

Abstract
The duplication of the eukaryotic genome is an intricate process that has to be tightly safe‐guarded. One of the most frequently occurring errors during DNA synthesis is the mis‐insertion of a ribonucleotide instead of a deoxyribonucleotide. Ribonucleotide excision repair (RER) is initiated by RNase H2 and results in error‐free removal of such mis‐incorporated ribonucleotides. If left unrepaired, DNA‐embedded ribonucleotides result in a variety of alterations within chromosomal DNA, which ultimately lead to genome instability. Here, we review how genomic ribonucleotides lead to chromosomal aberrations and discuss how the tight regulation of RER timing may be important for preventing unwanted DNA damage. We describe the structural impact of unrepaired ribonucleotides on DNA and chromatin and comment on the potential consequences for cellular fitness. In the context of the molecular mechanisms associated with faulty RER, we have placed an emphasis on how and why increased levels of genomic ribonucleotides are associated with severe autoimmune syndromes, neuropathology, and cancer. In addition, we discuss therapeutic directions that could be followed for pathologies associated with defective removal of ribonucleotides from double‐stranded DNA.
Keywords: DNA repair, ribonucleotide excision repair, RNA–DNA hybrid, RNase H2, topoisomerase 1
Subject Categories: DNA Replication, Repair & Recombination
Kellner and Luke review how RNase H2 or topoisomerase enzymes remove misincorporated ribonucleotides from genomic DNA, and the relevance of such mechanisms for both malignant and autoimmune diseases such as Aicardi–Goutières syndrome.
Glossary
- AGS
Aicardi–Goutières syndrome
- AOA1
Ataxia with oculomotor apraxia 1
- dNTP
deoxyribonucleotide triphosphate
- DSB
double‐strand break
- GCR
gross chromosomal rearrangements
- HDR
homology‐directed repair
- HR
homologous recombination
- INF
interferon
- LOH
loss of heterozygosity
- MMR
mismatch repair
- NER
nucleotide excision repair
- NHEJ
nonhomologous end‐joining
- PARP
(poly‐ADP)ribose polymerase
- PIP‐box
PCNA‐interacting motif
- RER
ribonucleotide excision repair
- RNAPII
RNA polymerase II
- RNase H
ribonuclease H
- rNMP
ribonucleoside monophosphate
- rNTP
ribonucleotide triphosphate
- SLE
systemic lupus erythematosus
- SSB
single‐strand break
- ssDNA
single‐stranded DNA
- Top1cc
topoisomerase 1 cleavage complex
- Top1
topoisomerase 1
Introduction
It is of critical importance that the duplication of human genomes is a tightly regulated and safe‐guarded process. The insertion of erroneous bases by replicative DNA polymerases can potentially manifest into heritable mutations with pathological consequences. Considerable emphasis has been placed on understanding the causes, consequences, and repair of faulty base incorporation, which leads to DNA mismatches. In some instances, the replicative polymerases still get it wrong despite the correct base being inserted and a Watson–Crick basepair being formed; this can occur when the inappropriate sugar moiety attached to the base is selected.
The replication of DNA utilizes deoxyribonucleotide triphosphates (dNTPs) to faithfully duplicate the genome. The synthesis of RNA via transcription, on the other hand, employs ribonucleotide triphosphates (rNTP), where the 2′‐carbon atom of the ribose sugar is hydroxylated. Intracellular concentrations of rNTPs highly exceed those of dNTPs (between 30‐ and 200‐fold in budding yeast depending on the base) (Nick McElhinny et al, 2010b), thereby increasing the likelihood that rNTPs are mistakenly used instead of dNTPs, despite the presence of the correct base attached. The three replicative DNA polymerases (Pol α, Pol ε, and Pol δ) harbor a highly conserved tyrosine residue adjacent to the active polymerization site that acts as “steric gate” to limit rNTP incorporation during DNA replication, by excluding the hydroxy group of the ribose (Brown & Suo, 2011). rNTP exclusion via the steric gate is however not flawless, and in vitro studies using endogenous concentrations of rNTPs and dNTPs suggest that in the Saccharomyces cerevisiae genome, approximately 13,000 ribonucleotides become incorporated into newly replicated DNA within a single round of replication (Nick McElhinny et al, 2010b). Similar frequencies of rNTP incorporation have been confirmed in vivo, resulting in an incorporation rate of approximately one rNMP per 6,500 bases (Lujan et al, 2013). In addition to rNTP mis‐incorporation, Pol α/Primase synthesizes short RNA primers to initiate Okazaki fragments that transiently make up ~ 5% of the nascent lagging strand (Zheng & Shen, 2011). Accordingly, in human cells the number of incorporated ribonucleotides per cell cycle is estimated to reach more than one million (Clausen et al, 2013). This makes ribonucleotides the most frequently incorporated non‐canonical nucleotide in duplex DNA, exceeding the combined total number of all abasic, oxidized, and otherwise modified nucleotides (Caldecott, 2014).
Once the rNTP is incorporated into the context of DNA, it exists as a ribonucleoside monophosphate (rNMP). The presence of rNMPs in a DNA template directly affects the processivity of DNA polymerization during semi‐conservative replication. Although budding yeast replicative polymerases can bypass a single rNMP, the in vitro efficiency of this bypass is strongly reduced (for Pol ε only about 66% bypass is achieved) and drops dramatically when replication has to transverse stretches of three or more consecutive rNMPs (Watt et al, 2011). As of yet, there is no direct in vivo evidence that DNA polymerases are affected by stretches of multiple rNMPs. Comparable in vitro observations have been made for the human replicative polymerases, Pol ε and Pol δ (Göksenin et al, 2012; Clausen et al, 2013). Therefore, the accumulation of rNMPs in genomic DNA likely induces replication stress and DNA damage signaling from yeast to human (Nick McElhinny et al, 2010a; Hiller et al, 2012; Lazzaro et al, 2012; Williams et al, 2013; Pizzi et al, 2015; Zimmermann et al, 2018) (discussed below). Thus, to preserve genome stability, rNMPs have to be efficiently removed from the genome. A dedicated repair pathway, referred to as ribonucleotide excision repair (RER), employs specialized enzymes to eliminate rNMPs.
Error‐free and error‐prone removal of ribonucleotides
DNA polymerases have a built‐in proofreading mechanism whereby faulty base incorporation is corrected through exonuclease‐mediated removal of the incorrect base (Burgers & Kunkel, 2017). Recognition and excision of rNMPs by DNA polymerases is only one‐third as effective as their proofreading of incorrect dNMP base pairing and in the case of Pol ε has been found to most likely not significantly contribute to rNMP removal (Shcherbakova et al, 2003; Williams et al, 2012). Instead, RNase H enzymes have the capacity to cleave DNA at sites of rNMP incorporation and thus to initiate rNMP removal. RNase H is conserved in prokaryotes, but we will not cover bacterial RNase H enzymes here and instead refer interested readers to a study by Kochiwa et al (2007) for an overview on them. The eukaryotic RNase H family consists of the monomeric RNase H1 enzymes and the trimeric RNase H2 enzymes, both of which eliminate RNA‐DNA hybrid structures occurring throughout the genome (Cerritelli & Crouch, 2009). While the accidentally incorporated rNMPs are found both as single bases and in longer consecutive stretches, RNA–DNA hybrids can also form when single‐stranded RNA molecules anneal to a complementary DNA strand, thereby displacing the second strand of the DNA double helix. This three‐stranded structure, termed R‐loop, can have detrimental consequences on genome stability if not removed in a timely manner (Santos‐Pereira & Aguilera, 2015). RNase H1 requires at least four consecutive rNMPs to recognize an RNA–DNA hybrid structure, a situation that occurs in the context of an R‐loop. RNase H2, on the other hand, can act both on rNMP stretches such as those found in R‐loops, as well as on single and consecutive rNMPs in the context of double‐stranded DNA, making it a more versatile enzyme. Consistently, RNase H2 activity accounts for the bulk of RNase H activity in the cell (Sparks et al, 2012). Given the enzymatic capabilities of RNase H2, one may expect the existence of tight regulatory mechanisms in order to prevent chromosomal nicking at inappropriate times, e.g., during DNA replication.
In S. cerevisiae, the trimeric RNase H2 enzyme consists of the catalytic subunit Rnh201 and the accessory subunits Rnh202 and Rnh203. Human RNase H2 shows strong conservation and comprises RNASEH2A, the catalytic subunit, as well as RNASEH2B and RNASEH2C (Crow et al, 2006). Loss of any of its subunits renders the enzyme complex inactive (Jeong et al, 2004). The dual activity of RNase H2 toward R‐loops and rNMPs can be largely, but not entirely, uncoupled by the use of a separation‐of‐function allele of the catalytic Rnh201 subunit, RNH201‐P45D‐Y219A (or RNH201‐RED for ribonucleotide excision defective). This point mutation within the substrate‐interacting pocket completely abolishes activity of RNase H2 toward single rNMPs but retains approximately 40% of wildtype enzymatic activity toward longer rNMP stretches and R‐loops (Chon et al, 2013). Although this allele was originally constructed in yeast, it has since been recapitulated in human and mouse cells, resulting in a similar separation‐of‐function phenotype (Pizzi et al, 2015; Uehara et al, 2018; Zimmermann et al, 2018).
RNase H2 promotes error‐free RER
RNase H2 is responsible for the primary pathway of rNMP removal from genomic DNA, i.e., error‐free RER. Upon recognizing an rNMP in the context of duplex DNA, RNase H2 incises the DNA backbone on the 5′ side of the ribonucleotide to allow its subsequent removal and repair (Eder et al, 1993; Rydberg & Game, 2002; Fig 1). In vitro reconstitution experiments have elucidated the RER mechanism in detail (Sparks et al, 2012): Initial incision at the DNA–RNA junction mediated by RNase H2 produces a single‐stranded DNA break flanked by a 3′ hydroxy (3′OH) group and a 5′ phosphate. Starting from the 3′OH, Pol δ or (less efficiently) Pol ε perform strand displacement DNA synthesis, thereby creating a flap structure harboring the rNMP (Fig 1). This flap is subsequently removed by flap endonuclease (yeast Rad27/human FEN1) or the exonuclease Exo1. Finally, the remaining single‐stranded nick is sealed by DNA ligase (Sparks et al, 2012). Crystal structures of bacterial, mouse, and human RNase H2 enzymes have allowed to further dissect their substrate recognition, binding, and hydrolysis mechanisms (Rychlik et al, 2010; Shaban et al, 2010; Figiel et al, 2011): RNase H2 recognizes rNMPs at the (5′)RNA–DNA(3′) junction. The 5′‐phosphate of the rNMP is positioned into the active site of the complex, while its 2′OH interacts with a glycine–arginine–glycine (GRG) motif and a conserved tyrosine residue within the catalytic subunit, thereby improving substrate selectivity (Rychlik et al, 2010). The catalytic step takes place in the active site consisting of four conserved carboxylates, which coordinate metal ions and water molecules to attack the phosphate bond 5′ of the rNMP (Rychlik et al, 2010; Shaban et al, 2010).
Figure 1. Overview of ribonucleotide excision repair (RER).
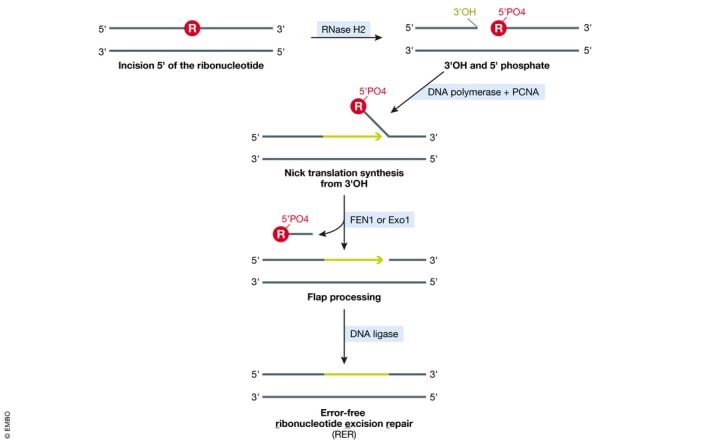
RNase H2 initiates RER by incising the DNA backbone at the rNMP (R in red circle). Nick translation DNA synthesis from the newly created 3′OH followed by FEN1/Exo1‐mediated flap processing and subsequent DNA ligation can efficiently repair the incised DNA, resulting in removal of the rNMP.
Error‐prone rNMP removal by Top1 in the absence of RNase H2
Prior to characterization of the RNase H2‐based RER mechanism in such intricate detail, in vitro work had demonstrated that topoisomerase 1 (Top1) can also process an rNMP‐containing DNA substrate (Sekiguchi & Shuman, 1997). More recently, this Top1‐dependent mechanism was also shown to remove rNMPs from DNA in vivo and thus to represent an important backup mechanism in RER‐defective cells lacking RNase H2 activity (Williams et al, 2013). In this reaction, the catalytic tyrosine residue of Top1 forms a Top1 cleavage complex (Top1cc), i.e., a covalent intermediate via transesterification at the 3′ terminal phosphate of the rNMP, in a similar manner to the reaction that Top1 initiates on pure supercoiled DNA lacking rNMPs. During the latter, canonical reaction, Top1 can readily catalyze religation of the created nick; when the nick occurs at an rNMP, however, the Top1‐phosphate bond is prone to attack from the adjacent 2′OH group of the ribose moiety, resulting in Top1 release and creation of an unligatable 2′,3′‐cyclic phosphate (Sekiguchi & Shuman, 1997; Fig 2A). The resulting nick flanked by the 2′,3′‐cyclic phosphate and a 5′OH group requires further processing before either ligation or extension is possible. Although Top1 cleavage can achieve error‐free repair of rNMPs, other more detrimental repair alternatives exist (Fig 2). In fact, Top1‐mediated processing of rNMPs greatly contributes to genome instability in the absence of RNase H2 (discussed below).
Figure 2. Topoisomerase 1 as backup for RER in rNMP removal from the genome.
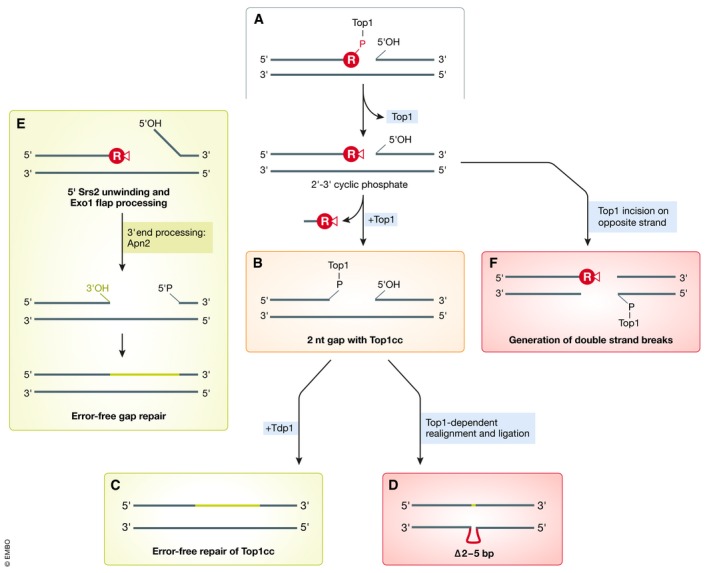
(A) In the absence of functional RNase H2, Top1 can act on accumulating rNMPs. Different outcomes have been characterized in budding yeast (see text for detailed descriptions), resulting either in error‐free repair or in repair that causes mutations or potentially lethal double‐strand breaks. (B–D) Secondary Top1‐mediated incision two basepairs upstream releases an rNMP‐dNMP dinucleotide and creates a Top1‐linked gap (B) that can be processed in an error‐free manner via Tdp1 (C), or in an error‐prone manner caused by Top1 realignment and religation (D). (E) Error‐free gap repair based on subsequent activities of Srs2 helicase, Exo1 exonuclease, and Apn2 abasic endonuclease, which prevent erroneous religation. (F) Secondary Top1 incision on the opposite strand creates DNA double‐strand breaks that require repair by homologous recombination.
Top1‐mediated removal has been described to be specific for rNMPs incorporated by the leading‐strand DNA polymerase Pol ε, and it appears to be resolved in different ways (Williams et al, 2013, 2015; Cho et al, 2015). In one scenario, Top1 can initiate a second cut on a dNMP two basepairs upstream of the initial cut, leading to the release of an rNMP‐dNMP dinucleotide (Sparks & Burgers, 2015; Fig 2B). In this case, the covalently bound Top1cc is processed and released by tyrosyl‐DNA phosphodiesterase Tdp1, leaving behind a two‐nucleotide gap that can be repaired in an error‐free manner (Fig 2C). As an alternative to this Tdp1‐dependent pathway, and especially within tandem repeat sequences, Top1 may realign the DNA backbone and ligate the nick (Huang et al, 2015; Sparks & Burgers, 2015). In this scenario, ligation by Top1 leads to characteristic slippage mutations consisting of two‐ to five‐basepair deletions (∆2–5 bp) (Nick McElhinny et al, 2010a; Kim et al, 2011; Fig 2D). Processing of the initial Top1‐created 5′OH via Srs2 helicase and Exo1 nuclease can disfavor direct religation, thereby reducing the risk of acquiring those ∆2–5 bp slippage mutations (Potenski et al, 2014). Here, the 3′–5′ helicase activity of Srs2 unwinds the DNA from this free 5′ end, followed by flap processing via Exo1. While creation of this DNA gap reduces the likelihood of Top1‐mediated ligation following a second Top1 incision, the 3′ DNA end still has to be processed to allow extension by Pol δ. Biochemical in vitro data supported by genetic interaction studies indicate that the abasic endonuclease Apn2 can process the 3′‐terminal 2′,3′‐cyclic phosphate and promote Pol δ extension (Li et al, 2019; Fig 2E).
Yet another scenario related to yeast Top1 activity at rNMP sites has been described: Following the first incision at the rNMP, Top1 can also cut on the DNA strand opposing the rNMP to create double‐strand breaks (DSBs), which then rely on homology‐directed repair (HDR) via Rad51 and Rad52 (Huang et al, 2015; Fig 2F). Consistently, RNase H2 loss in human cells results in synthetic lethality with the absence of either BRCA1 or BRCA2 HDR factors, highlighting the importance of HDR under conditions of rNMP accumulation (Zimmermann et al, 2018).
While rNMP removal via topoisomerase had initially only been described in S. cerevisiae, recent work employing various RER‐defective RNase H2‐mutant human cell lines has demonstrated that also human TOP1 can recognize an unrepaired rNMP and incise to create a DNA nick with the potential to compromise genome stability (Zimmermann et al, 2018). Whether TOP1 activity indeed removes rNMPs from human DNA as it does in yeast remains to be tested. In particular, it will be interesting to determine how Top1 recognizes rNMPs in the DNA, i.e., is Top1‐mediated rNMP processing a regulated process, or rather an accidental byproduct of Top1 action in relieving supercoiling? Another outstanding question is whether or not Top1 contributes to RER in the presence of RNase H2, and if so to what extent. Moreover, what determines which Top1 repair pathway (nicking out the rNMP, nicking and re‐ligating or DSB generation) will be employed at an rNMP? The latter question is particularly relevant in the context of RER‐defective cells, where the different Top1‐dependent processing mechanisms will have very different outcomes in terms of preserving genome integrity.
Alternative removal mechanisms and tolerance of rNMPs
Both yeast and mammalian cells lacking RNase H2 display signs of replication stress. In yeast mutants, the S‐phase checkpoint and the postreplicative repair pathway are constitutively activated, and accordingly, cells exhibit delayed cell cycle progression (Nick McElhinny et al, 2010a; Lazzaro et al, 2012; Williams et al, 2013; Zimmermann et al, 2018). The same defects are observed in RNase H2‐depleted human cells and cells from patients suffering from Aicardi–Goutières syndrome (AGS) associated with mutations in RNase H2 (discussed below) (Pizzi et al, 2015). Similarly, loss of RNase H2 in mouse cells results in altered cell cycle timing with accumulation of cells in G2/M phase, chronic activation of the DNA damage response, increase in single‐strand breaks (SSBs), and increased nuclear foci harboring the phosphorylated histone variant H2AX (γH2AX) (Hiller et al, 2012). In bacteria, nucleotide excision repair (NER) can serve as backup for RER (Cai et al, 2014), but a NER contribution to rNMP removal in yeast and human cells has been largely ruled out (Lazzaro et al, 2012; Lindsey‐Boltz et al, 2015). Mismatch repair (MMR), which is very efficient in recognizing and removing mismatched bases from dsDNA, also does not appear to contribute to rNMP removal in yeast (Lazzaro et al, 2012). Of note, RNase H2‐mediated incision at rNMPs has conversely been assigned a guiding role during MMR: As rNMPs are transiently inserted during DNA replication, nicks subsequently created by RNase H2 serve as a guide for strand determination and ensure that the MMR machinery specifically removes mismatches on the newly synthesized strand discriminated by the nicks (Ghodgaonkar et al, 2013; Lujan et al, 2013). Therefore, RNase H2 defects may increase the mutagenic load also in an indirect manner, by decreasing the efficiency of MMR.
Of note, the lethality of combined absence of both RNase H enzymes and Top1 in yeast (rnh1Δ rnh201Δ top1Δ) can be bypassed by expression of the RER‐deficient, but R‐loop processing‐proficient RNH201‐RED allele (Chon et al, 2013). This indicates that lethality in this strain is likely mediated by an accumulation of toxic R‐loops and implies that yeast cells can in principle tolerate the rNMPs that accumulate in the genome when both RER and Top1‐dependent repair are compromised. Therefore, either a yet unknown backup pathway for rNMP removal might exist, or yeast cells can survive high numbers of rNMPs in their genome despite a high load of DNA damage and replication stress. In cells that lack both RNase H1 and RNase H2, the postreplicative repair pathway has been found to be crucial (Lazzaro et al, 2012), but it has not been addressed to which extent this is due to either R‐loops or rNMPs, leaving the involvement of this pathway in the bypass of lethality allowed by the RNH201‐RED allele an interesting hypothesis to test. Another possibility to consider is that stretches of consecutive ribonucleotides, and not R‐loops, are responsible for the phenotypes observed in rnh1Δ rnh201Δ cells. This would also be consistent with a genetic rescue by the RNH201‐RED allele. Recent work has demonstrated that translesion polymerase eta (Pol η) can incorporate consecutive rNMP stretches at stalled replication forks in the presence of HU (Meroni et al, 2019). Consistently, the deletion of Pol η rescues the HU sensitivity of rnh1Δ rnh201Δ cells.
Given the importance of RER for maintaining genome integrity, it is surprising that DNA polymerases have evolved to permit rNTP usage at all. This may suggest that, pending their timely and controlled removal, rNMPs may also exert beneficial functions in certain situations (Potenski & Klein, 2014). As discussed above, MMR on the nascent leading strand is facilitated by RNase H2‐induced nicks at rNMPs (Ghodgaonkar et al, 2013; Lujan et al, 2013), and it is an intriguing possibility that other processes could be similarly affected by rNMPs incorporated by replicative DNA polymerases. Another source of rNMP incorporation with a beneficial role is in nonhomologous end‐joining (NHEJ) repair, where a critical role is played by DNA polymerase mu (Pol μ), which displays even weaker sugar selectivity than replicative DNA polymerases (Potenski & Klein, 2014). Indeed, transient incorporation of rNMPs at broken DNA ends effectively enhances their subsequent ligation (Pryor et al, 2018).
RER regulation
Though many aspects of the RER reaction have been well‐described, some rather fundamental aspects of the error‐free removal of rNMPs still require further investigation. The C‐terminal region of Rnh202 (hRNASEH2B) harbors a PCNA‐interacting peptide motif (PIP‐box) suggesting replisome association, the importance of which still remains unclear. While deletion of the PIP‐box affects localization of RNase H2 to sites of PCNA‐dependent DNA replication in human cells (Bubeck et al, 2011), an RNase H2 complex lacking the PIP‐box (RNASEH2B‐∆PIP) still retains residual co‐localization with PCNA (Kind et al, 2014). In budding yeast, Rnh202‐∆PIP‐mutant cells grow indistinguishably from cells with wildtype Rnh202, suggesting that recognition of the rNMP rather than interaction with PCNA is crucial for RER to function (Chon et al, 2013). One possibility is that, especially in human cells, PCNA might reinforce retention of RNase H2 at sites of DNA replication or repair synthesis but is not required for its recruitment per se. This is reminiscent of the recruitment mechanism reported for DNA methyltransferase Dnmt1, where ablation of the PIP‐box is compensated by direct interaction between the Dnmt1‐targeting sequence and DNA (Schneider et al, 2013), or the dual recruitment of poly(ADP‐ribose) glycohydrolase PARG through both its substrate poly(ADP‐ribose) (PAR) and its interaction with PCNA (Mortusewicz et al, 2011). Another simple interpretation of these results would be that RNase H2 interacts with additional replisome components other than PCNA, which too may assist RNase H2 delivery to rNMPs. Alternatively, RNase H2 may recruit PCNA and other repair factors to rNMPs to promote RER, instead of the other way around. In any case, the significance of the PIP‐box within the RNase H2 complex requires clarification.
While RNH201 mRNA expression peaks twice during the yeast cell cycle, during S phase and again during G2/M phase (Arudchandran et al, 2000), the protein accumulates progressively from G1 (where expression is very low) through to M phase (Lockhart et al, 2019). All other, non‐catalytic RNase H2 subunit proteins are constitutively expressed throughout the cell cycle, and the complex resides exclusively within the nuclear compartment (Reijns et al, 2012; Lockhart et al, 2019). Thus, RER could theoretically be initiated at any given time in the cell cycle. However, it is conceivable that RER may somehow be temporally regulated, or even coordinated with other DNA repair activities, in order to avoid untimely DNA nick generation and repair synthesis. Although the PIP‐box presence in RNase H2 may imply RER coupling to DNA replication, other DNA metabolism and repair processes (such as Okazaki fragment maturation and postreplicative repair) have been found to be postponable until late S/G2 phase without compromising their efficiency (Daigaku et al, 2010; Karras & Jentsch, 2010; Kahli et al, 2019). While RNase H2 did not localize to particular genomic regions in chromatin immunoprecipitations from asynchronously growing yeast cells (Zimmer & Koshland, 2016), it could be cross‐linked to telomeres in cells synchronized late in S phase, at times when the bulk of genomic DNA has been replicated (Graf et al, 2017). Consistently, upon fractionation of cell lysates, RNase H2 is more prominently chromatin‐associated in G2/M than in S phase, but to a lesser extent in G1 (Lockhart et al, 2019), suggesting that postreplicative chromatin association of RNase H2 may be a more general feature and not just restricted to telomeres. The same work further demonstrated that G2 phase‐restricted RNase H2 expression is sufficient to allow its functions both in R‐loop removal and RER; while restricting expression to S phase in fact causes defects in R‐loop processing as well as RER‐related toxicity (Lockhart et al, 2019). It may thus be crucial to limit the peak of RNase H2 activity to a postreplicative period, possibly to minimize generation of DNA double‐strand breaks arising from encounters of an oncoming replication fork with RNase H2‐induced nicks during S phase. Although risky, RNase H2 activity during S phase does likely still exist and may represent a tolerance pathway for dealing with rNMPs that have not been efficiently removed during the previous cell cycle, and for preventing Top1‐mediated genome instability.
Follow‐up studies to understand the mechanistic basis of RNase H2 cell cycle regulation will be an important next step. Chromatin localization could potentially be regulated by posttranslational modifications of RNase H2 or a cell cycle‐regulated RNase H2 inhibitory protein. With respect to the former, a yeast phosphoproteomics screen (Bodenmiller et al, 2010) yielded several phosphopeptides for Rnh202, incidentally the same RNase H2 subunit that also harbors the PIP‐box. The identification and characterization of RNase H2 posttranslational modifications could give valuable insights into the regulation of RER activity. In addition, cell cycle‐specific chromatin modifications could act as a recruitment signal for RNase H2 and need to be evaluated in this context (Fig 3).
Figure 3. Regulation of RER through RNase H2.
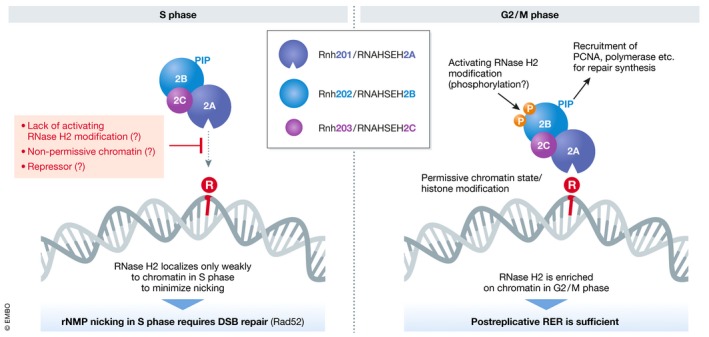
RNase H2 chromatin localization gradually increases throughout S phase but its activity may be kept in check to prevent creation of nicks during replication, where they would be converted into one‐ended DSBs by oncoming replication forks. Different regulatory mechanisms (in red) might account for RNase H2 inactivity during S phase. In G2/M, RNase H2 actively processes rNMPs to achieve successful RER.
Unrepaired rNMPs affect genome stability
Ribonucleotides that permanently reside in the genome can have detrimental consequences on genome stability. Due to the free 2′OH group of the ribose moiety, rNMPs are highly susceptible to spontaneous hydrolysis, thus creating genotoxic single‐stranded breaks in the DNA backbone (Li & Breaker, 1999). In yeast, the absence of RNase H2 increases spontaneous mutation rates as well as gene conversion events (Huang et al, 2003; Ii et al, 2011). Moreover, rNMP‐dependent gross chromosomal rearrangements (GCRs) are observed when homologous recombination (HR)‐mediated repair is compromised (Allen‐Soltero et al, 2014). The most characteristic type of mutation upon loss of RER manifests in short deletions in repetitive sequences, mostly between two and five basepairs in length (Nick McElhinny et al, 2010a). This mutational signature is generated by Top1 incision at rNMPs followed by faulty Top1‐dependent alignment of the nicked DNA strand and religation (see above) (Kim et al, 2011; Williams et al, 2017). Increased mutation rates are similarly Top1‐dependent (Potenski et al, 2014). Loss of RNase H2 activity in diploid yeast cells has been shown to increase both nonallelic HR and loss‐of‐heterozygosity (LOH) events (Conover et al, 2015; Zimmer & Koshland, 2016; Cornelio et al, 2017). Although the latter study attributed LOH largely to R‐loop misregulation (Zimmer & Koshland, 2016), parallel work found LOH and nonallelic HR increased due to Top1‐dependent processing of rNMPs in RER‐defective cells (Conover et al, 2015). Follow‐up work employing the RNH201‐RED separation‐of‐function allele however concluded that both the RER and the R‐loop removal function of RNase H2 contribute to genome stability in yeast (Cornelio et al, 2017). Similarly, the recent findings (discussed above) that TOP1 depletion in RNase H2‐deficient human cells reduces γH2AX‐marked damage foci and PARP inhibitor‐induced apoptosis to wildtype levels and relieves the S‐phase arrest (Zimmermann et al, 2018) indicate that compromised RER enhances TOP1‐dependent genome instability also in human cells.
Unrepaired rNMPs affect DNA structure
Many lines of in vitro evidence suggest that the presence of unrepaired rNMPs in the context of double‐stranded DNA affects the structure of the surrounding bases. Early studies analyzed short DNA molecules containing a single ribonucleotide by X‐ray diffraction (Ban et al, 1994; Egli et al, 1993; Wahl & Sundaralingam, 2000) or nuclear magnetic resonance (NMR) (Jaishree et al, 1993; Chou et al, 1991) and consistently report that the B‐form conformation usually adopted by DNA shifts either partially (i.e., locally) or completely to the A‐form typical of RNA molecules. More recent NMR work on a twelve‐basepair B‐form DNA molecule containing a single rNMP suggests that the ribose adopts an A‐form conformation, while surrounding dNMPs retain B‐conformations (Derose et al, 2012). These local disturbances of residues adjacent to the rNMP are consistent with atomic force microscopy (AFM)‐based studies revealing perturbed backbone elasticity in such rNMP‐containing DNA molecules (Chiu et al, 2014). Similar AFM analysis employing DNA molecules of up to one kilobase in length (i.e., more closely mimicking the in vivo context of rNMP sites) containing randomly incorporated rCTPs demonstrated shortening and increased elasticity of the DNA backbone (Meroni et al, 2017), suggesting that rNMPs can profoundly impact on the conformation of large DNA molecules beyond just their sites of incorporation.
Albeit difficult to prove experimentally, it is conceivable that these structural effects of rNMPs on the surrounding DNA may play an important role to enhance their recognition and subsequent removal. In the absence of functional RER, however, such structural perturbations may at the same time interfere with DNA transactions such as replication, repair, and nucleosome assembly. A plausible expectation is that DNA–protein interactions, especially those that occur in a sequence‐independent manner with proteins, would be disturbed to the greatest extent.
Effects of unrepaired rNMPs on chromatin and chromatin‐related processes
The most common sequence‐independent DNA–protein interaction is represented by DNA wrapping around histones at the level of the nucleosome. In this regard, in vitro studies point toward a strong negative impact of RNA‐containing DNA on nucleosome formation (Dunn & Griffith, 1980; Hovatter & Martinson, 1987). The consequential defects in nucleosome formation and stability could result in severe consequences on cellular fitness. Less stable nucleosomes may cause altered occupation patterns, decreased nucleosome density, and consequently a less densely packed genome. Such global effects are likely to influence cellular processes that are regulated by the state of chromatin, including gene expression, chromosome segregation, and DNA repair (Fig 4). Recent molecular dynamics simulations addressing the direct effect of ribonucleotides in the context of the nucleosome have modeled a single rNMP at different positions within a DNA‐bound histone octamer (Fu et al, 2018). Interestingly, nucleosome formation was differentially affected by both the translational setting (distance from nucleosome midpoint) and the rotational setting (facing outwards or toward the nucleosome) of the rNMP. Outward‐facing rNMPs, independent of their translational position, adopted a C2′‐endo conformation (associated with B‐DNA) and did not affect adjacent base pairs, rendering them indistinguishable from dNMPs in terms of their overall structure. In contrast, inward‐facing rNMPs, especially at a translational position that directly interacts with the nucleosome, were mostly found in a stable C3′‐endo conformation (associated with A‐DNA) and consequently increased the width of the DNA minor grove, disrupted Watson–Crick base pairing, and induced local unwinding of the DNA duplex (Fu et al, 2018). Besides demonstrating a direct effect of rNMPs on DNA structure, these findings also implicate that RER efficiency might depend on the position of the rNMP within the nucleosome. An altered DNA structure may serve as a prerequisite to signal to chromatin‐remodeling enzymes that subsequently create a chromatin environment that favors RER (Fig 4). Therefore, specific chromatin marks could support recruitment of RNase H2, either in addition to or redundant with the PIP‐box of RNase H2. It is interesting to note that inward‐facing rNMPs resulted in local DNA unwinding, which would also be more permissive to R‐loop formation. Thus, the accumulation of genomic rNMPs may be tightly linked to R‐loop accumulation in this respect.
Figure 4. Possible chromatin alterations associated with rNMPs.
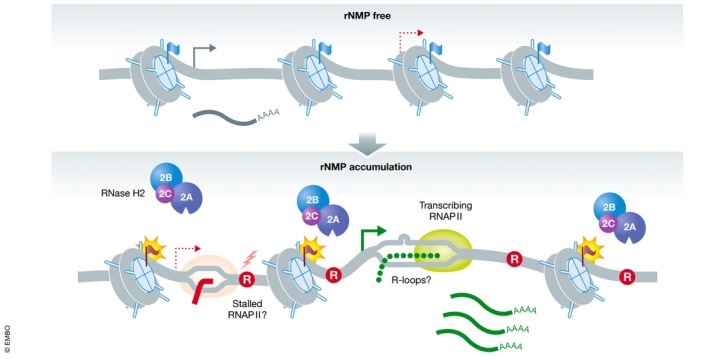
The accumulation or insertion of rNMPs (R in red circle) may affect the local chromatin environment by altering histone modifications on nucleosomes (flags) or even nucleosome stability and histone occupancy. These local chromatin changes may be important to allow RER by RNase H2. Such chromatin alterations may also lead to more open chromatin, increasing the expression of lowly expressed genes and thus enhancing formation of R‐loops. RNA polymerase stalling at persistent rNMPs may further impact on transcription processes.
A more open global chromatin environment due to decreased nucleosome stability would most likely affect transcription. Transcriptional analysis of yeast cells lacking RNase H2 function identified a subset of 349 differentially expressed genes, with approximately one‐third of them being upregulated and two‐thirds being downregulated (Arana et al, 2012). Loss of RNase H2 function leads to two phenotypic consequences: an accumulation of genomic rNMPs and of R‐loops (Cerritelli & Crouch, 2009). While the exact contribution of each RNase H2 function remains undefined in this transcriptional analysis, changes in gene expression have been correlated with an increased need for DNA repair and recombination factors, cell cycle‐regulatory proteins, and the induction of a general stress response (Arana et al, 2012). It cannot be discriminated whether accumulation of R‐loops or genomic instability induced by unrepaired rNMPs is causal for this transcriptional response. The ability of R‐loops to induce local chromatin compaction (Castellano‐Pozo et al, 2013) can also not be ruled out as contributing to the transcriptional changes observed. Therefore, future investigations using the RNH201‐RED separation‐of‐function mutant (Chon et al, 2013) may allow for clearer distinction between contributions of unrepaired rNMPs and R‐loops. Moreover, including spike‐in controls in RNA‐sequencing experiments (Lovén et al, 2012; Chen et al, 2016) would allow the detection of global changes in gene expression levels, as they could be caused by a more open chromatin structure.
In line with such transcriptomics studies, another crucial aspect to be tested is whether unrepaired rNMPs may impede the progression of RNA polymerases as much as progression of replicative DNA polymerases (Watt et al, 2011; Göksenin et al, 2012; Clausen et al, 2013). In this case, rNMPs could physically influence RNA transcription to varying degrees, depending on whether the rNMP resides in the transcribed or the non‐transcribed strand, whether a gene is expressed at high or low levels, and where within the gene the rNMP is located. Transcription‐associated mutagenesis can indeed be linked to unrepaired ribonucleotides (Cho & Jinks‐Robertson, 2017). The characteristic two‐ to five‐basepair slippage mutations generated by Top1 at unrepaired rNMPs are elevated within actively transcribed hotspot regions (Takahashi et al, 2011). At low levels of transcription, those mutation events are biased toward the leading strand (Cho et al, 2015), which harbors the majority of rNMPs (Nick McElhinny et al, 2010a,b). However, high transcription levels lead to loss of this strand bias during DNA replication, with mutations accumulating preferably in the non‐transcribed strand of a reporter gene (Cho et al, 2015). This led to the model that RNA polymerase II (RNAPII) might physically affect the ability of Top1 to religate the nicked DNA: RNAPII might push Top1 cross‐linked on the transcribed strand toward the nick, thus supporting alignment and error‐free ligation of the DNA, but conversely would push Top1 that has incised the non‐transcribed strand away from the nick (or alternatively shield the 5′OH opposite of the Top1cc), thereby hampering ligation and favoring mutagenesis. It is important to keep in mind that ribonucleotide‐mediated mutagenesis brought about by transcription can happen in both cycling and non‐cycling cells.
Compromised RER causes Aicardi–Goutières syndrome
RNase H2 is mutated in more than 50% of cases of the autosomal recessive genetic disorder Aicardi–Goutières syndrome (AGS) (Crow et al, 2006; Rice et al, 2007). This rare early‐onset autoinflammatory disease is reminiscent of both congenital viral infections and the autoimmune disease systemic lupus erythematosus (SLE), affecting brain and skin cells as well as the immune system (Crow & Manel, 2015). While unresolved R‐loops may contribute to disease pathology (Lim et al, 2015), DNA damage responses in RNase H2‐depleted human culture cells and mouse embryos, as well as in AGS patient‐derived cells, have demonstrated to be mainly caused by accumulation of rNMPs in DNA (Pizzi et al, 2015; Uehara et al, 2018). Similarly, some SLE patients exhibit mutations in RNase H2 that increase rNMP levels in their genome (Günther et al, 2015). Mice carrying homozygous deletions of any of the RNase H2 subunits display embryonic lethality (Hiller et al, 2012; Reijns et al, 2012; Uehara et al, 2018). Human RNase H2 is likely also essential for embryonic development, as all mapped AGS mutations in RNase H2 subunits are missense mutations that still allow the production of full‐length proteins. The in vitro analysis of RNase H2 complexes harboring AGS mutations revealed that most of them preserve enzymatic activity with the exception of RNASEH2A‐G37S, a mutation located in the substrate recognition motif of the catalytic subunit (Rychlik et al, 2010). Additionally, the disease‐linked mutant proteins are not defective in assembling the heterotrimeric enzyme complex (Crow et al, 2006; Rohman et al, 2008; Kind et al, 2014), albeit complex stability is reduced for some mutations (Nishimura et al, 2019). The variation of retained RNase H2 function of AGS mutants was similarly observed in budding yeast (Potenski et al, 2019). The introduction of conserved AGS mutations in the yeast RNase H2 genes revealed defects in terms of genetic interactions, genome instability, and RER proficiency, which ranged between what could be observed for the wildtype enzyme and a full deletion (Potenski et al, 2019).
Solving the crystal structure of human RNase H2 in complex with an rNMP‐containing DNA substrate allowed to make predictions on the in vivo effect of specific point mutations (Figiel et al, 2011). The identified AGS mutations either map to the hydrophobic core or to the surface of RNase H2 and could therefore impact on substrate recognition or positioning, or may affect protein–protein interactions between the subunits or with other proteins. In support of the latter, in vitro data and imaging‐based in vivo approaches showed reduced complex stability and diminished recruitment of the mutated RNase H2 complex to sites of PCNA‐dependent DNA replication and repair (Kind et al, 2014). Other enzymes involved in nucleic acid metabolism have also been found to be mutated in AGS patients, including: the 3′ to 5′ single‐stranded DNA exonuclease TREX1 (Crow et al, 2006), the 3′ to 5′ exonuclease and dNTP hydrolase SAMHD1 (Rice et al, 2009), the RNA adenosine deaminase ADAR1 (Rice et al, 2012), and the cytosolic double‐stranded RNA receptor gene IFIH1 (Rice et al, 2014). RNase H2 mutations make up more than half of all reported AGS cases, followed by TREX1 (22%) and SAMHD1 (13%) mutations (Crow et al, 2015). Although different genes are affected, the overall disease pathology is very similar.
The severity of symptoms as well as the time of disease onset varies between patients (Rice et al, 2007). The majority of patients with RNase H2 mutations go through an initial phase of normal development before presenting an increased type I interferon (IFN) response indicative of upregulated immune signaling, followed by loss of neurological function and in most cases death before they reach adulthood. In patients with mutations in the RNASEH2B subunit, disease onset was significantly delayed, neurological function largely preserved, and patients displayed reduced mortality (Crow et al, 2015). This observed variation may be explained by the recent observation, based on expression of different RNase H2 variants in mice, that unrepaired rNMPs in the genome can be tolerated to some extent (Uehara et al, 2018). If rNMP levels remain below a certain threshold, they induce expression of genes typical of an innate immune response, but are permissive to embryonic development. Above this threshold, however, cells die in a p53‐dependent manner (Fig 5A). Considering that different mutations in RNase H2 could result in different levels of retained RER activity, the overall rNMP load could differ greatly between patients. In line with this, IFN levels measured in patients dropped with increasing age (Rice et al, 2007), accompanied by loss of mental and physical abilities. Considering the rNMP threshold model proposed by Uehara et al, the decrease of IFN signaling and the concomitant increase of disabilities observed by Rice et al could reflect the transition from IFN signaling to p53‐mediated cell death once the rNMP load crosses the threshold.
Figure 5. Impact of AGS mutations and IFN gene activation in disease.
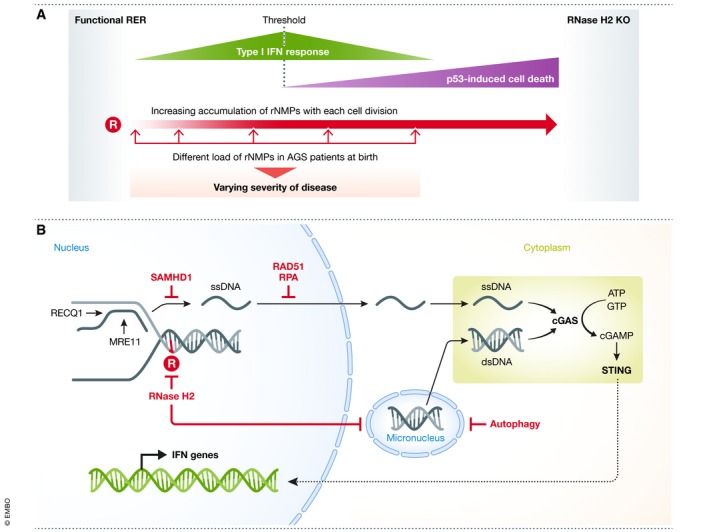
(A) With increasing rNMP load in the genome, IFN‐regulated genes are upregulated. When rNMP levels cross a threshold, p53‐dependent cell death leads to a decreased IFN response but an enhanced phenotype. Different RNase H2 mutations identified in AGS patients could result in different initial rNMP levels at birth and explain the observed differences in disease severity between patients. (B) Release of nuclear DNA into the cytoplasm triggers activation of cGAS, which in turn activates STING that subsequently induces IFN genes. SAMHD1 prevents ssDNA release from stalled replication forks processed by RECQ1 and MRE11. RAD51 and RPA bind to and retain ssDNA in the nucleus. RNase H2 removes rNMPs (R in red circle) from DNA and thereby prevents formation of micronuclei. Autophagy can clear micronuclei, preventing them from rupture.
Faulty RER leads to cGAS‐STING activation
Type I interferon signaling can be activated by the nucleic acid sensing cyclic GMP‐AMP synthase cGAS in conjunction with the adaptor protein STING. The cGAS‐STING axis elicits an innate immune response when the immune sensor cGAS recognizes free cytoplasmic DNA derived from pathogens, endogenous retroviral elements, mitochondrial DNA, or chromatinized nuclear DNA (Sun et al, 2013; Dhanwani et al, 2018). In addition, a type I interferon response can be activated by a nuclear fraction of cGAS that associates preferentially with centromeres (Gentili et al, 2019). In AGS, as revealed by work in patient‐derived cells and AGS mouse models, cytosolic cGAS‐STING mediates an upregulation of type I IFN genes and thus stimulates the autoimmune response (Mackenzie et al, 2016; Pokatayev et al, 2016). In RNase H2‐deficient cells, high levels of DNA damage (Hiller et al, 2012; Pizzi et al, 2015) increase the frequency of micronuclei, which have been found to colocalize with cGAS (Bartsch et al, 2017; MacKenzie et al, 2017). Upon breakdown of the unstable nuclear envelope encompassing a micronucleus, cGAS‐STING‐mediated recognition of the now cytoplasmic micronuclear DNA induces the expression of interferon‐induced genes (Fig 5B).
However, rNMP accumulation is likely not the only cause for cytoplasmic DNA species that trigger cGAS‐STING activity. AGS patients with SAMHD1 mutations do not display an accumulation of rNMPs (Lim et al, 2015), but the activation of an IFN response is similarly evoked by cGAS‐STING‐mediated sensing of endogenous DNA species in the cytoplasm (Coquel et al, 2018). SAMHD1 stimulates the exonuclease activity of MRE11 at stalled replication forks to facilitate ATR‐CHK1 activation and promote replication fork restart. In SAMHD1‐depleted cells, the RECQ1 helicase acts on stalled replication forks to displace ssDNA, which is then released by MRE11 and can become cytoplasmic (Coquel et al, 2018). Thus, altered processing of stalled forks can contribute to IFN induction. Since rNMPs lead to replication fork stalling and replication stress, it will be interesting to determine whether MRE11 also contributes to the accumulation of cytoplasmic DNA in the context of faulty RER (Fig 5B).
The involvement of cytoplasmic DNA in AGS etiology is further highlighted by the role of the exonuclease TREX1, another gene frequently mutated in AGS patients. TREX1 is located at the outer nuclear membrane, where it immediately degrades ssDNA that escapes into the cytoplasm (Wolf et al, 2016). The accumulation of ssDNA in the absence of TREX1 leads to an exhaustion of RAD51 and RPA, which in turn directly contributes to the generation of replication stress and p53‐dependent checkpoint signaling in addition to the activation of a cGAS‐STING‐mediated IFN response (Fig 5B). In line with this, depletion of RAD51 in both murine and human cell lines increases IFN signaling upon irradiating DNA damage (Bhattacharya et al, 2017). This suggests that the release of self‐derived DNA into the cytoplasm is a general phenomenon during DNA replication stress and is counteracted by mechanisms acting both at the stalled replication fork and in the cytoplasm. Characterizing in more detail how rNMPs interfere with DNA replication could allow for better understanding of how micronuclei and hence cytosolic nucleic acids arise, which eventually lead to autoimmunity in RNase H2‐mutated AGS patients.
Type I IFN signaling mediated by cGAS‐STING activates autophagy (Gui et al, 2019; Liu et al, 2019). At the same time, autophagy downregulates the IFN response by stimulating STING degradation (Prabakaran et al, 2018). Autophagy not only reduces IFN signaling in RNase H2‐deficient mouse embryonic fibroblasts (MEFs), but also clears accumulating micronuclei (Bartsch et al, 2017). Thus, inducing autophagy in AGS or SLE patients could offer a therapeutic approach based on two levels: (i) by downregulating IFN signaling through degradation of STING and dampening active autoimmunity in patients, and (ii) through clearance of immune‐stimulatory DNA species such as those contained in micronuclei (see Fig 5). Especially in early stages of disease, when autoimmune reactions are high and cell death has not yet occurred, patients might benefit from such therapy. Whether autophagy stimulation could prevent, or at least delay, the occurrence of the associated neurodegeneration and other symptoms attributed to cell death needs to be carefully evaluated. Recent work has demonstrated that replicative senescence due to telomere attrition induces cGAS‐STING activation and cell elimination through autophagy (Nassour et al, 2019). Therefore, although the stimulation of autophagy may lead to cGAS‐STING attenuation, it can also lead to enhanced cell elimination, which could exacerbate phenotypic onset, similar to what happens when p53 induces cell death if the rNMP threshold is reached (see above). Moreover, autophagy degrades nuclear lamina during cell senescence (Dou et al, 2015), thereby potentially exposing yet more endogenous DNA to cGAS. These concepts are still in their infancy, but require careful consideration in the context of AGS treatment.
The search for murine disease models of AGS has been challenging. Animals with full deletion of any RNase H2 subunit are inviable and die in utero unless p53 is co‐deleted (Hiller et al, 2012; Reijns et al, 2012). Cells from such embryos have enabled the characterization of DNA damage and genome instability phenotypes of AGS but could not give insight into the mechanism of IFN induction. Mechanistic dissection of IFN induction has instead been possible using expression of disease‐associated alleles in mice (Mackenzie et al, 2016; Pokatayev et al, 2016), but surprisingly, none of the mutant RNase H2‐expressing mice showed symptoms in their brains (Mackenzie et al, 2016), despite the fact that the brain is the organ most heavily affected in AGS and that patients display increased IFN activity in the cerebrospinal fluid (Crow et al, 2015). Brain‐specific conditional knockout of RNase H2 did not give rise to mice with a detectible brain phenotype either (Bartsch et al, 2018). Bartsch et al argue that a mouse brain undergoes fewer cell divisions than the human brain and might thus not reach the load of genomic rNMPs required to induce neuropathology (Uehara et al, 2018). In support of this notion, isolated astrocytes from those mouse brains recapitulated defects associated with AGS when cultured under mitogenic conditions, exhibiting high levels of DNA damage, increased type I IFN signaling, and limited proliferative capacity (Bartsch et al, 2018). As transgenic mice that overexpress type I IFN specifically in the brain display signs of neuropathology (Akwa et al, 1998), astrocytes from RNase H2‐deficient mouse brains could be a valuable tool to understand the transition between IFN signaling and the induction of cell death in AGS.
RER intermediates can also contribute to neurological disease
Even in the presence of a functional RER pathway, genomic rNMPs can still contribute to the development of another neurological disease, ataxia with oculomotor apraxia 1 (AOA1). This neurological syndrome is caused by mutations in APTX, the gene encoding the aprataxin enzyme (Moreira et al, 2001). Aprataxin promotes the repair of adenylated DNA ends (both at double‐strand breaks and at DNA nicks) that can arise due to abortive DNA ligation, but also upon premature ligation of nicked abasic sites (Ahel et al, 2006; Rass et al, 2007). By removing the covalently bound 5′‐adenosine monophosphate (5′AMP), aprataxin creates DNA ends that can be processed by DNA ligases. Structural studies of aprataxin proteins carrying mutations found in AOA1 patients suggest that the majority of mutations affect protein stability to various degrees, whereas one mutation directly interferes with the deadenylation reaction, and another variant causes allosteric changes of the active site conformation (Tumbale et al, 2018).
A nick flanked by a 3′OH and a 5′phosphate with a single rNMP located at its 5′‐side, a structure that would typically be created by RNase H2‐mediated incision (discussed above), is less‐efficiently ligated in vitro than nicks at dNMPs (Tumbale et al, 2014). Due to this inefficient ligation, the exposed 5′phosphate can become adenylated to yield a non‐repairable 5′AMP product. Without rapid processing, the RNase H2 action on rNMPs therefore creates substrates (5′AMP) for aprataxin. When aprataxin is mutated, the accumulation of irreparable 5′AMP consequently contributes to disease pathology in AOA1 patients. In line with this, S. cerevisiae cells lacking the aprataxin homolog Hnt3 are inviable when the incorporation of rNMPs is increased using a DNA Pol ε mutant (pol2‐M644G), but can be rescued by deletion of RNH201 (Tumbale et al, 2014).
RNase H2 mutations are implicated in cancer
Cells with dysfunctional RER experience high loads of DNA damage. Recent studies imply RER as an important tumor suppressor mechanism. When RNase H2 is disrupted in murine epidermal cells, a tissue with rapid cell turnover, type I IFN signaling and spontaneous DNA damage increase in the skin (Hiller et al, 2018). All RNase H2 epithelial knockout mice analyzed in this study developed squamous cell carcinoma (SCC) or precursor forms of skin cancer within less than a year. Concomitant loss of p53 led to enhanced skin inflammation and survival of more damaged cells, suggesting that the p53 DNA damage response limits oncogenic transformation of RER‐deficient epidermis through the elimination of highly damaged cells (Fig 6A).
Figure 6. Tumor suppressor functions of RNase H2 and RER .
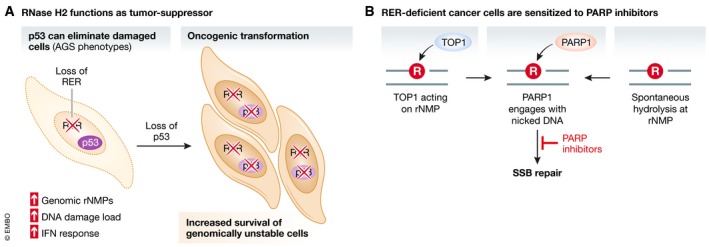
(A) Loss of RER leads to increased genomic rNMP accumulation and the indicated consequences, but damaged cells are eliminated as long as p53 is present. In the nervous system, cell elimination likely leads to the neurodegenerative phenotypes associated with AGS. Genome instability upon p53 loss can lead to oncogenic rearrangements and cancer development. (B) rNMPs that accumulate in the absence of RER are either cleaved by TOP1 or hydrolyzed spontaneously, creating single‐strand breaks (SSB). These nicks recruit PARP, sensitizing cells to PARP inhibitors that will create toxic PARP‐trapping lesions.
Similarly, deletion of RER in murine intestinal epithelial tissue, especially in the absence of p53, increased the development of intestinal and colorectal tumors (Aden et al, 2019). A potential link between non‐functional RER and human cancer is supported by analyses of publicly available transcriptomic (RNA‐seq) datasets from colorectal cancer (CRC) patients. Aden et al (2019) describe a correlation of decreased RNase H2 expression and poor survival rates, the importance of which remains to be evaluated.
A more direct analysis of the interconnection of cancer and defective RER in humans is difficult, as most AGS patients with hereditary RNase H2 mutations have a short lifespan (Crow et al, 2015). However, loss of RNase H2 function has been reported in chronic lymphocytic leukemia (CLL) and metastatic castration‐resistant prostate cancer (Zimmermann et al, 2018). Determination of RNASEH2B copy numbers in 100 patient‐derived CLL cell lines and 226 prostate cancers showed that at least one copy was lost in 57% and 36% of the respective cancer samples. Two loci on chromosome 13q14 frequently lost in those cancer types (microRNA cluster DLEU2‐mir‐15‐16 in CLL, and RB1 locus in prostate cancer) are located in close proximity to the RNASEH2B gene. Consequently, RNASEH2B is simultaneously lost upon deletion of these tumor suppressors.
The determination of RNASEH2B copy numbers in human cancer can become significant in the context of therapy, because RER‐deficient cells are sensitive to (poly‐ADP)ribose polymerase (PARP) inhibition. Mouse xenograft studies confirmed that tumors arising from implanted RER‐deficient CLL cells are similarly sensitized. Mechanistically, cell death is likely caused by PARP1‐trapping at DNA lesions that are created by TOP1‐mediated repair attempts at rNMPs (Zimmermann et al, 2018; Fig 6B). However, substrates for PARP1 could also arise in a TOP1‐independent manner through spontaneous hydrolysis of DNA at unrepaired rNMPs. This is supported by the notion that PARP1 can sense unligated Okazaki fragments due to the presence of DNA nicks between them, and signal for their repair (Hanzlikova et al, 2018). The loss of RNase H2 activity further renders cells hypersensitive to ATR inhibition, which is likely linked to replication stress when rNMPs are encountered by the replisome (Wang et al, 2018; Hustedt et al, 2019).
A detailed analysis of existing human cancer genomes could reveal other types of cancer that coincide with a loss of RNase H2 function. This is particularly important given the recent findings with regard to RNase H2 status and sensitivity to PARP or ATR inhibitors, as it may have important consequences with respect to therapeutic approaches in patients (Wang et al, 2018; Zimmermann et al, 2018; Hustedt et al, 2019). Therefore, it seems that the status of the p53 and/or pRb checkpoints may be critical in the fate of RER‐defective cells. Whereas p53‐mediated cell death appears to anticipate loss of neuronal function in AGS, it can nonetheless act as a tumor suppressor.
Finally, RNase H2 may be involved in cancer development and progression via additional, so far uncharacterized functions. One recent report identified reduced expression specifically of the RNASEH2C subunit, independent of enzymatic activity and not associated with an induction of the DNA damage response, as a driver of breast cancer metastasis in mice (Deasy et al, 2019). These authors also did not observe activation of cGAS‐STING signaling, but instead increased cytotoxic T‐cell‐mediated immune responses, highlighting the need to characterize the cancer roles of RNase H2 in more detail.
Conclusions
Ribonucleotide insertions into the genome cannot be avoided during DNA replication. How and when they are dealt with, however, can have significant consequences with respect to mutation frequency, genome stability, cell viability, and disease. In the best‐case scenario, RNase H2 excises the rNMP via the canonical RER pathway to achieve error‐free repair (Fig 1). New insights have illustrated the importance of limiting this reaction to a postreplicative phase to ensure that DNA nicking activity does not occur at the same time as DNA replication. How this temporal regulation is achieved still remains to be elucidated, as do many other questions with regard to how RER is achieved in vivo, including the relevance and nature of the interaction between RNase H2 and the replisome through PCNA (Fig 3).
When RER is impaired, there are two immediate consequences: (i) rNMPs accumulate to increased levels and (ii) Top1 takes over and removes rNMPs, but at a costly price to the integrity of the genome. The consequences of rNMP accumulation remain poorly described, and here, we have attempted to summarize the potential effects on chromatin state, transcriptome and RNAP II status and DNA repair by summarizing what is known from the literature (Fig 4). The Top1‐induced rNMP removal pathway, on the other hand, has been more extensively investigated and is associated with the induction of DNA damage and loss of genomic integrity (Fig 2). In AGS autoimmune disease, where neuronal RNase H2 function is compromised, genome instability can lead to the formation of micronuclei and activation of cGAS/STING, which then triggers an inflammatory response. The eventual activation of p53 will lead to cell elimination, which is responsible for the AGS phenotypes (Fig 5). In the absence of a checkpoint response, the mutator phenotype may drive oncogenic transformation (Fig 6). Indeed, loss of RNase H2 has recently been linked to different types of human cancer.
Due to the frequency of faulty rNTP insertions into the genome, RER defects lead to severe human pathologies. Nonetheless, our increased mechanistic understanding of this pathway now paves the way for possible new therapeutic strategies. In this respect, it is important to continue to understand RER and its regulations as well as the subsequent consequences when RER fails.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
We would like to thank Daniel Durocher and Duncan Smith for critically reading the manuscript and giving constructive feedback. Work in the laboratory of BL on RER is funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation)—project number 393547839—SFB 1361, sub‐project 9.
The EMBO Journal (2020) 39: e102309
See the Glossary for abbreviations used in this article.
Contributor Information
Vanessa Kellner, Email: vanessa.kellner@nyu.edu.
Brian Luke, Email: b.luke@imb-mainz.de.
References
- Aden K, Bartsch K, Dahl J, Reijns MAM, Esser D, Sheibani‐Tezerji R, Sinha A, Wottawa F, Ito G, Mishra N et al (2019) Epithelial RNase H2 maintains genome integrity and prevents intestinal tumorigenesis in mice. Gastroenterology 156: 145–159.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahel I, Rass U, El‐Khamisy SF, Katyal S, Clements PM, McKinnon PJ, Caldecott KW, West SC (2006) The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature 443: 713–716 [DOI] [PubMed] [Google Scholar]
- Akwa Y, Hassett DE, Eloranta M‐L, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL (1998) Transgenic expression of IFN‐α in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol 161: 5016–5026 [PubMed] [Google Scholar]
- Allen‐Soltero S, Martinez SL, Putnam CD, Kolodner RD (2014) A Saccharomyces cerevisiae RNase H2 interaction network functions to suppress genome instability. Mol Cell Biol 34: 1521–1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arana ME, Kerns RT, Wharey L, Gerrish KE, Bushel PR, Kunkel TA (2012) Transcriptional responses to loss of RNase H2 in Saccharomyces cerevisiae . DNA Repair (Amst) 11: 933–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arudchandran A, Cerritelli SM, Narimatsu SK, Itaya M, Shin DY, Shimada Y, Crouch RJ (2000) The absence of ribonuclease H1 or H2 alters the sensitivity of Saccharomyces cerevisiae to hydroxyurea, caffeine and ethyl methanesulphonate: implications for roles of RNases H in DNA replication and repair. Genes Cells 5: 789–802 [DOI] [PubMed] [Google Scholar]
- Ban C, Ramakrishnan B, Sundaralingam M (1994) A single 2′‐hydroxyl group converts B‐DNA to A‐DNA: crystal structure of the DNA‐RNA chimeric decamer duplex d(CCGGC)r(G)d(CCGG) with a novel intermolecular G‐C base‐paired quadruplet. J Mol Biol 236: 275–285 [DOI] [PubMed] [Google Scholar]
- Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, Spehlmann ME, Frey N, Saftig P, Chalaris A et al (2017) Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum Mol Genet 26: 3960–3972 [DOI] [PubMed] [Google Scholar]
- Bartsch K, Damme M, Regen T, Becker L, Garrett L, Hölter SM, Knittler K, Borowski C, Waisman A, Glatzel M et al (2018) RNase H2 loss in murine astrocytes results in cellular defects reminiscent of nucleic acid‐mediated autoinflammation. Front Immunol 9: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharya S, Srinivasan K, Abdisalaam S, Su F, Raj P, Dozmorov I, Mishra R, Wakeland EK, Ghose S, Mukherjee S et al (2017) RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res 45: 4590–4605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, Wanka S, Kraft C, Urban J, Campbell D, Pedrioli PG, Gerrits B, Picotti P, Lam H, Vitek O et al (2010) Phosphoproteomic analysis reveals interconnected system‐wide responses to perturbations of kinases and phosphatases in yeast. Sci Signal 3: rs4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Suo Z (2011) Unlocking the sugar ‘steric gate’ of DNA polymerases. Biochemistry 50: 1135–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bubeck D, Reijns MAM, Graham SC, Astell KR, Jones EY, Jackson AP (2011) PCNA directs type 2 RNase H activity on DNA replication and repair substrates. Nucleic Acids Res 39: 3652–3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgers PMJ, Kunkel TA (2017) The eukaryotic DNA replication fork. Annu Rev Biochem 86: 417–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Geacintov NE, Broyde S (2014) Ribonucleotides as nucleotide excision repair substrates. DNA Repair (Amst) 13: 55–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldecott KW (2014) Ribose – an internal threat to DNA. Science 343: 260–261 [DOI] [PubMed] [Google Scholar]
- Castellano‐Pozo M, Santos‐Pereira J, Rondón AG, Barroso S, Andújar E, Pérez‐Alegre M, García‐Muse T, Aguilera A (2013) R loops are linked to histone H3 S10 phosphorylation and chromatin condensation. Mol Cell 52: 583–590 [DOI] [PubMed] [Google Scholar]
- Cerritelli SM, Crouch RJ (2009) Ribonuclease H: the enzymes in eukaryotes. FEBS J 276: 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Hu Z, Xia Z, Zhao D, Li W, Tyler JK (2016) The overlooked fact: fundamental need for spike‐in control for virtually all genome‐wide analyses. Mol Cell Biol 36: 662–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SH, Flynn P, Wang A, Reid B (1991) High‐resolution NMR studies of chimeric DNA‐RNA‐DNA duplexes, heteronomous base pairing, and continuous base stacking at junctions. Biochemistry 30: 5248–5257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu HC, Koh KD, Evich M, Lesiak AL, Germann MW, Bongiorno A, Riedo E, Storici F (2014) RNA intrusions change DNA elastic properties and structure. Nanoscale 6: 10009–10017 [DOI] [PubMed] [Google Scholar]
- Cho JE, Kim N, Jinks‐Robertson S (2015) Topoisomerase 1‐dependent deletions initiated by incision at ribonucleotides are biased to the non‐transcribed strand of a highly activated reporter. Nucleic Acids Res 43: 9306–9313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho JE, Jinks‐Robertson S (2017) Ribonucleotides and transcription‐associated mutagenesis in yeast. J Mol Biol 429: 3156–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, Cerritelli SM (2013) RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 41: 3130–3143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen AR, Zhang S, Burgers PM, Lee MY, Kunkel TA (2013) Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase δ. DNA Repair (Amst) 12: 121–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conover HN, Lujan SA, Chapman MJ, Cornelio DA, Sharif R, Williams JS, Clark AB, Camilo F, Kunkel TA, Argueso JL (2015) Stimulation of chromosomal rearrangements by ribonucleotides. Genetics 201: 951–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva MJ, Técher H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz AL et al (2018) SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557: 57–61 [DOI] [PubMed] [Google Scholar]
- Cornelio DA, Sedam HNC, Ferrarezi JA, Sampaio NMV, Argueso JL (2017) Both R‐loop removal and ribonucleotide excision repair activities of RNase H2 contribute substantially to chromosome stability. DNA Repair (Amst) 52: 110–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul‐Hirji R et al (2006) Mutations in genes encoding ribonuclease H2 subunits cause Aicardi‐Goutières syndrome and mimic congenital viral brain infection. Nat Genet 38: 910–916 [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase D, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G et al (2015) Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet Part A 167A: 296–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Manel N (2015) Aicardi‐Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440 [DOI] [PubMed] [Google Scholar]
- Daigaku Y, Davies AA, Ulrich HD (2010) Ubiquitin‐dependent DNA damage bypass is separable from genome replication. Nature 465: 951–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deasy SK, Uehara R, Vodnala SK, Yang HH, Dass RA, Hu Y, Lee MP, Crouch RJ, Hunter KW (2019) Aicardi‐goutières syndrome gene rnaseh2c is a metastasis susceptibility gene in breast cancer. PLoS Genet 15: 1–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derose EF, Perera L, Murray MS, Kunkel TA, London RE (2012) Solution structure of the Dickerson DNA dodecamer containing a single ribonucleotide. Biochemistry 51: 2407–2416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanwani R, Takahashi M, Sharma S (2018) Cytosolic sensing of immuno‐stimulatory DNA, the enemy within. Curr Opin Immunol 50: 82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP et al (2015) Autophagy mediates degradation of nuclear lamina. Nature 527: 105–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn K, Griffith JD (1980) The presence of RNA in a double helix inhibits its interaction with histone protein. Nucleic Acids Res 8: 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eder PS, Walder RY, Walder JA (1993) Substrate specificity of human RNase H1 and its role in excision repair of ribose residues misincorporated in DNA. Biochimie 75: 123–126 [DOI] [PubMed] [Google Scholar]
- Egli M, Usman N, Rich A (1993) Conformational influence of the ribose 2′‐hydroxyl group: crystal structures of DNA‐RNA chimeric duplexes. Biochemistry 32: 3221–3237 [PubMed] [Google Scholar]
- Figiel M, Chon H, Cerritelli SM, Cybulska M, Crouch RJ, Nowotny M (2011) The structural and biochemical characterization of human RNase H2 complex reveals the molecular basis for substrate recognition and Aicardi‐Goutières syndrome defects. J Biol Chem 286: 10540–10550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu I, Smith DJ, Broyde S (2018) Rotational and translational positions determine the structural and dynamic impact of a single ribonucleotide incorporated in the nucleosome. DNA Repair (Amst) 73: 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentili M, Lahaye X, Nadalin F, Nader GFP, Puig Lombardi E, Herve S, De Silva NS, Rookhuizen DC, Zueva E, Goudot C et al (2019) The N‐terminal domain of cGAS determines preferential association with centromeric DNA and innate immune activation in the nucleus. Cell Rep 26: 2377–2393.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodgaonkar MM, Lazzaro F, Olivera‐Pimentel M, Artola‐Borán M, Cejka P, Reijns MA, Jackson AP, Plevani P, Muzi‐Falconi M, Jiricny J (2013) Ribonucleotides misincorporated into DNA act as strand‐discrimination signals in eukaryotic mismatch repair. Mol Cell 50: 323–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göksenin AY, Zahurancik W, LeCompte KG, Taggart DJ, Suo Z, Pursell ZF (2012) Human DNA polymerase ε is able to efficiently extend from multiple consecutive ribonucleotides. J Biol Chem 287: 42675–42684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf M, Bonetti D, Lockhart A, Serhal K, Kellner V, Maicher A, Jolivet P, Teixeira MT, Luke B (2017) Telomere length determines TERRA and R‐loop regulation through the cell cycle. Cell 170: 72–85.e14 [DOI] [PubMed] [Google Scholar]
- Gui X, Yang H, Li T, Tan X, Shi P, Li M, Du F, Chen ZJ (2019) Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 567: 262–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther C, Kind B, Reijns MAM, Berndt N, Martinez‐bueno M, Wolf C, Tüngler V, Chara O, Lee YA, Hübner N et al (2015) Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J Clin Invest 125: 413–424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzlikova H, Kalasova I, Demin AA, Pennicott LE, Cihlarova Z, Caldecott KW (2018) The importance of Poly(ADP‐ribose) polymerase as a sensor of unligated okazaki fragments during DNA replication. Mol Cell 71: 319–331.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A (2012) Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J Exp Med 209: 1419–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller B, Hoppe A, Haase C, Hiller C, Schubert N (2018) Ribonucleotide excision repair is essential to prevent squamous cell carcinoma of the skin. Cancer Res 78: 5917–5926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovatter KR, Martinson HG (1987) Ribonucleotide‐induced helical alteration in DNA prevents nucleosome formation. Proc Natl Acad Sci USA 84: 1162–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M‐E, Rio A‐G, Nicolas A, Kolodner RD (2003) A genomewide screen in Saccharomyces cerevisiae for genes that suppress the accumulation of mutations. Proc Natl Acad Sci USA 100: 11529–11534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SYN, Ghosh S, Pommier Y (2015) Topoisomerase I alone is sufficient to produce short DNA deletions and can also reverse nicks at ribonucleotide sites. J Biol Chem 290: 14068–14076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hustedt N, Álvarez‐Quilón A, McEwan A, Yuan JY, Cho T, Koob L, Hart T, Durocher D (2019) A consensus set of genetic vulnerabilities to ATR inhibition. Open Biol 9: 190156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ii M, Ii T, Mironova LI, Brill SJ (2011) Epistasis analysis between homologous recombination genes in Saccharomyces cerevisiae identifies multiple repair pathways for Sgs1, Mus81‐Mms4 and RNase H2. Mutat Res 714: 33–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaishree TN, Wang AHJ, van der Marel GA, van Boom JH (1993) Structural influence of RNA incorporation in DNA: quantitative nuclear magnetic resonance refinement of d(CG)r(CG)d(CG) and d(CG)r(C)d(TAGCG). Biochemistry 32: 4903–4911 [DOI] [PubMed] [Google Scholar]
- Jeong HS, Backlund PS, Chen HC, Karavanov AA, Crouch RJ (2004) RNase H2 of Saccharomyces cerevisiae is a complex of three proteins. Nucleic Acids Res 32: 407–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahli M, Osmundson JS, Yeung R, Smith DJ (2019) Processing of eukaryotic Okazaki fragments by redundant nucleases can be uncoupled from ongoing DNA replication in vivo . Nucleic Acids Res 47: 1814–1822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karras GI, Jentsch S (2010) The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 141: 255–267 [DOI] [PubMed] [Google Scholar]
- Kim N, Huang SYN, Williams JS, Li YC, Clark AB, Cho JE, Kunkel TA, Pommier Y, Jinks‐Robertson S (2011) Mutagenic processing of ribonucleotides in DNA by yeast topoisomerase I. Science 332: 1561–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind B, Muster B, Staroske W, Herce HD, Sachse R, Rapp A, Schmidt F, Koss S, Cardoso MC, Lee‐Kirsch MA (2014) Altered spatio‐temporal dynamics of RNase H2 complex assembly at replication and repair sites in Aicardi‐Goutières syndrome. Hum Mol Genet 23: 5950–5960 [DOI] [PubMed] [Google Scholar]
- Kochiwa H, Tomita M, Kanai A (2007) Evolution of ribonuclease H genes in prokaryotes to avoid inheritance of redundant genes. BMC Evol Biol 7: 128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaro F, Novarina D, Amara F, Watt DL, Stone JE, Costanzo V, Burgers PM, Kunkel TA, Plevani P, Muzi‐Falconi M (2012) RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol Cell 45: 99–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Breaker RR (1999) Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2′‐hydroxyl group. J Am Chem Soc 121: 5364–5372 [Google Scholar]
- Li F, Wang Q, Seol JH, Che J, Lu X, Shim EY, Lee SE, Niu H (2019) Apn2 resolves blocked 3′ ends and suppresses Top1‐induced mutagenesis at genomic rNMP sites. Nat Struct Mol Biol 26: 155–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim YW, Sanz LA, Xu X, Hartono SR, Chédin F (2015) Genome‐wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi‐Goutières syndrome. Elife 4: 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey‐Boltz LA, Kemp MG, Hu J, Sancar A (2015) Analysis of ribonucleotide removal from DNA by human nucleotide excision repair. J Biol Chem 290: 29801–29807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Wu H, Wang C, Li Y, Tian H, Siraj S, Sehgal SA, Wang X, Wang J, Shang Y et al (2019) STING directly activates autophagy to tune the innate immune response. Cell Death Differ 26: 1735–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockhart A, Pires V, Bento F, Kellner V, Luke‐Glaser S, Yakoub G, Ulrich HD, Luke B (2019) RNase H1 and H2 are differentially regulated to process RNA‐DNA hybrids. Cell Rep 29: 2890–2900.e5 [DOI] [PubMed] [Google Scholar]
- Lovén J, Orlando DA, Sigova AA, Lin CY, Rahl PB, Burge CB, Levens DL, Lee TI, Young RA (2012) Revisiting global gene expression analysis. Cell 151: 476–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan SA, Williams JS, Clausen AR, Clark AB, Kunkel TA (2013) Ribonucleotides are signals for mismatch repair of leading‐strand replication errors. Mol Cell 50: 437–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskait≐ Ž, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B et al (2016) Ribonuclease H2 mutations induce a cGAS/STING‐dependent innate immune response. EMBO J 35: 831–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie KJ, Carroll P, Martin CA, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A et al (2017) CGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meroni A, Mentegari E, Crespan E, Muzi‐Falconi M, Lazzaro F, Podestà A (2017) The incorporation of ribonucleotides induces structural and conformational changes in DNA. Biophys J 113: 1373–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meroni A, Nava GM, Bianco E, Grasso L, Galati E, Bosio MC, Delmastro D, Muzi‐Falconi M, Lazzaro F (2019) RNase H activities counteract a toxic effect of Polymerase η in cells replicating with depleted dNTP pools. Nucleic Acids Res 47: 4612–4623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira MC, Barbot C, Tachi N, Kozuka N, Uchida E, Gibson T, Mendonça P, Costa M, Barros J, Yanagisawa T et al (2001) The gene mutated in ataxia‐ocular apraxia 1 encodes the new HIT/Zn‐finger protein aprataxin. Nat Genet 29: 189–193 [DOI] [PubMed] [Google Scholar]
- Mortusewicz O, Fouquerel E, Amé JC, Leonhardt H, Schreiber V (2011) PARG is recruited to DNA damage sites through poly(ADP‐ribose)‐and PCNA‐dependent mechanisms. Nucleic Acids Res 39: 5045–5056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassour J, Radford R, Correia A, Fusté JM, Schoell B, Jauch A, Shaw RJ, Karlseder J (2019) Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 565: 659–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Kumar D, Clark AB, Watt DL, Watts BE, Lundström EB, Johansson E, Chabes A, Kunkel TA (2010a) Genome instability due to ribonucleotide incorporation into DNA. Nat Chem Biol 6: 774–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nick McElhinny SA, Watts BE, Kumar D, Watt DL, Lundstrom E‐B, Burgers PMJ, Johansson E, Chabes A, Kunkel TA (2010b) Abundant ribonucleotide incorporation into DNA by yeast replicative polymerases. Proc Natl Acad Sci USA 107: 4949–4954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T, Baba M, Ogawa S, Kojima K, Takita T, Crouch RJ, Yasukawa K (2019) Characterization of six recombinant human RNase H2 bearing Aicardi‐Goutiéres syndrome causing mutations. J Biochem 166: 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzi S, Sertic S, Orcesi S, Cereda C, Bianchi M, Jackson AP, Lazzaro F, Plevani P, Muzi‐Falconi M (2015) Reduction of hRNase H2 activity in Aicardi‐Goutières syndrome cells leads to replication stress and genome instability. Hum Mol Genet 24: 649–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, Crouch RJ (2016) RNase H2 catalytic core Aicardi‐Goutières syndrome–related mutant invokes cGAS–STING innate immune‐sensing pathway in mice. J Exp Med 213: 329–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenski CJ, Klein HL (2014) How the misincorporation of ribonucleotides into genomic DNA can be both harmful and helpful to cells. Nucleic Acids Res 42: 10226–10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenski CJ, Niu H, Sung P, Klein HL (2014) Avoidance of ribonucleotide‐induced mutations by RNase H2 and Srs2‐Exo1 mechanisms. Nature 511: 251–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potenski CJ, Epshtein A, Bianco C, Klein HL (2019) Genome instability consequences of RNase H2 Aicardi‐Goutières syndrome alleles. DNA Repair 84: 102614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran T, Bodda C, Krapp C, Zhang B, Christensen MH, Sun C, Reinert L, Cai Y, Jensen SB, Skouboe MK et al (2018) Attenuation of cGAS‐STING signaling is mediated by a p62/SQSTM 1‐dependent autophagy pathway activated by TBK1. EMBO J 37: e97858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryor JM, Conlin MP, Carvajal‐Garcia J, Luedeman ME, Luthman AJ, Small GW, Ramsden DA (2018) Ribonucleotide incorporation enables repair of chromosome breaks by nonhomologous end joining. Science 361: 1126–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rass U, Ahel I, West SC (2007) Actions of aprataxin in multiple DNA repair pathways. J Biol Chem 282: 9469–9474 [DOI] [PubMed] [Google Scholar]
- Reijns MAM, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F et al (2012) Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149: 1008–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B et al (2007) Clinical and molecular phenotype of Aicardi‐Goutières syndrome. Am J Hum Genet 81: 713–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA et al (2009) Mutations involved in Aicardi‐Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 41: 829–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA et al (2012) Mutations in ADAR1 cause Aicardi‐Goutières syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, del Toro Duany Y, Webb K, Desguerre I, Ariaudo G, Szybowska M, Lourenço CM, Vanderver A, Robinson RA, Latchman AR et al (2014) Gain‐of‐function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46: 503–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohman MS, Koga Y, Takano K, Chon H, Crouch RJ, Kanaya S (2008) Effect of the disease‐causing mutations identified in human ribonuclease (RNase) H2 on the activities and stabilities of yeast RNase H2 and archaeal RNase HII. FEBS J 275: 4836–4849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychlik MP, Chon H, Cerritelli SM, Klimek P, Crouch RJ, Nowotny M (2010) Crystal structures of RNase H2 in complex with nucleic acid reveal the mechanism of RNA‐DNA junction recognition and cleavage. Mol Cell 40: 658–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rydberg B, Game J (2002) Excision of misincorporated ribonucleotides in DNA by RNase H (type 2) and FEN‐1 in cell‐free extracts. Proc Natl Acad Sci USA 99: 16654–16659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos‐Pereira JM, Aguilera A (2015) R loops: new modulators of genome dynamics and function. Nat Rev Genet 16: 583–597 [DOI] [PubMed] [Google Scholar]
- Schneider K, Fuchs C, Dobay A, Rottach A, Qin W, Wolf P, Álvarez‐Castro JM, Nalaskowski MM, Kremmer E, Schmid V et al (2013) Dissection of cell cycle‐dependent dynamics of Dnmt1 by FRAP and diffusion‐coupled modeling. Nucleic Acids Res 41: 4860–4876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiguchi JA, Shuman S (1997) Site‐specific ribonuclease activity of eukaryotic DNA topoisomerase I. Mol Cell 1: 89–97 [DOI] [PubMed] [Google Scholar]
- Shaban NM, Harvey S, Perrino FW, Hollis T (2010) The structure of the mammalian RNase H2 complex provides insight into RNA·DNA hybrid processing to prevent immune dysfunction. J Biol Chem 285: 3617–3624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbakova PV, Pavlov YI, Chilkova O, Rogozin IB, Johansson E, Kunkel TA (2003) Unique error signature of the four‐subunit yeast DNA polymerase ε. J Biol Chem 278: 43770–43780 [DOI] [PubMed] [Google Scholar]
- Sparks JL, Chon H, Cerritelli SM, Kunkel TA, Johansson E, Crouch RJ, Burgers PM (2012) RNase H2‐initiated ribonucleotide excision repair. Mol Cell 47: 980–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks JL, Burgers PM (2015) Error‐free and mutagenic processing of topoisomerase 1‐provoked damage at genomic ribonucleotides. EMBO J 34: 1259–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ (2013) Cyclic GMP‐AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339: 786–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Burguiere‐Slezak G, Van der Kemp PA, Boiteux S (2011) Topoisomerase 1 provokes the formation of short deletions in repeated sequences upon high transcription in Saccharomyces cerevisiae . Proc Natl Acad Sci USA 108: 692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbale P, Williams JS, Schellenberg MJ, Kunkel TA, Williams RS (2014) Aprataxin resolves adenylated RNA‐DNA junctions to maintain genome integrity. Nature 506: 111–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumbale P, Schellenberg MJ, Mueller GA, Fairweather E, Watson M, Little JN, Krahn J, Waddell I, London RE, Williams RS (2018) Mechanism of APTX nicked DNA sensing and pleiotropic inactivation in neurodegenerative disease. EMBO J 37: 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara R, Cerritelli SM, Hasin N, Sakhuja K, London M, Iranzo J, Chon H, Grinberg A, Crouch RJ (2018) Two RNase H2 mutants with differential rNMP processing activity reveal a threshold of ribonucleotide tolerance for embryonic development. Cell Rep 25: 1135–1145.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Sundaralingam M (2000) B‐form to A‐form conversion by a 3’‐terminal ribose: crystal structure of the chimera d(CCACTAGTG)r(G). Nucleic Acids Res 28: 4356–4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Wang G, Feng X, Shepherd P, Zhang J, Tang M, Chen Z, Srivastava M, McLaughlin ME, Navone NM et al (2018) Genome‐wide CRISPR screens reveal synthetic lethality of RNASEH2 deficiency and ATR inhibition. Oncogene 38: 2451–2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt DL, Johansson E, Burgers PM, Kunkel TA (2011) Replication of ribonucleotide‐containing DNA templates by yeast replicative polymerases. DNA Repair (Amst) 10: 897–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Clausen AR, Nick McElhinny SA, Watts BE, Johansson E, Kunkel TA (2012) Proofreading of ribonucleotides inserted into DNA by yeast DNA polymerase ε. DNA Repair (Amst) 11: 649–656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Smith DJ, Marjavaara L, Lujan SA, Chabes A, Kunkel TA (2013) Topoisomerase 1‐mediated removal of ribonucleotides from nascent leading‐strand DNA. Mol Cell 49: 1010–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Clausen AR, Lujan SA, Marjavaara L, Clark AB, Burgers PM, Chabes A, Kunkel TA (2015) Evidence that processing of ribonucleotides in DNA by topoisomerase 1 is leading‐strand specific. Nat Struct Mol Biol 22: 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JS, Gehle DB, Kunkel TA (2017) The role of RNase H2 in processing ribonucleotides incorporated during DNA replication. DNA Repair (Amst) 53: 52–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick‐Mattheuer M, Kretschmer S, König N, Kurth T, Wieczorek D et al (2016) RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat Commun 7: 11752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Shen B (2011) Okazaki fragment maturation: nucleases take centre stage. J Mol Cell Biol 3: 23–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer AD, Koshland D (2016) Differential roles of the RNases H in preventing chromosome instability. Proc Natl Acad Sci USA 113: 12220–12225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Murina O, Reijns MAM, Agathanggelou A, Challis R, Tarnauskaite Ž, Muir M, Fluteau A, Aregger M, McEwan A et al (2018) CRISPR screens identify genomic ribonucleotides as a source of PARP‐trapping lesions. Nature 559: 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]