

Abstract
Formation of intrahepatic bile ducts (IHBDs) proceeds in accordance with their microenvironment. Particularly, mesenchymal cells around portal veins regulate the differentiation and ductular morphogenesis of cholangiocytes in the developing liver; however, further studies are needed to fully understand the arrangement of IHBDs into a continuous hierarchical network. This study aims to clarify the interaction between biliary and liver mesenchymal cells during IHBD formation. To identify candidate factors contributing to this cell–cell interaction, mesenchymal cells were isolated from embryonic day 16.5 matrix metalloproteinase 14 (MMP14)‐deficient (knockout [KO]) mice livers, in which IHBD formation is retarded, and compared with those of the wild type (WT). WT mesenchymal cells significantly facilitated the formation of luminal structures comprised of hepatoblast‐derived cholangiocytes (cholangiocytic cysts), whereas MMP14‐KO mesenchymal cells failed to promote cyst formation. Comprehensive analysis revealed that expression of vasoactive intestinal peptide (VIP) was significantly suppressed in MMP14‐KO mesenchymal cells. VIP and VIP receptor 1 (VIPR1) were mainly expressed in periportal mesenchymal cells and cholangiocytic progenitors during IHBD development, respectively, in vivo. VIP/VIPR1 signaling significantly encouraged cholangiocytic cyst formation and up‐regulated tight junction protein 1, cystic fibrosis transmembrane conductance regulator, and aquaporin 1, in vitro. VIP antagonist significantly suppressed the tight junction assembly and the up‐regulation of ion/water transporters during IHBD development in vivo. In a cholestatic injury model of adult mice, exogenous VIP administration promoted the restoration of damaged tight junctions in bile ducts and improved hyperbilirubinemia. Conclusion: VIP is produced by periportal mesenchymal cells during the perinatal stage. It supports bile duct development by establishing tight junctions and up‐regulating ion/water transporters in cholangiocytes. VIP contributes to prompt recovery from cholestatic damage through the establishment of tight junctions in the bile ducts.
VIP is produced by periportal mesenchymal cells during the perinatal stage. It supports bile duct development by establishing tight junctions and up‐regulating ion/water transporters in cholangiocytes. VIP also contributes to prompt recovery from cholestatic liver damage through the establishment of tight junctions in the bile ducts.
Abbreviations
- 3D
three dimensional
- Ab
antibody
- Alb
albumin
- ALT
alanine aminotransferase
- Aqp
aquaporin
- AST
aspartate transaminase
- CD
clusters of differentiation
- cDNA
complementary DNA
- CFTR
cystic fibrosis transmembrane conductance regulator
- CK
cytokeratin
- Ctrl
control
- CYP
cytochrome P450
- DAPI
4′,6‐diamidino‐2‐phenylindole
- D‐Bil
direct bilirubin
- DDC
3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine
- Des
desmin
- Dlk
delta like noncanonical Notch ligand 1
- DMEM
Dulbecco's modified Eagle's medium
- E
embryonic day
- EHS
Engelbreth‐Holm‐Swarm
- EpCAM
epithelial cell adhesion molecule
- FACS
fluorescence‐activated cell sorter
- Grhl2
grainyhead‐like transcription factor 2
- HNF
hepatic nuclear factor
- IHBD
intrahepatic bile duct
- iPS
induced pluripotent stem cell
- Jag1
Jagged1
- KO
knockout
- LMC
liver mesenchymal cell
- MACS
magnet activated cell sorter
- MMP14
matrix metalloproteinase 14
- P
postnatal day
- p75NTR
p75 neurotrophin receptor
- PBS
phosphate‐buffered saline
- PE
phycoerythrin
- PLC
phospholipase C
- qRT‐PCR
quantitative reverse‐transcription polymerase chain reaction
- Rab25
ras‐associated binding protein 25
- sh
short hairpin
- SLC4A2
solute carrier family 4 anion exchanger member 2
- T‐Bil
total bilirubin
- TJP1
tight junction protein1
- Vim
Vimentin
- VIP
vasoactive intestinal peptide
- VIPhyb
vasoactive intestinal peptide/neurotensin hybrid peptide
- VIPR
vasoactive intestinal peptide receptor
- WT
wild type
Intrahepatic bile ducts (IHBDs) are located downstream of the bile canaliculi and exhibit some characteristic functions in the adult liver. They secrete water and bicarbonate ions and provide a blood–bile barrier; these functions are attributed to orchestrated activities of ion and water transporters in cholangiocytes and firm intercellular tight junctions, respectively. Under chronic liver injury, IHBDs undergo dynamic remodeling (ductular reaction). The expanded bile duct branches benefit the injured parenchyma by accelerating the excretion of bile and toxic agents, providing liver stem/progenitor cells, and triggering further regeneration.1 However, the precise molecular mechanism remains unsolved.
In the fetal stage, IHBD development begins with commitment of hepatoblasts to the biliary lineage at embryonic day 13.5 (E13.5) in mice, followed by the formation of ductal plates, primitive bile duct‐like structures, and further rearrangement into mature three‐dimensional (3D) networks.2, 3, 4 This convoluted course proceeds in accordance with the microenvironment around the portal veins.5, 6 Previous reports have demonstrated that periportal mesenchymes regulate the differentiation of cholangiocytes and morphogenesis of IHBDs through transforming growth factor‐β (TGF‐β),7 Jagged1 (Jag1)‐Notch2 signaling,8, 9, 10 and some humoral factors.11 Although Notch signaling12 and increased bile flow13 trigger the dynamic rearrangement, the mechanism by which discontinuously scattered bile duct‐like structures are integrated into a hierarchical network is not fully understood.
Since the autonomic nervous system is recognized as an important participant of the microenvironment for liver development and regeneration, several roles of neurotransmitters in the liver have been reported. Norepinephrine from the synthetic nervous system and hepatic stellate cells suppress expansion of hepatic progenitor cells and attenuate liver regeneration.14, 15 Nerve growth factor from cholangiocytes and mesenchymes plays a crucial role in modulating the intrahepatic nerve network.16 Vasoactive intestinal peptide (VIP) is a neuropeptide secreted from a plexus of autonomic nerves surrounding the biliary tract17 and stimulates bile secretion in the adult liver.18 However, the expression and function of VIP during IHBD formation in the fetal and adult injured livers remain obscure.
This study aims to elucidate the molecular mechanisms of cell–cell interaction between liver mesenchymal cells (LMCs) and biliary cells during IHBD development. Our previous report19 showed that formation of bile duct‐like structures is retarted in the developing liver of matrix metalloproteinase 14‐deficient (MMP14‐knockout [KO]) mice. Analysis of fetal LMCs in MMP14‐KO livers revealed that VIP is a candidate humoral factor for regulating IHBD development. Our in vitro cholangiocyte differentiation model indicated that VIP promoted tubular morphogenesis and maturation of IHBDs by up‐regulating ion/water transporters and promoting tight junction establishment. Furthermore, our in vivo data demonstrated the potential of VIP to facilitate the establishment of intercellular tight junctions in the bile ducts during both development and recovery from cholestatic liver injury. These data demonstrate that VIP derived from LMCs promotes the tight junction assembly in IHBDs.
Materials and Methods
Animal Studies
C57B/6J WT mice were purchased from Nihon SLC (Shizuoka, Japan) in the experiments of primary hepatoblasts, VIP‐blockage, and 3,5‐diethoxycarbonyl‐1,4‐dihydrocollidine (DDC) treatment. Systemic MMP14‐KO mice with a C57BL/6J background have been reported by Oh et al.20 MMP14‐KO mice and wild‐type (WT) littermates were obtained by crossbreeding MMP14 heterozygous mice. In the VIP‐blockage experiments during embryogenesis, VIP/neurotensin hybrid peptide (VIPhyb; Bachem AG, Bubendorf, Switzerland), a VIP antagonist, was intraperitoneally injected into pregnant mice, as described.21, 22 Briefly, 2 μg per body weight (grams) VIPhyb diluted with 200 μL phosphate‐buffered saline (PBS) was injected into pregnant WT mice twice daily from E14. The control group was injected with 200 μL PBS. The embryos/neonates were analyzed at E16, E18, and postnatal day (P) 1. In DDC‐injured models, 7‐8‐week‐old female C57B/6J WT mice were fed with 0.1% DDC containing chow or normal chow (Oriental Yeast, Tokyo, Japan) for 5 weeks. In the recovery phase, mice were fed with normal chow. For the VIP administration experiment, mice were intraperitoneally injected with recombinant VIP (1 nmol per mouse; Sigma, St. Louis, MO) dissolved in PBS or PBS alone every other day during the recovery phase from DDC injury. Blood samples were analyzed by DRI‐CHEM 7000V (Fujifilm, Tokyo, Japan). All animals were treated according to the guidelines of Tokyo Medical and Dental University.
Purification and Culture of LMCs From Fetal Liver
We used the method to dissociate E13.5 liver23 to prepare single‐cell suspensions of E16.5 mouse liver. With some modifications, flow cytometry analysis was performed according to the method employed by Suzuki et al.24 Briefly, dissociated liver cells were incubated with biotin‐conjugated p75 neurotrophin receptor (p75NTR) antibody (Ab) (Medical and Biological Laboratories, Nagoya, Japan), phycoerythrin (PE)‐Cy7‐conjugated anti‐clusters of differentiation (CD)45 Ab (eBioscience, San Diego, CA), and PE‐Cy7‐conjugated anti‐Ter119 Ab (eBioscience) for 60 minutes at 4°C. The One‐Step Antibody Biotinylation Kit (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany) was used to biotinylate the p75NTR antibody in advance according to the manufacturer's protocol. In some experiments, the cells were costained with fluorescein isothiocyanate‐conjugated anti‐delta‐like noncanonical Notch ligand 1 (Dlk) Ab (Medical and Biological Laboratories), PE‐conjugated anti‐CD31 Ab (eBioscience), or PE‐conjugated anti‐CD71 Ab (eBioscience). After washing, the stained cells were incubated with anti‐Cy7 microbeads (Miltenyi Biotec) for 15 minutes at 4°C, followed by negative selection of Cy7+ cells by a magnetic activated cell sorter (MACS) (Miltenyi Biotec). After partial removal of CD45+ and Ter119+ cells, the remaining cells were further incubated with allophycocyanin‐conjugated streptavidin protein (Thermo Fisher Scientific, Waltham, MA). Cells were subjected to flow cytometry analysis and cell sorting by a fluorescence‐activated cell sorter (FACS) (FACS Aria; Beckon Dickinson, San Jose, CA). The CD45−Ter119−p75NTR+ fraction was sorted as LMCs. Purified cells were seeded to type IV collagen‐coated plates and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin. Detailed information on antibodies used for cell sorting is described in Supporting Table S1.
Cholangiocytic Differentiation Assay of Primary Hepatoblasts In Vitro
Primary mouse hepatoblasts were differentiated into cholangiocytic lineage in 3D culture preceded by 2D preculture, as described25 with some modifications. In brief, E13.5 liver was dissociated with collagenase, and the cells were incubated with anti‐Dlk Ab for 30 minutes at 4°C. Dlk+ cells were selected using MACS, and ~2 × 105 cells per well were seeded on a type I collagen‐coated 12‐well plate. Cells were cultured in hepatocyte culture media (DMEM supplemented with 10% FBS, 2 mM L‐glutamine, 1× insulin/transferrin/selenium, 1× nonessential amino acid, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10−7 M dexamethasone). Media was changed every other day. After a 7‐day culture, cells were suspended in gel consisting of a mixture of 50% biliary culture media, 40% Engelbreth‐Holm‐Swarm (EHS) gel, and 10% type I collagen at a density of 1 × 105 or 2 × 105 cells/mL, and cultured on cell culture inserts (Millipore, Billerica, MA). The biliary culture media was DMEM/F‐12 supplemented with 1× insulin/transferrin/selenium, 12.5 mM nicotinamide, 2.5 mM 4‐(2‐hydroxyethyl)‐1‐piperazine ethanesulfonic acid, 1× nonessential amino acid, 2% B27 supplement, 0.25 μM A‐83‐01, 10 μM Y‐27632, 20 ng/mL recombinant epidermal growth factor, 100 ng/mL recombinant R‐spondin 1, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10−7 M dexamethasone (suppliers are shown in the Supporting Information). When indicated, 10−7 M VIP was added to the culture media from the initiation of culture. The media was changed every 3 days. The resulting cholangiocytic cysts were analyzed at day 12 of 3D culture. The numbers of cysts larger than 100 μm in diameter with clear lumina were counted in five randomly selected fields for each well; total number is shown in Results. Independent analyses were performed in triplicate. In the experiment of coculture of the above‐mentioned cholangiocytes with LMCs, CD45−Ter119−p75NTR+ mesenchymal cells isolated by FACS were seeded on the bottom of the same well of cholangiocytic cysts from the beginning of 3D culture.
Knockdown Assay of VIP Receptors 1 and 2
For the knockdown assay of VIP receptor (R)1 and VIPR2, short hairpin RNA (shRNA) clones were constructed within the lentivirus plasmid vector pLKO.1‐CMV‐tGFP (Sigma). Two different sequences were prepared for each shRNA target, and shRNA for luciferase was used as control. The sequences of shRNA are shown in Supporting Table S2. Lentiviruses expressing shRNA were produced in 293T cells, as described.26 HPPL (a mouse hepatoblast cell line)27 or primary hepatoblasts were infected with the lentiviruses at five multiplicity‐of‐infection at day 3 of 2D preculture before cholangiocytic differentiation. Cells were collected on day 7 of preculture and subjected to 3D culture for cholangiocytic differentiation. The number of green fluorescent protein‐positive cysts larger than 50 μm with clear lumina was counted on day 12 of 3D culture.
Hepatic Maturation Assay of Primary Hepatoblasts In Vitro
Dlk+ primary hepatoblasts were isolated from E14.5 mouse liver and subjected to hepatic differentiation, as described.11, 28 Briefly, MACS‐sorted E14.5 primary hepatoblasts were seeded on 0.1% gelatin‐coated plates at a density of 6 × 104 cells/cm2. Cells were cultured in hepatocyte culture media supplemented with 10 ng/mL recombinant oncostatin M. The media was changed every other day. After 5 days of culture, the media was replaced by hepatocyte culture media containing 20% EHS gel; the cells were then cultured further for 2 days.
Further details of materials and methods are provided in the Supporting Information.
Statistics
Prism software (GraphPad Software, San Diego, CA) was used to calculate SDs and the statistical significance of differences between samples. The Student t test and Mann‐Whitney test were applied depending on assays. P < 0.05 was considered statistically significant.
Results
LMCs Promoted the Formation of Cholangiocytic Cysts In Vitro
First, we assessed the cell–cell interaction between cholangiocytes and LMCs during bile duct formation in vitro. p75NTR is a specific cell‐surface marker for LMCs in the developing liver.24 CD45−Ter119−p75NTR+ cells were isolated from E16.5 mouse liver using FACS (Fig. 1A). This fraction was distinguished from Dlkmidp75NTR− hepatoblasts (whose expression of Dlk is slightly lower than that in E12.5‐14.5 stages), CD31+ liver sinusoidal cells, and CD71+ erythroblasts (Supporting Fig. S1A). The p75NTR+ cells showed significantly higher expression of desmin (Des), actin alpha 2 (Acta2), and LIM homeobox 2 (Lhx2) compared to E16.5 whole‐liver lysates (Fig. 1B). Their production of desmin was maintained during the culture (Fig. 1C). On the other hand, the expression levels of hepatocyte, cholangiocyte, and sinusoidal endothelial cell markers were significantly lower in p75NTR+ cells (Supporting Fig. S1B).
Figure 1.
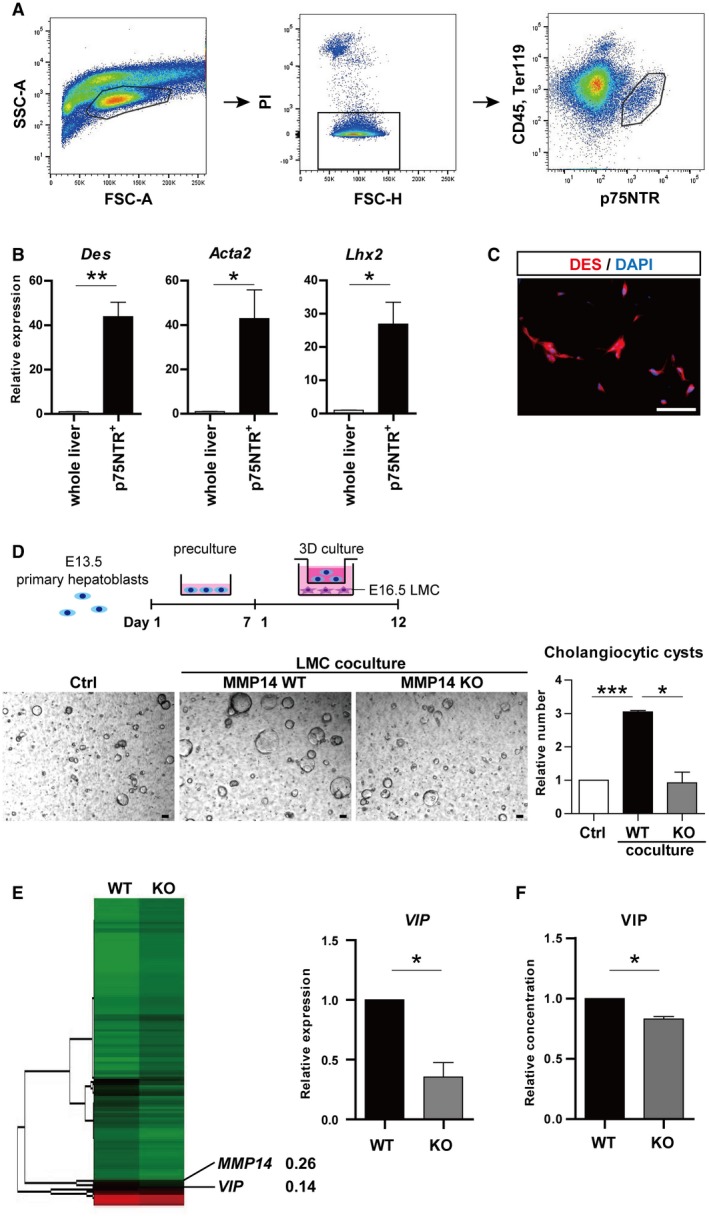
CD45−Ter119−p75NTR+ LMCs promoted the formation of cholangiocytic cysts in vitro. (A) Flowcytometric analysis of E16.5 liver cells with anti‐CD45, anti‐Ter119, and anti‐p75NTR antibodies. The CD45−Ter119−p75NTR+ fraction was sorted. (B) qRT‐PCR analysis of Des, Acta2, and Lhx2. (C) Immunostaining of DES (red). Nuclei were stained with DAPI (blue). (D) Upper panel: schema of cholangiocytic cyst‐induction strategy from E13.5 primary hepatoblasts. Two‐dimensional preculture was followed by 3D culture. In the coculture assay, E16.5 LMCs were seeded on the bottom well on day 1 of 3D culture. Lower panel: representative images and relative numbers of cholangiocytic cysts (diameter >100 µm). Number of cysts was significantly increased by trans‐well coculture with MMP14‐WT LMCs, whereas it was hardly affected by MMP14‐KO mesenchymal cells. (E) Hierarchical cluster analysis of gene expression based on microarray. Expression of VIP was decreased in MMP14‐KO LMCs (left panel). qRT‐PCR confirmed that VIP expression was significantly down‐regulated in MMP14‐KO LMCs (right panel). (F) VIP in supernatant derived from the cultured LMCs was analyzed by enzyme‐linked immunosorbent assay. Results represent the mean ± SD of at least three separate experiments (Student t test). Scale bars (C,D), 100 µm. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: Acta2, actin alpha 2; FSC‐A, forward scatter area; FSC‐H, forward scatter height; Lhx2, LIM homeobox 2; PI, propidium iodide; SSC‐A, side scatter area.
Cholangiocytic differentiation of primary hepatoblasts was assessed in combination with trans‐well coculture with LMCs. The number of WT cholangiocytic cysts was significantly increased when hepatoblasts were cocultured with WT mesenchymal cells, whereas the number was hardly affected by coculture with MMP14‐KO mesenchymal cells (Fig. 1D). Expression levels of Wnt5a, Jag1, and TGFβ1, which are known regulators of bile duct development,7, 8, 9, 10, 11 were equivalent in MMP14‐WT and MMP14‐KO mesenchymal cells (Supporting Fig. S1C). However, complementary DNA (cDNA) microarray analysis demonstrated that the expression of VIP decreased in MMP14‐KO mesenchymal cells compared to WT (Fig. 1E, left panel). No other humoral factors appeared in the top 10 down‐regulated genes in MMP14‐KO mesenchymal cells (Supporting Table S3). Both quantitative reverse‐transcription polymerase chain reaction (qRT‐PCR; Fig. 1E, right panel) and enzyme‐linked immunosorbent assay (Fig. 1F) confirmed that VIP production significantly decreased in MMP14‐KO mesenchymal cells. These data suggested that VIP derived from LMCs encourages the formation of cholangiocytic cysts from primary hepatoblasts.
VIP Promoted the Formation of Cholangiocytic Cysts Through VIPR1
Cysts were treated with VIP to investigate the role of VIP in cholangiocytic cyst formation. VIP promoted cyst formation in a dose‐dependent manner (Fig. 2A). The number of cholangiocytic cysts significantly increased with VIP treatment at 10−7 M (Fig. 2B). Immunohistologic analysis revealed that two types of VIP receptors, VIPR1 and VIPR2, were expressed on the cysts (Fig. 2C). To determine whether the receptors contribute to the promotive effect of VIP derived from mesenchymal cells, we assessed gene silencing of VIPR1 and VIPR2 in HPPL (a mouse hepatoblast cell line) (Fig. 2D). Immunoblot analysis validated the successful silencing of the target genes (Supporting Fig. S2A). The advantage of VIP treatment and coculture with LMCs in the formation of cholangiocytic cysts was canceled by gene silencing of VIPR1, whereas that of VIPR2 did not suppress the effects of VIP (Fig. 2E; Supporting Fig. S2B). Similarly, VIP failed to promote the formation of cysts from VIPR1 knocked‐down primary hepatoblasts (Supporting Fig. S2C). These data clearly indicated that VIP derived from LMCs enhances the formation of cholangiocytic cysts from hepatoblasts through VIPR1.
Figure 2.
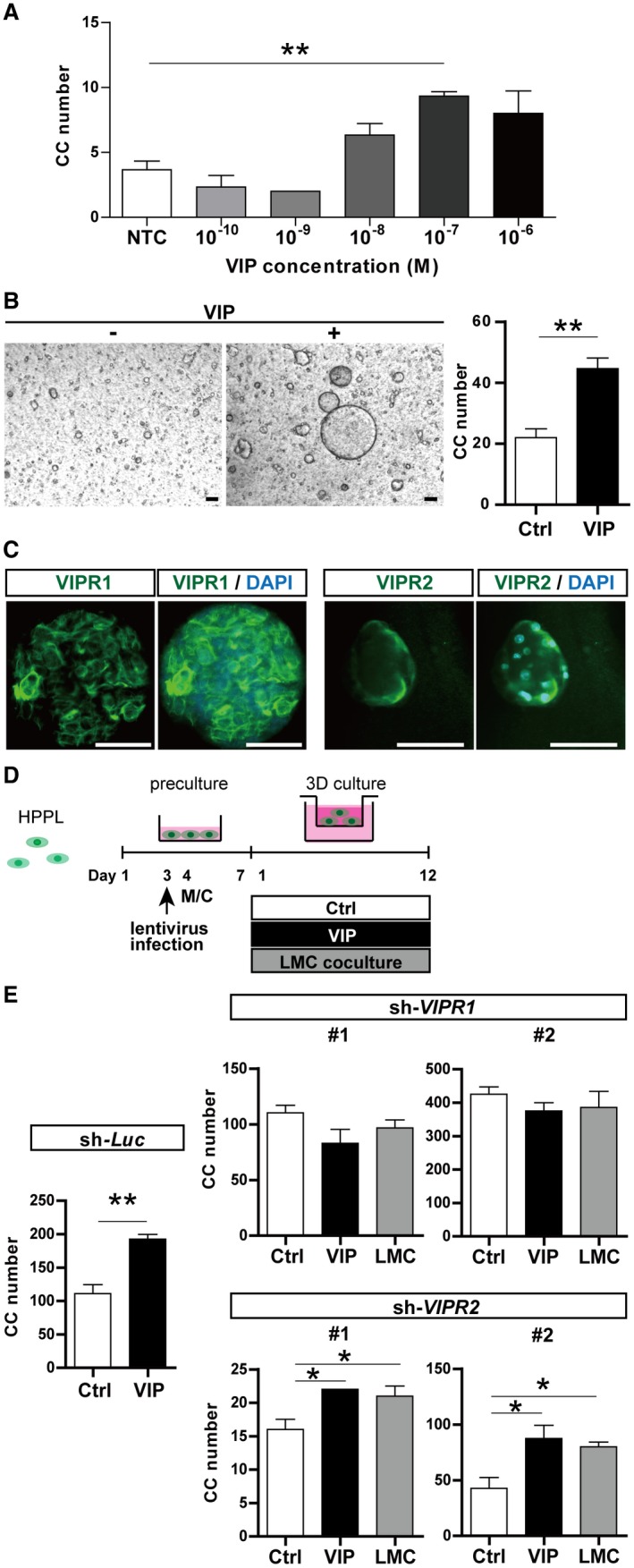
VIP enhanced formation of cholangiocytic cysts through VIPR1. (A) VIP promoted cholangiocytic cyst formation in a dose‐dependent manner. (B) Representative phase contrast views (left panel) and numbers (right panel) of cysts cultured with and without VIP. Number of cholangiocytic cysts (diameter >100 µm) was significantly increased by VIP treatment at 10−7 M. Scale bar, 100 µm. (C) Immunostaining of VIPR1 (green, left panel) and VIPR2 (right panel) in cholangiocytic cysts. Nuclei were stained with DAPI (blue). Scale bar, 100 µm. (D) Outline of the strategy for silencing of VIPR in HPPL (a hepatic progenitor cell line)27 and following induction of cholangiocytic cysts. HPPL cells were transduced with lentivirus expressing shRNA‐VIPR1, shRNA‐VIPR2, or shRNA‐Luc. Cells were subsequently subjected to 3D culture in combination with VIP treatment or coculture with LMCs. (E) Numbers of cholangiocytic cysts (diameter >50 µm) were counted on day 12 of 3D culture. The advantage of VIP treatment and LMC coculture was canceled by silencing of VIPR1 in HPPL but was maintained by silencing of Luc and VIPR2. Results represent the mean ± SD (Student t test). *P < 0.05, **P < 0.01. Abbreviations: CC, cholangiocytic cysts; Luc, luciferase; M/C, medium change; NTC, negative control.
Expression of VIP and VIPR1 During the Perinatal Stage In Vivo
The function of VIP in the formation of IHBDs remains unclear. Therefore, we studied in vivo localization of VIP and VIPR1 during IHBD development. The expression of VIPR1 increased from E13 to P1 in a time‐dependent manner and was maintained in the adult liver (Fig. 3A). VIP expression in whole liver increased from E18 to P1 (Fig. 3B, upper panel), and VIP expression in LMCs was elevated from as early as E16 and further in P1 (Fig. 3B, lower panel).
Figure 3.
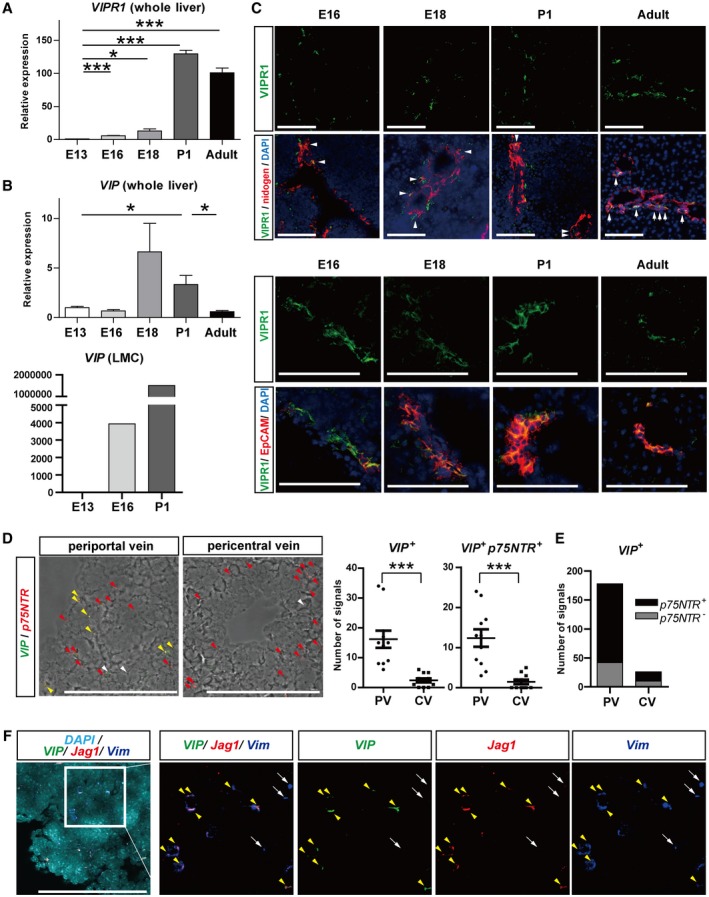
VIPR1 and VIP were detected in bile duct‐like structures and periportal mesenchymal cells, respectively, in the perinatal‐stage liver. (A,B) qRT‐PCR analysis of (A) VIPR1 and (B, upper panel) VIP. Both were increased in a time‐dependent manner during development in whole‐liver analysis. VIP peaked at the perinatal stage, whereas VIPR1 was maintained in adults. Expression of VIP was also markedly increased in E16 to P1 in LMCs (B, lower panel). (C) Upper panels: double immunostaining of VIPR1 (green) and nidogen (red). Nuclei were stained with DAPI (blue). During bile duct formation (E16‐P1), VIPR1 was detected in bile duct‐like structures (white arrowheads) and parenchyma in periportal areas. In the adult liver, VIPR1 was exclusively localized in bile ducts (white arrows). Lower panels: double immunostaining of VIPR1 (green) and EpCAM (red). Although VIPR1 was detectable in part of EpCAM− cells in periportal areas at E16, its expression was strictly localized to EpCAM+ cholangiocyte‐lineage cells at E18, P1, and adult livers. (D) In situ hybridization analysis of VIP and p75NTR in P1 liver. Left panels: fluorescent images layered with bright‐field images. Yellow, white, and red arrowheads represent VIP+p75NTR+, VIP+p75NTR−, and VIP−p75NTR+ signals, respectively. VIP signals (green dots) were observed in periportal areas, whereas p75NTR signals (red dots) were widely distributed in the liver. VIP signals colocalized with p75NTR signals represent VIP‐producing LMCs (yellow arrowheads). Right panels: numbers of p75NTR and/or VIP signals around PVs and CVs were counted in 11 randomly chosen fields from three different mice livers for each group. VIP signals, either colocalized with p75NTR signals or not, were significantly increased in PV areas compared to CV areas. (E) We detected 76.4% and 61.5% VIP signals in p75NTR + LMCs in PV and CV areas, respectively. (F) In situ hybridization analysis of VIP (green), Jag1 (red), and Vim (blue) in P1 liver. Nuclei were stained with DAPI (bright blue). VIP signals were observed in Vim + Jag1 + cells (yellow arrowheads). Alhough there were also Vim + Jag1− cells around PVs, which represent hepatic stellate cells, these cells were VIP negative (white arrows). Results represent the mean ± SD; (A,B) Student t test; (D) two‐tailed Mann‐Whitney test. Scale bars (C,D,F), 100 µm. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: CV, central vein; PV, portal vein.
Double immunostaining analysis of VIPR1 and nidogen (Fig. 3C, upper panels) showed that VIPR1 is expressed in most bile duct‐like structures from E16 to P1 (arrowheads), whereas VIPR1 was also detected in part of periportal parenchymal cells. In the adult liver, VIPR1 was clearly localized to IHBDs lined by nidogen (arrows) and was absent in the parenchyma. Moreover, production of VIPR1 was predominantly detected in epithelial cell adhesion molecule (EpCAM)+ cholangiocyte‐lineage cells (Fig. 3C, lower panels). Although VIPR1+EpCAM− cells were present around portal veins at the E16 stage, they gradually diminished as the development proceeded. VIPR1 was strictly localized to EpCAM+ cholangiocyte‐lineage cells after the P1 stage (Fig. 3C, lower panels).
To investigate the distribution of VIP‐expressing cells in the developing liver, in situ hybridization of VIP was analyzed in P1 livers (Fig. 3D‐F). At P1, nerve terminals do not reach liver parenchyma and p75NTR is a specific marker for LMCs.16 The number of VIP signals was significantly higher in the periportal area compared to the pericentral area (Fig. 3D). VIP signals were colocalized in p75NTR + LMCs (Fig. 3D, yellow arrowheads). The majority of VIP‐expressing cells were p75NTR + mesenchymal cells in P1 livers, whereas 23.6% and 38.5% of VIP signals in the periportal and pericentral areas, respectively, were detected in p75NTR − cells (Fig. 3E).
To determine whether the VIP‐expressing cells were portal mesenchymal cells, expression of VIP was assessed together with Vimentin (Vim, a common marker for LMCs) and Jag1 (a specific marker for portal mesenchymal cells) (Fig. 3F). All VIP signals were observed in Vim + Jag1+ cells (Fig. 3F, yellow arrowheads). Vim + Jag1 − cells, which represent hepatic stellate cells, were also observed; however, these cells did not express VIP (Fig. 3F, arrows).
These in vivo findings demonstrated that VIP and VIPR1 are predominantly expressed by perivascular mesenchymal cells in the portal area and by cholangiocyte‐lineage cells consisting of bile duct‐like structures, respectively, during the development of IHBDs. This is consistent with our in vitro data.
VIP Promoted Cholangiocytic Cyst Formation by Enhancing the Establishment of Tight Junctions and Up‐Regulating Ion/Water Transporters in the Cysts
To clarify the molecular mechanism of how VIP promotes the formation of bile ducts from hepatoblasts, three possible hypotheses were verified: (1) accelerated cell proliferation, (2) assembly of intercellular tight junctions, and (3) establishment of the water transportation system. Neither the number of cells consisting of cholangiocytic cysts (Fig. 4A) nor cyclin D1 expression (Fig. 4B) increased in VIP‐treated cysts, indicating that cell proliferation did not contribute to the promotion of cyst formation by VIP. As for tight junctions, the expressions of grainyhead‐like transcription factor 2 (Grhl2), ras‐associated binding protein 25 (Rab25), and tight junction protein 1 (Tjp1) significantly increased in VIP‐treated cysts compared with vehicle‐treated ones (Fig. 4B). TJP1 protein in VIP‐treated cholangiocytic cysts also increased (Fig. 4C‐E), demonstrating that VIP encourages the establishment of intercellular tight junctions in the cysts.
Figure 4.
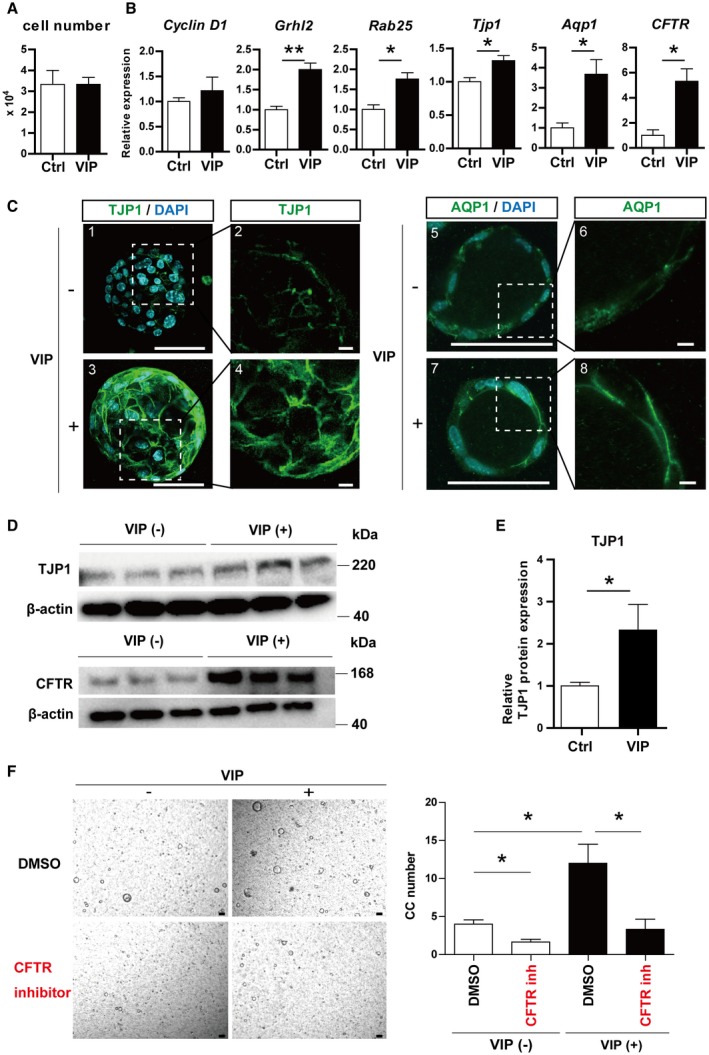
VIP promoted the establishment of tight junction and the production of CFTR and Aqp1 in cholangiocytic cysts. (A) Number of cells consisting of cholangiocytic cysts. (B) qRT‐PCR analysis of cholangiocytic cysts induced with or without VIP. Expression of cyclin D1 was not changed. Expression of genes related to tight junction (Grhl2, Rab25, and Tjp1) and ion/water transportation (Aqp1 and CFTR) was significantly increased in VIP‐treated cysts. (C) Immunostaining of TJP1 (ZO‐1; green, 1‐4) and AQP1 (green, 5‐8). Nuclei were stained with DAPI (blue). Production of TJP1 was increased by VIP and clearly lined the edge of cells. AQP1 was also clearly localized to the cell membrane in VIP‐treated cysts. Scale bars, 100 µm (1, 3, 5, 7), 10 µm (2, 4, 6, 8). (D) Immunoblot of TJP1 (upper panels) and CFTR (lower panels). Production levels in cholangiocytic cysts were both increased by VIP treatment. (E) Densitometry of TJP1 immunoblot shown in Fig. 4D, which was normalized by β‐actin. (F) Representative views (left panels) and numbers (right panels) of cholangiocytic cysts treated with VIP and/or CFTR inhibitor. Cholangiocytic cyst induction was impaired in the presence of CFTR inhibitor. Scale bars, 100 µm. Results represent the mean ± SD (Student t test). *P < 0.05, **P < 0.01. Abbreviations: CC, cholangiocytic cysts; DMSO, dimethyl sulfoxide; inh, inhibitor; ZO‐1, zonula occludens 1 (also known as TJP1).
To analyze the water transportation system in cholangiocytes, cystic fibrosis transmembrane conductance regulator (CFTR), aquaporin 1 (Aqp1), and solute carrier family 4 anion exchanger member 2 (SLC4A2) in the apical side and Aqp4 in the basolateral side were analyzed. The expression of Aqp1 significantly increased (Fig. 4B) and AQP1 protein was more clearly localized to the cell surface in VIP‐treated cysts compared to the control (Fig. 4C, right panels). CFTR was significantly up‐regulated in VIP‐treated cysts (Fig. 4B,D). Cholangiocytic cyst formation was significantly impaired by CFTR inhibitor, and the advantage of VIP treatment was also attenuated (Fig. 4F), indicating that VIP promotes cyst formation in a CFTR‐dependent manner. There was no significant change in the expression of Aqp4 or Slc4a2 (Supporting Fig. S3A). These data demonstrated that VIP promotes cholangiocytic cyst formation through the establishment of tight junctions and the up‐regulation of CFTR and Aqp1.
VIP Exhibited the Potential to Suppress the Commitment of EpCAM+ Hepatoblasts into the Hepatocyte Lineage
The effect of VIP on the fate of hepatoblasts was also investigated. Cholangiocytic markers in VIP‐treated cysts were equivalent to those in the controls (Supporting Fig. S3B). Moreover, neither the expression of hepatocyte nuclear factor 1β (HNF1β), HNF6, cytokeratin 7 (CK7), and CK19 nor the potential of cholangiocytic cyst formation was changed by VIP treatment during 2D preculture (Supporting Fig. S3C), indicating that VIP hardly affects fate determination of the hepatoblasts to the cholangiocyte lineage.
Next, the influence of VIP on hepatic maturation of hepatoblasts was investigated in vitro (Fig. 5A; Supporting Fig. S3D,E).28 Expression levels of hepatic nuclear factor 4α (HNF4α), alpha‐fetoprotein (Afp), and albumin (Alb) were not significantly changed (Fig. 5B), whereas maturation markers, such as tyrosine aminotransferase (TAT) and tryptophan oxygenase (TO), were significantly decreased by VIP treatment (Fig. 5C). VIP suppressed the expression of enzymes highly expressed in zone 1 (periportal) hepatocytes, which were arginase (Arg1) and carbamoyl‐phosphate synthetase 1 (CPS‐1). The expression of glutamine synthetase (GS), cytochrome P450, family 1, subfamily a, polypeptide 2 (CYP1A2), and CYP2E1, which have a distribution localized to zone 3 hepatocytes, was unchanged (Fig. 5D).
Figure 5.
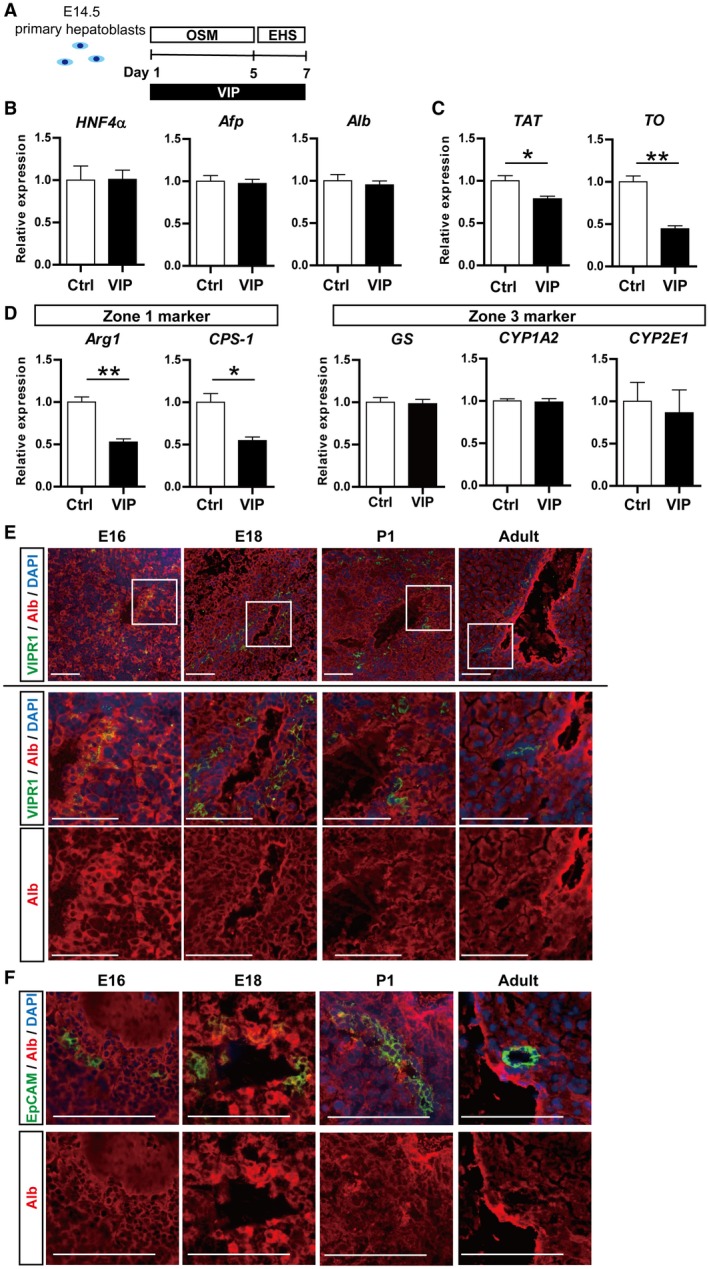
VIP showed the potential to suppress the commitment of EpCAM+ cholangiocytic‐lineage cells to hepatocyte lineage. (A) Outline of hepatic maturation experiment of E14.5 primary hepatoblasts. (B‐D) qRT‐PCR of hepatocyte markers. (B) While expression levels of HNF4α, Afp, and Alb were not affected, (C) those of TAT and TO were significantly suppressed by VIP. (D) VIP showed a specific inhibition of hepatic markers of zone 1 (Arg1 and CPS‐1), whereas it did not change those of zone 3 (GS, CYP1A2, and CYP2E1). (E) Double immunostaining of VIPR1 (green) and EpCAM (red). Nuclei were stained with DAPI (blue). Squared area in the top lane is shown in higher magnification in lower panels. VIPR1 was detected in part of Alb+ hepatoblasts in periportal parenchyma of E16 and E18 liver and in Albdull hepatoblasts in P1 liver. In the adult liver, Alb was undetectable in VIPR1+ cells. (F) Double immunostaining of EpCAM (green) and Alb (red). EpCAM+ cells around portal veins expressed Alb in E16, E18, and P1 liver. EpCAM and Alb were mutually exclusive in the adult liver. Scale bars (E,F), 100 µm. Results represent the mean ± SD (Student t test). *P < 0.05, **P < 0.01. Abbreviations: Afp, alpha‐fetoprotein; Arg1, arginase; CPS‐1, carbamoyl‐phosphate synthetase 1; GS, glutamine synthetase; OSM, oncostatin M; TAT, tyrosine aminotransferase; TO, tryptophan oxygenase.
Whereas VIPR1 was predominantly expressed in EpCAM+ cholangiocyte‐lineage cells in the perinatal stage (Fig. 3C), it was detected in part of Alb+ hepatoblasts in the periportal parenchyma of E16 and E18 liver and in Albdull hepatoblasts, which present relatively weak expression of Alb, in P1 liver in vivo (Fig. 5E). In E16, E18, and P1 liver, EpCAM+ cells around portal veins produced Alb, indicating the presence of EpCAM+VIPR1+Alb+~dull cells in zone 1 of perinatal‐stage livers (Fig5F). Moreover, EpCAM+ cholangiocyte‐lineage cells derived from neonatal liver exhibit a potential to differentiate into hepatocyte lineages in vitro.29 Taken together, these results suggest that VIP/VIPR1 signaling is a potential suppressor of hepatocytic differentiation of EpCAM+VIPR1+Alb+~dull cells during the perinatal stage.
VIP Promoted Cholangiocytic Cyst Formation by Human‐Induced Pluripotent Stem Cell‐Derived Cells
To study the effect of VIP on cholangiocytic cyst formation by human cells, human‐induced pluripotent stem cell‐derived hepatic progenitor cells (iPS‐HPCs) were differentiated into the cholangiocytic lineage30 with or without VIP. VIP treatment increased the number of cholangiocytic cysts derived from iPS‐HPCs, whereas its advantage was canceled by VIP antagonist (Supporting Fig. S4A). Expression of tight junction markers (Grhl2 and Rab25) and CFTR in the cysts was significantly increased by VIP treatment. Although there were some differences between species, VIP promoted the formation of human cholangiocytic cysts by encouraging the establishment of tight junctions and by up‐regulating CFTR expression, in accordance with mice data (Supporting Fig. S4B‐F).
Impaired VIP Signaling During Development Attenuated the Establishment of Tight Junctions in Bile Duct‐Like Structures In Vivo
To validate the functional significance of VIP in the development of IHBDs in vivo, WT pregnant mice were treated with VIPhyb, an antagonist of VIP effective in mice during embryogenesis, as described (Fig. 6A).21, 22, 31
Figure 6.
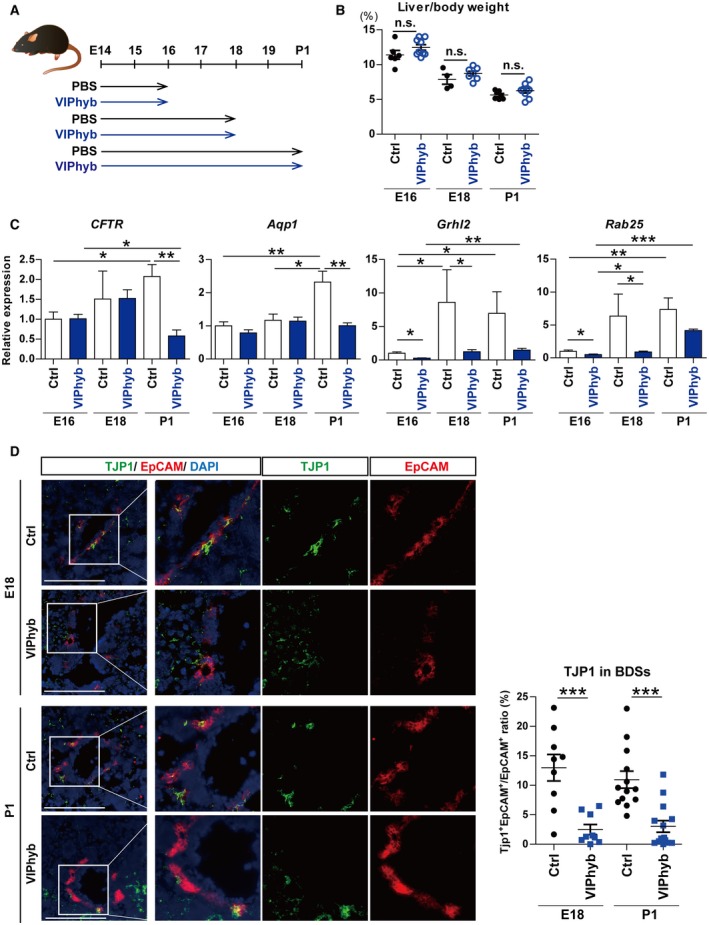
Impaired VIP signaling during development slowed the up‐regulation of ion/water transporters and the establishment of tight junction in bile duct‐like structures in vivo. (A) Outline of VIP blockage model in vivo. Pregnant WT mice were injected with 2 μg per body weight (g) of VIPhyb, a VIP antagonist, diluted with PBS twice daily from E14 to the day of analysis. The control group was injected with PBS. Embryos were analyzed at E16, E18, and P1. (B) Liver to body weight ratio at E16, E18, and P1. There was no significant change between control and VIPhyb groups. (C) qRT‐PCR of CFTR, Aqp1, Grhl2, and Rab25. All these genes increased as development proceeded in the control group. In the VIPhyb group, CFTR and Aqp1 were hardly elevated during development and were significantly lower than the control at the P1 stage. Grhl2 and Rab25 were also significantly down‐regulated compared to the control at E16 and E18. (D) Left panel: double immunostaining of TJP1 (green) and EpCAM (red). Nuclei were stained with DAPI (blue). Scale bars, 100 µm. Right panel: areas of EpCAM+ and TJP1+EpCAM+ cells, respectively, were quantified. TJP1+EpCAM+ to EpCAM+ ratio was significantly decreased by VIPhyb treatment. Nine to 12 randomly selected fields from three different E18 and P1 livers were analyzed in the control and VIPhyb groups. Results represent the mean ± SD. (B,D) Two‐tailed Mann‐Whitney test; (C) two‐tailed t test. *P < 0.05, **P < 0.01, ***P < 0.001. Abbreviations: BDS, bile duct‐like structure; n.s., not statistically significant.
There was no significant difference in liver to body weight ratio between the VIPhyb and the control (vehicle) groups (Fig. 6B). While the expression levels of CFTR and Aqp1 increased as the development proceeded in the control group, they were hardly elevated in the VIPhyb group, resulting in significantly lower expression than the control group in P1 liver (Fig. 6C). Grhl2 and Rab25 in the VIPhyb‐treated fetal livers were significantly down‐regulated compared to the control at E16 and E18 (Fig. 6C). To evaluate the effect of VIP blockage on TJP1 production in IHBDs alone, TJP1 protein in EpCAM+ cells was quantitatively examined by immunohistologic staining and image analyzer. The ratio of TJP1+EpCAM+ to EpCAM+ areas was significantly lower in the VIPhyb group compared with the control both in E18 and P1 liver (Fig. 6D).
These data clearly demonstrated that VIP is necessary for the up‐regulation of CFTR and Aqp1 and also plays an important role in the establishment of tight junctions during the development of IHBDs in vivo.
Tight Junction Is Impaired in DDC‐Injured IHBDs, Accompanied by Down‐Regulation of VIPR1 and VIP
Finally, the functional significance of VIP in the liver with IHBD damage was explored in an in vivo model. DDC‐injured liver is a well‐known experimental model of cholestatic injury and remodeling of IHBDs (ductular reaction). Moreover, the blood–bile barrier is impaired during DDC treatment because of the failure of tight junctions.32 Consistent with that report, Tjp1 expression in the liver, particularly in IHBDs, significantly decreased after 5 weeks of DDC treatment as ductular reaction was induced and increased during the recovery period (Supporting Fig. S5A,B). Although VIPR1 diminished by DDC treatment, its production was detectable as early as 1 week from DDC removal. It was further elevated in the later recovery phase (Supporting Fig. S5A,B).
VIP expression in nonparenchymal cells was significantly down‐regulated in DDC‐treated mice and was gradually elevated during the recovery (Supporting Fig. S5C,D). In situ hybridization assay of VIP, Vim, and Jag1 in the adult liver revealed that ~70% to 90% of VIP‐expressing Vim + mesenchymal cells were positive for Jag1 both in healthy normal livers and in the recovery phase from DDC damage in the adult liver (Supporting Fig. S5E, yellow arrowheads). Although a few Jag1 − Vim + cells expressing VIP (white arrows) were detected, this result indicated that the majority of VIP‐expressing cells are Jag1 + portal mesenchymal cells in the healthy and DDC‐injured adult livers as well as neonatal livers.
Reconstitution of Tight Junctions and Recovery From Hyperbilirubinemia Were Facilitated by VIP Treatment
Based on the above‐mentioned data, we assumed that restoration of impaired tight junctions in DDC‐injured IHBDs is facilitated by exogenous VIP supplement. To verify this hypothesis, VIP was intraperitoneally administered to DDC‐treated mice during the recovery phase (Fig. 7A). DDC treatment caused weight loss in mice (Fig. 7B) and marked elevation of aspartate transaminase (AST), alanine aminotransferase (ALT), total bilirubin (T‐Bil), and direct bilirubin (D‐Bil) in serum (Fig. 7C). There was no significant difference between the VIP‐treated and control groups regarding body weight changes, liver to body weight ratio, or serum AST, ALT, and T‐Bil levels (Fig. 7B,C). However, serum D‐Bil was significantly lower in the VIP‐treated group at the end of the recovery phase (Fig. 7C). The expression of EpCAM and CK19 (markers for ductular reaction) and VIPR1 was not affected by VIP treatment (Fig. 7D). Tjp1 was also not changed in whole liver. To evaluate the change of TJP1 in IHBDs alone, TJP1 protein in bile ducts (either original or remodeled) was determined by immunohistologic analysis and image analyzer. Compared to the control, TJP1 more clearly lined the EpCAM+ ductular structures in the liver of the VIP‐treated mice. The ratio of TJP1+EpCAM+ to EpCAM+ areas was significantly higher in the VIP group compared with the control (Fig. 7E), demonstrating that VIP improved the reconstruction of tight junctions in damaged bile ducts of DDC‐injured liver.
Figure 7.
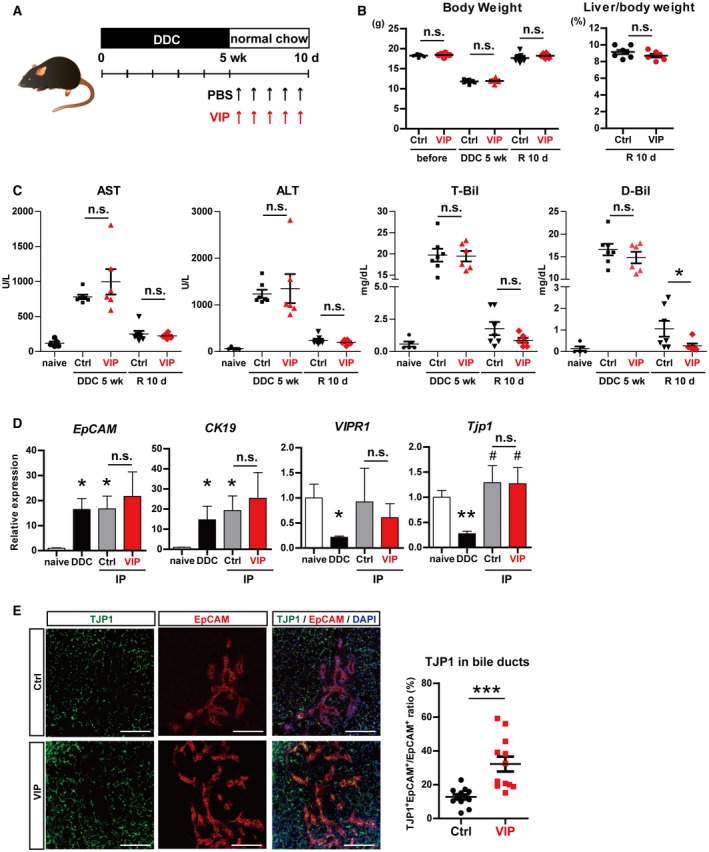
VIP enhanced the reconstruction of impaired tight junctions in DDC‐injured IHBDs and ameliorated hyperbilirubinemia. (A) Outline of DDC injury model. WT mice were fed DDC for 5 weeks. In the recovery phase, DDC was replaced with normal chow and mice were intraperitoneally injected with VIP (1 nmol per mouse) or PBS every other day. After 10 days of recovery period, liver samples were analyzed. (B) Left panel: body weights were markedly decreased by DDC feeding and recovered after the recovery phase. Right panel: liver to body weight ratio at the end of the recovery period was not changed by VIP treatment. (C) Serum AST, ALT, T‐Bil, and D‐Bil were increased by DDC injury compared to naive healthy controls and decreased after its removal. D‐Bil was significantly lower in the VIP‐treated group. (D) qRT‐PCR of EpCAM, CK19, Tjp1, and VIPR1. None were affected by VIP treatment in whole‐liver lysates. (E) Left panel: double immunostaining of TJP1 (green) and EpCAM (red). Nuclei were stained with DAPI (blue). Right panel: areas of EpCAM+ and TJP1+EpCAM+ cells were quantified. TJP1+EpCAM+ to EpCAM+ ratio was significantly increased by VIP treatment. Ten randomly selected fields from three different mice were analyzed in the control and VIP groups. Results represent the mean ± SD. (B,C) Two‐tailed Mann‐Whitney test; (D) two‐tailed t test. *P < 0.05, ***P < 0.001 compared to naive; #P < 0.05 compared to DDC. Abbreviations: d, days; IP, intraperitoneal injection; n.s., not statistically significant; R, recovery; wk, weeks.
There was no obvious improvement in fibrosis (Supporting Fig. S6A) or necrosis (Supporting Fig. S6B) by VIP treatment. CFTR was markedly up‐regulated by DDC feeding (Supporting Fig. S6C), suggesting that this model is not suitable for assessing VIP involvement in CFTR expression.
Discussion
This study reports the first evidence that VIP released from LMCs promotes the establishment of tight junctions and the expression of ion/water channels during the development of IHBDs both in vitro (Fig. 4) and in vivo (Fig. 6). Furthermore, the DDC injury model demonstrated that VIP facilitates the reconstruction of the impaired tight junction and further contributes to the improvement of hyperbilirubinemia during cholestatic liver injury (Fig. 7).
Tight junctions are essential for the establishment of a blood–bile barrier in bile canaliculi and IHBDs. Previous reports showed that several molecules regulated by VIP/VIPR1 signaling commit to the establishment of tight junctions; cyclic adenosine monophosphate (cAMP)/protein kinase A33 and protein kinase C pathways34 are activated downstream of G‐protein‐coupled receptors (GPCRs) and promote Tjp1 expression and tight junction integration. Peroxisome proliferator‐activated receptor (PPAR)34 also regulates tight junction assembly, which was up‐regulated in VIP‐treated cholangiocytic cysts along with GPCR signaling pathways in our data (Supporting Table S4). VIP has been reported to increase the expression of Tjp1 in intestinal epithelium35 and retina.36 More recently, it is argued that CFTR also regulates expression and distribution of tight junction‐related proteins, including various claudins, occuldins, and Tjp1.37, 38 Grhl2 and Rab25 cooperate to promote the expression and transportation of claudins to plasma membrane.39 Taken together, it is likely that VIP/VIPR1 signaling orchestrates tight junction assembly and its appropriate localization in IHBDs by directly increasing expression of Tjp1 and through various intracellular signaling that up‐regulates CFTR, Grhl2, and Rab25 (Fig. 8).
Figure 8.
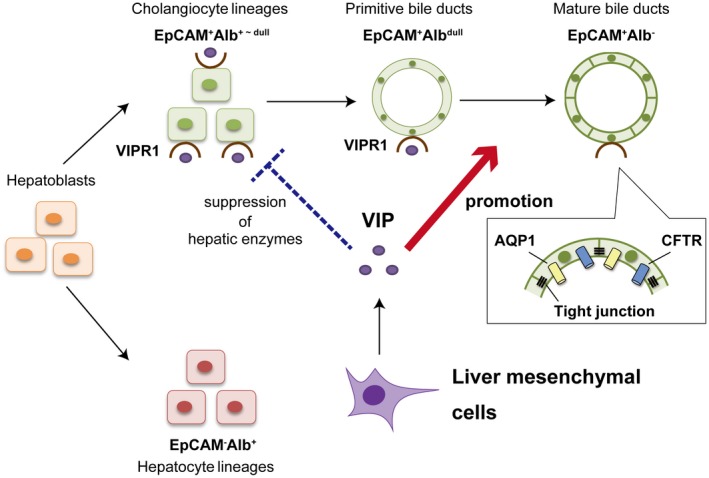
Schema of the function of VIP in regulation of IHBD formation in the developing livers. Bipotential hepatoblasts differentiate into EpCAM+Alb+~dull cholangiocyte and EpCAM−Alb+ hepatocyte lineages in midgestational fetal livers. VIP is produced from LMCs around portal veins in late gestational fetal and neonatal livers. VIP/VIPR1 signaling facilitates bile duct formation by promoting the establishment of intracellular tight junctions and a water transportation system of cholangiocytes (red arrow). VIP also demonstrates the potential to suppress the inadequate commitment of EpCAM+Alb+~dull cholangiocyte‐lineage cells into hepatocyte‐lineage cells in vitro (dashed blue bar).
Increased influx of bile is proposed as a major trigger for the structural rearrangement of IHBDs into a mature and hierarchical network, which takes place from E17 or later.13 As such, it is highly possible that VIP contributes to the dynamic reorganization of IHBDs in the perinatal stage by up‐regulating CFTR and Aqp1 and subsequently enforcing water transportation by cholangiocytes. VIP promotes the recruitment of CFTR to the cell‐surface membranes in a human lung cancer cell line, primary bronchial epithelial cells,40 and mouse salivary gland cells.41 In cholangiocytes, secretin, which shares common cAMP‐dependent signaling with VIP, enhances the trafficking of intracellular vesicular compartment including CFTR, Aqp1, and Slc4a2 to apical plasma membrane in rat models and as a consequence triggers ductular bile secretion.42, 43 Our data were distinctive from these reports regarding the point that VIP enhanced the total expression of CFTR and Aqp1 and did not only influence their distribution. Transcriptional regulation of these channels by VIP may be a unique mechanism in the developing stage and is lost in mature adult cells.
VIP suppressed the expression of mature and zone 1 hepatocyte markers, including key enzymes for the ornithine cycle and other amino acid metabolism, in the hepatic maturation assay in vitro (Fig. 5). Although EpCAM+ cells derived from neonatal liver show the characteristics of cholangiocyte‐lineage cells, they exhibit the potential to differentiate into hepatocytes and express these enzymes in vitro.29 Our in vivo data demonstrated that VIPR1 is detectable in EpCAM+Alb+~dull cells in zone 1 of perinatal‐stage livers (Fig. 5). Moreover, cDNA microarray analysis of cholangiocytic cysts revealed that genes and pathways highly expressed in mature hepatocytes were down‐regulated in the presence of VIP (Supporting Fig. S3F; Supporting Table S4). Taken together, our data and the previous report29 suggest that VIP derived from periportal mesenchymal cells contribute to the proper differentiation of EpCAM+Alb+~dull cholangiocyte‐lineage cells by preventing their commitment into hepatocyte lineages during perinatal stages, even though further study will be required to address this issue (Fig. 8).
Our data showed for the first time that LMCs have the potential to secrete VIP, a major neuropeptide that is widely distributed in the nervous system. Innervation of the liver is initiated no earlier than P2.16 Considering that LMCs exhibit some phenotypes similar to nerve cells,44, 45 it is conceivable that they secrete VIP on behalf of neurons in the perinatal stage. In congruence with the idea that VIP provides a favorable microenvironment for bile duct formation in developing liver, VIP expression was predominantly observed in the Jag1 + periportal mesenchymal cells (Fig. 3).
VIP expression in MMP14‐KO mesenchymal cells was significantly decreased compared to that in WT (Fig. 1). Gene ontology analysis of cDNA microarray data of MMP14‐WT and MMP14‐KO LMCs indicated that gene sets related to phospholipase C (PLC) were down‐regulated in MMP14‐KO cells (data not shown). PLC is essential for activating classical protein kinase C signals, which control the function of activator protein 1, one of the transcriptional factors regulating VIP gene expression.46 Thus, it can be assumed that suppressed activity of PLC resulted in the down‐regulation of VIP in MMP14‐KO LMCs. However, further investigation is required to fully understand the mechanism of how MMP14 regulates expression of VIP.
Failure of tight junctions is observed in various cholestatic diseases, such as primary biliary cholangitis and primary sclerosing cholangitis,47 as well as biliary tract cancer.48 Our DDC injury model demonstrated that VIP promotes the establishment of tight junctions not only in the developing liver but also in the liver with cholestatic diseases (Fig. 7). It is presumed that the reconstitution of the blood–bile barrier contributed to efficient bile drainage and prompt recovery from cholestasis, suggesting the therapeutic potential of VIP/VIPR1 regulation in cholestatic liver diseases. VIP failed to ameliorate fibrosis and necrosis in our experimental model. However, its therapeutic effect can be enhanced by up‐regulation of VIPR in cholangiocytes, which was decreased in DDC‐injured livers. Furthermore, recovering the impaired VIP/VIPR1 signaling in IHBDs could be a new therapeutic strategy for slowing the progression of cholestatic diseases.
Supporting information
Acknowledgment
We thank the FACS Core Laboratory of the Institute of Medical Science, University of Tokyo, for excellent technical assistance for flow cytometry and cell sorting. We also thank the Stem Cell Bank, Center for Stem Cell Biology and Regenerative Medicine, University of Tokyo, for the gift of the human iPS cell line TkDA3‐4.
Financial support: Supported in part by the Japan Society for the Promotion of Science/Ministry of Education, Culture, Sports, Science, and Technology Grants‐in‐Aid for Scientific Research (17K09407 to Y.I.; 17K19647 and 19H03635 to Y.A.; 18H02790 to S.K.; 18K07964 to S.A.; 19K08415 to M.N.), Japan Agency for Medical Research and Development (JP19fk0210060 to Y.A.; JP19fk0210018 to Y.A. and S.K.), and Research grants from Bristol Myers Squibb (37936225 to S.K.)
Potential conflict of interest: Dr. Kakinuma and Dr. Asahina receive funding from Gilead Sciences, AbbVie GK, Fujirebio, and Merck Sharp & Dohme. Dr. Kakinuma has received grants from Bristol Myers Squibb.
Contributor Information
Sei Kakinuma, Email: skakinuma.gast@tmd.ac.jp.
Yasuhiro Asahina, Email: asahina.gast@tmd.ac.jp.
Mamoru Watanabe, Email: mamoru.gast@tmd.ac.jp.
References
- 1. Kaneko K, Kamimoto K, Miyajima A, Itoh T. Adaptive remodeling of the biliary architecture underlies liver homeostasis. Hepatology 2015;61:2056‐2066. [DOI] [PubMed] [Google Scholar]
- 2. Raynaud P, Carpentier R, Antoniou A, Lemaigre FP. Biliary differentiation and bile duct morphogenesis in development and disease. Int J Biochem Cell Biol 2011;43:245‐256. [DOI] [PubMed] [Google Scholar]
- 3. Zong Y, Stanger BZ. Molecular mechanisms of bile duct development. Int J Biochem Cell Biol 2011;43:257‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gordillo M, Evans T, Gouon‐Evans V. Orchestrating liver development. Development 2015;142:2094‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ito K, Yanagida A, Okada K, Yamazaki Y, Nakauchi H, Kamiya A. Mesenchymal progenitor cells in mouse foetal liver regulate differentiation and proliferation of hepatoblasts. Liver Int 2014;34:1378‐1390. [DOI] [PubMed] [Google Scholar]
- 6. Miyajima A, Tanaka M, Itoh T. Stem/progenitor cells in liver development, homeostasis, regeneration, and reprogramming. Cell Stem Cell 2014;14:561‐574. [DOI] [PubMed] [Google Scholar]
- 7. Clotman F, Jacquemin P, Plumb‐Rudewiez N, Pierreux CE, Van der Smissen P, Dietz HC, et al. Control of liver cell fate decision by a gradient of TGF beta signaling modulated by Onecut transcription factors. Genes Dev 2005;19:1849‐1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kodama Y, Hijikata M, Kageyama R, Shimotohno K, Chiba T. The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology 2004;127:1775‐1786. [DOI] [PubMed] [Google Scholar]
- 9. Tanimizu N, Miyajima A. Notch signaling controls hepatoblast differentiation by altering the expression of liver‐enriched transcription factors. J Cell Sci 2004;117:3165‐3174. [DOI] [PubMed] [Google Scholar]
- 10. Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela‐Arispe ML. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development 2010;137:4061‐4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kiyohashi K, Kakinuma S, Kamiya A, Sakamoto N, Nitta S, Yamanaka H, et al. Wnt5a signaling mediates biliary differentiation of fetal hepatic stem/progenitor cells in mice. Hepatology 2013;57:2502‐2513. [DOI] [PubMed] [Google Scholar]
- 12. Sparks EE, Huppert KA, Brown MA, Washington MK, Huppert SS. Notch signaling regulates formation of the three‐dimensional architecture of intrahepatic bile ducts in mice. Hepatology 2010;51:1391‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tanimizu N, Kaneko K, Itoh T, Ichinohe N, Ishii M, Mizuguchi T, et al. Intrahepatic bile ducts are developed through formation of homogeneous continuous luminal network and its dynamic rearrangement in mice. Hepatology 2016;64:175‐188. [DOI] [PubMed] [Google Scholar]
- 14. Oben JA, Diehl AM. Sympathetic nervous system regulation of liver repair. Anat Rec A Discov Mol Cell Evol Biol 2004;280:874‐883. [DOI] [PubMed] [Google Scholar]
- 15. Soeda J, Mouralidarane A, Ray S, Novelli M, Thomas S, Roskams T, et al. The beta‐adrenoceptor agonist isoproterenol rescues acetaminophen‐injured livers through increasing progenitor numbers by Wnt in mice. Hepatology 2014;60:1023‐1034. [DOI] [PubMed] [Google Scholar]
- 16. Tanimizu N, Ichinohe N, Mitaka T. Intrahepatic bile ducts guide establishment of the intrahepatic nerve network in developing and regenerating mouse liver. Development 2018;145:pii:dev159095. [DOI] [PubMed] [Google Scholar]
- 17. Ueno T, Inuzuka S, Torimura T, Sakata R, Sakamoto M, Gondo K, et al. Distribution of substance P and vasoactive intestinal peptide in the human liver: light and electron immunoperoxidase methods of observation. Am J Gastroenterol 1991;86:1633‐1637. [PubMed] [Google Scholar]
- 18. Cho WK, Boyer JL. Vasoactive intestinal polypeptide is a potent regulator of bile secretion from rat cholangiocytes. Gastroenterology 1999;117:420‐428. [DOI] [PubMed] [Google Scholar]
- 19. Otani S, Kakinuma S, Kamiya A, Goto F, Kaneko S, Miyoshi M, et al. Matrix metalloproteinase‐14 mediates formation of bile ducts and hepatic maturation of fetal hepatic progenitor cells. Biochem Biophys Res Commun 2016;469:1062‐1068. [DOI] [PubMed] [Google Scholar]
- 20. Oh J, Takahashi R, Adachi E, Kondo S, Kuratomi S, Noma A, et al. Mutations in two matrix metalloproteinase genes, MMP‐2 and MT1‐MMP, are synthetic lethal in mice. Oncogene 2004;23:5041‐5048. [DOI] [PubMed] [Google Scholar]
- 21. Gressens P, Hill JM, Paindaveine B, Gozes I, Fridkin M, Brenneman DE. Severe microcephaly induced by blockade of vasoactive intestinal peptide function in the primitive neuroepithelium of the mouse. J Clin Invest 1994;94:2020‐2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu JY, Henins KA, Gressens P, Gozes I, Fridkin M, Brenneman DE, et al. Neurobehavioral development of neonatal mice following blockade of VIP during the early embryonic period. Peptides 1997;18:1131‐1137. [DOI] [PubMed] [Google Scholar]
- 23. Kakinuma S, Ohta H, Kamiya A, Yamazaki Y, Oikawa T, Okada K, et al. Analyses of cell surface molecules on hepatic stem/progenitor cells in mouse fetal liver. J Hepatol 2009;51:127‐138. [DOI] [PubMed] [Google Scholar]
- 24. Suzuki K, Tanaka M, Watanabe N, Saito S, Nonaka H, Miyajima A. p75 Neurotrophin receptor is a marker for precursors of stellate cells and portal fibroblasts in mouse fetal liver. Gastroenterology 2008;135:270‐281.e3. [DOI] [PubMed] [Google Scholar]
- 25. Anzai K, Chikada H, Tsuruya K, Ida K, Kagawa T, Inagaki Y, et al. Foetal hepatic progenitor cells assume a cholangiocytic cell phenotype during two‐dimensional pre‐culture. Sci Rep 2016;6:28283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaneko S, Kakinuma S, Asahina Y, Kamiya A, Miyoshi M, Tsunoda T, et al. Human induced pluripotent stem cell‐derived hepatic cell lines as a new model for host interaction with hepatitis B virus. Sci Rep 2016;6:29358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tanimizu N, Miyajima A, Mostov KE. Liver progenitor cells develop cholangiocyte‐type epithelial polarity in three‐dimensional culture. Mol Biol Cell 2007;18:1472‐1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kamiya A, Kojima N, Kinoshita T, Sakai Y, Miyaijma A. Maturation of fetal hepatocytes in vitro by extracellular matrices and oncostatin M: induction of tryptophan oxygenase. Hepatology 2002;35:1351‐1359. [DOI] [PubMed] [Google Scholar]
- 29. Tanimizu N, Nakamura Y, Ichinohe N, Mizuguchi T, Hirata K, Mitaka T. Hepatic biliary epithelial cells acquire epithelial integrity but lose plasticity to differentiate into hepatocytes in vitro during development. J Cell Sci 2013;126:5239‐5246. [DOI] [PubMed] [Google Scholar]
- 30. Tsunoda T, Kakinuma S, Miyoshi M, Kamiya A, Kaneko S, Sato A, et al. Loss of fibrocystin promotes interleukin‐8‐dependent proliferation and CTGF production of biliary epithelium. J Hepatol 2019;71:143‐152. [DOI] [PubMed] [Google Scholar]
- 31. Gozes I, McCune SK, Jacobson L, Warren D, Moody TW, Fridkin M, et al. An antagonist to vasoactive intestinal peptide affects cellular functions in the central nervous system. J Pharmacol Exp Ther 1991;257:959‐966. [PubMed] [Google Scholar]
- 32. Pradhan‐Sundd T, Vats R, Russell JO, Singh S, Michael AA, Molina L, et al. Dysregulated bile transporters and impaired tight junctions during chronic liver injury in mice. Gastroenterology 2018;155:1218‐1232.e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hopkins AM, Li D, Mrsny RJ, Walsh SV, Nusrat A. Modulation of tight junction function by G protein‐coupled events. Adv Drug Deliv Rev 2000;41:329‐340. [DOI] [PubMed] [Google Scholar]
- 34. Ogasawara N, Kojima T, Go M, Ohkuni T, Koizumi J, Kamekura R, et al. PPARgamma agonists upregulate the barrier function of tight junctions via a PKC pathway in human nasal epithelial cells. Pharmacol Res 2010;61:489‐498. [DOI] [PubMed] [Google Scholar]
- 35. Neunlist M, Toumi F, Oreschkova T, Denis M, Leborgne J, Laboisse CL, et al. Human ENS regulates the intestinal epithelial barrier permeability and a tight junction‐associated protein ZO‐1 via VIPergic pathways. Am J Physiol Gastrointest Liver Physiol 2003;285:G1028‐1036. [DOI] [PubMed] [Google Scholar]
- 36. Scuderi S, D'Amico AG, Castorina A, Imbesi R, Carnazza ML, D'Agata V. Ameliorative effect of PACAP and VIP against increased permeability in a model of outer blood retinal barrier dysfunction. Peptides 2013;39:119‐124. [DOI] [PubMed] [Google Scholar]
- 37. Ruan YC, Wang Y, Da Silva N, Kim B, Diao RY, Hill E, et al. CFTR interacts with ZO‐1 to regulate tight junction assembly and epithelial differentiation through the ZONAB pathway. J Cell Sci 2014;127:4396‐4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Castellani S, Favia M, Guerra L, Carbone A, Abbattiscianni AC, Di Gioia S, et al. Emerging relationship between CFTR, actin and tight junction organization in cystic fibrosis airway epithelium. Histol Histopathol 2017;32:445‐459. [DOI] [PubMed] [Google Scholar]
- 39. Tanimizu N, Mitaka T. Role of grainyhead‐like 2 in the formation of functional tight junctions. Tissue Barriers 2013;1:e23495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alshafie W, Chappe FG, Li M, Anini Y, Chappe VM. VIP regulates CFTR membrane expression and function in Calu‐3 cells by increasing its interaction with NHERF1 and P‐ERM in a VPAC1‐ and PKCepsilon‐dependent manner. Am J Physiol Cell Physiol 2014;307:C107‐C119. [DOI] [PubMed] [Google Scholar]
- 41. Nedvetsky PI, Emmerson E, Finley JK, Ettinger A, Cruz‐Pacheco N, Prochazka J, et al. Parasympathetic innervation regulates tubulogenesis in the developing salivary gland. Dev Cell 2014;30:449‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tietz PS, Marinelli RA, Chen XM, Huang B, Cohn J, Kole J, et al. Agonist‐induced coordinated trafficking of functionally related transport proteins for water and ions in cholangiocytes. J Biol Chem 2003;278:20413‐20419. [DOI] [PubMed] [Google Scholar]
- 43. Banales JM, Prieto J, Medina JF. Cholangiocyte anion exchange and biliary bicarbonate excretion. World J Gastroenterol 2006;12:3496‐3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Cassiman D, Denef C, Desmet VJ, Roskams T. Human and rat hepatic stellate cells express neurotrophins and neurotrophin receptors. Hepatology 2001;33:148‐158. [DOI] [PubMed] [Google Scholar]
- 45. Buniatian G, Hamprecht B, Gebhardt R. Glial fibrillary acidic protein as a marker of perisinusoidal stellate cells that can distinguish between the normal and myofibroblast‐like phenotypes. Biol Cell 1996;87:65‐73. [PubMed] [Google Scholar]
- 46. Symes A, Gearan T, Eby J, Fink JS. Integration of Jak‐Stat and AP‐1 signaling pathways at the vasoactive intestinal peptide cytokine response element regulates ciliary neurotrophic factor‐dependent transcription. J Biol Chem 1997;272:9648‐9654. [DOI] [PubMed] [Google Scholar]
- 47. Sakisaka S, Kawaguchi T, Taniguchi E, Hanada S, Sasatomi K, Koga H, et al. Alterations in tight junctions differ between primary biliary cirrhosis and primary sclerosing cholangitis. Hepatology 2001;33:1460‐1468. [DOI] [PubMed] [Google Scholar]
- 48. Nemeth Z, Szasz AM, Somoracz A, Tatrai P, Nemeth J, Gyorffy H, et al. Zonula occludens‐1, occludin, and E‐cadherin protein expression in biliary tract cancers. Pathol Oncol Res 2009;15:533‐539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials