

Abstract
Inhibitory codon pairs and poly(A) tracts within the translated mRNA cause ribosome stalling and reduce protein output. The molecular mechanisms that drive these stalling events, however, are still unknown. Here, we use a combination of in vitro biochemistry, ribosome profiling, and cryo‐EM to define molecular mechanisms that lead to these ribosome stalls. First, we use an in vitro reconstituted yeast translation system to demonstrate that inhibitory codon pairs slow elongation rates which are partially rescued by increased tRNA concentration or by an artificial tRNA not dependent on wobble base‐pairing. Ribosome profiling data extend these observations by revealing that paused ribosomes with empty A sites are enriched on these sequences. Cryo‐EM structures of stalled ribosomes provide a structural explanation for the observed effects by showing decoding‐incompatible conformations of mRNA in the A sites of all studied stall‐ and collision‐inducing sequences. Interestingly, in the case of poly(A) tracts, the inhibitory conformation of the mRNA in the A site involves a nucleotide stacking array. Together, these data demonstrate a novel mRNA‐induced mechanisms of translational stalling in eukaryotic ribosomes.
Keywords: disome, frameshift, ribosome collision, stalling, translation
Subject Categories: Structural Biology, Protein Biosynthesis & Quality Control
A combination of in vitro biochemistry, ribosome profiling, and cryo‐EM reveals that elongation‐slowing mRNA elements stall ribosomes in decoding‐incompatible RNA conformations in the ribosomal decoding center.
Introduction
Coding sequences for proteins in any genome (the open reading frames or ORFs) have evolved in the context of their full mRNA transcript to be expressed at the appropriate level. Interestingly, synonymous codon choice has been shown to have broad impact on many aspects of translation including translational efficiency (Tuller et al, 2010; Gingold & Pilpel, 2011), mRNA decay (Presnyak et al, 2015), and cotranslational protein folding (Pechmann & Frydman, 2013; Sander & Chaney, 2014; Buhr et al, 2016). The effects on translational efficiency are primarily mediated through the competition of cognate and near‐cognate tRNA interactions, as dictated by the pool of charged tRNAs available in the cell (Elf et al, 2003; Gingold & Pilpel, 2011; Dana & Tuller, 2014). Individual codons that are generally decoded by more abundant tRNAs and are associated with increased translation efficiency have been defined as “optimal” (Sharp & Li, 1987; dos Reis et al, 2004; Burgess‐Brown et al, 2008). Moreover, codon usage biases, codon context, and interactions between adjacent codons have all been suggested to play a role in translational efficiency (Quax et al, 2015; Brule & Grayhack, 2017), though their direct effects on elongation are still not fully understood.
A recent study in yeast defined a collection of 17 specific codon pairs that caused a substantial down‐regulation in protein output (Gamble et al, 2016). For 12 of these pairs, the order of the codons within the pair was critical for the observed inhibition. Despite the diverse nature of these pairs, there were some shared features. First, the proline codon CCG and the arginine codon CGA appeared frequently in the collection of inhibitory pairs. The CCG codon is decoded by a G‐U wobble base pair, while the CGA codon is the sole codon in yeast decoded by an obligate I:A wobble pair (Letzring et al, 2010). Notably, while the previous study (Gamble et al, 2016) concluded that these inhibitory codon pairs likely impacted the decoding step of elongation, there was little understanding of the molecular basis for these events.
Besides inhibitory codon pairs, poly(A) tracts represent perhaps the most abundant and potent stall‐inducing mRNA sequence in eukarya (reviewed in Arthur & Djuranovic, 2018). Translation of poly(A) sequences commonly occurs when ribosomes encounter an abnormal (premature) polyadenylation event within the ORF or when ribosomes read through a stop codon. Premature polyadenylation alone occurs in approximately 1% of yeast and human transcripts, highlighting the importance of this mechanism (Frischmeyer et al, 2002; Ozsolak et al, 2010). While translation of poly(A) tracts initially results in the synthesis of polylysine, long poly(A) tracts subsequently trigger quality control pathways that contribute to overall protein homeostasis (Brandman & Hegde, 2016; Joazeiro, 2019). The earliest studies suggested that this stalling was caused by electrostatic interactions between the poly‐basic nascent chain and the peptide exit tunnel of the ribosome (Lu & Deutsch, 2008). However, there are several lines of evidence suggesting that the stalling mechanism of poly(A) tracts is more complex. Interestingly, as few as two consecutive AAA codons were shown to cause ribosome sliding during translation in Escherichia coli (Koutmou et al, 2015). Moreover, the identity of the basic residue‐encoding codon is of particular importance for efficient stalling, as the CGA arginine‐encoding codon is most potent in yeast (Letzring et al, 2013), and AAA codons are more potent than AAG lysine‐encoding codons at inducing translational stalling (Arthur et al, 2015; Koutmou et al, 2015).
All of the inhibitory sequences described above result in partial or complete translational stalling in vivo. Considerable attention has been paid to the molecular consequences of the translating ribosomes encountering such mRNA sequences (i.e., the downstream quality control events that are triggered). In particular, recent work has suggested that ribosomal collisions with the leading stalled ribosome are a key event that triggers the quality control responses that include decay of the mRNA (“No Go Decay” or NGD) and the nascent peptide (ribosome‐associated quality control or RQC; Simms et al, 2017; Juszkiewicz et al, 2018; Ikeuchi et al, 2019). However, there has been little characterization of the molecular events on the ribosome that lead to such dramatic outcomes.
Here, we use a yeast in vitro reconstituted biochemical system to directly measure the rates of translation elongation that might be impacted by inhibitory codon pairs and poly(A) tracts. Use of this in vitro system allows for ready manipulation of mRNA coding sequence, tRNA identity, and concentration, as well as ribosome composition to reveal defects in the individual steps of translation elongation. Together with high‐resolution ribosome profiling, our results reveal clear defects in the decoding step as the primary determinant of ribosomal stalling on these inhibitory mRNA sequences. Cryo‐EM structures of ribosome complexes stalled at these mRNA sequences reveal detailed insights into the molecular basis for the translational stalling. Importantly, we observe decoding‐incompetent conformations of mRNA in the A sites of all stall‐inducing sequences that we studied, thus readily explaining the biochemically defined decoding defects. Moreover, structural characterization of poly(A)‐stalled disomes reveals a novel disome conformation with both ribosomes in the POST translocation state, suggesting a role for ribosome collisions in promoting frameshifting. Taken together, our data reveal a mRNA‐induced translational stalling mechanism of eukaryotic ribosomes.
Results
Inhibitory codon pairs slow elongation in vitro
To examine the impact of inhibitory codon pairs on translation elongation in vitro, we selected pairs that most potently reduced GFP expression in the in vivo experiments and those that contained codons which appeared in multiple inhibitory pairs (Fig 1A; Gamble et al, 2016). The strongest candidates were CGA–CGA and CGA–CCG encoding Arg–Arg and Arg–Pro, respectively. The arginine codon CGA is decoded by ICG tRNAArg where inosine forms a unique purine–purine I:A wobble pair. The proline codon CCG is found in many inhibitory codon pairs, likely because it is decoded by tRNA using a G‐U wobble pair, UGG tRNAPro (Fig 1A). The prevalence of and dependency on wobble base‐pairing in inhibitory codon pairs led Grayhack and co‐workers to conclude that elongation is blocked by non‐optimal codon–anticodon pairing at neighboring sites on the ribosome (i.e., the P and A sites). Furthermore, they showed that for these codon pairs, the order of the codons in the pair is critical; the reverse pair has little to no effect on protein output.
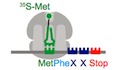
Figure 1. Inhibitory codon pairs slow elongation in vitro .
- Inhibitory pairs showing the inhibitory mRNA codons (red) and the optimal codons (green). Schematic representation of the in vitro elongation reactions performed using the reconstituted yeast translation system. Pelleted 80S initiation complexes (3 nM) are incubated with aminoacyl‐tRNAs (15–25 nM), elongation factors (1 μM), GTP, and ATP. Reactions are quenched with KOH and the products resolved by electrophoretic TLCs.
- Representative eTLCs (left) and corresponding elongation kinetics (right) for the CGA–CGA inhibitory pair (red) and the CGC optimal pair (green). Product formation is normalized to the fraction of Met ICs that form Met–Puro when reacted with puromycin (Fig EV1A).
- Representative eTLCs (left) and corresponding elongation kinetics (right) for the CGA–CCG inhibitory pair (red) and the CGC–CCA optimal pair (green). Product formation is normalized to the fraction of Met ICs that form Met–Puro when reacted with puromycin (Fig EV1A).
To monitor synthesis of tetrapeptides containing these inhibitory codon pairs, we employed an in vitro reconstituted yeast translation system (Eyler & Green, 2011; Schuller et al, 2017). Initiation complexes (ICs) were assembled using ribosome subunits, [35S]‐Met‐tRNAiMet, and mRNAs containing an AUG codon, the codon pair of interest, and an additional codon encoding Phe or Lys before or after the pair to enhance visualization of the products by electrophoretic thin‐layer chromatography (eTLC). Following purification, each IC was treated with puromycin (Pm) to release the nascent chain and determine the fraction of bound [35S]‐Met‐tRNAiMet that forms Met–Pm. Puromycin reacts with peptidyl‐tRNA bound to the ribosome when the peptidyl‐transferase center (PTC) of the large subunit is accessible and releases the polypeptide chain as peptidyl‐puromycin. As such, this assay reports on the overall competence and conformation of the peptidyl‐transferase center of the ICs. We consistently observed that ICs formed with the different mRNA transcripts formed Met–Pm products to a similar extent (Fig EV1A). Therefore, differences in the amount of peptide produced using ICs containing different mRNA templates were not due to the efficiency of IC formation or to the differential ability of the programmed ribosome to make peptide bonds.
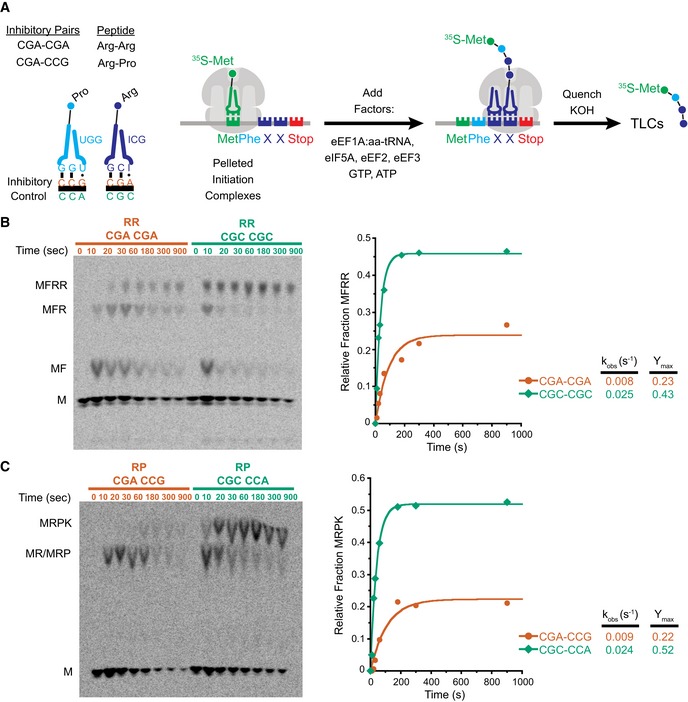
Figure EV1. Initiation complex test of Met–Pm activity and individual product analysis of MFRR elongation.
- Met–Pm activity for all the ICs formed with WT ribosomes on inhibitory mRNAs (red) and optimal mRNAs (green). There is no significant difference in activity at the last time point for any of the ICs.
- TLC showing peptidyl‐tRNA drop‐off using the PTH assay on MFRR ICs with the inhibitory (CGA–CGA) pair (red) and the optimal (CGC–CGC) pair (green). Time points were quenched with formic acid to assess drop‐off, and time points quenched with KOH were to monitor peptide formation as a control. There is no significant accumulation of peptidyl‐tRNA drop‐off products.
- TLC showing peptidyl‐tRNA drop‐off using the PTH assay on MRPK ICs with the inhibitory (CGA–CCG) pair (red) and the optimal (CGC–CCA) pair (green). Time points were quenched with formic acid to assess drop‐off. There is no significant accumulation of peptidyl‐tRNA drop‐off products.
- Elongation kinetics for the MFR and MFRR products together versus the final MFRR product alone for the inhibitory (CGA–CGA) pair (red) and the optimal (CGC–CGC) pair (green). The increased rate and amount of product formed for the MFR and MFRR data compared to the MFRR alone suggest that the addition of the second arginine is slower than the first.
- Elongation kinetics for the addition of a single arginine on an MFR (CGA) message in red and MFR (CGC) message in green. The addition of the first arginine is similar for CGA and CGC, again suggesting that the addition of the second arginine is the slower step.
For elongation reactions, the desired tRNAs were purified from bulk tRNA using biotinylated oligonucleotides (Yokogawa et al, 2010), charged with the corresponding aminoacyl‐tRNA synthetase, and the aminoacyl‐tRNAs were pre‐incubated with eEF1A and GTP to form ternary complexes. Ternary complexes were then mixed with purified ICs and elongation factors eEF2, eEF3, and eIF5A. Peptide formation was monitored by quenching time points of the reactions in KOH and resolving the formed products by eTLC (Fig 1A). The initial experiments were performed with ribosome complexes at ~3 nM and aa‐tRNAs at ~15–25 nM, where both binding and catalysis contribute to the observed rate (i.e., kcat/Km conditions). For each inhibitory codon pair, a control, optimal IC was prepared where the non‐optimal codons were replaced by synonymous codons that are decoded by the same tRNA, but without wobble base‐pairing. For example, the optimal codon CGC was used as a control for CGA because it is decoded by the same ICG tRNAArg via a pyrimidine‐purine C:I pair with a standard Watson–Crick geometry (Murphy & Ramakrishnan, 2004) instead of a purine–purine (A:I) wobble base pair (Fig 1A). These optimal controls were previously shown to have high‐to‐intermediate output in the initial study by Grayhack and co‐workers (Gamble et al, 2016), and their corresponding rates and endpoints in our in vitro experiments are comparable to values previously published (Schuller et al, 2017).
Visual examination of the reaction profiles for the inhibitory CGA–CGA codon pair (in red) relative to the optimal CGC–CGC codon pair (in green) reveals a clear defect in elongation (Fig 1B). First, the inhibitory Arg–Arg pair exhibits a significantly lower endpoint, with ~25% of the radiolabeled Met forming the final tetrapeptide product, MFRR, compared with ~45% for the optimal Arg–Arg sequence. In a similar fashion, there are clear elongation defects for the inhibitory Arg–Pro, CGA–CCG codon pair (in red) compared to the optimal CGC–CCA codon pair (in green; Fig 1C); the inhibitory Arg–Pro pair has only ~20% of the radiolabeled Met forming the final tetrapeptide product, MRPK, compared to ~50% for the optimal Arg–Pro sequence. As endpoint defects often suggest the existence of an off pathway reaction, we asked whether there were high levels of peptidyl‐tRNA drop‐off during elongation for the Arg–Arg or Arg–Pro reactions that might explain the observed defects. However, when we directly tested this possibility using an assay involving peptidyl hydrolase (Pth) that acts only on tRNAs not bound to the ribosome, we saw no evidence for drop‐off with any of the complexes (Fig EV1B and C; Shoemaker et al, 2010; Schuller et al, 2017).
In addition to the endpoint defects, we also observe a reduced rate of formation of the final peptide product for the complexes encoding both the Arg–Arg and Arg–Pro pairs; in each case, the observed rates were about threefold slower than those of their optimal counterparts (Fig 1B and C). For the Arg–Arg pair, where MFR and MFRR can be separately resolved, we see a substantial buildup of MFR intermediate peptide relative to the CGC–CGC dicodon control (Fig 1B). Quantification of both products (MFR and MFRR) of this inhibitory pair as well as elongation on a single arginine message (MFR) indicates that elongation through the first CGA codon is slightly slow, but that the subsequent elongation through the second CGA codon is the major inhibitory step (Fig EV1D and E).
Together, these data reveal in vitro defects in elongation reactions on the ribosome resulting from two distinct inhibitory codon pairs. These observations provide strong evidence that the initially observed effects in vivo (Gamble et al, 2016) reflect defects intrinsic to ribosome function rather than resulting from mRNA decay or other downstream cellular events.
Multiple defects in decoding caused by codon pairs
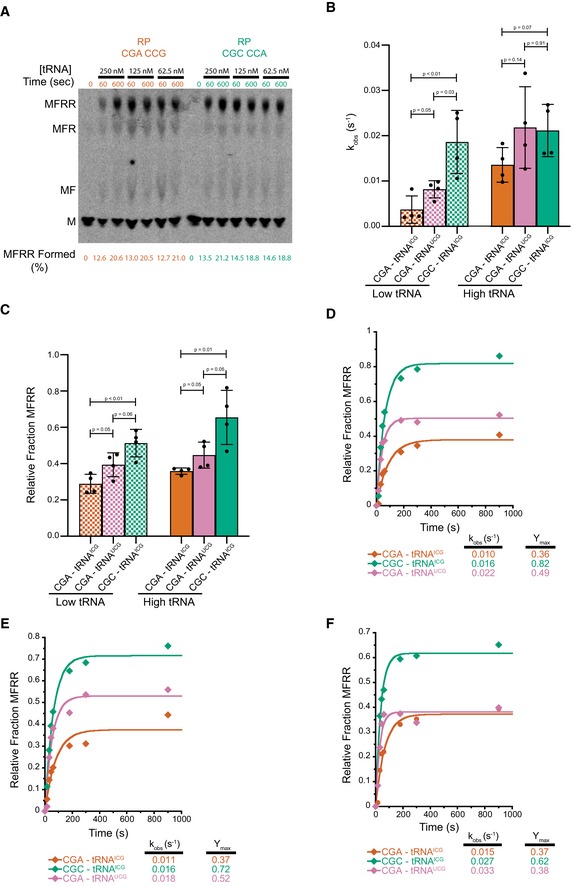
Assuming that one likely cause of elongation slowdown may be defects in decoding, we asked if the inhibition arises from simple defects in the kinetics of tRNA binding (a second‐order event) or instead from more downstream defects (i.e., in first‐order events) that follow including GTPase activation and accommodation (Gromadski & Rodnina, 2004; Zaher & Green, 2009). As the initial in vitro experiments were performed in a kcat/Km regime, we repeated the elongation assays at 10‐fold higher ternary complex concentrations that we established as saturating (Fig EV2A). For Arg–Arg, we see an approximately twofold rescue of the rate of the reaction with higher tRNA concentrations for the inhibitory pair (CGA–CGA) with only very modest changes in the rate of the reaction for the optimal pair (CGC–CGC; Fig 2A, left). Similarly, for the Arg–Pro combination, we see an approximately fourfold increase in the rate of the reaction with higher tRNA concentrations for the inhibitory pair (CGA–CCG) with only a modest, maximally 1.5‐fold increase, for the optimal pair (CGC–CCA; Fig 2A, right). These results suggest that tRNA binding contributes in part to the observed defects seen for the inhibitory pairs. Importantly, however, we observe that for both codon pairs (CGA–CGA and CGA–CCG), the endpoint defects are not overcome at high tRNA concentrations (Fig 2B). These latter data strongly suggest that a certain fraction of the complexes is unable to elongate independent of saturating levels of aminoacyl‐tRNA substrate.
Figure EV2. tRNA concentration titration and replicates with statistical analysis for UCG exact match tRNA experiments.
-
ATitration of aa‐tRNA to establish high concentration as saturating for elongation experiments. With this system, it is difficult to increase tRNA concentration, but going only twofold lower in concentration, we do not see differences in the amount of full‐length MFRR product being formed at early (60 s) time points as shown by the quantification below the TLC.
-
BComparison of observed rates of elongation for inhibitory CGA–CGA codon pair decoded by the native arginine ICGtRNA (red), inhibitory CGA–CGA codon pair decoded by the non‐natural arginine UCGtRNA (pink), and the optimal CGC–CGC codon pair decoded by the native arginine ICGtRNA (green). Data from experiments performed with limiting tRNA concentrations shown in hatched bars (25 nM aa‐tRNA) and saturating tRNA concentrations in solid bars (250 nM aa‐tRNA). Error bars represent standard deviations calculated from at least three experimental replicates (exact number of replicates indicated by the number of dots for each bar plotting the mean for the data). P‐values calculated using Student's t‐test and rounded to two decimal places.
-
CComparison of observed endpoints for inhibitory CGA–CGA codon pair decoded by the native arginine ICGtRNA (red), inhibitory CGA–CGA codon pair decoded by the non‐natural arginine UCGtRNA (pink), and the optimal CGC–CGC codon pair decoded by the native arginine ICGtRNA (green). Data from experiments performed with limiting tRNA concentrations shown in hatched bars (25 nM aa‐tRNA) and saturating tRNA concentrations in solid bars (250 nM aa‐tRNA). Error bars represent standard deviations calculated from at least three experimental replicates (exact number of replicates indicated by the number of dots for each bar plotting the mean for the data). P‐values calculated using Student's t‐test and rounded to two decimal places.
-
D–FElongation kinetics for the CGA–CGA inhibitory codon pair with the native arginine ICGtRNA (red) or the non‐native arginine UCGtRNA (pink) and for the CGC–CGC optimal control pair with the native arginine ICGtRNA (green) for the three other replicate experiments used to calculate the averages in (B and C).
Figure 2. Effects of tRNA concentration on elongation rates and endpoints.
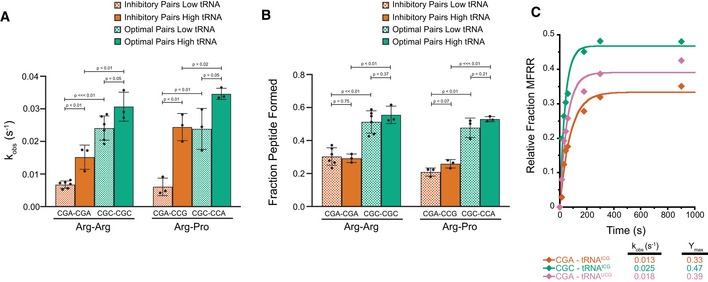
- Comparison of observed rates of elongation for inhibitory pairs (red) and their optimal controls (green) at limiting tRNA concentrations (hatched bars, 15–25 nM aa‐tRNA) and saturating tRNA concentrations (solid bars, 150–250 nM aa‐tRNA).
- Comparison of total peptide formation for inhibitory pairs (red) and their optimal controls (green) at limiting tRNA concentrations (hatched bars, 15–25 nM aa‐tRNA) and saturating tRNA concentrations (solid bars, 150–250 nM aa‐tRNA).
- Elongation kinetics for the CGA–CGA inhibitory codon pair with the native arginine ICGtRNA (red) or the non‐native arginine UCGtRNA (pink) and for the CGC–CGC optimal control pair with the native arginine ICGtRNA (green).
Given the unusual nature of the I:A wobble base pair found in the P site after incorporation of the first Arg in the codon pair, we also wondered whether the substantial defects that we observed might be rescued with the use of a non‐natural, exact match UCG tRNAArg as shown in vivo in the previous study (Gamble et al, 2016). We expressed the non‐natural tRNAArg on a CEN plasmid in yeast and purified it as above using a biotinylated oligonucleotide. In elongation reactions performed under kcat conditions (high tRNA concentrations), this non‐natural tRNA did partially rescue the endpoint defects in the elongation reaction associated with the CGA–CGA codon pair (Figs 2C and EV2B–F); these data suggest that the unusual I:A pairing in the P site at least partially contributes to the endpoint defects associated with these inhibitory codon pairs.
Increased 21‐nt RPFs on inhibitory pairs indicate an empty ribosomal A site
To further investigate the molecular mechanisms of inhibition underlying the inhibitory codon pairs, we turned to high‐resolution ribosome profiling (Wu et al, 2019). We recently reported that ribosome profiling using a cocktail of elongation inhibitors can trap ribosomes in their different functional states, distinguished by the size of ribosome‐protected footprints (RPFs). For example, when cycloheximide (CHX) and tigecycline (TIG) are added to yeast lysates to prevent ribosomes from translating post‐cell lysis, RPFs that are 21 nucleotides (nts) in length correspond to ribosomes in a “classical” or POST state waiting to decode the next aminoacyl‐tRNA, while RPFs that are 28 nts in length correspond primarily to ribosomes trapped in a “rotated” or PRE state (Wu et al, 2019). Building on an earlier study that showed an enrichment in ribosome density when the 17 inhibitory codon pairs are aligned (Gamble et al, 2016; Matsuo et al, 2017), we generated libraries using CHX and TIG to better distinguish the functional state of the paused ribosomes. In the plot shown in Fig 3A, the average ribosome density on the 17 inhibitory codon pairs (with the first codon in the P site and second in the A site) is shown as a function of the RPF length on the Y‐axis. We observe that while the density of 28‐nt RPFs is fairly constant across this region, there is a large accumulation of 21‐nt RPFs at the second codon of the problematic pair which is positioned in the A site (Fig 3A). These data indicate that for these 17 inhibitory pairs, elongation inhibition is caused by slow decoding of the second codon of the inhibitory pair, resulting in an empty A site that yields shorter footprints.
Figure 3. Increased 21‐nt RPFs on inhibitory pairs indicate an empty ribosomal A site.
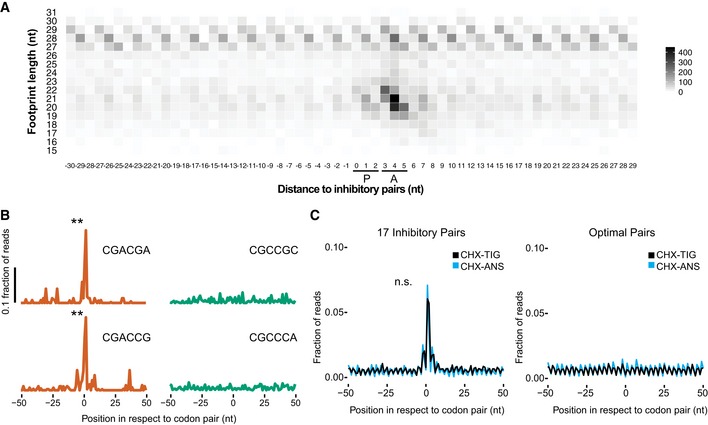
- Meta‐analysis of footprint size of all 17 inhibitory pairs identified by Grayhack and co‐workers (Gamble et al, 2016), aligning the first codon of the pair in the ribosomal P site.
- Metacodon analysis of 21‐nt RPFs centered at the first codon of each inhibitory pair (red) compared to their corresponding optimal pair (green). P‐values were determined by Student's t‐test between each inhibitory pair and the corresponding optimal pair (**P < 0.01, n = 2).
- Comparison of 21‐nt RPFs aligned at all 17 inhibitory codons from libraries made with CHX/ANS (blue) and CHX/TIG (black) (left) to their corresponding optimal pairs with the same antibiotic combination (right). No significant difference found (n.s.: P = 0.9961, n = 2) between CHX‐TIG and CHX‐ANS.
We can also look individually at the representative codon pairs studied above (CGA–CGA and CGA–CCG), and we see significant accumulation of 21‐nt RPFs in the A site relative to the amount observed for their optimal counterparts (red versus green; Fig 3B). These data provide direct evidence that elongation inhibition on these codon pairs results from slow decoding of the second codon of the inhibitory pair.
We also considered the possibility that for the inhibitory codon pairs, tRNAs are bound (or accommodated) but fail to undergo peptidyl transfer, perhaps because of a misalignment in the peptidyl‐transferase center of the 60S subunit. We observed previously that the addition of anisomycin (ANS), a peptidyl‐transferase inhibitor, together with CHX, blocks bound tRNAs from forming peptide bonds such that they eventually fall out of the A site during library preparation; in these libraries, 21‐nt RPFs represent a combination of two different ribosome populations, those in a pre‐accommodation and a pre‐peptidyl transfer state. Indeed, in samples prepared with CHX/ANS, we observe more 21‐nt RPFs at peptide motifs known to undergo slow peptidyl transfer relative to those motifs or codons enriched in the CHX/TIG samples (Fig EV3A; for problematic peptide motifs, see Schuller et al, 2017). If the chemistry of peptide bond formation were slow for the inhibitory codon pairs, we would expect to see an increase of 21‐nt RPFs at these sites in the CHX/ANS library relative to the CHX/TIG library. Instead, we see the same level of enrichment of 21‐nt RPFs at these sites (Fig 3C, left), arguing that the limiting step for the inhibitory base pairs is not peptide bond formation. These findings are consistent with the hypothesis that certain wobble pairs impact the decoding center in the 40S subunit, affecting decoding or accommodation, rather than activities in the peptidyl‐transferase center of the large subunit. For the optimal codon pairs for these same amino acid sequences, no pauses are seen in either sample indicating that the pausing at inhibitory codons is due to the codon/tRNA pairing in the A site rather than to the amino acid sequence per se (Fig 3C, right).
Figure EV3. Ribosome profiling analysis showing defects in peptide bond formation.
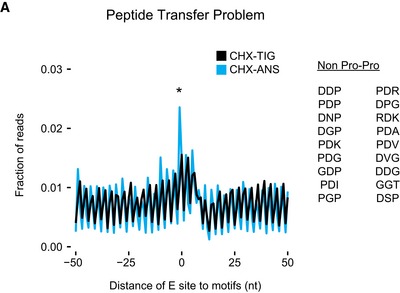
Metacodon analysis of 21‐nt RPFs in libraries prepared with CHX/ANS (blue) showing an increase in ribosome density at tripeptide motifs that undergo slow peptide bond formation (Schuller et al, 2017) compared to libraries prepared with CHX/TIG. P‐value was determined by Student's t‐test between CHX‐TIG and CHX‐ANS datasets on the tripeptide motifs indicated (*P = 0.0363, n = 3).
Decoding‐incompatible mRNA conformation causes the inhibitory codon pair‐mediated stalling
To investigate the molecular basis of the inhibitory codon pairs involving the problematic CGA codon, we turned to structural studies of complexes stalled at CGA–CCG and CGA–CGA codon pairs. We used a yeast cell‐free in vitro translation system in which we translated mRNA reporters containing these codon pairs. Translation extracts were prepared from yeast cells lacking Ski2p, a component of the 3′–5′ mRNA decay system, to enhance mRNA stability. Both mRNA reporters contained sequences coding for an N‐terminally His8‐HA‐tagged truncated uL4 (Knorr et al, 2019) followed by the stalling (CGA–CCG)2 or (CGA–CGA)2 codon pairs (Appendix Figs S1A and S2A). To avoid capturing read‐through products, the stalling sequences were followed by three UAA(A) stop codon quadruplets, one in each reading frame, which would lead to termination upon read‐through. Ribosome‐nascent chain complexes (RNCs) were affinity‐purified using magnetic beads and separated on a sucrose density gradient, and the 80S fractions were subjected to cryo‐EM (Appendix Figs S1 and S2).
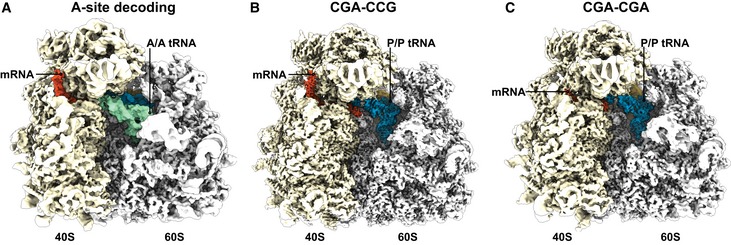
Classification of ribosomal particles for both stalling sequences (CGA–CCG and CGA–CGA) revealed the most abundant classes to be programmed ribosomes in the post‐translocation state (POST state) with tRNAs in the P/P and E/E state, but not in the A site (Appendix Figs S3 and S4). The structure of the CGA–CCG‐stalled ribosome was reconstructed to an average resolution of 2.6 Å, while the CGA–CGA‐stalled ribosome was reconstructed to an average resolution of 3.2 Å (Fig EV4B and C). To compare these structures on a molecular level with a canonical A‐site tRNA decoding situation, we refined our previously produced structure of cycloheximide‐stalled ribosomes in the pre‐translocation state (PRE state) with A/A and P/P tRNAs to 3.1 Å with focus on the mRNA decoding in the A site (Figs 4A and EV4A; preprint: Buschauer et al 2019). Molecular models were built and refined for all structures allowing for an in‐depth analysis (Fig 4A–I). The cryo‐EM density and model of the nascent chain allowed us to clearly identify the stall site at the first codon of the inhibitory codon pair in the P site of the small subunit and the second codon of the pair in the A site. As such, the first CGA‐encoded Arg has been incorporated into the peptidyl‐tRNA while the following CCG is in the A site waiting to be decoded (Fig EV5A). Structural analysis of the CGA–CCG‐ and CGA–CGA‐stalled RNCs revealed no perturbations of the peptidyl‐transferase center (PTC; Fig EV5B), in agreement with the puromycin reactivity of these stalled ribosomes (Appendix Fig S5). On the other hand, we saw a strikingly unusual conformation of the mRNA in the A site of these structures when compared with the canonical decoding situation (Fig 4A–I).
Figure EV4. Cryo‐EM structures of RNCs stalled on inhibitory codon pairs in comparison with the A‐site decoding situation.
-
A–CCryo‐EM density maps filtered according to local resolution used to build molecular models. (A) Cryo‐EM map of the pre‐state RNC with tRNA in the A site. (B) Cryo‐EM map of the CGA–CCG‐stalled RNC. (C) Cryo‐EM map of the CGA–CGA‐stalled RNC.
Figure 4. CGA–CCG and CGA–CGA induce stalling through decoding‐incompatible mRNA conformations in the A site.
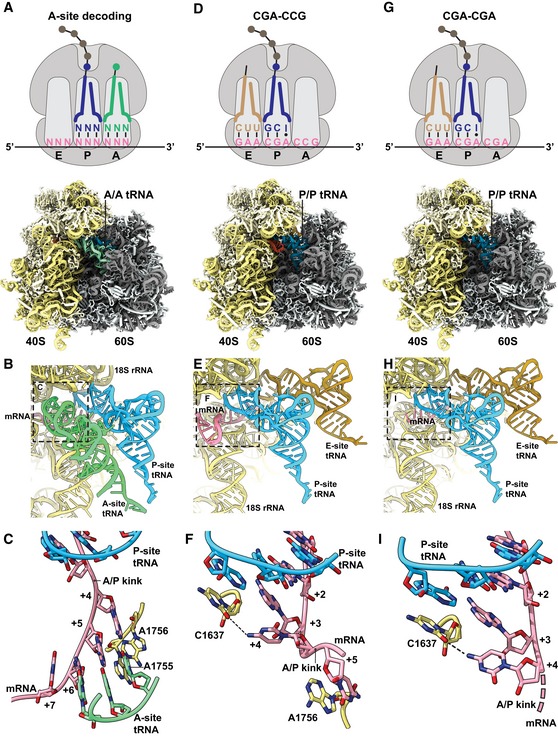
-
A–CCryo‐EM structural characterization of the pre‐state 80S RNC with A‐site tRNA in the decoding center. (A) Schematic representation of the decoding situation (top) and molecular model for the pre‐state RNC with A‐site tRNA in the decoding center. (B) General overview of the A, P, and E sites with A/A and P/P tRNAs and mRNA. (C) Detailed view of the mRNA in the A site using stick model with cartoon phosphate backbone representation. The 18S rRNA bases A1755 and A1756 recognize the minor groove of A‐site tRNA–mRNA interaction during tRNA decoding.
-
D–FCryo‐EM structural characterization of the CGA–CCG‐stalled 80S RNC. (D) Schematic representation of the stalling situation (top) and molecular model of the CGA–CCG‐stalled RNC (bottom). (E) General overview of the A, P, and E sites with P/P and E/E tRNAs and mRNA. (F) Detailed view of the mRNA in the A site using stick model with cartoon phosphate backbone representation. The mRNA positions +2 to +5 and their interactions are shown. The C +4 is flipped by approximately 95° degrees toward the wobble A:I base pair in the P site and stabilized by interaction with the C1637 of the 18S rRNA helix 44. The C +5 is stabilized by stacking interaction with the A1756 of the 18S rRNA which normally recognizes the minor groove of A‐site tRNA–mRNA interaction during decoding (C).
-
G–ICryo‐EM structural characterization of the CGA–CGA‐stalled 80S RNC. (G) Schematic representation of the stalling situation (top) and molecular model of the CGA–CGA‐stalled RNC (bottom). (H) General overview of the A, P, and E sites with P/P and E/E tRNAs and mRNA. (I) Detailed view of the mRNA in the A site as in (F). Downstream mRNA is indicated by the dotted line. Note the rotation of the C+4 base compared to the CGA–CCG mRNA.
Figure EV5. Structural details of the codon‐based stalling.
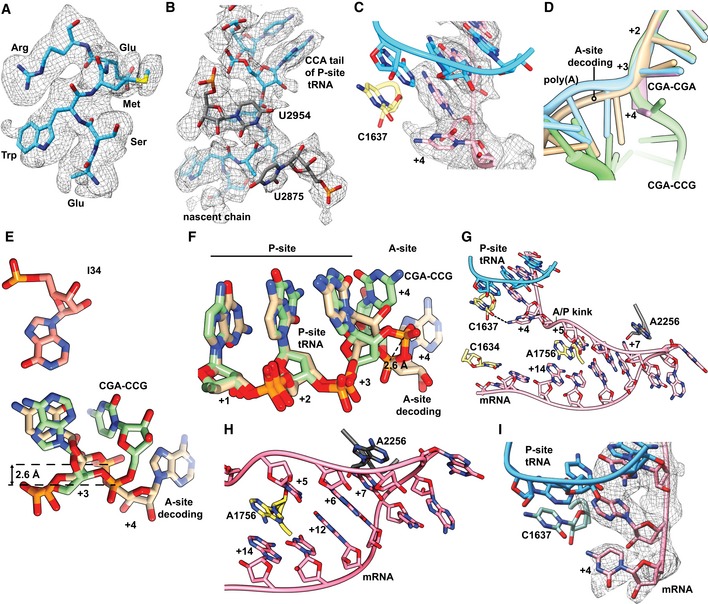
-
ACryo‐EM density (mesh) and stick model of the nascent chain residues modeled in the 80S ribosome stalled on the CGA–CCG inhibitory codon pair. The density of the CGA‐encoded Arg of the first codon pair is clearly distinguishable as well as the bulky density of Trp65 of the uL4 encoded upstream in the translated mRNA construct.
-
BCryo‐EM density (mesh) and stick model of the peptidyl‐transferase center as observed in the structure of ribosome stalled on the CGA–CCG inhibitory codon pair. Notably, for the U2875 (U2506 in E. coli), no clear density is observed, most likely due to enhanced flexibility. Yet, no directly observable major perturbation of the PTC components is apparent.
-
CCryo‐EM density (mesh) and stick model with cartoon phosphate backbone representing the mRNA positions +1 to +4 and their interactions in the CGA–CCG‐stalled ribosome.
-
DEffect of flipped C+4 on the general path of the mRNA in the A site. A cartoon representation of mRNAs in all four discussed 80S structures is compared.
-
E, FComparison between the A‐site tRNA decoding situation and the CGA–CCG‐stalled situation of the mRNA in positions +1 to +4. In the CGA–CCG A site, the C+4 is flipped by approximately 95° degrees toward the wobble A:I base pair and the mRNA backbone is shifted by 2.6 Å at the phosphate linking A+3 and C+4.
-
GOverview of the mRNA and its interactions in the A site of CGA–CCG reporter stalled ribosome using stick model with cartoon phosphate backbone representation. Sequence of mRNA positions +4 to +14 is CCGCGACCGUA.
-
HDetail of the tip of the hairpin from (G) with stabilizing stacking interactions between A2256 of the 25S rRNA and the C+7 of the mRNA and among A1756 of the 18S rRNA intercalated between the C+5 and the A+14 of the mRNA.
-
ICryo‐EM density (mesh) and stick model with cartoon phosphate backbone representing the mRNA positions +1 to +4 and their interactions in the CGA–CGA‐stalled ribosome.
The most striking mRNA structure is formed on the CGA–CCG reporter mRNA. In our 2.6 Å map, we can clearly identify the CGA codon in the P site and the anticodon of ICG tRNAArg making standard Watson–Crick interactions as observed before (Schmidt et al, 2016) at the first two positions of the codon and a purine:purine A:I base pair at the wobble position (Fig 4E). However, the first nucleotide in the A site (the C+4 of the CCG codon) is found in an unusual conformation that is well defined by the cryo‐EM density (Fig EV5C). Compared to the control canonical decoding situation (Fig 4A–C), C+4 is flipped by approximately 95° degrees toward the wobble A:I base pair in the P site. Stabilization of C+4 in this position appears to be facilitated by an H‐bond formed with C1637 of 18S rRNA helix 44 (C1400 in E. coli) which stacks on the I of the ICG tRNAArg in the P site (Fig 4F). Compared to the canonical decoding situation, accommodation of the purine:purine A:I wobble base pair at position +3 shifts the mRNA backbone by 2.6 Å at the phosphate linking +3 and +4, thus forcing the general path of the downstream mRNA into an unusual direction (Fig EV5D–F). Importantly, this alteration in the mRNA structure moves the crucial A/P kink to occur between positions +4 and +5 (Fig 4F). The A/P kink, normally positioned between positions +3 and +4, was shown to be crucial for A‐site interaction and proofreading activity, especially for difficult‐to‐decode near‐cognate tRNAs (Keedy et al, 2018). In the flipped‐out position seen here, the C+4 seems unlikely to be engaged by a canonical codon:anticodon interaction with the incoming aminoacyl‐tRNA (Fig 4C and F). This rearrangement of the mRNA itself could explain the previously proposed communication between ribosomal P and A sites (Gamble et al, 2016).
Moreover, following C+4, the mRNA folds into a stable mRNA hairpin structure that directly occludes tRNA binding in the A site. In the hairpin, the C+5 base‐pairs with G+12 and the G+6 base‐pairs with C+11, while nucleotides C+7–C+10 form a rather flexible tetraloop at the tip of the hairpin (Fig EV5G and H). Interestingly, this structure is stabilized by A1756 (A1493 in E. coli) of the 18S rRNA which flips out of helix h44 as well as the rearranged A2256 (A1913 in E. coli) of the 25S rRNA helix 69. Normally, A2256 forms a dynamic inter‐subunit bridge 2A by intercalating into the 18S rRNA helix 44. However, to support the observed mRNA secondary structure formation, A2256 rotates by 101 degrees and stacks with C+7 of the mRNA (Fig EV5I). Taken together, this structure rationalizes how accommodation of the UGG tRNAPro in the A site on the CGA–CCG inhibitory dicodon is prevented: (i) by positioning of C+4 in a conformation incompatible with decoding, (ii) by shifting the crucial mRNA A/P kink one position downstream, and (iii) by sterically blocking the tRNA binding site with an mRNA secondary structure.
Analogous to the CGA–CCG situation, we saw a specific inhibitory conformation of C+4 in the CGA–CGA mRNA cryo‐EM structure (Fig 4G–I). Again, well supported by cryo‐EM density, the conformation of C+4 is essentially the same as observed for the CGA–CCG reporter, with an 84° rotation of the cytosine base (Figs 4I and EV5F). After position +4, however, the mRNA density is weak and does not allow for reliable model building. These observations suggest a more flexible conformation of downstream mRNA in this structure. Nonetheless, the general path of mRNA seems to be shifted in the same direction as seen for the CGA–CCG case and the A/P kink in mRNA is also dislocated downstream as it cannot be observed between positions +3 and +4 (Figs 4I and EV5C). Taken together, these two structures show how rearrangement of the mRNA induced by the wobble‐decoded CGA codon in the P site causes perturbations in the A site that disfavor decoding.
Decoding‐incompatible mRNA conformation contributes to poly(A) tract‐mediated stalling
Next, we wondered whether the CGA‐dependent codon pair stalling mechanism is structurally related to poly(A)‐mediated stalling. First, using our in vitro system, we see slower elongation on a MFK5 AAA IC as compared to an AAG IC, consistent with earlier observations in E. coli (Koutmou et al, 2015). Despite resolution limitations of eTLC with multiple lysines, when we compare the earliest time points for AAA complexes with those for AAG complexes, the AAA complexes have only elongated to MF and MFK, whereas the AAG complexes are already making MK2 and larger products as indicated by the fast‐running smear (Fig EV6A). These data are consistent with earlier reports documenting differences in elongation on iterated AAA relative to AAG lysine codons in other systems (Arthur et al, 2015; Koutmou et al, 2015).
Figure EV6. Elongation of AAA is slower than AAG on MFK5 initiation complexes.
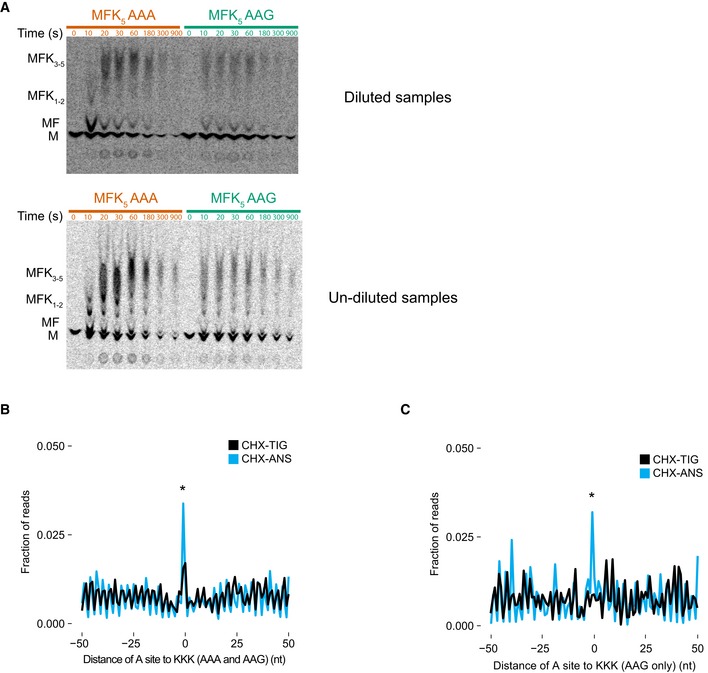
- TLC showing peptide bond formation of MFK5 messages on inhibitory AAA codons (red) and control AAG codons (green). Samples were diluted 1–4 μl in water (top) or undiluted (bottom) to better resolve individual products on TLC. MFK5 AAG complexes are making longer lysine peptides (indicated by higher bands on the TLC) than AAA at early time points.
- Metacodon analysis of 21‐nt RPFs in libraries prepared with CHX/ANS (blue) and CHX‐TIG (black) showing an increase in ribosome density at polylysine motifs (KKK) comprised of both AAA and AAG codons. P‐value was determined by Student's t‐test between CHX‐TIG and CHX‐ANS datasets on the tripeptide motifs indicated (*P = 0.0204, n = 3).
- Metacodon analysis of 21‐nt RPFs in libraries prepared with CHX/ANS (blue) showing an increase in ribosome density at polylysine motifs (KKK) comprised of only AAG codons indicative of slow peptide transfer compared to libraries prepared with CHX‐TIG (black). P‐value was determined by Student's t‐test between CHX‐TIG and CHX‐ANS datasets on the tripeptide motifs indicated (*P = 0.0117, n = 3).
We also analyzed the ribosome profiling data to ask whether we could define the elongation defects on iterated lysine residues, transcriptome‐wide. Genome‐wide alignment of KKK sequences (including both AAA and AAG lysine codons) revealed ribosome pausing both in libraries prepared with TIG/CHX and ANS/CHX (Fig EV6B). We attribute the increased ribosome occupancy that we observe in the TIG/CHX sample to ribosomes struggling to decode, and thus impeded in the binding of tRNAs to the small subunit decoding center (consistent with mRNA structural obstruction). We attribute the even higher ribosome occupancy in the ANS/CHX sample to the combined problems of decoding (seen in the TIG/CHX sample) and of peptidyl transfer on the large subunit. To separate the peptidyl transfer and decoding defects, we aligned KKK sequences composed entirely of AAG codons as these are not thought to be particularly problematic. Indeed, in the TIG/CHX sample, we see no evidence of ribosome accumulation on these AAG‐only KKK sequences, but in the ANS/CHX sample, we do see evidence of ribosome accumulation, consistent with defects in peptidyl transfer (Fig EV6C). Together, these data provide strong support for the specificity of mRNA‐driven decoding defects on iterated AAA codons, and of general peptidyl transfer defects in the PTC resulting from the iteration of lysine residues.
For cryo‐EM, we used an analogous approach to that used for CGA‐dependent codon pair‐mediated stalling with a modified mRNA reporter comprising a 49‐nucleotide‐long poly(A) tract (Appendix Fig S6). As for both inhibitory codon pairs (CGA–CCG and CGA–CGA) discussed above, classification of poly(A)‐stalled ribosomal particles revealed that a majority (78%) of programmed particles are in the POST state without A‐site tRNA (Appendix Fig S7). We reconstructed the poly(A)‐stalled ribosome structure to an overall resolution of 3.1 Å, which allowed for building and refinement of a molecular model (Fig 5A and B).
Figure 5. Ribosomes stalled on poly(A) stretches reveal alterations in both the peptidyl‐transferase and decoding centers.
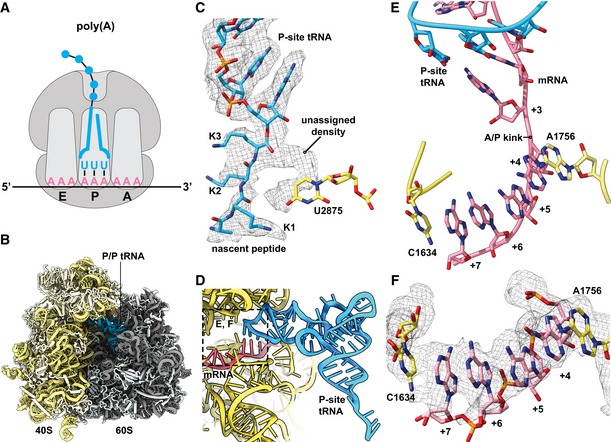
-
A, BSchematic representation (A) of the stalling situation on poly(A) tract mRNA. Cryo‐EM density map of the poly(A)‐stalled 80S RNC filtered according to local resolution and used to build the molecular model (B).
-
CCryo‐EM density (mesh) and stick model with cartoon phosphate backbone representing the peptidyl‐tRNA in the peptidyl‐transferase center (PTC).
-
DGeneral overview of the A, P, and E sites with the P/P tRNA and mRNA.
-
E, FDetailed view of the mRNA in the A site using stick model with cartoon phosphate backbone representation and cryo‐EM density (mesh). The poly‐adenine mRNA sequence forms a π‐stacking array between positions +4 and +7, which is stabilized from both sides by stacking of 18S rRNA bases C1634 and A1756.
In the resulting structure, we first analyzed the PTC to look for potential structural changes that might rationalize previous arguments that sequential lysines in the peptide tunnel lead to translational stalling due to their basic nature (Lu & Deutsch, 2008). We were able to model the last three C‐terminal residues of the nascent chain as lysines, consistent with the RNC being stalled on the poly(A) tract. In the PTC, we observed the terminal lysine side chain pointing toward the A site and an extra density not explained by the nascent peptide model (Fig 5C). Overall, however, the crucial catalytic bases (U2875 and U2954, corresponding to U2506 and U2585 in E. coli) did not seem to be hindered from moving into the induced state conformation upon tRNA binding in the A site, therefore hinting that any perturbations of the PTC geometry are relatively modest. Consistent with this hypothesis, these complexes are reactive to puromycin (Appendix Fig S5C). Moreover, these observations do not provide an explanation for the absence of A‐site tRNA in 93% of particles. Therefore, we investigated the mRNA conformation in the A‐site decoding center.
When we examined the molecular details in the decoding center, we clearly saw the structure of the codon–anticodon interaction between the AAA codon and UUU tRNALys in the P site with no apparent perturbations (Fig 5D). Strikingly, however, the four downstream adenosines in the A‐site decoding center are engaged in a π‐stacking array, adopting essentially the same single‐stranded helical conformation recently reported by Passmore and colleagues for isolated poly(A) sequence (Tang et al, 2019). This +4 to +7 π‐stack is stabilized on both sides by flipped‐out rRNA nucleotides A1756 and C1634. Indeed, C1634 (C1397 in E. coli) is found in an unusual, previously unobserved conformation (Fig 5E and F). In this arrangement, the AAA codon in the A site adopts what is clearly a decoding‐incompetent conformation that likely directly contributes to poly(A)‐mediated stalling, although the general path of mRNA does not seem to be as strongly affected as in the case of both inhibitory codon pairs (Fig EV5C). Taken together, for RNCs stalled on poly(A), we observe structural changes assumed by the mRNA in the A site that preclude canonical interactions with the decoding tRNA.
Ribosome collisions on poly(A) tracts affect disome formation
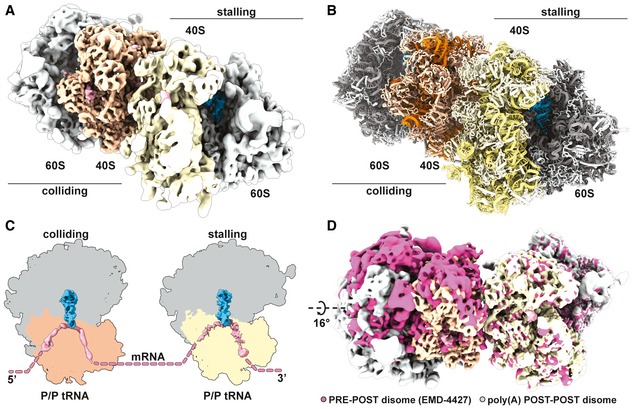
Given that ribosome collisions have been shown to produce crucial substrates for quality control pathways (Simms et al, 2017; Juszkiewicz et al, 2018; Ikeuchi et al, 2019), we wondered if poly(A) tracts in our system would generate a stable ribosome collision amenable to structural analysis. Therefore, we prepared a disome fraction of the poly(A)‐stalled RNCs as a minimal ribosome collision species and determined structural information by cryo‐EM (Appendix Fig S6). We processed the data using the 80S extension approach as described previously (Ikeuchi et al, 2019) and segregated classes of ribosomal particles stalled in the POST and PRE states (Appendix Fig S9). When we further sorted particles corresponding to the above‐described poly(A)‐stalled 80S POST state class, we observed disome structures as expected; however, these POST state ribosomes were found in both the first “stalled” and the second “colliding” positions. These collided disomes, which were composed of two POST state ribosomes, are thus strikingly different from previously characterized disomes in both mammalian and yeast systems (Juszkiewicz et al, 2018; Ikeuchi et al, 2019). In these previous structures, the second colliding ribosome was always present in a rotated PRE state, with tRNAs in the A/P and P/E states unable to translocate any further downstream. We refined the disome class containing the colliding 80S in the POST state to an overall resolution of 3.8 Å and clearly confirmed that both individual 80S ribosomes are present in the canonical POST state conformation in this disome assembly (Fig 6A–C). Direct comparison of POST–POST with the POST–PRE disome assemblies showed that the second colliding ribosome would have to rotate by 16° to structurally mimic the previously reported POST–PRE conformation (Fig 6D). Taken together, these data indicate that the second colliding ribosome is able to complete the translocation step along the mRNA, a step that would normally be prevented by the stable “roadblock” of the leading stalled ribosome. Therefore, we suggest that poly(A) tracts, which are known to be slippery and allow for sliding, can result in a less rigidly arrested first stalled ribosome.
Figure 6. Disomes stalled on poly(A) tracts form a novel POST–POST assembly.
-
A, BComposite cryo‐EM density map (A) of the POST–POST disome stalled on the poly(A) mRNA reporter filtered according to local resolution and used to build the molecular model (B).
-
CCut top views of both the first (stalling) and the second (colliding) ribosomes forming the disome. Observed ribosomal and tRNA translocation states are indicated.
-
DComparison of ribosomal assemblies between the previously described CGA–CCG‐stalled yeast disome in pink (EMD‐4427; Ikeuchi et al, 2019a) and the novel POST–POST assembly observed in poly(A) stalling. The EMD‐4427 density map was fitted into the density of the first stalling ribosome on the poly(A) reporter. The indicated rotation was calculated using the 60S subunit, as the compared colliding ribosomes are not in the same translocation state (PRE versus POST).
Discussion
Gene expression can be fine‐tuned by the selection of specific codons within the context of the degeneracy of the genetic code. While traditional metrics like the codon adaptation index or tRNA adaptation index take into account how commonly a codon is used or how abundant its cognate tRNA is, respectively, it is not well understood why specific codon pairs are underrepresented in genomes compared to their expected values based on the frequency of each individual codon in the pair (Yarus & Folley, 1985; Fedorov et al, 2002). The work of Grayhack and co‐workers (Gamble et al, 2016) identified 17 codon pairs in Saccharomyces cerevisiae that reduce protein expression, offering experimental insights into how codon pairs affect translation. In particular, they showed that the neighboring ribosomal A and P sites can interact to limit protein output in a codon pair‐mediated way, and hypothesized that wobble base‐pairing played a role in this inhibition.
Our results with an in vitro reconstituted translation system directly show that elongation rates of inhibitory codon pairs are slower than those of their optimal counterparts, confirming the hypothesis that inhibition is intrinsic to the ribosome and is likely to involve interactions with the tRNA substrates. These findings are broadly consistent with previously published in vivo data (Gamble et al, 2016) although direct comparisons of the magnitude of defects in these systems are likely not of particular value given their very different limitations. For both the Arg–Arg (CGA–CGA) and Arg–Pro (CGA–CCG) pairs, strong defects in the rates and endpoints of the reactions are observed (Fig 2A and B). The observation that the strong endpoint defects are not affected by increased tRNA concentration suggests that there are fundamental structural defects that preclude A‐site binding/reactivity for some fraction of the ribosome complexes. Consistent with previous work by Grayhack and co‐workers (Gamble et al, 2016), the unique I:A wobble associated with decoding CGA codons by the ICG tRNAArg has a strong effect on interactions in the P site that structurally extend into perturbations of the A site (Fig 4F and I). We additionally find that these defects are partially rescued by substitution of a UCG tRNAArg that no longer relies on I:A pairing (Figs 2C and EV2B–F), consistent with previous in vivo studies (Gamble et al, 2016).
The observation that the kinetics of decoding are retarded by inhibitory codon pairs in biochemical assays was corroborated by our high‐resolution ribosome profiling studies. We see an enrichment of 21‐nt RPFs, corresponding to ribosomes lacking a tRNA in the A site, when the first codon of the pair is in the ribosomal P site and the second codon is in the A site (Fig 3A and B). Comparing the results from the CHX/ANS library with the CHX/TIG library, we see the same level of these 21‐nt RPFs, indicating that peptide bond formation is not limiting the inhibitory codon pair‐stalled ribosomes (Fig 3C). This observation is consistent with the fact that the optimal codon pairs (which encode the same amino acid residues and use the same tRNAs) elongate at normal rates both in vivo and in vitro. These data indicate that the inhibitory codon pairs affect the decoding center of the 40S subunit rather than the peptidyl‐transferase center of the 60S subunit. Overall, our data are consistent with the idea that the major mechanism of inhibition on most of these inhibitory codon pairs is through impairment of tRNA binding/accommodation.
Detailed mechanistic insight into the origins of A‐site accommodation defects was ultimately provided by our structural analysis. In our cryo‐EM structures of ribosome‐nascent chain complexes stalled on the CGA–CGA or CGA–CCG codon pairs, we identified several structural details that likely directly affect tRNA binding/accommodation activity. Interestingly, in each case these alterations are mediated by the structure of the mRNA itself and readily explain the previously proposed communication between the ribosomal A and P sites (Figs 4 and EV5; Gamble et al, 2016). In particular, for both the CGA–CCG and CGA–CGA inhibitory pairs, the C+4 mRNA nucleotide is dramatically flipped away from the A‐site decoding center of the ribosome. The C+4 nucleotide instead makes contact with the P‐site codon and interacts with C1637 of 18S rRNA which stacks to the anticodon inosine decoding the wobble position (A+3) of mRNA in the P site. The path of the mRNA is also affected by the purine:purine A:I wobble base pair at position +3 and shifts toward C1637. This perturbation involving the A:I wobble interaction provides an immediate explanation for why the CGA codon in particular confers the strongest elongation defect. Moreover, the A/P kink of the mRNA, which was shown to be crucial for A‐site interaction and proofreading (Keedy et al, 2018), is moved downstream in these structures as a consequence (Fig 4F and I). This critical structure is typically stabilized by an ammonium ion in X‐ray structures (Rozov et al, 2019) and was proposed to be essential for frame maintenance by preventing slippage (Selmer et al, 2006). Finally, in the case of CGA–CCG, we observe a hairpin structure formed by mRNA nucleotides between positions +5 and +14 (Fig EV5D and E). This structure may be particular to this reporter mRNA sequence since no equivalent stable mRNA secondary structure is formed in the case of the CGA–CGA‐stalled RNC. Interestingly, a similar A‐site hairpin was observed previously in a structure implicated in translational bypassing (Agirrezabala et al, 2017).
Consistent with the earlier work (Gamble et al, 2016), we see a specific deleterious effect of I:A wobble decoding on translation efficiency in inhibitory codon pairs containing the 5′ CGA codon. Previously, the purine:purine Ianti:Aanti base pair was analyzed in the A site only, where its accommodation affects and alters mainly the anticodon of tRNA, due to its unique “wide” purine–purine geometry (Murphy & Ramakrishnan, 2004). In contrast, in our structure of the Ianti:Aanti wobble pair in the P site, we find that its accommodation affects not the anticodon of tRNA but rather the mRNA backbone (Fig 5E and H). This mRNA distortion apparently imposes allosteric effects on the neighboring region resulting in the unusual mRNA conformation in the A site. The modification of adenosine to inosine (Gerber & Keller, 1999) expands the decoding range of the ICG tRNAArg as inosine is able to base‐pair with cytidine, uridine, and even adenosine at the wobble position. It is intriguing to observe that this seemingly elegant evolutionary decoding mechanism has certain associated disadvantages as the non‐optimal CGA codon (decoded via the I:A interaction with the ICG tRNAArg) is slow to decode and leads to deleterious effects on mRNA stability (Presnyak et al, 2015). It is in principle possible that under normal conditions, an Isyn–Aanti conformation (Hoogsteen base pair with the hypoxanthine base of inosine rotated by 180° respective to its ribose) is adopted to minimize geometric strain and allow for further elongation. In this particular case, the inhibitory codon pairs studied here could prevent this Hoogsteen base pair formation and thereby create a dead‐end situation. Since the Isyn–Aanti conformation has not been observed in the context of decoding by ICG tRNAArg, this alternative would require closer investigation in the future.
Poly(A) tracts are known to be problematic sequences for the ribosome. Previous studies proposed that electrostatic interactions between the poly‐basic nascent chain and the peptide exit tunnel of the ribosome might be the primary features that elicit ribosomal stalling (Lu & Deutsch, 2008). Our cryo‐EM structures allow us to look at the peptidyl‐transferase center (PTC) on poly(A)‐stalled ribosomes where we see that the crucial catalytic bases do not seem to be hindered from moving into the induced state conformation for peptidyl transfer; however, we note the presence of an extra density which is not clearly interpretable (Fig 5C). This extra density adopts a defined shape next to the last nascent amino acid residue and could potentially be assigned to a mixed nascent chain state or even a small molecule. Importantly, this observed geometry of the PTC cannot explain the highly efficient stalling on poly(A) tracts and the absence of any A‐site tRNA in 93% of particles in the dataset, but it could reflect perturbations in the PTC associated with iterated lysine residues. Interestingly, we see an accumulation of 21‐nucleotide RPFs at KKK motifs on iterated AAG codons in ribosome profiling samples prepared with ANS/CHX, while there is no accumulation in samples prepared with TIG/CHX (Fig EV6C). These data are indicative of a modest deficiency in peptidyl transfer associated specifically with polylysine motifs.
In light of this limited contribution to stalling by PTC perturbation, we closely investigated the structural features of the decoding center in the small subunit of the ribosome. There we found that an mRNA‐mediated mechanism is directly contributing to stalling. Consecutive mRNA adenosines are engaged in a π‐stacking array in the A site, stabilized on both sides by rRNA base stacking interactions, and adopt a helical conformation typical for single‐stranded poly(A) stretches (Fig 5E and F; Tang et al, 2019). This π‐stacking array represents a decoding‐incompetent structure. These findings are consistent with the increase in 21‐nucleotide RPFs in ribosome profiling samples prepared with TIG/CHX when aligning KKK motifs comprising both AAA and AAG codons, but not when aligning KKK motifs from AAG alone (Fig EV6B and C). These ideas agree with previous observations that consecutive AAG codons are less efficient in stalling than AAA codons (Koutmou et al, 2015) despite encoding for the same amino acid residue and that the intrinsic π‐stacked helical structure of poly(A) single‐strand tract is efficiently disrupted by inclusion of guanosines (Tang et al, 2019). Together, we conclude that the inhibitory conformation of mRNA in the A site is the prominent contributor to the poly(A)‐mediated stalling mechanism.
Interestingly, when studying ribosomal collisions as a consequence of poly(A)‐mediated stalling, we found a large fraction of the disomes in a novel POST–POST state that was distinct from the previously characterized disome structures in both mammalian and yeast systems (Fig 6A and B; Juszkiewicz et al, 2018; Ikeuchi et al, 2019). In both previous structures, the second colliding ribosome is captured in a rotated PRE state unable to translocate further. Finding both collided ribosomes in the POST state indicates that the second colliding ribosome completed the translocation step, likely due to a weaker “roadblock” presented by the first stalled ribosome. Since poly(A) tracts were characterized as slippery (Koutmou et al, 2015), it is tempting to speculate that applying force on the first stalled ribosome by the colliding ribosome(s) could contribute to ribosome sliding on the mRNA and loss of reading frame. This model is consistent with recent findings that directly implicate ribosomal collisions in +1 frameshifting (Simms et al, 2019) and translational reprogramming by frameshifting in bacteria (Smith et al, 2019). Ribosomal collisions could, in principle, disrupt the interaction between the P‐site tRNA and the mRNA in the first ribosome and contribute to +1 frameshifting observed after ribosomal pausing (Dinman, 2012). We speculate that the loss of reading frame in the case of collisions on poly(A) tracts is facilitated by (i) the fact that the P‐site tRNA is the only one left on the stalled ribosome after the E‐site tRNA dissociates and (ii) the fact that the P‐site tRNA only interacts with the mRNA via relatively less stable A:U base pairs. These ideas are consistent with earlier studies arguing that reading frame maintenance is predominantly affected by the energetics of the P‐site codon–anticodon interaction (Baranov et al, 2004).
Taken together, our work combines in vitro and in vivo methods to study the effects of inhibitory mRNA sequences and shows for the first time detailed mechanistic insight into mRNA‐mediated translation stalling via decoding obstruction.
Materials and Methods
Ribosome preparation
WT Ribosomes were purified and isolated as subunits as previously described (Eyler & Green, 2011).
Purification of translation factors
Translation initiation factors eIF1, eIF1A, eIF5, and eIF5B were expressed and purified from E. coli and eIF2 was expressed and purified from S. cerevisiae as previously described (Acker et al, 2007; Eyler & Green, 2011). The translation elongation factor eIF5A was purified from E. coli as previously described (Gutierrez et al, 2013; Schuller et al, 2017). The translation elongation factors eEF2 and eEF3 were purified from S. cerevisiae as previously described (Schuller et al, 2017).
Purification of aminoacyl synthetases
Escherichia coli strain containing the phenylalanine synthetase gene (YLR060W) was purchased from Dharmacon. Plasmids containing the arginine and proline synthetase genes (YDR341C and YHR020W, respectively) were gifted from the Grayhack Lab. All plasmids were transformed into BY4741 yeast strain and grown initially in CSM –ura glucose media (Sunrise Science) and induced in –ura galactose media overnight. Harvested cells grown in small scale (500 ml) were lysed by vortexing with acid‐washed glass beads (sigma) in extraction buffer (50 mM Tris–Cl, pH 7.5, 1M NaCl, 1 mM EDTA, 4 mM MgCl2, 5 mM DTT, 10% glycerol). Larger scale preparations (2 l) were lysed by CryoMill, and lysate was flowed over 5‐ml Ni column (GE) and batch‐eluted in 5–10 ml (extraction buffer used for lysis with 5 mM BME rather than DTT). Lysates were then diluted in IPP0 buffer (10 mM Tris–Cl, pH 8, 0.1% NP‐40) and incubated for a minimum of 2 h with IgG sepharose beads (Sigma) at 4°C. Beads were spun down at low speed (2 krpm), and unbound supernatant was removed. The beads were then washed with multiple times with IPP150 buffer (10 mM Tris–Cl, pH 8, 150 mM NaCl, 0.1% NP‐40) to remove all unbound protein and washed subsequently with cleavage buffer (10 mM Tris–Cl, pH 8, 150 mM NaCl, 0.1% NP‐40, 0.5 mM EDTA, 1 mM DTT). The protein was then cleaved from the beads using 3C protease (Sigma) in cleavage buffer overnight at 4°C. Cleaved protein was removed from beads, flash‐frozen in small aliquots, and stored at ‐80°C for use.
Purification of bulk yeast tRNA
tRNA isolation protocol was derived from a protocol to isolate RNA from E. coli (Ehrenstein, 1967) with minor changes and an added LiCl precipitation to remove rRNA and mRNA. Briefly, 3L of BY4741 yeast alone or expressing a plasmid of interest was grown to an OD600 of 1 and harvested by centrifugation. Cell pellets were resuspended in 20 ml Buffer A (50 mM NaOAc, pH 7.5, 10 mM MgOAc). Phenol:chloroform extraction of RNA and DNA was performed using an equal volume of acid phenol:chloroform, pH 4.5 (VWR). rRNA and mRNA were then pelleted by LiCl precipitation, and tRNA and DNA were then ethanol‐precipitated. DNA was then removed by isopropyl alcohol precipitation. tRNA was then deacylated by incubation in 1 M Tris–Cl, pH 9, for 3 h at room temperature. Deacylated tRNA was then purified by ethanol precipitation and resuspended in water for acylation and use in in vitro assays.
Purification and charging of tRNAs
Initiator methionine and lysine tRNAs were purchased from tRNA probes (College Station, TX). Phenylalanine tRNA was purchased from Sigma. Arginine and proline tRNAs were isolated from bulk yeast tRNA using 3′ biotinylated oligonucleotides (IDT—listed below) as previously described (Yokogawa et al, 2010).
Oligo for A(I)CG‐tRNAArg: 5′ – CGC AGC CAG ACG CCG TGA CCA TTG GGC – 3′ Biotin
Oligo for UGG tRNAPro: 5′ – CCA AAG CGA GAA TCA TAC CAC TAG AC – 3′ Biotin
Leu‐2um plasmids for overexpressing native and exact match tRNAs were received from the Grayhack Lab (ECB0873 ACG‐tRNAArg, ECB0874 UCG tRNAArg). tRNA sequences were moved to pRS316 vector by Gibson cloning for lower level overexpression. The low‐copy CEN plasmids containing the tRNA sequences were transformed into the BY4741 yeast strain. Bulk tRNA was then purified by the protocol above, and the non‐native tRNA was then isolated by the same 3′ biotinylated oligonucleotide method previously (Yokogawa et al, 2010) using the specific oligonucleotides listed below (IDT):
Oligo for A(I)CG‐tRNAArg: 5′ – CGC AGC CAG ACG CCG TGA CCA TTG GGC – 3′ Biotin
Oligo for UCG tRNAArg: 5′ – CGA AGC CAG ACG CCG TGA CCA TTG GGC – 3′ Biotin
All isolated tRNAs were subjected to CCA addition as described previously (Gutierrez et al, 2013). Isolated tRNALys was charged using S100 extract; tRNAPhe, tRNAArg, and tRNAPro were charged using purified synthetases; and tRNAiMet was charged with EasyTag Methionine L‐[35S] (Perkin Elmer) and purified synthetase as previously described with minor changes (Eyler & Green, 2011). Briefly, reactions contained 1× buffer 517 (30 mM HEPES–KOH, pH 7.4, 30 mM KCl, 15 mM MgCl2), 4 mM ATP, 5 mM DTT, 10–20 μM amino acid, 3 μM CCA‐added tRNA, and a 1/5th volume of an S100 extract or 10 μM tRNA synthetase. Reactions were incubated at 30°C for 30 min and then extracted twice with acid phenol and once with chloroform. tRNA was precipitated with ethanol, resuspended in 20 mM KOAc and 2 mM DTT (pH 5.2), and stored in small aliquots at −80°C.
In vitro 80S initiation complex formation
80S initiation complexes were formed as previously described (Schuller et al, 2017) with minor differences. Briefly, 3 pmol of 35S‐Met‐tRNAiMet was mixed with 50 pmol of eIF2 and 1 mM GTP in 1× Buffer E (20 mM Tris, pH 7.5, 100 mM KOAc, pH 7.6, 2.5 mM Mg(OAc)2, 0.25 mM spermidine, and 2 mM DTT) for 10 min at 26°C. Next, a mixture containing 25 pmol 40S subunits, 200 pmol mRNA (purchased from IDT), 125 pmol eIF1, and 125 pmol eIF1A in 1× Buffer E was added for 5 min. To form the 80S complex, a mixture containing 25 pmol 60S subunits, 150 pmol eIF5, 125 pmol eIF5b, and 1 mM GTP in 1× Buffer E was added for 1 min. Complexes were then mixed 1:1 with Buffer E containing 17.5 mM Mg(OAc)2 to yield a final magnesium concentration of 10 mM. Ribosomes were then pelleted through a 600‐μl sucrose cushion containing 1.1 M sucrose in Buffer E with 10 mM Mg(OAc)2 using a MLA‐130 rotor (Beckmann) at 263,970 g for 1 h at 4°C. After pelleting, ribosomes were resuspended in 15–25 μl of 1× Buffer E containing 10 mM Mg(OAc)2 and stored at −80°C.
In vitro reconstituted translation elongation
Translation elongation reactions were performed as previously described (Eyler & Green, 2011; Schuller et al, 2017) with minor differences. Briefly, aa‐tRNA ternary complex was formed by incubating aa‐tRNA (1.5–2 μM), eEF1A (5 μM), and 1 mM GTP, in 1× Buffer E for 10 min at 26°C. Limited amounts of 80S initiation complexes (3 nM) were then mixed with aa‐tRNA ternary complex (15–25 nM for low concentrations and 150–250 nM for high concentrations), eEF2 (500 nM), eEF3 (1 μM), eIF5A (1 μM), ATP (3 mM), and GTP (2 mM). Reactions were incubated at 26°C and time points quenched into 500 mM KOH. Samples were diluted 1 into 3 μl water before monitoring peptide formation electrophoretic TLC (Millipore). TLC plates were equilibrated with pyridine acetate buffer (5 ml pyridine, 200 ml acetic acid in 1 l, pH 2.8) before electrophoresis at 1,200 V for 25 to 30 min. Plates were developed using a Typhoon FLA 9500 Phosphorimager system (GE Healthcare Life Sciences) and quantified using ImageQuantTL (GE Healthcare Life Sciences). Time courses were fit to single‐exponential kinetics using KaleidaGraph (Synergy software). Prism version 8 (GraphPad Software Inc.) was used for statistical analyses. Unpaired two‐tailed, parametric t‐tests were performed for all datasets. The resulting P‐values were reported on all graphs and rounded to two decimal places.
In vitro Met–puromycin assay
Reactions were set up as previously described (Schuller et al, 2017). Reactions were performed for each set of initiation complexes made and used to normalize peptide formation from elongation. Briefly, 2 nM initiation complexes and 1 μM eIF5A in 1× Buffer E (20 mM Tris, pH 7.5, 100 mM KOAc, pH 7.6, 2.5 mM Mg(OAc)2, 0.25 mM spermidine, and 2 mM DTT) were incubated at 26°C in the presence of 4 mM puromycin. Time points over the course of 120 min were quenched into 500 mM KOH and analyzed by electrophoretic TLC (Millipore). TLC plates were equilibrated with pyridine acetate buffer (5 ml pyridine, 200 ml acetic acid in 1 l, pH 2.8) before electrophoresis at 1,200 V for 25 min. Plates were developed using a Typhoon FLA 9500 Phosphorimager system (GE Healthcare Life Sciences) and quantified using ImageQuantTL (GE Healthcare Life Sciences).
In vitro Pth assay to access peptidyl‐tRNA drop‐off
Translation elongation reactions were performed in the presence of 27 μM peptidyl‐tRNA hydrolase (Pth) to monitor drop‐off of peptidyl‐tRNAs from translating ribosomes as described previously (Schuller et al, 2017). Time points for drop‐off products were quenched with 10% formic acid and were analyzed by electrophoretic TLC in pyridine acetate buffer (see above) at 1,200 V for 30 min.
Preparation of ribosome footprint libraries and analysis of aligned footprints
WT cells were grown to OD ~0.5 in 1 l of YPD media (sample 1) or transferred to YPGR media (2% galactose and 2% raffinose) for 6 h (sample 2) and harvested by fast filtration followed by flash‐freezing in liquid nitrogen. Cell pellets were ground with 1 ml footprint lysis buffer [20 mM Tris–Cl (pH 8.0), 140 mM KCl, 1.5 mM MgCl2, 1% Triton X‐100, 0.1 mg/ml CHX, 0.1 mg/ml TIG] in a Spex 6870 freezer mill. Lysed cell pellets were diluted to 15 ml in footprint lysis buffer and clarified by centrifugation. Polysomes were isolated from sucrose cushions for library construction as described previously (Wu et al, 2019).
3′ adapter (NNNNNNCACTCGGGCACCAAGGA) was trimmed, and 4 random nucleotides included in RT primer were removed from the 5′ end of reads (RNNNAGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC/iSP18/TTCAGACGTGTGCTCTTCCGATCTGTCCTTGGTGCCCGAGTG). Trimmed reads longer were aligned to yeast ribosomal and non‐coding RNA sequence. Unmapped reads were mapped to R64‐1‐1 S288C reference genome assembly (SacCer3) from the Saccharomyces Genome Database Project using STAR (Dobin et al, 2013) as described previously (Wu et al, 2019). Data shown in Fig 3 for WT are identical to those published previously (Wu et al, 2019). Relative ribosome occupancies for codon pairs were computed by taking the ratio of the ribosome density in a 3‐nt window at the dicodon over the density in the coding sequence (excluding the first and the last 15 nt).
Preparation of stalled ribosome‐nascent chain complexes
We generated a series of mRNA reporters containing three different stalling sequences (CGA–CCG)2, (CGA–CGA)2, and poly(A) (Appendix Figs S1A, S2A and S6A). These sequences were placed downstream of a sequence coding for TEV‐cleavable N‐terminal His‐ and HA‐tags and the first 64 amino acid residues of truncated uL4. Corresponding mRNAs were produced using the mMessage mMachine Kit (Thermo Fisher) utilizing an upstream T7 promoter and translated in a yeast cell‐free translation extract from ski2Δ cells.
This yeast translation extract was prepared, and in vitro translation was performed essentially as described before (Waters & Blobel, 1986). In brief, the cells were grown in YPD medium to OD600 of 1.5–2.0. Spheroplasts were prepared from harvested washed cells using 10 mM DTT for 15 min at room temperature and 2.08 mg zymolyase per 1 g of cell pellet for 75 min in 1 M sorbitol at 30°C. Spheroplasts were then washed and lysed in a Dounce homogenizer as described (Waters & Blobel, 1986) before using lysis buffer comprising 20 mM HEPES, pH 7.5, 100 mM KOAc, 2 mM Mg(OAc)2, 10% glycerol, 1 mM DTT, 0.5 mM PMSF, and complete EDTA‐free protease inhibitors (GE Healthcare). The S100 fraction of lysate supernatant was passed through PD10 column (GE Healthcare) and used for in vitro translation. In vitro translation was performed at 17°C for 75 min using great excess of template mRNA (38 μg per 415 μl of extract) to prevent degradation of resulting stalled ribosomes by endogenous response factors.
Respective stalled RNCs were affinity‐purified using the His6‐tag of the nascent polypeptide chain essentially as described before (Ikeuchi et al, 2019; Tesina et al, 2019). After in vitro translation, the extract was applied to Ni‐NTA Dynabeads™ (Invitrogen) and incubated while rotating for 15 min at 4°C. The beads were washed three times with excess of a wash buffer containing 50 mM HEPES/KOH (pH 7.5), 100 mM KOAc, 25 mM Mg (OAc)2, 250 mM sucrose, 0.1% Nikkol, and 5 mM ß‐mercaptoethanol and eluted in 400 μl of the same buffer containing 300 mM imidazole. The elution was applied to a 10–50% sucrose gradient in wash buffer, and ribosomal fractions were separated by centrifugation for 3 h at 172,000 g at 4°C in a SW40 rotor. For gradient fractionation, a Piston Gradient Fractionator™ (BIOCOMP) was used. The 80S (mono)ribosome (and for poly(A) also the disome) fractions were collected, applied onto 400 μl of sucrose cushion buffer, and spun at 534,000 g for 45 min at 4°C in a TLA‐110 rotor. The resulting ribosomal pellets were resuspended carefully on ice in 25 μl of grid buffer (20 mM HEPES/KOH, pH 7.2, 50 mM KOAc, 5 mM Mg(OAc)2, 125 mM sucrose, 0.05% Nikkol, 1 mM DTT, and 0.01 U/μl SUPERase‐IN™; Invitrogen).
Collected 80S fractions of CGA–CCG‐ and CGA–CGA‐stalled RNCs were also subjected to puromycin reactions with 1 mM puromycin at 20°C. Time point samples were heated for 5 min at 60°C with reducing sample buffer and analyzed by SDS–PAGE and Western blotting.
Electrophoresis and Western blotting
Protein samples of in vitro translation reactions and subsequent purifications were separated on SDS–PAGE at neutral pH condition (pH 6.8, for purified protein samples) and were transferred on PVDF membrane (Immobilon‐P; Millipore). After blocking with 5% skim milk in PBS‐T, the membranes were incubated with anti‐HA‐peroxidase antibody (1:5,000; Roche, Cat# 12013819001, clone 3F10) for 1 h at room temperature, followed by washing three times with PBS‐T. Chemiluminescence was detected using SuperSignal® substrate (Thermo Fisher) in a LAS4000 Mini (GE Healthcare).
Cryo‐EM
Freshly prepared samples (stalled monosomes or disomes) were applied to 2‐nm pre‐coated Quantifoil R3/3 holey carbon support grids and vitrified. Data were collected at Titan Krios TEM (Thermo Fisher) equipped with a Falcon II direct detector at 300 keV under low‐dose conditions of about 25e‐/Å2 for 10 frames in total and defocus range of −1.3 to −2.8 μm. Magnification settings resulted in a pixel size of 1.084 Å/pixel. In the case of CGA–CGA RNCs, a higher magnification was used, resulting in a pixel size of 0.847 Å/pixel. Original image stacks were summed and corrected for drift and beam‐induced motion at the micrograph level by using MotionCor2 (Zheng et al, 2017). The contrast transfer function (CTF) estimation and resolution range of each micrograph were performed with Gctf (Zhang, 2016).
Cryo‐EM data processing
All datasets were processed using standard procedures with programs GAUTOMATCH (http://www.mrc-lmb.cam.ac.uk/kzhang/) used for particle picking and RELION‐3 for data processing and 3D reconstruction (Zivanov et al, 2018). For each dataset, picked particles were extracted for 2D classification using a box of 400 pixels rescaled to 70 pixels. After selection of suitable 2D classes, particles were extracted for initial 3D refinement followed by 3D classification using a box of 400 pixel rescaled to 120 pixels and a mask diameter of 300 Å.
The CGA–CCG dataset was described before with focus on the Xrn1 factor bound (Tesina et al, 2019). We now re‐processed this dataset with focus on the ribosome itself. Individual translation states were separated as before with around 60% of the particles containing tRNAs in the P/P and E/E conformation (Appendix Fig S3). These classes were joined and separated into four subclasses sorting out low‐resolution particles. Further subclassification was performed using a mask covering tRNAs. This approach sorted out a population of particles without the E‐site tRNA. The cleaned population of particles was further processed using particle CTF refinement yielding a final resolution of 2.6 Å. This cryo‐EM density map was filtered according to local resolution and used for interpretation (Appendix Fig S8A).
For the CGA–CGA dataset, 840,234 particles were used after 2D classification and sorted into six classes in 3D classification. A vast majority of programmed ribosomal particles in the dataset were found in the post‐translocation state, while a single class containing tRNAs in P/P E/E conformation represented 39.9% of the whole dataset (Appendix Fig S4). As further classification of this class was mainly yielding volumes sorted based on position of expansion segment 27 on the periphery of the ribosome, the class was further processed as a whole. The final cryo‐EM density map reaching an overall resolution of 3.2 Å after particle CTF refinement was filtered according to local resolution and used for interpretation (Appendix Fig S8B).
For the poly(A) 80S dataset, 840,234 particles were used after 2D classification and sorted into six classes in 3D classification (Appendix Fig S7). Analogous to previous datasets, a vast majority of programmed ribosomal particles represented classes in the post‐translocation state. Class 3 containing tRNAs in P/P E/E conformation was subsorted based on tRNA presence into classes containing only P/P tRNA and a class containing both P/P and E/E tRNAs. The dominant classes of P/P tRNA state were joined and further processed using particle CTF refinement and Bayesian polishing. The resulting cryo‐EM density map reached an overall resolution of 3.1 Å. This volume was subjected to focused refinement using a mask covering the 60S subunit and the decoding center. This yielded a better resolved density map (3.0 Å) in the region of interest and was used for interpretation after filtering according to local resolution (Appendix Fig S8C).
Reconstruction of the poly(A) disome
The poly(A) disome dataset was collected as described above. The dataset was processed using the “80S extension” approach as described previously (Ikeuchi et al, 2019). Initial 3D classification yielded a class of ribosomes in the POST state with P/P tRNA as described above for the poly(A) monosome. Surprisingly, subsorting of this class revealed that approximately the same share of particles in this class represented the first stalling and the second colliding ribosome judging by the density of the neighboring ribosome close to the mRNA entry and exit site, respectively (Appendix Fig S9). Further processing of the leading POST state ribosome (with neighbor density at mRNA exit) yielded a standard POST–PRE hybrid disome assembly as observed for the CGA–CCG‐stalled disome (Ikeuchi et al, 2019). On the other hand, processing of the second colliding ribosome in the POST state (with neighbor density at mRNA entry) revealed a novel POST–POST disome assembly. Both these volumes were obtained by stepwise box extension and refinement with particle re‐centering (fist 500 pixels rescaled to 120 pixels, followed by 700 pixels rescaled to 506 pixels). Soft masks covering individual ribosomal bodies were used for multi‐body refinement to obtain more detailed information (Nakane et al, 2018). The resulting volumes were filtered according to local resolution (Appendix Fig S10) and fitted into the consensus refinement yielding a composite cryo‐EM density map at 3.8 Å overall resolution.
Model building
To generate molecular models for our structures, we used our previously refined models for stalled yeast ribosomes (Tesina et al, 2019, PDB ID: 6Q8Y; and Ikeuchi et al, 2019, PDB ID: 6I7O). First, individual subunits and tRNAs were fitted as rigid bodies into the densities. These models were then refined and remodeled in COOT (Brown et al, 2015) and Phenix (Adams et al, 2010). Cryo‐EM structures and models were displayed with UCSF Chimera (Pettersen et al, 2004) and ChimeraX (Goddard et al, 2018). Detailed statistics of model refinements and validations are listed in Appendix Table S1.
Author contributions
RG, RBe, TB, and ARB conceived the study and designed experiments. LNL performed yeast in vitro kinetic experiments and analyzed the corresponding data. CC‐CW performed experiments and data analysis for ribosome profiling. PT prepared cryo‐EM samples, performed cryo‐EM experiments, and processed the data. OB collected the data. RBu built atomic models, and JC, PT, and TB refined them. PT, LNL, ARB, TB, RBe, and RG wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
The authors would like to thank Elisabeth Heckel for help with sample preparation and Charlotte Ungewickell and Susanne Rieder for technical support. The authors would also like to thank members of the Green Lab for helpful discussions, Elizabeth Grayhack for the tRNA and synthetase plasmids, and David Mohr and the Johns Hopkins Genetic Resources Core Facility for sequencing assistance. This study was supported by a grant of the Deutsche Forschungsgemeinschaft (DFG) (DFG; BE1814/15‐1) to RBe and a Ph.D. fellowship from Boehringer Ingelheim Fonds (BIF) to RBu. Additionally, this work was supported by the NIH (2R37GM059425 to RG), T32 GM008403, and HHMI (RG and CW).
The EMBO Journal (2020) 39: e103365
Contributor Information
Thomas Becker, Email: becker@genzentrum.lmu.de.
Roland Beckmann, Email: beckmann@genzentrum.lmu.de.
Rachel Green, Email: ragreen@jhmi.edu.
Data availability
The cryo‐EM structures reported here have been deposited in the Protein Data Bank and in the Electron Microscopy Data Bank under accession codes 6T4Q and EMD‐10377 for the CGA–CCG‐stalled ribosome, 6T7I and EMD‐10396 for the CGA–CGA‐stalled ribosome, 6T7T and EMD‐10397 for the poly(A)‐stalled ribosome, and 6T83 and EMD‐10398 for the poly(A)‐stalled collided disome. Ribosome profiling datasets have been published previously and deposited under GSE115162 (Wu et al, 2019) https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115162
References
- Acker MG, Kolitz SE, Mitchell SF, Nanda JS, Lorsch JR (2007) Reconstitution of yeast translation initiation. Methods Enzymol 430: 111–145 [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse‐Kunstleve RW et al (2010) PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66: 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agirrezabala X, Samatova E, Klimova M, Zamora M, Gil‐Carton D, Rodnina MV, Valle M (2017) Ribosome rearrangements at the onset of translational bypassing. Sci Adv 3: e1700147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur L, Pavlovic‐Djuranovic S, Smith‐Koutmou K, Green R, Szczesny P, Djuranovic S (2015) Translational control by lysine‐encoding A‐rich sequences. Sci Adv 1: e1500154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur LL, Djuranovic S (2018) PolyA tracks, polybasic peptides, poly‐translational hurdles. WIREs RNA 9: e1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranov PV, Gesteland RF, Atkins JF (2004) P‐site tRNA is a crucial initiator of ribosomal frameshifting. RNA 10: 221–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandman O, Hegde RS (2016) Ribosome‐associated protein quality control. Nat Struct Mol Biol 23: 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Long F, Nicholls RA, Toots J, Emsley P, Murshudov G (2015) Tools for macromolecular model building and refinement into electron cryo‐microscopy reconstructions. Acta Crystallogr D Biol Crystallogr 71: 136–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brule CE, Grayhack EJ (2017) Synonymous codons: choose wisely for expression. Trends Genet 33: 283–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, Schwalbe H, Rodnina MV, Komar AA (2016) Synonymous codons direct cotranslational folding toward different protein conformations. Mol Cell 61: 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess‐Brown NA, Sharma S, Sobott F, Loenarz C, Oppermann U, Gileadi O (2008) Codon optimization can improve expression of human genes in Escherichia coli: a multi‐gene study. Protein Expr Purif 59: 94–102 [DOI] [PubMed] [Google Scholar]
- Buschauer R, Matsuo Y, Chen Y‐H, Alhusaini N, Sweet T, Sugiyama T, Ikeuchi K, Cheng J, Matsuki Y, Gilmozzi A et al (2019) The Ccr4‐Not complex monitors the translating ribosome for codon optimality. bioRxiv 10.1101/854810 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana A, Tuller T (2014) The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res 42: 9171–9181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinman JD (2012) Mechanisms and implications of programmed translational frameshifting. Wiley Interdiscip Rev RNA 3: 661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenstein G (1967) [76] Isolation of sRNA from intact Escherichia coli cells. Methods Enzymol 12: 588–596 [Google Scholar]
- Elf J, Nilsson D, Tenson T, Ehrenberg M (2003) Selective charging of tRNA isoacceptors explains patterns of codon usage. Science 300: 1718–1722 [DOI] [PubMed] [Google Scholar]
- Eyler DE, Green R (2011) Distinct response of yeast ribosomes to a miscoding event during translation. RNA 17: 925–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov A, Saxonov S, Gilbert W (2002) Regularities of context‐dependent codon bias in eukaryotic genes. Nucleic Acids Res 30: 1192–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischmeyer PA, van Hoof A, O'Donnell K, Guerrerio AL, Parker R, Dietz HC (2002) An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295: 2258–2261 [DOI] [PubMed] [Google Scholar]
- Gamble CE, Brule CE, Dean KM, Fields S, Grayhack EJ (2016) Adjacent codons act in concert to modulate translation efficiency in yeast. Cell 166: 679–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber AP, Keller W (1999) An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science 286: 1146–1149 [DOI] [PubMed] [Google Scholar]
- Gingold H, Pilpel Y (2011) Determinants of translation efficiency and accuracy. Mol Syst Biol 7: 481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, Ferrin TE (2018) UCSF ChimeraX: meeting modern challenges in visualization and analysis. Protein Sci 27: 14–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromadski KB, Rodnina MV (2004) Kinetic determinants of high‐fidelity tRNA discrimination on the ribosome. Mol Cell 13: 191–200 [DOI] [PubMed] [Google Scholar]
- Gutierrez E, Shin B‐SS, Woolstenhulme CJ, Kim J‐RR, Saini P, Buskirk AR, Dever TE (2013) eIF5A promotes translation of polyproline motifs. Mol Cell 51: 35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi K, Tesina P, Matsuo Y, Sugiyama T, Cheng J, Saeki Y, Tanaka K, Becker T, Beckmann R, Inada T (2019) Collided ribosomes form a unique structural interface to induce Hel2‐driven quality control pathways. EMBO J [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joazeiro CAP (2019) Mechanisms and functions of ribosome‐associated protein quality control. Nat Rev Mol Cell Biol 20: 368–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juszkiewicz S, Chandrasekaran V, Lin Z, Kraatz S, Ramakrishnan V, Hegde RS (2018) ZNF598 is a quality control sensor of collided ribosomes. Mol Cell 72: 469–515032704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keedy HE, Thomas EN, Zaher HS (2018) Decoding on the ribosome depends on the structure of the mRNA phosphodiester backbone. Proc Natl Acad Sci USA 115: E6731–E6740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr AG, Schmidt C, Tesina P, Berninghausen O, Becker T, Beatrix B, Beckmann R (2019) Ribosome‐NatA architecture reveals that rRNA expansion segments coordinate N‐terminal acetylation. Nat Struct Mol Biol 26: 35–39 [DOI] [PubMed] [Google Scholar]
- Koutmou KS, Schuller AP, Brunelle JL, Radhakrishnan A, Djuranovic S, Green R (2015) Ribosomes slide on lysine‐encoding homopolymeric A stretches. Elife 4: e05534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzring DP, Dean KM, Grayhack EJ (2010) Control of translation efficiency in yeast by codon‐anticodon interactions. RNA 16: 2516–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzring DP, Wolf AS, Brule CE, Grayhack EJ (2013) Translation of CGA codon repeats in yeast involves quality control components and ribosomal protein L1. RNA 19: 1208–1217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Deutsch C (2008) Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol 384: 73–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt C, Udagawa T, Sato F, Tsuchiya H, Becker T, Tanaka K et al (2017) Ubiquitination of stalled ribosome triggers ribosome‐associated quality control. Nat Commun 8: 159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy FVT, Ramakrishnan V (2004) Structure of a purine‐purine wobble base pair in the decoding center of the ribosome. Nat Struct Mol Biol 11: 1251–1252 [DOI] [PubMed] [Google Scholar]
- Nakane T, Kimanius D, Lindahl E, Scheres SH (2018) Characterisation of molecular motions in cryo‐EM single‐particle data by multi‐body refinement in RELION. Elife 7: e36861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, Milos PM (2010) Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143: 1018–1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechmann S, Frydman J (2013) Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat Struct Mol Biol 20: 237–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR et al (2015) Codon optimality is a major determinant of mRNA stability. Cell 160: 1111–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quax TE, Claassens NJ, Söll D, van der Oost J (2015) Codon bias as a means to fine‐tune gene expression. Mol Cell 59: 149–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis M, Savva R, Wernisch L (2004) Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32: 5036–5044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Khusainov I, El Omari K, Duman R, Mykhaylyk V, Yusupov M, Westhof E, Wagner A, Yusupova G (2019) Importance of potassium ions for ribosome structure and function revealed by long‐wavelength X‐ray diffraction. Nat Commun 10: 2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander IM, Chaney JL, Clark PL (2014) Expanding Anfinsen's principle: contributions of synonymous codon selection to rational protein design. J Am Chem Soc 136: 858–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt C, Kowalinski E, Shanmuganathan V, Defenouillere Q, Braunger K, Heuer A, Pech M, Namane A, Berninghausen O, Fromont‐Racine M et al (2016) The cryo‐EM structure of a ribosome‐Ski2‐Ski3‐Ski8 helicase complex. Science 354: 1431–1433 [DOI] [PubMed] [Google Scholar]
- Schuller AP, Wu CC, Dever TE, Buskirk AR, Green R (2017) eIF5A functions globally in translation elongation and termination. Mol Cell 66: 194–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmer M, Dunham CM, Murphy F, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V (2006) Structure of the 70S ribosome complexed with mRNA and tRNA. Science 313: 1935–1942 [DOI] [PubMed] [Google Scholar]
- Sharp PM, Li WH (1987) The codon Adaptation Index–a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res 15: 1281–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Eyler DE, Green R (2010) Dom34:Hbs1 promotes subunit dissociation and peptidyl‐tRNA Drop‐Off to initiate No‐Go decay. Science 330: 369–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms CL, Yan LL, Zaher HS (2017) Ribosome collision is critical for quality control during no‐go decay. Mol Cell 68: 361–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms CL, Yan LL, Qiu JK, Zaher HS (2019) Ribosome collisions result in +1 frameshifting in the absence of No‐Go decay. Cell Rep 28: 1679–1689 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Costello MS, Kettring AH, Wingo RJ, Moore SD (2019) Ribosome collisions alter frameshifting at translational reprogramming motifs in bacterial mRNAs. Proc Natl Acad Sci USA 116: 21769–21779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang TTL, Stowell JAW, Hill CH, Passmore LA (2019) The intrinsic structure of poly(A) RNA determines the specificity of Pan2 and Caf1 deadenylases. Nat Struct Mol Biol 26: 433–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesina P, Heckel E, Cheng J, Fromont‐Racine M, Buschauer R, Kater L, Beatrix B, Berninghausen O, Jacquier A, Becker T et al (2019) Structure of the 80S ribosome‐Xrn1 nuclease complex. Nat Struct Mol Biol 26: 275–280 [DOI] [PubMed] [Google Scholar]
- Tuller T, Waldman YY, Kupiec M, Ruppin E (2010) Translation efficiency is determined by both codon bias and folding energy. Proc Natl Acad Sci USA 107: 3645–3650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters MG, Blobel G (1986) Secretory protein translocation in a yeast cell‐free system can occur posttranslationally and requires ATP hydrolysis. J Cell Biol 102: 1543–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC, Zinshteyn B, Wehner KA, Green R (2019) High‐resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress. Mol Cell 73: 959–970 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarus M, Folley LS (1985) Sense codons are found in specific contexts. J Mol Biol 182: 529–540 [DOI] [PubMed] [Google Scholar]
- Yokogawa T, Kitamura Y, Nakamura D, Ohno S, Nishikawa K (2010) Optimization of the hybridization‐based method for purification of thermostable tRNAs in the presence of tetraalkylammonium salts. Nucleic Acids Res 38: e89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaher HS, Green R (2009) Fidelity at the molecular level: lessons from protein synthesis. Cell 136: 746–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K (2016) Gctf: real‐time CTF determination and correction. J Struct Biol 193: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, Agard DA (2017) MotionCor2: anisotropic correction of beam‐induced motion for improved cryo‐electron microscopy. Nat Methods 14: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH (2018) New tools for automated high‐resolution cryo‐EM structure determination in RELION‐3. Elife 7: e42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The cryo‐EM structures reported here have been deposited in the Protein Data Bank and in the Electron Microscopy Data Bank under accession codes 6T4Q and EMD‐10377 for the CGA–CCG‐stalled ribosome, 6T7I and EMD‐10396 for the CGA–CGA‐stalled ribosome, 6T7T and EMD‐10397 for the poly(A)‐stalled ribosome, and 6T83 and EMD‐10398 for the poly(A)‐stalled collided disome. Ribosome profiling datasets have been published previously and deposited under GSE115162 (Wu et al, 2019) https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115162