

Abstract
Variations in disease onset and/or severity have often been observed in siblings with cystic fibrosis (CF), despite the same CFTR genotype and environment. We postulated that genomic variation (modifier and/or pharmacogenomic variants) might explain these clinical discordances. From a cohort of patients included in the Wisconsin randomized clinical trial (RCT) of newborn screening (NBS) for CF, we identified two brothers who showed discordant lung disease courses as children, with one milder and the other more severe than average, and a third, eldest brother, who also has severe lung disease. Leukocytes were harvested as the source of DNA, and whole-genome sequencing (WGS) was performed. Variants were identified and analyzed using in-house-developed informatics tools. Lung disease onset and severity were quantitatively different between brothers during childhood. The youngest, less severely affected brother is homozygous for HFE p.H63D. He also has a very rare PLG p.D238N variant that may influence host–pathogen interaction during chronic lung infection. Other variants of interest were found differentially between the siblings. Pharmacogenomics findings were consistent with the middle, most severely affected brother having poor outcomes to common CF treatments. We conclude that genomic variation between siblings with CF is expected. Variable lung disease severity may be associated with differences acting as genetic modifiers and/or pharmacogenomic factors, but large cohort studies are needed to assess this hypothesis.
Keywords: bronchiectasis, chronic lung disease, diabetes mellitus, elevated sweat chloride, exocrine pancreatic insufficiency, meconium ileus, multiple bilateral pneumothoraces, recurrent Aspergillus infections, recurrent respiratory infections
INTRODUCTION
Variations in disease onset and/or severity have been observed in siblings with cystic fibrosis (CF) for decades (Macgregor and Rhaney 1948). The most notable differences relate to CF lung disease. Many factors have been suggested to explain discordances such as gender, meconium ileus, age of diagnosis, cystic fibrosis transmembrane conductance regulator (CFTR) genotype, acquisition of respiratory pathogens, treatment regimens, and, most recently, genetic modifiers (Orenstein et al. 1977; Kerem and Kerem 1996; Li et al. 2004; Cutting 2010; Sanders et al. 2014; O'Neal and Knowles 2018).
Prior to the onset of routine early diagnosis through newborn screening (NBS), it was proposed from an influential sibling pair study that diagnosis at a younger age leading to early treatment initiation might account for lessened severity in the younger sibling (Orenstein et al. 1977). Although NBS does lead to a reduction in the severity of outcomes for the majority of children (Farrell et al. 2001), there is subgroup known as “nonresponders,” approximately one-third of patients, who do not show the same degree of benefit from early diagnosis (Sanders et al. 2018). After discovery of the CFTR gene (Kerem et al. 1989), studies on genotype–phenotype relationships, although illuminating with regard to pancreatic functional variations (Kerem and Kerem 1996; Durno et al. 2002), did not succeed in explaining differences in the onset and severity of lung disease (Kerem et al. 1990; Kerem and Kerem 1996). In fact, observations on homozygous F508del patients revealed such great variability in respiratory disease outcomes that attention turned to the identification of genetic modifiers (Cutting 2010; Corvol et al. 2015; O'Neal and Knowles 2018). Application of genome-wide association studies (GWASs) has more recently facilitated identification of certain classes of modifiers resulting in phenotypic variation (Corvol et al. 2015).
Individual specific variable drug responses can present significant challenges for the effective and safe treatment of many human diseases. Pharmacogenomics (PGx) attempts to relate drug response to patient-specific genome in order to better customize therapeutic selections. We posited that the whole-genome sequencing (WGS) data generated for phenotypic modifier studies might simultaneously be useful for the extraction of PGx findings of relevance for patients with CF.
Here we report an investigation of non-CFTR genetic factors in three brothers with CF who showed considerable clinical variations in the course of disease manifestations, particularly during childhood. Their discordances led us to search for genetic modifiers and pharmacogenomics differences using a WGS-based approach. Our hypothesis is that genomic variation will help explain clinical differences among siblings.
RESULTS
Patient Phenotypes
All three brothers were confirmed by WGS to have a CFTR genotype of F508del/CFTRdele2,3 and no other CFTR variants. None had meconium ileus, each was diagnosed with a sweat chloride level of >100 mmol/L, and all had pancreatic insufficiency. Their clinical histories are summarized in Table 1. Quantitative longitudinal evaluations of lung disease were performed and data are shown in Figures 1 and 2.
Table 1.
Summary of clinical characteristics
| Characteristics | Eldest brother | Middle brother | Youngest brother |
|---|---|---|---|
| Gestational age (weeks) | 38 | 38 | 41 |
| Birth weight (g) | 2830 | 2410 | 3520 |
| Year of birth | 1984 | 1990 | 1992 |
| Age at diagnosis (weeks) | 12 | 7 | 5 |
| NBS (yes/no) | No | Yes | Yes |
| IRT level (ng/mL) | NAa | 234 | 251 |
| Sweat Chloride (mEq/L) | 104/98 | 111/110 | 110/107 |
| CFTR genotype | F508del/dele2,3 | F508del/dele2,3 | F508del/dele2,3 |
| Meconium ileus | No | No | No |
| Age at PERT (wk) | NAa | 36 | 38 |
| Age at 1st AF acquisition (yr) | 18 | 11 | 14.75 |
| % cultures revealing AF | 31 | 77 | 38 |
| Age of lung transplant (yr) | 29 | 20 | ____b |
| Other comorbidities | CFRD, CFLD | CFRD, pneumothoraces | ____b |
| Age of death (yr) | ____b | 24 | ____b |
aInformation not available or test not done.
bNone at time of publication.
(NBS) Newborn screening, (IRT) immunoreactive trypsinogen, (PERT) pancreatic enzyme replacement therapy, (AF) Aspergillus fumigatus, (CFRD) cystic fibrosis–related diabetes, (CFLD) cystic fibrosis liver disease.
Figure 1.
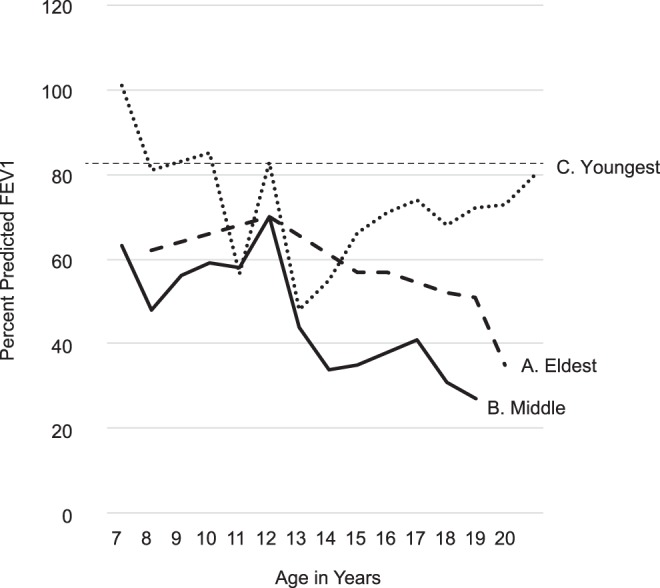
Pulmonary function test results in the three brothers (i.e., percent predicted FEV1 [ppFEV1] values as related to age). The data obtained on three brothers are shown as A (eldest), B (middle), and C (youngest) and demonstrated that the youngest had better pulmonary function than his brothers. After 20 yr, the eldest brother deteriorated and reached 20% at 28 yr of age.
Figure 2.

Wisconsin chest X-ray scores (Koscik et al. 2000) in the three brothers as compared to published data (Farrell et al. 2003) from the Wisconsin randomized clinical trial (RCT) of the newborn screening (NBS) cohort in which a thoracic radiologist and pediatric pulmonologist rated chest radiographs independently, with their scores being averaged. The data obtained on three brothers are shown as A (eldest), B (middle), and C (youngest) and demonstrated that the youngest had lower and therefore milder lung disease than his brothers throughout childhood. In the case of the eldest brother labeled A, the scores assigned by the same pediatric pulmonologist were adjusted to conform with the averaging method. The gray shaded area labeled D represents the mean ± 95% confidence interval for the entire RCT cohort. (CXR) Chest X-ray.
The eldest brother was diagnosed with CF at 3 mo of age because of failure to thrive. Despite initiation of pancreatic enzyme replacement therapy (PERT), he tracked under the fifth percentile for weight throughout childhood. After 12 yr of Pseudomonas aeruginosa (PA)-positive cultures and progressive lung disease, his sputum culture revealed Aspergillus fumigatus (AF) at 18 yr. He received a bilateral lung transplantation at 29 yr of age after his percent predicted forced expiratory volume in 1 sec (ppFEV1) had decreased to 20% and his Wisconsin chest X-ray (WCXR) score was rising dramatically (Fig. 2).
The middle brother was diagnosed with CF through NBS and began PERT at 36 wk of age when pancreatic insufficiency was demonstrated through protocol-guided studies (Farrell 2000). Although PA was not cultured until 16 yr of age, he was noted by WCXR scores to have developed irreversible lung disease (i.e., WCXR > 5) (Farrell et al. 2003) by 2 yr of age and had severely progressive lung disease between 5 and 14 yr (Fig. 2). His sputum culture first grew AF at age 11 yr. He was treated for AF with itraconazole, but eradication was unsuccessful. He underwent bilateral lung transplantation age 20 yr, and died at 24 yr because of multisystem organ failure.
The youngest brother was also diagnosed with CF through NBS. He had a relatively mild course of lung disease throughout most of childhood; this was clearly less severe than the cohort of other screened patients in the RCT (Fig. 2). After age 12 yr, he began to deteriorate in association with culturing AF. He has yet to acquire PA through his current age of 26 yr.
Modifiers of Cystic Fibrosis
Our WGS-based analysis identified five variants that might affect the brothers’ phenotypes (Table 2). All three brothers were found to have a common heterozygous nonsynonymous variant in EPHX1 previously shown to reduce enzyme activity by at least 20% (Hassett et al. 1994). A low activity of EPHX1 has been identified as a risk factor for chronic obstructive pulmonary disease (COPD), and this specific variant has been associated with altered risk of pulmonary disease (Smith and Harrison 1997; Vibhuti et al. 2007: Castaldi et al. 2009; Carlton et al. 2003). Although association between variant homozygosity and increased risk of COPD was recently confirmed via comprehensive meta-analysis, the findings were not supported for variant heterozygosity (Li et al. 2013).
Table 2.
Genetic modifiers identified in the brothers
| Gene | Chr | DNA change | Variant type | Variant | dbSNP ID/ClinVar ID | AF(%) | Brother | Phenotype associations | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Eldest | Middle | Youngest | ||||||||
| EPHX1 | 1 | g.226019633T > C | Missense |
NM_000120.3 c.337T > C p.Y113H |
rs1051740 | 31.65 | 10/24 Het |
25/52 Het |
21/35 Het |
COPD |
| HFE | 6 | g.26091179C > G | Missense |
NM_000410.3 c.187C > G p.H63D |
rs1799945 | 10.82 | 10/30 Het |
14/29 Het |
32/32 Hom |
Hemochromatosis Liver disease Type II diabetes |
| MBL2 | 10 | g.54531242G > A | Missense |
NM_000242.2 c.154C > T p.R52C |
rs5030737 | 5.48 | 15/23 Het |
– | 13/28 Het |
Respiratory tract infections Bronchiectasis Lung function |
| PLG | 6 | g.161137720G > A | Missense |
NM_000301.3 c.712G > A p.D238N |
rs756287836 | 0.0036 | – | – | 13/28 Het |
Chronic mucosal membrane inflammation Recurrent sinobronchial infections |
| SERPINA1 | 14 | g.94847262T > A | Missense |
NM_000295.4 c.863A > T p.E288V |
rs17580 | 2.34 | – | 18/40 Het |
21/35 Het |
COPD Increased risk for pulmonary and liver disease |
Sequence coverage for each variant is provided along with the call made.
(Chr) Chromosome, (COPD) chronic obstructive pulmonary disease, (AF(%)) % allele frequency in gnomAD shown in percentages, (Het) heterozygous, (Hom) homozygous.
A known deleterious missense variant in HFE (p.H63D) was found to be heterozygous in the eldest and middle brothers and homozygous in the youngest. This variant is pathogenic for a mild form of hereditary hemochromatosis (Feder et al. 1996). Although clinical hemochromatosis is mostly associated with p.C282Y homozygosity, there is evidence that p.H63D can act as a modifier for α1-antitrypsin deficiency liver and lung disease (Joly et al. 2017). Functional variation in HFE has been associated with risk of infection in general (Khan et al. 2007; Nairz et al. 2017; Gill et al. 2019) and deteriorating lung function specifically in CF (Smith et al. 2019).
A third variant of interest is the known deleterious variant in MBL2 (D variant; p.R52C), which was heterozygous in the eldest and youngest brothers. MBL2 plays a role in immune function, activating the lectin complement pathway and leading to clearance by phagocytes. This variant is pathogenic for mannose-binding protein deficiency (Heitzeneder et al. 2012). Variants in this gene have been associated with recurrent respiratory tract infections, bronchiectasis severity, and age-related decline in lung function (Horiuchi et al. 2005; Haerynck et al. 2012; Liu et al. 2012; Pifferi et al. 2015). The presence of functional variants in MBL2 has shown association with different aspects (lung function, infection, and survival) of CF in some studies (Dorfman et al. 2008; McDougal et al. 2010).
The youngest brother was heterozygous for an ultra-rare, conserved, nonsynonymous predicted to be damaging variant (p.D238N) in a known kringle-binding domain in the plasminogen protein (PLG) (Arai et al. 1998). Although not previously reported, this variant would be categorized as likely pathogenic based on American College of Medical Genetics and Genomics (ACMG) guidelines (Richards et al. 2015). Functional variation in PLG is associated with plasminogen deficiency, which has been causally linked to a rare chronic inflammatory disease of the mucous membranes. PLG deficiency also predisposes patients to recurrent sinobronchial AF infections in immunocompromised individuals (Zaas et al. 2008).
Finally, a known deleterious variant in SERPINA1 (PiS; p.E288V) was identified in the middle and youngest brothers. This variant is a known risk factor for COPD and a known pathogenic variant for α1-antitrypsin deficiency, which increases the risk for pulmonary and liver disease in both heterozygous and homozygous states (Pérez-Rubio et al. 2015; Greene et al. 2016). Other variants in this gene have been shown to modify CF-related disease (Debray et al. 2019).
Pharmacogenomic Findings
Pharmacogenomic variants categorized as level 1A, 1B, 2A, and 2B (Whirl-Carrillo et al. 2012) were identified for a number of drugs commonly used in the treatment of CF (Table 3). Whereas all three brothers are similar for a number of loci, there remain some notable differences. For example, several loci assessed (CRHR2, COL22A1, and ADRB2) have been associated with efficacy of the β2-adrenergic salbutamol (Bleecker et al. 2007; Poon et al. 2008; Duan et al. 2014), with each brother having unique combinations. Of these, the middle brother might have the least benefit from albuterol. Similarly, the middle brother was unique in his TMPT haplotype associated with thiopurine (e.g., azathioprine) inactivation that would put him at higher risk of toxicity when treated with thiopurine drugs.
Table 3.
Pharmacogenomic findings relevant to the management of cystic fibrosis
| Drugs | Eldest brother | Middle brother | Youngest brother |
|---|---|---|---|
| Tobramycin: Eldest − Middle + Youngest − |
MT-RNR1—patient is homoplastic (A) for this rs267606617 allele. Associated with decreased risk of aminoglycoside-induced hearing loss. | MT-RNR1—patient is homoplastic (A) for this rs267606617 allele. Associated with decreased risk of aminoglycoside-induced hearing loss. | MT-RNR1—patient is homoplastic (A) for this rs267606617 allele. Associated with decreased risk of aminoglycoside-induced hearing loss. |
| Salbutamol: Eldest − Middle − Youngest − |
CRHR2—patient is homozygous (A/A) for the rs7793837 allele; associated with a better response (increased acute bronchodilation). COL22A1—patient is homozygous (C/C) for the rs6988229 allele; associated with a decreased bronchodilator response (FEV1). ADRB2—patient is heterozygous (A/G) for the rs1042713 allele. Children with A/G genotype with asthma who are treated with salmaterol or salbutamol may have a decreased response to treatment than children with G/G genotypes, and better response than children with A/A genotype. |
CRHR2—patient is heterozygous (A/T) for the rs7793837 allele; associated with intermediate response. COL22A1—patient is homozygous (C/C) for the rs6988229 allele; associated with a decreased bronchodilator response (FEV1). ADRB2—patient is homozygous (A/A) for the rs1042713 allele. Children with the AA genotype with asthma who are treated with salmeterol or salbutamol may have a decreased response to treatment. |
CRHR2—patient is homozygous (A/A) for the rs7793837 allele; associated with a better response (increased acute bronchodilation). COL22A1—patient is homozygous (C/C) for the rs6988229 allele; associated with a decreased bronchodilator response (FEV1). ADRB2—patient is homozygous (A/A) for the rs1042713 allele. Children with the AA genotype with asthma who are treated with salmeterol or salbutamol may have a decreased response to treatment. |
| Omeprazole: Eldest − Middle − Youngest + Voricanazole: Eldest − Middle − Youngest − Othera |
CYP2C19*1B/*2B—intermediate-to-poor metabolizer. Patient may require lower doses of drugs metabolized by CYP2C19 enzyme compared to normal/rapid metabolizers to avoid toxicity or switching to a drug independent of CYP2C19 metabolism. | CYP2C19*17/*2B—intermediate metabolizer. Standard dosing guidelines generally apply. | CYP2C19*1B/*2B—intermediate-to-poor metabolizer. Patient may require lower doses of drugs metabolized by CYP2C19 enzyme compared to normal/rapid metabolizers to avoid toxicity or switching to a drug independent of CYP2C19 metabolism. |
| Tacrolimus: Eldest + Middle + Youngest − |
CYP3A5 (*3/*3)—patient is homozygous for the two alleles (rs776746 and rs28383479) that make up the CYP3A5*3 haplotype. Decreased metabolizer; may require a lower dose. |
CYP3A5 (*3/*3)—patient is homozygous for the two alleles (rs776746 and rs28383479) that make up the CYP3A5*3 haplotype. Decreased metabolizer; may require a lower dose. |
CYP3A5 (*3/*3)—patient is homozygous for the two alleles (rs776746 and rs28383479) that make up the CYP3A5*3 haplotype. Decreased metabolizer; may require a lower dose. |
| Azathioprine: Eldest − Middle − Youngest − |
TPMT (*1A/*1A)—associated with (1) increased inactivation of thiopurines due to normal TPMT activity; (2) a decreased risk for toxicity when receiving thiopurine drugs and purine analogs. Patients may still be at risk for toxicity when taking thiopurine drugs and purine analogs. | TPMT (*1/*3A)—associated with (1) decreased inactivation of thiopurines due to deficient TPMT activity; (2) an increased risk for toxicity when receiving thiopurine drugs and purine analogs. | TPMT (*1A/*1A)—associated with (1) increased inactivation of thiopurines due to normal TPMT activity; (2) a decreased risk for toxicity when receiving thiopurine drugs and purine analogs. Patients may still be at risk for toxicity when taking thiopurine drugs and purine analogs. |
aPrescribing information based on CYP2C19 haplotypes is available for a number of other drugs, including the following: amitriptyline, citalopram, escitalopram, esomeprazole, clomipramine, clopidogrel, doxepin, imipramine, lansoprazole, moclobermide, rabeprazole, sertraline, and trimipramine.
CYP2C19 is a highly polymorphic enzyme that contributes to the metabolism of many clinically relevant drugs (Wrighton et al. 1996; Gaedigk et al. 2018). Individuals can be categorized as ultra-rapid, rapid, intermediate, or poor metabolizers, depending on absent, decreased, normal, or increased function (Scott et al. 2013). As shown in Table 3, we were able to determine that all three brothers had a no function copy of this gene (*2B) and that both the eldest and youngest brothers have a *1B second copy, whereas the middle brother's second allele is *17. Thus, at best, the middle brother is an intermediate metabolizer, because the *17 gain-of-function allele does not completely compensate for the no function allele.
Multiple genes encode CYP3A enzymes, with CYP3A4 and CYP3A5 regarded as the most important (Williams et al. 2002). Expression is highly polymorphic with 52 CYP3A4 and 26 CYP3A5 star alleles cataloged (Gaedigk et al. 2018). All of the brothers were found to be homozygous for the two alleles that make up the CYP3A5 *3 haplotype (CYP3A5 *3/*3) and are thus considered to be CYP3A5 non-expressors who may have decreased metabolism of a large percentage of oxidatively metabolized drugs (Williams et al. 2002). This variation in expression is particularly important for drugs and substrates with narrow therapeutic indices, including the immunosuppressants cyclosporine and tacrolimus, and can result in clinically significant differences in toxicity and response (Ericson et al. 2017). Additional PGx related findings can be found in Supplemental Table 1.
DISCUSSION
O'Neal and Knowles (2018) stated that “[t]he not-too-distant future holds many more opportunities for discovery of CF gene modifiers … whole-genome sequencing will expand GWAS-type studies to rare variant analyses.” In support of their prediction, we suggest that our WGS assessment of the three brothers identified several variants that could predispose a patient to better or worse outcomes and that the combination of exploring for genetic modifiers plus pharmacogenomics variants could be valuable. It was particularly interesting to compare the mildly affected youngest brother with his siblings and observe notable differences in both potential genetic modifiers and pharmacogenomics factors.
We suggest that the variants identified in these brothers may contribute to their varying courses of CF lung disease, including a known disease-associated variant in SERPINA 1 (middle and youngest), a known CF modifier (Bartlett et al. 2009), and the HFE p.H63D variant found in a heterozygous state in the middle and eldest brother and in a homozygous state in the youngest brother. The former is known to increase risk of liver disease in CF (Bartlett et al. 2009) and COPD and liver disease in otherwise healthy individuals (Pérez-Rubio et al. 2015; Greene et al. 2016). HFE variants have been reported to be present at a surprisingly high frequency among CF patients (Devaney et al. 2003; Smith et al. 2019) and appear to be modifiers of lung disease severity based on accelerating pulmonary dysfunction (Smith et al. 2019). This effect might be attributable to altering susceptibility to infections with many bacteria, including PA (Khan et al. 2007).
The known deleterious MBL2 p.R52C variant, which was heterozygous in the eldest and youngest brothers, has been associated with recurrent respiratory tract infections and greater bronchiectasis severity in patients with ciliary dyskinesia (Pifferi et al. 2015). It has also been reported that mannose-binding lectin (MBL)-deficient CF patients are colonized with PA earlier compared to MBL-sufficient patients (Haerynck et al. 2012; Gravina et al. 2015), and MBL has been linked to the age-related decline in lung function in CF patients (Garred et al. 1999; Pifferi et al. 2015). Consistent with these data, the eldest brother colonized PA at the youngest age.
Only the youngest brother was found to be heterozygous for a very rare nonsynonymous variant in the plasminogen gene (PLG; p.D238N). Plasminogen has been shown to directly bind to AF and individuals with PLG deficiency can have tracheobronchial fibrin deposits, which have been shown to impair the ciliary system, thereby predisposing to recurrent sinobronchial infections (Schuster et al. 2007). In a patient with CF in which mucociliary clearance is already compromised (Chatelin et al. 2017), PLG deficiency may increase the risk of Aspergillus bronchitis or related symptoms (Zaas et al. 2008). The presence of the variant in the youngest brother is consistent with his deterioration after 12 yr of age, when he showed recurrent AF-positive cultures.
CYP2C19 status explains ∼40% of the variability in response to azole antifungal agents for systemic use (Berge et al. 2011). Given the potential severity of illness in CF patients with invasive fungal infections, it is critical that therapeutic antifungal concentrations be attained rapidly (Berge et al. 2011; Hamadeh et al. 2017). Variability in response complicates optimal dosing and can increase (dependent on genotype) either risk of toxicity or risk of undertreatment (Berge et al. 2011; Hamadeh et al. 2017). Of note, in this case, the youngest brother, who may be at increased risk for AF infection owing to modifiers, may also be at increased risk of toxicity at standard therapeutic antifungal dosage because of CYP2C19 genotype. Although a lower dose may be necessary to avoid toxicity in such individuals, it is important to note that the severity of fungal infection–related disease combined with high variability in antifungal response already necessitates a therapeutic drug monitoring (TDM) approach that would be expected to dramatically reduce toxicity associated risk in this individual (Billaud et al. 2010).
Conclusions
Our data show that there is notable genomic variation in CF related loci across these three siblings with CF. A complex interplay between the risk to disease or infection conferred by genetic modifiers and the response to drugs used to treat the disease and infection as the result of pharmacogenomics were likely contributory to the clinical outcomes in these patients.
To our knowledge, this is the first application of WGS to individual patients with CF applied in an effort to clarify (and eventually predict) the course of the disease in a strategy of personalized/precision medicine. However, additional cohort studies are needed to further assess this hypothesis. We also suggest that analysis of WGS data initially generated for identification of causal or modifier variation might also provide useful for the generation of pharmacogenomics data. Clearly, further evaluation with regards to specificity, sensitivity, and reproducibility would be required.
METHODS
Summary of RCT Protocol
The Wisconsin RCT of NBS was initiated in April 1985, and patients were eligible for enrollment until July 1994. The protocol for this unique project was conducted before mandatory registration and has been described in detail elsewhere (Farrell et al. 2001). Relevant to this study, each patient was managed with an Evaluation and Treatment Protocol (Farrell 2000) and had clinical outcome variables assessed systematically every 3 mo, along with regular nutritional status evaluations using anthropometry and biochemical markers. To evaluate lung disease, we used (Farrell et al. 2003) a combination of spirometry and longitudinal quantitative radiology annually and biannually, respectively, scoring the chest radiographs with the sensitive WCXR scoring method—a rating system that scores the severity of air trapping, peribronchial thickening, bronchiectasis, other indicators of infection, and atelectasis (Koscik et al. 2000). From this cohort of patients, we identified for WGS two brothers who as children showed lung disease course discordances—one, the youngest, was milder, whereas the middle brother appeared more severe than average—and a third, eldest brother (born before the RCT) also had severe lung disease.
Sample Handling and Whole-Genome Sequencing
After we obtained IRB approvals from the University of Wisconsin Human Subjects Committee and written informed consent obtained directly for research and publication, we collected whole blood samples from each brother in 4 mL volumes, anticoagulated specimens with EDTA, and then harvested leukocytes from the postcentrifugation buffy coats. The middle brother's DNA was isolated from the blood sample at the University of Wisconsin, where the sample underwent WGS using an Illumina HiSeq 4000 instrument. The harvested leukocyte samples from the eldest and youngest brothers were sent frozen to HudsonAlpha Institute for Biotechnology for on-site DNA isolation using the QIAsymphony DSP DNA Mini Kit. The extracted DNA was then quantified using a Qubit system as well as using agarose gel electrophoresis to ensure sufficient yield and quality for sequencing. The eldest and youngest brothers’ DNA samples were sequenced at HudsonAlpha Institute for Biotechnology's Genomic Services Laboratory using an Illumina HiSeqX paired-end 150-bp read methodology. Samples were sequenced to a mean depth of 35× coverage with >80% of bases covered at 20×. Specific coverage information for variants reported is provided in Table 2.
Secondary and Tertiary Analyses
Secondary and tertiary analyses followed standard methods (Worthey 2017). In brief, sequences for each of the brothers were aligned to reference genome hg19 using BWA-mem according to standard protocols (Li and Durbin 2009). The aligned sequences were processed using the GATK package for base quality score recalibration, insertion/deletion realignments, duplicates removal, and discovery of single-nucleotide substitutions and small insertions and deletions, as well as genotyping (DePristo et al. 2011). Quality control metrics and sample swap tests were applied, including blinded CFTR genotype determinations and sample relationship metrics. The resultant variant files were loaded into the tertiary analysis software, Codicem (http://envisiongenomics.com/codicem-analysis-platform/), developed by the Worthey Laboratory at the HudsonAlpha Institute for Biotechnology. Coverage information for variants reported is provided in Table 2.
Codicem was used to analyze and support interpretation of the variant data (described elsewhere (Holt et al. 2019). Preset filter steps were used to remove variants of poor quality as well as variants in genes on the American College of Medical Genetics list of 59 genetic conditions with no phenotype connection to CF (Kalia et al. 2017). This was used to reduce, if not eliminate, the potential for identifying incidental findings that should be returned, per the IRB-approved protocol. The filtering strategy used in Codicem also prioritized and excluded variants based on various in silico damage predictions, existing disease associations including CF modifiers (e.g., Corvol et al. 2015), phenotype terms, and population allele frequencies. This reduced the number of variants to be reviewed to those most likely to be significant. Variants passing these filters were reviewed by two variant analysts who further assessed variants based on their potential effect on gene function and CF pathogenesis/progression. Variants of interest were selected and passed to team review. Variants subsequently deemed likely contributory underwent Sanger confirmation. These analyses allowed us to create the list of potential genetic modifiers provided in Supplemental Table 2.
Pharmacogenomics Analysis
Pharmacogenomic definitions were downloaded from PharmGKB (Barbarino et al. 2018), an expert-curated database, in order to build a pharmacogenomics database containing simple (i.e., single-variant) to complex (i.e., many-variant) haplotype/diplotype associations along with drug metabolism, response, and adverse reaction data. The association of pharmacogenomic findings for a given patient followed a three-step process. First, the pharmacogenomics annotation pipeline, an in-house application written in Java, gathered all relevant variant information for a patient from a variant call format (VCF) file. Next, diplotypes and genotypes were inferred using this data. Finally, pharmacologically relevant curations for each patient were extracted by matching haplotype to association data. All curations selected to be of interest were manually rereviewed by two analysts. Passing curations were categorized by drug class, as defined by Drug Bank (Wishart et al. 2018). Variants for drug classes of interest (e.g., antibiotics and immunosuppressants) were manually reviewed to identify potentially significant variant-drug combinations. Pharmacogenomic variants present in each of the brothers were automatically annotated with their respective effects matched to this database and subject to additional manual curation.
ADDITIONAL INFORMATION
Data Deposition and Access
Deidentified variant data has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV001142519.1, SCV001142520.1, SCV001142521.1, SCV001142522.1, and SCV001142523.1. Raw sequencing data could not be deposited because of lack of consent.
Ethics Statement
This study was approved by the IRB at the University of Wisconsin. Appropriate written consent was obtained prior to participation in this study.
Author Contributions
E.A.W. and P.M.F. were responsible for the conception and design of this study and supervised the work. A.T.B., P.M.F., and A.L. were involved in gathering of clinical data and patient management. M.A.W., C.B., D.B., J.M.H., N.S., B.W., and E.A.W. contributed to genomic analysis and interpretation. M.A.W., C.B., D.B., A.T.B., N.S., B.W., P.M.F., and E.A.W. contributed to writing this work, and all authors participated in reviewing the manuscript.
Acknowledgments
We thank members of the Wisconsin Cystic Fibrosis Neonatal Screening Project team, especially the Madison CF Center; Dr. Michael Rock, Professor Michael Sussman, and Joshua Hyman at the Biotechnology Center of the University of Wisconsin–Madison; and Sangita Murali, PhD at the Department of Nutritional Sciences, of the University of Wisconsin–Madison.
Funding
This study was supported by the National Institutes of Health (NIH) grant R01DK034108 and funding from The Legacy of Angels Foundation. P.M.F. reports consulting work with and grant funding from the Cystic Fibrosis Foundation. M.A.W., A.L., C.B., D.B., J.M.H., N.S., B.W., and E.A.W. have no financial relationships relevant to this article to disclose.
Competing Interest Statement
The authors have declared no competing interest.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Arai K, Madoiwa S, Mimuro J, Asakura S, Matsuda M, Sako T, Sakata Y. 1998. Role of the kringle domain in plasminogen activation with staphylokinase. J Biochem 123: 71–77. 10.1093/oxfordjournals.jbchem.a021918 [DOI] [PubMed] [Google Scholar]
- Barbarino JM, Whirl-Carrillo M, Altman RB, Klein TE. 2018. PharmGKB: a worldwide resource for pharmacogenomic information. Wiley Interdiscip Rev Syst Biol Med 10: e1417 10.1002/wsbm.1417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett JR, Friedman KJ, Ling SC, Pace RG, Bell SC, Bourke B, Castaldo G, Castellani C, Cipolli M, Colombo C, et al. 2009. Genetic modifiers of liver disease in cystic fibrosis. J Amer Med Assn 302: 1076–1083. 10.1001/jama.2009.1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge M, Guillemain R, Trégouet DA, Amrein C, Boussaud V, Chevalier P, Lillo-Lelouet A, Le Beller C, Laurent-Puig P, Beaune PH, et al. 2011. Effect of cytochrome P450 2C19 genotype on voriconazole exposure in cystic fibrosis lung transplant patients. Eur J Clin Pharmacol 67: 253–260. 10.1007/s00228-010-0914-2 [DOI] [PubMed] [Google Scholar]
- Billaud EM, Guillemain R, Berge M, Amrein C, Lefeuvre S, Louet AL, Boussaud V, Chevalier P. 2010. Pharmacological considerations for azole antifungal drug management in cystic fibrosis lung transplant patients. Med Mycol 48(Suppl 1): S52–S59. 10.3109/13693786.2010.505203 [DOI] [PubMed] [Google Scholar]
- Bleecker ER, Postma DS, Lawrance RM, Meyers DA, Ambrose HJ, Goldman M. 2007. Effect of ADRB2 polymorphisms on response to longacting β2-agonist therapy: a pharmacogenetic analysis of two randomised studies. Lancet 370: 2118–2125. 10.1016/S0140-6736(07)61906-0 [DOI] [PubMed] [Google Scholar]
- Carlton VE, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL, Strauss KA, Shneider BL, Lim WA, Salen G, et al. 2003. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet 34: 91–96. 10.1038/ng1147 [DOI] [PubMed] [Google Scholar]
- Castaldi PJ, Hersh CP, Reilly JJ, Silverman EK. 2009. Genetic associations with hypoxemia and pulmonary arterial pressure in COPD. Chest 135: 737–744. 10.1378/chest.08-1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatelin R, Anne-Archard D, Murris-Espin M, Thiriet M, Poncet P. 2017. Numerical and experimental investigation of mucociliary clearance breakdown in cystic fibrosis. J Biomech 53: 56–63. 10.1016/j.jbiomech.2016.12.026 [DOI] [PubMed] [Google Scholar]
- Corvol H, Blackman SM, Boelle PY, Gallins PJ, Pace RG, Stonebraker JR, Accurso FJ, Clement A, Collaco JM, Dang H, et al. 2015. Genome-wide association meta-analysis identifies five modifier loci of lung disease severity in cystic fibrosis. Nat Commun 6: 8382 10.1038/ncomms9382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutting GR. 2010. Modifier genes in Mendelian disorders: the example of cystic fibrosis. Ann N Y Acad Sci 1214: 57–69. 10.1111/j.1749-6632.2010.05879.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debray D, Corvol H, Housset C. 2019. Modifier genes in cystic fibrosis-related liver disease. Curr Opin Gastroenterol 35: 88–92. 10.1097/MOG.0000000000000508 [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. 10.1038/ng.806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaney J, Maher JM, Smith T, Houghton JA, Glennon M. 2003. HFE alleles in an Irish cystic fibrosis population. Genet Test 7: 155–158. 10.1089/109065703322146876 [DOI] [PubMed] [Google Scholar]
- Dorfman R, Sandford A, Taylor C, Huang B, Frangolias D, Wang Y, Sang R, Pereira L, Sun L, Berthiaume Y, et al. 2008. Complex two-gene modulation of lung disease severity in children with cystic fibrosis. J Clin Invest 118: 1040–1049. 10.1172/JCI33754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan QL, Lasky-Su J, Himes BE, Qiu W, Litonjua AA, Damask A, Lazarus R, Klanderman B, Irvin CG, Peters SP, et al. 2014. A genome-wide association study of bronchodilator response in asthmatics. Pharmacogenomics J 14: 41–47. 10.1038/tpj.2013.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durno C, Corey M, Zielenski J, Tullis E, Tsui LC, Durie P. 2002. Genotype and phenotype correlations in patients with cystic fibrosis and pancreatitis. Gastroenterology 123: 1857–1864. 10.1053/gast.2002.37042 [DOI] [PubMed] [Google Scholar]
- Ericson JE, Zimmerman KO, Gonzalez D, Melloni C, Guptill JT, Hill KD, Wu H, Cohen-Wolkowiez M. 2017. A systematic literature review approach to estimate the therapeutic index of selected immunosuppressant drugs after renal transplantation. Ther Drug Monit 39: 13–20. 10.1097/FTD.0000000000000364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell PM. 2000. Improving the health of patients with cystic fibrosis through newborn screening. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Adv Pediatr 47: 79–115. [PubMed] [Google Scholar]
- Farrell PM, Kosorok MR, Rock MJ, Laxova A, Zeng L, Lai HC, Hoffman G, Laessig RH, Splaingard ML. 2001. Early diagnosis of cystic fibrosis through neonatal screening prevents severe malnutrition and improves long-term growth. Wisconsin Cystic Fibrosis Neonatal Screening Study Group. Pediatrics 107: 1–13. 10.1542/peds.107.1.1 [DOI] [PubMed] [Google Scholar]
- Farrell PM, Li Z, Kosorok MR, Laxova A, Green CG, Collins J, Lai HC, Makholm LM, Rock MJ, Splaingard ML. 2003. Longitudinal evaluation of bronchopulmonary disease in children with cystic fibrosis. Pediatr Pulmonol 36: 230–240. 10.1002/ppul.10336 [DOI] [PubMed] [Google Scholar]
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr, Ellis MC, Fullan A, et al. 1996. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet 13: 399–408. 10.1038/ng0896-399 [DOI] [PubMed] [Google Scholar]
- Gaedigk A, Ingelman-Sundberg M, Miller NA, Leeder JS, Whirl-Carrillo M, Klein TE. 2018. The pharmacogene variation (PharmVar) consortium: incorporation of the human cytochrome P450 (CYP) allele nomenclature database. Clin Pharmacol Ther 103: 399–401. 10.1002/cpt.910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garred P, Pressler T, Madsen HO, Frederiksen B, Svejgaard A, Høiby N, Schwartz M, Koch C. 1999. Association of mannose-binding lectin gene heterogeneity with severity of lung disease and survival in cystic fibrosis. J Clin Invest 104: 431–437. 10.1172/JCI6861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill D, Benyamin B, Moore LSP, Monori G, Zhou A, Koskeridis F, Evangelou E, Laffan M, Walker AP, et al. 2019. Associations of genetically determined iron status across the phenome: a mendelian randomization study. PLoS Med 16: e1002833 10.1371/journal.pmed.1002833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravina LP, Crespo C, Giugno H, Sen L, Chertkoff L, Mangano A, Castanos C. 2015. Mannose-binding lectin gene as a modifier of the cystic fibrosis phenotype in Argentinean pediatric patients. J Cyst Fibros 14: 78–83. 10.1016/j.jcf.2014.07.012 [DOI] [PubMed] [Google Scholar]
- Greene CM, Marciniak SJ, Teckman J, Ferrarotti I, Brantly ML, Lomas DA, Stoller JK, McElvaney NG. 2016. α1-antitrypsin deficiency. Nat Rev Dis Primers 2: 16051 10.1038/nrdp.2016.51 [DOI] [PubMed] [Google Scholar]
- Haerynck F, Van Steen K, Cattaert T, Loeys B, Van Daele S, Schelstraete P, Claes K, Van Thielen M, De Canck I, Mahachie John JM, et al. 2012. Polymorphisms in the lectin pathway genes as a possible cause of early chronic Pseudomonas aeruginosa colonization in cystic fibrosis patients. Hum Immunol 73: 1175–1183. 10.1016/j.humimm.2012.08.010 [DOI] [PubMed] [Google Scholar]
- Hamadeh IS, Klinker KP, Borgert SJ, Richards AI, Li W, Mangal N, Hiemenz JW, Schmidt S, Langaee TY, Peloquin CA, et al. 2017. Impact of the CYP2C19 genotype on voriconazole exposure in adults with invasive fungal infections. Pharmacogenet Genomics 27: 190–196. 10.1097/FPC.0000000000000277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassett C, Aicher L, Sidhu JS, Omiecinski CJ. 1994. Human microsomal epoxide hydrolase: genetic polymorphism and functional expression in vitro of amino acid variants. Hum Mol Genet 3: 421–428. 10.1093/hmg/3.3.421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitzeneder S, Seidel M, Förster-Waldl E, Heitger A. 2012. Mannan-binding lectin deficiency—Good news, bad news, doesn't matter? Clin Immunol 143: 22–38. 10.1016/j.clim.2011.11.002 [DOI] [PubMed] [Google Scholar]
- Holt JM, Wilk B, Birch CL, Brown DM, Gajapathy M, Moss AC, Sosonkina N, Wilk MA, Anderson JA, Harris JM, et al. 2019. VarSight: prioritizing clinically reported variants with binary classification algorithms. BMC Bioinformatics 20: 496 10.1186/s12859-019-3026-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi T, Gondo H, Miyagawa H, Otsuka J, Inaba S, Nagafuji K, Takase K, Tsukamoto Y, Koyama T, Mitoma H, et al. 2005. Association of MBL gene polymorphisms with major bacterial infection in patients treated with high-dose chemotherapy and autologous PBSCT. Genes Immun 6: 162–166. 10.1038/sj.gene.6364165 [DOI] [PubMed] [Google Scholar]
- Joly P, Vignaud H, Di Martino J, Ruiz M, Garin R, Restier L, Belmalih A, Marchal C, Cullin C, Arveiler B, et al. 2017. ERAD defects and the HFE-H63D variant are associated with increased risk of liver damages in α 1-antitrypsin deficiency. PLoS One 12: e0179369 10.1371/journal.pone.0179369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, Herman GE, Hufnagel SB, Klein TE, Korf BR, et al. 2017. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med 19: 249–255. 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- Kerem E, Kerem B. 1996. Genotype-phenotype correlations in cystic fibrosis. Pediatr Pulmonol 22: 387–395. [DOI] [PubMed] [Google Scholar]
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, Buchwald M, Tsui LC. 1989. Identification of the cystic fibrosis gene: genetic analysis. Science 245: 1073–1080. 10.1126/science.2570460 [DOI] [PubMed] [Google Scholar]
- Kerem E, Corey M, Kerem BS, Rommens J, Markiewicz D, Levison H, Tsui LC, Durie P. 1990. The relation between genotype and phenotype in cystic fibrosis–analysis of the most common mutation (delta F508). N Engl J Med 323: 1517–1522. 10.1056/NEJM199011293232203 [DOI] [PubMed] [Google Scholar]
- Khan FA, Fisher MA, Khakoo RA. 2007. Association of hemochromatosis with infectious diseases: expanding spectrum. Int J Infect Dis 11: 482–487. 10.1016/j.ijid.2007.04.007 [DOI] [PubMed] [Google Scholar]
- Koscik RE, Kosorok MR, Farrell PM, Collins J, Peters ME, Laxova A, Green CG, Zeng L, Rusakow LS, Hardie RC, et al. 2000. Wisconsin cystic fibrosis chest radiograph scoring system: validation and standardization for application to longitudinal studies. Pediatr Pulmonol 29: 457–467. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lai HJ, Kosorok MR, Laxova A, Rock MJ, Splaingard ML, Farrell PM. 2004. Longitudinal pulmonary status of cystic fibrosis children with meconium ileus. Pediatr Pulmonol 38: 277–284. 10.1002/ppul.20092 [DOI] [PubMed] [Google Scholar]
- Li H, Fu WP, Hong ZH. 2013. Microsomal epoxide hydrolase gene polymorphisms and risk of chronic obstructive pulmonary disease: a comprehensive meta-analysis. Oncol Lett 5: 1022–1030. 10.3892/ol.2012.1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Liu FL, Bai ZJ, Zhao N, Zhang LY, Lu X, Chen ZL. 2012. Defective activities, but not secretions, resulting from gene point mutations of human mannan-binding lectin. Mol Med Rep 5: 1121–1127. 10.3892/mmr.2012.782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor AR, Rhaney K. 1948. Congenital fibrocystic disease of the pancreas; a report of two proved cases of dissimilar clinical types in siblings. Arch Dis Child 23: 56–60. 10.1136/adc.23.113.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDougal KE, Green DM, Vanscoy LL, Fallin MD, Grow M, Cheng S, Cutting GR. 2010. Use of a modeling framework to evaluate the effect of a modifier gene (MBL2) on variation in cystic fibrosis. Eur J Hum Genet 18: 680–684. 10.1038/ejhg.2009.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairz M, Schroll A, Haschka D, Dichtl S, Tymoszuk P, Demetz E, Moser P, Haas H, Fang FC, Theuri I, et al. 2017. Genetic and dietary iron overload differentially affect the course of Salmonella typhimurium infection. Front Cell Infect Microbiol 7: 110 10.3389/fcimb.2017.00110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neal WK, Knowles MR. 2018. Cystic fibrosis disease modifiers: complex genetics defines the phenotypic diversity in a monogenic disease. Annu Rev Genomics Hum Genet 19: 201–222. 10.1146/annurev-genom-083117-021329 [DOI] [PubMed] [Google Scholar]
- Orenstein DM, Boat TF, Stern RC, Tucker AS, Charnock EL, Matthews LW, Doershuk CF. 1977. The effect of early diagnosis and treatment in cystic fibrosis: a seven-year study of 16 sibling pairs. Am J Dis Child 131: 973–975. 10.1001/archpedi.1977.02120220039005 [DOI] [PubMed] [Google Scholar]
- Pérez-Rubio G, Jiménez-Valverde LO, Ramirez-Venegas A, Camarena A, Sansores RH, Flores-Trujillo F, Reséndiz-Hernández JM, Falfán-Valencia R. 2015. Prevalence of α-1 antitrypsin high-risk variants in Mexican mestizo population and their association with lung function values. Arch Bronconeumol 51: 80–85. 10.1016/j.arbres.2014.09.010 [DOI] [PubMed] [Google Scholar]
- Pifferi M, Bush A, Michelucci A, Di Cicco M, Piras M, Caramella D, Mazzei F, Neri M, Pioggia G, Tartarisco G, et al. 2015. Mannose-binding lectin 2 gene polymorphism and lung damage in primary ciliary dyskinesia. Pediatr Pulmonol 50: 179–186. 10.1002/ppul.23026 [DOI] [PubMed] [Google Scholar]
- Poon AH, Tantisira KG, Litonjua AA, Lazarus R, Xu J, Lasky-Su J, Lima JJ, Irvin CG, Hanrahan JP, Lange C, et al. 2008. Association of corticotropin-releasing hormone receptor-2 genetic variants with acute bronchodilator response in asthma. Pharmacogenet Genomics 18: 373–382. 10.1097/FPC.0b013e3282fa760a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. 2015. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17: 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DB, Li Z, Laxova A, Rock MJ, Levy H, Collins J, Ferec C, Farrell PM. 2014. Risk factors for the progression of cystic fibrosis lung disease throughout childhood. Ann Am Thorac Soc 11: 63–72. 10.1513/AnnalsATS.201309-303OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders DB, Zhang Z, Farrell PM, Lai HJ; Wisconsin Cystic Fibrosis Neonatal Screening Group. 2018. Early life growth patterns persist for 12 years and impact pulmonary outcomes in cystic fibrosis. J Cyst Fibros 17: 528–535. 10.1016/j.jcf.2018.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster V, Hügle B, Tefs K. 2007. Plasminogen deficiency. J Thromb Haemost 5: 2315–2322. 10.1111/j.1538-7836.2007.02776.x [DOI] [PubMed] [Google Scholar]
- Scott SA, Sangkuhl K, Stein CM, Hulot JS, Mega JL, Roden DM, Klein TE, Sabatine MS, Johnson JA, Shuldiner AR, et al. 2013. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther 94: 317–323. 10.1038/clpt.2013.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CA, Harrison DJ. 1997. Association between polymorphism in gene for microsomal epoxide hydrolase and susceptibility to emphysema. Lancet 350: 630–633. 10.1016/S0140-6736(96)08061-0 [DOI] [PubMed] [Google Scholar]
- Smith DJ, Klein K, Hartel G, Wainwright CE, Bell SC, Anderson GJ, Reid DW. 2019. Mutations in the HFE gene can be associated with increased lung disease severity in cystic fibrosis. Gene 683: 12–17. 10.1016/j.gene.2018.10.002 [DOI] [PubMed] [Google Scholar]
- Vibhuti A, Arif E, Deepak D, Singh B, Qadar Pasha MA. 2007. Genetic polymorphisms of GSTP1 and mEPHX correlate with oxidative stress markers and lung function in COPD. Biochem Biophys Res Commun 359: 136–142. 10.1016/j.bbrc.2007.05.076 [DOI] [PubMed] [Google Scholar]
- Whirl-Carrillo M, McDonagh EM, Hebert JM, Gong L, Sangkuhl K, Thorn CF, Altman RB, Klein TE. 2012. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther 92: 414–417. 10.1038/clpt.2012.96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Ring BJ, Cantrell VE, Jones DR, Eckstein J, Ruterbories K, Wrighton SA. 2002. Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab Dispos 30: 883–891. 10.1124/dmd.30.8.883 [DOI] [PubMed] [Google Scholar]
- Wishart DS, Feunang YD, Guo AC, Lo Z, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, et al. 2018. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 46: d1074–d1082. 10.1093/nar/gkx1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthey EA. 2017. Analysis and annotation of whole-genome or whole-exome sequencing derived variants for clinical diagnosis. Curr Protoc Hum Genet 95: 9.24.1–9.24.28. 10.1002/cphg.49 [DOI] [PubMed] [Google Scholar]
- Wrighton SA, VandenBranden M, Ring BJ. 1996. The human drug metabolizing cytochromes P450. J Pharmacokinet Biopharm 24: 461–473. 10.1007/BF02353474 [DOI] [PubMed] [Google Scholar]
- Zaas AK, Liao G, Chien JW, Weinberg C, Shore D, Giles SS, Marr KA, Usuka J, Burch LH, Perera L, et al. 2008. Plasminogen alleles influence susceptibility to invasive aspergillosis. PLoS Genet 4: e1000101 10.1371/journal.pgen.1000101 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Deidentified variant data has been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV001142519.1, SCV001142520.1, SCV001142521.1, SCV001142522.1, and SCV001142523.1. Raw sequencing data could not be deposited because of lack of consent.