

SUMMARY
To understand the molecular mechanisms that mediate the anti-hepatitis B virus (HBV) effect of interferon (IFN) therapy, we conduct high-throughput bimolecular fluorescence complementation screening to identify potential physical interactions between the HBx protein and 145 IFN-stimulated genes (ISGs). Seven HBx-interacting ISGs have consistent and significant inhibitory effects on HBV replication, among which TRIM5γ suppresses HBV replication by promoting K48-linked ubiquitination and degradation of the HBx protein on the K95 ubiquitin site. The B-Box domain of TRIM5γ under overexpression conditions is sufficient to trigger HBx degradation and is responsible both for interacting with HBx and recruiting TRIM31, which is an ubiquitin ligase that triggers HBx ubiquitination. High expression levels of TRIM5γ in IFN-α-treated HBV patients might indicate a better therapeutic effect. Thus, our studies identify a crucial role for TRIM5γ and TRIM31 in promoting HBx degradation, which may facilitate the development of therapeutic agents for the treatment of patients with IFN-resistant HBV infection.
Graphical Abstract
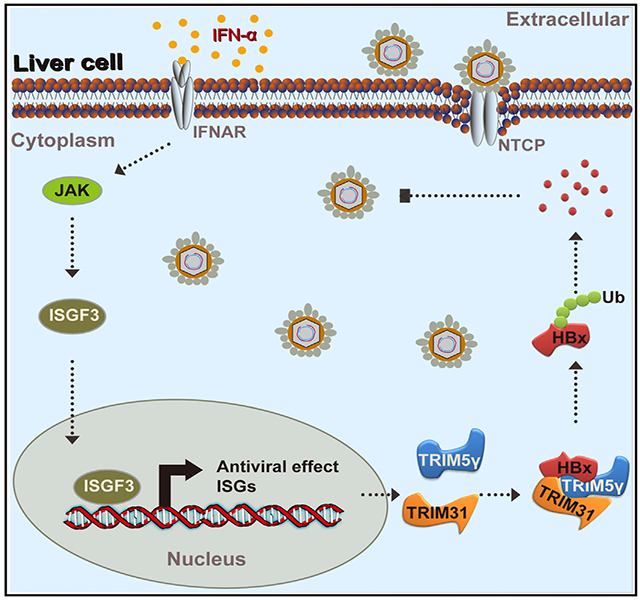
In Brief
In brief, Tan et al. find that IFN-induced TRIM5γ recruits TRIM31 to degrade HBx, resulting in suppression of hepatitis B virus replication.
INTRODUCTION
Chronic hepatitis B virus (HBV) infection remains a major public health threat. It is associated with increasing rates of hepatocellular carcinoma (HCC) worldwide. According to the World Health Organization (WHO), 2 billion people are infected with HBV, of which over 240 million are chronically infected, which places these people at a high risk for development of liver cirrhosis and eventually HCC. Although a vaccine has been available for HBV for almost 30 years, nearly 1 million people die each year due to HBV-related complications, including severe chronic hepatitis, cirrhosis, and primary liver cancer (Mutimer and Lok, 2012).
HBV is a partially double-stranded DNA virus belonging to the Hepadnaviridae family (Evans and Seeger, 2007). After entry into host cells, the viral genome translocates to the nucleus and is converted to covalently closed circular DNA (cccDNA), which is the transcription template for all HBV viral RNAs. Currently available HBV therapeutics (including INF-α and antiviral drugs) fail to eradicate the cccDNA reservoir from infected hepatocytes, despite suppressing replication of new viral DNA. Consequently, the persistent cccDNA results in post-treatment viral rebound (Nassal, 2015; Revill and Locarnini, 2016).
The HBV-encoded regulatory protein hepatitis B virus X protein (HBx) stimulates HBV gene expression from the cccDNA template; however, the mechanism by which HBx facilitates HBV replication is not clear (Keasler et al., 2007). HBx is a 154-amino-acid protein with an N-terminal negative regulatory domain and C-terminal transactivation or coactivation domain. HBx interacts with several cellular proteins, and its role in virus replication may be mediated through these interactions. The best-characterized HBx binding partner is the damage-specific DNA binding protein 1 (DDB1) (Sitterlin et al., 1997). The interaction between HBx and DDB1 is conserved among the HBx proteins from all mammalian hepadnaviruses and woodchuck hepatitis virus (WHV) X protein (Sitterlin et al., 1997). This binding is essential for HBV replication (Hodgson et al., 2012). It was recently reported that an HBx-DDB1-CUL4-ROC1 E3 ligase complex targets the SMC5/6 complex to enhance HBV gene expression from episomal cccDNA (Mitra and Guo, 2016; Murphy et al., 2016).
IFN therapy is an approved treatment modality for chronic HBV infection. However, the poor response and substantial side effects associated with this treatment impact its clinical utility (Tan et al., 2018b). HBV has been shown to inhibit IFN-stimulated genes (ISGs) and even the production of IFN in infected patients (Khan et al., 2016; Lim et al., 2017; Liu et al., 2015; Xu et al., 2016). However, the mechanism of HBV-mediated inhibition of ISGs is poorly understood. Therefore, unraveling the molecular mechanisms of the interaction between IFN and HBV during the course of viral infection is a key imperative, which may help improve the efficacy of IFN therapy.
The induction of the type I IFN family (IFN-α, -β, -ε, -κ, and -ω) is perhaps the most important innate immune response to viral infection. Since its discovery in 1957, type I IFN has been recognized as the major antiviral cytokine in vertebrates (Hayes and Chayama, 2017; Isaacs and Lindenmann, 1957). Most nucleated vertebrate cells are able to both produce and respond to type I IFN. The antiviral function of type I IFN is carried out through its binding to the type I IFN receptor (IFNAR1), activation of the JAK/STAT pathway, and subsequent induction of about 300 ISGs (Ivashkiv and Donlin, 2014), which can inhibit different stages of the viral life cycle (Chen et al., 2014; Liu et al., 2013). Some ISGs have already been shown to inhibit HBV replication. For example, TRIM22 was shown to confer antiviral immunity against HBV by its inhibitory effect on viral core promoter activity (Gao et al., 2009). We reported that TRIM14 inhibited HBV replication by blocking the formation of the DDB1-HBx-Smc complex (Xu et al., 2019), and APOBEC3A/B was shown to play a critical role in the degradation of nuclear HBV cccDNA (Lucifora et al., 2014). Unfortunately, there is currently no effective treatment for patients who respond poorly to IFN therapy. Therefore, characterization of the ISGs that specifically modulate HBV infection and replication is a key imperative. In the present study, we conducted a high-throughput bimolecular fluorescence complementation (BiFC) assay screen and identified 41 out of 145 ISG products that interact with the HBx protein. The interaction of 26 ISGs was further confirmed by co-immunoprecipitation (coIP). Seven HBx-interacting ISGs (GBP2, PVRL4, CBFβ, TRIM38, TRIM5γ, TRIM25, and Gadd45γ) showed strong inhibitory effects on HBV replication. The TRIM5 family was famous since the discovery of TRIM5α as a blocking factor of HIV infection by binding to the capsid that surrounds and protects the viral core (Stremlau et al., 2004). TRIM5 proteins share a common tripartite motif, which consists of RING, B-box 2, and coiled-coil domains, followed by SPRY, a C-terminal domain that is required for capsid recognition (Ganser-Pornillos and Pornillos, 2019). Interestingly, TRIM5γ does not have a SPRY domain. Here, we further narrowed our investigation to TRIM5γ anti-HBV function and showed that the BBox domain of TRIM5γ is sufficient for interaction with HBx and for triggering the HBx-K48 ubiquitination and degradation. Furthermore, we showed that this process likely requires the E3 ubiquitin ligase activity of TRIM31, recruited by TRIM5γ. More importantly, we found that high induction of TRIM5γ in the IFN-α-treated HBV patients predicts a much better outcome. Our study, therefore, highlights a critical role for TRIM5γ and TRIM31 in promoting HBx degradation and reveals another mechanism that might impact the effectiveness of IFN therapy in HBV-infected patients. In addition, our study suggests that TRIM5γ may serve as a biomarker to predict the response to IFN-α therapy and that BBOX may be a potential candidate for the development of therapeutic agents that target HBx.
RESULTS
Identification of 26 ISG Proteins That Interact with the HBx Protein by BiFC and CoIP Assays
We hypothesized that ISGs can inhibit HBV viral infection and replication through interactions with individual viral proteins. A recent study researched how ISGs inhibit viruses by overexpression of about 300 individual ISGs (Liu et al., 2012). HBx is an indispensable transcriptional factor in the HBV life cycle, which interacts with multiple proteins and promotes HBV replication (Keasler et al., 2007). In the present study, we used a BiFC assay to screen 145 ISG proteins for interaction with HBx. This assay involves transient expression of HBx fused with the N-terminal portion of the yellow fluorescent protein (YFP) and cellular ISGs fused to the C terminus of YFP (Figure 1A). Interaction between viral proteins and ISGs in living cells results in the emission of fluorescent signals (Figure 1B) that can be detected by flow cytometric analysis. The BiFC assay identified 41 ISGs that potentially interact with the HBx protein (Figure 1C). To further validate the BiFC results, we conducted a coIP assay. DDB1 was used as a positive control (Hodgson et al., 2012) (Figure S1A), and two BiFC-negative ISGs (UBA7 and TRIM62) were used as negative controls (Figure S1B). Under conditions of overexpression, the BiFC assay may yield some false-positive results; therefore, 15 ISGs in the BiFC-positive group were not consistent with our coIP experiment. Finally, the coIP assay confirmed the results of the BiFC asssay for 26 ISGs in 293T cells (Figure S1C). We further confirmed the interaction of HBx with seven ISGs (Figures S2F–S2K) that we found had an inhibitory effect on HBV replication (Figures S2A–S2D, gray bar; labeled in red in Figure 1C) in HepG2 cells.
Figure 1. Screening of ISGs That Interact with HBx by BiFC Assay.

(A) Schematic illustration of the BiFC assay. (B) Expression plasmids for YFPc-GBP2 and YFPc-TRIM62 were transfected individually with YFPn-HBx plasmid into 293T cells. YFP fluorescence intensity was measured 36 h post-transfection. Scale bars represent 100 μm. (C) A total of 145 YFPc-ISG plasmids were transfected individually with the YFPn-HBx plasmid into 293T cells, and YFP fluorescence was measured after 36 h by flow cytometry. YFP fluorescence mean intensity (MFI) of >2 was deemed to signify interaction with HBx (labeled in orange) and <2 was deemed to indicate absence of any interaction (labeled in green). The positive control (YFPc-58CUN+YFPn-FNCC) is marked in purple, and the negative control (YFPc-58CUN or YFPn-FNCC was transfected individually; MFI, 1) is in blue. Mean ± SD values from three independent experiments are shown.
High Induction of TRIM5γ in IFN-α Treatment of HBV Patients Indicates a Better Therapeutic Effect
As multiple potent anti-HBV ISGs belonging to the TRIM family of proteins (including TRIM5γ, TRIM38, and TRIM25) were identified in this study, we further investigated their anti-viral function. In a very recent study, we showed that TRIM25, which was induced by type I IFN in an interleukin-27 (IL-27)-dependent manner, might inhibit HBV replication by promoting IFN production (Tan et al., 2018a). Becuase TRIM5γ was found to have the highest induction in response to IFN-α treatment among the 7 potent anti-HBV ISGs tested (Figure 2A), we asked if the induction of TRIM5γ was important in IFN-α treatment of HBV patients. To answer this question, blood samples of 31 HBV-infected patients collected before or 17 weeks after IFN-α treatment were assessed for serum levels of ALT, AST, HBeAg and HBV DNA (Table S1). Our data indicated that IFN-α treatment significantly decreased ALT, AST, HBeAg, and HBV DNA levels (Figure 2B). Interestingly, after determining the TRIM5γ expression levels in patient peripheral blood mononuclear cells (PBMCs) at 15 min before or 96 h after IFN-α treatment, we divided the patients into 2 groups; one group (15 patients) had low TRIM5γ induction and the other (16 patients) had high induction (Figures 2C). Just as we expected, the high group responded well to the treatment and exhibited much better outcomes compared to the group with low TRIM5γ induction (Figure 2E). As a control, we also detected the expression of TRIM22 (Figure 2D), a protein that was shown to inhibit HBV replication and was associated with good response to IFN treatment in vitro (Lim et al., 2017); however, the induction of TRIM22 was not associated with patient outcomes (Figure 2E). These findings strongly indicate that high induction of TRIM5γ in patients on IFN-α therapy predicts better outcomes. Collectively, these results demonstrate the direct co-relation between TRIM5γ expression and efficacy of IFN therapy.
Figure 2. Higher TRIM5γ Induction Indicates a Better Therapeutic Outcome of HBV Patients Undergoing IFN-α Treatment.

(A) HepG2 cells were treated with IFN-α (10 ng/mL) or untreated; qPCR was performed to analyze the mRNA expression level of 7 ISGs, as indicated. (B) Blood samples of 31 HBV-infected patients before and 17 weeks after IFN-α treatment were collected for ALT, AST, HbeAg, and HBV DNA detection. (C and D) PBMCs from HBV patients before or after IFN treatment were isolated and subjected to RNA extraction; qPCR was performed to evaluate the TRIM5γ (C) or TRIM22 (D) mRNA level. (E) Seventeen weeks after IFN treatment, the outcome of the two HBV-infected patients groups (TRIM5γ high or low, TRIM22 high or low) were analyzed by comparing the AST, ALT, HbeAg, and HBV DNA levels. Mean ± SD values from three independent experiments are shown. *p < 0.05, **p < 0.01, ***p < 0.001, #p > 0.05.
TRIM5γ Inhibits HBV Infection and Replication by Targeting HBx
The above results have demonstrated that TRIM5γ interacted with HBx and inhibited HBV replication. Also, similar to IFNα treatment, the inhibition of HBV replication by TRIM5γ showed a kinetics correlation (Figures S3A–S3C). BMS-200475, an HBV replication inhibitor, significantly inhibited both wild type (WT) and ΔX HBV replication in HepG2 cells (Figure S3D). Interestingly, we found that the inhibitory effect of TRIM5γ on HBV replication was much less significant in the pHBV1.2-ΔX-plasmid-transfected HepG2 cells compared with that in pHBV1.2-WT-plasmid-transfected cells (Figure 3A), which indicates that HBx might be the target of TRIM5γ. To verify the results in the HBV infection system, we performed the infection assay in HepG2-NTCP cells, which can support HBV infection because of the high expression of NTCP (Figure S3E) (Yan et al., 2012). HBsAg was detected in HBV-infected HepG2-NTCP cells by immunofluorescence (Figure S3F). HepG2-NTCP cells were infected with lenti-ctrl or TRIM5γ; 16 h later, the cells were infected with HBV (as shown in Figure 3B), and as expected, HBV DNA, pgRNA, and HBsAg expressions in the TRIM5γ-overexpressed HepG2-NTCP cells were significantly inhibited in the cells infected with HBV (Figures 3C and S3G). Furthermore, we intended to verify the effect of TRIM5γ in the IFN-α-mediated inhibition of HBV in vitro and found that HepG2-NTCP WT or TRIM5α/γ knockout (KO) cells were infected by HBV with or without IFN-α treatment, as we expected, TRIM5α/γ KO enhanced HBV replication, and the inhibitory effects of IFN-α in HBV replication was significantly reduced in TRIM5α/γ KO HepG2-NTCP cells (Figures 3D, S3H, and S3I), which suggested the indispensiable role of TRIM5γ in IFN-α-mediated inhibition of HBV replication. These results indicated the essential role of TRIM5γ in inhibiting HBV infection and replication by targeting HBx.
Figure 3. TRIM5γ Inhibits HBV Infection and Replication by Targeting HBx.

(A) HepG2 cells were co-transfected with constructs expressing TRIM5γ proteins and either pHBV1.2 WT or DX, as indicated. After 72 h, cells and supernatant were harvested and subjected to qPCR or ELISA. (B) Scheme of HBV infection. (C) HepG2-NTCP cells were infected with lentiviral-ctrl or lentiviral-TRIM5γ for 16 h, and later, cells were washed with PBS and inoculated for 24 h with HBV. After infection, cells were washed three times with PBS and were maintained for another 9 days; the medium was changed every 2 days. The supernatant and cells were collected for detection of HBV DNA, pgRNA, and HBsAg by qPCR or ELISA. (D) HepG2-NTCP WT or TRIM5α/γ KO cells were infected with HBV for 24 h with or without IFN-α treatment (10 ng/mL) and analyzed as in (C). Mean ± SD values from three independent experiments are shown. *p < 0.05, **p < 0.01, ***p < 0.001.
TRIM5γ Specifically Promotes HBx Degradation by K48-Linked Ubiquitination
We further investigated the mechanisms that underlie the antiviral effect of TRIM5γ mediated through HBx. Most of the TRIM proteins are RING-type E3 ligases, which recruit ubiquitin to target proteins for degradation (Hatakeyama, 2011). Therefore, TRIM5γ was co-transfected with HBV protein expression plasmids (HBV Core, Pre-core, S, or X) into 293T cells. The results showed that TRIM5γ specifically and strongly promotes HBx degradation but not the other HBV proteins (Figure 4A). We also found that out of the proteins that were tested (including TRIM38), only TRIM5γ inhibited HBx expression (Figure 4B). For additional testing, we, therefore, selected TRIM5α, which is another well-characterized member of the TRIM5 family with known anti-viral function and has the same N-terminal region (1–298 amino acids) as that of TRIM5γ (Battivelli et al., 2011). We found that TRIM5γ but not TRIM5α promoted HBx degradation (Figure 4C). In addition, TRIM5γ-induced HBx degradation was confirmed by immunofluorescence (Figure 4D). To further confirm the results, HepG2 cells with KO of both TRIM5γ and TRIM5α were generated using the CRISPR/Cas9 system (Figure 4E; Figure S4A). As expected, HBx expression was significantly elevated in HepG2 cells that lacked TRIM5α/γ after transfection of HBx (Figure 4E, line 1 and 5). In addition, degradation of HBx by IFN-α stimulation was almost blocked in TRIM5α/γ KO HepG2 cells (Figure 4E). These results indicated that TRIM5γ could specifically trigger HBx degradation and that it plays an important role in IFN-induced HBx degradation. TRIM5 was reported to be involved in autophagy (Keown et al., 2018); however, TRIM5γ-mediated HBx degradation was not affected by 3-MA (3-Methyladenine), the autophagy inhibitor (Figure S4B). The TRIM family is a diverse family of RING-finger-domain-containing proteins that mediate post-translational modifications, such as protein ubiquitination, which are involved in a variety of cellular functions (Kawai and Akira, 2011). To further investigate the mechanisms involved in TRIM5γ-mediated HBx degradation, we evaluated the HBx ubiquitination by TRIM5γ. As expected, degradation of HBx by TRIM5γ was rescued by the proteasome inhibitor MG132 but not by Z-VAD, the caspase in hibitor (Figure 4F); furthermore, the HBx mRNA level was not affected by TRIM5γ (Figure S4C). In addition, the coIP assay confirmed that TRIM5γ promotes HBx ubiquitination; moreover, we found that enhanced TRIM5γ-mediated HBx ubiquitination was linked to K48 but not to K63 in both 293T and HepG2 cells (Figures 4G and 4H; Figure S4D). These findings suggested that TRIM5γ may promote HBx K48-linked ubiquitination and degradation by the proteasome pathway.
Figure 4. TRIM5γ Specifically Promotes HBx Degradation.

(A) 293T cells were co-transfected with constructs expressing FLAG-HBV proteins and either GFP-TRIM5γ or EV as indicated. After 36 h, cells were harvested and whole-cell lysates were subjected to immunoblotting using anti-FLAG, anti-GFP, or anti-Tubulin antibody. (B) 293T cells were co-transfected with FLAG-HBx and either GFP-TRIM5γ, -USP2, -TRIM38, -UBE3A, or -EV expression plasmids as indicated and were analyzed as in (A). (C) HepG2 cells were co-transfected with plasmids expressing FLAG-HBx and either HA-TRIM5γ or TRIM5α or hemagglutinin (HA) empty vector as indicated, and cells were collected 28 h after transfection. Whole-cell lysates were immunoblotted with antibodies as indicated. (D) FLAG-HBx and either HA-TRIM5γ or HA empty vector was transfected into HepG2 cells as indicated; 48 h later, cells were fixed in acetone-methanol and subjected to immunofluorescence staining analysis using FLAG and HA antibodies. Scale bars represent 20 μm. (E) TRIM5α and TRIM5γ were both knocked out in HepG2 cells by CRISPR/Cas9 technology; WT or TRIM5α/γ KO HepG2 cells were transfected with FLAG-HBx; and 24 h later, cells were treated with IFN-α as indicated. Post-treatment, whole-cell lysates were immunoblotted with the indicated antibodies. (F) HepG2 cells were co-transfected with HA-TRIM5γ and FlAG-HBx as indicated; 24 h later, cells were treated with 10 mM MG132 or Z-VAD for 8 h and collected for immunoblotting with antibodies as indicated. (G) 293T cells were co-transfected with HA-Ub and FLAG-HBx, with or without GFP-TRIM5γ; 24 h later, cells were treated with MG-132 as indicated for 8 h and subjected to coIP analysis. Cell lysates and precipitated samples were subjected to immunoblot analysis using the indicated antibodies. (H) 293T cells were co-transfected with K48 or K63-Ub and Flag-HBx, with or without GFP-TRIM5γ; 24 h later, cells were treated with MG132 (10 μM) for 8 h; and cells were collected and analyzed as in (G). Data are representative of at least three independent experiments.
TRIM5γ BBox Domain Is Responsible for the Interaction and Degradation of HBx
To identify the TRIM5γ domain that interacts with HBx, individual plasmids encoding truncated TRIM5γ (as shown in Figure 5A) were co-transfected with the HBx expression vector in 293T cells. The RING domain of TRIM proteins is known to confer E3 ligase activity by facilitating an interaction with E2 enzymes (Rajsbaum et al., 2014). Interestingly, deletion the TRIM5γ RING domain (ΔR) did not affect HBx degradation, whereas deletion of both the RING and BBox domains (ΔRB) abolished the ability of TRIM5γ to degrade HBx (Figure 5B). Consistent with this finding, the interaction between HBx and TRIM5γ was disrupted by RB deletion (ΔRB) but not by RING deletion (ΔR) (Figure 5C). In addition, we demonstrated that the BBox domain is sufficient to interact with HBx (Figure 5D) and induce HBx degradation by ubiquitination (Figures 5E and 5F). These results indicated that the TRIM5γ BBox domain is indispensable for the interaction between TRIM5 and HBx.
Figure 5. BBox Domain of TRIM5γ Is Indispensable for TRIM5γ-Induced HBx Degradation.

(A) Diagrams of mutant TRIM5γ constructs. (B) 293T cells were co-transfected with FLAG-HBx and TRIM5γ-derived constructs or EV as indicated, and whole-cell lysates were immunoblotted with anti-FLAG, HA, or GAPDH antibodies. (C) 293T cells were transfected as indicated; 28 h later, coIP was carried out, and cell lysates and precipitated samples were analyzed by immunoblotting using an anti-FLAG or anti-HA antibody. (D) 293T cells were transfected with glutathione S-transferase (GST)-BBox, flag-HBx, or EV as indicated and were subjected to coIP analysis as in (C). (E) 293T cells were co-transfected with FLAG-HBx and GST-BBox or HA-TRIM5γ plasmids and analyzed as in (B). (F) HepG2 cells were transfected with FLAG-HBx, HA-Ub, GST-Bbox, or GST-EV plasmids as indicated; 24 h later, cells were treated with MG132 for 8 h, collected, and subjected to coIP analysis. Data are representative of at least three independent experiments.
TRIM5γ Promotes HBx K95 Site Ubiquitinatin by Interacting with the C Terminus of HBx
It has been previously shown that the HBx region encompassing amino acid residues 88–100 and, particularly, Arg96 is critical for HBx interaction with some target proteins (Hodgson et al., 2012). To map the HBx region responsive for interaction with TRIM5γ, we deleted the amino acid residues 88–100 or made a R96E mutation (Figure 6A); surprisingly, we found that neither of these two mutations affect HBx interaction with TRIM5γ (Figure 6B). Therefore, we generated three truncated HBx constructs and peformed coIP of extracts from 293-T cells co-transfected with TRIM5γ and a truncated HBx construct. The results indicated that X3 strongly and X2 partialy interacts with TRIM5γ (Figure 6C). To further map the TRIM5γ interacting region of HBx, we truncated X2 by deleting amino acids 88–100 and divided X3 into two small peptides. CoIP results showed that X3b is likely responsible for interacting with TRIM5γ (Figure 6D). To further confirm these results, the X2b, X3b, or both fragments were deleted from the full-length HBx protein. The results indicated that deleting the X3b but not the X2b fragment of HBx disrupts its interaction with TRIM5γ (Figure 6E) and protects it from TRIM5γ-mediated degradation (Figure 6F). Interestingly, we noticed that after deletion of amino acid residues 88–100, HBx will not be degradated by TRIM5γ (Figure 6B), which indicated that this region might be responsible for ubiquitination. Thus, mutation was performed to the two lysines in this region, and interestingly, HBx was no longer degradated by TRIM5γ after K95 mutation but not K90 mutation (Figure 6G). In addition, TRIM5γ will not promote the ubiquitination of HBx after K95R mutation (Figure 6H). Taken together, these results clearly demonstrated that TRIM5γ interacted with the C terminus (129–155) of HBx and triggered its degradation by promoting HBx K95 site ubiquitinatin.
Figure 6. TRIM5γ Interacts with the C Terminus of HBx and Promotes HBx K95 Site Ubiquitination.

(A) Diagram of mutant HBx constructs. the numbers indicate the amino acids in the HBx constructs. (B–E) 293T cells were co-transfected with the expression vectors for Flag-TRIM5γ and GST-HBx WT or mutants (B, R96E, Del; C, X1, X2, X3; D, X2a, X2b, X3a, X3b; E, X3bD, DD, X2bD) or EV as indicated. After 28 h, cells were harvested, coIP with FLAG-HBx, and cell lysates and precipitated samples were analyzed by immunoblotting with the indicated antibodies. (F) HBx WT or truncations were co-transfected with TRIM5γ or EV into HepG2 cells; 28h later, cell lysates were subjected to immunoblot analysis using the indicated antibodies. (G) Plasmids of expression of HBx WT, K90R, or K95R were co-transfected with TRIM5γ or EV into HepG2 cells; 28h later, cell lysates were subjected to immunoblot analysis using the indicated antibodies. (H) HepG2 cells were co-transfected with the expression vectors for HA-TRIM5γ, HA-Ub, and FLAG-HBx WT or mutants as indicated; 28h later, cells were harvested and coIP with FLAG-HBx, and cell lysates and precipitated samples were analyzed by immunoblotting with the indicated antibodies. Data are representative of at least three independent experiments.
TRIM5γ Recruits TRIM31 for HBx Degradation
We showed that E3 ubiquitin ligase activity of TRIM5γ is not indispensable for degradation of HBx. Therefore, another E3 protein may be responsible for HBx degradation and TRIM5γ may possibly act as an adaptor in this process. Several TRIM proteins have been shown to inhibit HBV infection (Zhang et al., 2013), of which TRIM31 has recently emerged as an attractive candidate that possesses E3 ubiquitin ligase activity (Koza kova et al., 2015; Ra et al., 2016; Song et al., 2016). Consistent with this postulation, our data indicated that akin to TRIM5γ, TRIM31 also promoted HBx degradation and that MG132 inhibited this process (Figure 7A); furthermore, both TRIM5γ and TRIM31 promoted HBx ubiquitination in HepG2 cells (Figure 7B). In addition, C31 and H33 double mutation in the RING domain has been shown to block the E3 ligase activity of TRIM31 (Kozakova et al., 2015), We found that this double mutation abolished TRIM31-mediated HBx degradation (Figure 7C). This indicated that unlike TRIM5γ, TRIM31 functioned as an E3 ligase in HBx degradation. Therefore, TRIM31 could be the E3 ubiquitin ligase that causes the ubiquitination and degrdation of HBx. To investigate the role of TRIM31 in TRIM5γ-mediated HBx degradation, we used CRISPR/Cas9 technology and generated TRIM31-KO HepG2 cells (Figure 7D; Figure S5). Interestingly, we found that TRIM5γ-mediated HBx degradation was impaired in TRIM31 KO cells (Figure 7E) but was restored upon reconstitution of TRIM31 in KO cells by using a plasmid expressing WT TRIM31 (Figure 7F). Therefore, it appeared that TRIM31 is indispensable for TRIM5γ-mediated HBx degradation. Next, we investigated whether TRIM5γ recruits TRIM31 to degrade HBx. Our data indicated that both TRIM5γ and its BBox domain could interact with TRIM31 (Figures 7G and 7H) and form a TRIM31-TRIM5γ-HBx complex (Figure 7I). In addition, TRIM31 KO disrupted TRIM5γ-mediated inhibition of HBV (Figure 7J) and also inhibited the IFN-α-mediated inhibition of HBV replication (Figure 7K). Therefore, in response to HBV infection, TRIM5γ might recruit TRIM31 and form a complex with HBx, which leads to proteasomal degradation of HBx.
Figure 7. TRIM5γ Recruits TRIM31 to Degradate HBx.

(A) HepG2 cells were co-transfected with FLAG-HBx and HA-TRIM31 or HA empty vector plasmids; 36 h later, cells were left untreated or were treated with MG132 (10 μM) or Z-VAD (10 μM) as indicated. Cells were harvested after treatment, and whole-cell lysates were immunoblotted with the indicated antibodies. (B) HepG2 cells were transfected with FLAG-HBx, HA-Ub, HA-TRIM5γ, or TRIM31 expression plasmids as indicated; 24 h later, cells were treated with MG132 for 6 h and were collected and subjected to coIP analysis. (C) HepG2 cells were transfected with FLAG-HBx or co-transfected with TRIM31 plasmids (WT or mutant); 36 h later, cells were harvested and analyzed as in (A). (D) HepG2 WT or TRIM31 KO cell lysates were immunoblotted with TRIM31 or Tubulin antibody. (E) HepG2 WT orTRIM31KO cells were transfected withFLAG-HBxwithor without the TRIM5γ expression vector; 28h later, cells were collected and analyzed asin(A). (F) WT HepG2 cells or TRIM31 KO cells were transfected as indicated; 28 h later, cells were collected and analyzed as in (A). (G and H) Expression vectors for HA-TRIM5γ (G) or GST-BBox (H) were co-transfected into 293T cells with or without FLAG-TRIM31; 36 h later, cells were subjected to coIP using FLAG-TRIM31. Immunoblot analyses were carried out using anti-FLAG, anti-HA, or anti-GST antibody. (I) 293T cells were transfected with FLAG-HBx, HA-TRIM5γ, or HA-TRIM31 expression plasmids as indicated; 24 h later, cells were treated with MG132 (10 μM) for 8 h and subjected to coIP analysis. Immunoblotting was carried oout using an anti-FLAG or anti-HA antibodies. Data are representative of at least three independent experiments. (J) HepG2 WT or TRIM31 KO cells were transfected with pHBV1.3 plasmids or together with TRIM5γ or TRIM31 expression plasimds as indicated; 72 h later, the supernatant was tested for HBeAg and HBsAg content using ELISA. Mean ± SD values from three independent experiments are shown. **p < 0.01. (K) HepG2 WT or TRIM31 KO cells were transfected with pHBV1.3 plasmids; 24 h later, cells were treated with IFN-α (10ng/ml) or untreated. After another 48 h, qPCR was performed to evaluate the HBV pgRNA and DNA levels. Mean ± SD values from three independent experiments are shown. *p < 0.05, **p < 0.01.
DISCUSSION
Although IFN is widely used for treatment of HBV in clinical settings, the underlying mechanisms responsible for the antiviral effects of IFN against HBV are not fully understood. In the present study, we hypothesized that the anti-viral effect of IFN is at least partially dependent on the interaction between ISG products and HBV proteins. Therefore, we screened 145 ISGs that potentially interact with HBx to characterize the molecular mechanism of IFN-mediated inhibition of HBV through direct HBx-ISG protein-protein interactions. We identified 7 ISGs that significantly inhibited HBV replication by interacting with HBx. Moreover, we found that TRIM5γ inhibited HBV replication, likely by directing TRIM31 toward HBx K48 chain ubiquitination and degradation on the K95 ubiquitin site. An important finding of this study was that induction of high expression levels of TRIM5γ in HBV patients treated with IFN-α might indicate a better therapeutic effect and that BBOX may be a potential candidate for development of therapeutic agents that target HBx.
Activation of IFN production is one of the earliest transcriptional responses to viral infection. Interestingly, after HBV infection, INF signaling was reported to be impaired by HBx through upregulation of the suppressor of cytokine signaling 3 and protein phosphatase 2A (Tsunematsu et al., 2016). In addition, HBx was also shown to inhibit IFN-induced TRIM22 production by single CpG methylation in the 5′ UTR of TRIM22 (Lim et al., 2017), which demonstrates the importance of HBx in combating IFN signaling. In the data shown in Figure S1, several ISG proteins (e.g., Bach1 and IFITs) were much less expressed in the HBx-transfected cells, although we transfected the same amount of plasmids, which indicated that those proteins might be degradated by HBx. This finding is consistent with a report that showed lower expressions of IFIT families in HCC tissues than those in normal tissues (Yang et al., 2017). Moreover, HBx was recently shown to hijack the cellular DDB1-containing E3 ubiquitin ligase to target the Smc complex Smc5/6 for degradation, which, in turn, facilitates HBV replication (Murphy et al., 2016). As a key viral regulatory protein known to play a central role in HBV infection, replication, pathogenesis, and, possibly, carcinogenesis (Murakami, 2001), HBx is essential for maximal HBV replication and development of HCC (Berasain and Lechel, 2017; Keasler et al., 2007). HBV X region mutations were shown to be associated with clinical severity (Yeh et al., 2010). Direct interaction between ISGs and viral proteins plays an important role in restricting viral replication (Jäger et al., 2011; Stremlau et al., 2004). Therefore, identification of ISGs that directly interact with HBx may shed some light on the mechanism by which type I IFN inhibits HBV. We identified 7 ISGs (GBP2, PVRL4, CBFβ, TRIM38, TRIM5γ, TRIM25, and Gadd45γ) that interact with HBx and inhibit HBV replication. GBP2 has been previously shown to inhibit the replication of vesicular stomatitis virus (VSV) and encephalomyocarditis virus (EMCV) (Carter et al., 2005). The V domain of PVRL4 has been shown to be critical for cell entry and intercellular spread of canine distemper virus (CDV) (Delpeut et al., 2014). Interestingly, CBFβ was shown to be hijacked by HIV viral infectivity factor (VIF) to degrade APOBEC3G and promote HIV-1 infection (Jäger et al., 2011); conversely, it was found to play an opposite role with respect to HBV replication in the present study, and our further study on CBFβ suggested CBFβ inhibited HBV replication by blocking the HBx-Smc6 interaction Xu et al., 2019). Gadd45γ acts as a tumor suppressor gene by blocking S and G2/M cell cycle checkpoints (Vairapandi et al., 2002). To the best of our knowledge, there are only a few reports on the role of the above five proteins in HBV inhibition. Therefore, characterization of the mechanisms by which these ISGs combat HBV replication may facilitate the development of therapeutic strategies.
Interestingly, in addition to the four abovementioned ISGs, the rest of the most potent anti-HBV ISGs were Tripartite motif (TRIM) proteins, including TRIM5γ, TRIM38, and TRIM25. TRIM proteins are involved in multiple cellular functions, including antiviral activity (Nisole et al., 2005). TRIM E3 ubiquitin ligases, a subset of the TRIM family, have been shown to restrict viral replication by targeting viral proteins directly (Rajsbaum et al., 2014). TRIM25 has been shown to inhibit HBV replication (Zhang et al., 2013). In our recent study, TRIM25, which was induced by type I IFN in an IL-27-dependent manner, was shown to inhibit HBV replication by promoting IFN production (Tan et al., 2018a). In contrast, TRIM38 was shown to downregulate TLR-mediated immune responses by targeting TRIF and TNF-receptor-associated factor 6 (Zhao et al., 2012), which explains its adverse role in counteracting viral replication. In the present study, we found that TRIM5γ can specifically promote HBx degradation by ubiquitination on the K95 ubiquitin site; we also found that the Ring domain responsible for the ligase activity is not essential for degradation of HBx by TRIM5γ and that only the BBox of TRIM5γ, which is a 40 amino acid peptide, is enough to trigger HBx degradation. HBx has been shown to interact with multiple proteins, including the ubiquitin proteasome system (UPS) components (Minor and Slagle, 2014), and some deubiquitinases (DUBs) target HBx and protect it from proteasome degradation. It is possible that the interaction of TRIM5γ with other E3 ligase proteins results in HBx degradation. TRIM31, which is also reported to inhibit HBV replication (Zhang et al., 2013), has been shown to possess ubiquitin ligase activity (Liu et al., 2016; Song et al., 2016), and TRIM31 RING-finger mutations (C31A and H33A) have been shown to abolish the ligase activity of TRIM31 (Sugiura and Miyamoto, 2008). We have shown that overexpression of TRIM31 leads to HBx degradation. In addition, knocking out TRIM31 blocked the degradation of HBx by TRIM5γ, which indicated that TRIM31 was indispensable for TRIM5γ-induced HBx degradation. We further confirmed that TRIM31 interacts with TRIM5γ and HBx and forms a TRIM31-TRIM5γ-HBx complex. Our findings are consistent with a very recent report that showed that TRIM31 promotes NLRP3 K48-linked polyubiquitination and proteasomal degradation (Song et al., 2016). Nevertheless, further investigations are required to explore the exact mechanism underlying the HBx degradation by TRIM31.
In addition, we have shown that TRIM5γ but not TRIM5α can promote HBx degradation. TRIM5γ has a shorter and distinct C terminus than its isoform alpha, which may support the distinct molecular structure required for interaction with HBx. Interestingly, overexpression of TRIM5γ has been shown to be devoid of antiviral activity against N-MLV and HIV-1 by inhibiting the activity of TRIM5α (Stremlau et al., 2004) (Battivelli et al., 2011). Similarly, it will be easy to understand that the formation of the TRIM5α/γ dimer might inhibit the degradation of HBx by TRIM5γ and that overexpression of TRIM5α might promote HBV replication by offsetting the inhibitory effects of TRIM5γ. With sgRNA targeting the N terminus of TRIM5γ, the BBox region that we verified as the interaction domain with HBx could be destroyed, but TRIM5α will be also knocked out; thus it is difficult to a generate a cell line with TRIM5γ KO only. Nevertheless, overexpression of WT TRIM5γ but not TRIM5α in HepG2 cells resulted in HBx degradation and HBV replication inhibition, which confirmed the critical role of TRIM5γ in inhibiting HBV replication. It would be very interesting to fully understand the mechanism responsible for the differential function of these two molecules.
Collectively, we have demonstrated a crucial role for TRIM5γ and TRIM31 in promoting HBx degradation and have shown that a short peptide is sufficient to trigger HBx degradation. In addition, induction of high expression levels of TRIM5γ in HBV patients undergoing IFN-α therapy was associated with better outcomes. These findings may facilitate the development of therapeutic agents for the treatment of patients with IFN-resistant HBV infection, and TRIM5γ may serve as a useful biomarker to predict IFN-α therapeutic response. Nevertheless, because upregulation of the downstream ISGs is a key driver of the efficacy of IFN therapy, it will be interesting to investigate if HBV could regulate TRIM5γ and TRIM31 expression in response to IFN treatment, which may explain the mechanism of IFN resistance. Further investigation of the molecular mechanisms involved in the IFN-HBV interaction is required to fully resolve the problem of IFN resistance in HBV-infected patients.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Plasmids and cell lines generated in this study will be made available under a standard material transfer agreement. Requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Genhong Cheng (GCheng@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Thirty one HBV-infected patients were enrolled in this study (Table S1). Naive chronic hepatitis B patients (age: 18–65, serum HBsAg detectable for > 6 months) undergoing clinical IFNa2a treatment were enrolled into this study. Venous blood samples (5ml) were collected to obtain serum and PBMCs. HBV DNA level was detected using Rocheˈs COBAS TaqMan kit. Liver function, as well as biochemical parameters, were tested by an auto-matic biochemical instrument. Tests were conducted at the Hepatology Department of the First Hospital of Jilin University, Changchun, China. The study protocol was approved by the IRB of Jilin University, the First Hospital.
Human Embryonic Kidney 293T (HEK293T) cells, HepG2 cells, and HepG2-NTCP cells were maintained in DMEM containing 10% inactivated fetal bovine serum, penicillin (100 IU/mL) and streptomycin (100 mg/mL) and incubated at 37°C with 5% CO2.
METHOD DETAILS
Infusion cloning
The expression construct of TRIM5γ, TRIM5α, TRIM31, HBx, and TRIM5γ truncations were generated by cloning the coding region sequence of relative human genes into VR1012 vector with Flag, HA or GST tag according to the instruction of EASY-Uni Seamless Cloning kit (TransGen, Beijing, China). The YFP constructs were generated by inserting half of the YFP coding region sequence into the C-terminal of ISGs expression plasmids and the other half into the N-terminal of the HBx expression plasmid. Site directed muta-genesis of TRIM5γ and TRIM31 was generated by Quik-Change PCR (TransGen, Beijing, China). Primers are listed in Table S2.
Flow cytometry
Cells were collected by trypsinization, fixed in 2% paraformaldehyde, and analyzed on BD FACS Calibur flow cytometer (BD Biosciences, USA). HBV-YFP was quantified as the product of percent GFP-positive population and geometric mean of the fluorescence index (MFI), and normalized to that of the vector.
Total RNA and HBV DNA Extraction and Quantitative Real-time PCR
Total RNA was extracted using TRIZOl (Invitrogen, San Diego, CA, USA) and then converted to first-strand cDNA using Superscript III transcriptase (Invitrogen San Diego, CA, USA). HBV DNA was isolated from the whole cell-lysate or culture supernatant according to the kit instructions (TransGen, Beijing, China). GAPDH was used as an internal control; quantitative real-time PCR was carried out as described previously (Tan et al., 2018a). The sequences of gene-specific primer used for qPCR are shown in Table S3.
Co-immunoprecipitation and Western Blot Analysis
Cells were lysed 24–48 h after transfection of expression plasmids using 50 mM Tris–HCl, pH 8.0, 150 mM NaCl, 1% NP-40 containing cocktail inhibitors (Roche, USA). For immunoprecipitation, lysates were incubated overnight with ANTI-FLAG® M2 Affinity Gel (Sigma, USA). Immunoblotting was carried out as described previously (Tan et al., 2018a). Briefly, cells were collected and lysed by adding RIPA lysis buffer together with Protease/Phosphatase Inhibitor Cocktail on ice for 30 min and by tapping tubes every 10 min. Protein concentration was quantified by Coomassie Plus protein assay Reagent (Thermo Scientific, Rockford, IL, USA). The quantification of immunoblotting band intensity was carried out with ChemiDoc XRS+ Molecular Imager software (Bio-Rad, Philadelphia, PA, USA). Samples were separated by SDS-PAGE and transferred to PVDF membranes. After blocking in TBS containing 0.1% Tween-20 and 5% skim-milk, the blots were probed with relative antibodies.
Immunofluorescence
HepG2 cells were transfected with Flag-HBx plasmids or together with HA-TRIM5γ plasmids; 48 hours later, cells were fixed in acetone-methanol (1:1) at 37°C for 10 min. Subsequently, cells were washed with PBS and blocked with 5% BSA in PBST for 1 h, incubated with Flag(Mouse) and HA(Rabbit) antibody at 37°C for 1 h, washed in PBS, and then incubated with goat anti-mouse IgG conjugated with FITC or both Cy3(rabbit) and FITC(mouse) conjugated IgG (Proteintech). Cells were washed with PBS and observed under a fluorescence microscope.
ELISA
HepG2 Cells were mock transfected or transfected with ISG-expression plasmids together with pHBV1.3-HBV expression plasmids. Supernatant was collected after 72 hours, and the supernatant from HBV infected HepG2-NTCP cells was collected after 9 days for analysis by ELISA to detect the levels of HBeAg and HBsAg (Kehua Shengwu, China).
CRISPR / Cas9 Knockout
HepG2 or 293T cells were seeded in 24 well plates; 16 h later, two plasmids, one expressing Cas9 with TRIM5α/γ or TRIM31 sgRNA (FG-EH-Cas9–2F-PPW) and the other carrying a puromycin resistant gene (PL-GFP-IP), were co-transfected into HepG2 cells using ViaFect transfection reagent (Promega). At 36 h after transfection, cells were either selected by adding puromycin at a concentration of 2 μg/mL or collected for immunoblotting with specific TRIM5γ, TRIM5α or TRIM31 antibodies. Two days later, live cells were sorted into a 96-well plate at a density of 1 cell per well using a BD FACSAria. Immunoblotting was performed again to determine the level of gene editing efficiency after the clonal expansion and DNA sequencing was performed to verify the edited genes. SgRNAs are listed in the Table S2.
HBV infection assay
Serum from HBV patients with different virus strains (HBV DNA copies > 107) was collected and whole virus were concentrated using a PEG-it Virus Precipitation Solution (System Biosciences, USA). HepG2-NTCP cells were infected with lentiviral-ctrl (Lot#: GC12182K1701) or lentiviral-TRIM5γ (Lot#: GC05212K1804) for 16 hours, and later, cells were inoculated with the serum produced virus at an MOI (multiplicity of infection) of 1000 equivalents (Geq) per cell and were cultured in the presence of 4% PEG8000 and 2% DMSO for 24 h. After infection, cells were washed three times with PBS, and were maintained in DMEM medium for another 9 days; the medium was changed every two days. The supernatant and cells were collected for detection of HBV DNA, pgRNA and HBsAg by Q-PCR or Elisa; Expressions of TRIM5γ were detected by Q-PCR.
QUANTIFICATION AND STATISTICAL ANALYSIS
GraphPad Prism 5 software (GraphPad Software, San Diego, CA) was used for data analysis; a two-tail unpaired t test was used to assess between-group differences. p value < 0.05 was considered statistically significant.
DATA AND CODE AVAILABILITY
The published article includes all the datasets generated or analyzed during this study.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-TRIM5 gamma | Abcam | Cat# ab17203; RRID:AB_208592 |
| Anti-TRIM5 alpha | Abcam | Cat# ab59000; RRID:AB_2208591 |
| Anti-TRIM31 | Abcam | Abcam Cat# ab82680; RRID:AB_1860041 |
| Anti-GFP Tag Rabbit Polyclonal Antibody | Abbkine | Cat#A02021–2 |
| HA-tag Antibody | proteintech | Cat# 51064–2-AP; RRID:AB_11042321 |
| GST Tag Antibody | proteintech | Cat# 10000–0-AP; RRID:AB_11042316 |
| Monoclonal ANTI-FLAG® M2 antibody produced in mouse | Sigma | Cat# F1804; RRID:AB_262044 |
| GAPDH Antibody | proteintech | Cat# 10494–1-AP; RRID:AB_2263076 |
| β Tubulin Antibody (SAP.4G5) | Santa Cruz Biotechnology | Cat# sc-58884; RRID:AB_793548 |
| NTCP Polyclonal Antibody | Signalway Antibody | Cat#41759 |
| HBsAg antibody | lifespan biosciences | Cat# LS-C67520–1000; RRID:AB_10634869 |
| HRP-Goat Anti-Rabbit IgG(H+L) | proteintech | Cat# SA00001–2; RRID:AB_2722564 |
| HRP-Goat Anti-Mouse IgG(H+L) | proteintech | Cat# SA00001–1; RRID:AB_2722565 |
| Goat anti-mouse IgG (H+L), Cy3 conjugate | proteintech | Cat# SA00009–1; RRID:AB_2814746 |
| Goat anti-rabbit IgG (H+L), Cy3 conjugate | proteintech | Cat# SA00009–2 |
| Goat anti-rabbit IgG (H+L), FITC conjugate | proteintech | Cat#SA00003–2 |
| Goat anti-mouse IgG (H+L), FITC conjugate | proteintech | Cat#SA00003–1 |
| Biological Samples | ||
| HBV patient venous blood samples The First Hospital of Jilin University | N/A | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ANTI-FLAG M2 Affinity Gel | Sigma | A2220 |
| Z-VAD-FMK | promega | G7232 |
| Protease inhibitor | Roche | 11697498001 |
| MG132 | Sigma | M7449 |
| BMS-200475 | MedChemExprss | HY-13623A |
| 3-MA | MedChemExprss | HY-19312 |
| Recombinant Human IFN-α | biolegend | 592706 |
| Critical Commercial Assays | ||
| HBeAg detection Kit | Kehua shengwu | N/A |
| HBsAg detection Kit | Kehua shengwu | N/A |
| Viafect transfection reagent | promega | E4982 |
| PEG-it Virus Precipitation Solution | System Biosciences | BY-LV810A-1 |
| lentiviral-ctrl | Genecopoeia | Lot#: GC12182K1701 |
| lentiviral-TRIM5g | Genecopoeia | Lot#: GC05212K1804 |
| EasyPure® Viral DNA/ RNA Kit | Transgen | ER201–01 |
| EasyPure RNA Kit | Transgen | ER101–01 |
| TransScript® All-in-One First-Strand cDNA Synthesis SuperMix for qPCR(One-Step gDNA Removal) | Transgen | AT341 −02 |
| E.Z.N.A. Endo-Free Plasmid Mini Kit I | Omega | D6948–02 |
| pEASY-Blunt Cloning Kit | Transgen | CB101–02 |
| Faststart Universal SYBR Green Master(ROX) | Roche | #04913914001 |
| Cell Counting Kit-8 (CCK-8) | Bimake | B34302 |
| HBV patient venous blood samples The First Hospital of Jilin University | N/A | |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ANTI-FLAG M2 Affinity Gel | Sigma | A2220 |
| Z-VAD-FMK | promega | G7232 |
| Protease inhibitor | Roche | 11697498001 |
| MG132 | Sigma | M7449 |
| BMS-200475 | MedChemExprss | HY-13623A |
| 3-MA | MedChemExprss | HY-19312 |
| Recombinant Human IFN-a | biolegend | 592706 |
| Critical Commercial Assays | ||
| HBeAg detection Kit | Kehua shengwu | N/A |
| HBsAg detection Kit | Kehua shengwu | N/A |
| Viafect transfection reagent | promega | E4982 |
| PEG-it Virus Precipitation Solution | System Biosciences | BY-LV810A-1 |
| lentiviral-ctrl | Genecopoeia | Lot#: GC12182K1701 |
| lentiviral-TRIM5γ | Genecopoeia | Lot#: GC05212K1804 |
| EasyPure® Viral DNA/ RNA Kit | Transgen | ER201–01 |
| EasyPure RNA Kit | Transgen | ER101–01 |
| TransScript® All-in-One First-Strand cDNA Synthesis SuperMix for qPCR(One-Step gDNA Removal) | Transgen | AT341 −02 |
| E.Z.N.A. Endo-Free Plasmid Mini Kit I | Omega | D6948–02 |
| pEASY-Blunt Cloning Kit | Transgen | CB101–02 |
| Faststart Universal SYBR Green Master(ROX) | Roche | #04913914001 |
| Cell Counting Kit-8 (CCK-8) | Bimake | B34302 |
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Protease Inhibitor Cocktail (EDTA-Free, mini-Tablet) | Bimake | B14012 |
| FastDigest XbaI | Thermo Sci | FD0684 |
| Experimental Models: Cell Lines | ||
| HepG2 | ATCC | HB-8065 |
| HepG2-NTCP | This paper | |
| 293T | ATCC | ACS-4500 |
| Oligonucleotides | ||
| See Tables S2 and S3 | N/A | N/A |
| Recombinant DNA | ||
| HBx wt expression plasmid | This paper | N/A |
| HBx K90R expression plasmid | This paper | N/A |
| HBx K95R expression plasmid | This paper | N/A |
| HBx R96E expression plasmid | This paper | N/A |
| HBx 88–100 deletion expression plasmid | This paper | N/A |
| TRIM5γ and the truncations expression plasmids | This paper | N/A |
| pHBV1.3 plasmid | Lishan Su lab of University of North Carolina at Chapel Hill | N/A |
| pHBV1.2 WT and pHBV1.2 ΔX plasmids | Qiang Deng lab of Fudan University | N/A |
| GFP-ISGs expression plasmids | Cheng lab in UCLA | N/A |
| Software and Algorithms | ||
| Prism version 5 | GraphPad Software | https://www.graphpad.com |
Highlights.
Twenty-six in 145 interferon-stimulated gene products interact with HBx protein
Seven HBx-interacting ISGs significantly inhibited HBV replication
High induction of TRIM5γ indicated a better IFN-α therapeutic effect
B-Box domain of TRIM5γ is sufficient to trigger HBx degradation by recruiting TRIM31
ACKNOWLEDGMENTS
We thank Dr. Zhigang Wang and Yun Shao for analysis of data pertaining to HBV patients. The pHBV1.2 WT and DX plasmids were kindly provided by Professor Qiang Deng of Fudan University. This work was supported by the National Natural Science Foundation, China; Jilin Provincial Science and Technology Department of the Youth Fund Project; Jilin University Bethune training program and Jilin Province Education Department “13th Five-Year” Science and Technology Project; Jilin Province Medical and Health Personnel Special Project and Jilin University Excellent Young Teacher Training Program (grant no. 81401290, 20160520161JH, 470110000456, JJKH20190024KJ, and JLSCZD2019-75 to G.T.); the Ministry of Science and Technology of China (grant no. 2013CB911103); startup fund from The First Hospital of Jilin University to G.C.; and grants from US NIH (grant no. AI069120), the CAMS Initiative for Innovative Medicine (CAMS-I2M, 2016-I2M-1-005), the National Natural Science Foundation of China (81701567, 81773058, and 31800726), National Foreign Expert Fund (G20190001633 and G20190001639), Jiangsu Provincial Natural Science Foundation (BK20171232), and the Fund of Jiangsu Provincial Science and Technology Department (BM2016006).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.041.
DECLARATION OF INTERESTS
The authors declare that they have no conflict of interest.
REFERENCES
- Battivelli E, Migraine J, Lecossier D, Matsuoka S, Perez-Bercoff D, Saragosti S, Clavel F, and Hance AJ (2011). Modulation of TRIM5alpha activity in human cells by alternatively spliced TRIM5 isoforms. J. Virol 85, 7828–7835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berasain C, and Lechel A (2017). Targeting the correct target in HCC. Gut 66, 1352–1354. [DOI] [PubMed] [Google Scholar]
- Carter CC, Gorbacheva VY, and Vestal DJ (2005). Inhibition of VSV and EMCV replication by the interferon-induced GTPase, mGBP-2: differential requirement for wild-type GTP binding domain. Arch. Virol 150, 1213–1220. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang S, Yi Z, Tian H, Aliyari R, Li Y, Chen G, Liu P, Zhong J, Chen X, et al. (2014). Interferon-inducible cholesterol-25-hydroxylase inhibits hepatitis C virus replication via distinct mechanisms. Sci. Rep 4, 7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delpeut S, Noyce RS, and Richardson CD (2014). The V domain of dog PVRL4 (nectin-4) mediates canine distemper virus entry and virus cell-to-cell spread. Virology 454–455, 109–117. [DOI] [PubMed] [Google Scholar]
- Evans JD, and Seeger C (2007). Differential effects of mutations in NS4B on West Nile virus replication and inhibition of interferon signaling. J. Virol 81, 11809–11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganser-Pornillos BK, and Pornillos O (2019). Restriction of HIV-1 and other retroviruses by TRIM5. Nat. Rev. Microbiol 17, 546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao B, Duan Z, Xu W, and Xiong S (2009). Tripartite motif-containing 22 inhibits the activity of hepatitis B virus core promoter, which is dependent on nuclear-located RING domain. Hepatology 50, 424–433. [DOI] [PubMed] [Google Scholar]
- Hatakeyama S (2011). TRIM proteins and cancer. Nat. Rev. Cancer 11, 792–804. [DOI] [PubMed] [Google Scholar]
- Hayes CN, and Chayama K (2017). Interferon stimulated genes and innate immune activation following infection with hepatitis B and C viruses. J. Med. Virol 89, 388–396. [DOI] [PubMed] [Google Scholar]
- Hodgson AJ, Hyser JM, Keasler VV, Cang Y, and Slagle BL (2012). Hepatitis B virus regulatory HBx protein binding to DDB1 is required but is not sufficient for maximal HBV replication. Virology 426, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs A, and Lindenmann J (1957). Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci 147, 258–267. [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Kim DY, Hultquist JF, Shindo K, LaRue RS, Kwon E, Li M, Anderson BD, Yen L, Stanley D, et al. (2011). Vif hijacks CBF-b to degradeb APOBEC3G and promote HIV-1 infection. Nature 481, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, and Akira S (2011). Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol. Med 3, 513–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keasler VV, Hodgson AJ, Madden CR, and Slagle BL (2007). Enhancement of hepatitis B virus replication by the regulatory X protein in vitro and in vivo. J. Virol 81, 2656–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keown JR, Black MM, Ferron A, Yap M, Barnett MJ, Pearce FG, Stoye JP, and Goldstone DC (2018). A helical LC3-interacting region mediates the interaction between the retroviral restriction factor TRIM5α and mammalian autophagy-related ATG8 proteins. J. Biol. Chem 293, 18378–18386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M, Syed GH, Kim SJ, and Siddiqui A (2016). Hepatitis B Virus-Induced Parkin-Dependent Recruitment of Linear Ubiquitin Assembly Complex (LUBAC) to Mitochondria and Attenuation of Innate Immunity. PLoS Pathog. 12, e1005693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakova L, Vondrova L, Stejskal K, Charalabous P, Kolesar P, Leh-mann AR, Uldrijan S, Sanderson CM, Zdrahal Z, and Palecek JJ (2015). The melanoma-associated antigen 1 (MAGEA1) protein stimulates the E3 ubiquitin-ligase activity of TRIM31 within a TRIM31-MAGEA1-NSE4 complex. Cell Cycle 14, 920–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KH, Park ES, Kim DH, Cho KC, Kim KP, Park YK, Ahn SH, Park SH, Kim KH, Kim CW, et al. (2017). Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5′-UTR of TRIM22. Gut 67, 166–178. [DOI] [PubMed] [Google Scholar]
- Liu SY, Sanchez DJ, Aliyari R, Lu S, and Cheng G (2012). Systematic identification of type I and type II interferon-induced antiviral factors. Proc. Natl. Acad. Sci. USA 109, 4239–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu SY, Aliyari R, Chikere K, Li G, Marsden MD, Smith JK, Pernet O, Guo H, Nusbaum R, Zack JA, et al. (2013). Interferon-inducible cholesterol-25-hydroxylase broadly inhibits viral entry by production of 25-hydroxycholesterol. Immunity 38, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Peng N, Xie J, Hao Q, Zhang M, Zhang Y, Xia Z, Xu G, Zhao F, Wang Q, et al. (2015). Human hepatitis B virus surface and e antigens inhibit major vault protein signaling in interferon induction pathways. J. Hepatol 62, 1015–1023. [DOI] [PubMed] [Google Scholar]
- Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, Wang P, Zhao K, Hou J, Wang X, et al. (2016). The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat. Immunol 18, 212–224. [DOI] [PubMed] [Google Scholar]
- Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, et al. (2014). Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science 343, 1221–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor MM, and Slagle BL (2014). Hepatitis B virus HBx protein interactions with the ubiquitin proteasome system. Viruses 6, 4683–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra B, and Guo H (2016). Hepatitis B Virus X Protein Crosses Out Smc5/6 Complex to Maintain cccDNA Transcription. Hepatology 64, 2246–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami S (2001). Hepatitis B virus X protein: a multifunctional viral regulator. J. Gastroenterol 36, 651–660. [DOI] [PubMed] [Google Scholar]
- Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, Wu Y, Yu Y, Xiong Y, and Su L (2016). Hepatitis B Virus X Protein Promotes Degradation of SMC5/6 to Enhance HBV Replication. Cell Rep. 16, 2846–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutimer DJ, and Lok A (2012). Management of HBV- and HCV-induced end stage liver disease. Gut 61, i59–i67. [DOI] [PubMed] [Google Scholar]
- Nassal M (2015). HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 64, 1972–1984. [DOI] [PubMed] [Google Scholar]
- Nisole S, Stoye JP, and Saïb A (2005). TRIM family proteins: retroviral restriction and antiviral defence. Nat. Rev. Microbiol 3, 799–808. [DOI] [PubMed] [Google Scholar]
- Ra EA, Lee TA, Won Kim S, Park A, Choi HJ, Jang I, Kang S, Hee Cheon J, Cho JW, Eun Lee J, et al. (2016). TRIM31 promotes Atg5/Atg7-independent autophagy in intestinal cells. Nat. Commun 7, 11726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajsbaum R, García-Sastre A, and Versteeg GA (2014). TRIMmunity: the roles of the TRIM E3-ubiquitin ligase family in innate antiviral immunity. J. Mol. Biol 426, 1265–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revill P, and Locarnini S (2016). Antiviral strategies to eliminate hepatitis B virus covalently closed circular DNA (cccDNA). Curr. Opin. Pharmacol 30, 144–150. [DOI] [PubMed] [Google Scholar]
- Sitterlin D, Lee TH, Prigent S, Tiollais P, Butel JS, and Transy C (1997). Interaction of the UV-damaged DNA-binding protein with hepatitis B virus X protein is conserved among mammalian hepadnaviruses and restricted to transactivation-proficient X-insertion mutants. J. Virol 71, 6194–6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, Han L, Jiang G, Zhang L, Gao C, and Zhao W (2016). The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat. Commun 7, 13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, and Sodroski J (2004). The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427, 848–853. [DOI] [PubMed] [Google Scholar]
- Sugiura T, and Miyamoto K (2008). Characterization of TRIM31, upregulated in gastric adenocarcinoma, as a novel RBCC protein. J. Cell. Biochem 105, 1081–1091. [DOI] [PubMed] [Google Scholar]
- Tan G, Xiao Q, Song H, Ma F, Xu F, Peng D, Li N, Wang X, Niu J, Gao P, et al. (2018a). Type I IFN augments IL-27-dependent TRIM25 expression to inhibit HBV replication. Cell. Mol. Immunol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan G, Song H, Xu F, and Cheng G (2018b). When Hepatitis B Virus Meets Interferons. Front. Microbiol 9, 1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunematsu S, Suda G, Yamasaki K, Kimura M, Izumi T, Umemura M, Ito J, Sato F, Nakai M, Sho T, et al. (2016). Hepatitis B virus X protein impairs alpha-interferon signaling via up-regulation of suppressor of cytokine signaling 3 and protein phosphatase 2A. J. Med. Virol 89, 267–275. [DOI] [PubMed] [Google Scholar]
- Vairapandi M, Balliet AG, Hoffman B, and Liebermann DA (2002). GADD45b and GADD45g are cdc2/cyclinB1 kinase inhibitors with a role in S and G2/M cell cycle checkpoints induced by genotoxic stress. J. Cell. Physiol 192, 327–338. [DOI] [PubMed] [Google Scholar]
- Xu F, Song H, Li N, and Tan G (2016). HBsAg blocks TYPE I IFN induced up-regulation of A3G through inhibition of STAT3. Biochem. Biophys. Res. Commun 473, 219–223. [DOI] [PubMed] [Google Scholar]
- Xu F, Song H, Xiao Q, Li N, Zhang H, Cheng G, and Tan G (2019). Type III interferon-induced CBFβeta inhibits HBV replication by hijacking HBx. Cell. Mol. Immunol 16, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Zhong G, Xu G, He W, Jing Z, Gao Z, Huang Y, Qi Y, Peng B, Wang H, et al. (2012). Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1, e00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhou Y, Hou J, Bai C, Li Z, Fan J, Ng IOL, Zhou W, Sun H, Dong Q, et al. (2017). Hepatic IFIT3 predicts interferon-a therapeutic response in patients of hepatocellular carcinoma. Hepatology 66, 152–166. [DOI] [PubMed] [Google Scholar]
- Yeh CT, So M, Ng J, Yang HW, Chang ML, Lai MW, Chen TC, Lin CY, Yeh TS, and Lee WC (2010). Hepatitis B virus-DNA level and basal core promoter A1762T/G1764A mutation in liver tissue independently predict postoperative survival in hepatocellular carcinoma. Hepatology 52, 1922–1933. [DOI] [PubMed] [Google Scholar]
- Zhang S, Guo JT, Wu JZ, and Yang G (2013). Identification and characterization of multiple TRIM proteins that inhibit hepatitis B virus transcription. PLoS One 8, e70001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Wang L, Zhang M, Yuan C, and Gao C (2012). E3 ubiquitin ligase tripartite motif 38 negatively regulates TLR-mediated immune responses by proteasomal degradation of TNF receptor-associated factor 6 in macrophages. J. Immunol 188, 2567–2574. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all the datasets generated or analyzed during this study.