

Abstract
NAFLD is estimated to affect up to one third of the general population and is a major predisposing factor in the pathogenesis of hepatic insulin resistance, NASH, and T2D. While weight loss is highly effective at reversing NAFLD, insulin resistance and T2D, it is difficult to achieve and sustain; therefore, new therapies are urgently required. Our lab has recently developed a controlled-release mitochondrial protonophore (CRMP) that is functionally liver-targeted and promotes oxidation of hepatic triglycerides by promoting a subtle sustained increase in hepatic mitochondrial inefficiency. While we have previously demonstrated that CRMP safely reverses hypertriglyceridemia, fatty liver, and hepatic inflammation/fibrosis in diet-induced rodent models of obesity, there remains a critical need to assess the safety and efficacy in a model highly relevant to humans. Here, we evaluated the safety and impact of chronic CRMP treatment on hepatic mitochondrial oxidation and on the reversal of hypertriglyceridemia, NAFLD, and insulin resistance in two nonhuman primate models of metabolic syndrome. Using Positional Isotopomer NMR Tracer Analysis (PINTA), we demonstrate that acute CRMP treatment (single dose of 5 mg/Kg) increases rates of hepatic mitochondrial fat oxidation by 40%. Importantly, 6-weeks of CRMP treatment significantly reduced hepatic triglycerides in both nonhuman primate models independently of changes in body weight, food intake, body temperature or adverse reactions. CRMP treatment was also associated with a significant 20–30% reduction in fasting plasma triglycerides and LDL-C in spontaneously obese dysmetabolic nonhuman primates. Furthermore, oral administration of CRMP significantly reduced endogenous glucose production by 18%, which could be attributed to a ~20% reduction in hepatic acetyl-CoA content (as assessed by whole-body βOHB turnover) and pyruvate carboxylase flux. Collectively, these studies provide proof-of-concept data to support the development of novel liver-targeted mitochondrial uncouplers for the treatment of NAFLD/NASH and T2D in humans.
One sentence summary:
Liver-targeted mitochondrial uncoupling improves dyslipidemia and reduces hepatic triglyceride content in obese nonhuman primates.
INTRODUCTION
Caloric excess and a sedentary lifestyle have led to the global epidemic of obesity and metabolic syndrome (1), the hepatic consequence of which, non-alcoholic fatty liver disease (NAFLD), is estimated to affect up to one-third of the adult population in Western nations (2). This spectrum of liver disease is characterized by excessive cytoplasmic retention of triglycerides (3) and ranges from simple steatosis to nonalcoholic steatohepatitis (NASH), cirrhosis and hepatocellular carcinoma (HCC) (4). As the rates of obesity continue to rise, NAFLD has become the most common cause of liver disease worldwide (2, 5). Additionally, NAFLD increases the risk for developing insulin resistance, type 2 diabetes (T2D) and cardiovascular disease (CVD) (6–8).
Although the relationship between NAFLD and insulin resistance is complex and bidirectional (9), a substantial body of evidence in rodents and humans supports the role of ectopic lipid accumulation leading to impaired insulin action in the liver and skeletal muscle (10–16). In line with this, short-term caloric restriction reduces NAFLD and ectopic lipid accumulation and normalizes insulin sensitivity in rodent models of obesity and T2D (17). Indeed, lifestyle modifications remain the cornerstone of NAFLD and T2D management, with moderate weight reductions (<10%) causing a significant decrease in hepatic fat content and improvements in hepatic insulin sensitivity in T2D patients (18). Despite this, in many individuals these modifications cannot be implemented successfully or durably and new pharmacological agents are needed.
Liver-targeted mitochondrial uncoupling has recently gained attention as a potential therapeutic approach to increase hepatic fat oxidation and combat the life- and health-limiting consequences of NAFLD and T2D (19). Mitochondrial uncouplers (e.g. 2,4-dinitrophenol, DNP) shuttle protons across the inner mitochondrial membrane via a pathway that is independent of ATP synthase, thereby uncoupling nutrient oxidation from ATP production (20). DNP has been known to increase metabolic rates since the late 1800s and was an effective and widely used weight loss drug in the 1930s (21–24). Unfortunately, chronic ingestion of DNP is associated with a number of unwanted side effects, including fatal hyperthermia and death, and was banned by the FDA in 1938 due to its low therapeutic index (25). Nonetheless, given its ability to abrogate the development of NAFLD and hepatic insulin resistance in rats by promoting oxidation of hepatic triglycerides (26), we investigated whether DNP could be pharmacologically manipulated to improve its safety margin. In this regard, we recently developed a liver-targeted form of DNP (DNPME), as well as a version of DNP with lower peak plasma concentrations and sustained release pharmacokinetics (controlled-release mitochondrial protonophore, CRMP) and showed that these modified forms of DNP safely reverse hypertriglyceridemia, fatty liver, steatohepatitis and liver fibrosis in rodent models of NAFLD/NASH and T2D without inducing hyperthermia and associated hepatic/systemic toxicities (27–29). While these studies suggest that mitochondrial protonophores may be a novel therapy for the major components of metabolic syndrome, their therapeutic utility for the treatment of NAFLD and T2D in a model highly relevant to humans remains unknown. For example, rodents do not typically develop spontaneous diabetes and many are resistant to the development of diet-induced obesity-mediated T2D, even though they develop all symptoms of metabolic syndrome (30). Therefore, we aimed to determine the safety and efficacy of CRMP treatment (6 weeks) on the reversal of hypertriglyceridemia, NAFLD, and insulin resistance in diet-induced and spontaneous nonhuman primate models of metabolic syndrome, which develop all clinical features of diabetes, including obesity, insulin resistance, dyslipidemia and pancreatic pathology similar to humans. We hypothesized that CRMP treatment would reverse NAFLD and improve insulin sensitivity in obese nonhuman primates due to subtle sustained increases in rates of hepatic mitochondrial oxidation.
RESULTS
CRMP Reverses NAFLD in High-Fat Fructose Fed Cynomolgus Monkeys
To determine whether CRMP could safely and effectively reverse NAFLD in a relevant nonhuman primate model of fatty liver and insulin resistance (31, 32), we performed a pilot study in 6 high-fat fructose fed Cynomolgus monkeys. Briefly, Cynomolgus monkeys were fed a high-fat diet, along with fructose supplementation (in which 35% of calories were from fat) for 1 year. Following a baseline characterization, monkeys were treated with CRMP (0.8 mg/Kg body weight b.i.d.) for 6 weeks, at which point plasma and liver samples were collected under general anesthesia. Clinical chemistry and body temperature were also assessed throughout the course of the study to ensure animals remained in good health. A dose of 0.8 mg/Kg (b.i.d) was chosen based on previous pharmacokinetic studies which demonstrated that 0.8 mg/Kg CRMP treatment resulted in peak plasma DNP (the active moiety of CRMP) concentrations of 10–15 μM (Figure S1A). We have previously established that this is well below the lower plasma concentration threshold for DNP toxicity in rodents (~400 μmol/L), and slightly above the concentration needed to promote a sustained increase in hepatic mitochondrial fat oxidation (5 μmol/L) (27, 28). Importantly 6-weeks of CRMP treatment was not associated with any appreciable differences in body temperature [a known on-target side-effect of DNP (33)], blood pressure, heart rate, or food intake (Figure S1B–E). Consistent with our rodent studies, chronic CRMP treatment also showed a strong tendency to lower fasting plasma glucose concentrations (P = 0.15) and significantly reduced endogenous glucose production (P=0.05), without altering body weight (Figure 1B–D; Figure S1F). Interestingly, while CRMP had no effect on fasting plasma triglycerides in this fructose-supplemented NHP model of metabolic syndrome (Figure 1E), CRMP significantly increased plasma total cholesterol concentrations (P=0.02), which could be attributed to increases in HDL-cholesterol (HDL-C) (Figure 1F–H). Furthermore, there were no significant differences observed in the plasma AST, BUN or creatinine after chronic CRMP treatment (Figure 1I–L), indicating that 6-weeks of CRMP does not adversely affect liver or kidney function. Importantly, 6-weeks of CRMP treatment significantly reduced hepatic steatosis (P=0.04) in high-fat fructose fed Cynomolgus monkeys, as evidenced by the 30% reduction in hepatic triglyceride content (Figure 1M). Taken together, these data demonstrate that chronic CRMP treatment can safely and effectively reverse NAFLD in a diet-induced nonhuman primate model of fatty liver and insulin resistance.
Figure 1: CRMP Reduces Hepatic Triglycerides in High-Fat Fructose-Fed Cynomolgus Monkeys.
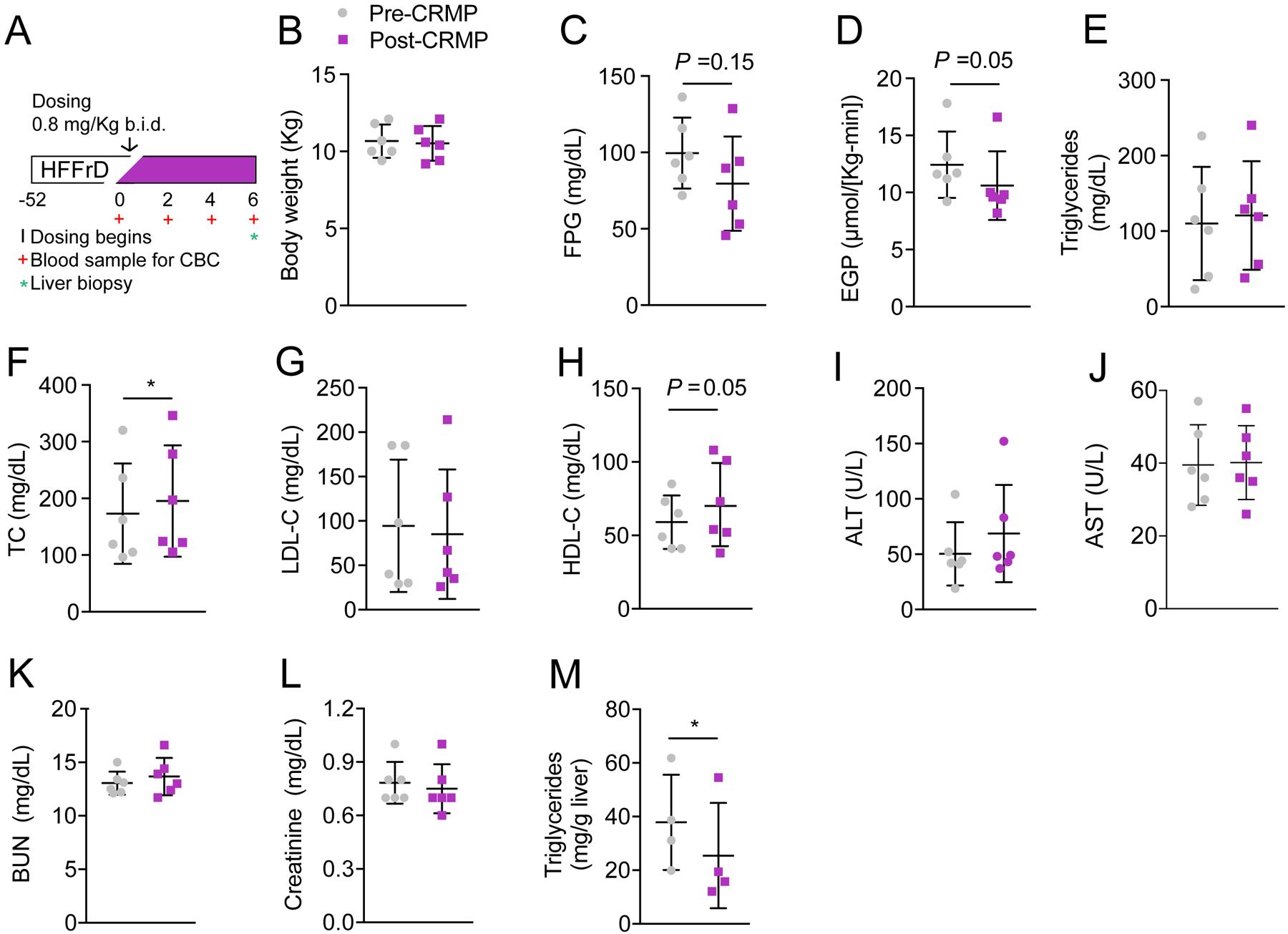
(A) Experimental outline of CRMP treatment in Cynomolgus monkeys. (B-L) Body weight (B), fasting plasma glucose (FPG) (C), endogenous glucose production (EGP) (D), plasma triglycerides (E), total cholesterol (TC) (F), LDL-C (G), HDL-C (H), ALT (I), AST (J), BUN (K), creatinine (L) and hepatic triglyceride content (M) in high-fat fructose fed cynomolgus monkeys before (pre-CRMP) and after CRMP (0.8 mpk b.i.d.) treatment for 6 weeks (post-CRMP). Data are mean ±SD. In panels (B-L), n = 6 per treatment group. In panel (L), n = 4 per treatment group. *P ≤ 0.05 by paired student’s t-test (baseline vs. treatment).
Tolerability of CRMP in Dysmetabolic Rhesus Monkeys
We next determined the tolerability of CRMP in a larger spontaneous nonhuman primate model of NAFLD and T2D (34). To this end, two dysmetabolic Rhesus monkeys were administered daily escalating doses of CRMP (1, 5, 10, and 25 mg/Kg CRMP) in banana mash (Figure S2A). Importantly, no significant differences in body temperature were detected throughout the study and both animals remained in good health (Figure S2B). We then examined the pharmacokinetic properties of CRMP after each dose. As shown in Figure S2C–D, oral dosing of CRMP (1–25 mg/Kg) led to peak plasma concentrations of 8–140 μmol/L DNP. Importantly, no changes in plasma AST, ALT, BUN or creatinine were observed at any dose of CRMP tested (Figure S2E–H), demonstrating that daily escalating doses of CRMP were well-tolerated in Rhesus monkeys.
CRMP Reduces Plasma and Hepatic Lipids in Dysmetabolic Rhesus Monkeys
To assess the safety and efficacy of 6-week CRMP treatment on rates of hepatic mitochondrial oxidation and the reversal of hypertriglyceridemia, NAFLD and insulin resistance in a highly relevant human model of metabolic syndrome, we performed a randomized control trial in spontaneously obese dysmetabolic Rhesus monkeys treated with 5 mg/(Kg-day) CRMP (n = 12) or vehicle control (n = 6) for six weeks (Figure 1A), a dose that leads to peak plasma DNP concentrations of 20–40 μmol/L and is effective at acutely increasing rates of hepatic mitochondrial oxidation (citrate synthase flux, Vcs) (Figure S3A–C; Figure 1B). As shown in Figure 2C–E, CRMP treatment significantly reduced total fat mass (P=0.001), independently of changes in body weight or lean mass. Nonhuman primates treated with CRMP also displayed a 30% reduction in plasma triglyceride concentrations (Figure 2F), and a significant reduction in plasma total cholesterol (P=0.00002), which could be attributed to a 30% reduction in VLDL-C and LDL-C concentrations; plasma HDL-C concentrations were unchanged (Figure 2G–J). Importantly, no apparent toxicity was associated with 6-week CRMP treatment as shown by the clinical chemistries and blood counts, which remained within normal limits throughout the study (Figure 2K–N; Table S1). Additionally, the hepatic GSH/GSSG ratio and protein carbonylation, markers of oxidative stress (35, 36), were not significantly altered after 6-weeks of CRMP treatment (Figure S4A–B).
Figure 2: CRMP Treatment Significantly Reduces Plasma Triglycerides, VLDL-C and LDL-C in Dysmetabolic Rhesus Monkeys.
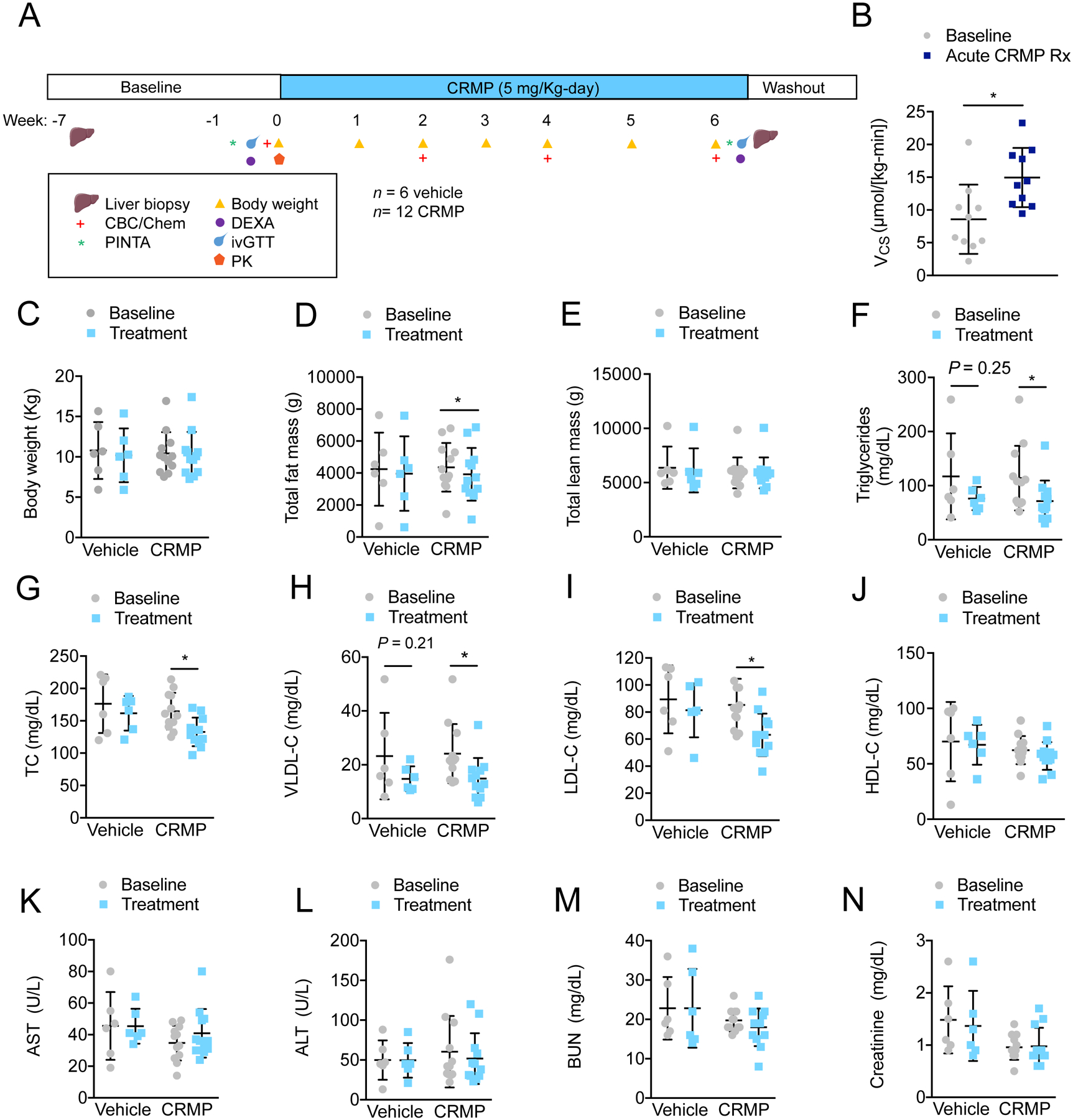
(A) Experimental outline of CRMP efficacy study in dysmetabolic Rhesus monkeys. (B) Citrate synthase flux (VCS) in dysmetabolic Rhesus monkeys after acute treatment with 5 mg/Kg CRMP (n = 10) or vehicle control (n = 6). *P ≤ 0.05 by unpaired student’s t-test. (C-N) Body weight (C), total fat mass (D) total lean mass (E), fasting plasma triglycerides (F), total cholesterol (TC) (G), VLDL-C (H), LDL-C (I), HDL-C (J), AST (K), ALT (L), BUN (M) and creatinine (N) in dysmetabolic Rhesus monkeys before (baseline) and after treatment with 5 mg/Kg CRMP (n =12) or vehicle control (n = 6) for 6 weeks (treatment). *P ≤ 0.05 by paired student’s t-test (baseline vs. treatment). In panels (B-N), data are mean ±SD.
CRMP Reduces Hepatic Triglycerides in Dysmetabolic Rhesus Monkeys
Spontaneously obese, dysmetabolic Rhesus monkeys often display hepatic histopathologic changes (fatty liver, inflammation, and fibrosis) that eventually progress to NAFLD/NASH (34). To evaluate the effect of CRMP treatment on hepatic steatosis, we measured the triglyceride content in liver biopsies from animals at baseline and after 6-weeks of CRMP treatment. Surprisingly, hepatic steatosis was not prominent in dysmetabolic Rhesus monkeys at baseline (Figure 3A, mean triglyceride content ≤ 25 mg/g liver); nonetheless, CRMP did significantly reduce hepatic triglycerides by ~30% (P=0.05, Figure 3A). Given the key role of ectopic lipids in promoting hepatic insulin resistance (26, 37), we next examined the effect of daily oral CRMP treatment on whole-body glucose metabolism and insulin resistance. While CRMP tended to reduce fasting plasma insulin concentrations and HOMA-IR, plasma glucose concentrations were unaltered (Figure 3B–D). Furthermore, no significant differences were observed in plasma glucose or insulin concentrations during an intravenous glucose tolerance test (Figure S5A–F).
Figure 3: CRMP Treatment Significantly Reduces Hepatic Triglycerides and Endogenous Glucose Production in Dysmetabolic Rhesus Monkeys.
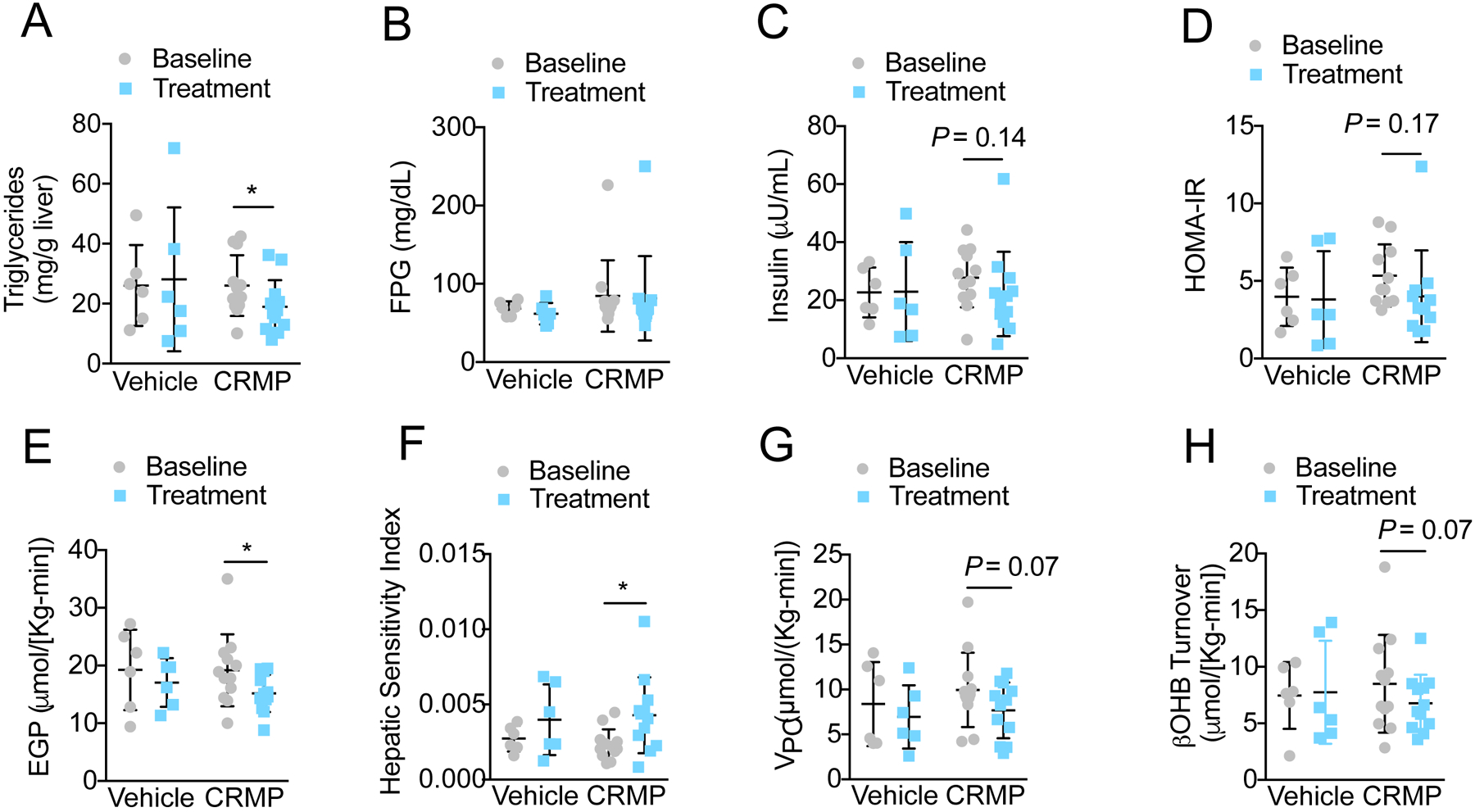
Hepatic triglycerides (A), fasting plasma glucose (FPG) (B), insulin (C), HOMA-IR (D), endogenous glucose production (EGP) (E), hepatic sensitivity index (F), pyruvate carboxylase flux (Vpc) (G), and βOHB turnover (H) in dysmetabolic Rhesus monkeys before (baseline) and after treatment with 5 mg/Kg CRMP (n =12) or vehicle control (n = 6) for 6 weeks (treatment). *P ≤ 0.05 by paired student’s t-test (baseline vs. treatment). In all panels, data are mean ±SD.
CRMP Increases Rates of Hepatic Mitochondrial Oxidation, Reduces Endogenous Glucose Production and Improves Hepatic Insulin Sensitivity in Dysmetabolic Rhesus Monkeys
We recently developed a non-invasive tracer method to model hepatic mitochondrial metabolism in vivo. This method, referred to as Positional Isotopomer NMR Tracer Analysis (PINTA), assesses hepatic glucose production, pyruvate carboxylase flux (Vpc) and citrate synthase flux (Vcs) based on NMR and GC/MS analysis of plasma following an infusion of [3-13C] lactate and [2H7] glucose (38). Using this approach, we examined VPC/VCS percent contributions of pyruvate to glucose and rates of gluconeogenesis in awake, overnight fasted Rhesus monkeys before and after 6-weeks of treatment with 5 mg/(Kg-day) CRMP. Additionally, we aimed to assess whether CRMP could produce chronic increases in hepatic mitochondrial oxidation rates (citrate synthase flux, VCS). Using this PINTA approach we found that acute CRMP treatment increased rates of VCS by 40% (Figure 2B). In contrast there were no changes in VCS 24-h following the last dose of chronic (6-weeks) CRMP treatment demonstrating that these effects are relatively short lived and not related to 6-week treatment; relative rates of VPC/VCS and VPC/VEGP were also unchanged after chronic CRMP treatment (Figure S6A–C). Consistent with this, DNP concentrations in chronically treated CRMP animals were not significantly different from vehicle controls (Figure S6D) and well below the concentration (<15 μmol/L) needed to obtain significant increases in hepatic mitochondrial oxidation (P=0.006, Figure 2B). Despite this washout effect, these acute increases in VCS (presumably occurring after each dose of CRMP over 6 weeks of treatment) were sufficient to increase hepatic insulin sensitivity and reduce EGP (Figure 3E–F). The reduction in rates of EGP could in turn be attributed to a 20% reduction in hepatic acetyl-CoA content [as reflected by reduced rates whole-body β-hydroxybutyrate (β-OHB) turnover and plasma β-OHB concentrations (39)] and pyruvate carboxylase flux (Figure 3G–H, Figure S6E). Indeed, linear regression analysis showed a strong correlation between percent reductions in EGP, VPC, and whole-body βOHB turnover (Figure S6F–H).
Validation of Mitochondrial Flux Measurements in Nonhuman Primates
Data from our lab demonstrates that [3-13C] lactate can be infused at a rate that provides sufficient label to measure hepatic mitochondrial fluxes without significantly altering hepatocellular mitochondrial metabolism (38, 40, 41). Specifically, the calculation of pyruvate carboxylase flux relative to citrate synthase flux (VPC/VCS) relies on the assumption that pyruvate cycling, due to fluxes through pyruvate kinase (PK) and/or malic enzyme (ME) relative to VPC is minimal and as such, would need to be adjusted for accordingly. While we have previously demonstrated that hepatic pyruvate cycling is relatively low in overnight fasted rodents, healthy control human subjects and insulin resistant human subjects with NAFLD (40, 42), this assumption has yet to be validated in nonhuman primates. As such, we next examined rates of hepatic mitochondrial oxidation and pyruvate cycling in a separate cohort of five dysmetabolic nonhuman primates following an infusion of [3-13C] lactate and liver biopsy (Figure 4A). We hypothesized that under fasting conditions, rates of pyruvate cycling (VPK) would be minimal relative to rates of VPC flux and not significantly affect our calculations of VPC/VCS. As shown in Figure 4B–C, rates of VPC/VCS and VPC/VEGP were similar across the infusion study regardless of whether [13C] glucose positional enrichments were measured in the liver (L) or adjacent plasma samples (plasma 1 and 2, P1 and P2) and correlated with VPC/VCS and VPC/VEGP rates identified in the cohort dysmetabolic Rhesus monkeys used in our efficacy study (Figure S5A–B). Furthermore, no significant differences were observed when we compared rates of VPC/VCS and VPC/VEGP calculated using PINTA or our previously validated ex vivo NMR method (Figure 4D–E), demonstrating the applicability of each method across species. Importantly, and as expected after an overnight fast, rates of pyruvate dehydrogenase flux (VPDH) relative to mitochondrial oxidation (VCS) were minimal (<6%) (Figure 4F–G). Moreover, based on our previous calculations of VPC (Figure 3G), rates of hepatic VPK flux were less than 40% that of VPC flux (Figure 4H–I), consistent with previous human studies. Taken together, these data suggest that in overnight fasted dysmetabolic Rhesus monkeys pyruvate cycling is minimal and does not significantly impact our VPC and VCS flux estimates.
Figure 4: Validation of PINTA to Assess Mitochondrial Metabolism in Nonhuman Primates.
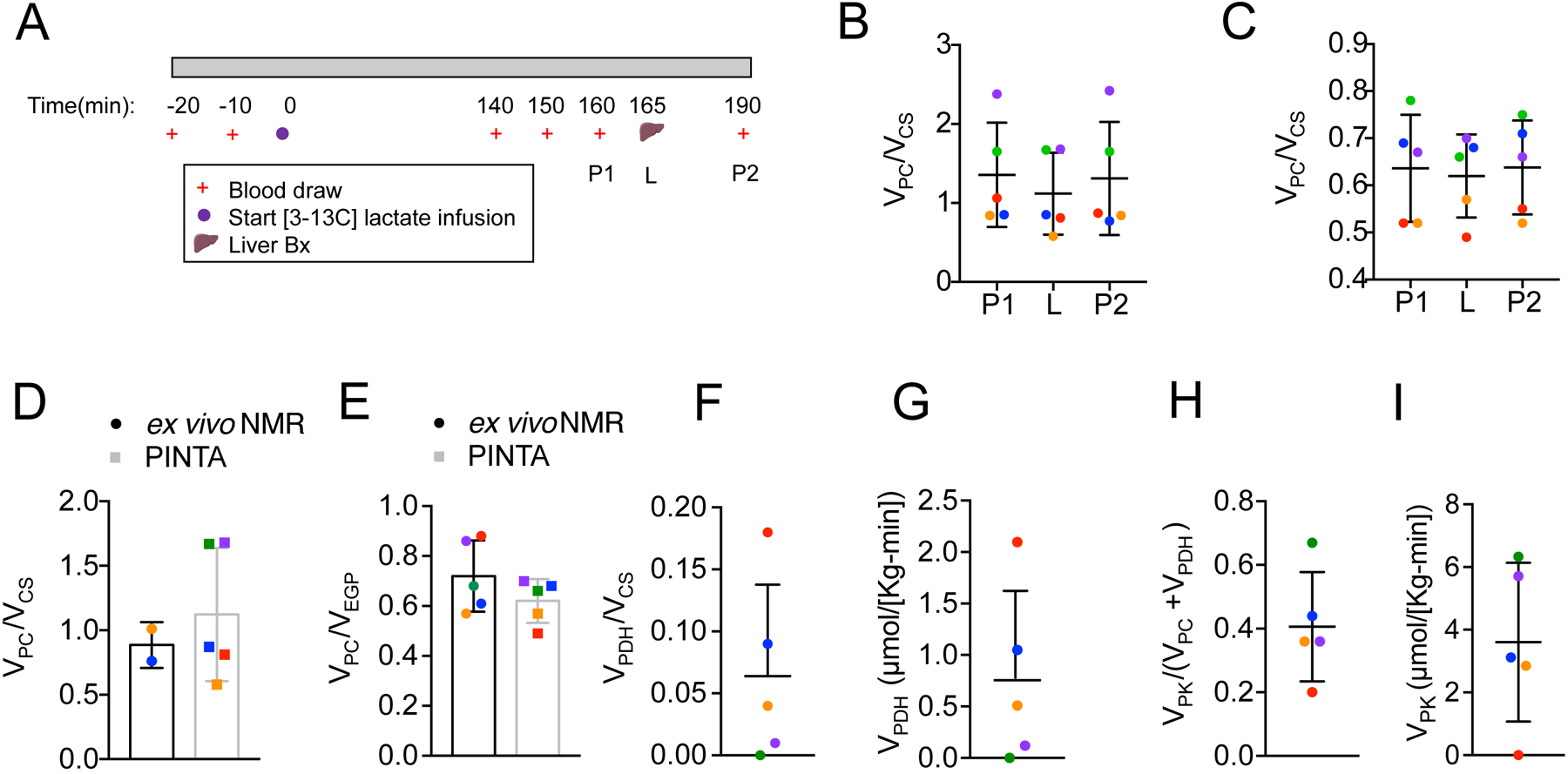
(A) Experimental outline of [3-13C] lactate infusion in dysmetabolic Rhesus monkeys (n = 5). (B-C) VPC/VCS (B) and VPC/VEGP in the plasma (P1 and P2) and livers of dysmetabolic Rhesus monkeys infused with 10 μmol/(Kg-min) [3-13C] lactate for 190 minutes as assessed by PINTA. (D-E) VPC/VCS (D) and VPC/VEGP (E) in the livers of dysmetabolic Rhesus monkeys infused with 10 μ mol/(Kg-min) [3-13C] lactate for 190 minutes as assessed by ex vivo NMR (black bars) and PINTA (grey bars). (F-I) VPDH/VCS (F), VPDH (G) VPK/VPC+VPDH (H), VPK (I) in the livers of dysmetabolic Rhesus monkeys infused with 10 μmol/(Kg-min) [3-13C] lactate for 190 minutes. In panels (B-I), data are mean ±SD.
DISCUSSION
Over the past years our understanding of the pathogenesis of NAFLD, insulin resistance and T2D has improved, and with that new pharmacological targets have emerged. Unfortunately, the complexity of insulin resistance and the presence of multiple feedback loops make it difficult to predict consequences of a particular intervention and as such, therapeutic interventions aimed at blocking biochemical pathways involved in glucose and lipid metabolism may prove difficult in practice (19). Promoting increased hepatic cellular energy expenditure through liver-targeted mitochondrial uncoupling may hold more promise. Indeed, multiple rodent studies support the use of small molecule mitochondrial uncouplers for the treatment of metabolic syndrome, despite their turbulent history in the clinic. Specifically, several groups have now demonstrated that when treated with DNP, mice fed a high-fat diet (HFD) showed reduced body weight, improved glucose tolerance, decreased plasma insulin, and decreased plasma and liver triglyceride content compared to control mice (43, 44). Our group has shown that a liver-targeted methyl-ether derivative of DNP and a slow-release, functionally liver-targeted formulation of DNP (CRMP) can safely reverse hepatic steatosis and improve insulin sensitivity in rodent models of NAFLD/NASH, T2D and lipodystrophy (27–29). Moreover, NEN, a salt form of the FDA approved drug, niclosamide, increased energy expenditure and lipid oxidation, dramatically improved hepatic steatosis and prevented diabetes in diet-induced obese and db/db mice (45). Collectively, these studies demonstrate the beneficial effects of promoting subtle sustained increases in hepatic mitochondrial uncoupling to promote fat oxidation and support this approach for treating hepatic steatosis, NASH, insulin resistance and T2D. However, to date no studies have investigated the effect of mitochondrial uncoupling to treat metabolic syndrome in a model that closely resembles humans.
Here, we examined the safety and efficacy of a functionally liver-targeted mitochondrial uncoupler (CRMP) on hypertriglyceridemia, hepatic steatosis and insulin resistance in two old-world nonhuman primate models of metabolic syndrome: high-fat fructose diet induced obese Cynolgmus monkeys and spontaneously obese dysmetabolic Rhesus monkeys. Importantly, 6-weeks of CRMP treatment (0.8 mg/Kg b.i.d. – 5 mg/Kg q.d.; peak plasma DNP concentrations of 40 μmol/L) did not cause any toxicity, inflammation, or other deleterious on-target side effects associated with systemic mitochondrial uncoupling (i.e. hyperthermia), demonstrating the feasibility of targeting DNP to the liver to improve its therapeutic index. Markers of liver function and oxidative stress also remained unchanged throughout the course of treatment in dysmetabolic Rhesus monkeys, suggesting that CRMP does not cause hepatic toxicity or promote oxidative stress in larger nonhuman primate models. Given that the 6-week no-observed-adverse-effect-level (NOAEL) of CRMP is 100 mg/kg in rodents (peak plasma concentrations of ~300 μmol/L) (28), this was not surprising, however the NOAEL of CRMP in nonhuman primates needs to be assessed in future studies.
Liver-targeted mitochondrial uncoupling has been shown to significantly reduce hypertriglyceridemia and improve hepatic steatosis in rodents by increasing fat oxidation in the liver. With this study, we confirm these findings in obese nonhuman primates. Specifically, CRMP treatment significantly increased rates of hepatic mitochondrial oxidation (citrate synthase flux, Vcs), reduced plasma and hepatic triglycerides by 30% and improved dyslipidemia. Previous studies have demonstrated that mitochondrial uncoupling reduces intracellular ATP concentrations, thereby increasing the ADP/ATP ratio and activating AMPK (46). Consequently, long term CRMP treatment may reduce circulating LDL-C by activating AMPK, phosphorylating and inhibiting HMGCR, and upregulating hepatic expression of the SREBP2-responsive gene, LDLR (47, 48). Alternatively, CRMP may activate the SREBP2 pathway by reducing hepatic acetyl-CoA content, the precursor of cholesterol biosynthesis. Indeed, here we show that 6-weeks of CRMP treatment is associated with a 20% reduction in hepatic acetyl-coA content [as assessed by βOHB turnover (39)] in dysmetabolic Rhesus monkeys. While the effect of CRMP treatment on the AMPK-ACC pathway and hepatic cholesterol synthesis remains to be determined, future studies are warranted to examine the therapeutic utility of CRMP to improve atherogenic dyslipidemia and treat cardiovascular disease.
In contrast with our rodent studies, 6-weeks of CRMP treatment did not affect fasting plasma glucose concentrations; however, plasma insulin and HOMA-IR tended to be reduced, suggesting a modest improvement in hepatic insulin sensitivity. Interestingly, a recent publication demonstrated that spontaneously obese nonhuman primates have only mild hepatic alterations in insulin signaling. Despite being classified as ‘dysmetabolic’, liver biopsies revealed that these monkeys had no sign of excess hepatic fat, suggesting that this non human primate model may not be ideal to examine lipid-induced insulin resistance (49). Indeed, in our cohort we observed minimal steatosis, with hepatic triglycerides only reaching ~25 mg/g liver tissue. In contrast, more pronounced hepatic steatosis was observed in high-fat fructose fed Cynomolgus monkeys and in this model, plasma glucose concentrations tended to be reduced with 6-weeks of CRMP treatment. Moving forward, the effect of CRMP on whole-body insulin sensitivity should be assessed in diet-induced nonhuman primate models of obesity, which develop NAFLD/NASH, hepatic insulin resistance and dysglycemia more similar to humans (50).
There is great interest in understanding the potential of therapies that modulate hepatic mitochondrial fat oxidation to reverse NAFLD. However, rates of hepatic mitochondrial fat oxidation have been difficult to assess in humans and results obtained using indirect approaches have been inconclusive. Moreover, using [13C3] propionate as a metabolic tracer, Sunny et al. recently found that rates of hepatic mitochondrial oxidation and hepatic pyruvate cycling were 2–3-fold greater in subjects with NAFLD compared to control subjects (51). In contrast, our lab has shown that rates of hepatic mitochondrial oxidation are not different between NAFLD and control subjects utilizing multiple independent biophysical approaches, in vivo 13C MRS and PINTA after an infusion of [1-13C]acetate and [3-13C] lactate (38, 42), respectively. While we and others have previously attributed these discrepancies to propionate-mediated disturbances in hepatic metabolism and/or pancreatic islet cell function (40, 52) the utility of our PINTA method to non-invasively assess rates of mitochondrial metabolism across species remained to be determined. In this study, we directly assessed rates of hepatic mitochondrial VPC flux and VCS flux using [3-13C] lactate in nonhuman primates for the first time. Importantly, in contrast to the results of Sunny et al. (51), we found that under fasting conditions, rates of hepatic pyruvate cycling (VPK) were minimal relative to rates of VPC flux and therefore do not significantly impact our calculations of VPC/VCS. Moreover, ratios of VPC/VCS were similar compared to previous studies conducted in humans and rodents, providing additional in vivo validation to the PINTA method.
Several limitations of our study require further discussion. We have previously demonstrated that CRMP is liver-targeted by first-pass metabolism in rodent models of obesity, NAFLD/NASH and T2D (28, 53), however the tissue distribution of DNP (active moiety of CRMP) was not measured in the current study. Given the on-target systemic toxicities of DNP (25), future studies using labeled CRMP and whole-body PET-MRI imaging should be conducted in nonhuman primates to assess whether DNP accumulates in the brain and/or heart before moving to Phase I clinical trials. Along these lines, long-term safety and efficacy studies with telemetry should also be conducted in nonhuman primates treated with CRMP. Another limitation is that the cohort of dysmetabolic Rhesus monkeys used in this study displayed little-to-no hepatic inflammation and/or fibrosis, which prevented us from assessing the effect of CRMP on NASH reversal. Previous studies have shown that CRMP can reduce inflammation and fibrosis in rodent models of NASH (28); therefore, studies examining the effect of CRMP to reverse hepatic fibrosis, inflammation, and/or stellate cell activation in nonhuman primates will be of great interest.
Despite these limitations, this study demonstrates that CRMP safely and effectively improves dyslipidemia and hepatic steatosis in two nonhuman primate models of metabolic syndrome, providing important proof-of-concept data for the development of novel liver-targeted mitochondrial uncoupling agents for the treatment of NAFLD/NASH and T2D and laying the groundwork for future nonhuman primate studies with liver-targeted mitochondrial uncouplers.
MATERIALS AND METHODS
Study Design.
The objectives of this study were to evaluate the safety and efficacy of CRMP treatment on hepatic mitochondrial oxidation and the reversal of hypertriglyceridemia, NAFLD and insulin resistance in dysmetabolic nonhuman primates. In one set of experiments, male Cynomolgus monkeys were fed a high fat diet (TAD 5L0P, TestDiet), along with fructose supplementation (in which 35% of calories were from fat) for 1 year. Animals were verified to be insulin-resistant and obese (body composition > 30% fat) for at least 12 months. Following a baseline characterization, monkeys were given 0.8 mg/Kg body weight CRMP (b.i.d) in ~1 tsp banana mash for 6 weeks. In another set of experiments, spontaneously obese, dysmetabolic Rhesus monkeys were fed a low fat, high complex carbohydrate diet (Labdiet 5037, Jumbo Monkey Diet) and randomized into two treatment groups: 5 mg/Kg-day CRMP or vehicle control (~ 1 tsp banana mash) for 6 weeks. The primary statistical outcome for this project was a reduction in hepatic triglyceride content assessed by liver biopsy before (baseline) and after 6 weeks of daily CRMP. We also calculated that there would be sufficient power to assess the following secondary outcomes: hepatic citrate synthase (CS) flux, ketogenesis and hepatic insulin sensitivity. Sample size was estimated to detect a difference in hepatic triglyceride content after 6-weeks of CRMP treatment and vehicle control based on the following assumptions: 1) type I error of 0.05; 2) 90% power; 3) a coefficient of variation of 40% for hepatic triglyceride content; and 4) an effect size of 1.00 (i.e. 27% of the expected mean of 3.66 prior to intervention). The n for individual experiments is indicated in the figure legends.
The following inclusion/exclusion criteria was used to enroll dysmetabolic Rhesus monkeys in the study:
Inclusion criteria: Nonhuman primates were 9–27 years of age at the pre-study (screening) evaluation; male or female gender; judged to be dysmetabolic on metabolic history and physical examination, specifically fulfilling at least 4 out of 7 metabolic parameters, which have previously been shown to correlate with increased NAFLD/NASH in dysmetabolic nonhuman primates (total fat > 20%, waist circumference > 44 cm, fasting plasma glucose > 85 mg/dL, plasma TAGs > 150 mg/dL, ALT >40 U/L males; >31 females, AST >37 U/L males; >31 females, H/R Ratio >1.4) (54).
Exclusion criteria: Nonhuman primates had a history of previous surgeries that might confound the results of the study or pose additional risk to the nonhuman primate by their participation in the study; had anemia, leukocytosis, thrombocytopenia, elevated liver and kidney values, and electrolyte disturbances; had history of pharmaceutical treatment within the past 3 months; or had a history of clinically significant endocrine, gastrointestinal, cardiovascular, hematological, hepatic, immunological, renal, respiratory, or genitourinary abnormalities or diseases.
While procedures were put in place to remove nonhuman primates from the study if an adverse event occurred (i.e. behavior deviated from normal [lethargy, discomfort, GI symptoms, reduced food intake], no monkeys had to be removed and no outliers were excluded. Blinding was performed during data collection and analysis. For each experiment, sample size reflects the number of independent biological replicates.
Rhesus Macaque Studies.
Spontaneously obese, dysmetabolic Rhesus macaques (Macaca mulatta; 9–27 years of age) of both sexes were singly housed at Crown Bioscience Louisiana Center (CBLA) under a 12 h light/dark cycle (6pm-6am) as previously described (49). Throughout the course of the study, monkeys were given ad lib access to water and food (Labdiet 5037, Jumbo Monkey Diet). In addition, each animal was given a variety of food treats (seasonal fruits and vegetables) 3 times a week. Unless otherwise specified, all studies were conducted in awake, chair-trained monkeys. For the dose escalation study, 2 dysmetabolic Rhesus monkeys were enrolled and given 1, 5, 10, and 25 mg/Kg CRMP in banana mash every 3 days for a period of 16 days. Blood was taken at 0 (pre-dose), 2, 4, 8, 12, and 24 h after each dose and immediately processed to plasma for subsequent analyses. For the efficacy study, 18 dysmetabolic Rhesus monkeys were enrolled and randomized into 2 treatment groups: vehicle (n = 6) and CRMP (n = 12). During the course of the study, animals received a voluntary, oral dose of 5 mg/Kg-day CRMP or vehicle control (banana mash). For the acute CRMP PINTA studies, 10 out of the 18 nonhuman primates used in the efficacy study were enrolled. For the PINTA validation studies, 5 nonhuman primates were enrolled (2 from the efficacy study plus an additional 3 dysmetabolic nonhuman primates).
The individual characteristics of the Rhesus monkeys utilized in each study are listed below:
| CRMP Dose Escalation Study | |||
|---|---|---|---|
| Animal ID | Sex | Age (years) | Body Weight (Kg) |
| M632 | F | 16 | 12.1 |
| M254 | F | 26 | 8.3 |
| CRMP Efficacy Study | ||||
|---|---|---|---|---|
| Animal ID | Sex | Age (years) | Body Weight (Kg) | Treatment Group |
| M956 | F | 9 | 13.74 | Vehicle |
| 75P | F | 16 | 10.38 | Vehicle |
| 88J | F | 19 | 5.92 | Vehicle |
| 35S | F | 16 | 10.68 | Vehicle |
| 38F | F | 22 | 8.34 | Vehicle |
| M767 | M | 13 | 15.65 | Vehicle |
| 031 | F | 20 | 9.30 | CRMP |
| 12K | F | 19 | 10.20 | CRMP |
| M619 | F | 17 | 10.20 | CRMP |
| 90J | F | 19 | 8.10 | CRMP |
| 98L | F | 18 | 10.08 | CRMP |
| AC94 | F | 18 | 9.40 | CRMP |
| AH78 | F | 15 | 11.74 | CRMP |
| AL33 | F | 13 | 11.06 | CRMP |
| M254 | F | 27 | 7.52 | CRMP |
| M632 | F | 17 | 13.30 | CRMP |
| M741 | M | 14 | 16.93 | CRMP |
| 68K | F | 19 | 7.92 | CRMP |
| Acute CRMP Treatment Study | |||
|---|---|---|---|
| Animal ID | Sex | Age (years) | Body Weight (Kg) |
| 031 | F | 21 | 9.49 |
| M649 | F | 18 | 10.97 |
| 90J | F | 20 | 8.5 |
| M741 | M | 15 | 17.58 |
| 98L | F | 19 | 10.47 |
| AL33 | F | 15 | 11.43 |
| M632 | F | 18 | 13.74 |
| M956 | F | 10 | 15.09 |
| 75P | F | 18 | 9.86 |
| M787 | M | 14 | 15.98 |
| PINTA Validation Study | |||
|---|---|---|---|
| Animal ID | Sex | Age (years) | Body Weight (Kg) |
| AH78 | F | 16 | 8.26 |
| 15G | F | 22 | 4.40 |
| 33720 | F | 17 | 8.04 |
| 68K | F | 21 | 5.90 |
| 37682 | M | 13 | 15.84 |
All procedures were approved by the Institutional Animal Care and Use Committee at University of Louisiana at Lafayette and guidelines set forth in the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals were followed. No animals were sacrificed or euthanized during the course of this study.
Cynomolgus Macaque Studies.
Male Cynomolgus macaques (Macaca fascicularis) were maintained on a high-fat diet (HFD; TestDiet 5L0P, Purina) with fructose supplementation (35% of calories were from fat) for more than 1 year by the Obese Resource at ONPRC and singly housed. For the efficacy study, 6 obese, insulin resistant Cynomolgus monkeys were enrolled and given 0.8 mg/Kg CRMP (b.i.d) in ~1 tsp banana mash for 6 weeks. The individual characteristics of the Cynomolgus monkeys utilized in each study are listed below:
| CRMP Efficacy Study | |||
|---|---|---|---|
| Animal ID | Sex | Age (years) | Body Weight (Kg) |
| A1 | M | 10 | 10.3 |
| A2 | M | 10 | 8.9 |
| A3 | M | 13 | 8.85 |
| A4 | M | 10 | 9.7 |
| A5 | M | 10 | 11.85 |
| A6 | M | 10 | 10.15 |
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the Oregon National Primate Research Center. No animals were sacrificed or euthanized during the course of this study.
Liver Tissue Biopsy.
Liver biopsies were obtained from dysmetabolic Rhesus monkeys at baseline, after 6 weeks of treatment with 5 mg/Kg CRMP (n = 12) or vehicle control (n = 6) and in a separate cohort of nonhuman primates used to validate PINTA for the assessment of hepatic mitochondrial metabolism. Briefly, overnight fasted (16 h) animals were sedated with ketamine (10 mg/Kg) and anesthetized with propofol and isoflurane prior to the initiation of the surgical procedure. A small midline abdominal incision was made and a piece of liver (~1 g) was excised using the guillotine method; livers were immediately flash frozen in liquid nitrogen. Before, during and after the surgery vital signs were monitored and checked to ensure they were within normal limits. All nonhuman primates were given at least 4 weeks rest before any other procedures were conducted. Liver biopsies (~50 mg) were obtained from high-fat fructose-fed Cynomolgus monkeys at baseline and after 6 weeks of treatment with CRMP (0.8 mg/Kg b.i.d) under isoflurane anesthesia per ONPRC standard operating procedures.
Body Fat Composition.
Total body and fat trunk composition were assessed in anesthetized nonhuman primates by a Dual Energy X-ray Absorptiometry (DEXA) as previously described (54, 55). Lunar Encore software (v13.6) was used to calculate fat, bone and other mass for different regions of the body.
Glucose Tolerance Tests.
Intravenous glucose tolerance tests were conducted in Rhesus monkeys as previously described (56). Briefly, after an overnight fast (16 h), animals were sedated with an intramuscular dose of 10 mg/Kg ketamine and given a bolus of 50% dextrose (250 mg/Kg) over 30 s through an intravenous cannula. Blood was then obtained through a separate intravenous cannula at 0 (pre-infusion), 3, 5, 7, 10, 15, 20 and 30-minutes post-infusion and centrifuged immediately. Plasma glucose and insulin concentrations were measured as previously described (56).
CRMP PK and Plasma DNP Measurements.
For the Rhesus CRMP dose escalation study and on the first (D1) and last day (D42) of dosing during the Rhesus efficacy study, blood samples were drawn from nonhuman primates at 0 (pre-dose), 2, 4, 8, 12 and 24 h after CRMP administration to measure DNP concentrations. For the Cynomolgus monkey CRMP PK study, unsedated blood samples were drawn at 0 (pre-dose 1), 1, 2, 3, 4, 5, 6, 7, 8, 9 (pre-dose 2), 10, 11, 24 (pre-dose 3), 25, 28, 30, 32 and 48 h after CRMP administration. Blood samples were immediately centrifuged and plasma was stored at −80°C. DNP concentrations were measured in the plasma by LC/MS-MS as previously described (28).
Clinical Chemistry, CBC and Observations.
Blood pressure, heart rate and core body temperature were collected continuously throughout the Cynomolgus monkey study with implanted telemetry units (TSE Stellar Telemetry) and data was analyzed via Notocord software. Unsedated blood samples were collected biweekly in overnight fasted Cynomolgus monkeys and immediately processed to plasma for assessment of glucose, triglycerides, total cholesterol, LDL-C, HDL-C, ALT, AST, BUN, and creatinine by COBAS as previously described (28). At Day 1, 14, 28 and 42 overnight fasted (16 h) dysmetabolic Rhesus monkeys were chaired, weighed and bled for specific metabolic parameters (CBC/clinical chemistry) as previously described (49). Hepatic sensitivity index was calculated using the following equation (Eq. 1):
| (1) |
where,
| (2) |
and
| (3) |
Hepatic Mitochondrial Flux Modeling.
Mitochondrial fluxes were measured in overnight (16 h) fasted Rhesus monkeys during baseline and after 6 weeks of CRMP treatment (5 mg/Kg-day) and in a separate cohort of monkeys 1 h post-CRMP (5 mg/Kg, p.o.) administration. Briefly, awake, chair-trained animals received a constant intravenous infusion of [3-13C] lactate (10 μmol/[Kg-min], 99% APE), [2H7] glucose (0.1 mg/[Kg-min] 99% APE), and [13C4] β-hydroxybutyrate (β-OHB) [0.01 mg/(Kg-min). Blood was drawn at −20, −10, 140, 150, 160, 170 and 180 min and processed to plasma for subsequent analysis. Whole-body β-OHB turnover and glucose turnover (endogenous glucose production, EGP) were calculated as previously described (57). Relative rates of VPC/VCS, and VPC/VEGP were determined by PINTA as previously described (17, 38) with minor modifications to correct for the possible contribution of [13C] bicarbonate to label the TcA cycle as follows. Liver [13c] bicarbonate enrichment was measured by Gc-MS (HP-1 column: 12m × 0.2mm × 0.33 μ m film, isothermal: 110 °C). Plasma (100 μl) or liver tissue (~25 μg) was placed in a GC-MS vial, purged with N2 gas, and sealed. To each sample, a saturated aqueous solution of citric acid (50 μl) was injected into the vial through the cap. After ten minutes, CO2 was sampled from the head-space and monitoring ions with m/z 44 and 45 daltons using electron impact ionization.
The fractional enrichment of glucose from 13CO2 can be increased from PC synthesis of [4-13C]OAA from 13CO2 and pyruvate. The labeling of glucose from 13-bicarbonate is dependent upon the relative flux of pyruvate to OAA with equilibration with fumarate and formation of PEP vs. flux of pyruvate to OAA to citrate (i.e. VPC/VCS). Only [1-13C]OAA (from the equilibration of [4-13C]OAA with fumarate) converted directly to PEP will label glucose (C3 and C4), since all 13CO2 of [4-13C]OAA is lost with flux through the TCA cycle. Hence, the correction of 13CO2 follows from:
13CO2 will label C4 of OAA to give [4-13C]OAA.
[4-13C]OAA randomizes to [1-13C]OAA and [4-13C]OAA. Enrichment in each position is 1/2 of the original [13CO2] enrichment.
[1-13C]OAA → [1–13C]PEP: Label of [4-13C]OAA is lost in OAA -> PEP.
[1-13C]PEP → [3-13C] glucose and [4-13C] glucose
13C of [4-13C]OAA and [1-13C]OAA is lost as 13CO2 with CS flux to 13C-citrate and first turn of the TCA cycle.
Therefore, the correction for [13CO2] is 2*[4-13C] glucose − 13CO2 × 1/2 × CF, where [13CO2] is the liver 13C-bicarbonate enrichment, and CF = (VPC + VCS)/VPC
VPC/VCS corrected for [13CO2] was determined iteratively.
In a separate set of experiments, lightly anesthetized, overnight (16 h) fasted Rhesus monkeys (n = 5) received a constant intravenous infusion of [3-13C] lactate (10 μmol/[Kg-min]) for 190 minutes. Blood was collected at −20, −10, 140, 150, 160, and 190 minutes and processed to plasma for subsequent analysis; liver tissue (~1 g) was obtained by laparoscopic surgery during the infusion and immediately snap-frozen at −80°C. Relative rates of hepatic and plasma Vpc/Vcs and Vpc/Vegp were determined by PINTA and ex vivo NMR as previously described (28). Relative rates of Vpk/Vpc+ Vpdh and Vpdh were calculated as previously described (40, 42). Pyruvate dehydrogenase flux (Vpdh) was calculated using the following equation (Eq. 4):
| (4) |
where, Vcs is the mean Vcs from the baseline nonhuman primate studies calculated in Figure S6C. Pyruvate kinase flux was calculated using the following equation (Eq. 5):
| (5) |
where, VPC is the mean VPC from the baseline nonhuman primate studies calculated in Figure 3G and VPDH denotes pyruvate dehydrogenase flux calculated using Eq. 5.
Hepatic Lipid Measurements.
Liver triglycerides were extracted by the method of Bligh and Dyer in overnight fasted nonhuman primates and quantified using the Sekisui Triglyceride-SL Kit (Sekisui) as previously described (10).
Oxidative Stress Measurements.
Protein carbonylation was determined in the livers (~50 mg) of dysmetabolic Rhesus monkeys at baseline and after 6 weeks of CRMP treatment using the Protein Carbonyl Colorimetric Assay Kit (Cayman Chemical) according to the manufacturer’s instructions. The hepatic ratio of GSH/GSSH was determined by LC-MS/MS as previously described (58).
Statistics.
All data are expressed as mean ± SD. Statistical differences were measured using an unpaired or paired two-sided Student’s t-test and one-way ANOVA with Bonferroni corrections for multiple comparisons when appropriate. A value of P <0.05 was considered statistically significant. Data analysis was performed using GraphPad Prism software Version 7 (Graphpad, San Diego, CA).
Supplementary Material
Figure S1: Safety and Efficacy Profile of CRMP Treated High-Fat Fructose-Fed Cynomolgus Monkeys
Figure S2: Oral CRMP Dose Escalation Study in Dysmetabolic Rhesus Monkeys
Figure S3: Plasma DNP Levels in Dysmetabolic Rhesus Monkeys
Figure S4: CRMP Treatment Does not Significantly Alter Oxidative Stress in Dysmetabolic Rhesus Monkeys
Figure S5: CRMP Treatment Does Not Significantly Alter Whole-body Glucose Tolerance in Dysmetabolic Rhesus Monkeys
Figure S6: Hepatic Mitochondrial Fluxes in Dysmetabolic Rhesus Monkeys Treated with CRMP
Table S1: Hematology and Clinical Chemistry in Dysmetabolic Rhesus Monkeys Before and After Treatment with CRMP
ACKNOWLEDGMENTS
We thank Keshia Toussaint, John Stack, Mario Khan, and Irina Smolgovsky for their expert technical assistance. These studies were funded by grants from the U.S. Public Health Service (R01 DK-113984, R01 DK119968, and P30 DK045735 to GIS, K99 CA-215315 to RJP and F32 DK-114954 to LG), Gilead Sciences, and the U.S. National Institutes of Health Office of the Director Grant (P51-OD-011092 for the operation of the ONPRC, support of the Obese Resource).
Footnotes
Competing Interests: RJP and GIS are inventors on Yale patents for liver-targeted mitochondrial uncoupling agents and controlled-release mitochondrial uncoupling agents for the treatment of NAFLD, NASH, T2D and related metabolic disorders (WO 2015/031756 A1). GIS serves on the advisory boards for Merck, Novo Nordisk, AstraZeneca, Gilead Sciences and Janseen Research and Development and he receives investigator-initiated support from AstraZeneca, Gilead Sciences and Merck. KFP receives investigator-initiated support from Merck. PK receives investigator-initiated support from Janssen Pharmaceuticals and Novo Nordisk. The other authors declare that they have no competing interests.
Data and Materials Availability: All data associated with this study are present in the paper or in the Supplementary Materials.
REFERENCES
- 1.Swinburn BA et al. , The global obesity pandemic: shaped by global drivers and local environments. Lancet 378, 804–814 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Younossi Z et al. , Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat Rev Gastroenterol Hepatol 15, 11–20 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Anstee QM, Reeves HL, Kotsiliti E, Govaere O, Heikenwalder M, From NASH to HCC: current concepts and future challenges. Nat Rev Gastroenterol Hepatol, (2019). [DOI] [PubMed] [Google Scholar]
- 4.Burt AD, Lackner C, Tiniakos DG, Diagnosis and Assessment of NAFLD: Definitions and Histopathological Classification. Semin Liver Dis 35, 207–220 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Younossi ZM et al. , Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Ballestri S et al. , Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J Gastroenterol Hepatol 31, 936–944 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Samuel VT, Shulman GI, Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab 27, 22–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Targher G, Day CP, Bonora E, Risk of cardiovascular disease in patients with nonalcoholic fatty liver disease. N Engl JMed 363, 1341–1350 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Farese RV Jr., Zechner R, Newgard CB, Walther TC, The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab 15, 570–573 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen MC et al. , Insulin receptor Thr1160 phosphorylation mediates lipid-induced hepatic insulin resistance. J Clin Invest 126, 4361–4371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Szendroedi J et al. , Role of diacylglycerol activation of PKCtheta in lipid-induced muscle insulin resistance in humans. Proc Natl Acad Sci U S A 111, 9597–9602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y et al. , Protein kinase C Theta inhibits insulin signaling by phosphorylating IRS1 at Ser(1101). J Biol Chem 279, 45304–45307 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Ter Horst KW et al. , Hepatic Diacylglycerol-Associated Protein Kinase Cepsilon Translocation Links Hepatic Steatosis to Hepatic Insulin Resistance in Humans. Cell Rep 19, 1997–2004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magkos F et al. , Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 142, 1444–1446 e1442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim JK, Gavrilova O, Chen Y, Reitman ML, Shulman GI, Mechanism of insulin resistance in A-ZIP/F-1 fatless mice. J Biol Chem 275, 8456–8460 (2000). [DOI] [PubMed] [Google Scholar]
- 16.Petersen KF et al. , Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 109, 1345–1350 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perry RJ et al. , Mechanisms by which a Very-Low-Calorie Diet Reverses Hyperglycemia in a Rat Model of Type 2 Diabetes. CellMetab 27, 210–217 e213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petersen KF et al. , Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54, 603–608 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goedeke L, Perry RJ, Shulman GI, Emerging Pharmacological Targets for the Treatment of Nonalcoholic Fatty Liver Disease, Insulin Resistance, and Type 2 Diabetes. Annu Rev Pharmacol Toxicol 59, 65–87 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD, Mitochondrial proton and electron leaks. Essays Biochem 47, 53–67 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tainter ML, Stockton AB, Cutting WC, Use of dinitrophenol in obesity and related conditions. A progress report. JAMA, 1472–1475 (1933). [Google Scholar]
- 22.Tainter ML, Stockton AB, Dinitrophenol in the treatment of obesity. Final report. JAMA 101, 322–336 (1935). [Google Scholar]
- 23.Cutting WC, Tainter ML, Metabolic actions of dinitrophenol with the use of balanced and unbalanced diets. JAMA 101, 2099–2102 (1933). [Google Scholar]
- 24.Simkins S, Dinitrophenol and desiccated thyroid in the treatment of obesity: a comprehensive clinical and laboratory study. JAMA 108, 2110–2118 (1937). [Google Scholar]
- 25.Grundlingh J, Dargan PI, El-Zanfaly M, Wood DM, 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J Med Toxicol 7, 205–212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samuel VT et al. , Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J Biol Chem 279, 32345–32353 (2004). [DOI] [PubMed] [Google Scholar]
- 27.Perry RJ et al. , Reversal of hypertriglyceridemia, fatty liver disease, and insulin resistance by a liver-targeted mitochondrial uncoupler. Cell Metab 18, 740–748 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI, Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347, 1253–1256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abulizi A et al. , A controlled-release mitochondrial protonophore reverses hypertriglyceridemia, nonalcoholic steatohepatitis, and diabetes in lipodystrophic mice. FASEB J 31, 2916–2924 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner JD, Cann JA, Zhang L, Harwood HJ, Diabetes and Obesity Research using Nonhuman Primates. Am Coll Lab, 699–732 (2012). [Google Scholar]
- 31.Cydylo MA, Davis AT, Kavanagh K, Fatty liver promotes fibrosis in monkeys consuming high fructose. Obesity (Silver Spring) 25, 290–293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harwood HJ Jr., Listrani P, Wagner JD, Nonhuman primates and other animal models in diabetes research. J Diabetes Sci Technol 6, 503–514 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch RA, Lee RC, Tainter ML, Dinitrophenol on Liver Function. Cal West Med 43, 337–339 (1935). [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y et al. , Hepatic Steatosis and Fibrosis in Obese, Dysmetabolic and Diabetic Nonhuman Primates Quantified by Noninvasive Echography. J Diabetes Metab 8, 767 (2017). [Google Scholar]
- 35.Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R, Protein carbonyl groups as biomarkers of oxidative stress. Clin Chim Acta 329, 23–38 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Zitka O et al. , Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett 4, 1247–1253 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Petersen MC, Shulman GI, Roles of Diacylglycerols and Ceramides in Hepatic Insulin Resistance. Trends Pharmacol Sci 38, 649–665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perry RJ et al. , Non-invasive assessment of hepatic mitochondrial metabolism by positional isotopomer NMR tracer analysis (PINTA). Nat Commun 8, 798 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perry RJ, Peng L, Cline GW, Petersen KF, Shulman GI, A Non-invasive Method to Assess Hepatic Acetyl-CoA In Vivo. Cell Metab 25, 749–756 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perry RJ et al. , Propionate Increases Hepatic Pyruvate Cycling and Anaplerosis and Alters Mitochondrial Metabolism. J Biol Chem 291, 12161–12170 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Befroy DE et al. , Direct assessment of hepatic mitochondrial oxidative and anaplerotic fluxes in humans using dynamic 13C magnetic resonance spectroscopy. Nat Med 20, 98–102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petersen KF, Befroy DE, Dufour S, Rothman DL, Shulman GI, Assessment of Hepatic Mitochondrial Oxidation and Pyruvate Cycling in NAFLD by (13)C Magnetic Resonance Spectroscopy. Cell Metab 24, 167–171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldgof M et al. , The chemical uncoupler 2,4-dinitrophenol (DNP) protects against diet-induced obesity and improves energy homeostasis in mice at thermoneutrality. J Biol Chem 289, 19341–19350 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ, Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell 7, 552–560 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Tao H, Zhang Y, Zeng X, Shulman GI, Jin S, Niclosamide ethanolamine-induced mild mitochondrial uncoupling improves diabetic symptoms in mice. Nat Med 20, 1263–1269 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhou G et al. , Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest 108, 1167–1174 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brown MS, Goldstein JL, The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell 89, 331–340 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Clarke PR, Hardie DG, Regulation of HMG-CoA reductase: identification of the site phosphorylated by the AMP-activated protein kinase in vitro and in intact rat liver. EMBO J 9, 2439–2446 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shang J et al. , Phenotyping of adipose, liver, and skeletal muscle insulin resistance and response to pioglitazone in spontaneously obese rhesus monkeys. Am J Physiol Endocrinol Metab 312, E235–E243 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Havel PJ, Kievit P, Comuzzie AG, Bremer AA, Use and Importance of Nonhuman Primates in Metabolic Disease Research: Current State of the Field. ILAR J 58, 251–268 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sunny NE, Parks EJ, Browning JD, Burgess SC, Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 14, 804–810 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tirosh A et al. , The short-chain fatty acid propionate increases glucagon and FABP4 production, impairing insulin action in mice and humans. Sci Transl Med 11, (2019). [DOI] [PubMed] [Google Scholar]
- 53.Wang Y et al. , Uncoupling Hepatic Oxidative Phosphorylation Reduces Tumor Growth in Two Murine Models of Colon Cancer. Cell Rep 24, 47–55 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lui Y et al. , Hepatic Steatosis and Fibrosis in Obese, Dysmetabolic, and Diabetic Nonhuman Primates Quantified by Noninvasive Echography. 8, 767 (2017). [Google Scholar]
- 55.Gu H et al. , Left ventricular diastolic dysfunction in nonhuman primate model of dysmetabolism and diabetes. BMC Cardiovasc Disord 15, 141 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Staup M, Aoyagi G, Bayless T, Wang Y, Chng K, Characterization of Metabolic Status in Nonhuman Primates with the Intravenous Glucose Tolerance Test. J Vis Exp, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goedeke L et al. , Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 68, 2197–2211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Madiraju AK et al. , Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature 510, 542–546 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Safety and Efficacy Profile of CRMP Treated High-Fat Fructose-Fed Cynomolgus Monkeys
Figure S2: Oral CRMP Dose Escalation Study in Dysmetabolic Rhesus Monkeys
Figure S3: Plasma DNP Levels in Dysmetabolic Rhesus Monkeys
Figure S4: CRMP Treatment Does not Significantly Alter Oxidative Stress in Dysmetabolic Rhesus Monkeys
Figure S5: CRMP Treatment Does Not Significantly Alter Whole-body Glucose Tolerance in Dysmetabolic Rhesus Monkeys
Figure S6: Hepatic Mitochondrial Fluxes in Dysmetabolic Rhesus Monkeys Treated with CRMP
Table S1: Hematology and Clinical Chemistry in Dysmetabolic Rhesus Monkeys Before and After Treatment with CRMP