform 2 (DL2) construct using the Q5 site directed mutagenesis kit (New England Biolabs). hnRNPDL isoform 3 (DL3) was obtained using pEGFP-C3-DL2 plasmid as a template and the Q5 site directed mutagenesis kit (New England Biolabs). The final vectors were transformed into One Shot TOP10 chemically competent E. coli cells (Thermo Fisher Scientific).
Mammalian cell culture and transfection
HeLa hnRNPDL KO (HeLaDL-KO) cell line was performed by CRISPR-Cas9 with following gRNA: gRNA1: ATTCTTGCTCGCGTTGATCT; gRNA2: ACAGAGTACTTGTCTCGATT. Both gRNA target the common exons in all 3 isoforms. pSpCas9(BB)-2A-Puro (PX459) V2.0 was a gift from Feng Zhang (Addgene plasmid # 62988; http://addgene.org/62988; RRID:Addgene_62988). HeLaDL-KO cells were grown and maintained in DMEM High Glucose medium (Hyclone) supplemented with 10% fetal bovine serum (FBS) (Hyclone) at 37°C and 5% CO2. Cells were passaged and plated using 1x TrypLE Express (Thermo Fisher Scientific).
For cellular transfection, HeLaDL-KO cells were seeded on 4-well or 8-well glass slides (Millipore) for immunofluorescence or 4-well lab-Tek chambered coverglass (Nunc) for FRAP. Cells were transfected 24h after seeding with 400 ng DNA using Lipofectamine2000 (Invitrogen). After 4 h of transfection, we changed cellular media to fresh one. Cellular stress was applied 24h post transfection.
Immunofluorescence
Transfected HeLaDL-KO cells were stressed or not with 5 μg/ml actinomycin D for 3 h. Cells were then fixed with 4% parafolmaldehyde (Electron Microscopy Sciences, #15713-S), permeabilized with 0,2% Triton X-100 and blocked with 1% bovine serum albumin (BSA). Primary antibodies used were against G3BP (611127; BD Biosciences), nucleolin (sc-8031; Santa Cruz) and hnRNPDL (ab183136; Abcam). For visualization, the appropriate host-specific Alexa Fluor 488 or 555 (Molecular Probes) secondary antibodies were used. Slides were mounted using ProLong Gold antifade reagent with DAPI (Invitrogen). Images were captured using a Leica TCS SP8 STED 3x confocal microscope (Leica Biosystems) with a 63x oil objective.
Fluorescence recovery after photobleaching (FRAP)
FRAP experiments were performed with the Opterra II Swept Field Confocal Microscope (Bruker) using Prairie View 5.5 Software. Immediately before imaging, the medium was changed to 500 μL complete phenol red-free DMEM medium (HyClone). During imaging, cells were maintained at 37°C and supplied with 5% CO2 using a Bold Line Cage Incubator (Okolabs) and an objective heater (Bioptechs). Imaging was performed using a 60x Plan Apo 1.40NA oil objective and Perfect Focus (Nikon) was engaged for the duration of the capture.
For imaging, cells were selected based on level of intensity. Time lapses were taken using the 488-nm imaging laser set at 100 power and 100-ms exposure with acquisition set at max speed (0.5 ms period) for 100 frames. Photobleaching of the nucleus occurred 2 s into capture, using the 488-nm FRAP laser to bleach the green channel. Data was repeated in triplicate for each condition, with each replicate having at least n = 10 cells. Data was opened in ImageJ 1.51J (NIH) using the Prairie Reader plugin. ROIs were generated in the photobleach region, a non-photobleached cell, and the background for each timelapse, and the mean intensity of each was extracted. These values were exported into Igor Pro 7.0 (WaveMetrics), where photobleach and background correction were performed, and fit FRAP curves using Hill’s equation were generated.
Size exclusion chromatography (SEC)
We analyzed individually the elution profile of the contents of cells expressing EGFP-hnRNPDL isoforms, one isoform at a time. A total of 3 dishes of 10 cm per each hnRNPDL isoform transfection were pooled for SEC analysis. Experiments were performed in triplicate for each isoform. HeLaDL-KO cells were transfected 24h after seeding with 3 μg DNA using Lipofectamine2000 (Invitrogen) and media was changed to fresh one after 5 h of transfection. Cells were collected 24 h post transfection, resuspended in 0.5 mL of 1xPBS supplemented with protease inhibitor cocktail (Roche) and lysed using 1 mL syringe (5-10 passes). Protein solubilization was confirmed by Bradford. Supernatant was collected by 5 min centrifugation at 1,000 g and 4°C, filtered with 0.45 μm PVDF membrane and loaded on a Superose 6 Increase 10/300 GL column (GE Healthcare) equilibrated with 1xPBS. The fractions were analyzed by Western Blot using a primary antibody against hnRNPDL (ab183136; Abcam) and the appropriate host-specific secondary antibody (IRDye 680RD Goat anti-Rabbit IgG (H + L), LI-COR, P/N 926-68071). The elution pattern of endogenous hnRNPDL in untransfected HeLa WT cells was also analyzed.
Drosophila stocks and culture
Gene sequences of hnRNPDL isoform 2 (DL2) WT, DL2 D259H, and DL2 D259N were synthesized and subcloned into the pUASTattB Drosophila expression vector (BioBasic Inc.). Flies carrying the transgenes were generated by performing a standard injection through the ϕC31 integrase-mediated transgenesis technique (BestGene Inc.). All Drosophila stocks were maintained in a 25°C incubator with a 12 h day/night cycle. Eye phenotypes were imaged by light microscopy. The w1118 line was used as control.
To prepare adult fly muscle for immunofluorescence, the Mhc-Gal4 driver was used to express the transgene in muscle at 25°C. Adult flies were embedded in a drop of OCT compound (Sakura Finetek) on a glass slide, frozen with liquid nitrogen and bisected sagitally by a razor blade. After fixing with 4% paraformaldehyde in PBS, hemithoraces were permeabilized with PBS containing 0.2% Triton X-100 and stained with anti-hnRNPDL antibody (ab183136; Abcam). Hemithoraces were additionally stained by Alexa Fluor 647 (Life Technologies) and DAPI according to manufacturer’s instructions. Stained hemi-thoraces were mounted in 80% glycerol, and the musculature was examined by STED (Leica SP8).
Quantification and Statistical Analysis
Statistical analyses were performed using GraphPad Prism v5. All data are shown as the mean ± standard error of the mean (SEM). The statistical significance of each isoform compared to unfused EGFP was investigated by ordinary two-way ANOVA followed by Dunnett’s multiple comparisons test. The number of samples analyzed per experiment is provided in the corresponding method details section. A p value of less than 0.05 was used to determine significance.
Data and Code Availability
This study did not generate any unique datasets or code.
Acknowledgments
We are grateful to Prof. T. Mittag, Dr. C. Mathieu, and Dr. M. Purice from St. Jude Children’s Research Hospital, USA, and UAB microscopy service of Barcelona for their technical advice. We acknowledge Leticia Nogueira, Antonio Fernando Ribeiro Jr., Mariz Vainzof, and Mayana Zatz from the Human Genome and Stem Cell Center, Biosciences Institute, University of São Paulo, Brazil, for their help with muscle samples. S.V. acknowledges funding from MINECO (BIO2016-78310-R) and ICREA (ICREA-Academia 2016). J.P.T. acknowledges the Howard Hughes Medical Institute, National Institutes of Health (R35NS097974), and the St. Jude Research Collaborative on Membrane-less Organelles. X.S. acknowledges funding from Obra Social “la Caixa,” AGAUR (2017 SGR 324), MINECO (BIO2012-31043 and BIO2015-70092-R), and the European Research Council (CONCERT, contract number 648201). IRB Barcelona is the recipient of a Severo Ochoa Award of Excellence from MINECO (government of Spain). C.B. acknowledges funding from “Ministerio de Educación y Formación Profesional.”
Author Contributions
C.B. designed and performed the experiments, analyzed the data, and wrote the manuscript. P.Y., M.C., J.M., M.P., and E.S. contributed to specific experiments. P.Y. supervised the experiments. H.J.K. designed the Drosophila experiments. M.C. and H.J.K. wrote the Drosophila section and composed Figure 7. P.Y., M.P., E.S., X.S., H.J.K., and J.P.T. edited the manuscript. J.P.T. and S.V. designed the project. S.V. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: January 28, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.080.
Contributor Information
J. Paul Taylor, Email: jpaul.taylor@stjude.org.
Salvador Ventura, Email: salvador.ventura@uab.cat.
Supplemental Information
References
- Akagi T., Kamei D., Tsuchiya N., Nishina Y., Horiguchi H., Matsui M., Kamma H., Yamada M. Molecular characterization of a mouse heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its tissue-specific expression. Gene. 2000;245:267–273. doi: 10.1016/s0378-1119(00)00032-9. [DOI] [PubMed] [Google Scholar]
- Arao Y., Kuriyama R., Kayama F., Kato S. A nuclear matrix-associated factor, SAF-B, interacts with specific isoforms of AUF1/hnRNP D. Arch. Biochem. Biophys. 2000;380:228–236. doi: 10.1006/abbi.2000.1938. [DOI] [PubMed] [Google Scholar]
- Babinchak W.M., Haider R., Dumm B.K., Sarkar P., Surewicz K., Choi J.K., Surewicz W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019;294:6306–6317. doi: 10.1074/jbc.RA118.007222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baralle F.E., Giudice J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017;18:437–451. doi: 10.1038/nrm.2017.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A., Martin M.J., O’Donovan C., Magrane M., Apweiler R., Alpi E., Antunes R., Arganiska J., Bely B., Bingley M., UniProt Consortium UniProt: a hub for protein information. Nucleic Acids Res. 2015;43:D204–D212. doi: 10.1093/nar/gku989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berardo A., Lornage X., Johari M., Evangelista T., Cejas C., Barroso F., Dubrovsky A., Bui M.T., Brochier G., Saccoliti M. HNRNPDL-related muscular dystrophy: expanding the clinical, morphological and MRI phenotypes. J. Neurol. 2019;266:2524–2534. doi: 10.1007/s00415-019-09437-3. [DOI] [PubMed] [Google Scholar]
- Biancalana M., Koide S. Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta. 2010;1804:1405–1412. doi: 10.1016/j.bbapap.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S., Bogaert E., Kovacs D., Konijnenberg A., Timmerman E., Volkov A., Guharoy M., De Decker M., Jaspers T., Ryan V.H. Phase Separation of C9orf72 Dipeptide Repeats Perturbs Stress Granule Dynamics. Mol. Cell. 2017;65:1044–1055.e5. doi: 10.1016/j.molcel.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018;28:420–435. doi: 10.1016/j.tcb.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogaert E., Boeynaems S., Kato M., Guo L., Caulfield T.R., Steyaert J., Scheveneels W., Wilmans N., Haeck W., Hersmus N. Molecular Dissection of FUS Points at Synergistic Effect of Low-Complexity Domains in Toxicity. Cell Rep. 2018;24:529–537.e4. doi: 10.1016/j.celrep.2018.06.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bounedjah O., Desforges B., Wu T.D., Pioche-Durieu C., Marco S., Hamon L., Curmi P.A., Guerquin-Kern J.L., Piétrement O., Pastré D. Free mRNA in excess upon polysome dissociation is a scaffold for protein multimerization to form stress granules. Nucleic Acids Res. 2014;42:8678–8691. doi: 10.1093/nar/gku582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady J.P., Farber P.J., Sekhar A., Lin Y.H., Huang R., Bah A., Nott T.J., Chan H.S., Baldwin A.J., Forman-Kay J.D., Kay L.E. Structural and hydrodynamic properties of an intrinsically disordered region of a germ cell-specific protein on phase separation. Proc. Natl. Acad. Sci. USA. 2017;114:E8194–E8203. doi: 10.1073/pnas.1706197114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke K.A., Janke A.M., Rhine C.L., Fawzi N.L. Residue-by-Residue View of In Vitro FUS Granules that Bind the C-Terminal Domain of RNA Polymerase II. Mol. Cell. 2015;60:231–241. doi: 10.1016/j.molcel.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R.K., Pappu R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA. 2013;110:13392–13397. doi: 10.1073/pnas.1304749110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfuss G., Kim V.N., Kataoka N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 2002;3:195–205. doi: 10.1038/nrm760. [DOI] [PubMed] [Google Scholar]
- El-Gebali S., Mistry J., Bateman A., Eddy S.R., Luciani A., Potter S.C., Qureshi M., Richardson L.J., Salazar G.A., Smart A. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47(D1):D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H., Bao S., Rahman M.A., Weyn-Vanhentenryck S.M., Khan A., Wong J., Shah A., Flynn E.D., Krainer A.R., Zhang C. Modeling RNA-Binding Protein Specificity In Vivo by Precisely Registering Protein-RNA Crosslink Sites. Mol. Cell. 2019;74:1189–1204.e6. doi: 10.1016/j.molcel.2019.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L., Teng P.K., Riek R., Eisenberg D. Identifying the amylome, proteins capable of forming amyloid-like fibrils. Proc. Natl. Acad. Sci. USA. 2010;107:3487–3492. doi: 10.1073/pnas.0915166107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueroussov S., Weatheritt R.J., O’Hanlon D., Lin Z.Y., Narula A., Gingras A.C., Blencowe B.J. Regulatory Expansion in Mammals of Multivalent hnRNP Assemblies that Globally Control Alternative Splicing. Cell. 2017;170:324–339.e23. doi: 10.1016/j.cell.2017.06.037. [DOI] [PubMed] [Google Scholar]
- Harrison A.F., Shorter J. RNA-binding proteins with prion-like domains in health and disease. Biochem. J. 2017;474:1417–1438. doi: 10.1042/BCJ20160499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Chen X., Wu B., Soler I.M., Chen S., Shen Y. Further defining the critical genes for the 4q21 microdeletion disorder. Am. J. Med. Genet. A. 2017;173:120–125. doi: 10.1002/ajmg.a.37965. [DOI] [PubMed] [Google Scholar]
- Iglesias V., Conchillo-Sole O., Batlle C., Ventura S. AMYCO: evaluation of mutational impact on prion-like proteins aggregation propensity. BMC Bioinformatics. 2019;20:24. doi: 10.1186/s12859-019-2601-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito D., Hatano M., Suzuki N. RNA binding proteins and the pathological cascade in ALS / FTD neurodegeneration. Sci. Transl. Med. 2017;9:eaah5436. doi: 10.1126/scitranslmed.aah5436. [DOI] [PubMed] [Google Scholar]
- Jobert L., Pinzón N., Van Herreweghe E., Jády B.E., Guialis A., Kiss T., Tora L. Human U1 snRNA forms a new chromatin-associated snRNP with TAF15. EMBO Rep. 2009;10:494–500. doi: 10.1038/embor.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei D., Yamada M. Interactions of heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its domains with high-affinity binding sites. Gene. 2002;298:49–57. doi: 10.1016/s0378-1119(02)00926-5. [DOI] [PubMed] [Google Scholar]
- Kamei D., Tsuchiya N., Yamazaki M., Meguro H., Yamada M. Two forms of expression and genomic structure of the human heterogeneous nuclear ribonucleoprotein D-like JKTBP gene (HNRPDL) Gene. 1999;228:13–22. doi: 10.1016/s0378-1119(99)00020-7. [DOI] [PubMed] [Google Scholar]
- Kato M., Yang Y.-S., Sutter B.M., Wang Y., McKnight S.L., Tu B.P. Redox State Controls Phase Separation of the Yeast Ataxin-2 Protein via Reversible Oxidation of Its Methionine-Rich Low-Complexity Domain. Cell. 2019;177:711–721.e8. doi: 10.1016/j.cell.2019.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura H., Tomozoe Y., Akagi T., Kamei D., Ochiai M., Yamada M. Identification of the nucleocytoplasmic shuttling sequence of heterogeneous nuclear ribonucleoprotein D-like protein JKTBP and its interaction with mRNA. J. Biol. Chem. 2002;277:2732–2739. doi: 10.1074/jbc.M108477200. [DOI] [PubMed] [Google Scholar]
- Kim H.J., Kim N.C., Wang Y.-D., Scarborough E.A., Moore J., Diaz Z., MacLea K.S., Freibaum B., Li S., Molliex A. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King O.D., Gitler A.D., Shorter J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Res. 2012;1462:61–80. doi: 10.1016/j.brainres.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster A.K., Nutter-Upham A., Lindquist S., King O.D. PLAAC: a web and command-line application to identify proteins with prion-like amino acid composition. Bioinformatics. 2014;30:2501–2502. doi: 10.1093/bioinformatics/btu310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.Z., Hou J., Wei Y., Luo X., Ye Y., Zhang Y. hnRNPDL extensively regulates transcription and alternative splicing. Gene. 2019;687:125–134. doi: 10.1016/j.gene.2018.11.026. [DOI] [PubMed] [Google Scholar]
- Liewluck T., Milone M. Untangling the complexity of limb-girdle muscular dystrophies. Muscle Nerve. 2018;58:167–177. doi: 10.1002/mus.26077. [DOI] [PubMed] [Google Scholar]
- Liu Y., Zhu X., Zhu J., Liao S., Tang Q., Liu K., Guan X., Zhang J., Feng Z. Identification of differential expression of genes in hepatocellular carcinoma by suppression subtractive hybridization combined cDNA microarray. Oncol. Rep. 2007;18:943–951. [PubMed] [Google Scholar]
- Mackenzie I.R., Nicholson A.M., Sarkar M., Messing J., Purice M.D., Pottier C., Annu K., Baker M., Perkerson R.B., Kurti A. TIA1 Mutations in Amyotrophic Lateral Sclerosis and Frontotemporal Dementia Promote Phase Separation and Alter Stress Granule Dynamics. Neuron. 2017;95:808–816.e9. doi: 10.1016/j.neuron.2017.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharana S., Wang J., Papadopoulos D.K., Richter D., Pozniakovsky A., Poser I., Bickle M., Guillén-boixet J., Franzmann T., Jahnel M. RNA buffers the phase separation behavior of prion-like RNA binding proteins. Science. 2018;360:918–921. doi: 10.1126/science.aar7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March Z.M., King O.D., Shorter J. Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease. Brain Res. 2016;1647:9–18. doi: 10.1016/j.brainres.2016.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marko M., Vlassis A., Guialis A., Leichter M. Domains involved in TAF15 subcellular localisation: dependence on cell type and ongoing transcription. Gene. 2012;506:331–338. doi: 10.1016/j.gene.2012.06.088. [DOI] [PubMed] [Google Scholar]
- Molliex A., Temirov J., Lee J., Coughlin M., Kanagaraj A.P., Kim H.J., Mittag T., Taylor J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133. doi: 10.1016/j.cell.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray D.T., Zhou X., Kato M., Xiang S., Tycko R., McKnight S.L. Structural characterization of the D290V mutation site in hnRNPA2 low-complexity-domain polymers. Proc. Natl. Acad. Sci. USA. 2018;115:E9782–E9791. doi: 10.1073/pnas.1806174115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy A.C., Dignon G.L., Kan Y., Zerze G.H., Parekh S.H., Mittal J., Fawzi N.L. Molecular interactions underlying liquid-liquid phase separation of the FUS low-complexity domain. Nat. Struct. Mol. Biol. 2019;26:637–648. doi: 10.1038/s41594-019-0250-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro S., Marinelli P., Diaz-Caballero M., Ventura S. The prion-like RNA-processing protein HNRPDL forms inherently toxic amyloid-like inclusion bodies in bacteria. Microb. Cell Fact. 2015;14:102. doi: 10.1186/s12934-015-0284-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelsky N.B., Taylor J.P. Bridging biophysics and neurology: aberrant phase transitions in neurodegenerative disease. Nat. Rev. Neurol. 2019;15:272–286. doi: 10.1038/s41582-019-0157-5. [DOI] [PubMed] [Google Scholar]
- Nigro V., Savarese M. Genetic basis of limb-girdle muscular dystrophies: the 2014 update. Acta Myol. 2014;33:1–12. [PMC free article] [PubMed] [Google Scholar]
- Patel A., Lee H.O., Jawerth L., Maharana S., Jahnel M., Hein M.Y., Stoynov S., Mahamid J., Saha S., Franzmann T.M. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. [DOI] [PubMed] [Google Scholar]
- Peskett T.R., Rau F., O’Driscoll J., Patani R., Lowe A.R., Saibil H.R. A Liquid to Solid Phase Transition Underlying Pathological Huntingtin Exon1 Aggregation. Mol. Cell. 2018;70:588–601.e6. doi: 10.1016/j.molcel.2018.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piñol-Roma S., Dreyfuss G. Transcription-dependent and transcription-independent nuclear transport of hnRNP proteins. Science. 1991;253:312–314. doi: 10.1126/science.1857966. [DOI] [PubMed] [Google Scholar]
- Piñol-Roma S., Dreyfuss G. Shuttling of pre-mRNA binding proteins between nucleus and cytoplasm. Nature. 1992;355:730–732. doi: 10.1038/355730a0. [DOI] [PubMed] [Google Scholar]
- Ryan V.H., Dignon G.L., Zerze G.H., Chabata C.V., Silva R., Conicella A.E., Amaya J., Burke K.A., Mittal J., Fawzi N.L. Mechanistic View of hnRNPA2 Low-Complexity Domain Structure, Interactions, and Phase Separation Altered by Mutation and Arginine Methylation. Mol. Cell. 2018;69:465–479.e7. doi: 10.1016/j.molcel.2017.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub V., Murphy A., Udd B., LGMD workshop study group ). 229th ENMC international workshop: Limb girdle muscular dystrophies – Nomenclature and reformed classification Naarden, the Netherlands, 17–19 March 2017. Neuromuscul. Disord. 2018;28:702–710. doi: 10.1016/j.nmd.2018.05.007. [DOI] [PubMed] [Google Scholar]
- Su H., Kodiha M., Lee S., Stochaj U. Identification of novel markers that demarcate the nucleolus during severe stress and chemotherapeutic treatment. PLoS ONE. 2013;8:e80237. doi: 10.1371/journal.pone.0080237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Chen H., Lu Y., Duo J., Lei L., OuYang Y., Hao Y., Da Y., Shen X.M. Limb girdle muscular dystrophy D3 HNRNPDL related in a Chinese family with distal muscle weakness caused by a mutation in the prion-like domain. J. Neurol. 2019;266:498–506. doi: 10.1007/s00415-018-9165-4. [DOI] [PubMed] [Google Scholar]
- Tannukit S., Wen X., Wang H., Paine M.L. TFIP11, CCNL1 and EWSR1 Protein-protein Interactions, and Their Nuclear Localization. Int. J. Mol. Sci. 2008;9:1504–1514. doi: 10.3390/ijms9081504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya N., Kamei D., Takano A., Matsui T., Yamada M. Cloning and characterization of a cDNA encoding a novel heterogeneous nuclear ribonucleoprotein-like protein and its expression in myeloid leukemia cells. J. Biochem. 1998;123:499–507. doi: 10.1093/oxfordjournals.jbchem.a021964. [DOI] [PubMed] [Google Scholar]
- Vernon R.M., Chong P.A., Tsang B., Kim T.H., Bah A., Farber P., Lin H., Forman-Kay J.D. Pi-Pi contacts are an overlooked protein feature relevant to phase separation. eLife. 2018;7:e31486. doi: 10.7554/eLife.31486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira N.M., Naslavsky M.S., Licinio L., Kok F., Schlesinger D., Vainzof M., Sanchez N., Kitajima J.P., Gal L., Cavaçana N. A defect in the RNA-processing protein HNRPDL causes limb-girdle muscular dystrophy 1G (LGMD1G) Hum. Mol. Genet. 2014;23:4103–4110. doi: 10.1093/hmg/ddu127. [DOI] [PubMed] [Google Scholar]
- Wang J., Choi J.-M., Holehouse A.S., Lee H.O., Zhang X., Jahnel M., Maharana S., Lemaitre R., Pozniakovsky A., Drechsel D. A Molecular Grammar Governing the Driving Forces for Phase Separation of Prion-like RNA Binding Proteins. Cell. 2018;174:688–699.e16. doi: 10.1016/j.cell.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegmann S., Eftekharzadeh B., Tepper K., Zoltowska K.M., Bennett R.E., Dujardin S., Laskowski P.R., MacKenzie D., Kamath T., Commins C. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018;37:e98049. doi: 10.15252/embj.201798049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C., Scott J., Shea J.E. Binding of Congo red to amyloid protofibrils of the Alzheimer Aβ(9-40) peptide probed by molecular dynamics simulations. Biophys. J. 2012;103:550–557. doi: 10.1016/j.bpj.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.Y., Li H., Lv X.Y., Wei Q., Li X., Liu X.Y., Zhou Q., Wei Y.Q. Overexpression of JKTBP1 induces androgen-independent LNCaP cell proliferation through activation of epidermal growth factor-receptor (EGF-R) Cell Biochem. Funct. 2008;26:467–477. doi: 10.1002/cbf.1468. [DOI] [PubMed] [Google Scholar]
- Yang L., Gal J., Chen J., Zhu H. Self-assembled FUS binds active chromatin and regulates gene transcription. Proc. Natl. Acad. Sci. USA. 2014;111:17809–17814. doi: 10.1073/pnas.1414004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying Y., Wang X.J., Vuong C.K., Lin C.H., Damianov A., Black D.L. Splicing Activation by Rbfox Requires Self-Aggregation through Its Tyrosine-Rich Domain. Cell. 2017;170:312–323.e10. doi: 10.1016/j.cell.2017.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Ji D., Hu X., Ni H., Ma W., Zhang X., Liao S., Zeng Z., Zhao Y., Zhou H. Oncogenic heterogeneous nuclear ribonucleoprotein D-like promotes the growth of human colon cancer SW620 cells via its regulation of cell-cycle. Acta Biochim. Biophys. Sin. (Shanghai) 2018;50:880–887. doi: 10.1093/abbs/gmy085. [DOI] [PubMed] [Google Scholar]
- Zhang T., Delestienne N., Huez G., Kruys V., Gueydan C. Identification of the sequence determinants mediating the nucleo-cytoplasmic shuttling of TIAR and TIA-1 RNA-binding proteins. J. Cell Sci. 2005;118:5453–5463. doi: 10.1242/jcs.02669. [DOI] [PubMed] [Google Scholar]
- Zhou H., Ge Y., Sun L., Ma W., Wu J., Zhang X., Hu X., Eaves C.J., Wu D., Zhao Y. Growth arrest specific 2 is up-regulated in chronic myeloid leukemia cells and required for their growth. PLoS ONE. 2014;9:e86195. doi: 10.1371/journal.pone.0086195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinszner H., Immanuel D., Yin Y., Liang F.X., Ron D. A topogenic role for the oncogenic N-terminus of TLS: nucleolar localization when transcription is inhibited. Oncogene. 1997;14:451–461. doi: 10.1038/sj.onc.1200854. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.