

Abstract
Alarmins and damage‐associated molecular patterns (DAMPs) are powerful inflammatory mediators, capable of initiating and maintaining sterile inflammation during acute or chronic tissue injury. Recent evidence suggests that alarmins/DAMPs may also trigger tissue regeneration and repair, suggesting a potential contribution to tissue fibrogenesis. High mobility group B1 (HMGB1), a bona fide alarmin/DAMP, may be released passively by necrotic cells or actively secreted by innate immune cells. Macrophages can release large amounts of HMGB1 and play a key role in wound healing and regeneration processes. Here, we hypothesized that macrophages may be a key source of HMGB1 and thereby contribute to wound healing and fibrogenesis. Surprisingly, cell‐specific deletion approaches, demonstrated that macrophage‐derived HMGB1 is not involved in tissue fibrogenesis in multiple organs with different underlying pathologies. Compared to control HMGB1Flox mice, mice with macrophage‐specific HMGB1 deletion (HMGB1ΔMac) do not display any modification of fibrogenesis in the liver after CCL4 or thioacetamide treatment and bile duct ligation; in the kidney following unilateral ureter obstruction; and in the heart after transverse aortic constriction. Of note, even under thermoneutral housing, known to exacerbate inflammation and fibrosis features, HMGB1ΔMac mice do not show impairment of fibrogenesis. In conclusion, our study clearly establishes that macrophage‐derived HMGB1 does not contribute to tissue repair and fibrogenesis.
Keywords: alarmin, DAMP, fibrosis, innate immunity
Abbreviations
- Acta2
alpha actin2
- ALT
alanine transaminase
- AST
aspartate transaminase
- BDL
bile duct ligation
- BW
body weight
- CCL4
carbon tetrachloride
- Col1a1
type1 alpha1 collagen
- Col1a2
type1 alpha2 collagen
- Col3a1
type3 alpha1 collagen
- DAMPS
damage‐associated molecular patterns
- DNA
deoxyribonucleic acid
- Emr1
EGF‐like module containing mucin‐like hormone receptor‐like 1
- FS
fractional shortening
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- HMGB1
high mobility group box 1
- HW/TL
heart weight to tíbia length ratio
- IL‐18
interleukin 18
- IL‐1β
interleukin 1β
- LPS
lipopolysaccharide
- LVEDD
left ventricular end‐diastolic dimension
- LVESD
left ventricular end‐systolic dimension
- PCR
polymerase chain reaction
- PRR
pattern recognition receptor
- RAGE
receptor for advanced glycation end products
- TAA
thioacetamide
- TAC
transverse aortic constriction
- TLR
toll‐like receptor
- TNFα
tumor necrosis factor α
- UUO
unilateral ureteral obstruction
- α‐SMA
α‐smooth muscle actin
1. INTRODUCTION
Sterile inflammation and regeneration are two biological processes that are tightly related to wound healing, but if uncontrolled could lead to scarring and fibrotic responses, contributing to the long term to organ loss of function.1, 2 Among cells involved in the modulation of these crucial functions, several types of macrophages play a central role in the clearance of dying/dead cells, and promote tissue repair by secreting a broad variety of molecules, such as cytokines, chemokines, or alarmins,1, 2, 3, 4 that either maintain the inflammation process and/or activate tissue myofibroblasts in charge of synthesizing extracellular matrix components to promote tissue architecture restoration.2, 5 Globally in the context of wound healing, immunostimulatory molecules originating either from macrophages (alarmins) or dying cells (such as damage‐associated molecular patterns [DAMPs]) have been characterized as both pro‐inflammatory but also as pro‐regenerating agents involved in progenitor cell proliferation and differentiation,1, 3, 4 thus potentially involve during chronic tissue injury, in inflammation and fibrosis processes which are now supported by several reports in the heart and lungs.6, 7 Among pro‐inflammatory and pro‐repair agents secreted by macrophages, high mobility group box 1 (HMGB1), also known for its alarmin and DAMP properties, has drawn a lot of attention as it is considered as an attractive target to treat acute and chronic inflammatory diseases8, 9, 10 and is also envisioned as a relevant biomarker of tissue injury11, 12 in humans. HMGB1 can reach the extracellular space, once released by necrotic epithelial cells and exerts its DAMPs function or when secreted by activated innate immune cells, where it mediates potent inflammatory effects.10, 13 Macrophages and other innate immune cells actively secrete HMGB1 upon an inflammatory challenge (interleukin 1β [IL1β], lipopolysaccharide [LPS], or tumor necrosis factor α [TNFα]) in vitro and in vivo14, 15, 16, 17 supporting a possible role of macrophage‐derived HMGB1 in tissue scarring. Recent publications documented a critical role of extracellular HMGB1 in regeneration and tissue repair processes in bone, muscle, or liver18, 19, 20, 21 suggesting a potential role in tissue fibrogenesis. Moreover, HMGB1 has been proposed to play a direct role in promoting fibrosis in the liver, lungs, kidney, and heart through different pathways such as inflammation or myofibroblast activation.22, 23, 24 However, in liver fibrosis, hepatocyte‐derived HMGB1 is likely not involved in fibrogenesis as shown in recent studies,19, 20 excluding any potential role of HMGB1 as a DAMP in liver scarring progression implying that non‐parenchymal cells, such as macrophages, could be the cellular source of HMGB1 during tissue fibrosis development.
To determine whether macrophage‐derived HMGB1 has a determinant role in fibrosis progression, we evaluated the impact of a macrophage‐specific HMGB1 genetic deletion on the liver, kidney, and cardiac fibrosis. Using very well‐established fibrosis models in three different organs, our anatomo‐pathological explorations revealed that unexpectedly, macrophage‐derived HMGB1 has no major role in fibrogenesis in mice.
2. MATERIALS AND METHODS
2.1. Animals
All experimental procedures were performed in accordance with institutional guidelines for animal studies and are approved by the Ethics Committee (US006 CREFRE ‐ CEEA‐122, 1710480320). Myeloid‐specific deletion of HMGB1 (HMGB1ΔMac) were generated crossing LysM‐CRE+/− (a generous gift from a gift from Dr Pierre Gourdy, Toulouse France) with HMGB1Flox mice (a generous gift from Dr Robert F. Schwabe, Columbia University, NY), littermates LysM‐CRE−/− HMGB1Flox/Flox (HMGB1Flox) were used as control. After randomization, 8 to 12‐week‐old male mice were used in this study. Mice were housed under specific pathogen‐free conditions at 20‐22°C and 50%‐60% humidity, with a 12 hour light/dark cycle and free access to water and food. For thermoneutrality studies, mice were housed at 30°C in a Noroit A‐Box (Noroit, Bouaye, France). Before running any experiments, mice were usually placed at 30°C for a 2‐week period of acclimation. At the time of sacrifice, tissues and organs were dissected, weighted and directly snap frozen in liquid nitrogen and stored at −80°C for gene expression and western blot analyses. For histological analysis, tissues were fixed in 10% formalin (Sigma‐Aldrich, St. Louis, MO, USA).
2.2. Genotyping
DNA extraction and polymerase chain reaction (PCR) were performed using Kapa mouse genotyping kit (Kapa Biosystems, Wilmington, MA) according to the manufacturer protocol. PCR reactions were performed using following primers: LysM‐CRE: 5′‐ACCGGTCGATGCAACGAGTGATGAG‐3′ (forward) and 5′‐AGTGCGTTCGAACGCTAGAGC‐3′ (reverse), LoxP1 5′‐TAAGAGCTGGGTAAACTTTAGGTG‐3′ (forward) and 5′‐GAAACAGACAAGCTTCAAACTGCT‐3′ (reverse), LoxP2 5′‐TGACAGGATACCCAGTGTTAGGGG‐3′ (forward) and 5′‐CCAGAGTTTAATCCACAGAAGAAA‐3′ (reverse). Deletion PCR was performed using LoxP1 forward and LoxP2 reverse primers.
2.3. Murine models of fibrosis
Liver chemically induced fibrosis was triggered by intraperitoneal injection (ip) of carbon tetrachloride (CCl4, 02671, Sigma‐Aldrich, St. Louis, MO, USA; 0.5 μL/g in corn oil, at a ratio of 1:3) for eight injections or by ip of thioacetamide (TAA, 163678, Sigma‐Aldrich, St. Louis, MO, USA) dissolved in NaCl 0.9% for 6 weeks (three injections per week) at increasing concentrations (first dose: 50 mg/kg, second dose: 100 mg/kg, third to sixth dose: 200 mg/kg, all following doses: 300 mg/kg) as previously described.25 Cholestatic‐induce liver fibrosis was induced in 8‐week‐old male mice by ligating the common bile duct for 21 days, as described.26 Kidney fibrosis was induced by unilateral ureteral obstruction (UUO) on 8‐week‐old mice. UUO was performed by ligating the ureter just below the renal pelvis for 7 days, as previously described.27 Transverse aortic constriction (TAC) was performed on 8 to 12‐week‐old male mice for 28 days. After anesthesia, the transverse aorta was isolated and TAC was performed by tying a waxed braided silk suture, to induce pressure overload and cardiac hypertrophy as previously described.28 For all surgical procedures, sham‐operated animals underwent the same operation except the suturing of inner organs.
2.4. Cardiac function assessment
The left ventricle dimensions were determined using echocardiography and (Time/Motion) mode acquisition from the parasternal short axis view at the level of the papillary muscles using a Vivid7 echograph and a 14 MHz transducer (i3L, GE Healthcare, Little Chalfont, UK). Images were transferred and analyzed offline with EchoPAC (GEHealthcare, Little Chalfont, UK).
2.5. Hepatic stellate cell isolation
Hepatic stellate cells were isolated from mice as described previously.25 Briefly, after cannulation of the inferior vena cava, the portal vein was cut, allowing retrograde step‐wise perfusion with pronase (Sigma‐Aldrich, St. Louis, MO, USA) and collagenase (Roche Diagnostics, Risch‐Rotkreuz, Switzerland) containing solutions, and subsequent 9.7% Nycodenz gradient centrifugation. Purity was assessed by vitamin A autofluorescence under a fluorescent microscope (Olympus 71IX). After 5 hours in dulbecco modified eagle medium (DMEM) 10% fetal calf serum (FCS), 1% Antibiotic‐Antimycotic (Gibco, Grand Island, NY, United States) and 0.1% Gentamicin (Gibco, Grand Island, NY, United States), stellate cells have been cultured with 1/1.74 diluted bone marrow‐derived macrophages (BMDM) conditioned media in DMEM 0.1% FBS for 24 hours.
2.6. Plasma analysis
Aspartate transaminase (AST) and alanine transaminase (ALT) levels were determined in plasma by the Phenotypage‐CREFRE facility using a Pentra400 biochemical analyzer (HORIBA Medical, Kyoto, Japan). HMGB1 circulating levels were assessed by ELISA (ST51011; IBL International, Hamburg, Germany) according to the manufacturer guidelines.
2.7. Bone marrow‐derived macrophages
Bone marrow cells were obtained by flushing the cavity of femurs and tibia of HMGB1Flox and HMGB1ΔMac mice (8 week old) in sterile DMEM F12 supplemented with 10% FCS and 1% penicillin/streptomycin (Sigma‐Aldrich, St. Louis, MO, USA). Cells were filtered on a 42 μm cell‐strainer, and after erythrocyte lysis, harvested cells are plated and treated with 10 ng/mL recombinant murine macrophage colony stimulating factor (M‐CSF) (PeproTech, Inc, Rocky Hill, NJ, USA) for 7 days. The medium was renewed every 2 days with DMEM 10% FCS with 10 ng/mL M‐CSF. Then, BMDM were treated with 10 ng/mL of LPS (055:B5, Sigma‐Aldrich, St. Louis, MO, USA) for 1 hour and 30 minutes, then washed two times to remove LPS using phosphate buffered saline (PBS), and conditioned media were generated in DMEM 0.1% FCS during 48 hours.
2.8. Immunocytofluorescence
Bone marrow‐derived macrophages were fixed after 7 days of differentiation, with 10% formalin. Cells were permeabilized with 0.1% Triton and stained with a primary antibody against HMGB1 (1:200, ab18256; Abcam, Cambridge, UK) and Alexa Fluor 546‐conjugated secondary antibody (1:800, A11010; Life Technologies, Carlsbad, CA, USA). Nuclei were stained with DAPI (Sigma‐Aldrich, St. Louis, MO, USA). Confocal microscopy was performed on an LSM 780 confocal laser microscope (Zeiss, Oberkochen, Germany) using a 63× oil immersion lens.
2.9. Stromal vascular fraction preparation
Mice were anesthetized with pentobarbital (50 mg/kg), then perfused with PBS to wash out blood from tissues. Mice were euthanized and tissues were harvested. Liver and lungs were digested with collagenase D (2.5 mg/mL; Roche Diagnostics, Risch‐Rotkreuz, Switzerland) under agitation for 20 minutes at 37°C. After centrifugation at 600 g for 10 minutes, stromal cells were separated in 30% percoll (GE Healthcare, 17‐0891‐01, Upsale, Sweden). After centrifugation at 600 g for 15 minutes, the pellet containing stromal cells were incubated with erythrocyte lysis buffer for 10 minutes followed by another centrifugation (600 g, 10 minutes) and resuspension in PBS.
2.10. Western blotting
Tissues were homogenized in RIPA buffer (TRIS 20 mmol/L, NaCl 150 mmol/L, EDTA 1 mmol/L, EGTA 1 mmol/L, TRITON X100 1%, Tetra‐Sodium Pyrophosphate 2.5 mmol/L, B‐Glycerophosphate 1 mmol/L, Sodium orthovanadate 1 mmol/L) containing proteases and phosphatases inhibitors (Sigma‐Aldrich, St. Louis, MO, USA) using Precellys sample lyzer (Bertin Technologies, Montigny le Bretonneux, France). Western blots were performed using standard procedures using antibodies against HMGB1 (1:1000, ab18256; Abcam, Cambridge, UK) and α‐smooth muscle actin (α‐SMA) (1:1000, ab5694; Abcam, Cambridge, UK). GAPDH (1:2000, ab181602; Abcam, Cambridge, UK), was used as a loading control.
2.11. Gene expression
RNA was extracted using GenJET RNA purification kit (ThermoScientific, Waltham, MA, USA) and DNAse treatment (Qiagen, Hilden, Germany). After dosage with Xpose (Trinean, Gentbrugge, Belgium), reverse transcription was performed using High Capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA, USA) according to the manufacturer protocol. Real‐Time‐qPCR (RT‐qPCR) was performed with indicated primer pairs gene expression is normalized using Rplp0 reference gene expression. Primer sequences were as follows: Acta2, 5′‐GTCCCAGACATCAGGGAGTAA‐3′ (forward) and 5′‐TCGGATACTTCAGCGTCAGGA‐3′ (reverse), Col1a1, 5′‐TGTGTGCGATGACGTGCAAT‐3′ (forward) and 5′‐GGGTCCCTCGACTCCTACA‐3′ (reverse), Col3a1, 5′‐AAGGCGAATTCAAGGCTGAA‐3′ (forward) and 5′‐TGTGTTTAGTACAGCCATCCTCTAGAA‐3′ (reverse), Emr1, 5′‐TGACAACCAGACGGCTTGTG‐3′ (forward) and 5′‐GCAGGCGAGGAAAAGATAGTGT‐3′ (reverse), Rplp0, 5′‐AGTCGGAGGAATCAGATGACGAT‐3′ (forward) and 5′‐GGCTGACTTGGTTGCTTTGG‐3′ (reverse), Tnfα 5′‐TGGGACAGTGACCTGGACTGT‐3′ (forward) and 5′‐TTCGGAAAGCCCATTTGAGT‐3′ (reverse), Il6, 5′‐GCCCACCAAGAACGATAGTCA‐3′ (forward) and 5′‐CAAGAAGGCAACTGGATGGAA‐3′ (reverse), Il1β, 5′‐CAACCAACAAGTGATATTCTCGATG‐3′ (forward) and 5′‐GATCCACACTCTCCAGCTGCA‐3′ (reverse), Ccn2, 5′‐GGCATCTCCACCCGAGTTAC‐3′ (forward) and 5′‐GATTTTAGGTGTCCGGATGCA‐3′ (reverse). For Col1a1, Col1a2, and Acta2 mRNA expression displayed in Figure S4, RNA were extracted using High Pure RNA Isolation kit (Roche Diagnostics, Risch‐Rotkreuz, Switzerland) and qPCR analysis was conducted using Taqman‐primer probes (Applied Biosystems, Foster City, CA, USA).
2.12. Histology and immunohistochemistry
10% formalin‐fixed samples were paraffin embedded and sliced at 5 µm. For picrosirius red staining, sections were dipped for 1.5 hours in picric acid containing 1% direct red 80 (365548; Sigma‐Aldrich, St. Louis, MO, USA) and 0.5% fast green (F7258; Sigma‐Aldrich, St. Louis, MO, USA). Immunohistochemistry was performed using anti‐mouse α‐SMA (ab5694; Abcam, Cambridge, UK) followed by a biotinylated goat anti‐rat antibody (BA‐9400; Vector Laboratories, Inc, Burlingame, CA) and streptavidin‐HRP (DY998; R&D systems, Minneapolis, MN) with 3‐3'‐Diaminobenzidine (DAB) as substrate (11718096001; Sigma‐Aldrich, St. Louis, MO, USA). Stained slides were scanned using a Nanozoomer scanner (Hamamatsu Photonics, Hamamatsu City, Japan). Quantification was performed using Photoshop (Adobe Systems, San Jose, CA, USA) software.
2.13. Statistics
Analyses are performed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA). All data are expressed as mean ± SEM, except otherwise indicated, statistical significance was determined by t test, Mann‐Whitney or two‐way ANOVA tests. P values <0.05 were considered significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
3. RESULTS
3.1. Liver HMGB1 levels are upregulated during liver fibrosis
As HMGB1 is thought to be a determinant player in fibrogenesis, we first characterized the expression levels of HMGB1 during fibrosis progression in the liver. Using two models of chemically (CCL4) or surgically (BDL)‐induced liver fibrosis, we show that HMGB1 protein levels were strongly upregulated in fibrotic livers compared to control livers (Figure 1A,B) suggesting a possible role of HMGB1 during fibrogenesis. To determine whether macrophage‐derived HMGB1 could be responsible for fibrosis progression, we generated mice specifically deleted for Hmgb1 gene in macrophages (HMGB1ΔMac) by crossing LysM‐Cre29 and HMGB1‐floxed transgenic mice.30 Besides getting a clear recombination in the Hmgb1 gene locus (Figure S1A,B), western blotting analysis demonstrated an efficient deletion of HMGB1, in vivo in naïve peritoneum macrophages or in resident macrophages from stromal vascular fraction of liver or lungs with 88%, 55%, and 98% of deletion, respectively (Figure 1C‐E) and in vitro in BMDM with 99% deletion using immunoblotting or immunocytofluorescence (Figure 1F). Upon basal conditions, compared to HMGB1Flox, HMGB1ΔMac mice did not show any detectable differences of fibrosis features with comparable protein levels of α‐SMA protein as measured by immunoblotting (Figure 1G) and normal picrosirius red staining surrounding liver vessels and staining of vascular smooth muscle cells (Figure 1H,I).
Figure 1.
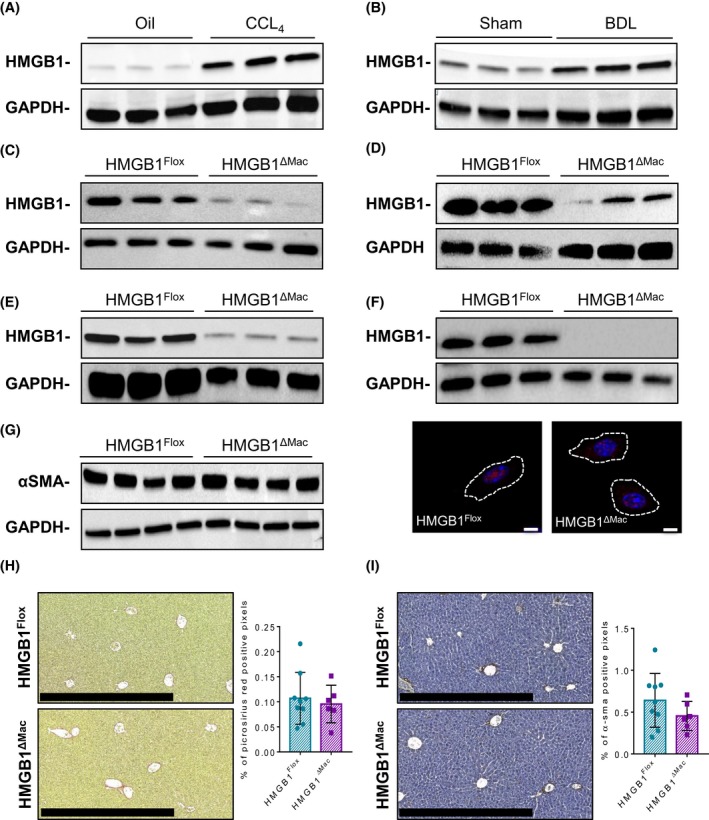
High mobility group B1 (HMGB1) regulation during fibrosis and validation and characterization of macrophage‐specific deletion of HMGB1. Representative immunoblotting against HMGB1 in oil (n = 9) or CCL4‐treated (n = 15) liver using glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) protein as a loading control (A). Representative immunoblotting against HMGB1 in liver extracts from sham (n = 9) or BDL (n = 4) animals using GAPDH protein as a loading control (B). Peritoneal macrophages isolated from HMGB1Flox (n = 3) and HMGB1ΔMac mice (n = 3) were analyzed by immunoblotting against HMGB1 and GAPDH (C). Cells from liver stromal vascular fraction, isolated from HMGB1ΔMac and HMGB1Flox mice, were analyzed by immunoblotting against HMGB1 and GAPDH (D). Cells from lung stromal vascular fraction, isolated from HMGB1Flox (n = 3) and HMGB1ΔMac mice (n = 3), were analyzed by immunoblotting against HMGB1 and GAPDH (E). Bone marrow‐derived macrophages (BMDM) isolated from HMGB1Flox (n = 3) and HMGB1ΔMac mice (n = 3) were analyzed by immunoblotting against HMGB1 and GAPDH (higher panel). BMDM isolated from HMGB1Flox and HMGB1ΔMac mice were analyzed by immunofluorescence confocal microscopy to detect HMGB1 (red) while nuclei were stained with DAPI (blue) (lower panel). Scale bar: 5 μm (F). Liver extracts of HMGB1Flox (n = 9) and HMGB1ΔMac (n = 6) mice were analyzed by western blotting directed against α‐smooth muscle actin (α‐SMA) (G). Representative images of liver fibrosis determined by picosirius Red staining from HMGB1Flox (n = 9) and HMGB1ΔMac (n = 6) mice. Scale bar: 500 μm (H). Representative images of myofibroblast activation determined by α‐SMA immunostaining from HMGB1Flox (n = 9) and HMGB1ΔMac (n = 6) mice liver sections. Scale bar: 500 μm (I)
3.2. HMGB1ΔMac mice do not have an increased liver fibrogenesis after CCL4 treatment
To determine whether macrophage‐derived HMGB1 is playing a key role in fibrogenesis, we first studied liver fibrosis. In this purpose, HMGB1Flox and HMGB1ΔMac mice were treated with eight i.p injections of carbon tetrachloride (CCL4) and were euthanized 2 days after the last injection. Hmgb1 gene deletion has been carefully monitored using deletion PCR on whole liver DNA extract from both HMGB1Flox and HMGB1ΔMac mice (Figure S1C). Following CCL4 treatment, HMGB1Flox mice exhibited a significant accumulation of collagen compared to oil‐treated mice as shown by picrosirius red staining (Figure 2A), but no differences were detected between HMGB1Flox and HMGB1ΔMac mice. Similarly, HMGB1Flox and HMGB1ΔMac mice displayed comparable levels of α‐SMA immunostaining (Figure 2B). Moreover, there were no differences in α‐SMA protein levels (Figure 2C) as well as similar mRNA expression levels of fibrogenic and pro‐inflammatory markers such as Acta2, Ccn2, Col1a1, Col3a1, Tnfα, and Il‐6 except for Emr1 and Il‐1β which were upregulated in HMGB1ΔMac mice (Figure 2D; Figure S2A). Finally, while CCL4 treatment induced a pronounced liver injury compared to oil‐treated mice, there were no differences in serum ALT or AST levels between HMGB1Flox and HMGB1ΔMac mice (Figure 2E). To rule out a possible overpowering effect of 8× CCL4 on HMGB1 potential fibrogenic effect, we used a less severe experimental setup, where HMGB1Flox and HMGB1ΔMac mice were subjected to only four injections of CCL4. Even on a shorter CCL4 model, HMGB1Flox and HMGB1ΔMac mice displayed the same extent of fibrosis in the liver (Figures S2F & S3A‐E), suggesting that macrophage‐derived HMGB1 does neither contribute to early fibrogenic events during fibrosis progression.
Figure 2.
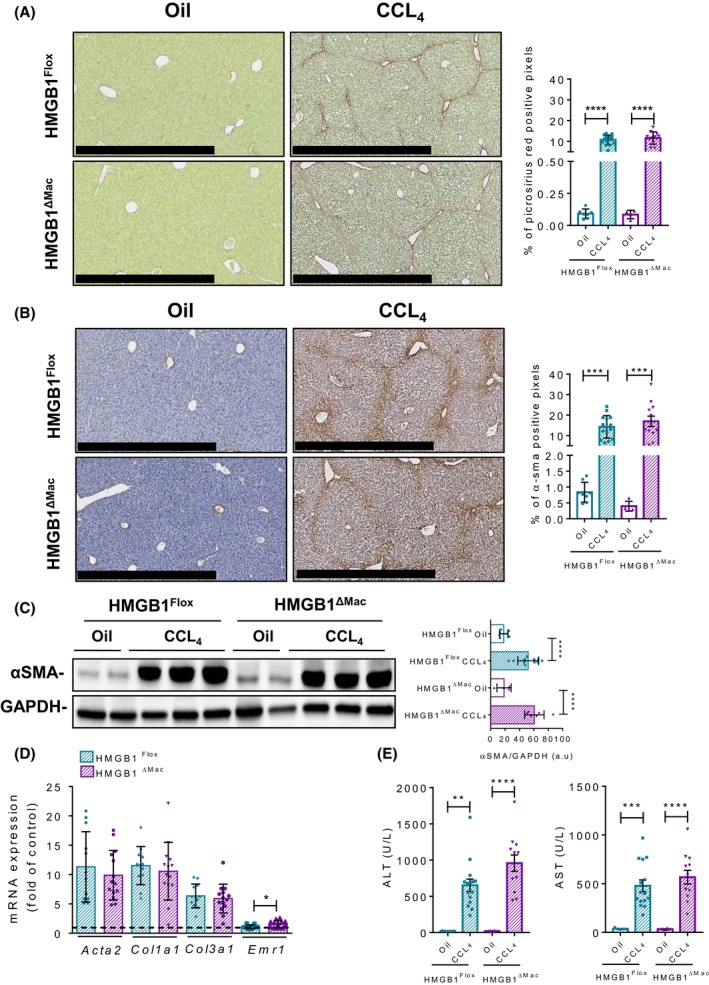
Macrophage‐specific deletion of high mobility group B1 (HMGB1) does not modify liver fibrogenesis after CCL4 treatment. Representative pictures of picrosirius red staining of HMGB1Flox mice and HMGB1ΔMac of liver section and quantification of positive pixels per liver section. Scale bar: 500 μm (A). Representative pictures of immunohistochemical staining with an antibody against α‐smooth muscle actin (α‐SMA) and quantification of positive pixels per liver section. Scale bar: 500 μm (B). Liver extracts from HMGB1Flox mice and HMGB1ΔMac were analyzed by western blotting directed against α‐SMA (C). Liver mRNA expression levels of classical fibrosis markers were detected using real‐time RT‐PCR, the dotted line indicates the baseline (D). Hepatic injury in HMGB1Flox mice and HMGB1ΔMac was determined by alanine transaminase (ALT) (left panel) and aspartate transaminase (AST) (right panel) levels (E). Statistical analysis was performed using Mann‐Whitney test. Data are expressed as means ± SEM. n = 9 in HMGB1Flox‐oil group; n = 6 in HMGB1ΔMac‐oil group; n = 15 in HMGB1Flox‐CCL4 group; n = 12 in HMGB1ΔMac‐CCL4 group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs oil/CCL4
3.3. HMGB1ΔMac mice do not display increased TAA‐induced liver fibrosis
To confirm these findings in a second well‐established model of chemical‐induced liver fibrogenesis, we next challenged mice using TAA. HMGB1ΔMac and HMGB1Flox mice were treated with 18 i.p injections of TAA. As performed on CCL4 model, Hmgb1 gene deletion was carefully monitored using deletion PCR on whole liver DNA extract from both HMGB1Flox and HMGB1ΔMac mice (Figure S1D). Similar to our results obtained in the CCL4 model, we did not detect any differences between HMGB1Flox and HMGB1ΔMac mice. After TAA treatment, HMGB1Flox mice exhibited a pronounced deposition of fibrillar collagen and α‐SMA staining compared to saline‐treated mice but the increase in fibrotic markers was identical between HMGB1Flox and HMGB1ΔMac mice (Figure 3A,B). In addition, α‐SMA (Figure 3C) protein levels, as well as mRNA expression of classical fibrosis and inflammatory markers such as Acta2, Ccn2, Col1a1, Col3a1, Emr1, and Il‐6 except for Tnfα, Il‐1β which were upregulated in HMGB1ΔMac mice (Figure 3D; Figure S2B) and serum levels of liver injury markers (Figure 3E), were comparable in HMGB1Flox and HMGB1ΔMac mice subjected to TAA treatment.
Figure 3.
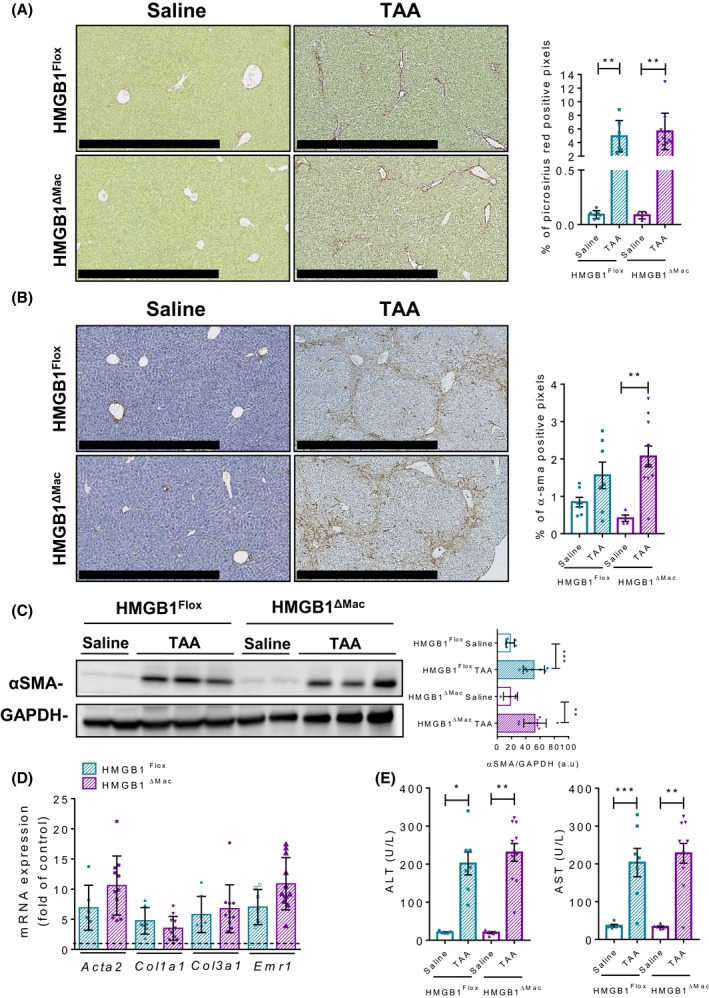
Macrophage‐specific deletion of high mobility group B1 (HMGB1) does not modify liver fibrogenesis after TAA treatment. Representative pictures of picrosirius red staining of HMGB1Flox mice and HMGB1ΔMac of liver section and quantification of positive pixels per liver section. Scale bar: 500 μm (A). Representative pictures of immunohistochemical staining with an antibody against α‐smooth muscle actin (α‐SMA) and quantification of positive pixels per liver section. Scale bar: 500 μm (B). Liver extracts from HMGB1Flox mice and HMGB1ΔMac were analyzed by western blotting directed against α‐SMA (C). Liver mRNA expression levels of classical fibrosis markers were detected using real‐time RT‐PCR, the dotted line indicates the baseline (D). Hepatic injury in HMGB1Flox mice and HMGB1ΔMac was determined by alanine transaminase (ALT) (left panel) and aspartate transaminase (AST) (right panel) levels (E). Statistical analysis was performed with Mann‐Whitney test. Data are expressed as means ± SEM. n = 6 in HMGB1Flox‐saline group; n = 6 in HMGB1ΔMac‐saline group; n = 7 in HMGB1Flox‐TAA group; n = 11 in HMGB1ΔMac‐TAA group. *P < 0.05, **P < 0.01, ***P < 0.001 vs saline/TAA
3.4. HMGB1ΔMac mice do not have an increased hepatic fibrogenesis after BDL
To corroborate these findings, we used a complementary model of cholestatic liver injury, bile duct ligation (BDL). As done previously, Hmgb1 gene deletion was carefully monitored using deletion PCR on whole liver DNA extracted from both HMGB1Flox and HMGB1ΔMac mice (Figure S1E). Twenty‐one days after BDL, HMGB1Flox mice displayed severe hepatic fibrosis as shown by picrosirius red staining but again no differences were detected between HMGB1Flox and HMGB1ΔMac mice (Figure 4A). Expression of α‐SMA was also increased in HMGB1Flox mice assessed by immunohistochemistry and immunoblotting but with no differences compared to HMGB1ΔMac mice (Figure 4B,C). mRNA expression of fibrogenic markers Acta2, Ccn2, Col1a1, Col3a1, Acta2, Col1a1, Col3a1, and inflammatory markers Emr1 and Il‐6, except for Tnfα, Il‐1β which were upregulated in HMGB1ΔMac mice (Figure 4D; Figure S2C) as well as serum ALT and AST levels, were also similar in HMGB1Flox and HMGB1ΔMac mice (Figure 4D,E).
Figure 4.
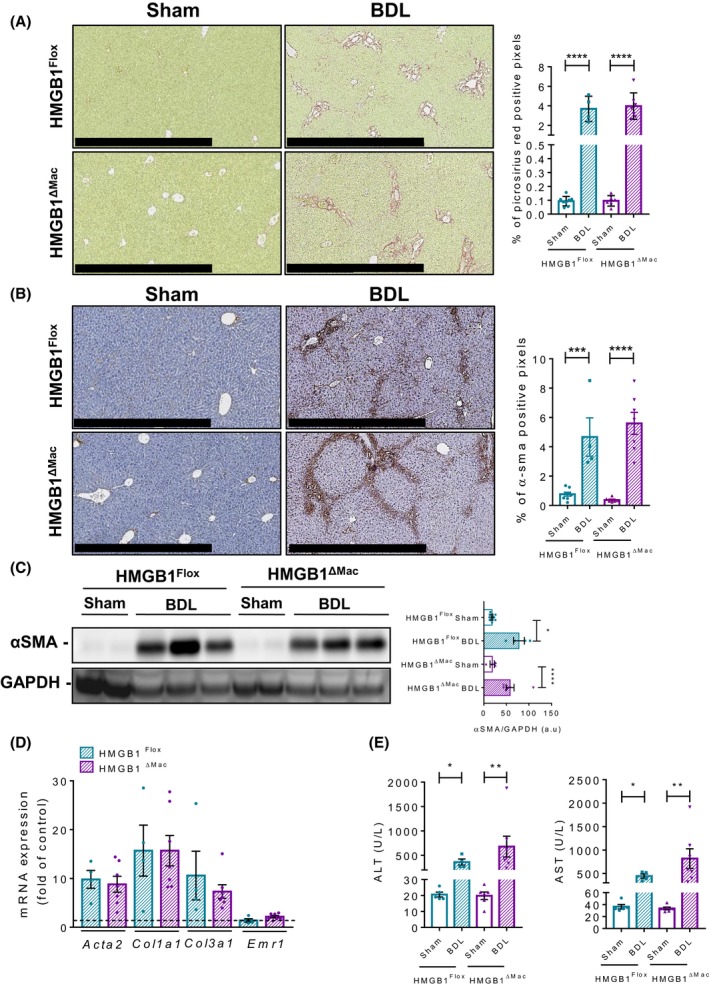
Macrophage‐specific deletion of high mobility group B1 (HMGB1) does not alter liver fibrogenesis after BDL. Representative pictures of picrosirius red staining of HMGB1Flox mice and HMGB1ΔMac of liver section and quantification of positive pixels per liver section. Scale bar: 500 μm (A). Representative pictures of immunohistochemical staining with an antibody against α‐smooth muscle actin (α‐SMA) and quantification of positive pixels per liver section. Scale bar: 500 μm (B). Liver extracts from HMGB1Flox mice and HMGB1ΔMac were analyzed by western blotting directed against α‐SMA (C). Liver mRNA expression levels of classical fibrosis markers were detected using real‐time RT‐PCR, the dotted line indicates the baseline (D). Hepatic injury in HMGB1Flox mice and HMGB1ΔMac was determined by alanine transaminase (ALT) (left panel) and aspartate transaminase (AST) (right panel) levels (E). Statistical analysis was performed with Mann‐Whitney test. Data are expressed as means ± SEM. n = 6 in HMGB1Flox‐sham group; n = 6 in HMGB1ΔMac‐sham group; n = 4 in HMGB1Flox‐BDL group; n = 7 in HMGB1ΔMac‐BDL group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs sham/BDL
As several studies suggested a direct effect of HMGB1 on hepatic stellate cells (HSCs) in the development of liver fibrosis,31, 32 we sought to further study the role macrophage derived‐HMGB1. Because (a) macrophages have a key role in promoting HSC activation, HSC survival, and liver fibrosis,26, 33 (b) LPS promotes liver fibrosis34 and (c) macrophages can release HMGB1 following LPS stimulation, we used LPS‐stimulated conditioned media from HMGB1Flox and HMGB1ΔMac macrophages to determine the role of macrophage‐derived HMGB1 in HSC activation. Of note, LPS strongly increased HMGB1 release over time, with a peak at 48 hours, which was almost completely blocked in HMGB1‐deleted macrophages (Figure S4A). Forty‐eight hour‐conditioned medium (CM) from LPS‐stimulated BMDM increased the activation of HSC compared to CM from unstimulated BMDM as shown by mRNA expression fibrogenic markers Col1a1, Col1a2, and Acta2 (Figure S4B‐D). However, HSC activation did not differ between CM prepared from bone marrow isolated from HMGB1Flox and HMGB1ΔMac mice, demonstrating that the pro‐fibrotic effects of BMDM are independent of HMGB1 (Figure S4B‐D).
3.5. Macrophage‐derived HMGB1 does not play a crucial role in UUO‐induced kidney fibrosis
As the results collected on three relevant injury models unexpectedly showed no effects of HMGB1 deletion on liver fibrosis, we next tested whether the role of macrophage HMGB1 in kidney fibrosis, where fibrotic mechanisms are slightly different compared to the liver. For this purpose, we employed a classical model of tubulointerstitial fibrosis induced by UUO.27 As performed in the liver, Hmgb1 gene deletion was carefully monitored using deletion PCR on whole kidney DNA extract from both HMGB1Flox and HMGB1ΔMac mice (Figure S1F). Seven days after UUO, HMGB1Flox mice displayed typical fibrosis features with a drastic deposition of fibrillar collagen and myofibroblasts activation (Figure 5A,B) compared to sham‐operated mice, as determined by immunohistochemistry. But similar to the liver fibrogenesis models, no differences were detected between HMGB1Flox and HMGB1ΔMac mice in UUO‐induced kidney fibrosis progression. Moreover, α‐SMA protein levels (Figure 5C), as well as mRNA expression of fibrogenic and inflammatory genes Acta2, Ccn2, Col1a1, Col3a1, Emr1, Il‐1β, and Il‐6 were comparable between HMGB1Flox and HMGB1ΔMac mice in the harvested kidney following UUO, except for Tnfα which was upregulated in HMGB1ΔMac mice (Figure 5D; Figure S2D).
Figure 5.
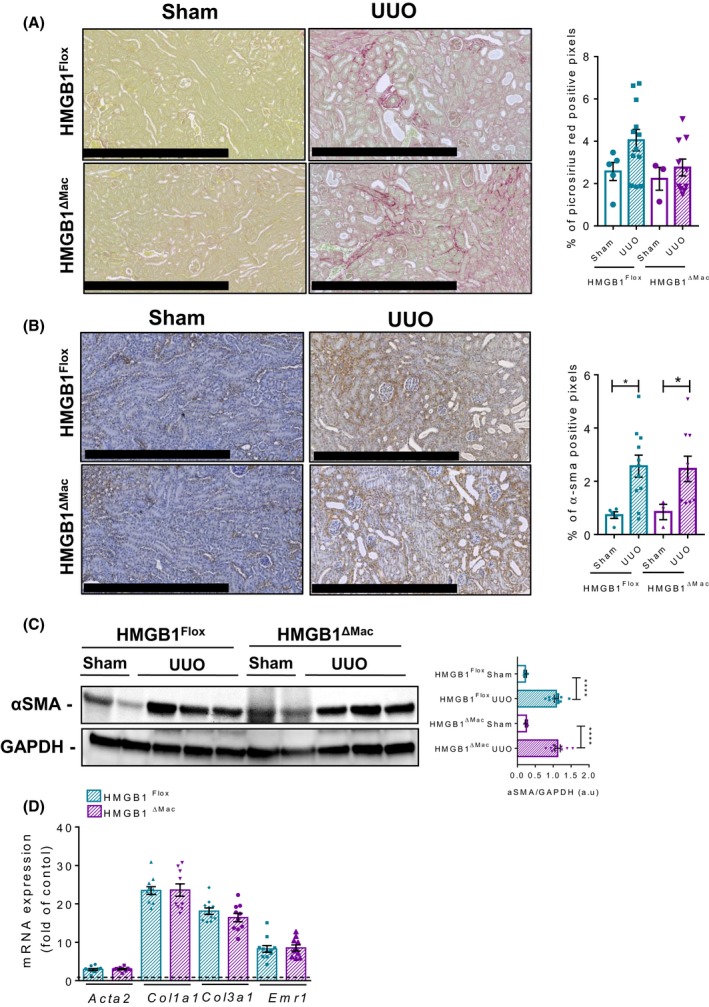
Macrophage‐specific deletion of high mobility group B1 (HMGB1) does not play a role in UUO‐induced kidney fibrosis. Representative pictures of picrosirius red staining of HMGB1Flox mice and HMGB1ΔMac of kidney section and quantification of positive pixels per kidney section. Scale bar: 500 μm (A). Representative pictures of immunohistochemical staining with an antibody against α‐smooth muscle actin (α‐SMA) and quantification of positive pixels per kidney section. Scale bar: 500 μm (B). Kidney extracts from HMGB1Flox and HMGB1ΔMac mice were analyzed by western blotting directed against α‐SMA (C). Kidney mRNA expression levels of classical fibrosis markers were detected using real‐time RT‐PCR, the dotted line indicates the baseline (D). Statistical analysis was performed with Mann‐Whitney test. Data are expressed as means ± SEM. n = 3 in HMGB1Flox‐sham group; n = 3 in HMGB1ΔMac‐sham group; n = 12 in HMGB1Flox‐UUO group; n = 10 in HMGB1ΔMac‐UUO group. *P < 0.05, **P < 0.01, ***P < 0.001 vs sham/UUO
3.6. HMGB1ΔMac mice do not display an increased cardiac fibrosis after TAC
In addition to the liver and kidney, we finally assessed the impact of HMGB1 deletion in macrophages on fibrosis progression in the heart as it has been reported recently that HMGB1 could play a key role in cardiac fibrosis,22 using TAC as a well‐established and clinically relevant model of cardiac fibrosis.35 As performed previously, Hmgb1 gene deletion was carefully monitored using deletion PCR on whole heart DNA extract from both HMGB1Flox and HMGB1ΔMac mice (Figure S1G). Twenty‐eight days after TAC, HMGB1Flox mice displayed pronounced impairments of cardiac function such as left ventricular end‐systolic and ‐diastolic dimension (Table1) compared to sham‐operated animals, accompanied with a strong collagen deposition (Figure 6A). But once again, HMGB1Flox and HMGB1ΔMac mice subjected to TAC showed a similar extent of functional alterations and extracellular matrix accumulation determined by picrosirius red staining or α‐SMA immunohistochemistry and immunoblotting (Table 1; Figure 6A‐C). In addition, similar levels of mRNA expression of classical fibrogenic markers such as Acta2, Ccn2, Col1a1, and Col3a1 mRNA, and inflammatory marker Emr1, Tnfα, and Il‐1b (except for Il‐6 which was upregulated in HMGB1ΔMac mice) mRNA were detected using real‐time qPCR in operated HMGB1Flox and HMGB1ΔMac mice (Figure 6D; Figure S2E).
Table 1.
Echocardiographic and anatomical analysis as a function of transverse aortic constriction (TAC). Statistical analysis was performed with a two‐way ANOVA analysis
| Time post surgery | 4 wk | |||
|---|---|---|---|---|
| Genotype | HMGB1Flox Sham | HMGB1ΔMac Sham | HMGB1Flox + TAC | HMGB1ΔMac + TAC |
| Echocardiographic parameters | ||||
| LVEDD (mm) | 3.443 ± 0.12 | 3.435 ± 0.11 | 4.018 ± 0.15** | 3.846 ± 0.12** |
| LVESD (mm) | 1.955 ± 0.09 | 1.98 ± 0.12 | 2.604 ± 0.22* | 2.39 ± 0.16* |
| FS (%) | 43.25 ± 1.93 | 42.25 ± 1.79 | 36.25 ± 3.29 | 38.36 ± 2.37 |
| Anatomical parameters | ||||
| BW (g) | 31.94 ± 0.54 | 30.49 ± 1.25 | 27.29 ± 0.90** | 28.73 ± 0.69** |
| Heart weight (mg) | 144.3 ± 7.14 | 152 ± 3.51 | 169.5 ± 11.21* | 174.3 ± 6.38* |
| HW/TL (mg/mm) | 78.27 ± 3.81 | 82.51 ± 1.70 | 97.8 ± 6.22** | 98.6 ± 3.64** |
Data are expressed as means ± SEM. n = 4 in HMGB1Flox/sham group; n = 4 in HMGB1ΔMac/sham group; n = 8 in HMGB1Flox/TAC group; n = 13 in HMGB1ΔMac/TAC group.
BW, body weight; FS, fractional shortening; HMGB1, high mobility group B1; HW/TL, heart weight to tíbia length ratio; LVEDD, left ventricular end‐diastolic dimension; LVESD, left ventricular end‐systolic dimension; TAC, transverse aortic constriction.
P < 0.05.
P < 0.01 vs sham/TAC.
Figure 6.
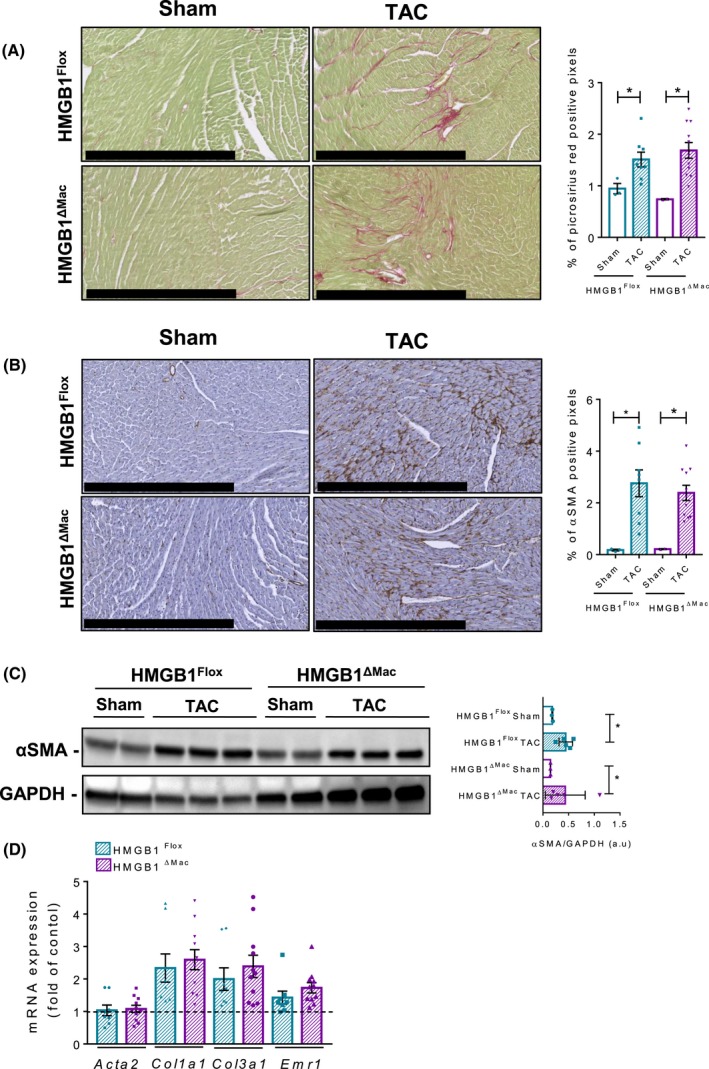
Macrophage‐specific deletion of high mobility group B1 (HMGB1) does not modify cardiac fibrogenesis after transverse aortic constriction (TAC). Representative pictures of picrosirius red staining of HMGB1Flox mice and HMGB1ΔMac of heart section and quantification of positive pixels per heart section. Scale bar: 500 μm (A). Representative pictures of immunohistochemical staining with an antibody against α‐smooth muscle actin (α‐SMA) and quantification of positive pixels per heart section. Scale bar: 500 μm (B). Heart extracts from HMGB1Flox mice and HMGB1ΔMac were analyzed by western blotting directed against α‐SMA (C). Heart mRNA expression levels of classical fibrosis markers were detected using real‐time RT‐PCR, the dotted line indicates the baseline (D). Statistical analysis was performed with Mann‐Whitney test. Data are expressed as means ± SEM. n = 3 in HMGB1Flox‐sham group; n = 3 in HMGB1ΔMac‐sham group; n = 8 in HMGB1Flox‐TAC group; n = 11 in HMGB1ΔMac‐TAC group. *P < 0.05, **P < 0.01, ***P < 0.001 vs sham/TAC
3.7. Thermoneutral housing has no incidence on fibrosis progression in HMGB1ΔMac mice compared to HMGB1Flox mice
In the recent years, several striking studies have demonstrated that for rodent models, as opposed to thermoneutral housing (30‐32°C), room temperature housing (20‐22°C) initiates and maintains a chronic cold stress that dramatically impairs physiology and immune response (eg, to inflammatory stimulus).36, 37, 38 As a severe alteration of immune responses has pronounced consequences on fibrosis progression,39, 40 we hypothesized that thermoneutral housing, rather than room temperature housing performed so far, might be a more relevant context to evaluate the impact of macrophage‐derived HMGB1 deletion on fibrosis development. For this purpose, after 2 weeks of acclimation at 30°C, HMGB1Flox and HMGB1ΔMac mice were subjected to two fibrosis models: BDL‐induced liver fibrosis and UUO‐induced kidney fibrosis. As done at room temperature conditions, Hmgb1 gene deletion has been carefully monitored using deletion PCR on whole liver or kidney DNA extract from both HMGB1Flox and HMGB1ΔMac mice (Figure 1H‐J). As expected, we found that compared to room temperature, thermoneutral housing affected global mouse physiology, and remarkably HMGB1 circulating levels were higher in mice housed at 30°C compare to 20°C housing conditions (Figure 5A). In addition, serum ALT and AST levels (Figure 5B‐C) and liver inflammation and fibrosis (Figure 5D‐E) were increased when mice are housed at 30°C compared to 20°C suggesting a higher hepatocyte turnover at 30°C. In both models and similar to what we observed at room temperature, HMGB1Flox mice exhibited a severe deposition of collagen and myofibroblast activation compared to sham‐operated animals (Figure 7A‐D). But similar to room temperature housing, HMGB1Flox, and HMGB1ΔMac mice displayed the same extent of fibrosis progression induced by either BDL or UUO procedures determined by picrosirius red staining and immuno‐detection (Figure 7A‐D). Expression of α‐SMA using immunoblotting and mRNA expression of fibrogenesis markers as Acta2, Ccn2, Col1a1, and Col3a1 displayed similar levels after BDL and UUO between HMGB1Flox and HMGB1ΔMac mice (Figure 7E‐H, Figure S2G‐H). Interestingly, most of the inflammatory markers Emr1, Tnfα, Il‐1b, and Il‐6 were decreased in HMGB1ΔMac compared to HMGB1Flox mice (Figure 7E‐H; Figure S2G,H) although no functional incidences have been emphasized. Finally, compared to sham‐operated mice, BDL induced a pronounced liver injury (Figure S6A,B) but there were no differences in serum ALT or AST levels between HMGB1Flox and HMGB1ΔMac mice. In parallel, BDL and UUO procedures provoked a marked increase of circulating levels of HMGB1 (Figure S6C), albeit not significant, compared to sham‐operated animals, and operated HMGB1Flox and HMGB1ΔMac mice displayed comparable amount of serum HMGB1 levels (Figure S6C). Altogether these results suggest that, despite a stronger level of fibrosis compared to room temperature conditions, thermoneutral housing did not reveal any role of macrophage‐specific HMGB1 in fibrosis progression. Therefore, conclusions drawn from room temperature experiments were fully confirmed under thermoneutral conditions.
Figure 7.
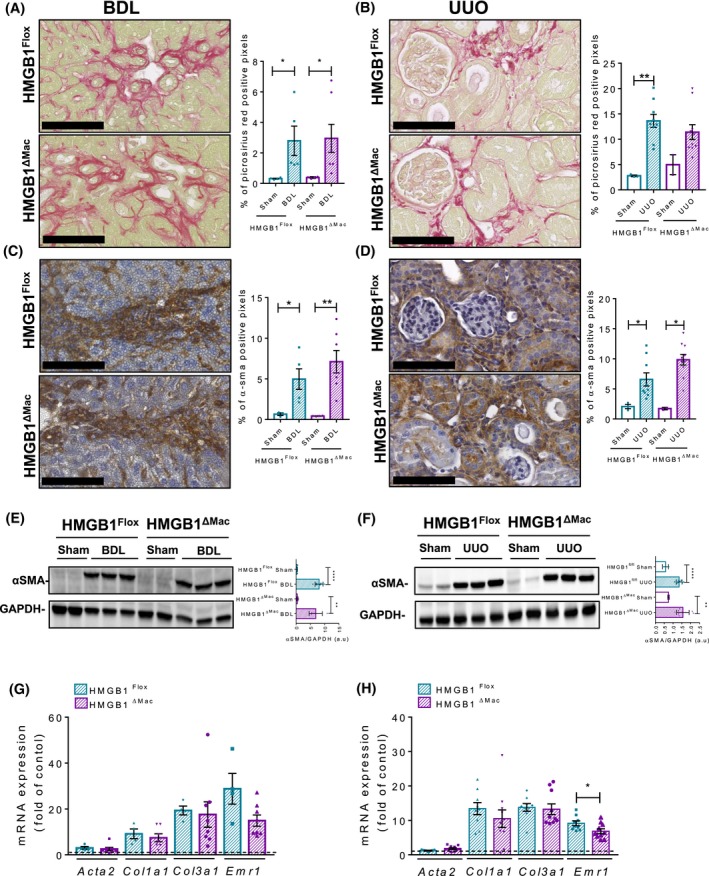
Thermoneutral housing has no incidence of fibrosis progression in mice with macrophage‐specific HMGB1 deletion (HMGB1ΔMac) compared to HMGB1Flox mice. Representative pictures of picrosirius red staining of sham and operated HMGB1Flox mice and HMGB1ΔMac of liver (A) and kidney (B) and quantification of positive pixels per tissue section. Scale bar: 100 μm (A,B). Representative pictures of immunohistochemical staining of sham and operated HMGB1Flox and HMGB1ΔMac mice with an antibody against α‐smooth muscle actin (α‐SMA) in the liver (C) and kidney (D) and quantification of positive pixels per tissue section. Scale bar: 100 μm (C,D). Liver (E) and kidney (F) extracts from sham and operated HMGB1Flox and HMGB1ΔMac mice were analyzed by western blotting directed against α‐SMA (E,F). Liver (G) and kidney (H) mRNA expression levels of classical fibrosis markers expressed in fold of sham‐operated animals (dotted line) were detected using real‐time RT‐PCR, the dotted line indicates the baseline (G,H). Statistical analysis was performed with Mann‐Whitney test. Data are expressed as means ± SEM. n = 4 (liver) n = 3 (kidney) in HMGB1Flox‐sham group; n = 3 (liver & kidney) in HMGB1ΔMac‐sham group; n = 5 in HMGB1Flox‐bile duct ligation (BDL) group; n = 7 in HMGB1ΔMac‐BDL group; n = 12 in HMGB1Flox‐unilateral ureteral obstruction (UUO) group; n = 12 in HMGB1ΔMac‐UUO group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 vs sham/BDL or sham/UUO
4. DISCUSSION
In the expanding field of sterile inflammation biology, many open questions remain in regard to the effects of alarmin/DAMPs in acute inflammation, tissue regeneration, scarring, and fibrosis. Here, using conditional deletion of HMGB1 protein specifically in macrophages, we clearly demonstrate that macrophage‐derived HMGB1 protein does not play a key role in fibrogenesis in three different organs using a wide range of well‐established models in vivo. In contrast, a recent study by Ge et al, using macrophage‐specific genetic ablation (LysM‐CRE) or neutralization strategies demonstrates that macrophage and hepatocyte‐derived HMGB1 rather participates in liver fibrosis.41 Employing cell‐specific knockout of HMGB1 in macrophages in vivo, our report questions the contribution of a prototypical DAMP or pro‐inflammatory cytokine HMGB1 in the pathogenesis of fibrosis. The opposite conclusions of both studies might be due to the use of different mice. While Ge et al used a conditional deletion of HMGB1 exons 2‐3, we employed mice expressing floxed exons 2‐4. It is hard to fully explain this obvious discrepancy but the presence of the exon 4 in the study of Ge et al may be a possible caveat as bioinformatics analysis tend to demonstrate the presence of an active open reading frame (ORF) even after CRE recombination, suggesting that exons 4 and 5 are still possibly translated and biologically active. These two different floxing strategies resulted in opposite results on HMGB1 and mitochondrial quality control and autophagy mechanism in vivo,30, 42 but similar outcomes in liver cancer progression19, 43 without really finding a rational molecular explanation.44, 45, 46 Further studies will be needed to decipher why both transgenic lines lead to similar conclusions in certain contexts and to opposite conclusions in specific settings.
The present work is clearly questioning the potential role of macrophage‐derived HMGB1 during fibrosis and its relevance in vivo. Numerous reports have been published in recent years, showing in vitro that innate immune cells might actively secrete HMGB1, after an inflammatory challenge such as LPS. In vivo, few reports suggesting that macrophage‐derived HMGB1 may be involved in acute inflammation diseases like in the colon or lungs.47, 48 But most of these in vivo studies used pharmacological agents such as ethyl‐pyruvate that has been identified as a potential blocker of the nuclear to cytoplasmic translocation47 typically observed in macrophages cell lines. In the meantime, other publications have demonstrated that epithelial cells might be capable of an active secretion of HMGB1 such as adipocytes, skin fibroblasts, or colon cancer cells49, 50, 51 implying that ethyl‐pyruvate may also prevent active secretion in these epithelial cells and would then not be specific of macrophages, undermining conclusions drawn from previous in vivo works using ethyl pyruvate. In this context, the in vivo role of macrophage‐derived HMGB1 is still elusive, and only cell‐specific deletion strategy might help to decipher precisely which processes are driven by macrophage‐derived HMGB1. To the best of our knowledge, only a few studies have been already published using a genetic model of macrophage‐specific invalidation of HMGB1. Among them, the work from Huebener et al demonstrated using two different CRE lines (MX1‐Cre and VAV1‐Cre) targeting bone marrow‐derived cells, that circulating levels of HMGB1 were severely blunted after an LPS challenge in vivo,52 meanwhile Yanai et al, using LysM‐Cre deleter targeting more specifically macrophages, showed no changes in HMGB1 circulating levels after the same LPS stimulation.53 Taken together these results tend to suggest that macrophages might be marginally involved, as opposed to in vitro studies, in active secretion process of HMGB1 after an LPS challenge questioning the relevance of HMGB1 active secretion by macrophages in vivo. It is possible that LPS is substantially different from stimuli encountered by macrophages during fibrogenic challenges such as CCL4 injections. But while we noted that HMGB1 circulating and liver levels were increased during all fibrogenic models used in our study, we found no changes in HMGB1 levels in plasma (Figure S7A‐E), but a slight decreases in fibrotic livers (Figure S7F,G) harvested from HMGB1ΔMac compared to HMGB1Flox mice supporting one more time a marginal contribution of macrophage‐derived HMGB1 in the whole circulating pool but a rather more significant role at the tissue level in the liver after CCL4 injection or following BDL (Figure S7F,G).
Despite being the best possible approach in vivo, HMGB1 cell‐specific deletion may also have potential caveats that need to be considered. As now well established, HMGB1 is within the cell, an abundant nuclear factor conserved among all eukaryotic cells, which regulates chromatin conformation and gene regulation and once outside the cells, a danger signal promoting inflammatory reactions. By deleting HMGB1 in the macrophages, we de facto blunted both intra and extracellular pools of HMGB1. And in their report, Yanai et al stated that HMGB1 macrophage‐specific deletion may have altered intracellular homeostasis and notably autophagy pathways,53 which would have serious consequences on macrophages physiology, polarizing them toward a pro‐inflammatory phenotype with notably a higher capacity to secrete IL1β and IL‐18 and promoting eventually macrophage cell death.53 In this context, we cannot rule out that deletion of Hmgb1 gene in macrophages may have altered macrophages function in our settings. Thus, we could hypothesize that generating such confounding factors (modifying macrophage global function and survival) could prevent us to properly assess the sole HMGB1 extracellular function in vivo and possible impact on fibrogenesis. Only sophisticated genetic strategies could help to discriminate between the role in macrophages of HMGB1 as a nuclear factor in one hand and as a secreted factor in the other hand.
Despite unexpected results on macrophage‐derived HMGB1 and fibrogenesis, our study does not question the overall role of DAMPs in fibrogenesis. Several publications demonstrated using well‐characterized DAMPs receptors, belonging to the pattern recognition receptor (PRR) family, that DAMPs and sterile inflammation players are definitely driving tissue scarring and fibrogenesis. Reports addressing potential HMGB1 receptors involvement in fibrogenesis clearly demonstrate that PRRs such as toll‐like receptors (TLRs) or receptor for advanced glycation end products (RAGE) are causally connected to tissue scarring in the lungs, kidney, heart, or liver. TLR4 knockout mice are protected against fibrogenesis upon chemically or surgically induced fibrosis in the liver,34 heart,54 and lungs,55 TLR2 knockout mice are preserved toward cardiac fibrosis,56 and TLR9 has been involved in liver fibrosis.57, 58 And RAGE has also been connected to fibrogenesis in the lungs59, 60 and kidneys.61 Thus, there is a consistent body of literature demonstrating that PRRs such as TLRs or RAGE are involved in fibrogenesis, therefore suggesting that DAMPs or other types of ligands besides HMGB1, may exert a pro‐fibrotic activity.62 In this context, HMGB1 certainly acts as DAMP in certain conditions or as an alarmin in other conditions depending on the cell source. Considering the complexity of sterile inflammation response, with such a variety of ligands and receptors, one could hypothesize that initiation of an inflammatory or pro‐fibrotic response may depend on an unknown threshold of necrosis, type of tissue injury combined with micro‐environmental conditions—not yet identified—which could affect and influence the channeling toward no response, or inflammatory and/or fibrotic response. Further in vivo work is needed, using sophisticated genetic tools, to clearly assess how on a molecular level, how nuclear and secreted HMGB1 may drive regeneration, inflammation, or fibrosis and to identify which cell source is specifically involved in each biological pathway.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
J. Personnaz and E. Piccolo designed research, performed experiments, analyzed data, and wrote the paper; M. Branchereau, A. Filliol, R. Paccoud, E. Moreau, D. Calise, E. Riant and C. Heymes performed experiments and analyzed data, P. Gourdy generously provided LysM‐CRE mice; R.F. Schwabe kindly provided HMGB1‐floxed mice, designed experiments, and revised the manuscript; C. Dray and P. Valet provided fundings and revised the manuscript; J.‐P. Pradère conceived the original hypothesis, designed all experiments, performed experiments, analyzed data, wrote the manuscript, provided fundings, and supervised the project.
Supporting information
ACKNOWLEDGEMENTS
The authors thank the phenotyping facility (Anexplo‐US006/CREFRE, Toulouse, France) for all plasma analysis and technical assistance, GenoToul Genome and Transcriptome core facility for technical assistance, Dr. Bernard Masri and the cellular imaging facility of the Institut des maladies métaboliques et cardiovasculaires (Toulouse, France) for helpful technical assistance. This study is supported by grants from INSERM, Paul Sabatier University, the Agence Nationale de la Recherche (ANR‐17‐CE14‐0016/Holistic, J‐P.P) and Association Française d'Etude et de Recherche sur l'Obésité. J.P. is supported by a scholarship from Paul Sabatier University, E.P. is supported by a scholarship from Agence Nationale de la Recherche (ANR‐17‐CE14‐0016/Holistic) and R.P. is supported by a scholarship from Région Midi‐Pyrénnées‐INSERM (n° 15050341).
Personnaz J, Piccolo E, Branchereau M, et al. Macrophage‐derived HMGB1 is dispensable for tissue fibrogenesis. FASEB BioAdvances. 2019;1:227–245. 10.1096/fba.2018-01033
Jean Personnaz and Enzo Piccolo contribute equally.
REFERENCES
- 1. Mack M. Inflammation and fibrosis. Matrix Biol. 2018;68-69:106-121. [DOI] [PubMed] [Google Scholar]
- 2. Weiskirchen R, Weiskirchen S, Tacke F. Organ and tissue fibrosis: molecular signals, cellular mechanisms and translational implications. Mol Aspects Med. 2018. S0098-2997(18)30038-4. [DOI] [PubMed] [Google Scholar]
- 3. Vannella KM, Wynn TA. Mechanisms of organ injury and repair by macrophages. Annu Rev Physiol. 2017;79:593‐617. [DOI] [PubMed] [Google Scholar]
- 4. Smigiel KS, Parks WC. Macrophages, wound healing, and fibrosis: recent insights. Curr Rheumatol Rep. 2018;20:17. [DOI] [PubMed] [Google Scholar]
- 5. Thannickal VJ, Zhou Y, Gaggar A, Duncan SR. Fibrosis: ultimate and proximate causes. J Clin Invest. 2014;124:4673‐4677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee J‐M, Yoshida M, Kim M‐S, et al. Involvement of alveolar epithelial cell necroptosis in IPF pathogenesis. Am J Respir Cell Mol Biol. 2018;59:215-224. rcmb.2017‐0034OC. [DOI] [PubMed] [Google Scholar]
- 7. Zhang W, Lavine KJ, Epelman S, et al. Necrotic myocardial cells release damage‐associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J Am Heart Assoc. 2015;4:e001993‐e001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vénéreau E, Ceriotti C, Bianchi ME. DAMPs from cell death to new life. Front Immunol. 2015;6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andersson U, Yang H, Harris H. High‐mobility group box 1 protein (HMGB1) operates as an alarmin outside as well as inside cells. Semin Immunol. 2018;38:40‐48. [DOI] [PubMed] [Google Scholar]
- 10. Kang R, Chen R, Zhang Q, et al. HMGB1 in health and disease. Mol Aspects Med. 2014;40:1‐116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dear JW, Clarke JI, Francis B, et al. Risk stratification after paracetamol overdose using mechanistic biomarkers: results from two prospective cohort studies. Lancet Gastroenterol Hepatol. 2018;3:104‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Venereau E, De Leo F, Mezzapelle R, Careccia G, Musco G, Bianchi ME. HMGB1 as biomarker and drug target. Pharmacol Res. 2016;111:534‐544. [DOI] [PubMed] [Google Scholar]
- 13. Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139‐162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andersson U, Wang H, Palmblad K, et al. High mobility group 1 protein (HMG‐1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med, Rockefeller University Press, 2000;192:565‐570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gil M, Kim YK, Hong SB, Lee, KJ . Naringin decreases TNF‐α and HMGB1 release from LPS‐stimulated macrophages and improves survival in a CLP‐induced sepsis mice. Mukhopadhyay P, editor. PLoS ONE, Public Library of Science. 2016;11:e0164186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. El Gazzar M. HMGB1 modulates inflammatory responses in LPS‐activated macrophages. Inflamm Res. 2007;56:162‐167. [DOI] [PubMed] [Google Scholar]
- 17. Gardella S, Andrei C, Ferrera D, et al. The nuclear protein HMGB1 is secreted by monocytes via a non‐classical, vesicle‐mediated secretory pathway. EMBO Rep. 2002;3:995‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tirone M, Tran NL, Ceriotti C, et al. High mobility group box 1 orchestrates tissue regeneration via CXCR18. J Exp Med. 2018;215:303‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hernandez C, Huebener P, Pradere J‐P, Antoine DJ, Friedman RA, Schwabe RF. HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J Clin Invest. 2018;128:2436‐2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Khambu B, Huda N, Chen X, et al. HMGB1 promotes ductular reaction and tumorigenesis in autophagy‐deficient livers. J Clin Invest. 2018;128:2419‐2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lee G, Espirito Santo AI, Zwingenberger S, et al. Fully reduced HMGB1 accelerates the regeneration of multiple tissues by transitioning stem cells to GAlert . Proc Natl Acad Sci. 2018;115:E4463‐E4472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu R‐N, Yu T‐Y, Zhou J‐C, et al. Targeting HMGB1 ameliorates cardiac fibrosis through restoring TLR2‐mediated autophagy suppression in myocardial fibroblasts. Int J Cardiol. 2018;267:156‐162. [DOI] [PubMed] [Google Scholar]
- 23. Hamada N, Maeyama T, Kawaguchi T, et al. The role of high mobility group box1 in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:440‐447. [DOI] [PubMed] [Google Scholar]
- 24. Tian S, Zhang L, Tang J, Guo X, Dong K, Chen S‐Y. HMGB1 exacerbates renal tubulointerstitial fibrosis through facilitating M1 macrophage phenotype at the early stage of obstructive injury. Am J Physiol Renal Physiol. 2015;308:F69‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Troeger JS, Mederacke I, Gwak G‐Y, et al. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology. 2012;143:1073-1083.e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pradere J‐P, Kluwe J, De Minicis S, et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology. 2013;58:1461-1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75:1145-1152. [DOI] [PubMed] [Google Scholar]
- 28. Bisserier M, Berthouze‐Duquesnes M, Breckler M, et al. Carabin protects against cardiac hypertrophy by blocking calcineurin, Ras, and Ca2+/calmodulin‐dependent protein kinase II signaling. Circulation. 2015;131:390‐400. [DOI] [PubMed] [Google Scholar]
- 29. Clausen BE, Burkhardt C, Reith W, Renkawitz R, Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. [DOI] [PubMed] [Google Scholar]
- 30. Huebener P, Gwak G‐Y, Pradere J‐P, et al. High‐mobility group box 1 is dispensable for autophagy, mitochondrial quality control, and organ function in vivo. Cell Metab. 2014;19:539‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arriazu E, Ge X, Leung T‐M, et al. Signalling via the osteopontin and high mobility group box‐1 axis drives the fibrogenic response to liver injury. Gut. 2017;66:1123‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. He Q, Fu Y, Ding X, et al. High‐mobility group box 1 induces endoplasmic reticulum stress and activates hepatic stellate cells. Lab Investig. 2018;98:1200‐1210. [DOI] [PubMed] [Google Scholar]
- 33. Duffield JS, Forbes SJ, Constandinou CM, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Seki E, De Minicis S, Österreicher CH, et al. TLR4 enhances TGF‐β signaling and hepatic fibrosis. Nat Med. 2007;13:1324‐1332. [DOI] [PubMed] [Google Scholar]
- 35. Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG. Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol. 2009;131:471‐481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giles DA, Moreno‐Fernandez ME, Stankiewicz TE, et al. Thermoneutral housing exacerbates nonalcoholic fatty liver disease in mice and allows for sex‐independent disease modeling. Nat Med, Nature Publishing Group. 2017;23:829‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ganeshan K, Chawla A. Warming the mouse to model human diseases. Nat Rev Endocrinol. 2017;13:458‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tian XY, Ganeshan K, Hong C, et al. Thermoneutral housing accelerates metabolic inflammation to potentiate atherosclerosis but not insulin resistance. Cell Metab, Elsevier. 2016;23:165‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wick G, Backovic A, Rabensteiner E, Plank N, Schwentner C, Sgonc R. The immunology of fibrosis: innate and adaptive responses. Trends Immunol, Europe PMC Funders. 2010;31:110‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest, American Society for Clinical Investigation. 2017;127:55‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ge X, Arriazu E, Magdaleno F, et al. High mobility group box‐1 drives fibrosis progression signaling via the receptor for advanced glycation end‐products in mice. Hepatology. 2018;68:2380‐2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tang D, Kang R, Livesey KM, et al. High‐mobility group box 1 is essential for mitochondrial quality control. Cell Metab. 2011;13:701‐711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen R, Zhu S, Fan X‐G, et al. High mobility group protein B1 controls liver cancer initiation through yes‐associated protein ‐dependent aerobic glycolysis. Hepatology. 2018;67:1823‐1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sun X, Tang D. Hepatocyte‐specific Hmgb1 deletion. Autophagy. 2015;11:1189‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Huebener P, Gwak GY, Schwabe RF. Comment on: HMGB1‐dependent and ‐independent autophagy. Autophagy. 2015;11:1187‐1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun X, Tang D. HMGB1‐dependent and ‐independent autophagy. Autophagy. 2014;10:1873‐1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davé SH, Tilstra JS, Matsuoka K, et al. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J Leukoc Biol. 2009;86:633‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Entezari M, Javdan M, Antoine DJ, et al. Inhibition of extracellular HMGB1 attenuates hyperoxia‐induced inflammatory acute lung injury. Redox Biol. 2014;2:314‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shimizu T, Yamakuchi M, Biswas KK, et al. HMGB1 is secreted by 3T3‐L1 adipocytes through JNK signaling and the secretion is partially inhibited by adiponectin. Obesity (Silver Spring). 2016;24:1913‐1921. [DOI] [PubMed] [Google Scholar]
- 50. Lee H, Park M, Shin N, et al. High mobility group box‐1 is phosphorylated by protein kinase C zeta and secreted in colon cancer cells. Biochem Biophys Res Commun. 2012;424:321‐326. [DOI] [PubMed] [Google Scholar]
- 51. Wang L, He L, Bao G, He X, Fan S, Wang, H . Ionizing radiation induces HMGB1 cytoplasmic translocation and extracellular release. Guo Ji Fang She Yi Xue He Yi Xue Za Zhi. 2016;40:91‐99. [PMC free article] [PubMed] [Google Scholar]
- 52. Huebener P, Pradere J‐P, Hernandez C, et al. The HMGB1/RAGE axis triggers neutrophil‐mediated injury amplification following necrosis. J Clin Invest. 2015;125:539‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yanai H, Matsuda A, An J, et al. Conditional ablation of HMGB1 in mice reveals its protective function against endotoxemia and bacterial infection. Proc Natl Acad Sci. 2013;110:20699‐20704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Li XX, Jiang DY, Huang XX, Guo SL, Yuan W, Dai HP. Toll‐like receptor 4 promotes fibrosis in bleomycin‐induced lung injury in mice. Genet Mol Res. 2015;14:17391‐17398. [DOI] [PubMed] [Google Scholar]
- 55. Dong R, Wang Z, Zhao C, et al. Toll‐like receptor 4 knockout protects against isoproterenol‐induced cardiac fibrosis: the role of autophagy. J Cardiovasc Pharmacol Ther. 2015;20:84‐92. [DOI] [PubMed] [Google Scholar]
- 56. Wang L, Li Y‐L, Zhang C‐C, et al. Inhibition of Toll‐like receptor 2 reduces cardiac fibrosis by attenuating macrophage‐mediated inflammation. Cardiovasc Res. 2014;101:383‐392. [DOI] [PubMed] [Google Scholar]
- 57. Watanabe A, Hashmi A, Gomes DA, et al. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll‐like receptor 9. Hepatology. 2007;46:1509‐1518. [DOI] [PubMed] [Google Scholar]
- 58. Gäbele E, Mühlbauer M, Dorn C, et al. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem Biophys Res Commun. 2008;376:271‐276. [DOI] [PubMed] [Google Scholar]
- 59. Englert JM, Hanford LE, Kaminski N, et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol. 2008;172:583‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ramsgaard L, Englert JM, Tobolewski J, et al. The role of the receptor for advanced glycation end‐products in a murine model of silicosis. Morty RE, editor. PLoS ONE. 2010;5:e9604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gasparitsch M, Arndt A‐K, Pawlitschek F, et al. RAGE‐mediated interstitial fibrosis in neonatal obstructive nephropathy is independent of NF‐κB activation. Kidney Int. 2013;84:911‐919. [DOI] [PubMed] [Google Scholar]
- 62. Chen L, Li J, Zhang J, et al. S100A4 promotes liver fibrosis via activation of hepatic stellate cells. J Hepatol. 2015;62:156‐164. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials