

Abstract
Icosabutate is a structurally engineered eicosapentaenoic acid derivative under development for nonalcoholic steatohepatitis (NASH). In this study, we investigated the absorption and distribution properties of icosabutate in relation to liver targeting and used rodents to evaluate the effects of icosabutate on glucose metabolism, insulin resistance, as well as hepatic steatosis, inflammation, lipotoxicity, and fibrosis. The absorption, tissue distribution, and excretion of icosabutate was investigated in rats along with its effects in mouse models of insulin resistance (ob/ob) and metabolic inflammation/NASH (high‐fat/cholesterol‐fed APOE*3Leiden.CETP mice) and efficacy was compared with synthetic peroxisome proliferator‐activated receptor α (PPAR‐α) (fenofibrate) and/or PPAR‐γ/(α) (pioglitazone and rosiglitazone) agonists. Icosabutate was absorbed almost entirely through the portal vein, resulting in rapid hepatic accumulation. Icosabutate demonstrated potent insulin‐sensitizing effects in ob/ob mice, and unlike fenofibrate or pioglitazone, it significantly reduced plasma alanine aminotransferase. In high‐fat/cholesterol‐fed APOE*3Leiden.CETP mice, icosabutate, but not rosiglitazone, reduced microvesicular steatosis and hepatocellular hypertrophy. Although both rosiglitazone and icosabutate reduced hepatic inflammation, only icosabutate elicited antifibrotic effects in association with decreased hepatic concentrations of multiple lipotoxic lipid species and an oxidative stress marker. Hepatic gene‐expression analysis confirmed the changes in lipid metabolism, inflammatory and fibrogenic response, and energy metabolism, and revealed the involved upstream regulators. In conclusion, icosabutate selectively targets the liver through the portal vein and demonstrates broad beneficial effects following insulin sensitivity, hepatic microvesicular steatosis, inflammation, lipotoxicity, oxidative stress, and fibrosis. Icosabutate therefore offers a promising approach to the treatment of both dysregulated glucose/lipid metabolism and inflammatory disorders of the liver, including NASH.
The aim of the present study was to evaluate the effects of icosabutate, a structurally engineered eicosapentaenoic acid (EPA) derivative, designed to overcome the inherent drawbacks of unmodified EPA for liver targeting. As a multi‐etiological disorder with limited success achieved to date with single target drugs, nonalcoholic steatohepatitis (NASH) is an attractive indication for drugs with pleiotropic targeting potential, such as ω‐3 fatty acids. However, a potentially important issue limiting their clinical efficacy is related to suboptimal liver targeting of ω‐3 fatty acids, which could be rectified through structural engineering. In this study, we investigated icosabutate in relation to liver targeting and used rodent models to evaluate the effects of icosabutate on glucose metabolism, insulin resistance, as well as NASH and fibrosis.
Abbreviations
- AA
arachidonic acid
- ALT
alanine aminotransferase
- AUC
area under the curve
- DAG
diacylglycerol
- EPA
eicosapentaenoic acid
- ET‐1
endothelin 1
- FFA
free fatty acid
- GSH
glutathione
- GSSG
oxidized glutathione
- H&E
hematoxylin and eosin
- HETE
hydroxyeicosatetraenoic acid
- HOMA‐IR
homeostasis model assessment of insulin resistance
- IL
interleukin
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PPAR
peroxisome proliferator‐activated receptor
- STAT1
signal transducer and activator of transcription 1
- TNFR
tumor necrosis factor receptor
Although treatment with high‐dose oral (4 g/day) eicosapentaenoic acid (EPA) ethyl ester was recently shown to markedly reduce major adverse cardiovascular events in subjects with elevated triglycerides,1 there is little evidence of beneficial effects of ω‐3 fatty acids in other metabolic and inflammatory conditions such as diabetes and nonalcoholic steatohepatitis (NASH). For example, in a recent 1‐year intervention study, EPA ethyl ester (1.8 or 2.7 g/day) had no effect on liver histology in NASH patients, and clinical trials examining the effects of high‐dose (>2 g/day) ω‐3 fatty‐acid supplementation on glycemic control are inconclusive.2
A potential explanation for the lack of efficacy of ω‐3 fatty acids in conditions beyond cardiovascular disease is that long‐chain fatty acids have absorption, distribution, and metabolism properties that prevent optimal liver targeting. First, regardless of the oral delivery form (free fatty acid, ethyl ester, triglyceride, or phospholipid), systemic absorption of long‐chain fatty acids occurs primarily through lymphatic vessels3, 4, 5 rather than the portal vein, resulting in systemic distribution into adipose tissue, skeletal muscle, and many other organs. The consequence of this absorption route is that only 8%‐16% of dietary fat reaches the liver after a meal.6 Second, as high doses of the more potent long‐chain fatty acids markedly increase their own oxidation as an energy source,7 the potential for achieving a dose‐dependent effect in the liver is severely limited. Finally, after reaching the liver, extracellular and intracellular receptor binding and eicosanoid pathway metabolism are mediated through nonesterified fatty acids (FFAs). Thus, the rapid incorporation of long‐chain fatty acids into complex lipids such as triglycerides and phospholipids (in addition to hepatic β‐oxidation for energy) may further decrease the availability of the most active free acid form in the liver.
Icosabutate, a structurally engineered EPA derivative in clinical development for NASH (NCT04052516), is designed to overcome the inherent drawbacks of unmodified EPA for liver targeting. It is structured (1) to remain in a free acid form by resisting incorporation into complex cellular lipids (through an ethyl group in the α‐position) and (2) to minimize metabolism through β‐oxidation (using an oxygen atom substitution in the β‐position), with a goal of achieving therapeutic efficacy beyond what is possible with oral dosing of unmodified ω‐3 fatty acids (Fig. 1). Icosabutate was initially developed as a hypolipidemic drug and demonstrated robust reductions in atherogenic lipids and apolipoproteins at a dose of 600 mg once daily in two recent clinical trials.8, 9 Interestingly, icosabutate also induced a marked and significant reduction in homeostasis model assessment of insulin resistance (HOMA‐IR),8 an effect not typically associated with unmodified ω‐3 fatty‐acid supplementation.10 This suggests that icosabutate's structural modifications not only facilitate quantitative advantages (i.e., low oral dose) but also confer qualitative differences in pharmacodynamic effects versus unmodified EPA.
Figure 1.
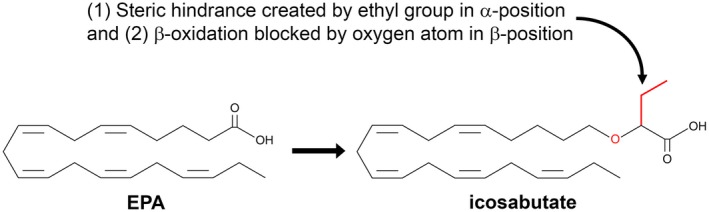
Chemical structure of icosabutate as compared with EPA. Icosabutate, a structurally engineered EPA derivative, is structured (1) to remain in a free acid form by resisting incorporation into complex cellular lipids (through an ethyl group in the α‐position) and (2) to minimize metabolism by way of β‐oxidation (through an oxygen atom substitution in the β position).
To further understand icosabutate's pharmacokinetic and pharmacodynamic profile, we carried out a range of preclinical studies addressing the absorption, distribution, and excretion characteristics of icosabutate. We also performed studies on its effects on glucose metabolism and insulin resistance, as well as steatosis, inflammation, lipotoxicity, oxidative stress, and fibrosis in relevant mouse models. In addition, bioinformatics analysis was used to study the underlying mechanisms involved in the observed hepatic effects.
Methods
Animals and Experimental Design
All animal care and experimental procedures were approved by the local Ethical Committee on Animal Care and Experimentation and were in compliance with European Community specifications regarding the use of laboratory animals. All animals were group‐housed in a temperature‐controlled room on a 12‐hour light‐dark cycle and had free access to food and water. Body weight, food intake (both between 8 and 9 am), and blood/plasma measures (after 4 hours of fasting) were regularly monitored during the studies. Animals were sacrificed by CO2 inhalation.
To analyze portal vein uptake of icosabutate, 8‐week old male Wistar rats (n = 4) were used and were pretreated with buprenorphine (0.01‐0.05 mg/kg, subcutaneously) and then anesthetized by pentobarbital (50 mg/kg, intraperitoneally). Rats received a single oral gavage of 100 mg/kg icosabutate, and blood was collected from portal vein and from mesenteric lymph duct over 24 hours.
To analyze tissue distribution and excretion of icosabutate, male albino Wistar rats (n = 7 for distribution, n = 3 for excretion) received a single oral gavage of 100 mg/kg [14‐C]‐icosabutate. Plasma concentrations and tissue distribution were measured by liquid scintillation counting and quantitative whole‐body autoradiography, respectively, at multiple time points (1, 2, 4, 8, and 24 hours, and 3 and 7 days). Routes and rates of excretion were measured by collection of urine and feces at corresponding time points.
To analyze the effects of icosabutate on obesity, hyperglycemia and insulin resistance, 6‐8‐week‐old male ob/ob mice were randomized into different groups (n = 10/group) and were left untreated (control group) or treated for 5 weeks with 135 mg/kg/d icosabutate through diet administration. As reference, groups mice treated for 5 weeks with 100 mg/kg/d fenofibrate or 30 mg/kg/d pioglitazone through diet administration were included. An oral glucose (2 g/kg) tolerance test was performed after 5 weeks, after 4‐hour fasting. First, baseline blood samples (t = 0) were collected from the tail vein into ethylene diamine tetraacetic acid (EDTA)‐coated tubes (Sarstedt, Nümbrecht, Germany). Subsequently, mice received a bolus (2 g/kg) of 10% (wt/vol) D‐glucose solution through gavage, and additional blood samples (30 µL) were drawn at 15, 30, 45, 60, and 120 minutes after injection.
To analyze the effects of icosabutate on hepatic steatosis, inflammation and fibrosis, 8‐15‐week‐old male APOE*3Leiden.CETP mice fed a high‐fat (24% wt/wt lard) and high cholesterol (1% wt/wt) diet14 were randomized into different groups (n = 12/group) and received daily vehicle (corn oil) gavages (control group) or were treated for 20 weeks with 112 mg/kg/d icosabutate through daily oral gavages. As a reference group, mice treated for 20 weeks with 13 mg/kg/d rosiglitazone through diet administration (receiving daily vehicle gavages) were included. NASH and fibrosis were scored in hematoxylin and eosin (H&E) or sirius red–stained cross sections using an adapted grading system of human NASH11, 12 and were analyzed by biochemical analysis, as described in more detail in the Supporting information.
At several time points during the studies, animals were fasted for 4 hours and blood was collected from the tail vein into EDTA‐coated tubes (Sarstedt) for different analytical measurements, as described in more detail in the Supporting information.
Liver samples were used for histology, hepatic lipid/lipidomics analysis, assessment of hepatic reduced glutathione (GSH), and oxidized glutathione (GSSG) and transcriptome analysis, all described in more detail in the Supporting information.
Statistical Analysis
All values shown represent the means ± SEMs. Statistical differences between groups were determined by using nonparametric Kruskal‐Wallis followed by Mann‐Whitney U test for independent samples using SPSS software (IBM Corp., Armonk, NY). A P value < 0.05 was considered statistically significant.
Results
Icosabutate Effectively Targets the Liver
To evaluate whether structural alterations of icosabutate enhanced uptake through the portal vein, absorption after single oral administration was analyzed in male Wistar rats with concurrent collection of blood from portal vein and lymph from mesenteric lymph duct. Area under the curve (AUC)0‐24h and maximal concentration (Cmax) values of 14C‐labeled icosabutate were approximately 2‐fold higher in the portal vein versus the mesenteric lymph (Table 1A). Accounting for the much higher flow rate of portal vein plasma (522 mL/h) versus mesenteric lymph (0.5 mL/h), the data demonstrate that icosabutate is almost entirely taken up through the portal vein (>99%) with only a small fraction of icosabutate being absorbed through the lymphatic pathway. This contrasts with the high uptake of unmodified long‐chain fatty acids by way of mesenteric lymph after incorporation into chylomicrons.3, 4, 5
Table 1.
Portal Vein and Mesenteric Lymph Absorption and Tissue Distribution of Icosabutate
| A | |||||||
|---|---|---|---|---|---|---|---|
| Portal Vein Plasma (Blood Flow Rate: 522 mL/h)* | Mesenteric Lymph (Lymph Flow Rate: 0.5 mL/h) | Portal Vein/Lymph Ratio | |||||
| Cmax (µg/mL) | Tmax (h) | AUC0‐24h (µgˑh/mL) | Cmax (µg/mL) | Tmax (h) | AUC0‐24h (µgˑh/mL) | ||
| Icosabutate in corn oil | 17.6 | 2 | 106 | 8.4 | 3 | 46 | 1,200:1 |
| B | |||||||
|---|---|---|---|---|---|---|---|
| Activity (µg/g tissue) | |||||||
| 1 Hour | 2 Hours | 4 Hours | 8 Hours | 1 Day | 3 Days | ||
| Liver | 77.7 | 134.0 | 174.0 | 145.0 | 25.5 | 2.7 | |
| Kidney cortex | 34.0 | 70.6 | 92.8 | 38.5 | 9.8 | 1.5 | |
| Kidney medulla | 47.3 | 148.0 | 102.0 | 76.6 | 34.5 | 0.5 | |
| Muscle | 3.6 | 5.2 | 10 | 4.8 | 1.2 | BLQ | |
| Subcutaneous fat | 9.0 | 14.6 | 25.3 | 15.4 | 7.1 | 4.0 | |
| Blood | 24.6 | 36.2 | 60.9 | 28.1 | 2.6 | BLQ | |
Assuming a volume ratio of plasma:blood of 1:2.
Abbreviations: BLQ, below the limit of quantification; h, hours.
Tissue distribution analysis showed peak concentrations of radioactivity in most tissues at 4‐8 hours after the dose (except the gastrointestinal tract) with highest concentrations in the liver and kidney (Table 1B). Most other tissues contained levels of radioactivity below that in plasma, indicating a limited distribution of absorbed radioactivity. There was a rapid decline in concentrations of drug‐related material over the study period, with excretion of radioactivity greater than 95% complete by 48 hours, and with 60% and 40% excreted through urine and feces, respectively (data not shown).
Icosabutate Improves Glucose Metabolism and Insulin Resistance
The effects of 4‐week treatment with icosabutate on glucose metabolism and insulin resistance were analyzed in a diabetic mouse model, ob/ob mice. We included fenofibrate to control for potential peroxisome proliferator‐activated receptor α (PPAR‐α)–mediated effects and added pioglitazone, a PPAR‐γ agonist (and to a lesser extent PPAR‐α activity13), as a positive control. As shown in Fig. 2, icosabutate significantly improved glucose metabolism. This was reflected by a significant decrease in blood glucose, blood hemoglobin A1c, plasma insulin, and HOMA‐IR (−50%, −47%, −76% and −87%, respectively; all p < 0.001). Fenofibrate and pioglitazone also improved glucose metabolism, although effects following plasma insulin were less pronounced with fenofibrate. Icosabutate markedly improved glucose tolerance after an oral glucose load (Fig. 2G) and significantly (p < 0.001) reduced AUC (0‐120 minutes) by 60% (Fig. 2H). Fenofibrate had a less potent effect on glucose tolerance and AUC, whereas pioglitazone reduced AUC to a similar degree as icosabutate. Pioglitazone demonstrated the largest reduction in HOMA‐IR (−97%, p < 0.001) and was the only compound to increase adiponectin (4.5‐fold, p < 0.001). In contrast to pioglitazone, neither fenofibrate nor icosabutate affected body weight (54.4 ± 1.5, 55.0 ± 1.1, 56.6 ± 1.4, and 62.3 ± 1.6 for control, icosabutate, fenofibrate and pioglitazone group, respectively, after 4 weeks of treatment) or adiponectin (Fig. 2E). Plasma alanine aminotransferase (ALT) levels were significantly decreased as compared with the control group by icosabutate treatment only (−33%, p < 0.01) (Fig. 2F).
Figure 2.
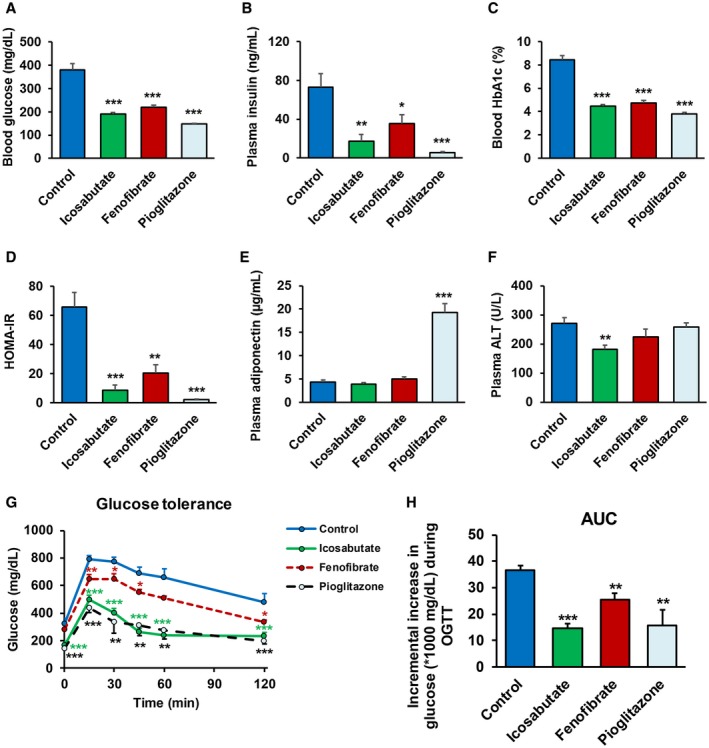
Icosabutate improves glucose metabolism and insulin sensitivity. Ob/ob mice were left untreated (control) or treated with icosabutate, fenofibrate, or pioglitazone for 5 weeks. Blood glucose (A), plasma insulin (B), blood hemoglobin A1c (C), HOMA‐IR (D), and plasma adiponectin (E) and plasma ALT (F) levels were measured after 4 weeks of treatment. Oral glucose tolerance test was performed in 4‐hour‐fasted ob/ob mice after 5 weeks of treatment. Blood glucose levels (G) were measured before (basal) and 15, 30, 45, 60, and 120 minutes after oral glucose load, and the AUC (H) was calculated. Values represent the mean ± SEM for 10 mice per group (*p < 0.05, **p < 0.01, ***p < 0.001 versus control). Abbreviations: HbA1c, hemoglobin A1c; OGTT, oral glucose tolerance test.
Icosabutate Improves Microvesicular Steatosis, Hepatic Inflammation, and Fibrosis
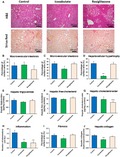
To assess the effects of icosabutate on NASH in a rodent model characterized by more severe hepatic inflammation and mild fibrosis, APOE*3Leiden.CETP mice14 were fed a high‐fat/cholesterol diet and treated with either icosabutate (112 mg/kg/d) or rosiglitazone (13 mg/kg/d) for 20 weeks. Icosabutate prevented microvesicular steatosis (−35%, p < 0.05) and hepatocellular hypertrophy (−82%, p < 0.01), but not macrovesicular steatosis (Fig. 3A‐D), whereas rosiglitazone did not significantly affect either parameter. Biochemical analysis of intrahepatic liver lipids (Fig. 3E‐G) revealed a significant reduction of hepatic cholesterol ester with icosabutate and rosiglitazone. Both icosabutate and rosiglitazone significantly prevented hepatic inflammation, as shown by the reduced number of inflammatory cell aggregates (Fig. 3H), by −62% and −67%, respectively (both p < 0.01). The anti‐inflammatory effect of icosabutate was confirmed by measurement of plasma inflammation markers. Plasma monocyte chemoattractant protein 1 was reduced by −30% and −46% for icosabutate and rosiglitazone (p < 0.01), respectively, although the reduction achieved by icosabutate was not significant (p = 0.123) (111.5 ± 17.1, 78.6 ± 6.4, and 65.1 ± 5.1 for control, icosabutate, and rosiglitazone‐treated group, respectively, after 20 weeks of treatment). For the liver‐derived biomarker serum amyloid A, icosabutate showed a more potent reduction (−68%, p < 0.001) versus control than rosiglitazone (−46%, p < 0.01) (29.8 ± 5.0, 9.7 ± 0.6, and 16.1 ± 3.3 for control, icosabutate, and rosiglitazone‐treated group, respectively, after 20 weeks of treatment). Despite comparable decreases in hepatic inflammatory cell aggregates, only icosabutate reduced hepatic collagen content (−32%, p < 0.01 versus control; Fig. 3J) and fibrosis surface area (−26%, p < 0.05; Fig. 3AI). Representative pictures of fibrosis (sirius red staining) and hepatic lipids (H&E stain) from individual mice from control, icosabutate, and rosiglitazone groups are shown in Fig. 3A. Rosiglitazone significantly increased food intake at multiple time points (p < 0.05) during the study but did not significantly increase body weight, whereas there was a small but significant decrease in body weight at week 16 and 20 versus control (−15%, p > 0.05) in the icosabutate group despite no change in food intake (data not shown).
Figure 3.
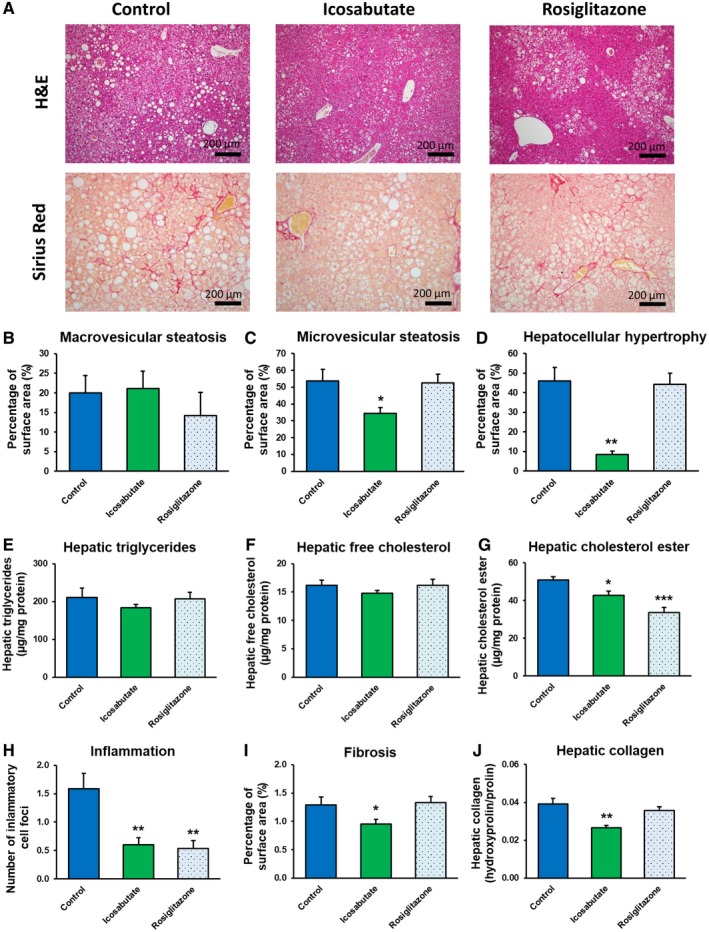
Icosabutate improves NASH and liver fibrosis. Histological photomicrographs of liver cross sections stained with H&E or sirius red (A) and quantitative analysis (B‐J) of NASH and liver fibrosis in APOE*3Leiden.CETP mice fed a high‐fat/cholesterol diet and left untreated (control) or treated with icosabutate or rosiglitazone for 20 weeks. Macrovesicular steatosis (B), microvesicular steatosis (C) and hepatocellular hypertrophy (D) as percentage of total liver area, intrahepatic triglycerides (E), free cholesterol (F) and cholesterol esters (G), inflammatory foci per millimeters‐squared microscopic field (H), and fibrosis as percentage of total liver area (I) or as hepatic collagen content (J) were analyzed. Values represent mean ± SEM for 12 mice per group (*p < 0.05, **p < 0.01, ***p < 0.001 versus control).
Icosabutate, but Not Rosiglitazone, Reduces Hepatic Concentrations of Multiple Lipotoxic Lipid Species and Oxidative Stress Markers
As icosabutate, but not rosiglitazone, reduced hepatic fibrosis despite comparable effects on influx of inflammatory cells, hepatic concentrations of NASH‐associated lipotoxic lipid species15 and oxidative stress were measured in liver tissue of APOE*3Leiden.CETP mice fed a high‐fat/cholesterol diet and treated for 20 weeks. As shown in Fig. 4, icosabutate significantly reduced hepatic concentrations of multiple lipotoxic lipid species, including FFAs, diacylglycerols (DAGs), bile acids, arachidonic acid, ceramides, and arachidonic acid–derived hydroxyeicosatetraenoic acids (HETEs, comprising 11[R]‐, 12‐, and 15[S] isomers). Icosabutate also reduced hepatic oxidative stress, as evidenced by the significant decrease in GSSG and the corresponding increase in the reduced GSH:GSSG ratio. In contrast to icosabutate, rosiglitazone had no effect on either lipotoxic lipid species or oxidative stress other than a significant increase in HETE concentrations.
Figure 4.
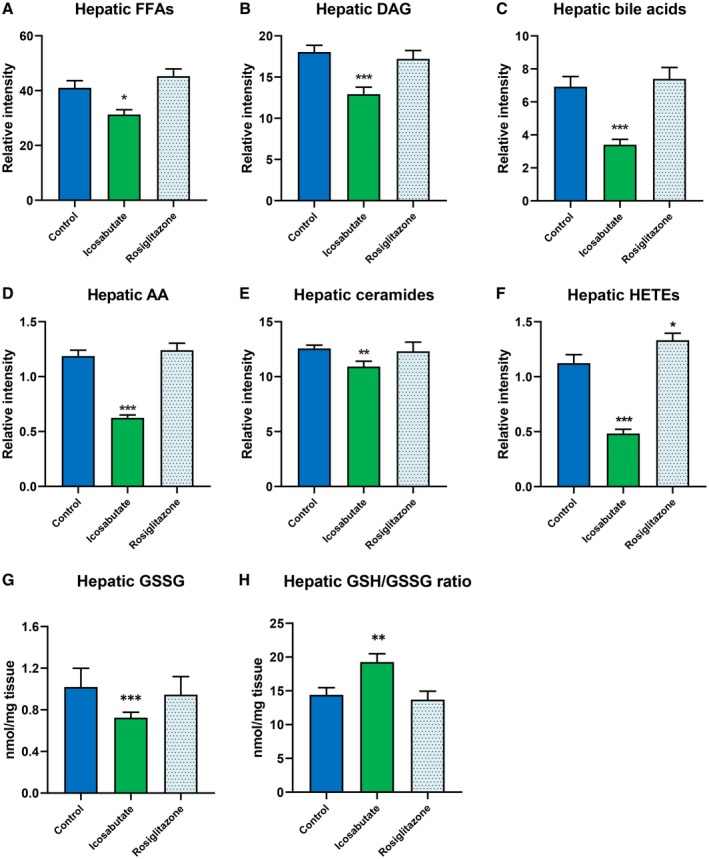
Icosabutate reduces the hepatic concentrations of lipotoxic lipid species and oxidative stress markers. Icosabutate reduces hepatic concentrations of lipotoxic lipid species: FFAs (A), DAGs (B), bile acids (C), arachidonic acid (D), ceramides (E), and HETEs (F) (comprising 11[R]‐, 12‐, and 15[S] isomers) in APOE*3Leiden.CETP mice fed a high‐fat/cholesterol diet and left untreated (control) or treated with icosabutate or rosiglitazone for 20 weeks. Icosabutate significantly improves the GSH/GSSG ratio (H) through a reduction in hepatic GSSG concentrations (G). No effect on any parameter was noted for rosiglitazone except a significant increase in HETEs. Data are presented as mean ± SEM for 12 mice per group (*p < 0.05, **p < 0.01, ***p < 0.001 versus control).
Icosabutate Activates Pathways Related to Hepatic Lipid and Energy Metabolism and Inhibits Inflammatory and Fibrogenic Response Pathways
To further investigate the mechanism by which icosabutate affects pathways involved in the development of NASH, transcriptome analysis of liver tissue was performed in the control and icosabutate group of APOE*3Leiden.CETP mice fed a high‐fat/cholesterol diet and treated for 20 weeks. An upstream regulator analysis was performed that predicts the activation state (z‐scores) of a protein, transcription factor, or cytokine based on the expression pattern of the genes downstream of this factor24 (Table 2). This analysis showed that inflammation was potently inhibited by icosabutate, as almost all inflammatory upstream regulators were found to be inhibited, including signal transducer and activator of transcription 1 (STAT1) and nuclear factor kappa B (NFκB) complex. Only two regulators were found to be up‐regulated (interleukin [IL]‐10RA and IL‐1RN), both of which have anti‐inflammatory roles. Several of the affected inflammatory regulators are also involved in liver necrosis, cell death or apoptosis, as indicated in Table 2. In addition, icosabutate activated regulators involved in lipid metabolism and energy production, and in general, the effects were directed at β‐oxidation, mitochondrial activation, and glucose metabolism. To investigate the effects of icosabutate on expression of genes pertaining to the sequence of events leading to fibrosis in more detail, a pathway analysis was performed that demonstrated the cell‐specific down‐regulation of fibrogenic genes in the liver. More specifically, the early signaling events in hepatic stellate cells (Fig. 5A), containing tumor necrosis factor receptor (TNFR), epidermal growth factor receptor, insulin‐like growth factor 1, IL‐6R, endothelin 1 (ET‐1), fibroblast growth factor receptor 1, and a few associated downstream factors, were down‐regulated by icosabutate. Furthermore, icosabutate was also able to down‐regulate genes involved in fibrogenesis in activated stellate cells (Fig. 5B), like IL‐6R, chemokine (C‐C motif) receptor 5, IL‐1β, TNFR, ET‐1, IL‐10, IL‐1α, and several downstream factors.
Table 2.
Upstream Regulators
| Upstream Regulator | Activation Z Score | P‐Value of Overlap | Function |
|---|---|---|---|
| Lipid metabolism | |||
| PPARA* | 7.782 | 2.65E‐86 | Major regulator of lipid metabolism; also regulator of cell death and apoptosis |
| PPARG | 7.287 | 7.19E‐11 | PPARG regulates fatty acid storage and glucose metabolism |
| SREBF2 | 5.048 | 1.77E‐16 | Controls cholesterol homeostasis by stimulating transcription of sterol‐regulated genes |
| SREBF1 | 4.822 | 4.53E‐18 | Involved in sterol biosynthesis |
| Nr1h/LXR | 4.286 | 1.00E‐05 | Important regulator of cholesterol, fatty acid, and glucose homeostasis |
| PPARGC1B | 3.996 | 1.50E‐06 | Involved in fat oxidation |
| PPARD | 3.740 | 2.78E‐22 | Regulates the peroxisomal beta‐oxidation pathway of fatty acids |
| ESRRA | 3.516 | 1.20E‐03 | Regulator of fatty acid metabolism |
| FGF21 | 3.129 | 2.17E‐04 | Involved in lipid metabolism and ketogenesis |
| Inflammatory response | |||
| STAT1* | −7.130 | 1.63E‐13 | Important for cell viability in response to different cell stimuli and pathogens; key modulator of cell death |
| IRF7 | −6.888 | 2.63E‐17 | Regulator of many interferon‐alpha genes |
| Interferon alpha | −6.710 | 2.93E‐15 | Subgroup of interferon proteins involved primarily in innate immune response against viral infection |
| IFNG* | −6.543 | 4.59E‐20 | Cytokine that induces an inflammatory response and apoptotic cell death |
| TNF* | −5.733 | 5.83E‐18 | Proinflammatory cytokine secreted primarily by macrophages; inducer of cell death, apoptosis |
| Ifnar | −5.682 | 3.98E‐17 | Receptor which binds endogenous type I interferon cytokines |
| IRF3* | −5.555 | 2.01E‐14 | Mediates cellular antiviral responses by both inducing antiviral genes and triggering apoptosis |
| NFkB (complex) | −4.873 | 2.39E‐06 | Key regulator of immune and inflammatory responses |
| IRF1* | −4.791 | 9.55E‐12 | Regulator of immune response, apoptosis, and DNA damage |
| TLR3 | −4.753 | 3.52E‐08 | A member of the toll‐like receptor family of pattern recognition receptors of the innate immune system |
| TLR9* | −4.722 | 4.88E‐05 | Receptor expressed in, e.g., dendritic cells, macrophages, natural killer cells; may activate apoptosis |
| TLR7* | −4.594 | 1.75E‐04 | Plays an important role in pathogen recognition and activation of innate immunity; may activate apoptosis |
| IL21 | −4.589 | 2.66E‐04 | Cytokine with immunoregulatory activity; may promote the transition between innate and adaptive immunity |
| CHUK* | −4.553 | 1.19E‐03 | Part of the IκB kinase complex; also plays a role in cell death and cell proliferation |
| NFATC2 | −4.409 | 3.74E‐04 | Pays a central role in inducing gene transcription during the immune response |
| IKBKB* | −4.344 | 6.79E‐04 | Protein subunit of IκB kinase; blocks NF‐κB activation; also plays a role in cell death and cell proliferation |
| IRF5* | −4.340 | 9.58E‐05 | Acts as a molecular switch to control whether macrophages promote or inhibit inflammation; promotes cell death |
| TLR4 | −4.329 | 7.39E‐06 | Activator of intracellular signaling pathway NF‐κB and inflammatory cytokine production |
| RELA* | −4.272 | 9.23E‐07 | Involved in NFκB heterodimer formation, nuclear translocation, and activation; also proto‐oncogene |
| IKBKG* | −4.136 | 2.06E‐04 | A subunit of the IκB kinase complex that activates NF‐κB; also plays a role in cell death and cell proliferation |
| Ifn | −3.968 | 9.97E‐12 | Group of cytokines produced by host cells in response to the presence of several pathogens |
| IFN alpha/beta | −3.715 | 2.64E‐07 | Cytokines involved primarily in innate immune response |
| IFN type 1 | −3.701 | 3.05E‐07 | Large subgroup of interferon proteins that help regulate the activity of the immune system |
| IL2 | −3.676 | 1.46E‐03 | Cytokine that stimulates the growth of T‐cell lymphocytes |
| IL1 | −3.629 | 1.35E‐02 | Group of cytokines that plays a central role in the regulation of immune and inflammatory responses |
| IL1A | −3.492 | 4.40E‐03 | A cytokine that plays one of the central roles in the regulation of the immune responses |
| IL6* | −3.485 | 5.83E‐06 | A pro‐inflammatory cytokine; associated with necrosis |
| JAK2* | −3.397 | 9.62E‐04 | Mediates essential signaling events in both innate and adaptive immunity; delays cell death |
| C3* | −3.332 | 4.92E‐04 | A protein of the immune system; associated with liver damage |
| IL1B* | −3.255 | 2.32E‐13 | An important mediator of the inflammatory response; also involved in cell proliferation, differentiation, and apoptosis |
| CD14* | −3.244 | 1.78E‐02 | A component of the innate immune system; expressed on monocytes/macrophage; plays a role in apoptototic cell clearance |
| CSF2 | −3.225 | 1.84E‐05 | Stimulates stem cells to produce granulocytes and monocytes |
| IL6R* | −3.219 | 2.64E‐03 | IL‐6 receptor; plays an important role in immune response |
| IL12 (complex) | −3.174 | 8.76E‐04 | A T cell–stimulating factor |
| C5 | −3.062 | 4.58E‐03 | Plays an important role in inflammatory and cell killing processes |
| CSF1 | −3.048 | 1.70E‐06 | Cytokine that influences differentiation into macrophages or other related cell types |
| OSM* | −3.023 | 1.24E‐07 | A pleiotropic cytokine that belongs to the IL‐6 group of cytokines; inducer of cell death |
| IL10RA | 3.366 | 3.46E‐14 | IL‐10 receptor; inhibits the synthesis of proinflammatory cytokines |
| IL1RN* | 3.834 | 7.40E‐08 | IL‐1 inhibitor |
| Energy production | |||
| INSR | 6.234 | 4.75E‐10 | Insulin receptor that plays a key role in the regulation of glucose homeostasis |
| PPARGC1A | 4.090 | 1.42E‐14 | Regulator of mitochondrial biogenesis and function |
| PPARGC1B | 3.996 | 1.50E‐06 | Involved in nonoxidative glucose metabolism and energy expenditure |
| ESRRA | 3.516 | 1.20E‐03 | Regulator of mitochondrial biogenesis, gluconeogenesis, and oxidative phosphorylation |
Effect of icosabutate on hepatic gene expression involved in lipid metabolism, inflammatory response, and energy production. High fat/cholesterol–fed APOE*3Leiden.CETP mice were left untreated or treated with icosabutate for 20 weeks. Data represent the predicted activation state (z score ≤ 3 or > 3) of the upstream regulators, based on the expression changes of known target genes. The overlap p value indicates the significance of the overlap between the known target genes of a transcription factor and the differentially expressed genes measured in an experiment. Red color indicates up‐regulation; green color indicates down‐regulation.
Indicates upstream regulators that are involved in liver necrosis, cell death, or apoptosis as well.
Abbreviations: C, complement; CD14, cluster of differentiation 14; CHUK, component of inhibitor of NFκB; CSF, colony stimulating factor; ESRA, estrogen receptor alpha; ESRRA, estrogen related receptor alpha; FGF21, fibroblast growth factor 21; IFN, interferon; Ifnar, interferon‐α/β receptor; IFNG, interferon gamma; IKBKB, inhibitor of NFκB subunit beta; IKBKG, inhibitor of NFκB subunit gamma; IL1RA, interleukin 1 receptor antagonist; IL10RA, interleukin 10 receptor subunit alpha; INSR, insulin receptor; IRF, interferon regulatory factor; JAK2, janus kinase 2; LXR, liver X receptor; NFATC2, nuclear factor of activated T‐cells, cytoplasmic 2; NFκB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; Nr1h: nuclear receptor family; OSM, oncostatin M; PPARA, PPAR‐alpha; PPARD, PPAR‐delta; PPARGC1B, PPARG Coactivator 1 Beta; RELA, REL‐associated protein involved in NFκB heterodimer formation; SREBF, sterol regulatory element binding transcription factor; TLR, toll‐like receptor; TNF, tumor necrosis factor.
Figure 5.
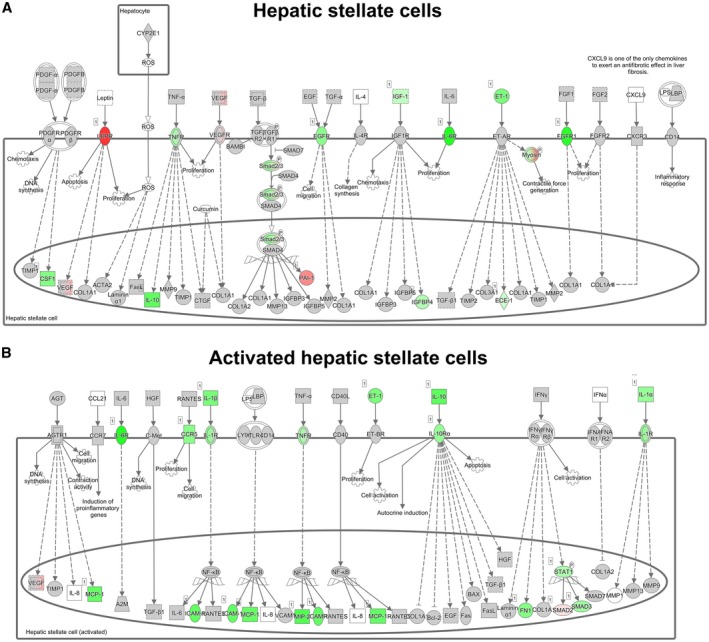
Hepatic fibrosis pathway analysis. Pathway analysis showing statistically significant gene‐expression changes in hepatic stellate cells (A) and activated hepatic stellate cells (B) of APOE*3Leiden.CETP mice fed a high‐fat/cholesterol diet and treated with icosabutate for 20 weeks relative to control group. Red color indicates up‐regulation and green color indicates down‐regulation. Abbreviations: BAX, B cell lymphoma 2–associated X protein; Bcl2, B cell lymphoma 2; CD, clusters of differentiation; CCL2, chemokine (C‐C motif) ligand 2; CCR9, chemokine (C‐C motif) receptor 9; CTGF, connective tissue growth factor; GSF1, growth stimulating factor 1; EGF, endothelial growth factor; FGF, fibroblast growth factor; FN1, fibroblast growth factor–inducible 1; HGF, hepatocyte growth factor; ICAM, intercellular cell adhesion molecule; IFN, interferon; IGF, insulin‐like growth factor; LBP, lipopolysaccharide‐binding protein; LPS, lipopolysaccharide; MCP‐1, monocyte chemoattractant protein 1; MMP, matrix metalloproteinase; NFκB, nuclear factor kappa B; PDGF, platelet‐derived growth factor; ROS, reactive oxygen species; SMAD, mothers against decapentaplegic homolog; TGF, transforming growth factor; TIMP, tissue inhibitor of metalloproteinase; VCAM1, vascular cell adhesion molecule 1; VEGF, vascular endothelial growth factor.
Discussion
We observed that icosabutate is absorbed from the gut almost entirely through the portal vein and accumulates rapidly in the liver. Icosabutate also induced marked improvements in glucose metabolism and insulin sensitivity in obese, type 2 diabetic ob/ob mice. In addition, in high‐fat/cholesterol‐fed APOE*3Leiden.CETP mice (a well‐established model for hyperlipidemia, atherosclerosis, and NAFLD/NASH14, 16, 17, 18), icosabutate significantly inhibited microvesicular steatosis, hepatocellular hypertrophy, hepatic inflammation, and fibrosis by beneficially regulating lipid and energy metabolism and inflammation. In addition, icosabutate significantly reduced the hepatic concentrations of multiple lipotoxic lipid species and oxidative stress.
As a multi‐etiological disorder with limited success achieved to date with single target drugs,15 NASH is an attractive indication for drugs with pleiotropic targeting potential, such as ω‐3 fatty‐acids.19 However, a potentially important issue limiting their clinical efficacy is related to suboptimal absorption, distribution, and metabolism properties of ω‐3 fatty acids for liver targeting, which in turn could be rectified using structural engineering.
The structural modifications of icosabutate confer effective targeting of the liver as evidenced by the more than 99% uptake through the portal vein. This lack of systemic distribution likely contributes to the potent pharmacological effects of icosabutate at translational doses. To translate the dosing used in our rodent studies (112 or 135 mg/kg/d ) to human dosing, the following simplified calculation based on body surface area20 can be used as a guide: mouse dose/12.3 × human body weight; thus, 112 mg/kg/d in mice is approximately 730 mg/day for an 80‐kg human. This corresponds to the highly efficacious 600‐mg/day dose used previously in the clinic.8, 9 This is markedly less than, for example, the 150 mg per mouse/day (versus approximately 4 to 7 mg/per mouse/day in current studies for APOE*3Leiden.CETP and ob/ob models, respectively) used in previous studies to achieve improvements in insulin resistance and inflammation with unmodified ω‐3 fatty‐acids.21 We used comparatively high doses of all positive controls to avoid inadequate dosing (e.g., rosiglitazone maximum dose in humans is 8 mg/kg, current study is 13 mg/kg; fenofibrate maximum dose in humans is 120 mg/d, present study is 100 mg/kg; pioglitazone maximum dose in humans is 15 mg/d, current study is 30 mg/kg). Icosabutate (300‐600 mg/day in humans versus 112‐135 mg/kg in current study) was therefore used at a lower dose in the current experiments in relation to the human dose versus all positive controls.
Resistance to incorporation into complex lipids likely also underlies the rapid decline in icosabutate and its metabolites, with excretion greater than 95% complete by 48 hours. This contrasts sharply to unmodified ω‐3 fatty acids, in which incorporation and accumulation in complex lipids such as cell membranes necessitates extensive wash‐out periods in cross‐over design studies.22
We tested icosabutate in high‐fat/cholesterol‐fed APOE*3Leiden.CETP mice that develop the characteristics of human NAFLD and NASH.12, 14, 16 Icosabutate significantly inhibited microvesicular steatosis, hepatocellular hypertrophy, and hepatic inflammation and fibrosis. Rosiglitazone also reduced inflammation, but no change was seen in fibrosis. A similar pattern was seen with another thiazolidinedione (pioglitazone) in the PIVENS study, with improvements in lobular inflammation without change in fibrosis.23 One possible explanation for this observation is the notable difference observed between icosabutate and rosiglitazone in the ability of icosabutate to lower the hepatic concentrations of multiple hepatic NASH‐associated lipotoxic lipid species (FFAs,24 ceramides,25 bile acids,26 DAG,24 arachidonic acid (AA),27 and AA‐derived HETEs28) in addition to hepatic oxidative stress—effects not seen with rosiglitazone. The decrease in hepatic bile acid concentrations can be explained by the PPARα‐agonistic activity of icosabutate, as PPAR‐α activators suppress bile acid synthesis.29
The substrate‐overload model, as illustrated by Sanyal et al.,15 proposes a stepwise path to fibrogenesis, with excess FFAs driving the generation of lipotoxic lipid species that in turn activate endoplasmic reticulum and oxidative stress. This induces an inflammatory response (and apoptosis/necrosis) in hepatocytes/macrophages that ultimately results in activation of stellate cells and fibrogenesis. The ability of icosabutate to reduce all components of this stepwise NASH model (i.e., from reduction in hepatic FFAs and lipotoxic lipid species to a decrease in oxidative stress, inflammation, and fibrosis) is promising with respect to the need for upstream and/or pleiotropic targeting in NASH. Of particular interest, icosabutate was highly effective in reducing the hepatic concentrations of HETEs, pro‐inflammatory arachidonic acid metabolites that have been associated with progression from normal to NAFLD to NASH30 and are associated with inflammation score in human NASH.28 The marked decrease in hepatic AA concentrations after icosabutate treatment suggests that substrate availability may explain the decreased HETE concentrations. In addition to the hepatic antifibrotic effects of PLA2 inhibitors in mice, the potential role of the arachidonic cascade in liver fibrosis is supported by recent findings that demonstrate the protective effect of aspirin on fibrosis progression in NASH patients.31 Finally, with respect to the reduction in hepatic oxidative stress after treatment with icosabutate, it has recently been proposed that oxidative stress drives NASH and fibrosis by activation of STAT1,32 which was the most inhibited upstream regulator in response to treatment with icosabutate.
Modified cholesterol (cholesterol crystals) in the liver is also believed to be involved in the pathogenesis of human NASH.33 The mechanism underlying the ability of icosabutate, but not rosiglitazone, to significantly reduce microvesicular steatosis in high‐cholesterol‐fed mice may be related to regulation of hepatic cholesterol metabolism, as cholesterol overload in mice induces microvesicular steatosis in association with increased oxidative stress,34 both of which are significantly diminished by icosabutate. However, both rosiglitazone and icosabutate significantly reduced hepatic cholesterol ester, so absolute concentrations of unmodified cholesterol do not explain the reduction in microvesicular steatosis after icosabutate treatment only.
It should be noted that although the high‐fat‐fed APOE*3Leiden.CETP mouse model is an established rodent NAFLD/NASH model that develops a more robust hepatic inflammatory and fibrotic response with additional cholesterol in the diet,12, 14, 16 the increase in fibrosis in this study was relatively mild due to the relatively short duration. Furthermore, in the current study, icosabutate was administered at the same time as the start of the high‐fat/cholesterol diet and not in a treatment design (i.e. in a situation of existing NASH and fibrosis). We evaluated the effects of a 6‐week treatment of icosabutate in APOE*3Leiden.CETP mice first fed a high‐fat diet (without supplemented cholesterol) for 20 weeks as well and found a significant reduction of −48% (p = 0.001) versus control in hepatic triglycerides (data not shown). As these dietary conditions were less suitable to induce hepatic inflammation and fibrosis, the latter study suggested that icosabutate could treat existing steatosis, but did not provide clues on treatment properties for inflammation and fibrosis. Although the results with icosabutate in the current study in the high‐fat/cholesterol‐fed APOE*3Leiden.CETP mouse model are encouraging, delayed treatment design studies in more severe inflammatory/fibrotic models are currently being evaluated.35
In addition to the effects on hepatic lipid species, inflammation and fibrosis, icosabutate demonstrated marked improvements in glucose metabolism and insulin sensitivity in ob/ob mice. These observations are in line with the clinical observations of icosabutate in hypertriglyceridemic subjects, in whom icosabutate significantly lowered both fasting plasma insulin and insulin sensitivity (HOMA‐IR).9 There are several possible mechanisms underlying these effects related to either direct effects following insulin signaling or indirectly through inhibition of inflammatory responses.36 The moderately positive effects of fenofibrate suggest that PPAR‐α may, at least partially, be involved in the improvements in insulin resistance in ob/ob mice. However, icosabutate elicited a more powerful effect than fenofibrate, particularly in response to an oral glucose load, and moreover also reduced plasma ALT. The comparable effect of pioglitazone and icosabutate on glucose tolerance in response to a glucose load is remarkable, given that icosabutate does not target PPAR‐γ (as evidenced by lack of effect on plasma adiponectin and body weight, in addition to lack of PPAR‐γ or PPAR‐δ activity in vitro [in vitro data not shown]). Reductions in hepatic lipid species known to aggravate insulin sensitivity such as AA metabolites,37 DAG,38 and ceramides39 could potentially explain icosabutate's potent effects on glycemic control, and/or activation of ω‐3‐sensitive G‐protein–coupled receptors.40
A potential limitation of these studies is the use of 2 different thiazolidinediones, pioglitazone and rosiglitazone, for the ob/ob and APOE*3Leiden.CETP studies, respectively. Indeed, a recent meta‐analysis demonstrated that their clinical efficacy in NASH with advanced liver fibrosis is differentiated.41 However, it is also important to emphasize that the data presented herein are primarily intended to establish pharmacokinetic and pharmacodynamic characteristics of a viable drug (i.e., icosabutate) rather than compare the relative efficacy of well‐established thiazolidinediones in different mouse models.
In conclusion, we have demonstrated that icosabutate, a structurally engineered EPA derivative, selectively targets the liver and elicits potent insulin‐sensitizing, hepatic anti‐inflammatory, and antifibrotic effects in preclinical models of type 2 diabetes or hepatic inflammation/metabolic dysregulation. Given its benign safety profile and established hypolipidemic effects in humans,8, 9 icosabutate offers a promising therapeutic approach to the treatment of both inflammatory/fibrotic liver disease and co‐existing glucose and lipid abnormalities.
Supporting information
Acknowledgments
We thank Martien P. M. Caspers, Wen Liang, and Anita van Nieuwkoop for their excellent technical assistance, and Juan M. Falcon‐Perez, Sebastiaan van Liempd, and Diana Cabrera from the Metabolomics Platform of CIC bioGUNE (Bizkaia, Spain) for their contributions toward UPLC‐MS/MS analysis of GSH and GSSG in liver tissue. We also thank R&D Pronova Biopharma/BASF for their extensive work in the development of icosabutate.
This work was supported by Pronova Biopharma AS/BASF and NorthSea Therapeutics and the TNO research program “Biomedical Health.” NorthSea Therapeutics was involved in the study design and preparation of the manuscript (D.A.F, T.S), but had no role in the data collection.
Potential conflict of interest: Dr. Alonso is employed by OWL Metabolomics. Dr. Fraser is employed by NorthSea Therapeutics, which owns the rights to icosabutate. Dr. Iruarrizaga‐Lejarreta is employed by OWL Metabolomics. Dr. Princen received grants from Pronova Biopharma AS/BASF and NorthSea Therapeutics. Dr. Skjaeret is employed by NorthSea Therapeutics, which owns the rights to icosabutate.
[Correction added 6 January 2020 to Table 1, Part A. The heading for Column 3 was incorrect and has been updated.]
References
Author names in bold designate shared co‐first authorship.
- 1. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med 2019;380:11‐22. [DOI] [PubMed] [Google Scholar]
- 2. Innes JK, Calder PC. The differential effects of eicosapentaenoic acid and docosahexaenoic acid on cardiometabolic risk factors: a systematic review. Int J Mol Sci 2018;19:532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schuchardt JP, Hahn A. Bioavailability of long‐chain omega‐3 fatty acids. Prostaglandins Leukot Essent Fatty Acids 2013;89:1‐8. [DOI] [PubMed] [Google Scholar]
- 4. Sigalet DL, Winkelaar GB, Smith LJ. Determination of the route of medium‐chain and long‐chain fatty acid absorption by direct measurement in the rat. J Parenter Enteral Nutr 1997;21:275‐278. [DOI] [PubMed] [Google Scholar]
- 5. Carlier H, Bernard A, Caselli C. Digestion and absorption of polyunsaturated fatty acids. Reprod Nutr Dev 1991;31:475‐500. [DOI] [PubMed] [Google Scholar]
- 6. Lambert JE, Parks EJ. Postprandial metabolism of meal triglyceride in humans. Biochim Biophys Acta 2012;1821:721‐726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Plourde M, Chouinard‐Watkins R, Rioux‐Perreault C, Fortier M, Dang MTM, Allard MJ, et al. Kinetics of 13C‐DHA before and during fish‐oil supplementation in healthy older individuals. Am J Clin Nutr 2014;100:105‐112. [DOI] [PubMed] [Google Scholar]
- 8. Bays HE, Hallén J, Vige R, Fraser D, Zhou R, Hustvedt SO, et al. Icosabutate for the treatment of very high triglycerides: a placebo‐controlled, randomized, double‐blind, 12‐week clinical trial. J Clin Lipidol 2016;10:181‐191.e2. [DOI] [PubMed] [Google Scholar]
- 9. Kastelein JJP, Hallén J, Vige R, Fraser DA, Zhou R, Hustvedt SO, et al. Icosabutate, a structurally engineered fatty acid, improves the cardiovascular risk profile in statin‐treated patients with residual hypertriglyceridemia. Cardiology 2016;135:3‐12. [DOI] [PubMed] [Google Scholar]
- 10. Gao H, Geng T, Huang T, Zhao Q. Fish oil supplementation and insulin sensitivity: a systematic review and meta‐analysis. Lipids Health Dis 2017;16:131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 12. Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS One 2014;9:e115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Smith U. Pioglitazone: mechanism of action. Int J Clin Pract Suppl 2001:13‐18. [PubMed] [Google Scholar]
- 14. Liang W, Verschuren L, Mulder P, van der Hoorn JW, Verheij J, van Dam AD, et al. Salsalate attenuates diet induced non‐alcoholic steatohepatitis in mice by decreasing lipogenic and inflammatory processes. Br J Pharmacol 2015;172:5293‐5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van den Hoek AM, van der Hoorn JW, Maas AC, van den Hoogen RM, van Nieuwkoop A, Droog S, et al. APOE*3Leiden.CETP transgenic mice as model for pharmaceutical treatment of the metabolic syndrome. Diabetes Obes Metab 2014;16:537‐544. [DOI] [PubMed] [Google Scholar]
- 17. Zadelaar S, Kleemann R, Verschuren L, de Vries‐Van der Weij J, van der Hoorn J, Princen HM, et al. Mouse models for atherosclerosis and pharmaceutical modifiers. Arterioscler Thromb Vasc Biol 2007;27:1706‐1721. [DOI] [PubMed] [Google Scholar]
- 18. Kuhnast S, Fiocco M, van der Hoorn JW, Princen HM, Jukema JW. Innovative pharmaceutical interventions in cardiovascular disease: focusing on the contribution of non‐HDL‐C/LDL‐C‐lowering versus HDL‐C‐raising: a systematic review and meta‐analysis of relevant preclinical studies and clinical trials. Eur J Pharmacol 2015;763:48‐63. [DOI] [PubMed] [Google Scholar]
- 19. Jump DB, Lytle KA, Depner CM, Tripathy S. Omega‐3 polyunsaturated fatty acids as a treatment strategy for nonalcoholic fatty liver disease. Pharmacol Ther 2018;181:108‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 2016;7:27‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, et al. GPR120 is an omega‐3 fatty acid receptor mediating potent anti‐inflammatory and insulin‐sensitizing effects. Cell 2010;142:687‐698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Allaire J, Vors C, Tremblay AJ, Marin J, Charest A, Tchernof A, et al. High‐dose DHA has more profound effects on LDL‐related features than high‐dose EPA: the ComparED Study. J Clin Endocrinol Metab 2018;103:2909‐2917. [DOI] [PubMed] [Google Scholar]
- 23. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med 2010;362:1675‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Neuschwander‐Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774‐788. [DOI] [PubMed] [Google Scholar]
- 25. Luukkonen PK, Zhou Y, Sadevirta S, Leivonen M, Arola J, Oresic M, et al. Hepatic ceramides dissociate steatosis and insulin resistance in patients with non‐alcoholic fatty liver disease. J Hepatol 2016;64:1167‐1175. [DOI] [PubMed] [Google Scholar]
- 26. Chiang JYL. Targeting bile acids and lipotoxicity for NASH treatment. Hepatol Commun 2017;1:1002‐1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanai S, Ishihara K, Kawashita E, Tomoo T, Nagahira K, Hayashi Y, et al. ASB14780, an orally active inhibitor of group IVA phospholipase A2, is a pharmacotherapeutic candidate for nonalcoholic fatty liver disease. J Pharmacol Exp Ther 2016;356:604‐614. [DOI] [PubMed] [Google Scholar]
- 28. Hall Z, Bond NJ, Ashmore T, Sanders F, Ament Z, Wang X, et al. Lipid zonation and phospholipid remodeling in nonalcoholic fatty liver disease. Hepatology 2017;65:1165‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Post SM, Duez H, Gervois PP, Staels B, Kuipers F, Princen HM. Fibrates suppress bile acid synthesis via peroxisome proliferator‐activated receptor‐alpha‐mediated downregulation of cholesterol 7alpha‐hydroxylase and sterol 27‐hydroxylase expression. Arterioscler Thromb Vasc Biol 2001;21:1840‐1845. [DOI] [PubMed] [Google Scholar]
- 30. Puri P, Wiest MM, Cheung O, Mirshahi F, Sargeant C, Min HK, et al. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009;50:1827‐1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Simon TG, Henson J, Osganian S, Masia R, Chan AT, Chung RT, et al. Daily aspirin use associated with reduced risk for fibrosis progression in patients with nonalcoholic fatty liver disease. Clin Gastroenterol Hepatol 2019;17:2776‐2784.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grohmann M, Wiede F, Dodd GT, Gurzov EN, Ooi GJ, Butt T, et al. Obesity drives STAT‐1‐dependent NASH and STAT‐3‐dependent HCC. Cell 2018;175:1289‐1306.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ioannou GN, Landis CS, Jin GY, Haigh WG, Farrell GC, Kuver R, et al. Cholesterol crystals in hepatocyte lipid droplets are strongly associated with human nonalcoholic steatohepatitis. Hepatol Commun 2019;3:776‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dominguez‐Perez M, Simoni‐Nieves A, Rosales P, Nuno‐Lambarri N, Rosas‐Lemus M, Souza V, et al. Cholesterol burden in the liver induces mitochondrial dynamic changes and resistance to apoptosis. J Cell Physiol 2019;234:7213‐7223. [DOI] [PubMed] [Google Scholar]
- 35. Fraser DA, Schuppan D, Wang X‐Y, Feigh M, Thorbek DD, Alonso C, et al. SAT‐346‐Icosabutate induces a potent reduction in hepatic oxidative stress in rodent models of metabolic stress and fibrosing NASH. J Hepatol 2019;70:e791. [Google Scholar]
- 36. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest 2006;116:1793‐1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med 2015;21:239‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kumashiro N, Erion DM, Zhang D, Kahn M, Beddow SA, Chu X, et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc Natl Acad Sci U S A 2011;108:16381‐16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Raichur S, Brunner B, Bielohuby M, Hansen G, Pfenninger A, Wang B, et al. The role of C16:0 ceramide in the development of obesity and type 2 diabetes: CerS6 inhibition as a novel therapeutic approach. Mol Metab 2019;21:36‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Oh DY, Olefsky JM. Omega 3 fatty acids and GPR120. Cell Metab 2012;15:564‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Musso G, Cassader M, Paschetta E, Gambino R. Thiazolidinediones and advanced liver fibrosis in nonalcoholic steatohepatitis: a meta‐analysis. JAMA Intern Med 2017;177:633‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials