

Abstract
Canonical transient receptor potential‐6 (TRPC6) channels have been implicated in the progression of several forms of kidney disease (1). While there is strong evidence that glomerular TRPC6 channels are dysregulated in diabetic nephropathy (DN), there is no consensus as to whether deletion or inactivation of TRPC6 is protective in animal models of DN. A previous study in Dahl salt‐sensitive rats suggests that TRPC6 knockout has a modest protective effect in streptozotocin (STZ)‐induced DN (2). In the present study, we examined whether inactivation of TRPC6 channels by CRISPR/Cas9 editing (Trpc6 del/del rats) affects progression of STZ‐induced DN in Sprague‐Dawley rats. Wild‐type littermates (Trpc6 wt/wt rats) were used as controls. We observed that a single injection of STZ resulted in severe hyperglycemia that was sustained over a 10‐week period, accompanied by a marked reduction in circulating C‐peptide, dyslipidemia, and failure to gain weight compared to vehicle‐treated animals. Those effects were equally severe in Trpc6 wt/wt and Trpc6 del/del rats. STZ treatment resulted in increased urine albumin excretion at 4, 8, and 10 weeks after injection, and this effect was equally severe in Trpc6 wt/wt and Trpc6 del/del rats. TRPC6 inactivation had no effect on blood urea nitrogen (BUN), plasma creatinine concentration, urine nephrin excretion, or kidney weight:body weight ratio measured 10 weeks after STZ injection. STZ treatment evoked modest and equivalent mesangial expansion in Trpc6 wt/wt and Trpc6 del/del rats. In summary, we observed no protective effect of TRPC6 inactivation on STZ‐induced DN in rats on the Sprague‐Dawley background.
Keywords: diabetic nephropathy, ion channel, nephrin, podocyte
Abbreviations
- BUN
blood urea nitrogen
- Dahl SS
dahl salt-sensitive
- DN
diabetic nephropathy
- ESRD
end-stage renal disease
- FSGS
focal and segmental glomerulosclerosis
- GBM
glomerular basement membrane
- GFR
glomerjlar filtration rate
- NTS
nephrotoxic serum
- PAN
puromycin aminonucleoside
- PAS
periodic acid-Schiff’s
- SMA
α-smooth muscle actin
- STZ
streptozotocin
- TRPC6
canonical transient receptor potential-6 channels
1. INTRODUCTION
Diabetic nephropathy (DN)(1, 2) is a major cause of end‐stage renal disease (ESRD).3, 4 Current therapies for DN entail intensive glycemic control and inhibition of renin‐angiotensin systems,3, 5 but in many cases, these therapies fail to prevent progression to ESRD.6 Consequently, it is important to identify new and plausible therapeutic targets for DN. The mechanisms driving progression of DN are complex, and treatment outcomes appear to be affected by genetic background.7 This is also reflected in animal models where the severity of DN is highly strain‐dependent.8, 9, 10, 11
Podocytes are among the earliest cell types affected in DN,12 and the proteinuria associated with DN is correlated with foot process effacement and subsequent detachment of podocytes from the glomerular basement membrane (GBM).13 Podocytes are highly differentiated polarized cells that do not readily regenerate, and therefore, podocyte loss affects the integrity of the glomerular filter and is thought to contribute to progression of glomerulosclerosis and renal dysfunction in a wide range of renal diseases.14, 15 While podocytes appear to be especially sensitive, diabetes induces functional changes in mesangial cells, glomerular endothelial cells, tubular cells, and vascular smooth muscle, and within the renal interstitium.16, 17
Canonical transient receptor potential‐6 channels are non‐selective Ca2+‐permeable cation channels expressed in many different cell types. We have recently reviewed the current understanding of the role of TRPC6 and related cationic channels in the pathophysiology of kidney disease, and their status as potential therapeutic targets.1 Podocyte TRPC6 channels are expressed at the slit diaphragm domains of foot processes18, 19 and along major processes and in the cell body.1, 20 They are also present in mesangial cells.1, 21 Gain‐of‐function mutations in the Trpc6 gene are associated with familial forms of focal segmental glomerulosclerosis (FSGS).19, 22 TRPC6 dysregulation is also linked to progression of acquired forms of proteinuric kidney diseases.1, 23, 24, 25, 26 We have recently reported that podocyte TRPC6 channels are dysregulated in the chronic puromycin aminonucleoside (PAN) nephrosis model of acquired FSGS in Sprague‐Dawley rats,1, 25 as well as in response to circulating factors implicated in primary and recurrent FSGS.24, 25 Moreover, we have observed that TRPC6 inactivation exerts a marked renoprotective effect in chronic PAN nephrosis25 and, to a lesser extent, in the nephrotoxic serum (NTS) model of autoimmune glomerulonephritis.27
It has been widely reported that glomerular TRPC6 channels are substantially more abundant in type 1 and type 2 diabetes and in podocytes cultured in the presence of elevated external glucose.28, 29, 30, 31, 32, 33 This is due at least in part to oxidative stress that can be driven by hyperglycemia, and by the surrounding pro‐inflammatory milieu.1 In addition, a protective effect of Trpc6 knockout has been reported in animal models of type 1 diabetes, although the outcomes varied substantially depending on which animal model was used. For example, a protective effect of Trpc6 knockout was observed in the Akita mouse model of type 1 diabetes at 12 and 16 weeks of age. However, the protective effects declined after that, and by 20 weeks of age, the Trpc6 knockout mice actually had more severe mesangial expansion than wild‐type controls.34 The gradual decline in protection conferred by Trpc6 knockout was attributed to several factors, including progressive insulin resistance and increased renal expression of pro‐inflammatory signaling systems that occurred as Trpc6 knockout animals became older.34 In a different study, a modest renoprotective effect was reported in the streptozotocin (STZ) model of type 1 diabetes in Dahl salt‐sensitive rats maintained on a normal diet (0.4% NaCl) in which Trpc6 was deleted using CRISPR/Cas9 gene editing.2 In those experiments, Trpc6 knockout rats exhibited a reduction in urine nephrin excretion, which suggests attenuation of diabetes‐induced podocyte detachment compared to wild‐type controls. These authors also reported a reduction in foot process effacement (although that effect was not quantified). On the other hand, they did not observe any reductions in albumin excretion or any change in light microscopic histology in diabetic Trpc6 knockout rats.2
In the present study, we have investigated whether TRPC6 channels play a role in the progression of DN in STZ‐treated Sprague‐Dawley rats, a strain that has been widely used in studies on renal physiology and pathophysiology. In these experiments, we used Sprague‐Dawley rats in which TRPC6 channels were inactivated by a global constitutive deletion in exon 2 of the Trpc6 gene generated by CRISPR/Cas9, which we have described previously.25 Rats homozygous for this deletion, hereafter referred to as Trpc6 del/del, exhibited marked protection from chronic PAN nephrosis compared to wild‐type littermate controls (Trpc6 wt/wt),25 and we hypothesized that these animals would also be protected from STZ‐induced nephropathy. However, in marked contrast to the predictions of that hypothesis, we were not able to discern either a protective or exacerbating effect of TRPC6 inactivation on renal function or on the diabetic phenotype at any time point that we examined following the initial STZ injection.
2. MATERIALS AND METHODS
2.1. Animals
All animal procedures were conducted according to protocols approved by the University of Houston Institutional Animal Care and Use Committee (IACUC) following National Institutes of Health and Animal Research: Reporting of In Vivo Experiments guidelines. These studies used male Trpc6 del/del rats at 8‐9 weeks of age and male Trpc6 wt/wt littermates, which we have described previously.25 Briefly, CRISPR/Cas9 methods were used to produce a 239‐bp deletion within exon 2 of the Trpc6 gene, which encodes an essential portion of the ankyrin repeat domain of the Trpc6 gene. As a consequence of this deletion, all of exon 2 was spliced out of the Trpc6 transcripts, resulting in non‐functional channels.25
2.2. Streptozotocin (STZ)‐induced diabetes
Rats were weighed and placed in metabolic cages for collection of 12‐hr urine samples, which were used to obtain baseline measures of renal function. Two days later, rats were administered a single i.p. injection of STZ (65 mg/kg in 0.1 mol/L Na‐citrate buffer, pH 4.5) or 0.1 mol/L Na‐citrate vehicle (pH 4.5). Animals did not receive any exogenous insulin after the STZ injection. Blood was collected via the lateral tail vein five days after injections, and STZ‐treated rats with hyperglycemia >450 mg/dL, and all of the vehicle‐treated animals, were monitored over the next 10 weeks. Additional blood samples were collected at four and ten weeks following STZ or vehicle injections to assess the progression of diabetes and to monitor renal function by measurements of blood urea nitrogen (BUN) and plasma creatinine. Urine samples were also collected at various times following the injections, and urine albumin and nephrin levels were quantified by ELISAs (Ethos Biosciences Inc), whereas creatinine was quantified using a colorimetric assay based on the Jaffe reaction (Ethos Biosciences). At the end of the 10‐week protocol, animals were euthanized by CO2 inhalation followed by cervical dislocation, kidneys were excised and weighed, and the left renal cortex was used for various immunoblot analyses. The right kidneys were used for histological analysis as described previously25 and further below.
2.3. Immunoblot analysis and enzyme‐linked immunosorbent assays
The cortex of the left kidney was diced into small pieces, lysed in 1 mL of M‐PER™ mammalian protein extraction buffer (Thermo Fisher Scientific) containing Protease Inhibitor Cocktail (Sigma‐Aldrich), and homogenized by sonication on ice. Homogenates were clarified by centrifugation at 15 115 g for 15 minutes at 4°C, and immunoblot analyses of the supernatants were carried out using standard methods as described previously.24 Antibodies used for immunoblot were mouse monoclonal anti‐TRPC6 (sc‐515837, Santa Cruz Biotechnology, used at 1:200); mouse monoclonal anti‐TRPC5 (NeuroMAB, N67/15, used at 1:200); rabbit polyclonal anti‐podocin (PA5‐79757, Thermo Fisher Scientific, used at 1:500); mouse monoclonal anti‐β‐actin (AC004, ABclonal, used at 1:5000); and mouse monoclonal anti‐α‐smooth muscle actin (α‐SMA) (clone 1A4, A2547, Sigma‐Aldrich, used at 1:500). Immunoblot experiments were performed in triplicate and quantified by densitometry using ImageJTM software. Plasma C‐peptide was measured by ELISA (#80‐COTRT‐E01; ALPCO, Salem, NH), and plasma creatinine was measured using an enzymatic assay (Crystal Chem, Elk Grove Village, IL). Urine creatinine was measured using a colorimetric assay based on the Jaffe reaction (Ethos Biosciences). BUN, glucose, triglycerides, and total cholesterol were measured using standard clinical methods at the Baylor College of Medicine Metabolic and Phenotyping Core Facility.
2.4. Histopathology
The right kidney was fixed in 10% buffered formalin, embedded in paraffin, and 4‐μmol/L sections were cut and stained with periodic acid‐Schiff (PAS).25 To assess the glomerular injury in PAS‐stained slides, 50 glomeruli per animal for all animals in all groups (N = 7‐9 rats per group) were picked at random and mesangial matrix expansion was scored by a blind observer on a scale of 0‐3 as described previously.35 Briefly, 0 = normal glomeruli; 1 = glomeruli with 25%‐50% of their volume occupied by mesangial matrix; 2 = glomeruli with 51%‐75% of their volume occupied by mesangial matrix; and 3 = glomeruli with over 76% of their volume occupied by mesangial matrix. For each animal, at least 50 glomeruli were evaluated and used to acquire a mean value for that animal. Statistical analyses were then carried out using the animal means for all animals of a given genotype and treatment group.
2.5. Statistical analyses
All statistical analyses were carried out using the online computational tools at (http://www.vassarstats.net) with α = .05. Data from urine and plasma measurements were analyzed by two‐way ANOVA. The two independent variables were genotype (Trpc6 wt/wt vs Trpc6 del/del) and drug treatment (STZ vs vehicle). To assess whether the magnitude of any of the STZ effects was affected by TRPC6 inactivation, the key statistical parameters are the F and P values for the interaction between drug effects and genotype. Immunoblot data in bar graphs are presented as mean ± SEM of triplicate independent measurements, with the ordinate normalized to the lowest value observed in a control group.
3. RESULTS
Experiments in this study were carried out on Trpc6 del/del rats that were created on a Sprague‐Dawley background, which have been described previously.25 Briefly, a 239‐bp deletion was introduced into exon 2 of the Trpc6 gene, resulting in deletion of the entire exon after transcription. The resulting truncated TRPC6 channels (which can be detected in glomeruli at very low levels) are non‐functional.25 Wild‐type (Trpc6 wt/wt) littermates were used as controls. Experiments were carried out on four groups of animals: vehicle‐treated Trpc6 wt/wt rats; STZ‐treated Trpc6 wt/wt rats; vehicle‐treated Trpc6 del/del rats; and STZ‐treated Trpc6 del/del rats. Diabetes was induced by a single intraperitoneal injection of STZ in both Trpc6 wt/wt and Trpc6 del/del rats. Plasma glucose levels were monitored at 5 days, 4 weeks, and 10 weeks following STZ injections. STZ‐treated rats exhibited severe hyperglycemia by 5 days after STZ injection, and this was apparent throughout the period of the experiment, whereas vehicle‐treated control animals exhibited stable blood glucose (Figure 1A). STZ‐treated rats of both genotypes failed to gain weight following STZ injection, whereas vehicle‐treated controls exhibited a >50% increase in body weight over the 10 weeks following injection (Figure 1B). Consistent with this, C‐peptide levels were reduced in all STZ‐treated rats (Figure 1C and Figure S1). All of the vehicle‐treated rats, regardless of their genotype, exhibited a noticeable increase in C‐peptide between 4 and 10 weeks after initial injections, and we do not know why this occurred. STZ‐treated rats exhibited a significant increase in plasma triglycerides and total cholesterol concentrations at 4 and 10 weeks after STZ administration (Figure 1D,E and Figure S1), and again, this did not depend on genotype. The absence of functional TRPC6 channels had no effect on any of these measurements, and two‐way ANOVA based on measurements made 10 weeks after injections did not discern any interaction between the effects of genotype and responses to STZ (Figure S1).
Figure 1.
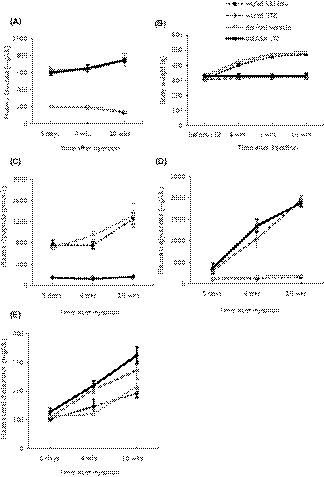
Characteristics of streptozotocin (STZ)‐induced diabetes in Sprague‐Dawley rats. Rats were given a single injection of STZ or vehicle and then followed over time. A, STZ‐injected rats exhibited hyperglycemia throughout the course of the experiment, and this was equivalent in Trpc6 wt/wt and Trpc6 del/del rats. Vehicle‐injected rats of both genotypes exhibited normal and stable blood glucose concentration. B, Vehicle‐injected rats continued to gain weight over the next 10 wk, but STZ‐injected rats did not gain weight. There was no discernible difference between Trpc6 wt/wt and Trpc6 del/del rats. C, Plasma C‐peptide was very low in STZ‐injected rats but not in vehicle‐injected rats. For unknown reasons, plasma C‐peptide levels increased between 4 and 8 wk of age, but this happened in both Trpc6 wt/wt and Trpc6 del/del rats. D, STZ treatment resulted in increased plasma triglycerides in Trpc6 wt/wt and Trpc6 del/del rats. E, STZ treatment also increased plasma total cholesterol in Trpc6 wt/wt and Trpc6 del/del rats. In this and all subsequent figures, data are presented as mean ± SEM
Streptozotocin‐treated rats exhibited changes in renal function. A baseline 12‐hr urine sample was collected from all animals two days prior to STZ or vehicle injections. STZ‐treated animals had marked increases in 12‐hr urine albumin excretion at 4, 8, and 10 weeks following injection compared to vehicle‐treated controls, and this pattern appeared virtually identical in Trpc6 wt/wt and Trpc6 del/del rats (Figure 2A). A similar pattern was observed in measurements of BUN (Figure 2B,C). Plasma creatinine (Figure 2D), urine nephrin excretion (Figure 2E), and kidney weight: body weight ratios (Figure 2F) measured at 10 weeks following injections were also increased in STZ‐treated animals. However, two‐way ANOVA revealed no significant interaction between effects of genotype and STZ on any of these parameters. In summary, STZ treatment resulted in a severe diabetic phenotype accompanied by declines in renal function, but there was no evidence of a protective effect of TRPC6 inactivation at any point during the 10‐week course of the experiment.
Figure 2.
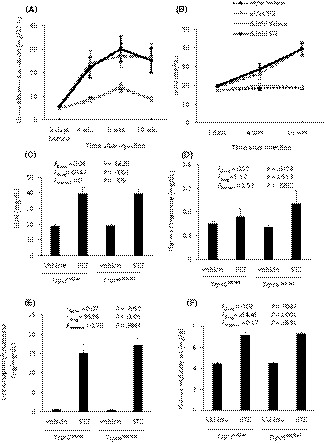
Changes in renal function during streptozotocin (STZ)‐induced diabetes in Sprague‐Dawley rats. A, Urine albumin excretion increased at 4 to 10 wk following STZ injection. This effect was equally severe in Trpc6 wt/wt and Trpc6 del/del rats. B, STZ treatment resulted in gradual increase in blood urea nitrogen (BUN) in STZ‐injected Trpc6 wt/wt and Trpc6 del/del rats, but there was no change in vehicle‐treated controls. C, Analysis of BUN in Trpc6 wt/wt and Trpc6 del/del rats at 10 wk following STZ or vehicle injection revealed a robust effect of the drug treatment but no effect of genotype. Two‐way ANOVA revealed no interaction between effects of STZ and genotype, indicating no protective effect of TRPC6 inactivation. D, A similar pattern was discerned from measurements of plasma creatinine. E, STZ treatment evoked an increase in urine nephrin excretion measured at 10 wk following injections by ELISA and normalized to urine creatinine, but two‐way ANOVA revealed no protective effect of TRPC6 inactivation. F, STZ injection resulted in increased kidney weight: body weight ratio at the end of the experiment, but two‐way ANOVA revealed no protective effect of TRPC6 inactivation
Animals were euthanized after completion of the 10‐week protocol, and mesangial expansion was quantified by an observer blind to the treatment group from PAS‐stained sections. The effects of STZ on renal histology were relatively minor compared to those that we have seen in other kidney disease models in these rat strains.25, 27 STZ treatment resulted in mesangial matrix expansion and modest interstitial hypercellularity in all of the STZ‐treated animals (Figure 3A). Glomerulosclerosis was rare. There were occasional regions where tubular atrophy and hyalinization could be detected in STZ‐treated animals. Glomeruli were scored on a semi‐quantitative scale of 0‐3 based on the severity of mesangial matrix expansion.35 These analyses revealed a modest but statistically significant effect of STZ, consistent with previous reports.4, 28, 36, 37, 38 However, there was no evidence of a protective effect of TRPC6 inactivation on glomerular pathology, and indeed, there was a trend toward an increase in mesangial matrix expansion simply as a result of TRPC6 inactivation (Figure 3A,B). In these animals, fibrosis was minimal as assessed by immunoblot analysis of α‐smooth muscle actin (α‐SMA) content (Figure 3C). Consistent with previous reports, we observed that STZ treatment resulted in an increase in the overall abundance of TRPC6 protein in renal cortex in Trpc6 wt/wt rats (Figure 4A). Note that low levels of non‐functional TRPC6 protein are detectable in Trpc6 wt/wt rats due to expression of Trpc6 transcripts lacking exon 2. Podocin levels were not affected by STZ treatment or by TRPC6 inactivation (Figure 4B). Given that STZ treatment resulted in increased urine nephrin excretion (Figure 2E), this suggests that the remaining glomerular podocytes were hypertrophic.39 It bears noting that TRPC5 channels have also been implicated in the pathogenesis of chronic kidney disease.1, 40, 41 Here, we note that there was no change in renal cortical TRPC5 abundance as a result of TRPC6 inactivation, STZ treatment, or both (Figure 4C).
Figure 3.
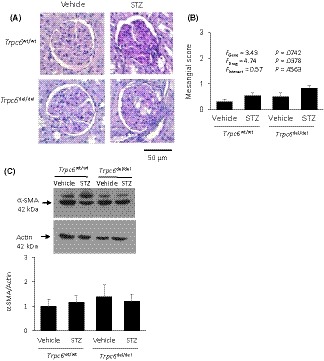
Canonical transient receptor potential‐6 (TRPC6) inactivation did not affect course of renal pathology. A, Typical examples of PAS‐stained glomeruli from streptozotocin (STZ)‐ or vehicle‐treated Trpc6 wt/wt and Trpc6 del/del rats as indicated. B, Semi‐quantitative analysis of mesangial expansion in glomeruli carried out by an observer blind to treatment group or genotype. Two‐way ANOVA indicates increase in mesangial expansion in STZ‐treated animals with no interaction between drug treatment and genotype. There is a marked trend toward increased mesangial expansion in Trpc6 del/del rats. C, Immunoblot analysis of α‐SMA revealed no effect of either STZ treatment or genotype, suggesting that fibrosis in these animals was minimal
Figure 4.
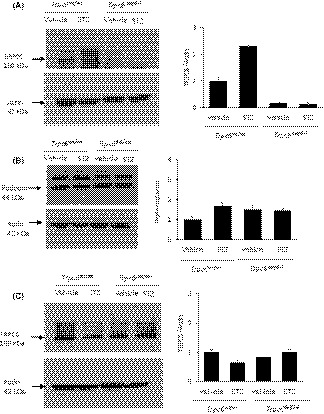
Abundance of Canonical transient receptor potential‐6 (TRPC6) and podocin in renal cortex. A, Immunoblot analysis showing increase in TRPC6 content at 10 wk after streptozotocin (STZ) injection in Trpc6 wt/wt rats, and extremely low abundance of truncated TRPC6 in Trpc6 del/del rats as described previously.25 A typical immunoblot showing results from representative animals is shown to the left, whereas densitometric analysis from the groups of animals is shown to the right. Note that those remaining TRPC6 proteins are not functional.25 B, There was no consistent effect of drug treatment or genotype on podocin abundance of renal cortex. A typical immunoblot showing results from representative animals is shown to the left, whereas densitometric analysis from the groups of animals is shown to the right. C, There was no consistent effect of drug treatment or genotype on TRPC5 abundance of renal cortex. A typical immunoblot showing results from representative animals is shown to the left, whereas densitometric analysis from the groups of animals is shown to the right
4. DISCUSSION
It has previously been shown that glomerular TRPC6 channels are upregulated in animal models of diabetes28, 31 and following exposure of podocytes to high glucose in vitro.29 It is now well established that sustained upregulation of TRPC6 can drive the progression of glomerular disease. Thus, gain‐of‐function mutations of TRPC6 result in severe familial FSGS that most typically occurs with an adult onset in humans.19, 22 Mice selectively over‐expressing either wild‐type or mutant TRPC6 in podocytes similarly exhibit albuminuria, foot process effacement, and glomerulosclerosis.42 Conversely, inactivation of TRPC6 using CRISPR/Cas9 methods reduces all aspects of kidney disease in the chronic PAN nephrosis model of FSGS in Sprague‐Dawley rats,25 and reduces glomerular disease caused by sustained angiotensin II infusions lasting up to three weeks in mice.43, 44 TRPC6 inactivation also reduced glomerulosclerosis in anti‐GBM autoimmune glomerulonephritis in rats.27 The purpose of the present study was to examine whether TRPC6 inactivation produces a similar protective effect in a rat model of diabetes.
A possible role for TRPC6 in driving DN has been examined by other investigators, but a consensus has not yet emerged, probably because of differences in experimental models. For example, using a standard low‐dose STZ model in mice, it was observed that triple knockout of the known diacylglycerol‐responsive TRPC channels (TRPC3, TRPC6, and TRPC7) reduced glomerular hypertrophy, albuminuria, and podocyte loss in diabetic animals, and also attenuated pro‐inflammatory TGF‐β signaling in glomeruli.29 Hyperglycemia was comparable in STZ‐treated triple knockout and wild‐type animals. However, triple knockout animals exhibited marked reductions in body weight (by 15%‐18%), and this effect was additive with the reductions in weight gain that typically occur in STZ diabetes in mice. With this triple knockout model, it is not possible to infer that protective effects are due to TRPC6 deletion.
A more recent study examined the effects of a single global and constitutive Trpc6 knockout in the Akita mouse model of type I diabetes.34 These workers observed reductions in albuminuria in Akita mice with Trpc6 knockout at 12 and 16 weeks of age compared to Akita mice that expressed wild‐type TRPC6 channels. However, this protective effect was no longer detected by 20 weeks of age, at which point mesangial expansion was actually more severe in Trpc6 knockout Akita mice. These authors reported enhanced p38 and cyclooxygenase 2 signaling along with gradual development of insulin resistance in Trpc6 knockout mice, which overcame the initial protective effects observed when the animals were younger.34
A modest protective effect of global Trpc6 knockout has also been reported in Dahl salt‐sensitive (Dahl SS) rats treated with a single injection of STZ.2 It is important to note that in that study, there were no improvements in urine albumin excretion, glomerular injury, glomerular fibrosis, tubular protein casts, or glomerular filtration rate (GFR) in Trpc6 knockout Dahl SS rats. However, these workers observed a reduction in foot process effacement (although this was not quantified) along with a reduction in urine nephrin content in STZ‐treated Trpc6 knockout animals.2 The later measurement can be used to infer podocyte detachment, and this pattern suggests a modest protective effect of the Trpc6 knockout. The animals in that study were maintained on a 0.4% NaCl diet and were therefore likely to be normotensive, although blood pressures were not reported.
Our experiments were carried out in rats on a Sprague‐Dawley background, which has been more extensively used in previous studies of glomerular function. Our experiments were carried out at nearly the same age as those performed by Spires et al,2 as they followed their animals for 11 weeks following STZ, whereas we monitored animals for 10 weeks. Compared to the studies of Spires et al,2 hyperglycemia in our animals was somewhat more severe during the first 28 days following STZ injection but was similar by the end of the experiment (10 weeks after STZ injection). In this study, we did not see any protective effect of TRPC6 inactivation by any measure, notably including quantitative analyses of urine nephrin levels, which were significantly elevated in STZ‐treated Trpc6 wt/wt and Trpc6 del/del rats by 10 weeks after STZ. The main experimental difference between our studies and that of Spires et al2 is the genetic background of the rats used for study. In both animal models, the CRISPR/Cas9 procedures resulted in complete loss of TRPC6 function. In both cases, TRPC6 inactivation had no impact on urine albumin excretion or light‐level histology of renal cortex. The principle difference is in our conclusions regarding podocyte detachment based on analyses of urine nephrin levels. Here, we will note that we measured nephrin using a commercial ELISA that was recently optimized for use in rats, whereas Spires et al2 used immunoblot analyses of urine. We do not think that this can explain the different conclusions, although it is somewhat surprising that Spires et al observed significant changes in urine nephrin without corresponding changes in urine albumin excretion.2
Genetic background is a major issue in the context of DN, but it has been most extensively and quantitatively studied in mice.8, 9, 10, 11 It is now clear that there is large variability in susceptibility of widely used inbred mouse strains to albuminuria and glomerulosclerosis in DN. Moreover, susceptibility loci for DN have been identified in humans.45 It is possible that the genetic changes that make Dahl SS rats a good model for studies of hypertension38 result in enhanced TRPC6 signaling somewhere within the animal (possibly but not necessarily within glomeruli). In this regard, Dahl SS rats typically exhibit slightly increased urine albumin excretion, even on low‐salt diets.46
In contrast to the pattern observed in Akita mice,34 there was no point in the course of our analyses where a protective effect of TRPC6 inactivation on urine albumin excretion or BUN could be discerned. Thus, urine albumin excretion was elevated at all of the time points following STZ treatment that we measured, (4, 8, and 10 weeks) and urine albumin excretion was indistinguishable in Trpc6 wt/wt or Trpc6 del/del rats. We cannot exclude the possibility that Trpc6 del/del rats are at least partially insulin‐resistant by the time we started our experiments, which would negate protective effects in diabetes to a greater extent than in other disease models we have examined.25, 27
In summary, in the present study we did not obtain any evidence, suggesting that TRPC6 channels are useful targets for pharmacotherapy of DN. While TRPC6 inhibitors remain plausible targets for some glomerular diseases, for example in certain forms of FSGS,1, 25, 27 the data from animal models of DN are much less encouraging. The most robust result has been obtained by simultaneous knockout of all known diacylglycerol‐responsive TRPC channels, as might be expected to occur with a less selective channel inhibitor.29 However, the effect of combined inhibition of those channels on body weight essentially precludes consideration of that approach as a therapeutic strategy. The possibility that TRPC6 inhibition might promote insulin resistance34 is especially problematic in the context of DN. Finally, we note that while there are some similarities between the pathophysiology of DN and secondary FSGS, diabetes is a complex and systemic disease that simultaneously affects multiple cell types, including virtually every cell type in the kidney. This may also explain why we observed a protective effect of TRPC6 inactivation in chronic PAN nephrosis25 but failed to see any protection in STZ‐induced diabetes.
CONFLICT OF INTEREST
The authors have nothing to disclose.
AUTHOR CONTRIBUTIONS
N. Hassanzadeh Khayyat, E.Y. Kim, and S.E. Dryer designed the research. N. Hassanzadeh Khayyat performed the research. N. Hassanzadeh Khayyat analyzed data and prepared figures. N. Hassanzadeh Khayyat, S.E. Dryer, and E.Y. Kim wrote the paper.
Supporting information
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grant R01‐DK104708.
Hassanzadeh Khayyat N, Kim EY, Dryer SE. TRPC6 inactivation does not protect against diabetic kidney disease in streptozotocin (STZ)‐treated Sprague‐Dawley rats. FASEB BioAdvances. 2019;1:773–782. 10.1096/fba.2019-00077
REFERENCES
- 1. Dryer SE, Roshanravan H, Kim EY. TRPC channels: regulation, dysregulation and contributions to chronic kidney disease. Biochim Biophys Acta. 2019;1865:1041‐1066. [DOI] [PubMed] [Google Scholar]
- 2. Spires D, Ilatovskaya DV, Levchenko V, et al. Protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am J Physiol Renal Physiol. 2018;315:F1091‐F1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Molitch ME, DeFronzo RA, Franz MJ, et al. Nephropathy in diabetes. Diabetes Care. 2004;27:S79‐S83. [DOI] [PubMed] [Google Scholar]
- 4. Tesch GH, Allen TJ. Rodent models of streptozotocin‐induced diabetic nephropathy. Nephrology (Carlton). 2007;12:261‐266. [DOI] [PubMed] [Google Scholar]
- 5. Johnson SA, Spurney RF. Twenty years after ACEIs and ARBs: emerging treatment strategies for diabetic nephropathy. Am J Physiol Renal Physiol. 2015;309:F807‐F820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruggenenti P, Cravedi P, Remuzzi G. Mechanisms and treatment of CKD. J Am Soc Nephrol. 2012;23:1917‐1928. [DOI] [PubMed] [Google Scholar]
- 7. Pezzolesi MG, Poznik GD, Mychaleckyj JC, et al. Genome‐wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes. 2009;58:1403‐1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Breyer MD, Bottinger E, Brosius FC 3rd, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27‐45. [DOI] [PubMed] [Google Scholar]
- 9. Brosius FC 3rd, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503‐2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gurley SB, Clare SE, Snow KP, Hu A, Meyer TW, Coffman TM. Impact of genetic background on nephropathy in diabetic mice. Am J Physiol Renal Physiol. 2006;290:F214‐F222. [DOI] [PubMed] [Google Scholar]
- 11. Qi Z, Fujita H, Jin J, et al. Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes. 2005;54:2628‐2637. [DOI] [PubMed] [Google Scholar]
- 12. Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626‐1634. [DOI] [PubMed] [Google Scholar]
- 13. Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74:22‐36. [DOI] [PubMed] [Google Scholar]
- 14. Kriz W, LeHir M. Pathways to nephron loss starting from glomerular diseases‐insights from animal models. Kidney Int. 2005;67:404‐419. [DOI] [PubMed] [Google Scholar]
- 15. Kriz W, Lemley KV. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int. 2017;91:1283‐1286. [DOI] [PubMed] [Google Scholar]
- 16. Vallon V, Komers R. Pathophysiology of the diabetic kidney. Compr Physiol. 2011;1:1175‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vallon V, Thomson SC. Renal function in diabetic disease models: the tubular system in the pathophysiology of the diabetic kidney. Annu Rev Physiol. 2012;74:351‐375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huber TB, Schermer B, Müller RU, et al. Podocin and MEC‐2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci USA. 2006;103:17079‐17086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reiser J, Polu KR, Möller CC, et al. TRPC6 is a glomerular slit diaphragm‐associated channel required for normal renal function. Nat Genet. 2005;37:739‐744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dryer SE, Reiser J. TRPC6 channels and their binding partners in podocytes: role in glomerular filtration and pathophysiology. Am J Physiol Renal Physiol. 2010;299:F689‐F701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sours S, Du J, Chu S, Ding M, Zhou XJ, Ma R. Expression of canonical transient receptor potential (TRPC) proteins in human glomerular mesangial cells. Am J Physiol Renal Physiol. 2006;290:F1507‐F1515. [DOI] [PubMed] [Google Scholar]
- 22. Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801‐1804. [DOI] [PubMed] [Google Scholar]
- 23. Ilatovskaya DV, Staruschenko A. TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am J Physiol Renal Physiol. 2015;309:F393‐F397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim EY, Roshanravan H, Dryer SE. Changes in podocyte TRPC channels evoked by plasma and sera from patients with recurrent FSGS and by putative glomerular permeability factors. Biochim Biophys Acta. 2017;1863:2342‐2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim EY, Yazdizadeh Shotorbani P, Dryer SE. Trpc6 inactivation confers protection in a model of severe nephrosis in rats. J Mol Med. 2018;96:631‐644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Möller CC, Wei C, Altintas MM, et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18:29‐36. [DOI] [PubMed] [Google Scholar]
- 27. Kim EY, Yazdizadeh Shotorbani P, Dryer SE. TRPC6 inactivation does not affect loss of renal function in nephrotoxic serum glomerulonephritis in rats, but reduces severity of glomerular lesions. Biochem Biophys Rep. 2019;17:139‐150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ilatovskaya DV, Blass G, Palygin O, et al. A NOX4/TRPC6 pathway in podocyte calcium regulation and renal damage in diabetic kidney disease. J Am Soc Nephrol. 2018;29:1917‐1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu B, He X, Li S, Xu B, Birnbaumer L, Liao Y. Deletion of diacylglycerol‐responsive TRPC genes attenuates diabetic nephropathy by inhibiting activation of the TGFβ1 signaling pathway. Am J Transl Res. 2017;9:5619‐5630. [PMC free article] [PubMed] [Google Scholar]
- 30. Ma R, Liu L, Jiang W, Yu Y, Song H. FK506 ameliorates podocyte injury in type 2 diabetic nephropathy by down‐regulating TRPC6 and NFAT expression. Int J Clin Exp Pathol. 2015;8:14063‐14074. [PMC free article] [PubMed] [Google Scholar]
- 31. Sonneveld R, van der Vlag J, Baltissen MP, et al. Glucose specifically regulates TRPC6 expression in the podocyte in an AngII‐dependent manner. Am J Pathol. 2014;184:1715‐1726. [DOI] [PubMed] [Google Scholar]
- 32. Yao XM, Liu YJ, Wang YM, et al. Astragaloside IV prevents high glucose‐induced podocyte apoptosis via downregulation of TRPC6. Mol Med Rep. 2016;13:5149‐5156. [DOI] [PubMed] [Google Scholar]
- 33. Zhang X, Song Z, Guo Y, Zhou M. The novel role of TRPC6 in vitamin D ameliorating podocyte injury in STZ‐induced diabetic rats. Mol Cell Biochem. 2015;399:155‐165. [DOI] [PubMed] [Google Scholar]
- 34. Wang L, Chang JH, Buckley AF, Spurney RF. Knockout of TRPC6 promotes insulin resistance and exacerbates glomerular injury in Akita mice. Kidney Int. 2019;95:321‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roshanravan H, Kim EY, Dryer SE. NMDA receptors as potential therapeutic targets in diabetic nephropathy: increased renal NMDA receptor subunit expression in Akita mice and reduced nephropathy following sustained treatment with Memantine or MK‐801. Diabetes. 2016;65:3139‐3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ilatovskaya DV, Levchenko V, Lowing A, Shuyskiy LS, Palygin O, Staruschenko A. Podocyte injury in diabetic nephropathy: implications of angiotensin II‐dependent activation of TRPC channels. Sci Rep. 2015;5:17637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kitada M, Ogura Y, Koya D. Rodent models of diabetic nephropathy: their utility and limitations. Int J Nephrol Renovasc Dis. 2016;9:279‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Slaughter TN, Paige A, Spires D, et al. Characterization of the development of renal injury in type‐1 diabetic Dahl salt‐sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2013;305:R727‐R734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim NH, Rincon‐Choles H, Bhandari B, Choudhury GG, Abboud HE, Gorin Y. Redox dependence of glomerular epithelial cell hypertrophy in response to glucose. Am J Physiol Renal Physiol. 2006;290:F741‐F751. [DOI] [PubMed] [Google Scholar]
- 40. Schaldecker T, Kim S, Tarabanis C, et al. Inhibition of the TRPC5 ion channel protects the kidney filter. J Clin Invest. 2013;123:5298‐5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhou Y, Castonguay P, Sidhom EH, et al. A small‐molecule inhibitor of TRPC5 ion channels suppresses progressive kidney disease in animal models. Science. 2017;358:1332‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krall P, Canales CP, Kairath P, et al. Podocyte‐specific overexpression of wild type or mutant trpc6 in mice is sufficient to cause glomerular disease. PLoS ONE. 2010;5:e12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Eckel J, Lavin PJ, Finch EA, et al. TRPC6 enhances angiotensin II‐induced albuminuria. J Am Soc Nephrol. 2011;22:526‐535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang L, Jirka G, Rosenberg PB, et al. Gq signaling causes glomerular injury by activating TRPC6. J Clin Invest. 2015;125:1913‐1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sandholm N, Groop PH. Genetic basis of diabetic kidney disease and other diabetic complications. Curr Opin Genet Dev. 2018;50:17‐24. [DOI] [PubMed] [Google Scholar]
- 46. Garrett MR, Dene H, Rapp JP. Time‐course genetic analysis of albuminuria in Dahl salt‐sensitive rats on low‐salt diet. J Am Soc Nephrol. 2003;14:1175‐1187. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials