

Abstract
Phosphorylation is a post-translational modification with a vital role in cellular signaling. Isobaric labeling-based strategies, such as tandem mass tags (TMT), can measure the relative phosphorylation states of peptides in a multiplexed format. However, the low stoichiometry of protein phosphorylation constrains the depth of phosphopeptide analysis by mass spectrometry. As such, robust and sensitive workflows are required. Here we evaluate and optimize high-Field Asymmetric waveform Ion Mobility Spectrometry (FAIMS) coupled to Orbitrap Tribrid mass spectrometers for the analysis of TMT-labeled phosphopeptides. We determined that using FAIMS-MS3 with three compensation voltages (CV) in a single method (e.g., CV = −40/−60/−80 V) maximizes phosphopeptide coverage while minimizing inter-CV overlap. Furthermore, consecutive analyses using MSA-CID (multistage activation collision-induced dissociation) and HCD (higherenergy collisional dissociation) fragmentation at the MS2 stage increases the depth of phosphorylation analysis. The methodology and results outlined herein provide a template for tailoring optimized FAIMS-based methods.
Keywords: phosphorylation, FAIMS, TMTpro0, SL-TMT, SPS-MS3, MSA
Graphical Abstract
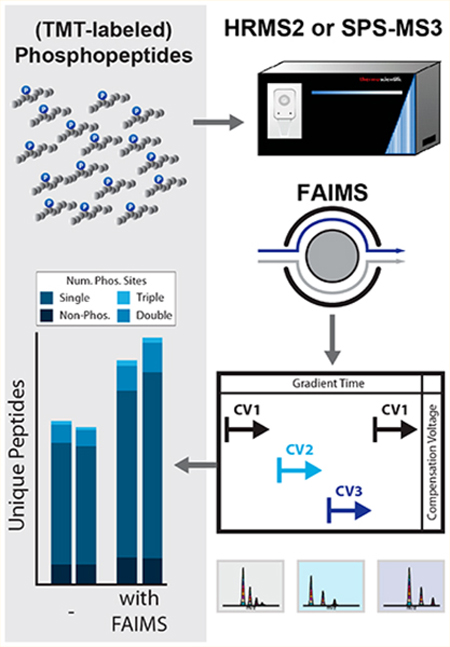
INTRODUCTION
The comprehensive analysis of post-translational modifications (PTM) by mass spectrometry-based proteomics is challenging. Protein phosphorylation is a PTM of central importance in cellular signaling and is essential to numerous cellular functions. Dysregulation of phosphorylation-associated signaling cascades has often been implicated in diseases, most notably in cancer.1 Moreover, kinases and phosphatases constitute about 2% of the human genome.2 Three in every four proteins are estimated to be phosphorylated in their life cycle; however, the overall phosphorylation stoichiometry at any given time is typically low.3 Due to low stoichiometry, the abundance of phosphorylation is orders of magnitude below that of the unphosphorylated peptide, and as such, improvements in sample preparation, data collection, and analysis are needed to increase phosphopeptide coverage.4 The adaptation of novel methods and technologies to enhance phosphopeptide analysis is imperative to expand our understanding of phosphorylation-related cellular signaling.
Isobaric tag-based proteome profiling is a leading technology for multiplexed proteomic and phosphoproteomic analysis of complex peptide mixtures.5,6 Sample multiplexing allows for improved statistical power by including replicates within a single experiment and limiting missing values which often occur across experiments. The introduction of the SL-TMT (streamlined tandem mass tag) protocol simplified isobaric tag-based proteome profiling.7 A “mini-phos” analysis is integrated in the SL-TMT workflow, which facilitates a single, small-scale phosphopeptide enrichment as part of any TMT experiment.8 Such analysis permits the survey of several thousand phosphorylation sites with nominal cost and minimal effort. The adoption of “mini-phos” analyses is hampered by lack of depth due to the relatively low amount of analyte compared to larger-scale studies that enrich from several milligrams of protein lysate.9,10 As such, a need exists for analytically deep, yet highly efficient, phosphorylation analysis workflows. Here we aim to increase the depth of phosphopeptide analysis at the data collection level when using ion mobility separation prior to synchronous precursor selection (SPS)-MS3 analysis.
We apply and optimize high-field asymmetric waveform ion mobility spectrometry (FAIMS) for the analysis of isobaric tag-labeled, phosphorylated peptide-enriched samples using the recently introduced FAIMS-Pro interface.11–13 FAIMS separates on the basis of differences in mobility in high and low electric fields, here generated from an asymmetric waveform as they transit between an inner and outer electrode.14–16 This asymmetric waveform applies a short high positive voltage— the peak which is termed “dispersion voltage” or “DV”—and a longer low negative voltage. The drift of an ion toward electrodes can be stopped by the application of a tunable small dc voltage (“compensation voltage” or “CV”) to either of the plates to compensate for the ion drift. A peptidic ion travels through the plates at a specific characteristic CV value, so alternating CVs can rapidly separate different types of ions.17 Ion mobility is dependent upon an ion’s mass, charge, size, and shape as it traverses the electrode and collides with the buffer gas molecules. This segregation is orthogonal to that of liquid chromatography-based separation.
Advantages of FAIMS include reduced chemical noise and enhanced detection.18 We previously leveraged these benefits to measure interference for whole proteome analysis using the TKO (triple knock out) standard.19,20 Those data showed lower interference for FAIMS-MS3 compared to the traditional SPS-MS3 method.11 Here, we investigated (1) the effect of different CVs and combinations thereof and (2) the advantages of using collision-induced dissociation (CID) or higher-energy collisional dissociation (HCD) activation energy in the MS2 stage for SPS-MS3 analyses. We optimized the FAIMS-based analysis of phosphopeptides derivatized with both classic TMT and the recently released TMTpro chemistry for single shot analyses. Our data demonstrate how FAIMS can be used to improve the depth of multiplexed phosphorylation site analysis and provide a foundation for other investigators studying phosphorylation or any other post-translational modification.
METHODS
Sample Preparation
The samples used were immobilized metal affinity-chromatography-enriched phosphopeptides from human cell lines (HCT116) or mouse brain tissue,21 as processed using the SL-TMT protocol.7 Phosphopeptides were enriched using the High-Select Fe-NTA Phosphopeptide Enrichment Kit8 and desalted (C18 SepPak, Waters). Enriched phosphopeptides were labeled as described previously with TMT or TMTpro0.7 For TMT10, the labeled peptides were mixed 1:1 across all channels, and desalted (C18 SepPak, Waters) prior to use. Further details are available in the Supporting Information.
Mass Spectrometric Data Acquisition and Data Analysis
Labeled peptides were resuspended in 5% ACN/5% formic acid at 1 mg/mL and loaded at 1 μg, unless noted otherwise, on an in-house pulled C18 column (30−35 cm, 2.6 μm Accucore (Thermo Fisher), 100 μm ID), and eluted using a linear gradient from 100% buffer A (5% ACN, 0.125% formic acid) to 30% buffer B (95% ACN, 0.125% formic acid) using an Easy-nLC 1000 (Thermo Scientific). Eluted peptides were analyzed by an Orbitrap Fusion or Fusion Lumos mass spectrometer over a 1–3 h gradient. Instrument parameter details, corresponding to a given figure, are outlined in Table S1. In all cases, the dispersion voltage is set at −5000 V. MS1 features were determined using RawFileReader (https://planetorbitrap.com/rawfilereader).
Data Were Processed via a SEQUEST v 28 (rev. 12)-Based Pipeline Searches and Linear Discriminant Analysis10,22
Tags on lysine residues and peptide N-termini (TMT10: +229.163 Da, TMT0: +224.152 Da, or TMTpro0: +295.190 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) and phosphorylation (+79.966 Da) were set as variable modifications. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR).23,24 Further details are available in the Supporting Information.
Data Access
RAW files will be made available upon request. In addition, the data have been deposited to the ProteomeXchange Consortium via the PRIDE25 partner repository with the data set identifier PXD014593. Table S2 includes tables of the data represented in the figures.
RESULTS AND DISCUSSION
The potential for analytically deeper data acquisition and improved quantitative accuracy in isobaric tag-based workflows has greatly expanded by the use of FAIMS.11,13 In the current work, we aimed to determine if FAIMS could be employed to improve multiplexed phosphoproteome analysis. We have shown previously that optimizing method compensation voltages (CV) within a single method can improve proteome coverage.11 We note that the physical properties of phosphorylated peptides differ from their nonphosphorylated counterparts; in addition, the chemical properties of multiplex labeled peptides vary from metabolically labeled or label-free peptides. As such, here, we sought to optimize FAIMS methods for multiplexed phosphopeptide analysis.
To generate a diverse phosphoproteome standard we employed two sample types in this work. The first was a phospho-enriched (enrichment >95%) tryptic digest of HCT116 lysate (human, Figure 1A), and the second was primary murine phosphopeptides derived from whole brain lysates. We chose these samples as they represent two core areas of interest, the first being in vitro analysis of dynamic phosphoproteomic signaling and the second the profiling of phosphoproteomic signatures in primary tissues.21,26,27 In both cases, phosphopeptides were enriched prior to TMT labeling to more efficiently use the labeling reagent on only the phosphopeptides rather than the whole proteome (Figure 1A).
Figure 1.

Optimizing the FAIMS compensation voltage (CV) for phosphopeptide analysis. A. General FAIMS workflow for phosphopeptide analysis. Samples were processed using a modified SL-TMT protocol in which phosphopeptides were enriched prior to TMT labeling. The resulting peptides were analyzed by a FAIMS-Pro-equipped Orbitrap Fusion or Fusion Lumos mass spectrometer. B. MS1 features (z = +2) detected at varying compensation voltages (−110 V to −10 V). C. Unique peptides (divided by degree of phosphorylation) identified by various data acquisition methods. All data were collected with high-resolution MS2 (HRMS2) without FAIMS (in triplicates) or with CVs ranging from −100 to −30 V (no peptides were identified at −110 V or −20 V). Dotted line represents the proportion of non-phosphorylated peptides.11 D. Comparison of unique peptides identified as label-free (left), and derivatized with TMT reagent, (i.e., TMT, middle or TMTpro, right). Analyses were performed in triplicate. Error bars are standard deviations.
Initially we tested the effect of derivatization on phosphopeptide distributions across an array of CVs (−110 V to −10 V). The instrument (Orbitrap Lumos) acquired MS1 scans at each of the 21 CV values in series for each duty cycle. We then measured and compared the total MS1 features and their charge observed for each CV. We observed that for the three chemistries tested (label-free, TMT10, and TMTpro), the distribution of MS1 features was noticeably shifted depending on the labeling strategy, particularly for the z = +2 features (Figures 1B and S1A). We concluded that specific FAIMS optimization would be necessary depending on the chemistry used.
Previous work has reported the utility of FAIMS for phosphoproteomics as either better than or worse than non-FAIMS methods (i.e., offline fractionation).12,28 Thus, we were interested in establishing whether FAIMS would benefit multiplexed phosphoproteomic workflows. To optimize the identification of labeled phosphorylated peptides, we designed individual LC-MS/MS methods with CVs ranging from −20 V to −120 V in 10 V increments (FAIMS-Pro, Orbitrap Lumos). We compared the total identifications observed across this broad CV range (no identifications were observed at CV = −110 V or −120 V), using HRMS2-based analysis of replicate sample injections (Figure 1C). The maximal identification rate for unique peptides was observed at CV = −80 V, which contrasts that of nonphosphorylated samples from a previous study (CV = −50 V).11 This finding confirms our speculation that due to the phosphorylation event, phosphopeptides were stable in the FAIMS device under different electric fields compared to bulk peptide samples. We also note that by using FAIMS at a single CV of −80 V, the total unique peptide identification was similar to the replicate non-FAIMS HRMS2 analyses. We next examined the charge state distribution of the phosphopeptides with respect to CV (Figure S1D). Precursors with a 3+ charge state dominated the phosphopeptide population, as expected. Lower CVs, i.e. < −80 V, exhibited a predominance of 3+ ions, while higher CVs demonstrated a prominent increase in the number of 2+ and change states greater than 3+.
One benefit of the new FAIMS-Pro device was the ability to quickly switch between different CVs within a single method (dwell time ~25 ms).29 We and others previously showed that combining multiple CVs within a single run or between runs improved whole proteome coverage.11–13 Therefore, we next tested if increasing the number of CVs within a single method (multi-CV method) increased the unique peptide identifications for nonderivatized (label-free) and derivatized (TMT and TMTpro) samples. We chose CVs for multi-CV methods that either included the maxima observed in Figure 1B (CV =−80 V) or CVs near this value. We note that the low resolution of the FAIMS-Pro device enabled detection of the same precursors across nearby CVs.11 As the relative maxima of unique peptide identifications were observed at CV = −70/− 80 V, we initially chose CVs near these values for the two-CV method (Figures 1B and S2A,B). As expected, for all of the peptide chemistries we tested, this two-CV method improved identification rates (Figure 1D).
Expanding on this work to determine the optimal CVs for a triple-CV method, we tested a combination of CVs over a 45 V range including the method previously observed to improve multiplexed nonenriched proteome coverage and quantitation (CV = −40/−60/−80 V).11 We examined the overlap among CVs using an upset plot.30 The CVs −80, −70, and −60 V generated the highest number of peptides; however, the overlap between these CVs was the highest of the binary comparisons (Figure S2A). As such, we selected a lower CV (−40 V or −45 V or −55 V), with a smaller intersection of unique peptide identifications. We further designed an LC-MS/MS data acquisition strategy that cycled through 19 different CVs from −10 V to −100 V in 5 V increments in a single run (Figure S3A). We aimed to maximize the peptide diversity and total unique peptide identifications of different CV combinations by comparing the intersection of identified peptides between any two CVs, (e.g., CV = −65 V and all 18 other CVs). We noted that the parallelized FT (Fourier transform) cycle time plus CV dwell time for all 19 CVs was less than the average peak width (Figure S3B). We observed the maximum total peptides (with low peptide overlap between CVs) when the ΔCV was 15–20 V for both low(Figure S3C) and high-resolution (Figure S3D) MS2 fragmentation spectra, in keeping with previous studies.11 Taking a CV in the middle of the phosphopeptide distribution (CV = −65 V), we observed the maximum total peptides across two CVs at CV = −40/−75 V and CV = −45/−80 V for low- and high-resolution MS2 spectra, respectively. Furthermore, we observed that beyond a 10 V difference in CV, FAIMS peptide separation was commensurate with what we typically obtained with off-line fractionation7,31 (i.e., an intersection <10% unique peptides, Figure S3E). A direct comparison thereof was not performed here, but merits future consideration. Based on these data, we tested replicate triple-CV methods using (1) CV = −40/−60/−80, (2) CV = −45/− 65/−85, and (3) CV = −55/−65/−85 (Figure 1D). Combining triple-CV methods resulted in 30% more unique peptide identifications than HRMS2 analysis without FAIMS for the three peptide label chemistries tested (Figure 1D). Interestingly, for both the classic TMT and TMTpro chemistries, we observed no significant difference between the triple CV methods tested (p-value <0.05), suggesting that the low resolution FAIMS device enables these methods to capture a diverse population of phosphopeptides even with moderate shifts in the CV.
To explore further, we examined the proportions of peptides with a given charge state across CVs (Figure S2C). The charge state distributions remained relatively constant for SPS-MS3 (no FAIMS), HRMS2 (no FAIMS), as well as methods with 2 or 3 CVs. We observed differences in the charge state distribution in the single CV analyses, where the proportion of 2+ and 3+ ions increases with higher (less negative) CVs, as a result of FAIMS separation. Strikingly, we noted that the optimized FAIMS CVs (e.g., −80/−60/−40 V) for phosphoproteomic analysis were like those observed previously for TMT-tagged, but nonphosphorylated, peptides.11 These data (coupled to those highlighted in Figure S1) suggest that the TMT tag (whether classic TMT or TMTpro) may exert an equal or greater effect on the FAIMS stability of the observed peptides than the phosphorylation event. Furthermore, we observed that the stability of TMT-labeled phosphopeptides diverged from nonderivatized phosphopeptides reinforcing the need to optimize and validate methods specifically for labeled phosphopeptides.
Next, we examined peptide spectral match metrics to determine if FAIMS introduced any inherent biases in the phosphoproteomic analyses. The SEQUEST XCorr value was higher in the SPS-MS3 method which used CID-activation at the MS2 stage, as well as low resolution mass analysis (Figure S2D). However, the remaining methods which all used HCDactivation at the MS2 stage and high-resolution (Orbitrap) fragment analysis generated lower, but acceptable, XCorr values. We concluded that using FAIMS and different CVs (or combinations of CVs) did not influence the quality of the MS2 spectra. Our data also showed relatively constant observed mass-to-charge ratios for SPS-MS3 (no FAIMS), HRMS2 (no FAIMS), as well as methods with 2 or 3 CVs (Figure S2E). However, when scanning CVs from −10 V to −100 V, we observed a downward trend in peptide m/z as the CVs decreased (i.e., become more negative). Likewise, an identical pattern was observed for peptide length, as expected (Figure S2F). These data illustrated that CV-specific differences exist which may be used to identify specific sets of phosphopeptides. Similarly, we noted that deviations from HRMS2 regarding the distribution of m/z and peptide length were rectified by using multiple CVs in a single method. Specifically, methods with multiple CVs allowed for diverse populations of ions to be interrogated while increasing depth of phosphopeptide analysis.
We also tested if FAIMS biased phosphoproteomics for either of the most common methods of SPS-MS3-based phosphorylation analysis that use multistage activation-enhanced CID (MSA-CID, hereafter “MSA”)32 or lower energy HCD (NCE = 30–32) activation.7 We compared the number of unique peptides identified with these activation modalities in quadruplicate analyses using CV = −80/−60/− 40 V on an Orbitrap Fusion Lumos mass spectrometer (Figure 2A). We also tested the utility of phosphorylation-specific (PS) neutral loss ion fragments for improving peptide identification, which is an option available in the SEQUEST search engine (Figure S4A).33 CID activation without MSA resulted in the lowest number of phosphopeptide identifications while CID-MSA consistently generated the highest number of identifications. We observed an increase in the number of identified phosphopeptides when using phosphorylation-specific search with HCD or MSA data, as these methods are more likely to produce such fragments compared to standard CID.33 No effect was observed for fragmentation or search strategies for the nonphosphorylated peptides in the same runs (Figure S4A).
Figure 2.

Benchmarking the selection of optimal FAIMS compensation voltages (CV) for phosphoproteome analysis. A. Enriched-phosphopeptides from human cell lysate are TMT0-labeled, fragmented with different fragmentation schemes. B. Matrix of binary comparisons of the unions of phosphopeptides (total phosphopeptides identified from combining two runs) found in A. C. Intersection of unique phosphopeptides identified using either HCD or MSA fragmentation. Error bars: standard deviation. ANOVA, Tukey HSD: * p-value <0.05; ** p-value <0.01. D. Proteins extracted from mouse brain tissue, digested, enriched for phosphopeptides, and labeled with TMT. The data were collected with different methods (MSA or HCD, various CV combinations). Bar graphs illustrating the unique quantifiable phosphopeptides (unique, with summed TMT reporter ion signal-to-noise >200) with varying acquisition energy and compensation voltages (CV).
To analyze these data further, we compared the overlap among methods with different activation types and replicates there of (Figure 2A,B). Our findings show that the overlap was lower, and a modest increase in unique peptides was observed when MSA fragmentation is complemented with a second, separate analysis using HCD fragmentation as opposed to two back-to-back analyses with MSA (3−4% improvement, ANOVA, Tukey HSD). Moreover, for the murine phosphoproteome sample we observed an increase in the number of unique peptides and phosphopeptides when FAIMS-MS3 was used versus non-FAIMS-MS3 for both fragmentation strategies (Figure S4B). Therefore, as with the cell line study, FAIMS-MS3 improved the number of singly, doubly, and triply phosphorylated peptides compared to standard SPS-MS3.
We, and others, have shown the advantages of SPS-MS3 over HRMS2 for reducing ion interference-related ratio compression.19,20,34–37 Here, we used TMT-labeled phosphopeptides from a tryptic digest of mouse whole brain lysate to optimize FAIMS for SPS-MS3 data acquisition. Such samples are not as readily available as those from cell culture, but developing methods for effective isolation, enrichment, and analysis of primary tissue phosphoproteomes continues to be one of the most challenging for quantitative proteomic methods.21,26,27 We compared MSA and HCD as the MS2 collision type, with no FAIMS voltage and three different triple CVs methods on both the Orbitrap Fusion and Fusion Lumos mass spectrometers (Figure 2C). Marked improvement in the number of phosphopeptides was observed when the FAIMS electric field was applied, compared to without, for both instruments. As with the cell line data, we observed more quantifiable phosphopeptides when we employed FAIMS compared to the same analyses without applying FAIMS voltages (Figures 2D and S4C).
We observed a slight difference in the number of phosphopeptides between HCD and MSA-CID. Thus, we recommend that samples, such as those prepared using the “mini-phos” protocol,7 should be analyzed once by each method to ensure robust identification of unique phosphopeptides, if the sample amount permits.
CONCLUSIONS
We have presented FAIMS as a strategy to increase the depth of TMT-labeled phosphopeptide analysis. We present an optimized workflow using the recently released FAIMS-Pro coupled with a Tribrid mass spectrometer11 that provided consistent improvement in identified phosphopeptides for both cell line- and primary tissue-derived samples. Inclusion of two separate sample injections with different collision energy types (HCD and MSA-CID) further improved the diversity of phosphopeptide identifications. Concerning the choice of CVs, we obtained similar identification rates (within 10%) with different CV combinations so long as three CVs were used for multiplexed analyses. Thus, the potential exists that specific CV combinations, as well as our comparison of HCD versus CID-MSA activation, may vary on a given instrument and sample type, as displayed herein by differences in Fusion versus Lumos instruments and cell lysate versus brain tissue samples, respectively. Nonetheless, we have presented a template as a starting point that may be followed to best optimize a given system for multiplexed phosphoproteome analysis. Furthermore, different combinatorial CV methods across replicate injections may further increase phosphoproteome depth, as demonstrated by Pfammatter et al.13 Finally, to maximize the utility of both HCD- and MSA-based analyses, we recommend the inclusion of phosphorylation-specific ion searching to improve scoring and downstream site localization.33 In summary, we have introduced, assessed, and optimized FAIMS for TMT-labeled phosphoproteome analysis and have highlighted the advantages of FAIMS for increasing the analytical depth of multiplexed phosphoproteomics.
Supplementary Material
Supplemental Methods: Extended sample preparation and mass spectrometry data analysis. Figure S1: MS1 features across CVs for differentially derivatized peptide populations. Figure S2: Extended optimization of FAIMS for phosphorylation analysis. Figure S3: Determining the combination of compensation voltages (CV) resulting in the highest number of CV-exclusive peptides. Figure S4: Extended comparison of methods using HCD and MSA fragmentation for MS2 analysis. (PDF)
Table S1: Summary of data acquisition method parameters (XLSX)
Table S2: Tables of data arranged by figure (XLSX)
ACKNOWLEDGMENTS
We would like to thank members of the Gygi Lab at Harvard Medical School. This work was funded in part by an NIH/ NIDDK grant R01GM132129 (J.A.P.) and GM67945 (S.P.G).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.9b00759.
The authors declare no competing financial interest.
REFERENCES
- (1).Singh V; Ram M; Kumar R; Prasad R; Roy BK; Singh KK Phosphorylation: Implications in Cancer. Protein J. 2017, 36 (1), 1–6. [DOI] [PubMed] [Google Scholar]
- (2).Manning G; Whyte DB; Martinez R; Hunter T; Sudarsanam S The protein kinase complement of the human genome. Science 2002, 298 (5600), 1912–34. [DOI] [PubMed] [Google Scholar]
- (3).Sharma K; D’Souza RC; Tyanova S; Schaab C; Wisniewski JR; Cox J; Mann M Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014, 8 (5), 1583–94. [DOI] [PubMed] [Google Scholar]
- (4).Possemato AP; Paulo JA; Mulhern D; Guo A; Gygi SP; Beausoleil SA Multiplexed Phosphoproteomic Profiling Using Titanium Dioxide and Immunoaffinity Enrichments Reveals Complementary Phosphorylation Events. J. Proteome Res. 2017, 16 (4), 1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Hogrebe A; von Stechow L; Bekker-Jensen DB; Weinert BT; Kelstrup CD; Olsen JV Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9 (1), 1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rauniyar N; Yates JR 3rd Isobaric labeling-based relative quantification in shotgun proteomics. J. Proteome Res. 2014, 13 (12), 5293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Navarrete-Perea J; Yu Q; Gygi SP; Paulo JA Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res. 2018, 17 (6), 2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Paulo JA; Navarrete-Perea J; Erickson AR; Knott J; Gygi SP An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies. J. Am. Soc. Mass Spectrom. 2018, 29 (7), 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mertins P; Tang LC; Krug K; Clark DJ; Gritsenko MA; Chen L; Clauser KR; Clauss TR; Shah P; Gillette MA; Petyuk VA; Thomas SN; Mani DR; Mundt F; Moore RJ; Hu Y; Zhao R; Schnaubelt M; Keshishian H; Monroe ME; Zhang Z; Udeshi ND; Mani D; Davies SR; Townsend RR; Chan DW; Smith RD; Zhang H; Liu T; Carr SA Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography−mass spectrometry. Nat. Protoc. 2018, 13 (7), 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Huttlin EL; Jedrychowski MP; Elias JE; Goswami T; Rad R; Beausoleil SA; Villen J; Haas W; Sowa ME; Gygi SP A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143 (7), 1174–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Schweppe DK; Prasad S; Belford MW; Navarrete-Perea J; Bailey DJ; Huguet R; Jedrychowski MP; Rad R; McAlister G; Abbatiello SE; Woulters ER; Zabrouskov V; Dunyach JJ; Paulo JA; Gygi SP Characterization and Optimization of Multiplexed Quantitative Analyses Using High-Field Asymmetric-Waveform Ion Mobility Mass Spectrometry. Anal. Chem. 2019, 91 (6), 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pfammatter S; Bonneil E; McManus FP; Thibault P Accurate Quantitative Proteomic Analyses Using Metabolic Labeling and High Field Asymmetric Waveform Ion Mobility Spectrometry (FAIMS). J. Proteome Res. 2019, 18 (5), 2129–2138. [DOI] [PubMed] [Google Scholar]
- (13).Pfammatter S; Bonneil E; McManus FP; Prasad S; Bailey DJ; Belford M; Dunyach JJ; Thibault P A Novel Differential Ion Mobility Device Expands the Depth of Proteome Coverage and the Sensitivity of Multiplex Proteomic Measurements. Mol. Cell. Proteomics 2018, 17 (10), 2051–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Hebert AS; Prasad S; Belford MW; Bailey DJ; McAlister GC; Abbatiello SE; Huguet R; Wouters ER; Dunyach JJ; Brademan DR; Westphall MS; Coon JJ Comprehensive Single-Shot Proteomics with FAIMS on a Hybrid Orbitrap Mass Spectrometer. Anal. Chem. 2018, 90 (15), 9529–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Prasad S; Belford MW; Dunyach JJ; Purves RW On an aerodynamic mechanism to enhance ion transmission and sensitivity of FAIMS for nano-electrospray ionization-mass spectrometry. J. Am. Soc. Mass Spectrom. 2014, 25 (12), 2143–53. [DOI] [PubMed] [Google Scholar]
- (16).Purves RW; Prasad S; Belford M; Vandenberg A; Dunyach JJ Optimization of a New Aerodynamic Cylindrical FAIMS Device for Small Molecule Analysis. J. Am. Soc. Mass Spectrom. 2017, 28 (3), 525–538. [DOI] [PubMed] [Google Scholar]
- (17).Saba J; Bonneil E; Pomies C; Eng K; Thibault P Enhanced sensitivity in proteomics experiments using FAIMS coupled with a hybrid linear ion trap/Orbitrap mass spectrometer. J. Proteome Res. 2009, 8 (7), 3355–66. [DOI] [PubMed] [Google Scholar]
- (18).Bonneil E; Pfammatter S; Thibault P Enhancement of mass spectrometry performance for proteomic analyses using high-field asymmetric waveform ion mobility spectrometry (FAIMS). J. Mass Spectrom. 2015, 50 (11), 1181–95. [DOI] [PubMed] [Google Scholar]
- (19).Gygi JP; Yu Q; Navarrete-Perea J; Rad R; Gygi SP; Paulo JA Web-Based Search Tool for Visualizing Instrument Performance Using the Triple Knockout (TKO) Proteome Standard. J. Proteome Res. 2019, 18 (2), 687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Paulo JA; O’Connell JD; Gygi SP A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass Spectrom. 2016, 27 (10), 1620–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Paulo JA; McAllister FE; Everley RA; Beausoleil SA; Banks AS; Gygi SP Effects of MEK inhibitors GSK1120212 and PD0325901 in vivo using 10-plex quantitative proteomics and phosphoproteomics. Proteomics 2015, 15 (2−3), 462–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Beausoleil SA; Villen J; Gerber SA; Rush J; Gygi SP A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24 (10), 1285–92. [DOI] [PubMed] [Google Scholar]
- (23).Elias JE; Gygi SP Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Elias JE; Gygi SP Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4 (3), 207–14. [DOI] [PubMed] [Google Scholar]
- (25).Perez-Riverol Y; Csordas A; Bai J; Bernal-Llinares M; Hewapathirana S; Kundu DJ; Inuganti A; Griss J; Mayer G; Eisenacher M; Perez E; Uszkoreit J; Pfeuffer J; Sachsenberg T; Yilmaz S; Tiwary S; Cox J; Audain E; Walzer M; Jarnuczak AF; Ternent T; Brazma A; Vizcaino JA The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019, 47 (D1), D442–D450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Mertins P; Tang LC; Krug K; Clark DJ; Gritsenko MA; Chen L; Clauser KR; Clauss TR; Shah P; Gillette MA; Petyuk VA; Thomas SN; Mani DR; Mundt F; Moore RJ; Hu Y; Zhao R; Schnaubelt M; Keshishian H; Monroe ME; Zhang Z; Udeshi ND; Mani D; Davies SR; Townsend RR; Chan DW; Smith RD; Zhang H; Liu T; Carr SA Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc. 2018, 13 (7), 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Schweppe DK; Rigas JR; Gerber SA Quantitative phosphoproteomic profiling of human non-small cell lung cancer tumors. J. Proteomics 2013, 91, 286–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhao H; Cunningham DL; Creese AJ; Heath JK; Cooper HJ FAIMS and Phosphoproteomics of Fibroblast Growth Factor Signaling: Enhanced Identification of Multiply Phosphorylated Peptides. J. Proteome Res. 2015, 14 (12), 5077–87. [DOI] [PubMed] [Google Scholar]
- (29).Humphrey P; Hassell K; Blackburn M; Belford M; Volny M; Peterman S; Huguet R Developing the Research to Routine Workflows with FAIMS: Automating Large-scale SRM Method Creation for Routine HeLa Peptide Screening In Annual Congress in Clinical Mass Spectrometry; Palm Springs, CA, 2019. [Google Scholar]
- (30).Lex A; Gehlenborg N; Strobelt H; Vuillemot R; Pfister H UpSet: Visualization of Intersecting Sets. IEEE Trans Vis Comput. Graph 2014, 20 (12), 1983–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Paulo JA; O’Connell JD; Everley RA; O’Brien J; Gygi MA; Gygi SP Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J. Proteomics 2016, 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Schroeder MJ; Shabanowitz J; Schwartz JC; Hunt DF; Coon JJ A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal. Chem. 2004, 76 (13), 3590–8. [DOI] [PubMed] [Google Scholar]
- (33).Payne SH; Yau M; Smolka MB; Tanner S; Zhou H; Bafna V Phosphorylation-specific MS/MS scoring for rapid and accurate phosphoproteome analysis. J. Proteome Res. 2008, 7 (8), 3373–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Paulo JA; Navarrete-Perea J; Guha Thakurta S; Gygi SP TKO6: A Peptide Standard To Assess Interference for Unit-Resolved Isobaric Labeling Platforms. J. Proteome Res. 2018, 18 (1), 565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Muntel J; Kirkpatrick J; Bruderer R; Huang T; Vitek O; Ori A; Reiter L Comparison of Protein Quantification in a Complex Background by DIA and TMT Workflows with Fixed Instrument Time. J. Proteome Res. 2019, 18 (3), 1340–1351. [DOI] [PubMed] [Google Scholar]
- (36).Savitski MM; Mathieson T; Zinn N; Sweetman G; Doce C; Becher I; Pachl F; Kuster B; Bantscheff M Measuring and managing ratio compression for accurate iTRAQ/TMT quantification. J. Proteome Res. 2013, 12 (8), 3586–98. [DOI] [PubMed] [Google Scholar]
- (37).Dayon L; Sonderegger B; Kussmann M Combination of gasphase fractionation and MS(3) acquisition modes for relative protein quantification with isobaric tagging. J. Proteome Res. 2012, 11 (10), 5081–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods: Extended sample preparation and mass spectrometry data analysis. Figure S1: MS1 features across CVs for differentially derivatized peptide populations. Figure S2: Extended optimization of FAIMS for phosphorylation analysis. Figure S3: Determining the combination of compensation voltages (CV) resulting in the highest number of CV-exclusive peptides. Figure S4: Extended comparison of methods using HCD and MSA fragmentation for MS2 analysis. (PDF)
Table S1: Summary of data acquisition method parameters (XLSX)
Table S2: Tables of data arranged by figure (XLSX)