

SUMMARY
The mechanisms by which feeding and fasting drive rhythmic gene expression for physiological adaptation to daily rhythm in nutrient availability are not well understood. Here we show that, upon feeding, the RNA binding protein NONO accumulates within speckle-like structures in liver cell nuclei. Combining RNA-immunoprecipitation and sequencing (RIP-seq), we find that an increased number of RNAs are bound by NONO after feeding. We further show that NONO binds and regulates the rhythmicity of genes involved in nutrient metabolism post-transcriptionally. Finally, we show that disrupted rhythmicity of NONO target genes has profound metabolic impact. Indeed, NONO-deficient mice exhibit impaired glucose tolerance and lower hepatic glycogen and lipids. Accordingly, these mice shift from glucose storage to fat oxidation, and therefore remain lean throughout adulthood. In conclusion, our study demonstrates that NONO post-transcriptionally coordinates circadian mRNA expression of metabolic genes with the feeding/fasting cycle, thereby playing a critical role in energy homeostasis.
eTOC blurb
Benegiamo et al. identify a metabolic role for the nuclear RNA-binding protein NONO. Upon feeding, NONO binds to pre-mRNAs of genes involved in glucose and fat metabolism in the liver, regulating their rhythmic expression post-transcriptionally. NONO-deficient mice exhibit disruptions in this temporal regulation and shift from glucose storage to fat oxidation.
Graphical Abstract
INTRODUCTION
Diurnal rhythms in gene expression are crucial for metabolic homeostasis. Such rhythms synchronize the transcription of genes necessary for anabolic and catabolic metabolism with periods of feeding and fasting, respectively (Panda, 2016). These oscillations are driven in part acutely in response to feeding and fasting, in part by an endogenous circadian oscillator present in nearly all cells and tissues, and in part systemically (Brown, 2016). Indeed, genetic disruption of the molecular clock or an erratic eating pattern can each disrupt the temporal coordination between metabolic demand and gene expression, leading to metabolic disease (Baron and Reid, 2014; Bass and Takahashi, 2010; Zarrinpar et al., 2016). However, mechanisms by which the feeding/fasting cycle and gene expression are temporally coordinated have not been fully elucidated.
The liver plays an important role in metabolic homeostasis, as it is the primary site for the daily metabolism of macronutrients (e.g. carbohydrates, lipids and amino-acids). In turn, glycogen is predominantly stored there during feeding, and during fasting this glycogen is largely returned to glucose each day. Consistent with this rhythmic function, ~24h rhythms in both mRNA and protein levels are associated with a large fraction of the liver coding genome (Hughes et al., 2009; Reddy et al., 2006; Vollmers et al., 2012; Zhang et al., 2014). Several studies have shown that the feeding/fasting cycle is the main driver of daily rhythm in mRNA levels for the liver (Atger et al., 2015; Damiola et al., 2000; Sobel et al., 2017; Vollmers et al., 2009). While a number of these genes have daily expression patterns driven primarily by transcriptional rhythms, cycling of most mature mRNAs is not driven by underlying rhythms in transcription (Koike et al., 2012; Menet et al., 2012), suggesting the importance of as yet unknown post-transcriptional mechanisms.
Circadian rhythms in cellular function are mostly driven by a circadian oscillator composed of a cell-autonomous transcriptional-translational feedback loop, in which heterodimers of the activator proteins CLOCK and BMAL bind to E-box promoters and drive the expression of the repressors CRYs (Cry1, Cry2) and PERs (Per1, Per2, Per3). CRY and PER heterodimers then translocate back into the nucleus, and repress activities of the BMAL-CLOCK complex, thus establishing circadian rhythmicity in gene expression (Lowrey and Takahashi, 2011; Panda et al., 2002).
Post-transcriptional regulatory mechanisms (such as splicing, localization, polyadenylation, stabilization and degradation) are driven by RNA binding proteins (RBPs) that often mediate complex activities involving interactions with other proteins and RNA species (Uren et al., 2012). The RBP non-POU domain-containing octamer binding (NONO) belongs to the Drosophila Behavior Human Splicing (DBHS) family that includes the paralogues SFPQ and PSPC1. These predominantly nuclear proteins have two RNA-recognition motives (RRMs) and are defined as ‘multifunctional’, as they regulate gene expression in numerous ways, affecting transcriptional activation and inhibition, as well as RNA splicing, stabilization, and export (Knott et al., 2016). NONO, SFPQ, and PSPC1, together with other pre-mRNA splicing factors, transcription factors, and heterogeneous nuclear ribonucleoproteins (hnRNPs), are also components of paraspeckles - subnuclear bodies assembled on the long non-coding RNA Neat1 (Fox and Lamond, 2010; Fox et al., 2002; Yamazaki and Hirose, 2015). Interestingly, NONO interacts with PER proteins and affects circadian rhythmicity in flies and in mammalian cell lines (Brown et al., 2005; Kowalska et al., 2012, 2013). A second, and likely not mutually exclusive, aspect of DBHS family proteins is their role in regulating RNA expression in response to stress stimuli (Prasanth et al., 2005).
Here we find that feeding increases the number of NONO-containing, speckle-like structures in nuclei of liver cells. NONO interacts with RNA processing factors in the liver and the number of NONO-bound RNAs increases upon feeding. NONO primarily binds promoter-proximal introns of transcripts. A large fraction of NONO-bound RNAs encodes proteins implicated in glucose uptake and macronutrient metabolism. Furthermore, we demonstrate that NONO post-transcriptionally regulates mRNA levels of these genes in response to a nutritional stimulus. The absence of NONO-mediated regulation of target RNAs profoundly impacts metabolic health. Indeed, NONO-deficient mice exhibit impaired glucose tolerance, reduced capacity to store glycogen and lipids in the liver, and a lean phenotype. Together these results indicate that NONO coordinates the pre-mRNA processing of metabolic genes in response to nutritional stimuli. We therefore propose that post-transcriptional mechanisms directed by NONO represent an important regulatory node at the intersection between cycles of feeding/fasting and rhythmic gene expression.
RESULTS
Feeding increases the number of NONO-containing speckle-like structures in nuclei of liver cells
Since NONO plays a role in the circadian clock and is involved in the response to stress stimuli, we analyzed NONO protein levels in the mouse liver in response to fasting and feeding. Mice were fed a normal chow only during the dark phase of a 12 h light:12 h dark cycle (i.e., a 12:12 LD cycle) for one week. Livers were then harvested after 12 h of fasting, after 2 h of re-feeding (i.e., 2 h of feeding following the 12 h fast) or 60 minutes after an intraperitoneal glucose injection following light-phase fasting. In these conditions, NONO protein levels remained unchanged, as shown by immunoblotting and nuclear immunostaining (Figure S1A, S1B and S1E). Using super-resolution microscopy, however, we observed dramatic changes in NONO subnuclear localization (Figure 1A and 1D). In general, NONO was diffusely distributed throughout liver-cell nuclei after fasting. After feeding, or an intraperitoneal administration of glucose, the number of NONO-containing subnuclear speckle-like structures increased (Figure 1B, 1C, 1E, and 1F). This increase in the number of speckle-like structures was not due to an increase in nuclear volume (Figure S1C, S1D, S1F, S1G). Round-the-clock immunofluorescence in liver of ad-libitum fed mice kept in constant darkness did not show statistically significant circadian variation in NONO-containing speckle-like structures, likely due to uncontrolled feeding (data not shown). Feeding-induced speckle-like structures localized to the interchromatin space of the nucleus, spatially separated from heterochromatin rich DAPI foci. The increased number of NONO-containing speckle-like structures in liver nuclei after feeding or an acute glucose challenge suggests that NONO may play a role in hepatic responses to nutritional stimuli.
Figure 1. NONO-containing speckle-like structures increase in the nucleus of the hepatocyte upon feeding.

(A) NONO immunostaining on liver cryosections in fasted mice or 2h after re-feeding. (B) Distribution of the number of speckle-like structures per nucleus in fasted mice or 2h after re-feeding. (C) Average number of speckle-like structures per nucleus. (D) NONO immunostaining on liver cryosections in fasted mice or 1h after glucose injection. (E) Distribution of the number of speckle-like structures per nucleus in fasted mice or 1h after glucose injection. (F) Average number of speckle-like structures per nucleus. (B), (C), (E), and (F), n>170 nuclei. In (C) and (F) results are represented as box and whiskers: 10–90 percentile range, ‘+’ sign represents mean. Statistical analysis student t test with Welch’s correction, ***p<0.0001. Scale bar=3μm. See also Figure S1.
NONO interacts with RNA processing factors
Most RBPs interact with each other and with other proteins in macromolecular complexes to regulate every step of the RNA life cycle, such as transcription, splicing, transport, and translation (Gehring et al., 2017). To characterize NONO-containing macromolecular complexes in different nutritional conditions, we performed immunoprecipitation followed by mass spectrometry (IP-MS) of NONO from mouse liver nuclei collected at three different times of the day. Again, mice were fed only during the dark phase of a 12:12 LD cycle, and samples were collected after 10 h of fasting (ZT10), 2 h after re-feeding (ZT14), and towards the end of the feeding period (ZT22) (Figure S2A and S2B). We found that the most abundant interactions were common to all the three time points (Figure S2C). Among NONO interacting proteins were the two known paralogues and direct NONO interactors: SFPQ and PSPC1. We also found that several HNRNPs, some splicing factors, RNA-binding proteins and one of the main components of the DBC1–ZIRD complex (DBIRD) were NONO interactors (Figure S2D and Table S1). Several of these interacting factors also localize to paraspeckles (Naganuma et al., 2012; Yamazaki and Hirose, 2015) and/or were previously identified as components of messenger ribonucleoprotein complexes (mRNPs), involved in mRNA metabolism and post-transcriptional gene regulation (Close et al., 2012; Mannen et al., 2016; Sánchez-Jiménez and Sánchez-Margalet, 2013; Weidensdorfer et al., 2009). Although NONO was previously shown to interact with the PER protein complex (Brown et al., 2005), no circadian clock proteins were identified in our IP-MS analysis, suggesting that the circadian sub-complex may not be a major component of the NONO complex in the liver. This detailed characterization of NONO complexes suggests that NONO interacts with ribonucleoprotein complexes in the liver and may play a role in mRNPs assembly, RNA metabolism and post-transcriptional gene regulation.
The number of transcripts bound by NONO increases upon feeding
To identify RNAs bound by NONO complexes in different nutritional conditions, we performed RNA immunoprecipitation followed by sequencing (RIP-seq) of NONO from mouse liver nuclei collected after 10 h of fasting (ZT10), 2 h after re-feeding (ZT14) and towards the end of the feeding period (ZT22) (for mice fed during the dark phase of a 12:12 LD cycle) (Figure 2A and 2B). We identified a total of 1140 transcripts that bound NONO during at least one of the three time points. Importantly, NONO bound more transcripts at ZT14 (2h after re-feeding), compared with the two other time points; NONO bound 373 transcripts at ZT10, 833 at ZT14, and 452 at ZT22 (Figure 2C and Table S2). Furthermore, at ZT14 the average size of NONO binding peaks (i.e., the number of base pairs encompassed by each peak) also increased; average peak sizes were 2203 ± 1611 bp at ZT10, 3060 ± 1723 bp at ZT14 and 2202 ± 1141 bp at ZT22 (Figure 2D and 2E). This difference was confirmed when we compared only the 134 transcripts that were common to the three time-points (Figure S3A). This suggests that NONO binds a higher number of RNAs at the beginning of the feeding phase and it likely binds these RNAs in bigger macromolecular complexes. Among NONO targets at ZT10 was the non-coding RNA NEAT1, which serves as a scaffolding RNA for the formation of paraspeckles (Fox et al., 2002). About 80% of NONO binding sites were within introns, in particular we found enrichment for peaks mapping to intron 1 (Figure 2F and 2G). Indeed, relative distance of the identified intron peaks from the transcription start site (TSS) normalized to gene size showed a tendency for NONO to bind regions of the RNA proximal to the TSS (Figure S3B). Moreover, NONO did not bind uniformly throughout each intron, but instead was enriched within specific regions of each bound intron (Figure 2I and Figure S3C–E). In silico analyses identified 6 putative binding-site motifs significantly enriched at introns (Figure 2H). (It should be noted that although the composition of these binding site motifs was consistent across time points, more stringent mapping techniques and mutation analyses would be needed to establish more precise consensus sequences). Together, these data suggest that NONO primarily binds to pre-mRNA and the number of NONO-bound RNAs increases at the beginning of the feeding phase.
Figure 2. NONO preferentially binds introns and the number of bound transcripts increases upon feeding.

(A) Experimental design indicating the time points at which liver were collected for the RIP-seq (see text for details). (B) Outline of the experimental methods for the NONO RIP-seq in liver nuclei (see also STAR METHODS). (C) Number of genes bound at each time point and overlap among time points. (D) Peak size distribution at each time point, number of peaks = 380 (ZT10), 986 (ZT14), 497 (ZT22). (E) Average peak size for all the peaks identified at each time point. (F) Annotation of the NONO RIP-seq peaks, n=3. (G) Intron position distribution of the RIP-seq peaks, n=3; (H) Significantly enriched RNA motifs among the RIP-seq intron peaks; (I) UCSC genome browser view of the RIP-seq reads mapping to Gck gene, the right panel highlights the NONO RIP-seq peaks in Gck intron 4. Each time point is a pool of 2 independent RIP experiments. In (E) results are represented as box and whiskers: 10–90 percentile range, ‘+’ sign represents mean value. In (F) and (G) results are represented as mean ± SEM. Statistical analysis one-way ANOVA, ***p<0,0001. See also Figure S3 and Table S2.
NONO regulates the rhythmicity of its target RNAs post-transcriptionally
The liver’s circadian transcriptome results from the combined actions of the circadian clock and feeding/fasting rhythms (Damiola et al., 2000; Vollmers et al., 2009). Since: 1) the number of NONO-containing speckle-like structures increased in response to feeding, 2) NONO interacted with RNA processing factors, and 3) NONO bound to a different number of genes at different times of the day; we hypothesized that NONO may contribute to the daily rhythm of hepatic gene expression. To address whether NONO contributes to rhythmic gene expression in response to feeding/fasting, diurnal liver transcriptomes were assessed in nonogt mice (i.e. mice lacking Nono mRNA and protein expression; Kowalska et al., 2012) and wildtype (WT) littermates. Nonogt mice exhibit a slight change in circadian period length when placed in constant darkness, but under a 12:12 LD cycle their daily activity:rest cycle is indistinguishable from WT littermates (Kowalska et al., 2012). Mice were habituated to have food available only at lights off (ZT12–24) for one week. During the second week, mice were collected every 2 h throughout the 24h day (Figure 3A). Both total and nuclear RNAs from a pool of two biological replicates were sequenced at two-hour resolution (Figure 3B). (For statistical reasons to determine accurate circadian phase, we favored a higher density of time points over a lower one with conventional identically-timed replicates (Li et al., 2015)). In these conditions NONO mRNA and protein levels showed constant amount across the 24 h (Figure S4A and S4B).
Figure 3. NONO regulates the rhythmicity of its target RNA post-transcriptionally.

(A) Experimental design indicating the time points at which livers were collected for transcriptome analysis. (B) Schematic of the experimental methods for the liver transcriptome (see also STAR METHODS). (C) Distribution of high throughput sequencing signal within exon (blue), intron (green) and intergenic sequences (yellow) in the nuclear RNA-Seq (left panel) and in the total RNA-Seq (right panel) datasets. (D) Percentage of genes cycling at the mature mRNA level (tExon dataset) in the whole liver transcriptome (left panel) and among the NONO target genes (right panel) calculated by combining the RIP-seq and WT diurnal transcriptome datasets. (E) Normalized profile of expression of NONO-bound cycling genes (mature mRNA, tExon dataset) in WT and nonogt mice at the indicated time points. High expression is displayed in orange, low expression in blue. Each time point is a pool of 2 mice. (F) Peak phase distribution of the same genes as in (E) separated by bins of 1h: right panel mature mRNA peak phases (tExon, n=231), left panel corresponding pre-mRNA peak phases (nIntron, n=58). (G) Distribution of the mature mRNA (tExon) peak phase difference between WT and nonogt mice. Dashed line indicates mean phase difference (1.7h). See also Figure S4, S5, Table S3 and S4.
In the total-RNA fraction the majority of reads (71%) mapped to exons, whereas in the nuclear-RNA fraction, 76% of reads mapped to introns, suggesting that the nuclear RNA is enriched in pre-mRNAs and newly transcribed RNAs (Figure 3C). Thus, for further analysis we considered the exon FPKM (Fragments Per Kilobase per Million mapped reads) from the total-RNA fraction (tExon) as an estimation of mature mRNA levels, and the intron FPKM from the nuclear-RNA fraction (nIntron) as an approximation of pre-mRNA levels and transcriptional activity (Gaidatzis et al., 2015).
In the experimental conditions described above, 22% (3152 out of 14527) of the genes expressed in the liver of WT mice showed a 24 h rhythm in mature mRNA levels (tExon dataset, Figure 3D left), with peak mRNA levels coinciding with the end of fasting or feeding time periods (Figure S4E), in agreement with other recent studies (Atger et al., 2015; Vollmers et al., 2012). However, only 32% of these cycling mRNAs were transcribed in a cyclical fashion (nIntron dataset; Table S3). This suggests that post-transcriptional mechanisms likely account for the cycling of most mature mRNAs, as has been suggested by other recent reports (Koike et al., 2012; Menet et al., 2012). Comparing the NONO RIP targets that resulted to be expressed in the liver (n=943) with the cycling transcriptome revealed that 36% (338) of these NONO targets exhibited circadian rhythm in mature mRNA levels (Figure 3D right and Table S4). Importantly, daily fluctuations in the mature mRNA levels for these genes were altered in nonogt mice, such that mature mRNA levels for 107 genes were no longer rhythmic, and peak levels of mature mRNA for 231 genes were delayed by 2h on average (Figure 3E and 3G). This delay is likely induced post-transcriptionally: for the subset of genes that were rhythmic both transcriptionally (nIntron dataset) and at the level of mature mRNA accumulation (tExon dataset), 58 genes in total, the delay in peak expression between nonogt and WT mice was only observed in the tExon dataset (Figure 3F, right), whereas timing within the nIntron dataset remained identical between the two genotypes (Figure 3F, left).
Even at the genome-wide level, for genes that cycled in both WT and nonogt mice, irrespective of whether or not they were NONO targets, peak levels of mature mRNAs were also delayed by 1h on average, but peak levels of transcription were not (Figure S4C–F). The phase delay observed when exon sequences from the total RNA were compared with intron sequences from the nuclear RNA did not result from differences in RNA preparation, since we did not observe any delay when comparing the intron phases of expression from the nuclear and total RNA fractions (nIntron vs tIntron, Figure S4F). Together, these data suggest that: 1) for NONO direct targets, the loss of NONO severely impacts both rhythmicity and phase of their mature mRNA, but not transcription of the genes themselves, and 2) the loss of NONO also indirectly affects the phase of oscillation for mature mRNAs associated with other cycling genes in the liver.
Daily patterns of both mature mRNA levels (tExon) and transcription (nIntron) of known core circadian clock components did not show large alterations in their oscillation profile in nonogt liver (Figure S5H–N). All core clock genes showed strong rhythmicity in transcription, which preceded rhythms in mature mRNA levels by 1–2 h in both genotypes. Thus, the effects we document are likely independent of oscillations in core clock components.
NONO regulates glucose-induced gene expression post-transcriptionally and is required for normal glucose homeostasis
We have shown that the number of NONO-bound RNAs increases upon feeding and that NONO regulates the oscillation of its target mRNAs post-transcriptionally. To test the physiological relevance of NONO function, we examined the predicted functions of its rhythmic target genes. NONO target genes were significantly enriched in gene ontology terms related to metabolism (Table S5). KEGG functional annotation of the same genes specifically revealed carbohydrate and amino-acid metabolism as major functional clusters (Figure 4A, 4B and Table S5).
Figure 4. NONO regulates glucose induced gene expression post-transcriptionally.

(A) Enriched KEGG pathways (Z-score >2.5) among the NONO-bound cycling genes. (B) Glucose metabolism related pathways enriched among the NONO-bound cycling genes. Grey squares represent pathways; pathways names are indicated in blue. NONO target genes are represented as yellow circles, for each NONO-target gene the gene name is indicated in black. Grey arrows connect each gene to the pathways it belongs to. (C) GCK and GLUT2 protein expression during the dark/feeding phase and protein quantification (right panels) at the beginning of the dark phase (ZT10–16) and at the end of the dark phase (ZT18–22). ZT10–16 n=4 per group, ZT18–22 n=3 per group. (D) Experimental design for (E), in order to acutely induce Gck and Glut2 expression mice were fasted for 12 hours and injected intraperitoneally with glucose (2g/kg). WT and nonogt liver were collected either at T0 or 60 minutes after glucose injection (T60); (E) qPCR of Gck and Glut2 intron or spliced mRNA levels at T0 and T60, n=3–4 per group per time point. Statistical analysis for (E), two-way ANOVA, Bonferroni posttest. Statistical analysis for (C), student t test. *p<0.05. Results are represented as mean ± SEM. See also Table S5.
Consistent with the mechanistic role we propose for NONO, under a 12-h fasting:12-h feeding cycle, peak levels of Glucokinase (Gck) and Glucose transporter-2 (Glut2) mRNAs were delayed in nonogt mice (Figure S5A and S5E). Correspondingly, delays in these mRNA peaks affected GCK and GLUT2 protein levels in the liver, as levels of both proteins were reduced at the beginning of the feeding phase in nonogt mice (Figure 4C).
To further test and confirm our hypothesis that NONO is important for pre-mRNA processing of these transcripts, we measured their expression before and after an acute intraperitoneal injection of glucose into WT and nonogt mice (Figure 4D). Sixty minutes (T60) after glucose injection, the fold induction (compared to time 0 (T0)) for Gck and Glut2 spliced mRNA was significantly lower in nonogt mice compared to the WT littermates. In contrast, no differences in intron induction were seen between the two genotypes, demonstrating that NONO regulates these genes post-transcriptionally in response to glucose (Figure 4E).
Both Glut2 and Gck bind glucose with low affinity and act as glucose sensors for the hepatocytes during the fed state, when glucose levels rise in the blood (Figure 5A; Massa et al., 2011; Thorens and Mueckler, 2010). Thus, reduced levels of GLUT2 and GCK in nonogt mice would be predicted to cause physiological changes in glucose utilization. This is exactly what we find: nonogt mice showed increased post-prandial blood glucose levels when administered a glucose tolerance test (GTT) (Figure 5B), whereas their insulin sensitivity was unaffected (Figure 5C). In addition, nonogt and WT mice exhibited similar fasting glucose and insulin levels (Figure 5D and 5E). These data suggest that the impaired glucose tolerance in nonogt mice reflects the role played by NONO in regulating glucose uptake in the hepatocytes. In the liver, glucose-6-phosphate is used in glycogen synthesis, glycolysis, and the pentose phosphate pathway, and is eventually stored as fat (Figure 5A). Accordingly, we measured lower levels of hepatic glycogen at the end of the feeding period in nonogt mice compared to WT controls (Figure 5F).
Figure 5. NONO expression in the liver is required for normal glucose homeostasis.

(A) Illustration of the role of GCK and GLUT2 in glucose uptake in the hepatocyte. (B) Glucose tolerance test. Right panel: area under the curve (AUC), n=10 per group. (C) Insulin tolerance test. Right panel: AUC, WT n=9, nonogt n=8. (D) Fasting glucose and (E) fasting insulin measured in the same group of mice after 12h of light-phase fasting, WT n=10, nonogt n=8. (F) Hepatic glycogen content at the end of the fasting phase (fasted: ZT8–12) and at the end of the feeding phase (fed: ZT18–22), n=6 per group. (G) Scheme of NONO protein main domains, in red are indicated the mutations in the NONO protein sequence that disrupt the ability of NONO to bind RNA used for the experiment in (L). (H) Scheme of the AAV8 viral constructs used for the experiment in (K) and (L). (I) Experimental scheme for (K) and (L): on week 1 glucose tolerance was measured in nonogt mice and on week 2 the same mice were injected with either AAV8.NONO (blue) or AAV8.NONOmut (red). After 30 days from the injection glucose tolerance was measured again in the same mice. (J) NONO immunostaining on liver cryosections of WT, nonogt mice or nonogt mice infected with either AAV8.NONO or AAV8.NONOmut, scale bar=3μM. (K) and (L) Glucose tolerance test in nonogt mice before and 30 days after injection with AAV8.NONO (K) or AAV8.NONOmut (L), histograms on the right represent AUC, AAV8.NONO n=5, AAV8.NONOmut n=7. Statistical test for (B), (C), (K) and (L) two-way ANOVA, Bonferroni posttest, on the bottom left of each panel is indicated the ANOVA p value for time-group interaction. Statistical test for (D), (E), (F) and AUC histograms student’s t test. *p<0.05, **p<0.01, ***p<0.001.
Changes in glucose metabolism and glycogen storage could theoretically derive either from alterations in the liver (the site of this metabolism and storage) or in the pancreas (secreting the insulin and glucagon that regulate it). To test the extent to which loss of NONO from adult mouse liver cells contributes to glucose intolerance, we restored NONO expression in nonogt mice using adeno-associated viruses (AAVs) that expressed NONO under the control of the liver-specific TBG promoter. We injected two different AAV2/8 viral vectors, one that expressed wildtype NONO, and one that expressed a mutant version of NONO with 4 point mutations (2 in each of the RRM domains) (Figure 5G and 5H). These mutations disrupt the ability of NONO to bind RNA (Kuwahara et al., 2006). Levels of AAV-derived NONO protein in liver cells were comparable to those observed in WT mice (Figure 5J). Glucose tolerance was tested in nonogt mice before and 4 weeks after AAV injection (Figure 5I). Liver-specific re-expression of WT NONO significantly improved glucose tolerance (Figure 5K), whereas this was not the case for mice expressing the NONO RRM-mutant (Figure 5L). This demonstrated that NONO functions in the liver to influence whole-body glucose homeostasis, and that the ability of NONO to bind RNA is required for this function. Together these data suggest that NONO binds and post-transcriptionally regulates the expression of glucose-responsive genes in the liver, and that NONO is important for whole-body glucose homeostasis and glycogen storage in hepatocytes.
Nonogt mice store less fat and exhibit increased fat catabolism
To further characterize the role of NONO in liver metabolism we analyzed the metabolomes of fasted and re-fed animals, both nonogt and WT. Livers were collected toward the end of the fasting phase (ZT8, ZT10, and ZT12) and toward the beginning of the feeding phase (ZT14, ZT16, and ZT18). Principal component analysis of the 12 samples showed their separation along genotype and feeding condition. Consistent with our RNA analyses, we found more separation between WT and nonogt mice when the two re-fed groups were compared (Figure S6A). We found 182 significantly affected bio-chemicals when comparing WT and nonogt fed groups, whereas only 86 metabolites changed in comparisons between WT and nonogt fasted groups (Figure S6B). In the fed state, nonogt livers contained significantly lower levels of the glycolytic intermediates 3-phosphoglycerate, and phosphoenol pyruvate (PEP), as well as the pentose phosphate pathway intermediate 6-phosphogluconate (Figure S6C–E). In contrast, the mitochondrial TCA cycle intermediates were elevated (Figure S6F–I). In the transcriptome dataset, the cycling NONO target ATP-citrate-lyase (Acly) exhibited reduced level of expression and a phase delay (Figure S5D). ACLY uses citrate (an intermediate in the TCA cycle) and coenzyme A (CoA) to synthesize cytoplasmic Acetyl CoA. Accordingly, we found increased amounts of citrate, aconitate, succinate, and CoA in nonogt livers during the fed state (Figure S6F–I). Cytoplasmic acetyl CoA is the building block for many molecules, including fatty acids. Another NONO target gene that is important for lipid metabolism is mitochondrial glycerol-3-phosphate acyltransferase (Gpam). GPAM catalyzes the initial and committing step in glycerolipid biosynthesis and plays a pivotal role in the regulation of cellular triacylglycerol levels. Gpam is expressed at high level during the feeding phase, however nonogt mice showed delayed and reduced levels of Gpam mature mRNA (Figure S5B). Accordingly, nonogt livers had reduced levels of triglycerides, reduced overall liver lipid content, and increased levels of glycerol-3-phospate (Figure 6A, 6B and S6J). Increases in glycerol-3-phosphate in the fasted and fed states might be due to increased breakdown of triglycerides. Indeed, we also found high levels of triglyceride catabolism intermediates (diacylglycerols, monoacylglycerols and fatty acids) in the livers of nonogt mice during the fed state (Figure S6J).
Figure 6. Nonogt mice have reduced body fat and body weight.

(A) Representative image of hepatic lipid staining by Oil-Red-O (ORO) in WT and nonogt mice at 45 weeks of age and quantification of the ORO staining signal (right panel), n=3 per group. (B) Hepatic triglycerides content quantification, WT n=11, nonogt n=8. (C) Body weight of WT and nonogt mice form 6 to 42 weeks of age measured weekly, WT n=14, nonogt n=13. (D) Cumulative food consumption of the same mice as in (C), calculated from the cage average food consumption measured weekly, n=4 cages per group. (E) Cage activity (15 minutes bin) average of 4 consecutive days and 24h average cage activity (right panel), n=4 per group. (F) Representative images of WT and nonogt mice. (G) Representative H&E staining images of the epididymal fat pads at 42 weeks of age, scale bar=200μm. (H) and (I) Fat mass and lean mass of the same mice as in (C) measured every 3 weeks starting from 14 weeks of age, WT n=14, nonogt n=13. (J) Left panel: respiratory exchange ratio (15 minutes bin), average of 4 consecutive days. Right panel: histogram representing day and night average RER, n=4 per group. (K) Adipocyte size distribution and average adipocyte diameter (right panel) calculated from the epididymal fat H&E staining images, n=5. Statistical analysis for (A), (B), (E), (J) and (K) Student t-test. Statistical analysis for (C), (D), (H), and (I) 2-way ANOVA, on each panel is indicated the ANOVA p value for time-group interaction. *p<0.05, **p<0.01, ***p<0.001 (for B), ***p<0.0001 (for C and H). Results are represented as mean ± SEM. See also Figure S6 and S7.
To assess the physiological consequence of NONO deficiency, we evaluated the metabolic phenotype of nonogt mice. Nonogt mice are born at Mendelian ratio and survive normally into adulthood. At 10 weeks of age, nonogt mice weighed 10% less than WT littermates (Figure S7A). When fed a normal chow ad libitum, nonogt mice and WT littermates consumed equivalent amounts of food, and exhibited comparable daily activity levels (Figure 6D, 6E, and S7B). Over the next 32 weeks, nonogt mice gained less weight, such that at 42 weeks they weighed 20% less than their WT littermates (Figure 6C and S7A). Body composition analysis revealed that nonogt mice accumulated less fat mass, whereas their gains in lean mass were similar to those seen in WT mice (Figure 6H and 6I). Both leptin and ghrelin levels were reduced in nonogt mice (Figure S7C and S7D). However, since food consumption in any case was unaltered, we turned to energy utilization as the more likely source of body weight differences.
To examine whole-body energy utilization, we assessed nonogt and WT littermates using indirect calorimetry. During the fed state, the respiratory exchange ratio (RER) for both genotypes was equivalent and close to 1, indicating that carbohydrates were used as the predominant energy source. However, during the light period, when mice are usually at rest and have reduced food intake, the RER slowly dropped to ~0.8 in WT mice, whereas in nonogt mice, the RER rapidly dropped to 0.7 (Figure 6J), indicating increased reliance on fatty acids as an energy source for nonogt mice. The relatively quick decline in fasting RER in nonogt mice may also indicate reduced energy storage during the feeding phase. nonogt mice also exhibited reduced abdominal fat deposits and smaller adipocytes (Figure 6F, 6G and 6K).
When nonogt mice were fed a normal chow (60% of the energy coming from carbohydrates), they weighed less and had less body fat. This reduced adiposity could have resulted from a primary defect in fat absorption and storage. To test this hypothesis, we subjected nonogt and WT littermates to ad libitum feeding of a high fat diet (HDF; 60% of the energy coming from fat) for 12 weeks. The two genotypes consumed equivalent amounts of food (Figure S7F). After 12 weeks of a HFD, the nonogt and WT mice had equivalent body weights and whole-body fat mass (Figure S7E and S7G). As anticipated, nonogt mice fed a HFD were more glucose intolerant compared to their WT littermates (Figure S7H). This demonstrates that a HFD can compensate for the inefficient glucose uptake seen in nonogt mice fed a normal chow. Together, these data indicate that nonogt mice have three inter-related metabolic defects in the liver: 1) reduced glucose utilization in glycolytic and glycogen synthesis pathways, 2) impaired triglycerides synthesis, and 3) increased reliance on fatty acids as an energy source during fasting.
DISCUSSION
Rhythms in gene transcription are the first step toward generating oscillations in mRNA levels, but recent studies have revealed that a wide range of factors contribute to this oscillations. Regulators of transcription elongation, as well as RNA processing, transport, translation, and degradation may affect steady-state mRNA levels at any given time (Benegiamo et al., 2016). It has been suggested that the dynamic regulation of transcription, mRNA processing, and mRNA degradation are all required to generate high-amplitude oscillation in mRNA levels (Rabani et al., 2014).
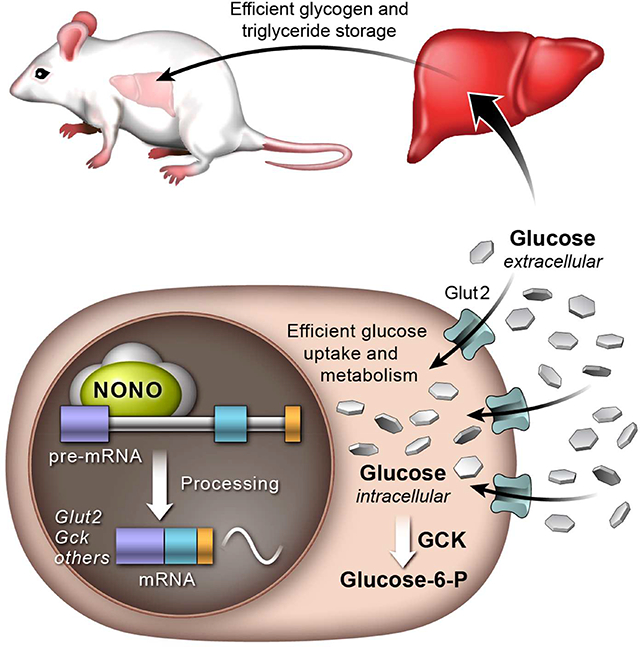
Here we show that the RNA-binding protein NONO plays an important role in coordinating the rhythmic expression of genes involved in glucose and fat metabolism with the feeding/fasting cycle. Furthermore, NONO regulates these genes post-transcriptionally. We propose a mechanism in which NONO enhances the processing of target RNAs to allow robust in-phase oscillations (Figure 7). Further studies are required to understand precisely how NONO affects this process. Previous studies have shown that NONO and SFPQ interact with the carboxy-terminal domain of RNA-polymerase II, leading to the hypothesis that these factors couple transcription to post-transcriptional events (Emili et al., 2002). Our mass spectrometry data further support this idea, and we speculate that NONO may function as a molecular scaffold to assemble the mRNA processing machinery on target RNAs. For example, levels of Gck mature mRNA peak toward the end of the night, yet NONO binds to Gck pre-mRNA at all time points tested (ZT10, ZT14 and ZT22). Thus, we hypothesize that NONO primes its target pre-mRNAs for processing. In the absence of NONO, this recruitment is less efficient, leading to delays in the phase of oscillation or loss of rhythmicity.
Figure 7. Proposed model of how NONO regulates gene expression in the liver and its physiological consequences.

(A) In the presence of NONO, NONO-target RNAs involved in glucose metabolism and other metabolic pathways are efficiently processed to allow robust and in phase oscillation and normal glucose uptake and storage in the hepatocytes. (B) When NONO is absent, pre-mRNA processing of its target genes is less efficient and this leads to loss of oscillation or delay in the phase of oscillation. Altered expression pattern of NONO-target genes has a profound metabolic impact; inefficient glucose uptake that leads to impaired glucose tolerance, reduced glucose and fat storage and increased fat breakdown.
In agreement with what has been demonstrated for NonA, the Drosophila homolog of NONO (McMahon et al., 2016), we found that NONO bound primarily to introns. Since our data further point to a role for NONO in post-transcriptional processing, it might therefore play a role in Intron-Mediated Enhancement (IME), an unknown mechanism by which introns may increase gene expression through recruitment of splicing and mRNA export machineries (Reed and Hurt, 2002; Gallegos and Rose, 2015). Consistent with this idea, our data show that NONO preferentially bound promoter-proximal introns, and promoter-proximal introns are more likely to contain sequences important for IME (Rose et al., 2008).
One major unanswered question is how metabolic signals result in the accumulation of NONO in speckle-like structures. This question also remains for all other subnuclear structures containing NONO. One possibility is that increased transcription itself may play a role: not only is transcription required for the formation of Neat1-containing paraspeckles (Mao et al., 2011), but also chromatin modifying complexes associated with transcriptional activation (Kawaguchi et al., 2015). Since feeding in the liver is associated with increase in cellular RNA content (Sinturel et al., 2017), the increase in speckle-like structures may be due simply to increases in transcription. Alternatively, specific phosphorylation events could play a role, as suggested in the case of MNK kinases (Buxadé et al., 2008) or during mitosis (Proteau et al., 2005).
NONO, SFPQ, and PSPC1 often function as heterodimers, and have been described as multifunctional proteins whose specific role in cellular processes may depend on the molecular and cellular context (Knott et al., 2016). Several other roles have been proposed for these factors. As the principal components of nuclear paraspeckles, NONO has been postulated to play a role in nuclear RNA retention by binding to stem-loop structures in the 3’ untranslated regions (UTRs) of messages (Prasanth et al., 2005). Such UTR regions can even heterologously confer circadian rhythmicity in transcript oscillations (Torres et al., 2016). Yet again, NONO and related factors have been implicated as transcriptional cofactors, binding to histone modifiers to regulate chromatin structure and circadian function (Brown et al., 2005; Duong et al., 2011). Recent research suggests that several different classes of NONO-containing nuclear speckle-like structures may exist (Li et al., 2017), and gel filtration analyses suggest that only a subset of NONO-containing speckle-like structures are circadian in nature (Brown et al., 2005). It is tempting to speculate that these different functions may exist in spatially distinct structures such as the feeding-induced speckle-like structures we describe here. Indeed, our mass spectrometric analyses did not detect clock proteins in these feeding-induced NONO complexes, nor were chromatin-modifying complexes abundant, suggesting that they are entities different from the circadian structures described previously. It is also unclear whether these structures contain the long noncoding RNA Neat1 that defines bona-fide paraspeckles.
Further studies are required to assess whether the other two members of the DBHS family function with NONO to regulate metabolic homeostasis. Recent studies have shown that PSPC1 plays a role in adipose tissue development (Wang et al., 2017). It is therefore likely that DBHS proteins play complementary roles in different organs to regulate different aspects of metabolism. In this respect, NONO itself has been shown to: 1) regulate phosphodiesterase mRNA splicing and degradation in human adrenocortical cells to affect glucocorticoids production (Lu and Sewer, 2015), 2) positively regulate lipogenesis in breast cancer cells through SREBP1 (Zhu et al., 2015), and 3) act as an mTOR cofactor (Amelio et al., 2007). We predict that each member of the DBHS family will have unique and common targets in each tissue, and mediate both unique and redundant functions. Here we demonstrate that NONO functions in the liver to maintain whole-organism glucose homeostasis. However, we cannot exclude the possibility that NONO plays important roles in other organs (e.g. the pancreas and muscle) to regulate responses to nutritional stimuli.
In conclusion, we report a novel role for the nuclear RNA-binding protein NONO in metabolic homeostasis. Specifically, NONO binds to pre-mRNAs encoding several key regulators of liver glucose and fat metabolism. NONO enhances the processing of these pre-mRNAs, allowing for robust daily oscillation in mRNA levels, which is necessary to store excess nutrients (as glycogen and triglycerides) during the postprandial period. NONO-deficient mice exhibit disruptions in this temporal regulation, such that animals shift from storing glucose as glycogen and fat to fat oxidation. This results in reduced glucose tolerance, reduced levels of glycogen and triglycerides in the liver, and reduced levels of body fat and body weight (Figure 7). Disruption of the temporal coordination between metabolic demand and gene expression leads to the development of metabolic diseases, like obesity and diabetes. The role of RNA-binding proteins in regulating gene expression under diverse environmental conditions is only now beginning to be elucidated. Our findings help to better understand how metabolic homeostasis is maintained in mammals, and identify novel therapeutic targets for treating diabetes and other associated metabolic dysfunctions.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for reagents and resources may be directed to satchin@salk.edu.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
All animal experiments were carried out in accordance with the guidelines of the IACUC of the Salk Institute, and the approval of the Zürich Cantonal Veterinary Authority. All mice were maintained in a pathogen-free environment and housed in clear shoebox cages in groups of five animals per cage with constant temperature (20–22°C) and humidity and 12h:12h light-dark cycle. All animals had access to water at all times. Unless otherwise indicated, mice were fed ad libitum with a normal chow diet (LabDiet-5010). For the fasting/feeding experiments mice were fasted for 12h during the light phase (ZT0-ZT12) and fed for 12h during the dark phase (ZT12-ZT24). For the HFD experiments mice were fed ad libitum with a diet containing 60% fat (58Y1). Nonogt mice were obtained as described previously (Kowalska et al., 2012) and were backcrossed against the C57BL/6J background for more than 10 generations. Unless otherwise indicated in the figures and figure legends, all experiments were performed in adult mice (10–20 weeks of age). Older mice were used for Figure 6A, 6F, 6G and 6K (40–50 weeks of age). In all experiments wild type and mutant male littermates were compared.
METHOD DETAILS
Liver immunostaining
The livers were perfused with 2 ml of ice-cold PBS through the spleen and immediately collected. Pieces of the main liver lobes were embedded in optimal cutting temperature (O.C.T.) compound (Tissue-Plus, Scigen, 4583) and frozen at −80°C. For cryosectioning the embedded liver was equilibrated at −20°C in a cryos tat chamber and 10μm thick sections were cut. Liver slices were collected on 25×75×1.0mm microscope slides (Superfrost Plus, Fisherbrand) and stored at −80°C. For immunostainin g frozen sections were thawed in a sealed environment to avoid damage by frozen water crystals. Liver sections were fixed at room temperature for 10 minutes in 4% PFA diluted in PBS. Sections were then washed 3 times in TBS-T (50mM Tris, 150mM NaCl, 0.05% Tween-20). The tissue sections were permeabilized in 1% Triton-X 100 diluted in TBS (50mM Tris, 150mM NaCl) for 30 minutes at room temperature. The sections were then incubated in blocking buffer (1% BSA, 0.5% Triton-X 100 in TBS) for 30 minutes. After the blocking step, liver sections were incubated for 1h with rabbit anti-NONO antibody (Kowalska et al., 2012) diluted 1:500 in blocking buffer. After 3 washes in TBS-T, the sections were incubated with the secondary antibody Alexa Fluor 488 conjugate anti-rabbit IgG (Thermo Fisher) diluted 1:1000 in blocking buffer for 30 minutes. Sections were washed three times with TBS-T. At the first wash DAPI was added to the washing buffer at a concentration of 0.5μg/ml. Sections were then mounted and imaged. Images were acquired with a 63x/1.4 NA Apochromat oil objective on an LSM 880 using the Airyscan detector (Carl Zeiss Microscopy). The images were acquired in “SR mode”, which configures the 32 GaAsP detectors on the Airyscan detector as equivalent to 0.2 Airy Units. The 32-channel data was then processed using Zen software (Carl Zeiss Microscopy) (Huff, 2015). All images were acquired with a 63X objective, using 1.8% laser, 750 gain, and 0.97μs pixel dwell time, and they were all processed with an “Airyscan parameter” (Wiener filter strength) of 4.1.
Speckle-like structures and fluorescence quantification
For image analysis Imaris software (Bitplane) was used. For the speckle-like structures quantification we used the ‘spots’ option. We considered ‘speckle-like structures’ objects within the nuclei (identified by DAPI stained areas) with an estimated diameter of 0.3μm, ‘quality’ above 900 and ‘intensity’ above 924. The same parameters were applied to all images. Image analysis was performed automatically by Imaris software, for this reason no blinding was needed. For fluorescence intensity quantification, average intensity within the nuclei was normalized to average intensity outside the nuclei (background).
Immunoprecipitation from liver nuclear lysates
Immunoprecipitation of NONO complexes was performed from mouse liver nuclear extracts. The livers were perfused with 2 ml of ice-cold PBS through the spleen and immediately collected. Liver nuclei were isolated from an entire mouse liver as previously described (Ripperger and Schibler, 2006). Briefly the dissected mouse liver was rapidly homogenized in 5ml of ice cold PBS and mixed to 25ml of 2.4M sucrose buffer (2.4M sucrose, 150mM glycine, 10mM Hepes pH7.6, 15mM KCl, 2mM EDTA, 0.5mM spermidine, 0.15mM spermine, 1mM PMSF, 5mM β-mercaptoethanol and protease inhibitors (pepstatin A, leupeptin, aprotinin, trypsin inhibitor)). The homogenate was layered on top of a 10ml cushion of 2M sucrose buffer (2M sucrose, 10% glycerol, 125mM glycine, 10mM Hepes pH7.6, 15mM KCl, 2mM EDTA, 0.5mM spermidine, 0.15mM spermine, 1mM PMSF, 5mM β-mercaptoethanol and protease inhibitors) and centrifuged for 45min at 24,000rpm at 4°C in a Beckmann SW28 rotor. Isolated liver nuclei were resuspended in 800μl of Nuclei Dilution Buffer (100mM KCl, 0.2mM EDTA, 20% glycerol, 20mM Hepes pH7.6, 1mM PMSF, 5mM β-mercaptoethanol and protease inhibitors) and centrifuged at 5000rpm for 30 seconds. The nuclei pellet was lysed in nuclei lysis buffer (25mM Tris:HCl pH7.4, 187.5mM NaCl, 2.5mM EDTA, 10% NOG and protease inhibitors) and incubated for 20 minutes on ice. Lysates were centrifuged at 10000rpm for 10 minutes at 4°C and the protein concentration in the supernatant was measured with Pierce BCA assay (Thermofisher) following manufacturer’s instructions. To immunoprecipitate NONO complexes, 600μg of nuclear protein lysate were incubated with 10ul of protein A agarose beads cross-linked to polyclonal rabbit anti-NONO antibody (Kowalska et al., 2012) for 2h at 4°C. Imm unoprecipitated NONO complexes were washed 3 times with IP washing buffer (75mM Tris:HCl pH7.4, 150mM NaCl, 1mM EDTA, 10% glycerol, 0.1% NOG and protease inhibitors) and eluted with 0.1M glycine pH 2.5. After elution the acidic glycine was neutralised with tris-base.
Mass spectrometry analysis
Mass spectrometry analysis was performed as described previously (Ma et al., 2016). Samples were precipitated by methanol/chloroform. Dried pellets were dissolved in 8M urea/100mM TEAB, pH 8.5. Proteins were reduced with 5mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP, Sigma-Aldrich) and alkylated with 50mM chloroacetamide (Sigma-Aldrich). Proteins were digested overnight at 37°C in 2M urea/100mM TEAB, pH 8.5, with trypsin (Promega). Digestion was quenched with formic acid, 5% final concentration. The digested samples were analyzed on a Fusion Orbitrap tribrid mass spectrometer (Thermo). The digest was injected directly onto a 30cm, 75um ID column packed with BEH 1.7μm C18 resin (Waters). Samples were separated at a flow rate of 200nl/min on a nLC 1000 (Thermo). Buffer A and B were 0.1% formic acid in water and acetonitrile, respectively. A gradient of 1–25%B over 90min, an increase to 40%B over 30min, an increase to 90%B over another 10min and held at 90%B for a final 10min of washing was used for 140min total run time. Column was re-equilibrated with 20ul of buffer A prior to the injection of sample. Peptides were eluted directly from the tip of the column and nanosprayed directly into the mass spectrometer by application of 2.5kV voltage at the back of the column. The Orbitrap Fusion was operated in a data dependent mode. Full MS1 scans were collected in the Orbitrap at 120K resolution with a mass range of 400 to 1600m/z and an AGC target of 5e5. The cycle time was set to 3 seconds, and within this 3 seconds the most abundant ions per scan were selected for CID MS/MS in the ion trap with an AGC target of 1e4 and minimum intensity of 5000. Maximum fill times were set to 50ms and 100ms for MS and MS/MS scans respectively. Quadrupole isolation at 1.6m/z was used, monoisotopic precursor selection was enabled and dynamic exclusion was used with exclusion duration of 5 seconds. Protein and peptide identification were done with Integrated Proteomics Pipeline – IP2 (Integrated Proteomics Applications). Tandem mass spectra were extracted from raw files using RawConverter (He et al., 2015) and searched with ProLuCID (Xu et al., 2015) against mouse UniProt database. The search space included all fully-tryptic and half-tryptic peptide candidates. Carbamidomethylation on cysteine was considered as a static modification. Data was searched with 50ppm precursor ion tolerance and 600ppm fragment ion tolerance. Data was filtered to 10ppm precursor ion tolerance post search. Identified proteins were filtered using DTASelect (Tabb et al., 2002) and utilizing a target-decoy database search strategy to control the false discovery rate to 1% at the protein level. Interactors were sorted by average emPAI (Ishihama et al., 2005). The top 50 proteins (excluding contaminants and ribosomal proteins) were selected.
Native RNA-immunoprecipitation (RIP)
RIP was performed in native conditions as previously described (Keene et al., 2006; Zhao et al., 2008, 2010), using one mouse liver nuclear lysate per IP. The livers were perfused with 2 ml of ice-cold PBS through the spleen and immediately collected. Liver nuclei isolation from mouse liver has been previously described (Ripperger and Schibler, 2006); briefly the dissected mouse liver was rapidly homogenized in 5ml of ice cold PBS and mixed to 25ml of 2.4M sucrose buffer (2.4M sucrose, 150mM glycine, 10mM Hepes pH7.6, 15mM KCl, 2mM EDTA, 0.5mM spermidine, 0.15mM spermine, 1mM PMSF, 1mM DTT, 400μM VRC and protease inhibitors (pepstatin A, leupeptin, aprotinin, trypsin inhibitor)). The homogenate was layered on top of a 10ml cushion of 2M sucrose buffer (2M sucrose, 10% glycerol, 125mM glycine, 10mM Hepes pH7.6, 15mM KCl, 2mM EDTA, 0.5mM spermidine, 0.15mM spermine, 1mM PMSF, 1mM DTT, 400μM VRC and protease inhibitors) and centrifuged for 45min at 24,000rpm at 4°C in a Beckmann SW28 rotor. Isolated liver nuclei were resuspended in Nuclei Dilution Buffer (100mM KCl, 0.2mM EDTA, 20% glycerol, 20mM Hepes pH7.6, 1mM PMSF, 1mM DTT, 400μM VRC, 80U/ml RNAse OUT and protease inhibitors). Liver nuclei were lysed in Polysome lysis buffer (100mM KCl, 5mM MgCl2, 10mM HEPES pH 7.0, 0.5% NP40, 1mM DTT, 80U/ml RNAse OUT, 400uM VRC and protease inhibitors) and incubated 20 minutes on ice. Lysate was centrifuged and protein concentration in the supernatant was measured with Pierce BCA assay (Thermofisher). 0.5mg of protein were incubated with 40ul of protein A agarose beads pre-coated with 10μg of polyclonal rabbit anti-NONO antibody (Kowalska et al., 2012) or normal rabbit IgG (sc-2027) for 2h at 4°C. Before the incubation, 1/10 of the supernatant was put aside to be used as input. After incubation samples were washed 4 times with NT2 buffer (50mM Tris-HCl pH 7.4, 150 mM NaCl, 1mM MgCl2, 1% Triton-X 100). Immunoprecipitated RNA and input RNA were extracted using Trizol reagent (Invitrogen).
RNA extraction
For RNA extraction the livers were perfused with 2 ml of ice-cold PBS through the spleen and immediately collected. For total liver RNA extraction pieces from the main liver lobes were ground to a powder in liquid nitrogen. About 10mg of tissue were used for RNA extraction using Trizol reagent (Invitrogen). For nuclear RNA extraction liver nuclei were isolated as described above (see Native RNA-immunoprecipitation section). The nuclei pellet was rapidly resuspended in Trizol reagent (invitrogen) and purified.
High-throughput RNA sequencing (RNA-seq)
Liver total, nuclear and RIP purified RNA were used for RNA-seq. RNA from two biological replicates per time point was pooled prior to library preparation. Libraries were prepared using Illumina’s TruSeq Stranded Total RNA Library Prep Kit with Ribo-Zero Gold according to manufacturer’s instructions. In brief, rRNA was depleted from total RNA (1μg) by using subtractive hybridization. The RNA was then fragmented by metal-ion hydrolysis and subsequently converted to cDNA using SuperScript II. The cDNA was then end-repaired, adenylated and ligated with Illumina sequencing adapters. Finally, the libraries were enriched by PCR amplification. All sequencing libraries were then quantified, pooled and sequenced at single-end 50 base-pair (bp) on Illumina HiSeq 2500 at the Salk NGS Core. Each library was sequenced on average 20 million reads. Raw sequencing data was demultiplexed and converted into FASTQ files using CASAVA (v1.8.2).
RNA-seq and RIP-seq data analysis
Sequenced reads were quality-tested using FASTQC (available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and aligned to the mm10 mouse genome using the STAR aligner (Dobin et al., 2013) version 2.4.0k. Mapping was carried out using default parameters (up to 10 mismatches per read, and up to 9 multi-mapping locations per read). For transcriptome data analysis, nuclear RNA-Seq and total RNA-Seq normalized gene expression (FPKM) was quantified across all gene exons and introns separately, using the top-expressed isoform as a proxy for gene expression. For the RIP-seq analysis, reads uniquely mapping to the genome were used for peak calling with Homer (Heinz et al., 2010) assuming the size of 500, extending the peaks to cover the full enriched region, assuming a fold enrichment of at least 2 over input reads, a poisson p-value threshold relative to input count of 1e-4. The peaks that had less then 1.5 fold enrichment over IgG control and less the 200 tag count were filtered out. Homer was also used to generate normalized read count density tracks for visualization of peaks, motif enrichment analysis, and peak annotation. Motif enrichment analysis was carried out for the peak regions found in introns, searching for motif lengths of 8, 10, and 12, or using a set of all vertebrate motifs known to Homer and using default values for all other parameters.
Statistical analysis of rhythmic gene expression
All RNA-Seq samples exhibited a relatively similar expression profile, reflecting the low variation in sequencing depth between the samples (total RNA-seq samples 17.4×106±4.1×106 uniquely mapped reads, nuclear RNA-seq samples 19.7×106±4.4×106 uniquely mapped reads). We compared FPKM values from nuclear Introns and Exons as well as total Introns and Exons separately in both WT and nonogt animals based on a common list of 20770 genes. Only genes with an average FPKM value ≥0.3 over the 12 time points were considered as expressed. We then used meta2d, a function of the R package MetaCycle, to evaluate periodicity in the RNA seq data (Wu et al., 2016). Briefly, meta2D incorporates ARSER (Yang and Su, 2010), JTK_CYCLE (Hughes et al., 2010), and Lomb-Scargle (Glynn et al., 2006) and it implements N-version programming concepts to integrate their results (p and q values, period, phase and amplitude, see Table S3). Transcripts were considered to be rhythmically expressed when the integrated p value was <0.05. The different analyses were run on homemade MATLAB (MathWorks) programs.
Gene ontology and pathway over-representation analysis (ORA)
For gene ontology and pathway ORA analysis GO-Elite was used as previously described (Zambon et al., 2012). 2000 permutations, Z-score cut-off of 1.96, and permuted p-value cut-off of 0.05 were used. Ontology terms were sorted by Z-score.
Gene Expression Analysis by RT-qPCR
Total RNA was prepared from WT and nonogt livers as described above and quantified using Nanodrop 2000 spectrophotometer (ThermoFisher). 1μg of RNA was reverse transcribed using qScript cDNA SuperMix (QuantaBio) in a final volume of 20μl, according to manufacturer’s instructions. Reverse transcribed RNA was diluted 1:6 and 25ng of cDNA were used in qPCR reactions. qPCR reactions were prepared using the FastStart Universal SYBR Green Master Mix (Roche). The sequences of the primers used are listed in Table S6. Reactions were run on a 7900HT Fast Real-Time PCR System (ThermoFisher) by the Functional Genomics Core Facility of the Salk Institute. Gene expression was normalised to 18S RNA and quantified using the comparative CT method.
Western Blotting
For protein extraction, 10–20mg of frozen liver powder were homogenised in RIPA lysis buffer (10mM Tris-HCl pH8.0, 1mM EDTA, 1% Triton-X 100, 0.1% SDS, 140mM NaCl and protease inhibitor cocktail). Samples were incubated with agitation for 30 minutes at 4°C and clarified by centrifugation and at 13000rpm for 10 minutes at 4°C. The protein concentration in the supernatant was determined using the BCA assay (Pierce). Equal amounts of protein (40 μg) were heat-denaturated in Bolt LDS Sample Buffer (after addition of Bolt Sample Reducing Agent, ThermoFisher), resolved by SDS-PAGE using Bolt 4–12% Bis-Tris Plus Gels (ThermoFisher), and transferred to a nitrocellulose membrane using the iBlot Dry Blotting system (ThermoFisher). The membranes were blocked in 1XPBS 1% Casein Blocker (BioRad) diluted 1:10 for 1h at room temperature and then incubated with antibodies against NONO (rabbit polyclonal (Kowalska et al., 2012)), GCK (rabbit polyclonal, sc-7908), GLUT2 (rabbit polyclonal, ThermoFisher 720238) and TBP (rabbit polyclonal, sc-273). Alexa Fluor 680 conjugate anti-Rabbit IgG (ThermoFisher, A-10043) was used as secondary antibody. Membrane-bound immune complexes were detected by Odyssey Imaging Systems (LI-COR Biosciences). Quantification was performed using Image Studio software (LI-COR Biosciences). Data were normalized to TBP protein expression.
GTT and ITT
For GTT and ITT mice were fasted for 12h during the light phase (ZT0-ZT12). Glucose (2g/kg for mice on normal chow, 1g/kg for mice on HFD) or insulin (0.5U/kg body weight) were injected intraperitonally. Blood glucose level was measured using Nova Max plus glucose meter prior to injection and after 15, 30, 60, 90 and 120 minutes after injection. In case of a technical problem with i.p. injection (i.e. no increase or decrease in blood glucose after glucose or insulin injection respectively), mice were excluded from the analysis or re-tested.
Blood measurements
For all blood measurements mice were fasted for 12h during the light phase (ZT0-ZT12). Blood glucose content was measured through tail vein bleeding using the Nova Max plus glucose meter. Serum insulin levels were determined using Ultra Sensitive Mouse Insulin ELISA Kit (Crystal Chem), according to manufacturer’s instructions. Whole blood was withdrawn from the tail vein, and serum was separated by centrifugation. 5μl of serum were used for quantification. Leptin and ghrelin levels were determined using Bio-Plex Pro Mouse Diabetes 8-Plex Assay (Bio-rad) following manufacturer’s instructions. Whole blood was withdrawn from the tail vein, and serum was separated by centrifugation and diluted 4-fold for quantification.
Liver glycogen quantification
Liver glycogen content was assayed as previously described (Passonneau and Lauderdale, 1974). Liver samples were ground in liquid nitrogen and 15–20mg of liver powder was placed in 0.5ml of 2M HCl and incubated at 100°C for 1h. After neutralization with an equal volume of 2M NaOH, the liberated glucose units were assayed enzimatically using the glucose (hk) assay kit (GAHK20, sigma). Glycogen content was expressed as micromoles of glucosyl units liberated per gram wet liver weight.
Adeno-associated Viruses (AAV) Strains, Propagation and Injection
Viruses were derived from pENN.AAV.TBG.PI; an adeno-associated virus, serotype 8 with a TBG promoter (Hogan et al., 2015). Briefly, 1–2μg of pENN.AAV.TBG.PI were digested with enzymes MluI and SalI (New England Biolabs). Meanwhile, the same amount of pUC57 constructs expressing either WT NONO (pUC57.NONO) or NONO with mutations F113A, F115A, K192A and I194A (pUC57.NONOmut) were also digested with MluI and SalI. The pUC57.NONO and pUC57.NONOmut constructs were produced by GenScript. The digested pENN.AAV.TBG.PI was ligated with the WT or mutant NONO inserts using T4 ligase (New England Biolabs) and transformed in One Shot TOP10 Chemically Competent E. coli (Invitrogen). The vectors were amplified using endotoxin-free maxi prep kit (Qiagen). The amplified vector squences were verified by sequencing and checked for recombination prior to virus packaging. The resulting vectors (pENN.AAV.TBG.PI.NONO and pENN.AAV.TBG.PI.NONOmut) were packaged by the Salk Institute Gene Transfer Targeting and Therapeutics Core. The amplified viruses are referred to as AAV8.NONO and AAV8.NONOmut. Mice received a single tail vein injection of 1011 genome copies of either one of the described viral vectors.
Oil-Red-O staining
Oil-red-o staining was performed as previously described (Mehlem et al., 2013). Briefly, mice were sacrificed and the liver was perfused with ice-cold 1XPBS to remove the excess of blood. Pieces from the main liver lobes were embedded in optimal cutting temperature (O.C.T.) compound (Tissue-Plus, Scigen, 4583) and frozen at −80°C. For cryosectioning the embedded liver was equilibrated at −20°C in a cryos tat chamber and 14μm thick sections were cut. Unfixed tissue slides were stained with ORO and images were taken with a Zeiss VivaTome microscope at a 20X magnification. 10 images were taken per mouse and ImageJ was used for quantification of liver lipid content.
Liver triglycerides quantification
Livers were collected from ad libitum fed mice. The livers were perfused with 2 ml of ice-cold PBS through the spleen and immediately collected. For triglycerides quantification, Triglyceride Quantification kit (abcam) was used, following manufacturer’s instructions. Briefly 100mg of liver powder were homogenized in 5% NP-40 using a dounce homogenizer. The samples were then heated to 80–100°C for 2–5 mi nutes and cooled down to room temperature. The heating step was repeated one more time and the samples were centrifuged at top speed for 2 minutes. Supernatants were diluted 1:10 before proceeding with the assay.
Body composition
Magnetic resonance imaging scans for fat and lean mass were performed using an Echo MRI-100 instrument according to the manufacturer’s instructions.
Indirect calorimetry
Mice were held in standard temperature conditions (20–22°C) and individually housed for at least 3 days before calorimetry experiments. Food intake, locomotor activity, oxygen consumption and carbon dioxide production were simultaneously measured for individually housed mice with a LabMaster system (TSE Systems). Data were collected for 3–4 days and analysed.
Histology
Mouse abdominal fat tissue was fixed in 10% formalin and paraffin embedded. Sections (6μm) were used for haematoxylin and eosin staining.
Metabolomics analysis
Frozen liver powder aliquots were used for detection and relative quantification of metabolites by Metabolon as described (Evans et al., 2009). The dataset comprises a total of 588 compounds of known identity (named bochemicals). An identical mass-equivalent of each liver was extracted and run across the platform; no additional normalization was applied prior to statistical analysis. Following log transformation and imputation of missing values, if any, with the minimum observed value for each compound, ANOVA contrasts were used to identify biochemicals that differed significantly between experimental groups. Only biochemicals with p≤0.05 were considered statistically significant. Analysis by two-way ANOVA identified biochemicals exhibiting significant interaction and main effects for experimental parameters of genotype and treatment.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters, including the exact value of n, descriptive statistics and statistical significance are reported in the method details, figures and the figure legends. All samples represent biological replicates. Unless otherwise specified in figure legends, all values shown in graphs are represented as mean±SEM. For statistical significance of the differences between the means of two groups, we used Student’s t test. F test was used to assess equal variances between groups, if variances differed significantly an unpaired t test with Welch’s correction was used. Statistical significance of differences among multiple groups (≥3) was calculated by performing ANOVA multiple comparisons. Statistical tests were performed using Graph Pad Prism 5. Unless otherwise indicated in the method details, no inclusion or exclusion criteria were applied to any data. All image analyses were performed using automatic programs applying the same analysis parameters to all images.
DATA AND SOFTWARE AVAILABILITY
The data discussed in this study have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE98042 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98042).
Supplementary Material
Table S1. Identified NONO interactors (related to Figure S2)
Table S2. NONO RIP-seq identified peaks (related to Figure 2)
Table S3. WT and nonogt liver diurnal transcriptomes (related to Figure 3)
Table S4. Diurnal expression of identified NONO RIP-seq targets (from liver transcriptome) (related to Figure 3)
Table S5. Gene Ontology of NONO RIP-seq targets (related to Figure 4)
Table S6. Primer Sequences (related to STAR METHODS)
HIGHLIGHTS.
Upon feeding, NONO-containing speckle-like structures increase in liver cell nuclei
The number of target RNAs bound by NONO increases upon feeding
NONO binds and regulates the rhythmicity of metabolic genes post-transcriptionally
NONO is required for normal glucose and lipid homeostasis in mice
ACKNOWLEDGEMENTS
GB is a member of the Molecular Life Sciences Program of the Life Sciences Zurich graduate School. GB was partially supported by fellowships from Glenn Center for Aging Research and the Swiss National Science Foundation (SNF). This work was partially supported by Paul F. Glenn Center for Biology of Aging Research at the Salk Institute, the Helmsley Foundation, the American Federation for Aging Research grant M14322 (to SP). This work was partially supported by the Waitt Advanced Biophotonics Core, the GT3 Core, The Razavi Newman Integrative Genomics and Bioinformatics Core, the Mass Spectrometry Core and the NGS Core of the Salk Institute with funding from NIH-NCI CCSG: P30 014195, NINDS Neuroscience Core Grant: NS072031, the Waitt Foundation, NINDS R24 Core Grant, NEI, the Chapman Foundation and the Helmsley Center for Genomic Medicine. SAB and EM were supported by SNF and the Hartmann-Mueller Foundation. We thank Dr. Marc Montminy for sharing equipment and reagents. We thank Sam Van De Velde for reagents. We thank Uri Manor, Puifai Santisakultarm, Manching Ku, Max Shokhirev, James Moresco and Jolene Diedrich for technical support; Amandine Chaix for technical support and comments on the manuscript; Emily Manoogian, Gabriele Sulli and David O’Keefe for comments and editing the manuscript. We are grateful to Jamie Simon for the graphical abstract and Figure 7 artwork.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Amelio AL, Miraglia LJ, Conkright JJ, Mercer BA, Batalov S, Cavett V, Orth AP, Busby J, Hogenesch JB, and Conkright MD (2007). A coactivator trap identifies NONO (p54nrb) as a component of the cAMP-signaling pathway. Proc. Natl. Acad. Sci. U. S. A 104, 20314–20319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atger F, Gobet C, Marquis J, Martin E, Wang J, Weger B, Lefebvre G, Descombes P, Naef F, and Gachon F (2015). Circadian and feeding rhythms differentially affect rhythmic mRNA transcription and translation in mouse liver. Proc. Natl. Acad. Sci. U. S. A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron KG, and Reid KJ (2014). Circadian misalignment and health. Int. Rev. Psychiatry Abingdon Engl 26, 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass J, and Takahashi JS (2010). Circadian integration of metabolism and energetics. Science 330, 1349–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benegiamo G, Brown SA, and Panda S (2016). RNA Dynamics in the Control of Circadian Rhythm. Adv. Exp. Med. Biol 907, 107–122. [DOI] [PubMed] [Google Scholar]
- Brown SA (2016). Circadian Metabolism: From Mechanisms to Metabolomics and Medicine. Trends Endocrinol. Metab. TEM 27, 415–426. [DOI] [PubMed] [Google Scholar]
- Brown SA, Ripperger J, Kadener S, Fleury-Olela F, Vilbois F, Rosbash M, and Schibler U (2005). PERIOD1-associated proteins modulate the negative limb of the mammalian circadian oscillator. Science 308, 693–696. [DOI] [PubMed] [Google Scholar]
- Buxadé M, Morrice N, Krebs DL, and Proud CG (2008). The PSF.p54nrb complex is a novel Mnk substrate that binds the mRNA for tumor necrosis factor alpha. J. Biol. Chem. 283, 57–65. [DOI] [PubMed] [Google Scholar]
- Close P, East P, Dirac-Svejstrup AB, Hartmann H, Heron M, Maslen S, Chariot A, Söding J, Skehel M, and Svejstrup JQ (2012). DBIRD complex integrates alternative mRNA splicing with RNA polymerase II transcript elongation. Nature 484, 386–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damiola F, Le Minh N, Preitner N, Kornmann B, Fleury-Olela F, and Schibler U (2000). Restricted feeding uncouples circadian oscillators in peripheral tissues from the central pacemaker in the suprachiasmatic nucleus. Genes Dev. 14, 2950–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinforma. Oxf. Engl 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong HA, Robles MS, Knutti D, and Weitz CJ (2011). A molecular mechanism for circadian clock negative feedback. Science 332, 1436–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emili A, Shales M, McCracken S, Xie W, Tucker PW, Kobayashi R, Blencowe BJ, and Ingles CJ (2002). Splicing and transcription-associated proteins PSF and p54nrb/nonO bind to the RNA polymerase II CTD. RNA N. Y. N 8, 1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AM, DeHaven CD, Barrett T, Mitchell M, and Milgram E (2009). Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem 81, 6656–6667. [DOI] [PubMed] [Google Scholar]
- Fong K-W, Li Y, Wang W, Ma W, Li K, Qi RZ, Liu D, Songyang Z, and Chen J (2013). Whole-genome screening identifies proteins localized to distinct nuclear bodies. J. Cell Biol 203, 149–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, and Lamond AI (2010). Paraspeckles. Cold Spring Harb. Perspect. Biol 2, a000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AH, Lam YW, Leung AKL, Lyon CE, Andersen J, Mann M, and Lamond AI (2002). Paraspeckles: a novel nuclear domain. Curr. Biol. CB 12, 13–25. [DOI] [PubMed] [Google Scholar]
- Gaidatzis D, Burger L, Florescu M, and Stadler MB (2015). Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat. Biotechnol 33, 722–729. [DOI] [PubMed] [Google Scholar]
- Gallegos JE, and Rose AB (2015). The enduring mystery of intron-mediated enhancement. Plant Sci. Int. J. Exp. Plant Biol 237, 8–15. [DOI] [PubMed] [Google Scholar]
- Gehring NH, Wahle E, and Fischer U (2017). Deciphering the mRNP Code: RNA-Bound Determinants of Post-Transcriptional Gene Regulation. Trends Biochem. Sci [DOI] [PubMed] [Google Scholar]
- Glynn EF, Chen J, and Mushegian AR (2006). Detecting periodic patterns in unevenly spaced gene expression time series using Lomb-Scargle periodograms. Bioinforma. Oxf. Engl 22, 310–316. [DOI] [PubMed] [Google Scholar]
- He L, Diedrich J, Chu Y-Y, and Yates JR (2015). Extracting Accurate Precursor Information for Tandem Mass Spectra by RawConverter. Anal. Chem 87, 11361–11367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan MF, Ravnskjaer K, Matsumura S, Huising MO, Hull RL, Kahn SE, and Montminy M (2015). Hepatic Insulin Resistance Following Chronic Activation of the CREB Coactivator CRTC2. J. Biol. Chem 290, 25997–26006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff J (2015). The Airyscan detector from ZEISS: confocal imaging with improved signal-to-noise ratio and super-resolution. Nat Meth 12. [Google Scholar]
- Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, Panda S, and Hogenesch JB (2009). Harmonics of circadian gene transcription in mammals. PLoS Genet. 5, e1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB, and Kornacker K (2010). JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J. Biol. Rhythms 25, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihama Y, Oda Y, Tabata T, Sato T, Nagasu T, Rappsilber J, and Mann M (2005). Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol. Cell. Proteomics MCP 4, 1265–1272. [DOI] [PubMed] [Google Scholar]
- Kawaguchi T, Tanigawa A, Naganuma T, Ohkawa Y, Souquere S, Pierron G, and Hirose T (2015). SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc. Natl. Acad. Sci. U. S. A 112, 4304–4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keene JD, Komisarow JM, and Friedersdorf MB (2006). RIP-Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc 1, 302–307. [DOI] [PubMed] [Google Scholar]
- Knott GJ, Bond CS, and Fox AH (2016). The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 44, 3989–4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike N, Yoo S-H, Huang H-C, Kumar V, Lee C, Kim T-K, and Takahashi JS (2012). Transcriptional architecture and chromatin landscape of the core circadian clock in mammals. Science 338, 349–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska E, Ripperger JA, Muheim C, Maier B, Kurihara Y, Fox AH, Kramer A, and Brown SA (2012). Distinct roles of DBHS family members in the circadian transcriptional feedback loop. Mol. Cell. Biol 32, 4585–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalska E, Ripperger JA, Hoegger DC, Bruegger P, Buch T, Birchler T, Mueller A, Albrecht U, Contaldo C, and Brown SA (2013). NONO couples the circadian clock to the cell cycle. Proc. Natl. Acad. Sci. U. S. A 110, 1592–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara S, Ikei A, Taguchi Y, Tabuchi Y, Fujimoto N, Obinata M, Uesugi S, and Kurihara Y (2006). PSPC1, NONO, and SFPQ are expressed in mouse Sertoli cells and may function as coregulators of androgen receptor-mediated transcription. Biol. Reprod. 75, 352–359. [DOI] [PubMed] [Google Scholar]
- Li J, Grant GR, Hogenesch JB, and Hughes ME (2015). Considerations for RNA-seq analysis of circadian rhythms. Methods Enzymol. 551, 349–367. [DOI] [PubMed] [Google Scholar]
- Li R, Harvey AR, Hodgetts SI, and Fox AH (2017). Functional dissection of NEAT1 using genome editing reveals substantial localisation of the NEAT1_1 isoform outside paraspeckles. RNA N. Y. N. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, and Takahashi JS (2011). Genetics of circadian rhythms in Mammalian model organisms. Adv. Genet 74, 175–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JY, and Sewer MB (2015). p54nrb/NONO regulates cyclic AMP-dependent glucocorticoid production by modulating phosphodiesterase mRNA splicing and degradation. Mol. Cell. Biol 35, 1223–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Diedrich JK, Jungreis I, Donaldson C, Vaughan J, Kellis M, Yates JR, and Saghatelian A (2016). Improved Identification and Analysis of Small Open Reading Frame Encoded Polypeptides. Anal. Chem 88, 3967–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannen T, Yamashita S, Tomita K, Goshima N, and Hirose T (2016). The Sam68 nuclear body is composed of two RNase-sensitive substructures joined by the adaptor HNRNPL. J. Cell Biol 214, 45–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao YS, Sunwoo H, Zhang B, and Spector DL (2011). Direct visualization of the co-transcriptional assembly of a nuclear body by noncoding RNAs. Nat. Cell Biol. 13, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa ML, Gagliardino JJ, and Francini F (2011). Liver glucokinase: An overview on the regulatory mechanisms of its activity. IUBMB Life 63, 1–6. [DOI] [PubMed] [Google Scholar]
- McMahon AC, Rahman R, Jin H, Shen JL, Fieldsend A, Luo W, and Rosbash M (2016). TRIBE: Hijacking an RNA-Editing Enzyme to Identify Cell-Specific Targets of RNA-Binding Proteins. Cell 165, 742–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlem A, Hagberg CE, Muhl L, Eriksson U, and Falkevall A (2013). Imaging of neutral lipids by oil red O for analyzing the metabolic status in health and disease. Nat. Protoc. 8, 1149–1154. [DOI] [PubMed] [Google Scholar]
- Menet JS, Rodriguez J, Abruzzi KC, and Rosbash M (2012). Nascent-Seq reveals novel features of mouse circadian transcriptional regulation. eLife 1, e00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naganuma T, Nakagawa S, Tanigawa A, Sasaki YF, Goshima N, and Hirose T (2012). Alternative 3’-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J. 31, 4020–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S (2016). Circadian physiology of metabolism. Science 354, 1008–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S, Hogenesch JB, and Kay SA (2002). Circadian rhythms from flies to human. Nature 417, 329–335. [DOI] [PubMed] [Google Scholar]
- Passonneau JV, and Lauderdale VR (1974). A comparison of three methods of glycogen measurement in tissues. Anal. Biochem 60, 405–412. [DOI] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, and Spector DL (2005). Regulating gene expression through RNA nuclear retention. Cell 123, 249–263. [DOI] [PubMed] [Google Scholar]
- Proteau A, Blier S, Albert AL, Lavoie SB, Traish AM, and Vincent M (2005). The multifunctional nuclear protein p54nrb is multiphosphorylated in mitosis and interacts with the mitotic regulator Pin1. J. Mol. Biol 346, 1163–1172. [DOI] [PubMed] [Google Scholar]
- Rabani M, Raychowdhury R, Jovanovic M, Rooney M, Stumpo DJ, Pauli A, Hacohen N, Schier AF, Blackshear PJ, Friedman N, et al. (2014). High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 159, 1698–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AB, Karp NA, Maywood ES, Sage EA, Deery M, O’Neill JS, Wong GKY, Chesham J, Odell M, Lilley KS, et al. (2006). Circadian orchestration of the hepatic proteome. Curr. Biol. CB 16, 1107–1115. [DOI] [PubMed] [Google Scholar]
- Reed R, and Hurt E (2002). A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 108, 523–531. [DOI] [PubMed] [Google Scholar]
- Ripperger JA, and Schibler U (2006). Rhythmic CLOCK-BMAL1 binding to multiple E-box motifs drives circadian Dbp transcription and chromatin transitions. Nat. Genet 38, 369–374. [DOI] [PubMed] [Google Scholar]
- Rose AB, Elfersi T, Parra G, and Korf I (2008). Promoter-proximal introns in Arabidopsis thaliana are enriched in dispersed signals that elevate gene expression. Plant Cell 20, 543–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salton M, Lerenthal Y, Wang S-Y, Chen DJ, and Shiloh Y (2010). Involvement of Matrin 3 and SFPQ/NONO in the DNA damage response. Cell Cycle Georget. Tex 9, 1568–1576. [DOI] [PubMed] [Google Scholar]
- Sánchez-Jiménez F, and Sánchez-Margalet V (2013). Role of Sam68 in post-transcriptional gene regulation. Int. J. Mol. Sci. 14, 23402–23419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinturel F, Gerber A, Mauvoisin D, Wang J, Gatfield D, Stubblefield JJ, Green CB, Gachon F, and Schibler U (2017). Diurnal Oscillations in Liver Mass and Cell Size Accompany Ribosome Assembly Cycles. Cell 169, 651–663.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobel JA, Krier I, Andersin T, Raghav S, Canella D, Gilardi F, Kalantzi AS, Rey G, Weger B, Gachon F, et al. (2017). Transcriptional regulatory logic of the diurnal cycle in the mouse liver. PLoS Biol. 15, e2001069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, McDonald WH, and Yates JR (2002). DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J. Proteome Res. 1, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenzer S, Moro A, Kuharev J, Francis AC, Vidalino L, Provenzani A, and Macchi P (2013). Proteome-wide characterization of the RNA-binding protein RALY-interactome using the in vivo-biotinylation-pulldown-quant (iBioPQ) approach. J. Proteome Res. 12, 2869–2884. [DOI] [PubMed] [Google Scholar]
- Thorens B, and Mueckler M (2010). Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 298, E141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres M, Becquet D, Blanchard M-P, Guillen S, Boyer B, Moreno M, Franc J-L, and François-Bellan A-M (2016). Circadian RNA expression elicited by 3’-UTR IRAlu-paraspeckle associated elements. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uren PJ, Bahrami-Samani E, Burns SC, Qiao M, Karginov FV, Hodges E, Hannon GJ, Sanford JR, Penalva LOF, and Smith AD (2012). Site identification in high-throughput RNA-protein interaction data. Bioinforma. Oxf. Engl 28, 3013–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmers C, Gill S, DiTacchio L, Pulivarthy SR, Le HD, and Panda S (2009). Time of feeding and the intrinsic circadian clock drive rhythms in hepatic gene expression. Proc. Natl. Acad. Sci. U. S. A 106, 21453–21458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmers C, Schmitz RJ, Nathanson J, Yeo G, Ecker JR, and Panda S (2012). Circadian oscillations of protein-coding and regulatory RNAs in a highly dynamic mammalian liver epigenome. Cell Metab. 16, 833–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Rajbhandari P, Damianov A, Han A, Sallam T, Waki H, Villanueva CJ, Lee SD, Nielsen R, Mandrup S, et al. (2017). RNA-binding protein PSPC1 promotes the differentiation-dependent nuclear export of adipocyte RNAs. J. Clin. Invest [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidensdorfer D, Stöhr N, Baude A, Lederer M, Köhn M, Schierhorn A, Buchmeier S, Wahle E, and Hüttelmaier S (2009). Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA N. Y. N 15, 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Anafi RC, Hughes ME, Kornacker K, and Hogenesch JB (2016). MetaCycle: an integrated R package to evaluate periodicity in large scale data. Bioinforma. Oxf. Engl 32, 3351–3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Park SK, Venable JD, Wohlschlegel JA, Diedrich JK, Cociorva D, Lu B, Liao L, Hewel J, Han X, et al. (2015). ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J. Proteomics 129, 16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, and Hirose T (2015). The building process of the functional paraspeckle with long non-coding RNAs. Front. Biosci. Elite Ed 7, 1–41. [DOI] [PubMed] [Google Scholar]
- Yang R, and Su Z (2010). Analyzing circadian expression data by harmonic regression based on autoregressive spectral estimation. Bioinforma. Oxf. Engl 26, i168–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambon AC, Gaj S, Ho I, Hanspers K, Vranizan K, Evelo CT, Conklin BR, Pico AR, and Salomonis N (2012). GO-Elite: a flexible solution for pathway and ontology over-representation. Bioinformatics 28, 2209–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrinpar A, Chaix A, and Panda S (2016). Daily Eating Patterns and Their Impact on Health and Disease. Trends Endocrinol. Metab. TEM 27, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, and Carmichael GG (2001). The fate of dsRNA in the nucleus: a p54(nrb)containing complex mediates the nuclear retention of promiscuously A-to-I edited RNAs. Cell 106, 465–475. [DOI] [PubMed] [Google Scholar]
- Zhang R, Lahens NF, Ballance HI, Hughes ME, and Hogenesch JB (2014). A circadian gene expression atlas in mammals: implications for biology and medicine. Proc. Natl. Acad. Sci. U. S. A 111, 16219–16224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Sun BK, Erwin JA, Song J-J, and Lee JT (2008). Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science 322, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, and Lee JT (2010). Genome-wide identification of polycombassociated RNAs by RIP-seq. Mol. Cell 40, 939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Zhao X, Zhao L, Yang H, Liu L, Li J, Wu J, Yang F, Huang G, and Liu J (2015). p54(nrb)/NONO regulates lipid metabolism and breast cancer growth through SREBP-1A. Oncogene. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Identified NONO interactors (related to Figure S2)
Table S2. NONO RIP-seq identified peaks (related to Figure 2)
Table S3. WT and nonogt liver diurnal transcriptomes (related to Figure 3)
Table S4. Diurnal expression of identified NONO RIP-seq targets (from liver transcriptome) (related to Figure 3)
Table S5. Gene Ontology of NONO RIP-seq targets (related to Figure 4)
Table S6. Primer Sequences (related to STAR METHODS)
Data Availability Statement
The data discussed in this study have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE98042 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98042).