

Abstract
Diabetic kidney disease (DKD) is a life‐limiting condition characterized by progressive and irreversible loss of renal function. Currently, the estimated glomerular filtration rate (eGFR) and albuminuria are used as key markers to define DKD. However, they may not accurately indicate the degree of renal dysfunction and injury. Current therapeutic approaches for DKD, including attainment of blood pressure goals, optimal control of blood glucose and lipid levels, and the use of agents to block the renin‐angiotensin‐aldosterone system (RAAS) can only slow the progression of DKD. Hence, early diagnosis and innovative strategies are needed to both prevent and treat DKD. In recent years, a novel class of noncoding RNA, microRNAs (miRNAs) are reported to be involved in all biological processes, including cellular proliferation, apoptosis, and differentiation. miRNAs are small noncoding RNAs that regulate gene expression by posttranscriptional and epigenetic mechanisms. They are found to be in virtually all body fluids and used successfully as biomarkers for various diseases. Urinary miRNAs correlate with clinical and histologic parameters in DKD and differential urinary miRNA expression patterns have been reported. Kidney fibrosis is the common end stage of various CKD including DKD. Transforming growth factor‐β(TGF‐β) is regarded as the master regulator of kidney fibrosis, which is likely at least in part through regulating miRNA expression. miRNA are widely involved in the progression of DKD via many molecular mechanisms. In this review, the involvement of miRNA in fibrosis, inflammation, hypertrophy, autophagy, endoplasmic reticulum (ER) stress, oxidative stress, insulin resistance, and podocyte injury will be discussed, as these mechanisms are believed to offer new therapeutic targets that can be exploited to develop important treatments for DKD over the next decade.
Keywords: biomarker, diabetic kidney disease, fibrosis, microRNA, therapy
1. INTRODUCTION
Diabetic kidney disease (DKD), characterized by glomerular hypertrophy, proteinuria, decreased glomerular filtration, and kidney fibrosis, is a major microvascular complication of diabetes.1, 2 Due to the high prevalence of diabetes, DKD has emerged as an important public health concern as more than a half of patients with type 2 diabetes (T2DM) and one third of those with type 1 diabetes (T1DM) develop DKD.3 Consequently, DKD is a major cause of end‐stage kidney disease (ESKD), resulting in death or requiring renal replacement therapy, either kidney transplantation or dialysis.4 It thus represents a significant burden on the health system and cost to the community.
Currently, eGFR and albuminuria are used as key markers to define DKD at a specific point in time.5, 6 eGFR is generally calculated from the serum creatinine measurement with equations that variously also require age, body size, and assigned values based upon sex and race. The two most common equations are Chronic Kidney Disease Epidemiology Collaboration (CKD‐EPI) creatinine equation (2009) and Modification of Diet in Renal Disease Study (MDRD) equation. More recently, calculation of eGFR using other laboratory biomarkers such as cystatin C has emerged with apparent greater accuracy and a different set of CKD‐EPI calculators was established using the result of a cystatin C test. However, equations based on cystatin C overestimate directly measured GFR, while equations based on serum creatinine underestimate GFR.7 Also, in elderly persons, the variables affecting creatinine tend to be more pronounced because of comorbid conditions.8 Albuminuria strongly predicts progression of DKD, but it lacks specificity and sensitivity for ESKD and progressive decline in eGFR. In T1DM, only about one third of those with microalbuminuria had progressive renal function decline.9 In T2DM, a large proportion of those who have renal disease progression are normoalbuminuric.10, 11, 12 To conclude, both eGFR and albuminuria have their limitations as predictors of kidney injury.13 Thus, a sensitive and easily detectable biomarker is needed to monitor the decline in kidney function and to separate “progressors” from “non‐progressors” in those with DKD.
Numerous non‐modifiable risk factors for DKD, including ethnicity and inherited genetic difference, have been identified.14 Hyperglycemia, advanced glycation end products (AGEs), inflammation via cytokines/chemokines, and aberrant hemodynamics contribute to the above pathological changes.15 Hence, current therapies for DKD focus on blood pressure control with inhibitors of the RAAS, on glycemic and lipid control, and lifestyle changes.16, 17, 18 Despite tight control of blood glucose levels with glucose lowing medications, of hyperlipidaemia with statins and of blood pressure with angiotensin‐converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs),19, 20, 21 a large proportion of patients develop ESKD.22 Therefore, some novel therapeutic strategies are required. Development of novel therapeutic options requires identification of new molecular mechanisms underlying the development of DKD and then targeted development of therapeutics.
Taken together, novel molecular mechanisms underlying DKD should be investigated to design both future biomarkers of disease progression and optimal therapies. With the development of high‐throughput technologies, the important role of epigenetic mechanisms, especially the miRNAs have been explored.23, 24 Circulating miRNAs are present in body fluids and are known to influence gene expression and regulation.24 Abundant expression, lower complexity, tissue specificity, stability, and evolutionary conservation are some of the qualities that make circulating and urinary miRNAs attractive as noninvasive biomarkers to reflect pathophysiological conditions and disease states.25 More importantly, a variety of studies have reported the role of miRNAs in the pathology of DKD, thus these small molecules present new possibilities for therapeutic intervention.23 Currently, many attempts to downregulate or upregulate miRNAs using one of several delivery approaches in animal models of DKD in vivo have been achieved.2 In the future, the control of the expression of miRNA might be developed for patient use.
2. miRNA
miRNAs (19‐28 nucleotides in length) comprise a novel class of endogenous short noncoding single‐stranded RNA that regulate various cellular processes such as cell death, differentiation, proliferation, metabolism, and pathophysiology of many diseases via the regulation of target gene expression.23 Initially, miRNAs are transcribed from DNA into primary‐miRNAs (Pri‐miRNAs), which contain hairpin‐like structures. RNase III Drosha and its binding partner, DiGeorge syndrome critical region gene 8 (DGCR8), bind to the hairpin structures in Pri‐miRNAs and process them into precursor miRNAs (Pre‐miRNAs). Through Exportin 5, Pre‐miRNAs are transferred into the cytoplasm and are processed by another RNase III enzyme, Dicer, in collaboration with transactivating response RNA‐binding protein (TRBP) to generate the mature miRNA duplex. One strand of the duplex goes into RNA‐induced silencing complex (RISC), while the other is degraded. In RISC, mature miRNA recognizes target mRNAs through complete sequence complementarity, resulting in degradation of the target mRNA or more frequently, through incomplete sequence complementarity, resulting in inhibition of translation and protein synthesis (Figure 1).23
Figure 1.
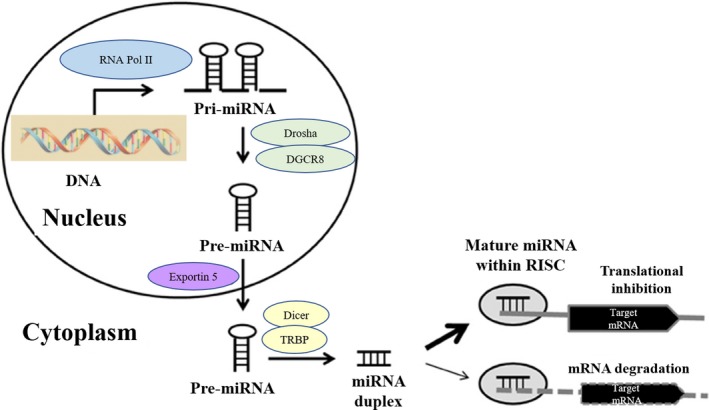
miRNA biogenesis and repression of gene expression
To date, more than 2500 mature miRNAs have been identified in humans (http://www.miRbase.org), which regulate at least 60% of protein‐coding genes.26 Moreover, one specific target gene can be regulated by many different miRNAs, and one single miRNA may alter the expression of a large number of target genes at different levels in a signaling cascade of a particular biological pathway.27 Hence, miRNAs can modulate the expression of numerous genes to alter key cellular functions and influence the course of various diseases.
3. QUANTIFICATION OF URINARY miRNAs AS BIOMARKERS FOR DKD
3.1. Existing biomarkers for DKD
Biomarkers are becoming increasingly important for predicting disease prognosis, enabling personalized therapy (precision medicine), and detecting early therapeutic and adverse responses to drugs. However, the identification, validation, and application of biomarkers are challenging, with several aspects, including understanding the biology of the biomarker and its relevance to disease, and the technological characteristics of the assay used for biomarker measurement.28 For DKD, biomarkers are increasingly being investigated for their utility in predicting patients most at risk of decline in kidney function to rationalize and target care.29
Clinically, eGFR and albuminuria are used to define the severity of DKD and to loosely identify those at risk of progression to ESKD. However, they may not accurately indicate the kidney function and injury in DKD.13, 30
Hence, some studies include other risk factors routinely captured in clinical records to predict kidney injury.31 These established clinical risk factors involve age, diabetes duration, HbA1c, systolic BP (SBP), albuminuria, prior eGFR and retinopathy status.31 However, there have been relatively few attempts to build and validate predictive equations using clinical data that would form the basis for evaluating the marginal improvement in prediction with these biomarkers.32, 33
Currently, an increasing number of novel biomarkers has emerged to identify both those at risk of DKD and early DKD with the aim of preventing the occurrence of ESKD. These novel biomarkers can be classified as: (a) Glomerular biomarkers: Transferrin, immunoglobulin G, ceruloplasmin, type IV collagen, laminin, glycosaminoglycans(GAGs), lipocalin‐type prostaglandin D synthase (L‐PGDS), fibronectin, podocytes‐podocalyxin, and vascular endothelial growth factor (VEGF); (b) Tubular biomarkers: neutrophil gelatinase‐associated lipocalin (NGAL), α‐1‐microglobulin, kidney injury molecule 1(KIM‐1), N‐acetyl‐β‐D‐glucosaminidase (NAG), cystatin C, and liver‐type fatty acid‐binding protein (L‐FABP); (c) Inflammatory biomarkers: TNF‐α, IL‐1β, IL‐18, interferon gamma induced protein (IP‐10), monocyte chemoattractant protein‐1 (MCP‐1), granulocyte colony‐stimulating factor (G‐CSF), eotaxins, RANTES (regulated on activation, normal T cell expressed and secreted) or CCL‐5; (d) Biomarkers of oxidative: 8‐oxo‐7,8‐dihydro‐2‐deoxyguanosine (8oHdG); (e) Others: podocalyxin, nephrin, AGEs.13 Although these biomarkers are potentially useful for the evaluation of DKD, none have been validated to improve clinical decision‐making.34 For example, neutrophil gelatinase‐associated lipocalin (NGAL) is a lipocalin iron‐carrying protein of 25 kDa which belongs to the super family of lipocalins. Urinary NGAL concentration has been found to be increased in diabetic subjects compared with healthy controls35 and to correlate negatively with eGFR, and positively with CysC, serum creatinine, and urea in patients with T2DM.36 Significant increases in urinary NGAL concentration have been demonstrated from normo‐ to micro‐to macroalbuminuric groups of patients with both T1DM and T2DM.37 Urinary NGAL correlates positively with glomerular hyperfiltration early in the clinical course of diabetes. However, after adjustment for factors including systolic blood pressure, HbA1c, and diabetes duration, there is no significant correlation between the urinary NGAL concentration and enhanced decline of eGFR in T2D with proteinuria.37
A number of “omics” studies have been performed in moderately sized cohorts to identify protein and metabolite biomarkers of DKD. CKD273 is a mass spectrometry‐based method combining data on 273 urinary peptides into a score that has high accuracy in the cross‐sectional classification of eGFR status.38 A study of 737 samples obtained at baseline in the Diabetic Retinopathy Candesartan Trials (DIRECT)‐Protect 2 indicated that the CKD273 score was strongly associated with incident microalbuminuria independently of baseline AER, eGFR, and other variables.39 Higher CKD273 score at baseline was associated with a larger reduction in ACR in the spironolactone group vs placebo.40 However, the interaction between treatment and CKD273 was not statistically significant and the concept that CKD273 will be useful in determining risk of disease progression and may also stratify treatment response is being more definitively tested in the ongoing PRIORITY trial of 3280 participants with T2DM.41 Other main “omics” studies include the SUMMIT study using mass spectrometry to measure low‐molecular‐weight metabolites, peptide and proteins (144 in all) as well as 63 proteins by ELISA and Luminex in a prospective design. However, in these global discovery studies, prediction has not been properly assessed on top of available clinical data.42
Hence, although these new biomarkers are promising, further studies are needed before establishing in clinical practice and overcoming the problems of specificity and technical variability.34 Therefore, it is necessary to explore novel biomarkers to allow accurate identification of kidney injury in DKD and “at risk” individuals.
3.2. Advantages of urinary miRNA measurement for kidney function
At present, biopsy is the gold standard diagnostic and prognostic test for kidney disease, but this is a highly invasive and expensive procedure with up to a 3% risk of complications.43 Clinically, the majority of patients who develop DKD do not undergo renal biopsy. Given there is an increasing enthusiasm to introduce treatments to reverse metabolic cytopathology before structural pathology develops, early identification of markers of pathology is necessary.
Urine is clearly a biological fluid that can reflect renal pathology.44 It can be collected easily, noninvasively, and at low cost. Urinary biomarkers may be elevated in diabetic patients even before the appearance of microalbuminuria and can be used as useful marker for detecting kidney impairment in patients with normoalbuminuria (early DKD).32, 33, 34, 35
Circulating miRNAs are relatively stable under different storage conditions such as extreme PH and long‐term room temperature storage, and resistant to RNase activity and repeated freeze‐thaw cycles.45, 46 Currently available commercial kits provide rapid extraction of total miRNAs from urine, based on the binding of small RNAs to specific material packed in columns.45 Up to now, researchers have developed many signal‐amplification strategies for miRNA detection such as hybridization chain reaction, nuclease amplification, rolling circle amplification, catalyzed hairpin assembly amplification, and nanomaterials‐based amplification.47 With the development of new technologies, it is now possible to quantify miRNA expression.
These observations, together with the fact that urinary miRNA concentrations have been found to associate with important clinical characteristics, including histopathological diagnosis strongly suggest that urinary miRNAs could be a promising pool of noninvasive biomarkers in DKD.48, 49, 50, 51
3.3. Urinary miRNAs signature in DKD
miRNAs in urine are released by cells of the nephron and downstream in the urinary tract. They are packed with membrane‐bound extracellular vesicles such as microvesicles, formed by outward budding of the plasma membrane; exosomes, secreted from multivesicular endosomes formed in the endocytic tract, and apoptotic bodies. Urinary miRNAs can also bind to Argonaute proteins and other proteins including high‐density lipoproteins (HDL).52 In a study including 80 T2DM patients with normoalbuminuria (n = 30), microalbuminuria (n‐30), or macroalbuminuria (n = 20), Jia et al showed that urinary miRNA‐192 was positively correlated with albuminuria levels and TGF‐β1 expression in patients with normoalbuminuria and microalbuminuria. Receiver operating characteristic (ROC) curve analysis revealed that miRNA‐192 had an area under the curve (AUC) of 0.802, which was better than miRNA‐194 with an AUC of 0.703 and miRNA‐215 with an AUC of 0.757 in discriminating the normoalbuminuric group from the microalbuminuric group, indicating the potential use of urinary miRNA‐192 as a biomarker of the early stage of DKD.51
Exosomes in urine originate from most kidney cells and thus are the best‐studied vehicles.53 Because exosomes can carry miRNAs to distant target cells, they represent an important mechanism for cell‐to‐cell communication.54, 55 miRNA‑29 levels in urine exosomes have been proposed as a biomarker of kidney fibrosis 56 but their expression can be dynamic. In a recent study, Mohan et al57 reported that urinary exosomal miRNA‐451‐5p level was increased in diabetic rats 3 weeks prior to significant albuminuria and 3 weeks before histological changes of kidney fibrosis. In contrast, renal expression of miRNA‐451‐5p declined and was negatively associated with the indices of renal pathology during the progression of DKD. Thus, urinary exosomal miRNA‐451‐5p may be secreted into the urine by the injured nephron in the early stage of DKD and hold prognostic value as an early and sensitive noninvasive indicator of renal damage.
However, DKD is a multifactorial progressive disease where the pathogenesis is extremely complex involving many different cells, molecules, and factors.1, 2, 4 Also, as many miRNAs have regulatory roles in both DKD and non‐DKD, single miRNA expression might exhibit poor specificity. Hence, there is an increase in studies utilizing urinary miRNA “biomarker panels”—multiple miRNAs measured in combination, to optimize diagnostic sensitivity and specificity as well as being more widely informative of disease processes.58, 59, 60 In a study by Argyropoulos et al,61 urine samples were assessed from a historical prospective cohort involving 906 eligible participants recruited from Children's Hospital of Pittsburgh in USA with a diagnosis of T1DM from 1950 to 1980. The urinary miRNA profile was assessed from 30 patients using a Bayesian procedure to normalize and convert raw signals to expression ratios. Urinary miRNA profile of 723 unique miRNAs in the urine of normoalbuminuric T1DM patients who did not develop DKD relative to patients who subsequently developed microalbuminuria were analyzed by qPCR. Eighteen miRNAs were found to be strongly associated with the subsequent development of microalbuminuria, while 15 miRNAs exhibited gender‐related differences in expression. A miRNA signature (miRNA‐105‐3p, miRNA‐1972, miRNA‐28‐3p, miRNA‐30b‐3p, miRNA‐363‐3p, miRNA‐424‐5p, miRNA‐486‐5p, miRNA‐495, miRNA‐5480‐3p and for women miRNA‐192‐5p, miRNA‐720) achieved high internal validity for the future development of microalbuminuria.61 However, there are also some limitations in this study. First, the relatively small number of patients makes the precise quantification of changes in expression rather challenging, which is reflected in the large confidence intervals for some of the microRNAs and their apparent lack of prognostic significance. Second, patients in the study have never had renal biopsies so that it is impossible to correlate the urinary miRNA expression with specific tissue pathology.
Recently, there are many studies performing meta‐analysis to screen out potential urinary miRNA biomarker candidates in DKD. For example, Park et al62 searched PubMed, Web of Science, and Cochrane Library for the meta‐analysis and suggested that miRNA‐126 and miRNA‐770 family miRNA were significantly dysregulated in both blood and urine from patients with DKD. Hence, they may have important diagnostic and pathogenetic implications for DKD. Another study performed a systematic review of the literature and bioinformatic analyses, indicating six consistently dysregulated miRNAs in DKD patients compared to controls: miRNA‐21‐5p, miRNA‐29a‐3p, miRNA‐126‐3p, miRNA‐192‐5p, miRNA‐214‐3p, and miRNA‐342‐3p. These six miRNAs are involved in pathways related to DKD pathogenesis and may constitute potential biomarkers (Table 1).63
Table 1.
Examples of miRNAs with potential as biomarkers in DKD
| miRNAs | Source | Study population | Sample size | Platform | Outcome/DKD stage | Reference |
|---|---|---|---|---|---|---|
| miR‐192 | Urinary extracellular vesicles | T2DM patients | 80 | Real‐time PCR | miR‐192 (AUC = 0.802)/Early stage | 51 |
| miR‐29c | Urinary exosome | CKD patients | 32 | Real‐time PCR | miR‐29c (r = −0.359; P < 0.05, AUC = 0.738)/ Tubulointerstitial fibrosis | 56 |
| miR‐451‐5p | Urinary exosome | Diabetic rats | 43 | Pilot small RNA sequencing, Real‐time PCR | Increased miRNA‐451‐5p (>1000‐fold)/ 3‐6 weeks | 57 |
| miR‐133b, miR‐342, MiR‐30 | Urinary exosome | T2DM patients | 156 | Bioinformatics analysis, Real‐time PCR | Elevated miR‐133b, miR‐342, and miR‐30a (P < 0.001) | 58 |
| miR‐2861, miR‐1915‐3p, miR‐4532 | Urine | DM patients | 145 | miRNA profiling, final selection approach, urine miRNA expression analysis, and in situ hydridization | Reduced miR‐2861, miR‐1915‐3p, and miR‐4532 (>10‐fold, P < 0.0001) | 60 |
| miR‐126, miR‐770 | Urine | DM patients | 2747 | Meta‐analysis | Upregulated miR‐126 (95% CI: 9.96‐862623.21) and miR‐770 (95% CI: 2.37‐44.25) | 62 |
To conclude, urinary miRNAs represent biomarkers with potential not only to augment the utility of albuminuria as a surrogate marker of glomerular filtration barrier (GFB) integrity but may also predict the progression of DKD before the onset of GFB breakdown. However, these studies still have limitations, including the relatively low number of patients recruited and inconsistencies between preclinical and clinical studies. Further investigations that use combined discovery/validation approaches or include larger cohorts are necessary to determine more accurate urinary miRNA expression profiles for early diagnosis and risk stratification in patients with diabetes mellitus.
4. THE ROLE OF miRNAs IN THE PATHOGENESIS OF DKD
Strong evidence for the involvement of miRNAs in kidney diseases has come from observations of mice with podocyte specific deletion of Dicer, an essential nuclease involved in miRNA biogenesis, indicating the involvement of miRNAs in kidney diseases.64, 65, 66 In an early study, at least five miRNAs (miRNA‐192, miRNA‐194, miRNA‐204, miRNA‐215, and miRNA‐216a) were considered to have an impact on kidney function as they were identified to be enriched in the kidney compared to other organs.67 Thus, the earliest studies to investigate the role of miRNA in kidney dysfunction focused on these miRNAs.
Over the past 10 years, our understanding of the molecular mechanisms by which diabetic conditions result in damage to the kidney has increased. Diabetic conditions induce inflammation, fibrosis, and hypertrophy in renal cells through various cytokines and growth factors such as TGF‐β1.5 The engagement of cytokines and growth factors with their receptors triggers signal transduction cascades that result in the activation of transcription factors to increase expression of inflammatory and fibrotic genes as well as epigenetic states including DNA methylation, chromatin histone modifications, and noncoding RNAs (lncRNAs and miRNAs).23 miRNAs induced by diabetic conditions can promote the expression of pathological genes via various posttranscriptional and post‐translational mechanisms.23 Kato et al68 were the first to describe involvement of a specific miRNA in DKD. They showed that miRNA‐192 is upregulated in vitro in mesangial cells (MCs) and in vivo in glomeruli from type 1 streptozotocin (STZ)‐induced and type 2 db/db mouse models of DKD. Repression of miRNA‐192 may promote collagen deposition in response to TGF‐β.68 During the last few years, various miRNAs have emerged as important members in the pathogenesis and progression of DKD through participation in fibrosis, inflammation, hypertrophy, autophagy, ER stress, oxidative stress, insulin resistance, and podocyte injury as summarized in Figure 2.
Figure 2.
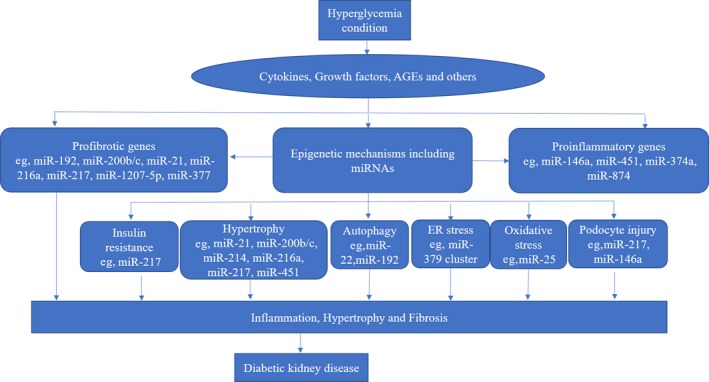
Mechanisms whereby miRNAs influence the pathogenesis of diabetic kidney disease (DKD). Hyperglycemia induces cytokines, growth factors, and dysregulation of miRNAs. miRNAs are involved in the progression of DKD by targeting genes related to fibrosis, inflammation, hypertrophy, autophagy, ER stress, oxidative stress, insulin resistance, and podocyte injury
4.1. Effect of miRNAs on kidney fibrosis in the progression of DKD
Kidney fibrosis, characterized by progressive tissue scarring that leads to glomerular and tubulointerstitial fibrosis, is the major pathological feature of ESKD.69 Although the precise sequence of molecular events that result in kidney fibrosis has not been completely elucidated, present data indicate that TGF‑β is the master regulator of this process as it acts as the major driver of matrix synthesis, inhibition of matrix degradation, and myofibroblast activation.69
TGF‐β isoform, TGF‑β1 acts through a canonical signaling pathway that involves the phosphorylation and activation of Smad2 and Smad3 by the TGF‐β receptor 1(TGFR1, also known as ALK5). Smad4 then binds activated Smad2/3, which enables this complex to translocate to the nucleus and transcribe specific genes. However, TGF‑β1 activation of TGFR1 can also activate a wide variety of Smad‐independent pathways (known as noncanonical signaling) to modify cell function. These non‐Smad pathways include those involving TGF‐β activated kinase 1, PI3K‐AKT, and Rho‐like GTPase signaling pathways.69
Recent studies show that TGF‐β1 regulates many miRNAs during the progression of DKD. Compared with nondiabetic control mice, miRNA‐192, miRNA‐200b/c, miRNA‐21, miRNA‐216a, miRNA‐217, and miRNA‐1207‐5p are upregulated in TGF‐β1‐treated murine kidney cells or in glomeruli of mouse models of diabetes.70 Functional studies of miRNA‐192 reveal that it can upregulate Collagen 1α (Col 1α) in MCs, which are key genes associated with the pathogenesis of DKD.68 miRNA‐192 can also regulate other miRNAs such as miRNA‐216a and miRNA‐217, which are related to cellular hypertrophy in DKD.71 miRNA‐200b/c, which are a subset of larger members of the miRNA200 family (miRNA‐200a, miRNA‐200b, miRNA‐200c, and miRNA‐141), are also downstream of miRNA‐192. Moreover, miR200b/c significantly upregulates Col 1α and Col 4α in MCs and diabetic mice.72 Wang el al showed that miRNA‐377 contributes to inhibition of p21‐activated kinase and superoxide dismutase and enhances fibronectin (FN) accumulation in mouse models of DKD.73 miRNA‐1207‐5p, which is upregulated by TGF‐β1, can increase expression of TGF‐β1, PAI‐1, and FN in MCs.74
In contrast, miRNA‐200a and miRNA‐141, which are inhibited by TGF‐β1, protect kidneys from fibrosis by suppressing the deposition of ECM and preventing epithelial to mesenchymal transition (EMT), respectively.70 Let‐7 family members are also downregulated in cultured human proximal tubular epithelial (HK2) cells treated with TGF‐β1, which induces fibrosis through the TGF‐β1R1 and upregulated collagen expression.75, 76
Moreover, TGF‐β1 expression can be regulated by miRNAs.2 In diabetic mice (STZ and db/db), TGF‐β1 levels are found to be upregulated by miRNA‐192 and miRNA‐200b/c.72 In addition, recent studies also demonstrated miRNA‐22 inhibited bone morphogenetic protein‐6 (BMP‐6) and BMP‐7 and further increased TGF‐β1 signaling.77 miRNA‐433 increases TGF‐β1 signaling and fibrosis by targeting antizyme inhibitor 1, a regulator of polyamine synthesis.78 These miRNA‐regulated circuits may amplify TGF‐β1 signaling to contribute to the progression in DKD. On the other hand, in vitro functional studies have identified type II TGF‐β receptor (TGFRII), Smad3, and TGFβ‐1 itself as miRNA‐23b targets, implying a negative feedback loop regulating TGF‐β‐1 signaling.79 miRNA‐29b can also suppress TGF‐β1 expression and thus inhibit TGF‐β1‐induced kidney fibrosis.80
4.2. Effect of miRNAs on inflammation in the progression of DKD
Augmented inflammation is a hallmark of diabetes,81 and this proinflammatory state plays a critical role in the development and progression of DKD.82, 83 Inflammatory factors such as infiltration of macrophage and inflammatory cytokines and chemokines (MCP‐1, IL‐6, TNF‐α, PAI‐1, CXCR4 etc) can activate myofibroblasts at injury sites in the kidney whilst inducing the differentiation of MCs, glomeruli, and renal tubular epithelial cells into fibroblasts, resulting in enhanced ECM production and deposition, which in turn promote fibrosis.84, 85, 86, 87
miRNA‐146a is a known anti‐inflammatory miRNA.88 miRNA‐146a expression increased in both peritoneal and intrarenal macrophages in diabetic mice. Mechanistic studies in the miRNA‐146a‐deficient mice showed that miRNA‐146a‐deficiency led to increased expression of M1 activation markers and suppression of M2 markers in macrophages as well as increased expression of proinflammatory cytokines, indicating that miRNA‐146a plays a crucial protective and anti‐inflammatory role during the pathogenesis of DKD.89 Activation of nuclear factor‐kappa B (NF‐κB) is associated with inflammation in the progression of DKD.90, 91, 92 miRNA‐451, which is downregulated in the kidneys of diabetic mice and MCs cultured in high glucose conditions, inhibits NF‐κB activity, and downregulated transcription of proinflammatory molecules in MCs.93 Yang et al94 showed that miRNA‐374a is downregulated in kidney samples from DKD patients. Functional studies indicated that miRNA‐374a suppressed inflammatory response in DKD by inhibition of IL‐6, TNF‐α, and MCP‐1.94 Recently, Yao et al95 reported that miRNA‐874 was downregulated in STZ‐induced DKD rats. Overexpressing miRNA‐874 with mimics attenuated the inflammatory response by decreasing IL‐6, L‐1β, and TNF‐α.
4.3. Effect of miRNAs on hypertrophy in the progression of DKD
Hypertrophy is one key feature of DKD1 and the hypertrophy‐related miRNAs have been identified. miRNA‐216a and miRNA‐217 activate Akt (a hypertrophy‐related kinase) by targeting PTEN in the STZ and db/db diabetic mouse model of DKD and MCs treated with TGF‐β1.71 In diabetic mice, miRNA‐21 also targets PTEN and activates Akt to contribute to renal hypertrophy.96, 97 PI3K is the upstream activator of Akt. miRNA‐200b/c activates Akt by targeting the PI3K inhibitor FOG2.98 Zhang et al99 investigated the role of miRNA‐451 in mesangial hypertrophy and found that miRNA‐451 negatively regulated the expression of Ywhaz (a protein related to activation of p38 MAPK signaling) through binding the Ywhaz 3′UTR. In db/db diabetic mice, miRNA‐451 is downregulated to induce hypertrophy through Ywhaz. miRNA‐34a is increased in MCs under high glucose conditions and db/db mice.100 Downregulation of miRNA‐34a can alleviate glomerular hypertrophy through targeting of growth arrest specific 1(GAS1).100 Recently, increased expression of miRNA‐214 has been found to be associated with decreased levels of PTEN and enhanced Akt phosphorylation to contribute to renal hypertrophy in renal glomerular MCs and proximal tubular epithelial cells.101 miRNA‐181a is upregulated in MCs exposed to high glucose and it downregulates the mammalian target of rapamycin (mTOR) inhibitor Deptor and activates mammalian target of rapamycin complex 2 (mTORC2) to mediate TGF‐β1‐induced glomerular MC hypertrophy.102
4.4. Effect of miRNAs on autophagy in the progression of DKD
Autophagy, a lysosomal degradation pathway, plays a crucial role in removing protein aggregates and damaged or excess organelles, including mitochondria, to maintain intracellular homeostasis.103 Therefore, autophagy may promote cellular health against various stress conditions, including hypoxia, ER stress, or oxidative stress.104 Cellular stresses and excess nutrition induced by metabolic dysfunction such as diabetes, impairs autophagy through activation of mammalian target of rapamycin complex 1 (mTORC1) and the reduction of AMP‐activated kinase (AMPK) and Sirt1 activity.105, 106 Thus, autophagy plays a crucial role in maintaining homeostasis in several organs, especially metabolic organs, and the impairment of autophagy is involved in the pathogenesis of metabolic diseases including DKD.106, 107
Recently, several miRNAs have been found to influence DKD by regulating autophagy. miRNA‐22 is found to suppress autophagy and inhibition of the endogenous miRNA‐22 increased autophagy and alleviated high glucose‐induced Col 4 and α‐SMA expression in NRK‐52E cells.108 Deshpande et al109 reported that the expression of autophagy genes was decreased in kidneys of STZ‐induced and db/db diabetic mice. miRNA‐192 inhibitors can reverse the reduction of autophagy genes in these mice. In kidneys of diabetic miRNA‐192‐KO mice, downregulation of autophagy genes was also attenuated. In mouse glomerular MCs with TGF‐β1 stimulation, miRNA‐192 mimic oligonucleotides decreased the expression of autophagy genes, indicating miRNA‐192 as a potential therapeutic target for DKD.109
In high glucose‐stimulated podocytes, miRNA‐217 expression was elevated and inhibition of miRNA‐217 can protectively antagonize high glucose‐induced podocyte damage and insulin resistance by restoring the defective autophagy pathway via targeting PTEN.110 Several miRNAs have also been implicated in oxidative stress and ER stress in DKD pathogenesis. For example, about 40 miRNAs are included in the miRNA‐379 cluster, which is regulated by ER stress in DKD.111 NOX4 is a major catalytic subunit of NADPH oxidase under hyperglycemia. miRNA‐25 inhibitor increased NOX4 mRNA and protein level in STZ‐induced diabetic rats, suggesting that decreased miRNA‐25 expression may upregulate NOX4 to promote oxidative stress and renal dysfunction in rats.112
5. THE UTILITY OF miRNAs AS NEW THERAPEUTIC TARGETS
5.1. Existing therapeutic targets of DKD
The etiology of DKD includes environmental insults, genetic susceptibility, and metabolic and hemodynamic factors resulting in poor glycemic control, hypertension, albuminuria, and excess cardiovascular risk factors.5 Thus, the treatment for DKD is mainly aimed at controlling metabolic and hemodynamic abnormalities. The agents for treatment includes the use of traditional anti‐hyperglycemic agents (AHAs) such as metformin or insulin, and RAAS inhibitors including angiotensin‐converting enzyme inhibitor (ACEI), angiotensin receptor blockers (ARBs) or aldosterone antagonists.
Sodium‐glucose linked transporters‐2 (SGLT2) inhibitors represent a new class of AHAs for the treatment of T2DM. The SGLT2 antagonists block the sodium‐coupled energy‐dependent glucose proximal tubular reabsorption, increase glucose excretion, and lower blood glucose levels. They have been shown to decrease glomerular hyperfiltration and albuminuria and thus reduce the progression of diabetic kidney disease.113 Although SGLT2 inhibitors are generally well tolerated, there are potential adverse events that healthcare providers and patients should be aware of. Most of these are related to glucosuria and osmotic diuresis.114, 115 GLP‐1 (glucagon‐like peptide‐1) receptor agonists are also a novel class of AHAs with a glucose‐dependent effect on pancreatic secretion of insulin and glucagon. They mimic the effects of the incretin hormone GLP‐1, which is released from the intestine in response to food intake. There are currently four approved GLP‐1 receptor agonists in the United States: exenatide, liraglutide, albiglutide, and dulaglutide. Evidence from animal studies indicates that GLP‐1 receptor agonists exert protective role in DKD with mechanisms that seem to be independent of their glucose‐lowering effect. Clinical studies support GLP‐1‐mediated renal protection,116 but there are some limitations. The most common adverse effect is gastrointestinal in nature, which include diarrhea, nausea, and vomiting.117
To conclude, despite numerous attempts to develop more effective drugs for the treatment of DKD, not many treatments have reached clinical use. Therefore, some novel therapeutic strategies are required.
5.2. Development of miRNA‐based therapies for DKD treatment
Many drugs targeting the pathogenic signaling such as inflammation and TGF‐β1 for treatment of DKD (mostly through protein‐coding genes) are under development.118 However, because of the limited number of protein‐coding genes, noncoding RNAs including miRNAs are attracting more attention as potential new drug targets.119 Distinct features of miRNAs including short sequence and their high homology across multiple vertebrate species make them potentially suitable as therapeutic agents.27 Many reports have shown that miRNAs are dysregulated in DKD. As listed in Table 2, the therapeutic potential of miRNAs has been well investigated in functional and mechanistic studies using methods to inhibit DKD‐inducing miRNAs or increase kidney‐protective miRNAs.2
Table 2.
Examples of miRNAs as therapeutic targets in DKD
| miRNAs | Targets | Study model | Pathological output | Reference |
|---|---|---|---|---|
|
miR‐192 |
SIP1, Zeb1 miR‐216a, miR‐217 miR‐200b/c |
MCs, STZ‐mice, db/db mice MCs, STZ‐mice, db/db mice MCs, STZ‐mice, db/db mice |
↑Col1α1 and Col1α2 ↑MC survival, Hypertrophy ↑TGF‐β1, Col1α, Col4α |
|
| miR‐216a, miR‐217 | PTEN | MCs, STZ‐mice, db/db mice | ↑MC survival, Hypertrophy | 71 |
| miR‐200b/c | Zeb1 | MCs, STZ‐mice, db/db mice | ↑TGF‐β1, Col1α, Col4α | 72 |
| miR‐377 | PAK1, SOD | MCs, STZ‐mice | ↑FN | 73 |
| miR‐1207‐5p | G6PD, PMEPA1, PDPK1, SMAD7 | MCs, RPTEC | ↑TGF‐β1, PAI‐1, FN | 74 |
| miR‐146a | Traf6, Irak‐1 | STZ‐mice |
↓TNF‐α, MCP‐1, IL‐1β, IL‐18 ↓Col1a2, Col4a2, PAI‐1, TGF‐β1 |
89 |
| miR‐451 | Ywhaz, p38 MAPK | db/db mice | ↓MC proliferation, mesangial hypertrophy | 93 |
| miR‐21 | PTEN | MCs, db/db mice |
↑ Hypertrophy, COL1α2, FN ↑TGF‐β1, NF‐κB |
96, 97 |
| miR‐22 | PTEN | NRK‐52E cells, STZ‐rats | ↓Autophage, Col4, α‐SMA | 108 |
SIP1: Smad‐interacting protein 1; ZEB1: zinc finger E‐box‐binding homeobox 1; PTEN: phosphatase and tensin homolog; PAK1: p21‐activated kinase; SOD: superoxide dismutase; G6PD: glucose‐6‐phosphate dehydrogenase; PMEPA1: prostate transmembrane protein, androgen induced 1; PDPK1: 3‐phosphoinositide‐dependent protein kinase‐1; SMAD7: SMAD family member 7; Traf6: TNF receptor‐associated factor 6; Irak‐1: Interleukin‐1 receptor‐associated kinase 1; Ywhaz: tyrosine 3‐monooxygenase/tryptophan 5‐monooxygenase activation protein zeta; p38 MAPK: p38 mitogen‐activated protein kinases.
Basically, manipulation of the activity of these miRNAs can be achieved by in vivo delivery of mimics to restore miRNA levels or inhibitors to block miRNA function.120 miRNA mimics are double‐stranded synthetic oligonucleotides (oligos) that effect the endogenous functions of the miRNA of interest but following chemical modification have increased stability and are efficiently taken up by cells. The most widely adopted strategy so far to block miRNA function is with chemically modified anti‐miRNA oligos (AMOs) designed against the mature miRNA sequence that are stable in circulation and are cell permeable. Kato et al111 have reported that a chemically modified oligo inhibits a cluster of nearly 40 miRNAs, in association with a reduction in glomerular ECM and hypertrophy in diabetic mice.
In addition to AMOs, miRNA inhibition can be achieved by expression of miRNA‐target sequences able to capture pathogenic miRNAs (miRNA sponge), short hairpin RNA plasmids to abrogate miRNA expression via RNA interference, miRNA knockout, or using oligonucleotides complementary either to the 3' untranslated region of the target mRNA binding site sequence (masking approach) or to the sequence of the miRNA (erasers).
Locked nucleic acid (LNA)‐modified oligos are one of the most potent modifications for inhibiting miRNA activity specifically.121 LNA‐modified anti‐miRNA‐192 specifically and effectively inhibited miRNA‐192, as well as downstream miRNAs (miRNA‐216a, miRNA‐217, and miRNA‐200 family) and p53 and reduced the gene expression of collagen, TGF‐β, and FN in kidneys of diabetic mice.122 Transfer of miRNA‐21 knockdown plasmids, which contained LNA‐anti‐miRNA‐21 into the diabetic kidneys of db/db mice at 10 weeks significantly attenuated microalbuminuria, kidney fibrosis, and inflammation at 20 weeks.123
Currently, some miRNA‐based therapeutic strategies are being assessed in clinical trials. The first one is a LNA‐anti‐miRNA‐122 (Miravirsen), which targets hepatitis C virus RNA.124, 125 In a phase 2 study, Miravirsen demonstrated dose‐dependent antiviral activity maintained over 4 weeks (289). Analyses of kidney biopsies showed that the inhibitor of miRNA‑21 (RG‑012) had positive effects in kidney fibrosis based on data from animal models of Alport syndrome although it was paused recently in clinical trial due to the chronic toxicity. Overexpression of miRNA‑29 seems to be a promising anti‐fibrotic approach. miRNA‑29 mimic (MRG‑201) is being assessed in a Phase II trial for the treatment of patients with a predisposition for keloid formation.126 Of note, the anti‐fibrotic effect of miRNA‑29 mimic is not specific to skin fibrosis but might be applicable to ESKD with kidney fibrosis including DKD.
The major obstacle to the therapeutic use of miRNAs is an efficient delivery method. Naked miRNAs are quickly degraded by nucleases and cleared via renal excretion.127, 128 Moreover, miRNA administration may induce innate immune responses, leading to unwanted toxicities.129 Because miRNAs are designed to target multiple pathways, they may cause off‐target gene silencing that can induce toxic effects. To avoid any off‐target effects and nonspecific immune response, miRNA should be delivered both efficiently and specifically.
One of the approaches used to overcome the poor stability and immune responses of miRNAs is based on chemical modifications including (a) ribose 2′‐OH group modification, (b) LNA modification, (c) backbone modification, and (d) peptide nucleic acids (PNA) modification, which can reduce the off‐target effects.130 Another strategy consists of conjugation with small transport domains such as aptamers and cell‐penetrating peptides. Aptamers are small single‐stranded oligonucleotides with a three‐dimensional structure that can bind to surface receptors with high affinity and specificity to act as drug delivery agents. Aptamers are suitable for targeting as they are noncytotoxic, non‐immunogenic, with superior tissue penetration, easy to modify and cheap.131 Conjugation of miRNAs to aptamers has been used to specifically target the nucleic acid to cells expressing the ligands recognized by the aptamer.132, 133, 134 Conjugation to cell‐penetrating peptides enables crossing of cell and endosomal membranes. With the development of nanotechnology, nanocarriers have recently become popular for miRNA delivery to enhance cellular uptake and delivery effectiveness as well as reduce toxicity.135 Nanocarriers are safe and require simple manufacturing. Moreover, they are characterized by their low immunogenicity, low cost, and versatility.136 In the future, novel therapies might be based on effective and specific delivery of combined miRNA modulators to complement current treatment for DKD.
6. CONCLUSIONS
Diabetic kidney disease is one of the most prevalent and life‐threatening complications of diabetes. Early diagnosis of DKD and identification of those likely to progress to ESKD has become highly important as it enables early treatment of patients before overt pathology presenting with proteinuria is evident. Since DKD is a complex, multifactorial disease, a miRNA profile, that includes multiple miRNAs representing distinct biologic pathways may have a better predictive value than a single miRNA. Although miRNA profiling enables researchers to more understand the complex pathways in the progression of DKD, it is still highly controversial if miRNA expression reflects the cause or consequence of the development of DKD. If they represent the consequence, they can serve as biomarkers. If they represent the cause they can serve as treatment targets. A combination of conventional (eGFR and albuminuria) and novel biomarkers (multiple miRNA to reflect the inflammation, fibrosis, hypertrophy, autophagy, ER stress, oxidative stress, insulin resistance, and podocyte injury) is potentially promising in accurately predicting the risk of ESKD. If miRNAs function as the cause of DKD, targeted multi‐miRNA‐based therapies that either restore or block miRNA expression and activity are very attractive.
CONFLICT OF INTEREST
None.
AUTHOR CONTRIBUTIONS
Q. Cao: wrote, edited and revised the manuscript; X.‐M. Chen: reviewed, commented and revised the manuscript; C. Huang: reviewed, commented and revised the manuscript; C.A. Pollock: reviewed, commented, revised and finalized the manuscript.
ACKNOWLEDGEMENTS
This work has been supported by Australian Postgraduate Award (APA) and Australian National Health and Medical Research Council Early Career Fellowships (NHMRC APP1110560).
Cao Q, Chen X‐M, Huang C, Pollock CA. MicroRNA as novel biomarkers and therapeutic targets in diabetic kidney disease: An update. FASEB BioAdvances. 2019;1:375–388. 10.1096/fba.2018-00064
REFERENCES
- 1. Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124:2333‐2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kato M, Natarajan R. MicroRNAs in diabetic nephropathy: functions, biomarkers, and therapeutic targets. Ann N Y Acad Sci. 2015;1353:72‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gnudi L, Coward R, Long DA. Diabetic nephropathy: perspective on novel molecular mechanisms. Trends Endocrinol Metab. 2016;27:820‐830. [DOI] [PubMed] [Google Scholar]
- 4. Fineberg D, Jandeleit‐Dahm KA, Cooper ME. Diabetic nephropathy: diagnosis and treatment. Nat Rev Endocrinol. 2013;9:713‐723. [DOI] [PubMed] [Google Scholar]
- 5. Kanwar YS, Sun L, Xie P, Liu FY, Chen S. A glimpse of various pathogenetic mechanisms of diabetic nephropathy. Annu Rev Pathol. 2011;6:395‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Levey AS, Becker C, Inker LA. Glomerular filtration rate and albuminuria for detection and staging of acute and chronic kidney disease in adults: a systematic review. JAMA. 2015;313:837‐846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stevens LA, Coresh J, Schmid CH, et al. Estimating GFR using serum cystatin C alone and in combination with serum creatinine: a pooled analysis of 3,418 individuals with CKD. Am J Kidney Dis. 2008;51:395‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Odden MC, Shlipak MG, Tager IB. Serum creatinine and functional limitation in elderly persons. J Gerontol A Biol Sci Med Sci. 2009;64:370‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Krolewski AS. Progressive renal decline: the new paradigm of diabetic nephropathy in type 1 diabetes. Diabetes Care. 2015;38:954‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Macisaac RJ, Jerums G. Diabetic kidney disease with and without albuminuria. Curr Opin Nephrol Hypertens. 2011;20:246‐257. [DOI] [PubMed] [Google Scholar]
- 11. Nosadini R, Velussi M, Brocco E, et al. Course of renal function in type 2 diabetic patients with abnormalities of albumin excretion rate. Diabetes. 2000;49:476‐484. [DOI] [PubMed] [Google Scholar]
- 12. Araki S‐i, Haneda M, Sugimoto T, et al. Factors associated with frequent remission of microalbuminuria in patients with type 2 diabetes. Diabetes. 2005;54:2983‐2987. [DOI] [PubMed] [Google Scholar]
- 13. Uwaezuoke SN. The role of novel biomarkers in predicting diabetic nephropathy: a review. Int J Nephrol Renovasc Dis. 2017;10:221‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Parving HH, Lewis JB, Ravid M, Remuzzi G, Hunsicker LG. Prevalence and risk factors for microalbuminuria in a referred cohort of type II diabetic patients: a global perspective. Kidney Int. 2006;69:2057‐2063. [DOI] [PubMed] [Google Scholar]
- 15. Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol Rev. 2013;93:137‐188. [DOI] [PubMed] [Google Scholar]
- 16. Nathan DM, Genuth S, Lachin J, et al; Diabetes Control and Complications Trial Research Group . The effect of intensive treatment of diabetes on the development and progression of long‐term complications in insulin‐dependent diabetes mellitus. N Engl J Med. 1993;329:977‐986. [DOI] [PubMed] [Google Scholar]
- 17. Patel A, MacMahon S, Chalmers J, et al; Advance Collaborative Group . Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. 2008;358:2560‐2572. [DOI] [PubMed] [Google Scholar]
- 18. Perkovic V, Heerspink HL, Chalmers J, et al. Intensive glucose control improves kidney outcomes in patients with type 2 diabetes. Kidney Int. 2013;83:517‐523. [DOI] [PubMed] [Google Scholar]
- 19. Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin‐converting‐enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med. 1993;329:1456‐1462. [DOI] [PubMed] [Google Scholar]
- 20. Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin‐receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851‐860. [DOI] [PubMed] [Google Scholar]
- 21. Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861‐869. [DOI] [PubMed] [Google Scholar]
- 22. Nathan DM, Bayless M, Cleary P, et al. Diabetes control and complications trial/epidemiology of diabetes interventions and complications study at 30 years: advances and contributions. Diabetes. 2013;62:3976‐3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kato M, Natarajan R. Diabetic nephropathy–emerging epigenetic mechanisms. Nat Rev Nephrol. 2014;10:517‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Thomas MC. Epigenetic Mechanisms in Diabetic Kidney Disease. Curr Diab Rep. 2016;16:31. [DOI] [PubMed] [Google Scholar]
- 25. Saikumar J, Ramachandran K, Vaidya VS. Noninvasive micromarkers. Clin Chem. 2014;60:1158‐1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kozomara A, Griffiths‐Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Asadullah K, Busch A, Gottwald M, Reinke P, Landeck L. Industry–academia collaborations for biomarkers. Nat Rev Drug Discovery. 2015;14:805. [DOI] [PubMed] [Google Scholar]
- 29. Wong MG, Pollock CA. (2014) Biomarkers in kidney fibrosis: are they useful? Kidney Int Suppl. 2011;4:79‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Colhoun HM, Marcovecchio ML. Biomarkers of diabetic kidney disease. Diabetologia. 2018;61:996‐1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Radcliffe NJ, Seah JM, Clarke M, MacIsaac RJ, Jerums G, Ekinci EI. Clinical predictive factors in diabetic kidney disease progression. J Diabetes Investig. 2017;8:6‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Elley CR, Robinson T, Moyes SA, et al. Derivation and validation of a renal risk score for people with type 2 diabetes. Diabetes Care. 2013;36:3113‐3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jardine MJ, Hata J, Woodward M, et al. Prediction of kidney‐related outcomes in patients with type 2 diabetes. Am J Kidney Dis. 2012;60:770‐778. [DOI] [PubMed] [Google Scholar]
- 34. Papadopoulou‐Marketou N, Kanaka‐Gantenbein C, Marketos N, Chrousos GP, Papassotiriou I. Biomarkers of diabetic nephropathy: a 2017 update. Crit Rev Clin Lab Sci. 2017;54:326‐342. [DOI] [PubMed] [Google Scholar]
- 35. Woo KS, Choi JL, Kim BR, Kim JE, An WS, Han JY. Urinary neutrophil gelatinase‐associated lipocalin levels in comparison with glomerular filtration rate for evaluation of renal function in patients with diabetic chronic kidney disease. Diabetes Metab J. 2012;36:307‐313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu J, Shao X, Lu K, et al. Urinary RBP and NGAL levels are associated with nephropathy in patients with type 2 diabetes. Cell Physiol Biochem. 2017;42:594‐602. [DOI] [PubMed] [Google Scholar]
- 37. Mahfouz MH, Assiri AM, Mukhtar MH. Assessment of neutrophil gelatinase‐associated lipocalin (ngal) and retinol‐binding protein 4 (rbp4) in type 2 diabetic patients with nephropathy. Biomark Insights. 2016;11:31‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Argiles A, Siwy J, Duranton F, et al. CKD273, a new proteomics classifier assessing CKD and its prognosis. PLoS ONE. 2013;8:e62837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lindhardt M, Persson F, Zurbig P, et al. Urinary proteomics predict onset of microalbuminuria in normoalbuminuric type 2 diabetic patients, a sub‐study of the DIRECT‐Protect 2 study. Nephrol Dial Transplant. 2017;32:1866‐1873. [DOI] [PubMed] [Google Scholar]
- 40. Lindhardt M, Persson F, Oxlund C, et al. Predicting albuminuria response to spironolactone treatment with urinary proteomics in patients with type 2 diabetes and hypertension. Nephrol Dial Transplant. 2018;33:296‐303. [DOI] [PubMed] [Google Scholar]
- 41. Lindhardt M, Persson F, Currie G, et al. Proteomic prediction and Renin angiotensin aldosterone system Inhibition prevention Of early diabetic nephRopathy in TYpe 2 diabetic patients with normoalbuminuria (PRIORITY): essential study design and rationale of a randomised clinical multicentre trial. BMJ Open. 2016;6:e010310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Looker HC, Colombo M, Hess S, et al. Biomarkers of rapid chronic kidney disease progression in type 2 diabetes. Kidney Int. 2015;88:888‐896. [DOI] [PubMed] [Google Scholar]
- 43. Lopez‐Giacoman S, Madero M. Biomarkers in chronic kidney disease, from kidney function to kidney damage. World J Nephrol. 2015;4:57‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rysz J, Gluba‐Brzozka A, Franczyk B, Jablonowski Z, Cialkowska‐Rysz A. Novel biomarkers in the diagnosis of chronic kidney disease and the prediction of its outcome. Int J Mol Sci. 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Papadopoulos T, Belliere J, Bascands JL, Neau E, Klein J, Schanstra JP. miRNAs in urine: a mirror image of kidney disease? Expert Rev Mol Diagn. 2015;15:361‐374. [DOI] [PubMed] [Google Scholar]
- 46. Glinge C, Clauss S, Boddum K, et al. Stability of circulating blood‐based MicroRNAs ‐ pre‐analytic methodological considerations. PLoS ONE. 2017;12:e0167969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen YX, Huang KJ, Niu KX. Recent advances in signal amplification strategy based on oligonucleotide and nanomaterials for microRNA detection‐a review. Biosens Bioelectron. 2018;99:612‐624. [DOI] [PubMed] [Google Scholar]
- 48. Peng H, Zhong M, Zhao W, et al. Urinary miR‐29 correlates with albuminuria and carotid intima‐media thickness in type 2 diabetes patients. PLoS ONE. 2013;8:e82607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhou J, Peng R, Li T, et al. A potentially functional polymorphism in the regulatory region of let‐7a‐2 is associated with an increased risk for diabetic nephropathy. Gene. 2013;527:456‐461. [DOI] [PubMed] [Google Scholar]
- 50. Eissa S, Matboli M, Aboushahba R, Bekhet MM, Soliman Y. Urinary exosomal microRNA panel unravels novel biomarkers for diagnosis of type 2 diabetic kidney disease. J Diabetes Complications. 2016;30:1585‐1592. [DOI] [PubMed] [Google Scholar]
- 51. Jia Y, Guan M, Zheng Z, et al. miRNAs in urine extracellular vesicles as predictors of early‐stage diabetic nephropathy. J Diabetes Res. 2016;2016:7932765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barreiro K, Holthofer H. Urinary extracellular vesicles. A promising shortcut to novel biomarker discoveries. Cell Tissue Res. 2017;369:217‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Pomatto M, Gai C, Bussolati B, Camussi G. Extracellular vesicles in renal pathophysiology. Front Mol Biosci. 2017;4:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. van Balkom BW, Pisitkun T, Verhaar MC, Knepper MA. Exosomes and the kidney: prospects for diagnosis and therapy of renal diseases. Kidney Int. 2011;80:1138‐1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tkach M, Thery C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226‐1232. [DOI] [PubMed] [Google Scholar]
- 56. Lv LL, Cao YH, Ni HF, et al. MicroRNA‐29c in urinary exosome/microvesicle as a biomarker of renal fibrosis. Am J Physiol Renal Physiol. 2013;305:F1220‐1227. [DOI] [PubMed] [Google Scholar]
- 57. Mohan A, Singh RS, Kumari M, et al. Urinary exosomal microRNA‐451‐5P is a potential early biomarker of diabetic nephropathy in rats. PLoS ONE. 2016;11:e0154055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Eissa S, Matboli M, Bekhet MM. Clinical verification of a novel urinary microRNA panal: 133b, ‐342 and ‐30 as biomarkers for diabetic nephropathy identified by bioinformatics analysis. Biomed Pharmacother. 2016;83:92‐99. [DOI] [PubMed] [Google Scholar]
- 59. Xie Y, Jia Y, Cuihua X, Hu F, Xue M, Xue Y. Urinary exosomal MicroRNA profiling in incipient type 2 diabetic kidney disease. J Diabetes Res. 2017;2017:6978984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cardenas‐Gonzalez M, Srivastava A, Pavkovic M, et al. Identification, confirmation, and replication of novel urinary microRNA biomarkers in lupus nephritis and diabetic nephropathy. Clin Chem. 2017;63:1515‐1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Argyropoulos C, Wang K, Bernardo J, et al. Urinary microrna profiling predicts the development of microalbuminuria in patients with type 1 diabetes. J Clin Med. 2015;4:1498‐1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Park S, Moon S, Lee K, Park IB, Lee DH, Nam S. Urinary and BLood MicroRNA‐126 and ‐770 are potential noninvasive biomarker candidates for diabetic nephropathy: a meta‐analysis. Cell Physiol Biochem. 2018;46:1331‐1340. [DOI] [PubMed] [Google Scholar]
- 63. Assmann TS, Recamonde‐Mendoza M, de Souza BM, Bauer AC, Crispim D. MicroRNAs and diabetic kidney disease: Systematic review and bioinformatic analysis. Mol Cell Endocrinol. 2018;477:90‐102. [DOI] [PubMed] [Google Scholar]
- 64. Harvey SJ, Jarad G, Cunningham J, et al. Podocyte‐specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol. 2008;19:2150‐2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ho J, Ng KH, Rosen S, Dostal A, Gregory RI, Kreidberg JA. Podocyte‐specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol. 2008;19:2069‐2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shi S, Yu L, Chiu C, et al. Podocyte‐selective deletion of dicer induces proteinuria and glomerulosclerosis. J Am Soc Nephrol. 2008;19:2159‐2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sun Y, Koo S, White N, et al. Development of a micro‐array to detect human and mouse microRNAs and characterization of expression in human organs. Nucleic Acids Res. 2004;32:e188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kato M, Zhang J, Wang M, et al. MicroRNA‐192 in diabetic kidney glomeruli and its function in TGF‐beta‐induced collagen expression via inhibition of E‐box repressors. Proc Natl Acad Sci U S A. 2007;104:3432‐3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Meng XM, Nikolic‐Paterson DJ, Lan HY. TGF‐beta: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12:325‐338. [DOI] [PubMed] [Google Scholar]
- 70. Wu H, Kong L, Zhou S, et al. The role of microRNAs in diabetic nephropathy. J Diabetes Res. 2014;2014:920134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kato M, Putta S, Wang M, et al. TGF‐beta activates Akt kinase through a microRNA‐dependent amplifying circuit targeting PTEN. Nat Cell Biol. 2009;11:881‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kato M, Arce L, Wang M, Putta S, Lanting L, Natarajan R. A microRNA circuit mediates transforming growth factor‐beta1 autoregulation in renal glomerular mesangial cells. Kidney Int. 2011;80:358‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang Q, Wang Y, Minto AW, et al. MicroRNA‐377 is up‐regulated and can lead to increased fibronectin production in diabetic nephropathy. FASEB J. 2008;22:4126‐4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Alvarez ML, Khosroheidari M, Eddy E, Kiefer J. Role of microRNA 1207–5P and its host gene, the long non‐coding RNA Pvt1, as mediators of extracellular matrix accumulation in the kidney: implications for diabetic nephropathy. PLoS ONE. 2013;8:e77468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Brennan EP, Nolan KA, Borgeson E, et al; GENIE Consortium . Lipoxins attenuate renal fibrosis by inducing let‐7c and suppressing TGFbetaR1. J Am Soc Nephrol. 2013;24:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang B, Jha JC, Hagiwara S, et al. Transforming growth factor‐beta1‐mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let‐7b. Kidney Int. 2014;85:352‐361. [DOI] [PubMed] [Google Scholar]
- 77. Long J, Badal SS, Wang Y, Chang BH, Rodriguez A, Danesh FR. MicroRNA‐22 is a master regulator of bone morphogenetic protein‐7/6 homeostasis in the kidney. J Biol Chem. 2013;288:36202‐36214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Li R, Chung AC, Dong Y, Yang W, Zhong X, Lan HY. The microRNA miR‐433 promotes renal fibrosis by amplifying the TGF‐beta/Smad3‐Azin1 pathway. Kidney Int. 2013;84:1129‐1144. [DOI] [PubMed] [Google Scholar]
- 79. Hagiwara S, McClelland A, Kantharidis P. MicroRNA in diabetic nephropathy: renin angiotensin, aGE/RAGE, and oxidative stress pathway. J Diabetes Res. 2013;2013:173783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li J, Cen B, Chen S, He Y. MicroRNA‐29b inhibits TGF‐beta1‐induced fibrosis via regulation of the TGF‐beta1/Smad pathway in primary human endometrial stromal cells. Mol Med Rep. 2016;13:4229‐4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27:813‐823. [DOI] [PubMed] [Google Scholar]
- 82. Tuttle KR. Linking metabolism and immunology: diabetic nephropathy is an inflammatory disease. J Am Soc Nephrol. 2005;16:1537‐1538. [DOI] [PubMed] [Google Scholar]
- 83. Mora C, Navarro JF. Inflammation and diabetic nephropathy. Curr Diab Rep. 2006;6:463‐468. [DOI] [PubMed] [Google Scholar]
- 84. Sayyed SG, Hagele H, Kulkarni OP, et al. Podocytes produce homeostatic chemokine stromal cell‐derived factor‐1/CXCL12, which contributes to glomerulosclerosis, podocyte loss and albuminuria in a mouse model of type 2 diabetes. Diabetologia. 2009;52:2445‐2454. [DOI] [PubMed] [Google Scholar]
- 85. Navarro‐Gonzalez JF, Mora‐Fernandez C, Muros de Fuentes M, Garcia‐Perez J. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327‐340. [DOI] [PubMed] [Google Scholar]
- 86. Lee IJ, Hilliard BA, Ulas M, et al. Monocyte and plasma expression of TAM ligand and receptor in renal failure: Links to unregulated immunity and chronic inflammation. Clin Immunol. 2015;158:231‐241. [DOI] [PubMed] [Google Scholar]
- 87. Vidakovic M, Grdovic N, Dinic S, Mihailovic M, Uskokovic A, Arambasic Jovanovic J. The importance of the CXCL12/CXCR87 axis in therapeutic approaches to diabetes mellitus attenuation. Front Immunol. 2015;6:403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. O'Connell RM, Rao DS, Baltimore D. microRNA regulation of inflammatory responses. Annu Rev Immunol. 2012;30:295‐312. [DOI] [PubMed] [Google Scholar]
- 89. Bhatt K, Lanting LL, Jia Y, et al. Anti‐inflammatory role of MicroRNA‐146a in the pathogenesis of diabetic nephropathy. J Am Soc Nephrol. 2016;27:2277‐2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mezzano S, Aros C, Droguett A, et al. NF‐kappaB activation and overexpression of regulated genes in human diabetic nephropathy. Nephrol Dial Transplant. 2004;19:2505‐2512. [DOI] [PubMed] [Google Scholar]
- 91. Ohga S, Shikata K, Yozai K, et al. Thiazolidinedione ameliorates renal injury in experimental diabetic rats through anti‐inflammatory effects mediated by inhibition of NF‐kappaB activation. Am J Physiol Renal Physiol. 2007;292:F1141‐1150. [DOI] [PubMed] [Google Scholar]
- 92. Zhang H, Sun SC. NF‐kappaB in inflammation and renal diseases. Cell Biosci. 2015;5:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Sun Y, Peng R, Peng H, et al. miR‐451 suppresses the NF‐kappaB‐mediated proinflammatory molecules expression through inhibiting LMP7 in diabetic nephropathy. Mol Cell Endocrinol. 2016;433:75‐86. [DOI] [PubMed] [Google Scholar]
- 94. Yang Z, Guo Z, Dong J, et al. miR‐374a regulates inflammatory response in diabetic nephropathy by targeting MCP‐1 expression. Front Pharmacol. 2018;9:900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Yao T, Zha D, Gao P, Shui H, Wu X. MiR‐874 alleviates renal injury and inflammatory response in diabetic nephropathy through targeting toll‐like receptor‐4. J Cell Physiol. 2018;234:871‐879. [DOI] [PubMed] [Google Scholar]
- 96. Dey N, Das F, Mariappan MM, et al. MicroRNA‐21 orchestrates high glucose‐induced signals to TOR complex 1, resulting in renal cell pathology in diabetes. J Biol Chem. 2011;286:25586‐25603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. McClelland AD, Herman‐Edelstein M, Komers R, et al. miR‐21 promotes renal fibrosis in diabetic nephropathy by targeting PTEN and SMAD7. Clin Sci (Lond). 2015;129:1237‐1249. [DOI] [PubMed] [Google Scholar]
- 98. Park JT, Kato M, Yuan H, et al. FOG2 protein down‐regulation by transforming growth factor‐beta1‐induced microRNA‐200b/c leads to Akt kinase activation and glomerular mesangial hypertrophy related to diabetic nephropathy. J Biol Chem. 2013;288:22469‐22480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Zhang Z, Luo X, Ding S, et al. MicroRNA‐451 regulates p38 MAPK signaling by targeting of Ywhaz and suppresses the mesangial hypertrophy in early diabetic nephropathy. FEBS Lett. 2012;586:20‐26. [DOI] [PubMed] [Google Scholar]
- 100. Zhang L, He S, Guo S, et al. Down‐regulation of miR‐34a alleviates mesangial proliferation in vitro and glomerular hypertrophy in early diabetic nephropathy mice by targeting GAS1. J Diabetes Complications. 2014;28:259‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bera A, Das F, Ghosh‐Choudhury N, Mariappan MM, Kasinath BS, Ghosh Choudhury G. Reciprocal regulation of miR‐214 and PTEN by high glucose regulates renal glomerular mesangial and proximal tubular epithelial cell hypertrophy and matrix expansion. Am J Physiol Cell Physiol. 2017;313:C430‐C447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Maity S, Bera A, Ghosh‐Choudhury N, Das F, Kasinath BS, Choudhury GG. microRNA‐181a downregulates deptor for TGFbeta‐induced glomerular mesangial cell hypertrophy and matrix protein expression. Exp Cell Res. 2018;364:5‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147:728‐741. [DOI] [PubMed] [Google Scholar]
- 104. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self‐digestion. Nature. 2008;451:1069‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Gonzalez CD, Lee MS, Marchetti P, et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7:2‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Ding Y, Choi ME. Autophagy in diabetic nephropathy. J Endocrinol. 2015;224:R15‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Choi A, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med. 2013;368:651‐662. [DOI] [PubMed] [Google Scholar]
- 108. Zhang Y, Zhao S, Wu D, et al. MicroRNA‐22 promotes renal tubulointerstitial fibrosis by targeting PTEN and suppressing autophagy in diabetic nephropathy. J Diabetes Res. 2018;2018:4728645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Deshpande S, Abdollahi M, Wang M, Lanting L, Kato M, Natarajan R. Reduced autophagy by a microRNA‐mediated signaling cascade in diabetes‐induced renal glomerular hypertrophy. Sci Rep. 2018;8:6954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Sun J, Li ZP, Zhang RQ, Zhang HM. Repression of miR‐217 protects against high glucose‐induced podocyte injury and insulin resistance by restoring PTEN‐mediated autophagy pathway. Biochem Biophys Res Commun. 2017;483:318‐324. [DOI] [PubMed] [Google Scholar]
- 111. Kato M, Wang M, Chen Z, et al. An endoplasmic reticulum stress‐regulated lncRNA hosting a microRNA megacluster induces early features of diabetic nephropathy. Nat Commun. 2016;7:12864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Fu Y, Zhang Y, Wang Z, et al. Regulation of NADPH oxidase activity is associated with miRNA‐25‐mediated NOX4 expression in experimental diabetic nephropathy. Am J Nephrol. 2010;32:581‐589. [DOI] [PubMed] [Google Scholar]
- 113. Wanner C, Inzucchi SE, Lachin JM, et al. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med. 2016;375:323‐334. [DOI] [PubMed] [Google Scholar]
- 114. Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2015;373:2117‐2128. [DOI] [PubMed] [Google Scholar]
- 115. Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644‐657. [DOI] [PubMed] [Google Scholar]
- 116. Mann J, Orsted DD, Brown‐Frandsen K, et al. Liraglutide and renal outcomes in type 2 diabetes. N Engl J Med. 2017;377:839‐848. [DOI] [PubMed] [Google Scholar]
- 117. Muskiet M, Tonneijck L, Smits MM, et al. GLP‐1 and the kidney: from physiology to pharmacology and outcomes in diabetes. Nat Rev Nephrol. 2017;13:605‐628. [DOI] [PubMed] [Google Scholar]
- 118. Breyer MD, Susztak K. The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov. 2016;15:568‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Witasp A, Van Craenenbroeck AH, Shiels PG, Ekstrom TJ, Stenvinkel P, Nordfors L. Current epigenetic aspects the clinical kidney researcher should embrace. Clin Sci (Lond). 2017;131:1649‐1667. [DOI] [PubMed] [Google Scholar]
- 120. Trionfini P, Benigni A, Remuzzi G. MicroRNAs in kidney physiology and disease. Nat Rev Nephrol. 2015;11:23‐33. [DOI] [PubMed] [Google Scholar]
- 121. Grünweller A, Hartmann KR. Locked nucleic acid oligonucleotides: the next generation of antisense agents? BioDrugs. 2007;21:235‐243. [DOI] [PubMed] [Google Scholar]
- 122. Putta S, Lanting L, Sun G, Lawson G, Kato M, Natarajan R. Inhibiting microRNA‐192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2012;23:458‐469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhong X, Chung AC, Chen HY, et al. miR‐21 is a key therapeutic target for renal injury in a mouse model of type 2 diabetes. Diabetologia. 2013;56:663‐674. [DOI] [PubMed] [Google Scholar]
- 124. Luna JM, Scheel TK, Danino T, et al. Hepatitis C virus RNA functionally sequesters miR‐122. Cell. 2015;160:1099‐1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Ottosen S, Parsley TB, Yang L, et al. In vitro antiviral activity and preclinical and clinical resistance profile of miravirsen, a novel anti‐hepatitis C virus therapeutic targeting the human factor miR‐122. Antimicrob Agents Chemother. 2015;59:599‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Harmanci D, Erkan EP, Kocak A, Akdogan GG. Role of the microRNA‐29 family in fibrotic skin diseases. Biomedical reports. 2017;6:599‐604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Raemdonck K, Vandenbroucke RE, Demeester J, Sanders NN, De Smedt SC. Maintaining the silence: reflections on long‐term RNAi. Drug Discov Today. 2008;13:917‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Yu B, Zhao X, Lee LJ, Lee RJ. Targeted delivery systems for oligonucleotide therapeutics. AAPS J. 2009;11:195‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Meng Z, Lu M. RNA interference‐induced innate immunity, off‐target effect, or immune adjuvant? Front Immunol. 2017;8:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Lennox KA, Behlke MA. Chemical modification and design of anti‐miRNA oligonucleotides. Gene Ther. 2011;18:1111‐1120. [DOI] [PubMed] [Google Scholar]
- 131. Sriramoju B, Kanwar R, Veedu RN, Kanwar JR. Aptamer‐targeted oligonucleotide theranostics: a smarter approach for brain delivery and the treatment of neurological diseases. Curr Top Med Chem. 2015;15:1115‐1124. [DOI] [PubMed] [Google Scholar]
- 132. Rohde JH, Weigand JE, Suess B, Dimmeler S. A universal aptamer chimera for the delivery of functional microRNA‐126. Nucleic Acid Ther. 2015;25:141‐151. [DOI] [PubMed] [Google Scholar]
- 133. Iaboni M, Russo V, Fontanella R, et al. Aptamer‐miRNA‐212 conjugate sensitizes NSCLC cells to TRAIL. Mol Ther Nucleic Acids. 2016;5:e289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Esposito CL, Catuogno S, de Franciscis V. Aptamer‐MiRNA conjugates for cancer cell‐targeted delivery. Methods Mol Biol. 2016;1364:197‐208. [DOI] [PubMed] [Google Scholar]
- 135. Chaudhary V, Jangra S, Yadav NR. Nanotechnology based approaches for detection and delivery of microRNA in healthcare and crop protection. J Nanobiotechnology. 2018;16:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Riley MK, Vermerris W. Recent advances in nanomaterials for gene delivery‐a review. Nanomaterials (Basel). 2017;7:94. [DOI] [PMC free article] [PubMed] [Google Scholar]