

Abstract
The aim of this review is to highlight the rich chemistry of α‐haloamides originally mainly used to discover new C−N, C−O and C−S bond forming reactions, and later widely employed in C−C cross‐coupling reactions with C(sp3), C(sp2) and C(sp) coupling partners. Radical‐mediated transformations of α‐haloamides bearing a suitable located unsaturated bond has proven to be a straightforward alternative to access diverse cyclic compounds by means of either radical initiators, transition metal redox catalysis or visible light photoredox catalysis. On the other hand, cycloadditions with α‐halohydroxamate‐based azaoxyallyl cations have garnered significant attention. Moreover, in view of the important role in life and materials science of difluoroalkylated compounds, a wide range of catalysts has been developed for the efficient incorporation of difluoroacetamido moieties into activated as well as unactivated substrates.
Keywords: amides, electrochemistry, photocatalysis, radical reactions, reaction mechanisms
What can α‐haloamides do? The α‐haloamide functional group is an efficient platform for a lot of transformations. Its framework, surprisingly rich of chemically “touchable” atoms, has been used for α‐aminations, radical cyclizations to β‐ and γ‐lactams, intra‐ and intermolecular transition metal catalyzed α‐alkylations, α‐alkenylations, α‐alkynylations, α‐arylations, as well as a three‐atom unit for heterocyclic formation.
1. Introduction
α‐Halocarbonyl compounds are useful synthetic intermediates in a number of widely different organic transformations. Among them, α‐haloamides have been attracting increasing attention in view of their high reactivity as building blocks for the preparation of a variety of intermediates for the synthesis of biologically active molecules and pharmaceuticals. These compounds are used as starting materials for the preparation of a myriad of compounds including α‐,1 β‐ and γ‐lactams,2 dioxopiperazines,3a–3b 2,5‐dioxopyrrolidines,4 oxazolidinones,5a–5f thiazolidin‐4‐ones,5b tetramic acids,6a–6b glycoside amides,7 and peptidomimetics.8a–8g
Thus, halogen substitution by a variety of nitrogen and oxygen nucleophiles allows the synthesis of aminoamides and alkoxyamides respectively, the site‐selective substitution of the bromine with O‐nucleophiles being promoted by silver oxide.9 Notably, the C−X bond polarization can be inverted by electroreduction generating enolates useful for the formation of a new C−C bond,10 while α‐halo‐7‐azaindoline amides behaving as halogen‐bearing enolate precursors take part to catalytic asymmetric Mannich‐Type reaction affording compounds with halogens on a stereogenic carbon.11
A variety of α‐chloro amides bearing a suitable located double bond has been utilized as convenient substrate for atom transfer radical cyclization.12a–12b The formation of cyclic systems by carbon‐carbon bond formation by the use of free radical cyclization protocols, originally mediated by organostannane or organosilane reagents (e. g. Bu3SnH or HSi(SiMe3)3), has been later more conveniently performed using copper complexes13 or visible light photoredox catalysis.14 Moreover, the development of transition metal‐catalyzed cross‐coupling reactions as C−C bond forming reactions between an organic electrophile and an organometallic reagents further expanded the synthetic usefulness of α‐haloamides. Notably, the potential of using transition metal‐catalyzed C−C bond formation to prepare enantioenriched molecules was immediately recognized by the synthetic chemistry community and α‐haloamides have been widely employed as organic electrophiles. In 2005, Fisher and Fu15 described the Ni‐catalyzed Negishi cross‐coupling of racemic α‐bromo amides with organozinc reagents in the presence of the chiral ligand (i‐Pr)‐Pybox as an exceptionally useful method to obtain enantiopure α‐chiral amides.
At present, information about the synthesis and the chemistry of α‐halogenated amides are just scattered throughout literature, if one excludes a minireview article published by Comesse et al.16 while this manuscript was under peer review process.
The aim of this survey is to focus new attention on the broad potential of the α‐haloamides as versatile synthons in organic synthesis; the number of papers dealing with the chemistry of α‐haloamides has been partitioned within different Sections giving the rightful emphasis to the reaction mechanisms.
A brief introduction is in Section 1.; methods for their preparations in racemic as well as in enantio‐enriched forms have been summarized in Section 2. The collected papers showcase a multivalent nature for the alpha‐carbon (αC), de facto α‐haloamides can act as suitable electrophiles for carbon‐ or heteroatom‐centered nucleophiles in reactions proceeding with displacement of the α‐halogen atom. Alternatively, the latter can be abstracted from the αC under reductive conditions, the single electron transfer process giving the corresponding carbon‐centered radical as the highly reactive species. On the other hand, the αC can turn to a nucleophile center both by removal of a proton under basic conditions or as a result of a two‐electron transfer process coming with removal of the α‐halogen. That is why most of chemical reactions occurs at this crucial site of the α‐haloamide substrates.
Our choice has been grouping articles according to the nature of the bond generated by the various reaction. Thus, Section 3. is dedicated to base‐promoted conversions of α‐haloamides into aziridinones, unusual bidentate electrophiles. Processes for α‐aminations are collected in Section 4., whereas processes for αC−C bond formation found accommodation in Sections 5–10, substantially ordered according to both the hybridization of the carbon atom that results connected to αC and the nature of processes involved in C−C bond construction. Collected in Section 11. is the copious number of papers dealing with reactions occurring simultaneously at the αC and N/O atoms where α‐haloamides act as three‐atom units for concise synthesis of heterocycles. Eventually, Section 12. is dedicated to synthetic processes not properly suited for inclusion in previous Sections.
For the sake of clarity the caption of each Scheme includes both the reference number and the Section to which the paper refers. In the text, as well as in the Schemes, bold capital letters denote reagents and products, reacting α‐haloamides being A. Synthetic intermediates and pivotal species in catalytic cycles are designated by Roman numerals.
2. Preparations
2.1. Preparation of Achiral and Chiral Racemic α‐halo Amides
The classical approach to the synthesis of α‐halo amides involves substitution at the carbonyl group of an α‐haloacetyl halide (acyl halide or ester) with a nucleophilic amine (Scheme 1). Such processes suffer the low reactivity displayed by sterically hindered or electron‐poor amines. The reaction of aniline derivatives and α‐bromoacetyl bromide in the presence of Et3N afforded the desired products in moderate to good yields (80‐90 %) however, NaH was required for reacting the 2,4,6‐tri‐tert‐butylaniline, the expected amide being formed in 55 % yield.17a–17c
Scheme 1.
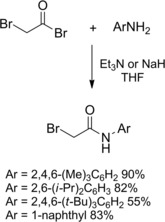
Preparation of N‐aryl‐2‐bromoacetamides (ref. [17a–c] – Section 2.1).
Recently,18 the 2‐bromo‐2‐methyl‐N‐o‐tolylpropanamide was prepared in good yield reacting o‐toluidine with 2‐bromo‐2‐methylpropanoyl bromide in boiling dioxane in the presence of K2CO3 (Scheme 2).
Scheme 2.
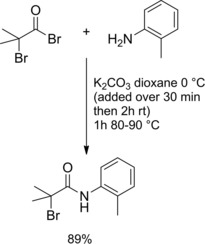
Preparation of 2‐bromo‐2‐methyl‐N‐o‐tolylpropanamide (ref. [18] – Section 2.1).
A variety of functionalized α‐chloroamides has been obtained by reaction between chloroacetyl chloride and mono‐ or bis‐aliphatic or aromatic amines in water under basic or neutral conditions.19
Interestingly, chloroacetyl chlorides reacted with N‐benzylidene imines in the presence of stoichiometric triethylamine to give high yields of 3‐chloro‐β‐lactams (Scheme 3)20 which were promptly dehalogenated by electroreduction in the presence of proton donors. Alternatively, electroreduction in the presence of electrophiles such as Ac2O or CO2 gave functionally 3‐substituted β‐lactams.21a–21b
Scheme 3.
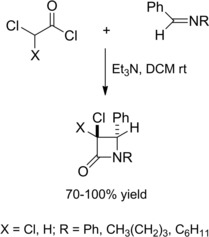
Preparation of 3‐chloro‐β‐lactams (ref. [20] – Section 2.1).
α‐Bromo amides can also be synthesized from the corresponding α‐bromo acid using N,N’‐diisopropylcarbodiimide (DIC) for the coupling with amines (Scheme 4.)22
Scheme 4.
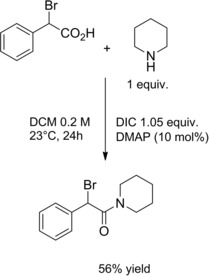
Amidation of α‐bromo acid using DIC (ref. [22] – Sction 2.1).
The intrinsic reactivity of ethyl chloroacetate could be altered in the presence of 2 mol% of La(OTf)3, the reaction with benzylamine providing the expected α‐chloroacetamide in 90 % yield (Scheme 5).23
Scheme 5.
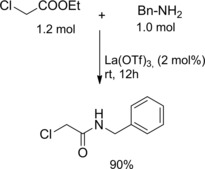
Amidation of ethyl chloroacetate using La(OTf)3 (ref. [23] – Section 2.1).
Alternatively, α‐halo amides are formed in good to moderate yields in a one‐pot multistep reaction between a variety of α,α‐dicyano epoxides (monosubstituted alkyl, aryl; disubstituted alkyl‐aryl) and amines (primary alkyl, aryl, amino esters, amino amides) in the presence of hydrohalides (Scheme 6).24
Scheme 6.
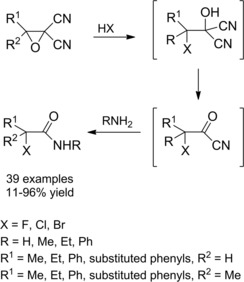
Preparation of α‐haloamides from dicyano epoxides (ref. [24] – Section 2.1).
In 2007, Vora and Rovis25a–25b reported the amidation of aldehydes with an α‐reducible centre using catalytic amounts (20 mol%) of N‐heterocyclic carbenes (NHCs) in conjunction with 1‐hydroxy‐7‐azabenzotriazole (HOAt) as the co‐catalyst (20 mol%) (Scheme 7).
Scheme 7.
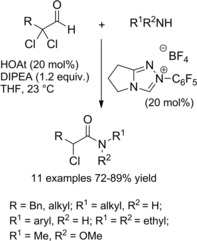
Preparation of α‐chloroamides using the NHC/HOAt system (ref. [25a] – Section 2.1).
In fact, by utilizing 2,2‐dichloro‐3‐phenyl propanal as the redox substrate, a variety of secondary and tertiary α‐chloro amides could be prepared in good yields.
It was hypothesized that the nucleophilic carbene (I) adds to the aldehyde generating the acyl azolium intermediate (II) which transferring the acyl moiety to HOAt (III) gives the activated carboxylate intermediate (IV). The latter, following nucleophilic attack by the amine provides the amide and regenerates the co‐catalyst (III) (Scheme 8).
Scheme 8.
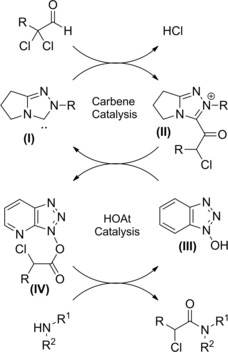
Mechanistic hypothesis for the NHC/HOAt‐promoted amidation of aldehydes with an α‐reducible centre (ref. [25a‐b] – Section 2.1).
β‐Carbonyl‐α‐bromoamides could be prepared in quantitative yields by chemoselective monobromination of the corresponding active methylene compounds using potassium bromide, hydrochloric acid and 30 % hydrogen peroxide in toluene (Scheme 9).26
Scheme 9.
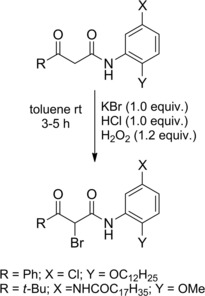
Preparation of α‐bromoamides via monobromination of β‐ketoamides (ref. [26] – Section 2.1).
The introduction of fluorine atoms at the α‐position of amides deserves special attention because it may lead to unprecedented or modified biological properties of the target molecule. Noel and co‐workers first exploited organic electrochemistry as an environmentally friendly method for achieving the α‐fluorination of N‐alkyl and N,N‐dialkyl phenylacetamides.27 Specifically, it was observed that under potentiostatic conditions the electrolysis led to predominantly side chain fluorination at the active methylene group, while nuclear fluorination was predominant under galvanostatic conditions (Scheme 10).
Scheme 10.
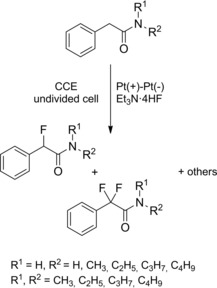
Anodic fluorination of phenylacetamides under CCE conditions (ref. [27] – Section 2.1).
It was also observed that the fluorination of N,N‐dialkyl phenylacetamides produced variety of products when compared to N‐alkyl phenylacetamides. Importantly, the solvent‐free anodic fluorination proceeded smoothly using Et3N ⋅ 4HF as the fluorinating ionic liquid medium in an undivided cell equipped with two platinum electrodes under current constant electrolysis (CCE) conditions.
Pace et al. introduced an efficient strategy to prepare a diverse array of α‐halo and α,α‐dihaloacetamides based on the reaction of widely available isocyanates with monohalolithium and dihalolithium carbenoids (Scheme 11).28a–28b
Scheme 11.
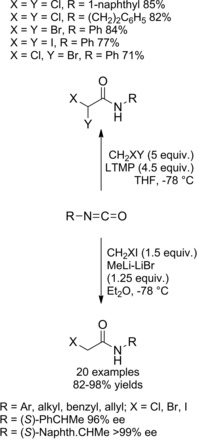
Preparation of α‐halo and α,α‐dihaloacetamides via homologation of isocyanates with lithium carbenoids (ref. [28a–b] – Section 2.1).
While monohalolithium carbenoids could be prepared by a smooth lithium‐iodine exchange, the preparation of the corresponding dihalo compounds proved to be highly dependent on the base used to realize the deprotonation, with lithium 2,2,6,6‐tetramethylpiperidine (LTMP) emerging as optimal. Notably, chloro‐, bromo‐ and iodomethyllithiums reacted equally to give the corresponding α‐haloamides in high yields. Moreover, halo‐acetamides with chiral frameworks on the amide nitrogen atom could be easily achieved using chiral isocyanates as starting materials.28b
A diazoacetate bearing an amide group acted as an effective acceptor‐acceptor substrate in a chloroarylation reaction with phenyl boronicacid and N‐chlorosuccinimide (NCS) (Scheme 12).29
Scheme 12.
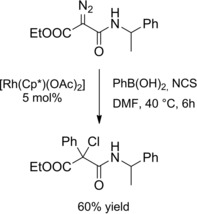
Synthesis of an α‐chloroamide by Rh(III)‐catalyzed cascade difunctionalization of an α‐diazocarbonyl compound (ref. [29] – Section 2.1).
The arylation/chlorination cascade reaction occurred in the presence of a [RhIII(Cp*)]‐catalyst. Principal steps in the catalytic cycle should be the formation of an arylrhodium(III) complex that evolves to an arylrhodium‐carbene complex by reaction with the diazocarbonyl compound. Then, a migratory insertion reaction should produce a rhodium(III)‐diketonate which coordinates NCS. Eventually, nucleophilic displacemnent of the N−Cl group by the diketonate ligand affords the α‐chloroamide with a quaternary stereocenter.
Ts‐ or Ms‐protected ynamides underwent Lewis acid‐catalyzed transformation into α‐halo amides through an alkyne oxidation‐halogenation tandem process with 2‐halopyridine N‐oxide (2 equiv.) serving both as the oxidant and the halogen source (Scheme 13).30a–30b
Scheme 13.
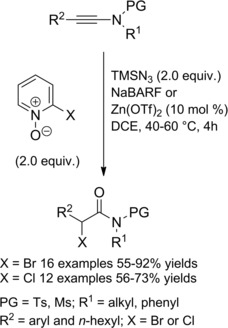
Preparation of α‐haloamides via ynamides oxidation‐halogenation tandem process (ref. [30a‐b] – Section 2.1).
The reaction occurred in few hours (DCE at 40–60 °C) in the presence of TMSN3 (2 equiv.) and the Lewis acid (10 mol%). Either Zn(OTf)2 or NaBARF (sodium tetrakis[3,5‐bis(trifluoromethyl)phenyl]borate) could be effective catalysts for activating the ynamides substrates to the nucleophilic attack of 2‐halopyridine N‐oxides, the ancillary event in the proposed reaction mechanism.
Thus, various aryl‐ substituted ynamides were converted in good to excellent yields both to α‐bromo and α‐chloro amides by employing 2‐bromo‐ and 2‐chloropyridine N‐oxide respectively. Moreover, albeit less efficiently, the protocol could also be applied to an alkyl‐substituted ynamide.
2.2. Preparation of Chiral Non‐Racemic α‐halo Amides
From chiral sources: The acyl nucleophilic substitution reaction between methyl (S)‐(‐)‐2‐chloropropionate and diethylamine performed in the presence of a Lewis acid (AlCl3 or ZrCl4) allowed the preparation of the corresponding (S)‐(+)‐N,N‐diethyl‐2‐chloropropionamide in high chemical (77 %) and optical (99 %) yield (Scheme 14).31
Scheme 14.
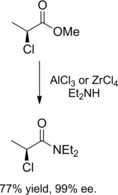
Methyl 2‐(S)‐chloropropionate as the chiral source (ref. [31] – Section 2.2).
2‐(S)‐Bromoamides could be prepared by amidation reaction of the corresponding 2‐(S)‐bromoacyl chloride in turn easily prepared from natural amino acids (α‐A.A) through diazotization‐halogenation and SOCl2‐mediated carboxy‐activation (Scheme 15).32 As the one‐pot two step process replacing the primary amino group for bromine atom occurred through vicinal carboxyl group assistance, the resulting 2‐bromoacids as well as the corresponding amides were produced with retention of configuration.
Scheme 15.
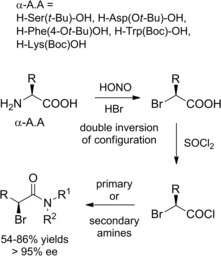
Natural amino acids as the chiral sources (ref. [32] – Section 2.2).
Using chiral auxiliaries: In 1995 Ward and co‐workers33 found out that diastereomeric α‐bromoamides derived from the Oppolzer's (−)‐camphorsultam34 underwent epimerization when treated with KBr (20 equiv.) in polar aprotic solvents (Scheme 16). De facto, the diastereomeric excess (de) increased from 0 to 70 % after 1 hr in refluxing MeCN (or DMSO at 60 °C). Specifically, the (R)‐isomers with the bromine located on the less hindered face were the preferred diastereomers.
Scheme 16.
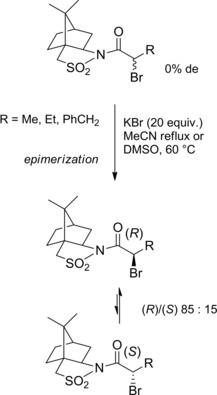
Bromide anion promoted epimerization of α‐bromoamides incorporating the Oppolzer's camphorsultam chiral auxiliary (ref. [33] – Section 2.2).
Interestingly, the diastereomeric population could also be controlled by favoring the conversion of the more soluble diastereomer to the less soluble one (Scheme 17). Caddick and Jenkins35 reported the crystallization‐induced dynamic resolution (CIDR) of α‐bromoacyl imidazolidinone where tetrabutylammoniumbromide (TBAB) was involved in the halogenated stereogenic center epimerization.
Scheme 17.
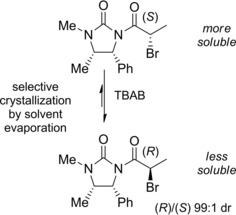
CIDR of the α‐bromopropanamide incorporating an imidazolidinone chiral auxiliary under TBAB epimerization conditions (ref. [35] – Section 2.2).
Park and co‐workers36 discovered that (S)‐1‐phenylethylamine was an effective chiral auxiliary for the higly stereoselective CIDR of α‐chloro‐α‐aryl acetamides (Scheme 18).
Scheme 18.
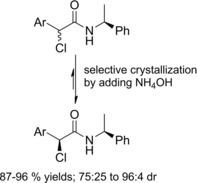
CIDR of α‐chloro‐α‐aryl acetamides incorporating the (S)‐1‐phenylethylamine chiral auxiliary under base‐catalyzed epimerization conditions (ref. [36] – Section 2.2).
Thus, the everyday (for 7 days) addition of NH4OH to the (αRS)‐diastereomeric mixtures in THF led the (αS)‐diastereomers to selectively crystallize. Notably, the chlorinated (S)‐stereogenic center was configurationally stable in the absence of base.
Using enzymes as chiral catalysts: Optically enriched α‐haloamides with concomitant enrichment of the starting α‐haloesters could be efficiently obtained using lipases as catalysts, Candida Antartica lipase (CAL) in anhydrous diisopropyl ether (DIPE) being the best choice for this approach (Scheme 19).37
Scheme 19.
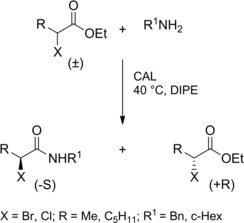
Preparation of enantio‐enriched α‐haloamides using CAL (ref. [37] – Section 2.2).
Haloalkane dehalogenases from five sources were heterologously expressed in Escherichia coli, isolated, and tested for their ability to achieve kinetic resolution of fourteen racemic α‐bromoamides with different α‐C and N substituents.38 Seven of these compounds could be converted with a high enantioselectivity (E value>200). In all cases, the (R)‐α‐bromoamides were the preferred substrates in the nucleophilic substitution reaction providing the (S)‐α‐hydroxyamides (Scheme 20). Conversions on a preparative scale with a catalytic amount of enzyme (enzyme:substrate ratio less 1 : 50 w/w) were all completed within 17–46 h and optically pure α‐bromoamides and α‐hydroxyamides were isolated with good yields.
Scheme 20.
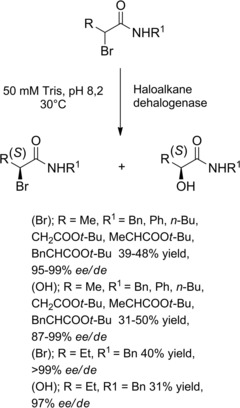
Preparation of enantio‐enriched α‐bromoamides using haloalkane dehalogenase (ref. [38] – Section 2.2).
Interestingly, by combining the haloalkane dehalogenase‐mediated enantioselective kinetic resolution with the bromide anion‐promoted epimerization of the halogenated stereogenic center, Janssen and co‐workers39 established a process for a dynamic kinetic resolution (DKR) of racemic N‐phenyl‐2‐bromopropionamide (Scheme 21). A polymer‐based phosphonium bromide in the form of a finely powdered water‐insoluble solid was entrusted with the rapid (αS) to (αR) conversion of the α‐bromoamide while the haloalkane dehalogenase catalyzed the enantioselective bromine substitution. In the overall the DKR provided the N‐phenyl‐2‐(S)‐hydroxypropionamide in 63 % yield and 95 % ee (or 78 % yield and 88 % ee).
Scheme 21.
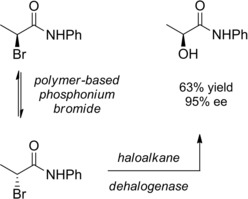
DKR of the N‐phenyl‐α‐bromopropionamide (ref. [39] – Section 2.2).
Using non‐ezymatic chiral catalysts: Vora and Rovis in their NHC‐redox amidation of α‐reducible aldehydes (Scheme 7, Section 2.1.)25a–25b reported a single asymmetric reaction. Precisely, the N‐benzyl‐2(R)‐chloro‐3‐phenyl propanamide was prepared reacting 2,2‐dichloro‐3‐phenyl propanal with benzylamine by using the chiral triazolium (I) as the NHC‐precatalyst (Scheme 22). Later on, they found that α‐fluoro‐α,β‐unsaturated aldehydes were a lot more gratifying substrates (Scheme 23).40 In fact, the NHC‐redox amidation protocol worked well in the conversion of α‐fluoroenals into enantioenriched α‐fluoroamides. The optimized conditions entailed reacting α‐fluoroenals with amine hydrochloride salts in toluene using the organocatlyst system composed of the chiral triazolium precatalyst (I) and the HOAt co‐catalyst. Excellent yields and selectivities were achieved provided that sodium pivalate (NaOPv) was the base; the presence of 4 A° molecular sieves was also important in order to preclude hydration.
Scheme 22.
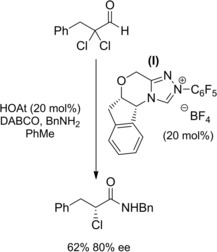
Preparation of the enantio‐enriched N‐benzyl‐2‐chloro‐3‐phenyl propanamide using the chiral NHC‐precursor (I) in the redox amidation (ref. [25a–b] – Section 2.2).
Scheme 23.
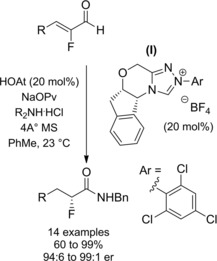
Preparation of enantio‐enriched α‐fluoroamides using the chiral NHC‐precursor (I) in the redox amidation (ref. [40] – Section 2.2).
The method showed to tolerate functional groups such as esters, silyl ethers, and carbamates providing a variety of secondary and tertiary α‐fluorinated amides including Weinreb amide derivatives.
In 2015 Sun and co‐workers41 reported the N‐fluorobenzenesulfonimide (NFSI) mediated asymmetric α‐fluorination of azolium enolates resulting from the addition of the Bode NHC catalyst onto the carbonyl group of α‐chloro aldehydes (Scheme 24). Indeed, various nucleophiles (NuH) including amines participated to the reaction with azolium enolates and NFSI. A suitable combination of precatalyst, oxidant, base, fluorination reagent and the additive pyrazole (as an acyl transfer reagent), converted racemic 4‐phenyl‐2‐chloro butanal and amines (aniline and benzyl amine) into the corresponding highly enantioenriched α‐fluoro amides. Notably, simple aliphatic aldehydes were suitable substrates too, with NFSI acting both as the oxidant and the fluorine source (bottom of Scheme 24).
Scheme 24.
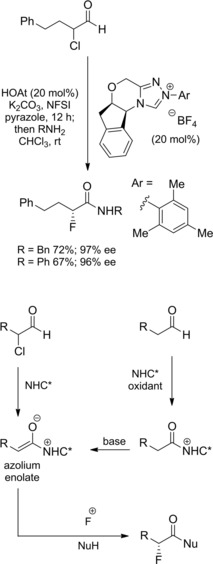
NFSI‐mediated asymmetric α‐fluorination of azolium enolates derived from the Bode NHC catalyst (ref. [41] – Section 2.2).
3. Aziridinones Ring‐Forming/Ring‐Opening Processes
Interest in α‐lactams dates back to 1962 when Baumgarten42a–42b first isolated and characterized the 1‐t‐butyl‐3‐phenylaziridinone B resulting from the base (t‐BuOK) mediated dehydrochlorination of N‐chloro‐N‐t‐butyl phenylacetamide A (Scheme 25). The physical data, together with the dual mode of ring cleavage following attack with O‐nuclephiles, supported the α‐lactam structure even though the alternative structures (valence tautomers) could not be ruled out.
Scheme 25.
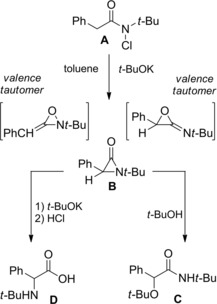
Baumgarten's preparation of aziridinone B and ring cleavage reactions with O‐nucleophiles (ref. [42a–b] – Section 3).
Specifically, while the recrystallized α‐lactam appeared to be a quite stable compound when kept dry in the freezer, it promptly reacted with t‐BuOH providing N‐t‐Bu‐2‐t‐BuO‐phenylacetamide C as well as with t‐BuOK/t‐BuOH affording N‐t‐Bu phenylglycine D after acid hydrolysis.
Later, Sheehan and Lengyel43 reported the synthesis of 1‐t‐butyl‐3,3‐dimethylaziridinone B by t‐BuOK (10.9 mmol) mediated dehydrobromination of N‐t‐Bu 2‐bromo‐2‐methylpropanamide A (20 mmol) in Et2O at −25 °C (Scheme 26).
Scheme 26.
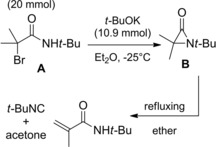
Preparation of aziridinone B by Sheehan and Lengyel (ref. [43] – Section 3).
The α‐lactam decomposed into N‐t‐butylmethacrylamide and smaller amounts of t‐butylisonitrile and acetone following a short reflux period in ether. Even more unstable resulted α‐lactams with less encumbered N‐substituents.
Scrimin and co‐workers44 found convenient to carry out the dehydrohalogenation reaction by treatment of the α‐haloamide substrates with potassium hydroxide in benzene or toluene in the presence of 18‐crown‐6 ether as a phase‐transfer catalyst.
In 2002, a further improvement has been described making use of sodium hydride in dichloromethane in the presence of 15‐crown‐5 ether at room temperature.45
Sheehan and Lengyel43 underlined a dichotomous outcome for the reactions between nucleophilic species with the electrophilic α‐lactam (aziridinone) A, the latter either as an isolated compound (Scheme 27) or a putative transient intermediate in the reaction medium (Scheme 28). Specifically, the aziridinone A reacted with nonionic nucleophiles (e. g., alcohol, amine, thiol) at room temperature affording N‐t‐Bu 2‐substituted‐isobutyramides B (Scheme 27).
Scheme 27.
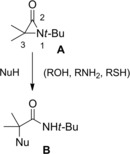
Ring opening reactions of the isolated aziridinone A with nonionic nucleophiles (ref. [43] – Section 3).
Scheme 28.
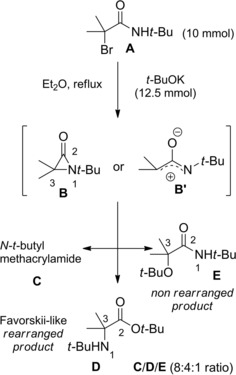
Ring opening reactions of the putative aziridinone B with the tert‐butoxy anion (ref. [43] – Section 3).
On the other hand, the dehydrobromination of the starting α‐bromoamide A (10 mmol) performed with t‐BuOK (12.5 mmol) in diethyl ether at reflux, returned the N‐t‐butylmethacrylamide C, the t‐Bu 2‐t‐butylamino‐2‐methyl propanoate D, and the N‐t‐Bu 2‐t‐BuO‐isobutyramide E in 8 : 4 : 1 ratio (Scheme 28). The formation of the isomeric products D and E was rationalized in terms of the acyl‐nitrogen cleavage and the alkyl‐nitrogen cleavage respectively of the in situ formed α‐lactam B when attacked by the tert‐butoxy anion.
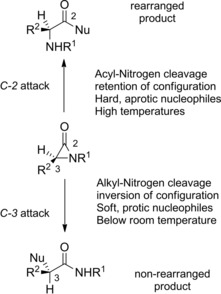
Thus, the preferential nucleophilic attack at C2 with 2–1 sigma bond heterolysis provided the Favorskii‐like “rearranged product” D whereas attack at C3 with 3–1 sigma bond heterolysis yielded the “non‐rearranged product” E.
As an alternative mechanism explaining the dual reactivity of α‐lactams toward nucleophiles it was advanced the involvement of a highly reactive aza‐oxyallylic cation B’ (Scheme 28). However, a set of experiments by Hoffman and co‐workers46 using enantiopure α‐lactams established that attack at the C‐2 position consistently produced an amino acid derivative with retention in stereochemistry, while attack at the C‐3 position produced an amide with inversion in stereochemistry. The latter finding ruled out the intermediacy of a planar aza‐oxyallylic cation intermediate, which would instead result in product racemization.
Despite numerous theoretical and mechanistic studies tried enlightening the outcome in nucleophilic aziridinone ring opening reaction, it remained difficult to predict whether nucleophilic attack would be favoured at the C‐2 or C‐3 position of the α‐lactam.47
Summing up, protic or ‘‘soft’’ nucleophiles reacting below approximately room temperature produce the “non‐rearranged products”. Conversely, aprotic or ‘‘hard’’ nucleophiles at elevated temperatures afford the “rearranged products” (Scheme 29).
Scheme 29.
Dichotomous outcome for the aziridinones ring opening reactions (ref. [47] – Section 3).
To complete the scenario D'Angeli and co‐workers in 196448 disclosed a rearrangement reaction competing with the Favorskii‐like one, occurring when the N‐cyclohexyl‐α‐chloro‐α,α‐diphenylacetamide A reacted with NaNH2 in liquid ammonia. They reported that the reaction furnished the N1‐cyclohexyl‐N1‐benzhydrylurea C as the predominant product (Scheme 30). Its formation plausibly deriving via C2−C3 sigma bond heterolysis of the incipient α‐lactam B in turn formed by base mediate dehydrochlorination of the starting α‐chloroamide. Importantly, the unusual aziridinone ring‐opening reaction occurred, provided that two phenyl substituents were present at the halogenated α‐carbon atom of the amide.
Scheme 30.
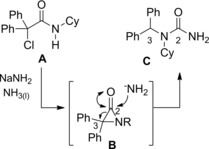
Unusual aziridinone ring opening reaction (ref. [48] – Section 3).
4. α‐Amination of α‐Haloamides
α‐Amino amides are a class of organic compounds exhibiting a variety of biological activity that makes them potential candidates for drug development and discovery. These compounds represent the smallest subunit of naturally occurring peptides and proteins; they have also been used as building blocks for the synthesis of different heterocycles and ligands for the metal‐catalysts.
Replacement of the α‐halogen atom by nitrogen nucleophiles in the presence of inorganic or organic bases is among the available methods for the synthesis of α‐amino amides using α‐chloro, α‐bromo and α‐iodoamides as starting materials. Lidocaine (15.7 g) has been recently produced by Ley and co‐workers under flow conditions within two working days (85 % overall yield).49 The second reaction of the two‐step synthesis consisted of the diethylamine alkylation with 2‐chloro‐N‐(2,6‐dimethylphenyl)acetamide in the presence of Et3N. Optimized conditions featured DMF as the solvent, a reactor temperature of 99 °C, residence time of 17.8 min, and 3.9 equiv. of amine to chloroacetamide.
2‐(Alkylamino)acetamides have been prepared by reaction of 2‐chloro‐N,N‐dimethylacetamide with amines and β‐aminoalcohols in the presence of sodium bicarbonate or triethylamine in benzene at reflux50 while potassium bicarbonate in refluxing acetonitrile has been used when 2‐bromoacetamide was the substrate.51
The reaction of 2‐bromo‐N‐cyclohexyl‐2‐methylpropanamide with 1‐cyclopentylpiperazine in the presence of solid sodium carbonate/potassium iodide in ethanol at reflux produced the “usual α‐aminoamide”. However, changing the reaction conditions to aqueous potassium hydroxide in ethanol at 50–70 °C led to the formation of a Favorskii‐like rearranged product: the “reversed α‐aminoamide” (Scheme 31).52
Scheme 31.
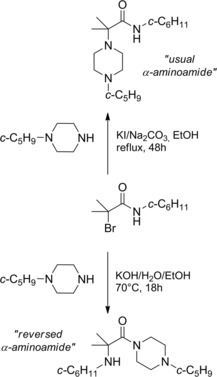
Dual reactivity for 2‐bromo‐N‐cyclohexyl‐2‐methylpropanamide toward 1‐cyclopentylpiperazine in the presence of inorganic bases (ref. [52] – Section 4).
In spite of several examples in literature showing a dual reactivity for the α‐haloamides intercepting amine nucleophiles,52, 53a–53b exclusive formation of “usual α‐aminoamides” has been observed by Guziec and Torres,54 Lai,55 and Scrimin and co‐workers.56 Tested reaction conditions entailed treating the α‐haloamide substrates in neat amine with powdered NaOH at 0–5 °C or their dropwise addition to amines in NaH/THF at room temperature. Additionally, the NaOH/H2O/DCM/TBAB biphasic system was also effective to produce “usual α‐aminoamides”.
The chemistry of α‐bromoamides toward bromine substitution by a variety of nucleophiles (primary, secondary and tertiary amines, carboxylic acids, alcohols, saccharides, aminoacids) has been deeply investigated by D'Angeli and co‐workers57 and summarized in a short review.58 Interestingly, they found that (S)‐2‐bromopropanamides reacted with amine nucleophiles either in the presence of soluble Ag(I) salts or insoluble Ag2O as promoters (Scheme 32).
Scheme 32.
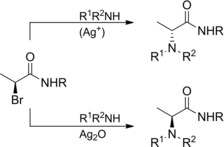
Stereospecific bromine replacement by amine nucleophiles in the presence of soluble Ag(I) salts or insoluble Ag2O (ref. [57] – Section 4).
In particular, the first ones afforded (R)‐2‐aminopropanamides, whereas substitution products with retention of configuration were formed by employing Ag2O. In the latter case, an early stage formation of aziridine‐lactams from the α‐bromo secondary amides was postulated as the pivotal step.57
The effectiveness of stoichiometric silver oxide in promoting direct functionalization of encumbered alkyl bromides including α‐tert‐alkyl bromides of primary, secondary and tertiary amides with O‐, N‐, and C‐nucleophiles was later improved by Vachal and co‐workers.59 Indeed, they were able to increase both the reaction rate and selectivity performing the substitution reaction in MeCN/H2O (95 : 5). The glad finding was explained assuming that the presence of water as a co‐solvent favors a tight association of all reaction partners in the bond‐forming event of the reaction coordinate.
Alternatively, stereoselectivity in asymmetric nucleophilic halogen substitution could be achieved using chiral α‐haloamides equipped with a chiral auxiliary so as one of the two α‐epimers could react faster than the other one, due to the difference in diastereomeric transistion state energies.60 Moreover, suitable DKR processes have been developed by ensuring a fast diastereoisomer conversion could take place in situ. A variety of chiral amines have been used as chiral auxiliaries for the DKR of α‐haloamides. Nunami et al. succeeded in efficient DKR of (αRS)‐epimers A incorporating the chiral auxiliary t‐butyl (4S)‐1‐methyl‐2‐oxoimidazolidin‐4‐carboxylate (Scheme 33).61
Scheme 33.
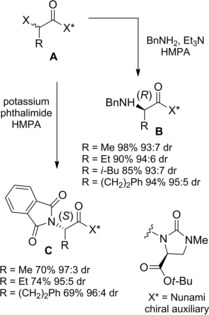
DKR of α‐haloamides A incorporating the Nunami chiral auxiliary in reactions with nitrogen nucleophiles under base‐catalyzed epimerization conditions (ref. [61] – Section 4).
Interestingly, the chiral auxiliary served to prepare both enantiomers B and C of the amino acid derivatives simply by shifting from benzylamine to the Gabriel reagent (potassium phthalimide) as nitrogen nucleophile in the stereospecific amination processes. The DKR processes occurring at room temperature in the polar solvent hexamethylphosphoramide (HMPA) gave high levels of stereocontrol under base‐catalyzed epimerization conditions. As the chiral auxiliary could be easily removed without racemization (BnOLi/THF) the process established a facile access to a range of optically active L‐ and D‐α‐amino acid derivatives.62a–62b
It is notable that the Nunami chiral auxiliary was also effective in the stereoselective alkylation of α‐bromo amides with malonic ester enolates, the DKR processes leading to carbon‐carbon bond formation.8f
Caddick and co‐workers introduced the (4R,5S)‐1,5‐dimethyl‐4‐phenylimidazolidin‐2‐one as the suitable chiral environment for efficient stereo‐differentiation of chiral α‐bromo amides A in halogen substitution reaction with a variety of nitrogen, sulfur and carbon nucleophiles (Scheme 34).63
Scheme 34.
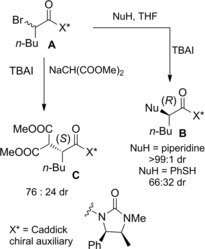
DKR of α‐haloamide A incorporating the Caddick chiral auxiliary in reactions with carbon and heteroatom nucleophiles using TBAI as the epimerizing agent (ref. [63] – Section 4).
Thus, high levels of selectivity were observed by using TBAI as the epimerizing agent. Interestingly, the SN2 reactions occurred with an unusual dichotomy of diastereoselection whereby a metalated nucleophile afforded mainly the (αS)‐epimeric product C whereas H‐bonding amine nucleophiles gave the (αR)‐epimeric product B; disappointing levels of stereo‐differentiation were achieved with sulfur nucleophiles. Importantly, both the coupling and the removal of the ephedrine‐derived chiral auxiliary required mild conditions.
Theoretical rationalization of the collected experimental results from the literature led Santos et al.64 to design a new imidazolidinone‐based chiral auxiliary effective for the DKR of diastereomeric 2‐bromopropanamides reacting with benzylamine (Scheme 35). The halogen substitution reaction onto amide A, conducted in the presence of TBAI and Et3N, provided the α‐aminated compound B in 92 % yield with >99 : 1 dr.
Scheme 35.
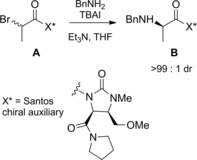
Santos chiral auxiliary effective for the DKR of diastereomeric 2‐bromopropanamides reacting with benzylamine using TBAI as the epimerizing agent (ref. [64] – Section 4).
Ward et al. obtained brilliant results using Oppolzer's chiral (−)‐camphorsultam, the aminated (αR)‐epimer B being formed as the single diastereomer in quantitative yield when an (αRS) diastereomeric mixture of the α‐bromo amide A reacted with dibenzylamine in refluxing acetonitrile (Scheme 36).65
Scheme 36.
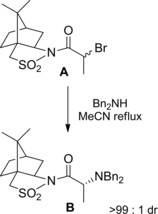
DKR of α‐bromoamide A incorporating the Oppolzer chiral auxiliary reacting with dibenzylamine under base‐catalyzed epimerization conditions (ref. [65] – Section 4).).
Asymmetric nucleophilic substitution reactions between dibenzylamine and α‐bromoamides A derived from L‐amino acids provided di‐ and tripeptide analogues B incorporating optically pure N‐protected (αR)‐unnatural amino acids in up to 95 % yield and >99 : 1 dr.8e, 8g The actual pathway for the asymmetric process in which the chiral information of an amino acid precursor is efficiently transferred to the new C−N bond formation at α‐halo carbon center, is a dynamic kinetic resolution requiring the TBAI/DIEA couple for the fast epimerization of the substrate (Scheme 37).
Scheme 37.
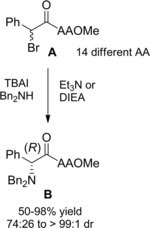
DKR of α‐bromoamides A derived from L‐amino acids reacting with dibenzylamine under TBAI/DIEA epimerization conditions (ref. [8e, 8g] – Section 4).
Instead, none or disappointing levels of stereo‐differentiation (51 : 49 to 77 : 23 dr) were achieved using benzylamine. However, the less sterically demanding amine nucleophile reacted with the N‐Bn α‐bromo phenylacetyl L‐AlaOMe A affording the 2(S),5(S)‐disubstituted diketopiperazine B with 98 : 2 dr (Scheme 38). Thus, the switching from secondary to tertiary amides (N‐Bn derivatives) favored the (αS)‐selectivity in the αC−N bond forming step.3b
Scheme 38.
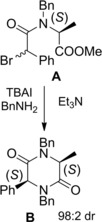
DKR of N‐Bn α‐bromoamide A reacting with benzylamine under TBAI epimerization conditions (ref. [3b] – Section 4).
In the following three examples, a copper catalyst is entrusted with the replacement of the α‐halogen atom by nitrogen nucleophiles.
A general method for the construction of α‐amino carbonyl compounds containing sterically hindered aniline moiety is based on reaction of α‐bromocarbonyl substrates (amides in this context) with phenylhydroxylamine using CuCl2/PMDTA as the optimal catalyst system (PMDTA=pentamethyldiethylenetriamine) (Scheme 39).66
Scheme 39.
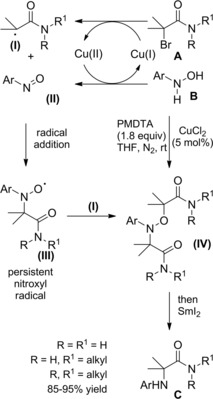
Copper‐catalyzed α‐amination of α‐bromoamides using phenylhydroxylamine (ref. [66] – Section 4).
The process results copper‐catalyzed as a Cu(I) single electron transfer (SET) to the α‐bromoamide A matches the N‐aryl hydroxylamine B oxidation that is in turn a Cu(II)‐mediated step. It has been proposed that the nitroso compound (II), formed in the Cu(II)‐mediated oxidation step, intercepts the α‐radicalamide (I) in turn formed in the Cu(I) SET‐step. The resulting nitroxyl radical (III) then captures a second radical species (I) giving the N−O adduct (IV) which undergoes Sm‐mediated reduction to the sterically hindered α‐anilinoamides C.
Fu and co‐workers established an elegant and convenient way for reacting racemic tertiary alkyl α‐chloroamides A with either carbazoles and indoles nucleophiles B (Scheme 40).67
Scheme 40.
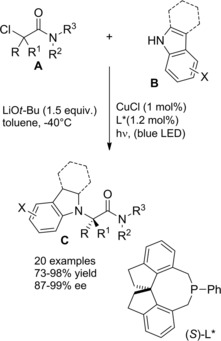
Asymmetric copper‐catalyzed C−N cross‐couplings induced by visible light (ref. [67] – Section 4).
Specifically, it was found that irradiation of the partners at −40 °C for 16 hours in the presence of CuCl, a chiral phosphine (L*), and lithium tert‐buthoxide (LiOt−Bu) generated the aimed C−N cross‐coupling products C featuring fully substituted stereocenters with good to excellent enantioselectivity. Notably, the enantioconvergent process transforming the racemic starting material into a single product enantiomer required a copper‐based chiral photocatalyst derived from commercially available components. As a possible mechanism, the irradiation of a copper‐nucleophile complex (I) leads to an excited‐state (II) that adds oxidatively to C−Cl bond of the α‐haloamide A (Scheme 41).
Scheme 41.
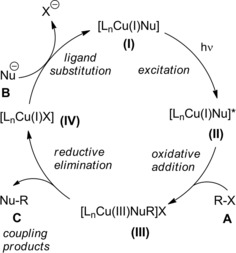
Mechanistic hypothesis for the copper‐catalyzed C−N cross‐couplings induced by visible light (ref. [67] – Section 4).
The resulting Cu(III)‐complex (III) delivers the cross‐coupled product C via a reductive elimination step. In such a way, the Cu(I)‐catalyst (IV) is restored for the subsequent ligand substitution with the nitrogen nucleophile giving (I).
A year later Nishikata et al. reported their achievements of a copper‐catalyzed amination of hindered tertiary alkyl α‐bromoamides with either amines or ammonia (top Scheme 42).68
Scheme 42.
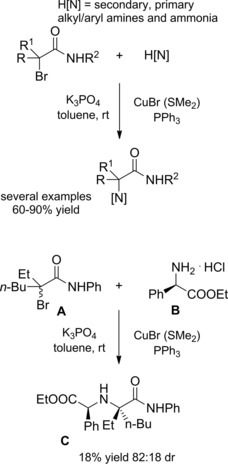
Copper‐catalyzed amination of hindered tertiary alkyl α‐bromoamides with either amines or ammonia (ref. [68] – Section 4).
Importantly, the method allowed for the facile preparation of congested and highly functionalized α‐aminoamides through C−N bond formation at room temperature with no overalkylation detectable when using ammonia. Interestingly, the chiral racemic α‐bromoamide A coupled with (R)‐phenylglycine ethyl ester B affording the corresponding aminated product C with a chiral quaternary carbon center with high diastereoselectivity (dr=82 : 18) (bottom Scheme 42). In addition, substrates possessing two different NH2 groups reacted chemoselectively (the aromatic amine undergoing preferential alkylation over the aliphatic one) and a primary amine was a better nucleophile with respect to a secondary one. Although β‐hydride elimination from an α‐bromoamide was problematic, it was found that the presence of PPh3 inhibited the undesired side reaction. On the basis of a serie of control experiments, the authors advanced a reaction mechanism overlapping the one proposed by Fu and co‐workers67 for the asymmetric copper‐catalyzed C−N cross‐couplings induced by visible light.
5. Intramolecular α‐Alkylation of α‐Haloamides: Syntheses of β‐Lactams
Variously decorated azetidin‐2‐ones are endowed with important biomedical activities. Moreover, enantiopure β‐lactams are versatile chiral intermediates in organic synthesis reason why, a great deal of work has been devoted to the development of efficient synthetic approaches to their preparation in a stereocontrolled fashion.
In‐depth studies by González‐Muñiz and co‐workers69a–69e esthablished that α‐haloamides derived from α‐amino acids were convenient precursors for acceding to this key structural motif in non‐racemic form. Thus, the simple base‐promoted (Cs2CO3 in MeCN) cyclization of some N‐benzyl‐N‐chloroacetyl amino acids provided the expected 1,4,4‐trisubstituted azetidin‐2‐ones in good yields and with ee up to 58 %. The moderate enantioselectivities during the 4‐exo‐tet cyclization process could be explained appealing to the memory of chirality phenomenon.70a–70d Intriguingly, the chirality of the α‐amino acid stereogenic center, missed upon proton abstraction is on the other hand transferred (partly in this case) to the final compound without using any external chiral source.
The same research group71 reported that the hydrogen‐bonding organocatalyst TADDOL along with the phosphazene base (tert‐butylimino)tris(pyrrolidino)phosphorane (BTPP), was a helpful additive to enhance memory of chirality while transforming α‐chloroamides A into the amino acid derived β‐lactams B (top Scheme 43). Surprisingly, the TADDOL additive was effective regardless of its configuration and enantiopurity. The degree of the selectivity improvement in favor of the (S)‐product was dependent on the nature of the amino acid aromatic side chain [Δee due to TADDOL: 36 % for Phe and Tyr, 8 % for Trp(Boc)] (bottom Scheme 43).
Scheme 43.
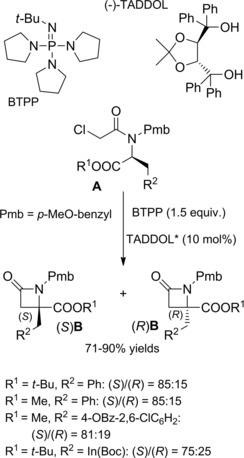
β‐Lactams via 4‐exo‐tet cyclization of α‐chloroamides derived from α‐amino acids in the presence of BTTP and TADDOL additives to enhance memory of chirality (ref. [71] – Section 5).
The base‐promoted 4‐exo‐tet‐cyclization strategy was also applied to enantiomerically pure N‐benzyl‐N‐chloropropanoyl amino acid derivatives A in order to accede to valuable chiral 1,3,4,4‐tetrasubstituted β‐lactams B (Scheme 44).72
Scheme 44.
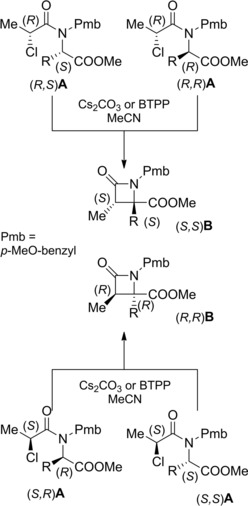
Diastereo‐ and enantioselective 4‐exo‐tet cyclization of α‐chloroamides derived from α‐amino acids (ref. [72] – Section 5).
Intriguingly, a diastereo‐ and enantioselective process yielded exclusively 3,4‐cis β‐lactam derivatives regardless of the absolute configuration of the α‐amino acid center, a stereochemical outcome de facto abolishing the memory of chirality phenomenon.
In fact, asymmetric construction of the quaternary stereogenic center was entirely dependent upon configuration of the 2‐chloropropionyl moiety.
Theoretical calculations on the transition states explained the high stereoselectivity of the process and prompted the authors to extend the protocol to the preparation of a 1,3,4‐trisubstituted 2‐azetidinone, a Gly‐derived β‐lactam, in enantiomeric pure form.73
β‐Lactams B were prepared in moderate yields by an electrochemical process where the electrogenerated base (EGB) diethyl malonate anion reacted with α‐haloacetanilides A (Scheme 45).74a–74b
Scheme 45.
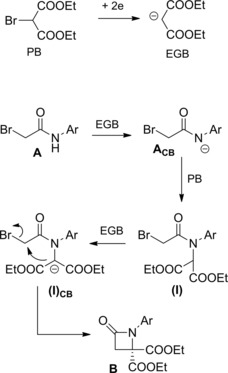
Electrochemical process converting α‐bromoamides and diethyl bromomalonate into β‐lactams (ref. [74a–b] – Section 5).
The formal insertion of a malonate framework into the amide skeleton could be rationalized in terms of halide displacement by an internal carbon centered nucleophile leading to C3−C4 β‐lactam bond formation. Presumably, diethyl bromomalonate acted both as a probase (PB) and an alkylating agent.
Thus, N‐alkylation of the α‐haloamide conjugated base ACB with the PB formed N‐bis(ethoxycarbonyl)methylhaloacetanilides (I) which provided azetidin‐2‐ones B prior deprotonation to (I)CB and the ensuing intramolecular alkylation reaction.
13 years later a simple diastereoselective electrosynthesis of cis‐3‐alkyl‐1‐benzyl‐4‐ethoxycarbonyl‐β‐lactams B was reported by the same research group (Scheme 46).75a–75b The process entailed the 4‐exo‐tet‐cyclization of N‐(ethoxycarbonyl)methyl‐N‐benzyl‐2‐bromoalkylcarboxamides A with cyanomethyl anion acting as EGB.
Scheme 46.
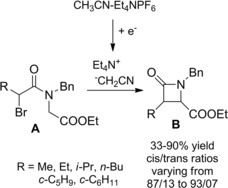
Diastereoselective electrosynthesis of β‐lactams B via 4‐exo‐tet cyclization of α‐bromoamides A (ref. [75a–b] – Section 5).
Indeed, galvanostatic cathodic reduction of a solution of acetonitrile and tetraethylammonium hexafluorophospate (Et4NPF6) (supporting electrolyte) conducted at room temperature and under a nitrogen atmosphere, provided tetraalkylammonium cyanomethyl anion. The latter, acting as a naked anion turned out to be a very reactive EGB.
The above strategy could be successfully extended to a variety of α‐bromoamides bearing a chiral auxiliary or chiral protective groups paving the way to an easy, high‐yielding, and stereocontrolled synthesis of chiral cis‐β‐lactams (Scheme 47).76
Scheme 47.
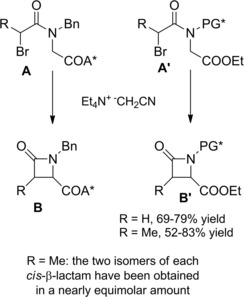
Stereocontrolled electrosynthesis of cis‐β‐lactams via 4‐exo‐tet cyclization of α‐bromoamides incorporating chiral auxiliaries or N‐protective groups (ref. [76] – Section 5).
The electrochemically induced cyclization of bromoamides to β‐lactams also occurred in room‐temperature ionic liquids (RTILs) (Scheme 48).77 The eco‐friendly process avoided the use of volatile organic solvents (VOCs) and supporting electrolytes, affording the desired β‐lactams in good to elevated yields via C3−C4 bond formation. The reaction protocol entailed the addition of the haloamide A to an electrolyzed solution of probases (PB) azobenzene or tetraethyl ethenetetracarboxylate in the RTIL (1‐butyl‐3‐methylimidazolium exafluorophospate). Cyclization to the corresponding β‐lactams B did take place via initial deprotonation at C4 by the EGB followed by intramolecular SN reaction.
Scheme 48.
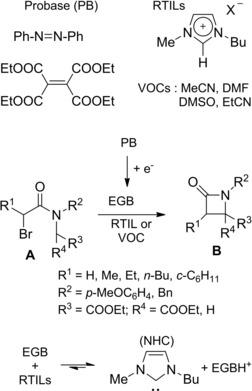
EGB/NHC‐promoted 4‐exo‐tet cyclization of α‐bromoamides in imidazolium‐based RTILs (ref. [77] – Section 5).
Thus, in accord to the reaction pathway, the reactivity of the bromoamides resulted affected mainly by the acidity of the proton at C4 and secondarily by the nature of the substituents on N and C3 atoms. As the N‐heterocyclic carbene (NHC) is a weaker base than the EGB, a proton‐exchange reaction between the EGB and the imidazolium cation was conjectured possible (bottom Scheme 48).
The “non‐innocent” nature of imidazolium‐based RTILs was then established succeeding in the azetidin‐2‐one synthesis just by exposure of α‐bromo amides to NHCs in situ formed by cathodic cleavage of the C2/hydrogen bond of RTILs.78 Indeed, the electrogenerated NHC acted as a basic promotor of the cyclization of bromo amides to β‐lactams (Scheme 49).
Scheme 49.
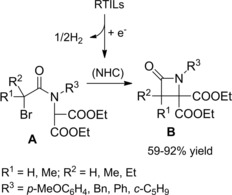
Electrogenerated NHC as basic promotor for the 4‐exo‐tet cyclization of α‐bromoamides (ref. [78] – Section 5).
Owing to the high chemoselectivity, mild reaction conditions and the elimination of toxic and harmful chemicals, electrochemical processes are of great interest. We confide that convenient devices standardized for both in batch and flow electrosynthesis which are nowadays commercially available, will direct even more synthetic chemists to contemplate electrochemical methods for acceding designed compounds also in a large‐scale production.
In 2014 Cramer and co‐workers79 disclosed an intriguing Pd(0)‐catalyzed synthesis of β‐lactams B from readily accessible N‐benzyl tertiary chloroacetamides A (Scheme 50). To work well the reaction required tertiary amide functional group with bulky and branched substituents on the amide nitrogen atom, the methyl group being not suitable. Undesired nucleophilic substitution at the α‐chloro position of the chloro amide by the carboxylate co‐catalyst, as well as aryl C(sp 2)‐H functionalization, were mitigated via co‐catalyst and ligand structure optimization.
Scheme 50.
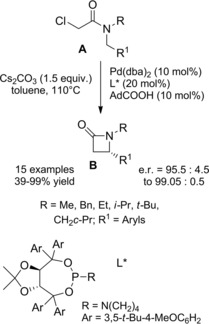
Pd(0)‐catalyzed synthesis of β‐lactams B from N‐benzyl tertiary chloroacetamides A (ref. [79] – Section 5).
It was proposed that a concerted metallated deprotonative (CMD) process, with activation of chloroacetamides at their benzylic C−H bond, was involved in the formation of a five‐membered palladacycle (I) (Scheme 51). The latter intermediate underwent a strain‐building reductive elimination to the four‐membered ring of the β‐lactam B. In this step the challenging C(sp 3)−C(sp 3) bond formation goes with the regeneration of the active Pd(0) species. Notably, in the presence of a chiral ligand (a bulky TADDOL‐derived phosphoramidite) in combination with AdCOOH (adamantyl carboxylic acid) the process resulted in an asymmetric C−H functionalization.
Scheme 51.
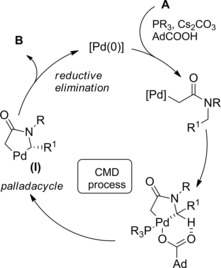
Mechanistic hypothesis for the Pd(0)‐catalyzed synthesis of β‐lactams from N‐benzyl tertiary chloroacetamides (ref. [79] – Section 5).
Thus, the enantiodiscriminating CMD‐step provided β‐lactams B in excellent yields and enantioselectivities (up to 99 % ee). The preferred absolute configuration of the chiral carbon was (S) as established by X‐ray crystallographic analysis. Intriguingly, when tertiary N‐cyclopropyl chloroacetamides A were the substrates, the palladium‐mediated enantiodiscriminating CMD‐process, proceeding with the Taddol phosphonite ligand L* and 1‐adamantane carboxylic acid cooperation, provided homo‐chiral (R,R‐configurated when R1=H) cyclopropane‐fused γ‐lactams B (Scheme 52).80
Scheme 52.
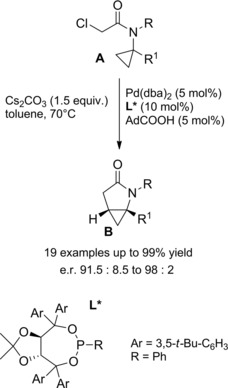
Pd(0)‐catalyzed synthesis of cyclopropane‐fused γ‐lactams B from N‐cyclopropyl chloroacetamides A (ref. [80] – Section 5).
A reductive elimination process onto a six‐membered palladacycle was advanced as the pivotal step forging enantioselectively the pyrrolidine ring through C(sp 3)−C(sp 3) bond formation. To note, the enantioselective process was found to be not limited to the use of cyclopropanes as source of activatable C−H bonds.
In 2016 Clark and co‐workers2 reported a fruitful method for achieving four‐membered lactams based on a Cu(0) (copper wire)‐mediated stereoelectronically favored81 4‐exo‐trig ring closure of the unsaturated α‐bromo amide A (Scheme 53).
Scheme 53.
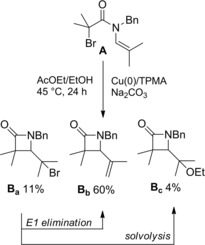
Cu(0)‐mediated 4‐exo‐trig ring closure of the α‐bromo enamide (ref. [2] – Section 5).
The cyclization reaction performed in the presence of the ligand complex tris(2‐pyridylmethyl)amine) (TPMA) and the base Na2CO3 afforded the β‐lactam derivatives Ba‐c with compound Bb arising from E1 elimination of the labile tertiary bromide functional group of Ba, and Bc by solvolysis of the corresponding tertiary cation intermediate.
6. Intramolecular α‐Alkylation of α‐Haloamides: Syntheses of γ‐Lactams
6.1. γ‐Lactams via 5‐exo‐trig Radical Cyclization
Organotin hydrides have been widely employed as radical reducing agents to convert unsaturated organohalides to various carbocycles or heterocycles. In this context haloamides provided with internal olefin group played a privileged role as useful precursors of electrophilic carboradicals undergoing cyclization to a wide range of pyrrolidin‐2‐ones.
In 1991 Ikeda and Ishibashi's research group82 reported the stereoselective syntheses of the Sceletium alkaloid (±)‐mesembranol (Scheme 54) and the Amaryllidaceae alkaloid (±)‐elwesine (scheme 55) through processes whose featuring step is a tributyltin hydride (Bu3SnH) mediated radical cyclization. Thus, dichloroacetamide A, following exposure to the action of Bu3SnH (1 equiv.) and a catalytic amount of azobis(isobutyronitrile) (AIBN) in boiling toluene, provided the expected cis‐3a‐aryloctahydroindolones B in 51 % yield along with product C (26 %), the latter resulting by the carbamoyl radical intermediate reduction. The lactam B was converted to (±)‐mesembranol according to an already described protocol.83
Scheme 54.
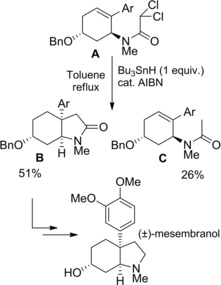
Synthesis of the Sceletium alkaloid (±)‐mesembranol by Bu3SnH‐mediated radical cyclization of the unsaturated dichloroacetamide A (ref. [82] – Section 6.1).
Scheme 55.
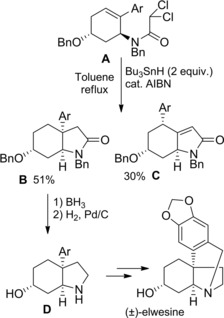
Synthesis of the Amaryllidaceae alkaloid (±)‐elwesine by Bu3SnH‐mediated radical cyclization of the unsaturated dichloroacetamide A (ref. [84] – Section 6.1).
On the other hand, dichloroacetamide A treated with 2 molar equiv. of Bu3SnH gave lactam B in 51 % yield along with the rearrangement product C (30 % yield) (Scheme 55). The former compound reduced with borane and deprotected by hydrogenolysis gave the amine D which was an advanced intermediate along the synthesis of the alkaloid (±)‐elwesine.84
As a further example, classical synthetic targets such as the kainic acid family were obtained from tri‐substituted pyrrolidin‐2‐ones B in turn derived by Bu3SnH mediated cyclization of suitable α‐chloroamides A incorporating a α,β‐unsaturated ester moiety (Scheme 56).85a–85b
Scheme 56.
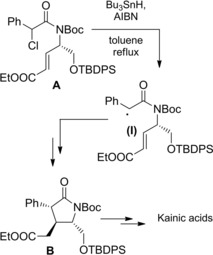
Synthesis of the Kainic acids by Bu3SnH‐mediated radical cyclization of the unsaturated chloroamide A (ref. [85a–b] – Section 6.1).
Atom transfer radical cyclizations (ATRC) represent a popular synthetic tool for the simultaneous formation of a C−C and C−X bond across an alkene or alkyne. They are de facto non‐reductive alternatives to organotin hydrides in mediating radical cyclization reactions in organic synthesis.
Homolitic Mn−Mn bond cleavage of dimanganese decacarbonyl [Mn2(CO)10], achieved by heating or photolysis, generates the manganese pentacarbonyl radical which can form carbon‐centered radical by halogen‐atom abstraction from suitable organic substrates. Thus, photolysis of iodide A with 0.1 equivalents of dimanganese decacarbonyl afforded pyrrolidinone B via iodine‐atom transfer 5‐exo‐trig cyclization (Scheme 57).86 Similarly the ATRC of A′ was the salient step in an enantioselective synthesis of the pyrrolizidine alkaloid (−)‐trachelanthramidine (Scheme 57).
Scheme 57.
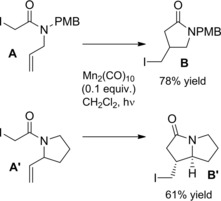
Dimanganese decacarbonyl promoted ATRC (ref. [86] – Section 6.1).
To note, the trapping of the intermediate cyclic primary radical with TEMPO has proved to be an effective method for the preparation of cyclic hydroxylamines B from α‐haloamides A (Scheme 58).86
Scheme 58.
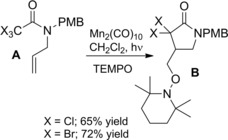
5‐exo‐trig cyclization of A with Mn2(CO)10 in the presence of TEMPO (ref. [86] – Section 6.1).
In transition‐metal‐catalyzed atom‐transfer radical cyclizations (TMC‐ATRC) it is a redox catalyst (typically a complex of copper or ruthenium) which is entrusted with the generation of carbon‐centered radicals. In this context, N‐allyl α‐haloamides A are de facto converted into γ‐lactams B through intramolecular addition of the C−X bond (X=Cl, Br, I) across the tethered olefin group (Scheme 59).
Scheme 59.
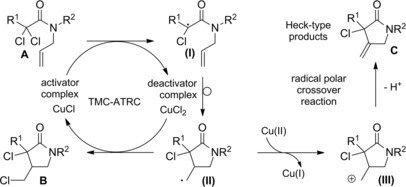
Transition‐metal‐catalysed atom‐transfer radical cyclization and radical‐polar crossover reaction–Section 6.1
The reaction mechanism features three basic steps: I) the transition‐metal‐catalyst (TMC‐ activator complex) increases its oxidation state by one abstracting a halogen atom from the amide substrate to give the TMC‐deactivator complex; II) the so formed N‐allyl‐carbamoyl methyl radical (I) cyclizes through a regioselective 5‐exo intramolecular attack onto the C=C bond; III) the new formed radical intermediate (II) is quenched by halogen transfer from the TMC‐deactivator complex. In the last step, the halogenated lactam B is formed while the transition‐metal‐catalyst complex is restored in the reduced state (TMC‐activator complex).
As a side reaction, the combination of two radicals leads to the TMC‐deactivator complex accumulation and to a decrease in catalytic rate.
Alternatively, in a radical‐polar crossover reaction, radical (II) oxidation by the TMC‐deactivator complex generates a carbenium ion (III) which is converted into Heck‐type products C through loss of a proton from the adjacent atom (Scheme 59). The radical oxidation step occurs with the TMC‐activator complex restoration. Ultimately, radical‐polar crossover reactions lead to replace the halogen atom of α‐haloamides with an alkenyl goup. Therefore, papers dealing with the topic are commented in the specific Section 8.
A variety of N‐allyl‐α‐haloamides has been successfully used as 5‐exo‐trig cyclization substrates to yield lactam scaffolds and different catalytic systems have been developed over 40 years. [Cu(I)87a–87k and Ru(II)87g, 88a–88m].
Nagashima and co‐workers prepared γ‐butyrolactams via copper or ruthenium‐catalyzed cyclization of N‐allyl trichloroacetamides A (Scheme 60). While RuCl2(PPh3)3 was an effective catalyst both for secondary and tertiary unsaturated amides, CuCl gave higher yields with the tertiary ones. The carbon‐carbon bond forming reactions, performed in sealed tubes at 140 °C, furnished regioselectively the α,α,γ‐trichloro‐γ‐lactams B, no δ‐lactams being usually detectable.89 The products could be transformed into various nitrogen‐heterocycles through selective dechlorination steps with Bu3SnH.90
Scheme 60.
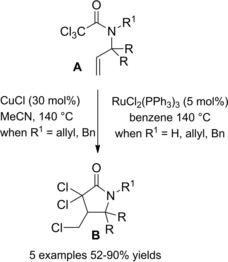
TMC 5‐exo‐trig ATRC (ref. [89] – Section 6.1).
The introduction of electron‐withdrawing groups to the nitrogen atom of the amide substrates together with the use of the CuX(bipy)‐catalyst (bipy=bipyridine) in dichloroethane led the cyclization processes to occur under mild conditions.91a–91b As a relevant application, the TMC‐ATRC of trichloroacetamide A gave easy access to cis‐3a‐aryloctahydroindole derivatives B which could be converted to racemic mesembrane and crinane alkaloids (Scheme 61).
Scheme 61.
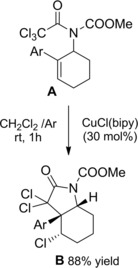
CuCl(bipy)‐promoted ATRC of trichloacetamide A (ref. [91a–b] – Section 6.1).
The Nagashima research group applied its efficient ATRC strategy to various N‐allyl‐N‐tosylhalodifluoroacetamides.92 They established the role of the N‐tosyl group was to lower the rotational barriers of the fluorinated amides leading to facile access of the rotamer favorable for the cyclization, namely that arranging CF2‐X and allyl group in the s‐cis configuration (Scheme 62).
Scheme 62.
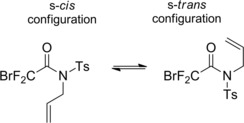
N‐allyl‐N‐tosylbromodifluoroacetamide rotamers (ref. [92] – Section 6.1).
Thus, α,α‐difluoro‐γ‐halo‐γ‐lactam derivatives B were achieved in good to high yields using CuX(bipy) catalysts, the reactivity of the halodifluoroacetamides A decreasing in the order I>Br≫Cl (Scheme 63). Interestingly, the 5‐exo‐trig cyclisation onto N‐allyliodoamide substrates could be triggered by fluorescent light at room temperature with the aid of catalytic amount of either PPh3 or Pd(PPh3)4 in THF.
Scheme 63.
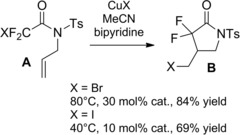
CuX(bipy)‐promoted ATRC of halodifluoroacetamides A (ref. [92] – Section 6.1).
N‐Allyl‐N‐tosyldichloroacetamides were less reactive substrates for the ATRC process and when standard catalysts, such as CuCl(bipy) or RuCl2(PPh3)3 were used then high reaction temperatures and/or high catalyst loadings were required.93 However, the cyclization of these reticent substrates has been later reported by Severin and co‐workers94 by using 5 mol% of the air‐stable Ru(III)‐complex [RuCl2Cp*‐(PPh3)] as a pre‐catalyst (Cp*=pentamethylcyclopentadienyl), in combination with Mg(0) in a “supplemental activator and regenerating agent” (SARA)‐ATRC (Scheme 64).2
Scheme 64.
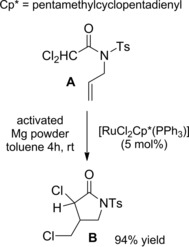
(SARA)‐ATRC of dichloroacetamide A promoted by [RuCl2Cp*−(PPh3)] in combination with Mg(0) (ref. [94] – Section 6.1).
In fact, the zero‐oxidation‐state Mg was the co‐catalyst committed with the in situ generation and re‐generation of the catalytically active ruthenium(II) species (activator complex) by reduction of ruthenium(III) species. The combined catalytic species converted dichloroacetamide A into the γ‐lactam derivative B with efficiency paralleling the one of the air and moisture sensitive methoxy‐bridged dimer [{RuCp*(OMe)}2] used in combination with pyridine as an activating ligand.
Clark and Wilson found AIBN (10 mol%) was an effective SARA additive in their copper mediated ATRC of N‐allyl tertiary and secondary alkyl bromoacetamides.95 The relatively slow 5‐exo‐trig cyclization reactions could be mediated both by CuBr or the more oxidatively stable CuBr2 (1 mol%) in conjunction with the TPMA ligand (1 mol%). Either heating at 50 °C in CH2Cl2 or at 110 °C in toluene furnished the atom transfer cyclized products in excellent yields without the need of an inert atmosphere.
Auto‐tandem catalysis is an atom‐economical and environmentally benign synthetic method involving two or more mechanistically distinct reactions promoted by a single catalyst only.
In 2013 Onitsuka and co‐workers96 disclosed an asymmetric auto‐tandem reaction catalyzed by a planar‐chiral cyclopentadienyl‐ruthenium complex able to promote both the allylic amidation and the ATRC (Scheme 65).
Scheme 65.
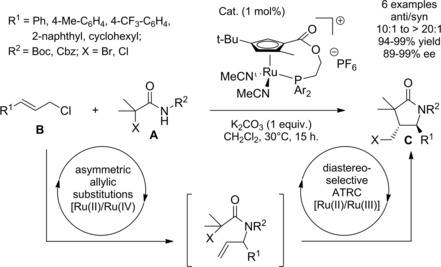
Asymmetric auto‐tandem allylic amidation/atom‐transfer radical cyclization catalyzed by a planar‐chiral cyclopentadienyl‐ruthenium complex (ref. [96] – Section 6.1).
In detail, the first reaction involved a [Ru(II)/Ru(IV)]‐cycle whereas the second one proceeded by a [Ru(II)/Ru(III)]‐cycle.
Thus, a sequential regio‐, enantio‐, and diastereoselective reaction of α‐haloamide derivatives A (including an α‐dichoroamide) with allylic chlorides B provided synthetically useful γ‐lactams C with multiple stereogenic centers through one‐pot sequential allylic amidation/atom‐transfer radical cyclization.
Interestingly, the concept of generating and re‐generating the active catalytic species in ATRC processes94 was also fulfilled electrochemically. Ozaki et al. studied the indirect electroreduction of N‐allyl and N‐propargyl‐α‐haloamides A by electrogenerated Ni(I) complexes in DMF or CH3CN containing a supporting electrolyte (Et4NClO4).97a–97b The electrochemical radical‐type cyclizations proceeding at room temperature required Ni(II) complexes which underwent reduction at a graphite electrode (Scheme 66). As single‐electron‐transfer catalyst the so formed Ni(I) species, reduced the haloamide substrates A to the corresponding carbamoyl radicals (I) which afforded the expected γ‐lactams via radical intermediate (II) in turn generated through a 5‐exo‐trig cyclization process.
Scheme 66.
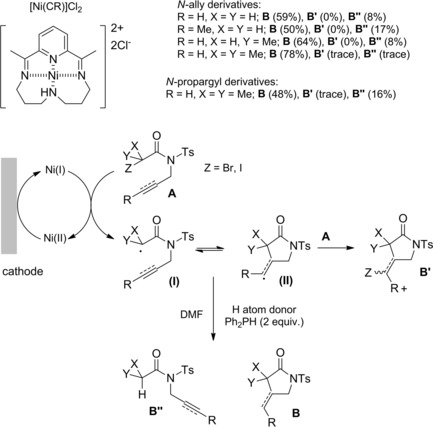
Radical‐type cyclization of α‐haloamides A by electrogenerated Ni(I) complexes (ref. [97a–b] – Section 6.1).
To close the cycle, the formed Ni(II) returned to the active Ni(I) through cathodic electroreduction. Notably, the method converted into cyclized products B, B′, substrates that tended to yield only the simple reduction products B′′ in attempted tin hydride‐mediated cyclizations. Moreover, the selective formation of halogenated and non‐halogenated pyrrolidinones could be achieved making use of the difference in the ability of the solvent to donate a hydrogen atom to the cyclic carbon‐centered radicals.
Later, Medeiros and co‐workers obtained better results in terms of yields and selectivities by conducting the electrosyntheses in “green” solvents such as EtOH and EtOH‐H2O mixtures.98
More recently, a copper‐catalyzed electrochemically mediated atom transfer radical cyclization (eATRC) of A has been developed as an easy and clean method allowing the synthesis of dichlorinated γ‐lactams B through the intermediacy of the radical species (I) and (II).99
In detail, the electrosynthesis conducted in acetonitrile used the [Cu(II)TPMA] catalyst which was activated and continuously regenerated to its active copper(I) form by reduction at a platinum (Pt) electrode (Scheme 67).
Scheme 67.
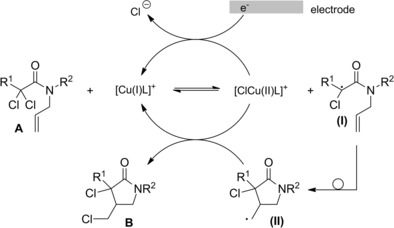
Copper‐catalyzed electrochemically mediated atom transfer radical cyclization of A (ref. [99] – Section 6.1).
The previously discussed (Scheme 53, Section 5.) Clark's method2 for achieving four‐membered lactams was also efficient to promote 5‐exo‐trig ATRC of unsaturated α‐halogeno amides. Thus, the Cu(0)/TPMA catalyst‐system converted dimethylsulfamoyl‐N‐protected‐N‐allyl carboxamides A into cis/trans mixture of five‐membered lactams B and C in good to excellent yield (Scheme 68). Precisely, the thermodynamically more stable and synthetically useful cis‐γ‐lactams B were the prevailing diastereomers. As a key feature the Cu(0) acted as a supplementary activator and reducing agent in a SARA‐ATRC process. To note, the Cu(0)/TPMA catalyst‐system gave disappointing results when tested using N‐propargyl‐N‐tosyl‐α‐bromoisobutanamide as a suitable substrate for the corresponding 5‐exo‐dig ATRC process.
Scheme 68.
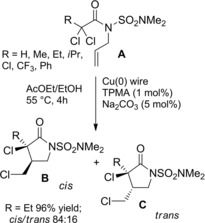
(SARA)‐ATRC of dichloroamides A promoted by Cu(0)/TPMA catalyst‐system (ref. [2] – Section 6.1).
6.2. γ‐Lactams via 5‐endo‐trig Radical Cyclization
General and productive synthetic protocols achieving γ‐lactams have also been developed looking at the alternative 5‐endo‐trig cyclization of αC‐centered radicals generated from N‐vinyl‐α‐haloamides, otherwise named α‐haloenamides.100a–100e In 2001, this topic has been authoritatively reviewed by Pearsons101 while more recently a short review on the radical cyclization of trichloroacetamides to lactams has appeared in the literature.102 For sake of completenesss we will report here only significative classical examples as well as the uncovered most recent findings.
Ikeda, Ishibashi, and co‐workers were the first to show that a variety of halo‐enamides under free‐radical reducing conditions (Bu3SnH) cyclized in a “disfavored” 5‐endo‐trig manner to give five‐membered lactams with high degree of efficiency (Scheme 69).103a–103b However, some exceptions (α‐bromo amides bearing sulfur and/or aryl substituent(s) at the terminus of the N‐vinylic bond) where β‐lactams resulted from preferential 4‐exo‐trig cyclization were also described.104a–104b
Scheme 69.
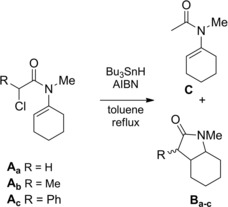
5‐endo‐trig radical cyclization of α‐ haloenamides under free‐radical reducing conditions (ref. [103a–b] – Section 6.2).
When a boiling solution of Aa in toluene was treated with 1.1 equiv. of Bu3SnH in the presence of a catalytic amount of azobisisobutyronitrile (AIBN), the octahydroindol‐3‐one Ba was obtained in 63 % yield along with the reduction product C (8 %).
The cyclizations of Ab and Ac occurred more cleanly to give the corresponding lactams Bb (3α‐Me : 3β‐Me=6 : 1) and Bc (3α‐Ph : 3β‐Ph=2 : 3) in 73 and 75 % yields, respectively (Scheme 69).
The 5‐endo‐trig radical cyclization of the α‐halo amides A generated the radical intermediate (I) from which the tetracyclic perhydroerythrinane skeleton B was built up via a successive 6‐endo‐trig cyclization onto the N‐tethered olefin group (Scheme 70).105
Scheme 70.
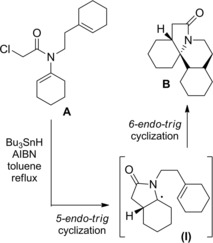
Radical cascade process providing the tetracyclic perhydroerythrinane skeleton B (ref. [105] – Section 6.2).
A similar radical cascade process providing the tricyclic indolizidinone B entailed an initial 5‐endo cyclization onto the dehydroamino ester group of A to generate the α‐amino ester radical (I) which in turn underwent a 6‐endo cyclization onto the N‐tethered cyclohexene moiety (Scheme 71).106a–106b
Scheme 71.
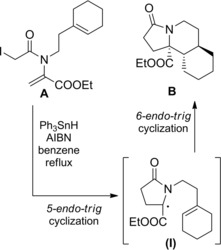
Radical cascade process providing the tricyclic indolizidinone B (ref. [106a–b] – Section 6.2).
Ishibashi and co‐workers107 made unusual observations in comparing radical cyclizations of α‐chloro‐, α‐bromo‐, and α‐iodoenamide Aa‐c which were precursors of the same radical species (Scheme 72).
Scheme 72.
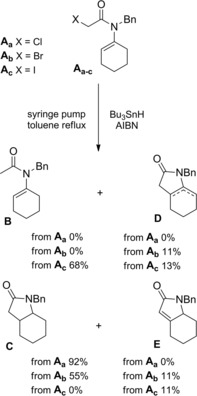
5‐endo‐trig radical cyclization of α‐haloenamides Aa‐c: comparison between α‐Cl, α‐Br, and α‐I substrates (ref. [107] – Section 6.2).
Slow syringe pump addition of Bu3SnH to α‐haloenamides at reflux in toluene provided a mixture of the expected cyclized/reduced product C along with cyclized/non‐reduced products D and E and directly reduced (non‐cyclized) product B in very different ratio. Surprisingly, the α‐iodoenamide was by far the poorest performer of the three.
Later, Curran and co‐workers108 established that the ability of the α‐iodoenamide Ac to provide cyclized products could be largely restored, with cyclized/reduced ratio up to >99 : 1, by adding K2CO3 as a base. Therefore, α‐chloroenamide Aa and α‐iodoenamide Ac behaved as complementary precursors, with chloride Aa giving largely cyclized/reduced product C and iodide Ac giving largely cyclized/non‐reduced products D and E.
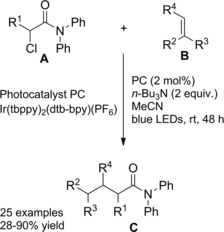
In 2016 Rueping and co‐workers14 described the first intramolecular version of their previously established method exploiting the electron‐rich Ir‐complex [Ir(tbppy)2(dtb‐bpy)PF6] to catalyze the photoredox intermolecular radical addition of α‐chloro amides onto alkenes (Scheme 78, Section 7.1.). Thus, the visible light mediated 5‐endo‐trig cyclization of α‐chloroenamides A led to the synthesis of valuable substituted γ‐lactams B (Scheme 73). The mild and tin‐free method requiring tributylamine to mediate the C−Cl bond single‐electron reduction generally proceeded in moderate to good yields with a broad range of substrates with different substitution patterns. Remarkably, in almost all cases, only one diastereoisomer was formed.
Scheme 78.
Visible light mediated reductive radical addition of the chloroamides A onto olefins B with [Ir(tbppy)2(dtb‐bpy)PF6] catalyst (ref. [112] – Section 7.1).
Scheme 73.
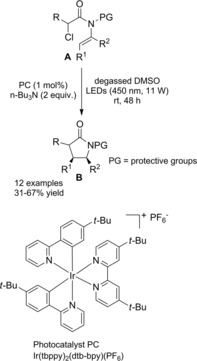
Visible light mediated 5‐endo‐trig cyclization of α‐chloroenamides A with [Ir(tbppy)2(dtb‐bpy)PF6] catalyst (ref. [14] – Section 6.2)
Regarding the photoreductive reaction mechanism (Scheme 74), it was assumed that irradiation with visible light triggers the photoredox catalytic cycle of Ir(III), whose stable and long‐lived excited state *Ir(III) acts as an oxidant towards trialkylamines generating the aminium radical cation I..+ The so formed Ir(II) can reduce the α‐chloroenamide A to the α‐carbonyl radical (I) which can partition between the directly reduced product C and the 5‐endo cyclized product B via the radical intermediate (II).
Scheme 74.
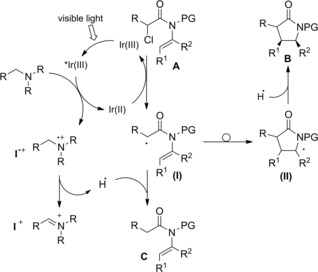
Mechanistic hypothesis for the photoreductive reaction mechanism (ref. [14] – Section 6.2).
7. Intermolecular α‐Alkylation of α‐Haloamides
7.1. From αC(Hal) to αC(Alk)
Oshima and co‐workers109 found that allylic gallium reagents gave cross‐coupling reactions with α‐iodo (or bromo) carbonyl compounds in the presence of triethylborane (Et3B) and oxygen. In particular, α‐haloamides A smoothly reacted with B (3 equiv.) at room temperature furnishing 4,5‐unsaturated amides C via C2−C3 bond formation (Scheme 75). An ethyl radical, formed in situ by Et3B oxidation, was assumed as the reactive species triggering the coupling reaction.
Scheme 75.
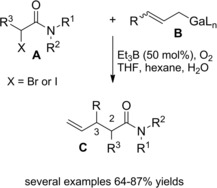
Cross‐coupling reactions of α‐haloamides with allylic gallium reagents in the presence of Et3B and oxygen (ref. [109] – Section 7.1).
Cross‐coupling racemic secondary alkyl α‐bromoamides A with freshly prepared organozinc reagents B in the presence of the catalyst system consisting of NiCl2 .glyme and (R)‐(i‐Pr)‐Pybox ligand, paved the way to a stereoconvergent synthesis of chiral amides C (Scheme 76).15, 110
Scheme 76.
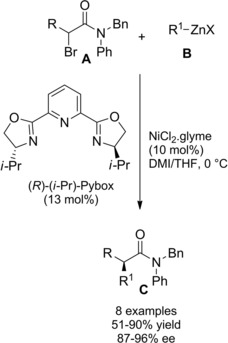
Asymmetric Negishi cross‐coupling reaction of α‐haloamides A with organozinc reagents B (ref. [15, 110] – Section 7.1).
The asymmetric Negishi reaction conducted in a solvent mixture of 1,3‐dimethyl‐2‐imidazolidinone (DMI) and THF at 0 °C well tolerated organozinc reagents with a variety of functional groups, such as olefins, ethers, imides, and nitriles. Notably, simple reduction of the resulting α‐chiral amides with LiAlH4 furnished the corresponding chiral non‐racemic primary alcohols in good yields.
Silver‐promoted ionization of α‐bromo amides has been conveniently used as source of unexpectedly stabile carbocations which gave rise to α‐alkylated amides by intercepting carbon‐based nucleophiles (Scheme 77). Thus, allyl‐ and methallyltrimethylsilane B took part to Ag(I)‐promoted carbon‐carbon bond forming reactions with α‐bromoamide A producing the corresponding allylated products C in moderate yields.111 To note, α‐bromo amides lacking the carbocation‐stabilizing aryl group didn't take part in α‐alkylation reactions.
Scheme 77.
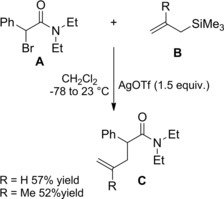
Ag(I)‐promoted carbon‐carbon bond forming reactions between α‐bromoamide A and allyl silanes B (ref. [111] – Section 7.1).
In 2014, Rueping and co‐workers112 established a tin‐free protocol to cross‐couple α‐chloro amides and alkenes where a regioselective photocatalyzed radical pathway produced α‐alkylated amides (Scheme 78). The reductive intermolecular radical addition process smoothly proceeded upon visible‐light irradiation of an acetonitrile solution of the chloroamides A and the olefins B (1 : 3 equiv. ratio) in the presence of tributylamine and the electron‐rich Ir catalyst [Ir(tbppy)2(dtb‐bpy)PF6]. The photoredox protocol being compatible with a broad spectrum of reagent partners served to achieve a series of alkylated amides C in good yields under mild conditions. With regard to the reaction mechanism it was advanced a photoredox catalytic cycle similar to the one previously described when discussing ref. [14] (Scheme 74, Section 6.2.).
Yasuda and co‐workers reported a visible‐light‐induced radical coupling reaction of silyl enol ethers with α‐bromoketones, α‐bromoesters, and α‐bromoamides to give 1,4‐dicarbonyls.113 Thus, the reaction between the bromoacetamide A and trimethylsilyl propen‐2‐yl ether B led to the γ‐ketoamide C via C2−C3 bond formation (Scheme 79).
Scheme 79.
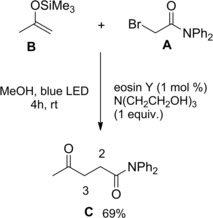
Visible‐light‐induced radical coupling reaction of silyl enol ethers B with α‐bromoamides A (ref. [113] – Section 7.1).
Interestingly, the inexpensive organic dye eosin Y together with triethanolamine (probably acting as a redox mediator in the eosin Y catalytic cycle) were an effective catalyst system to carry out the visible‐light‐induced, regioselective, non‐reductive radical addition process. In detail, a first SET from the photocatalyst to the α‐bromoamide generated the pivotal carbamoyl methyl radical (I) which, following addition onto the silyl enol ether B generated the siloxy‐substituted carbon radical (II) (Scheme 80). The latter, through a second SET‐step with the triethanolamine radical cation originated the siloxy‐substituted carbocation (III) from which the aimed γ‐ketoamide C resulted by bromide‐triggered trimethylsilyl group elimination.
Scheme 80.
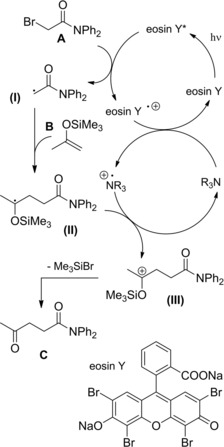
Mechanistic hypothesis for the visible‐light‐induced, non‐reductive radical addition process (ref. [113] – Section 7.1).
At almost the same time Cheng and co‐workers published their thiyl‐radical‐catalyzed photoreductive hydrodifluoroacetamidation of alkenes with Hantzsch ester.114 Irradiation of alkenes B and bromodifluoroacetamides A with blue light‐emitting diodes (LEDs) in acetonitrile at room temperature and in the presence of both the Hantzsch ester HE and diphenyldisulfide (Ph2S2) afforded hydrodifluoroacetamidation products C in moderate to good yields (Scheme 81). Terminal alkenes, cycloakenes, alkenes with heterocyclic moieties or alkenes bearing free as well as protected functional groups were, without distinction, good substrates for the hydrodifluoroacetamidation process. As regard the partners, tertiary, secondary and primary amides were well tolerated, the latter being inferior substrates.
Scheme 81.
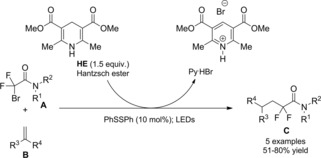
Visible‐light‐induced hydrodifluoroacetamidation of alkenes with Hantzsch ester (ref. [114] – Section 7.1).
Mechanistic investigations showed that the photoreductive radical addition reaction involved the phenyl thiyl radical PhS . as the hydrogen atom transfer (HAT) and the multifunctional reagent HE as the hydrogen donor and electron donor (Scheme 82). In the presence of visible light, the diphenyldisulfide undergoes homolysis, giving PhS . entrusted with a hydrogen atom transfer (HAT) with the activated Hantzsch ester. Subsequent single electron transfer (SET) from the resulting Hantzsch ester radical HE . to α‐bromofluoroacetamide A generates the difluoroamide radical (I) together with the pyridinium derivative Py . HBr. Subsequent addition of (I) to the alkene B affords the radical intermediate (II) that, by way of a second HAT reaction involving thiophenol PhSH furnishes the difluoroalkylation product C and regenerates the phenyl thiyl radical.
Scheme 82.
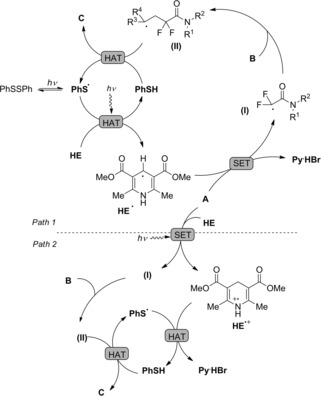
Mechanistic hypothesis for the visible‐light‐induced hydrodifluoroacetamidation of alkenes with Hantzsch ester (ref. [114] – Section 7.1).
An alternative plausible reaction pathway starts with a SET reaction from the excited Hantzsch ester HE and bromodifluoroacetamide A giving the cationic radical HE .+ and amide radical (I) as intermediates.
At this stage, a HAT reaction between HE .+ and PhS . yields the pyridinium species Py . HBr together with PhSH. Eventually, the HAT between (II) and PhSH provides the hydrodifluoroacetamidation product C with recover of the thiyl radical species.
Zhou et al.115 disclosed a fruitful, mild process to achieve a range of chiral α‐alkylalkanoic amides C by way of a transition‐metal‐catalyzed cross‐coupling reaction of racemic α‐bromoamides A (1.5 equiv.) and olefins B (Scheme 83).
Scheme 83.
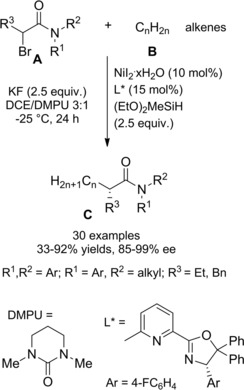
Ni‐catalyzed cross‐coupling reaction of racemic α‐bromoamides A and olefins B (ref. [115] – Section 7.1).
The method was versatile as either a variety of N‐aryl α‐bromoamides and alkenes with a diverse spectrum of functional groups were compatible. The enantioconvergent process using a Ni/Pyrox chiral catalyst provided enantioenriched compounds C with excellent terminal regioselectivity, regardless both of the starting position as well as of the number of substituents and of the geometry of the C=C bond in the olefinic substrates.
The reaction entailed a nickel‐hydride‐catalyzed alkene isomerization proceeding via hydrometalation‐chainwalking (Scheme 84). Thus, the first formed alkylnickel complex (I) is converted to the migrated alkylnickel complex (II) featuring the terminal C(sp3)‐H suitable activated for the alkylation process. The chiral ligand Pyrox bearing a methyl substituent at the C6‐position de facto promoted an asymmetric hydroalkylation reaction. Specifically, the stereochemical information generated after the oxidative addition of (II) onto the C−Br bond of the α‐haloamides A was retained in the products C in turn delivered by the Ni(III)‐complex (III) reductive elimination. Eventually, to close the catalytic circle, the L*NiH was regenerated in situ by reduction of L*NiBr with the hydrosilane reagent.
Scheme 84.
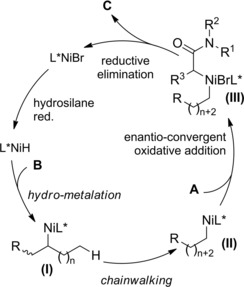
Mechanistic hypothesis for the Ni‐catalyzed cross‐coupling reaction of racemic α‐bromoamides A and olefins B (ref. [115] – Section 7.1).
Recently, by merging visible‐light photoredox catalysis and Lewis acid catalysis, Xu and co‐workers succeded in the coupling reaction of 2‐acyl imidazoles B with commercially available bromodifluoroacetamides A (Scheme 85).116 Thus, a variety of chiral γ‐keto amides C containing a gem‐difluoromethylene group was successfully obtained with high stereoselectivities via intermolecular αC−C(sp3) bond formation. Optimized conditions entailed visible‐light irradiation of the reaction mixture in dichloromethane and at room temperature with a 23 W compact fluorescent lamp (CFL).
Scheme 85.
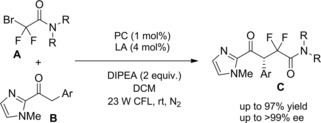
Coupling reaction of 2‐acyl imidazoles B with bromodifluoroacetamides A in the presence of the chiral Lewis acid (LA) and photocatalyst PC (ref. [116] – Section 7.1).
Notably, the above reaction conditions could be scaled up to 1.0 mmol. In addition to γ‐ketoamides bearing gem‐difluoroalkyl groups, the reaction could also be applied to synthesize monofluorinated adducts.
The coupling reactions used the dual catalyst consisting of the at‐metal Λ‐RhS as the chiral Lewis acid (LA) and Ir(ppy)2(dtbbpy)(PF6) as the photocatalyst (PC) (Scheme 86). Moreover, stoichiometric amounts of diisopropylethylamine (DIPEA) were required. Several control experiments indicated that the reaction involved a radical process where both an α‐carbonyl carbon radical from the imidazole substrate (II .+) and the difluoroacetyl radical (III) were generated in the dual catalytic process (Scheme 87). On these bases it was proposed the 2‐acyl imidazole substrate B first binds the LA‐catalyst to give intermediate (I) which upon deprotonation provides a rhodium enolate complex (II). The latter, engaged in a redox process with the excited photocatalyst [PC*]n+ generates (II .+) together with the highly reducing agent [PC](n−1)+ which is in turn involved in the SET step converting the bromodifluoroacetamide A into the radical species (III). The regio and stereoselective addition of radical (III) onto the complex (II) provides the reducing radical intermediate (IV) which is the precursor of the rhodium‐coordinated difluoroalkylated product (V). Either (II .+) or [PC*]n+ could be involved in the SET with (IV). Eventually, a ligand exchange onto (V) generates the fluorine‐containing γ‐keto acid derivatives C while restoring the intermediate (I).
Scheme 86.
The dual catalyst for the coupling reaction of 2‐acyl imidazoles with bromodifluoroacetamides (ref. [116] – Section 7.1).
Scheme 87.
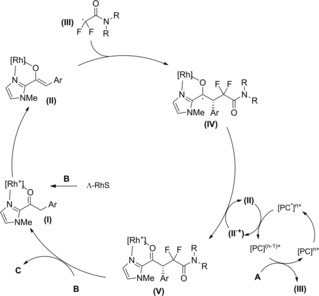
Mechanistic hypothesis for the coupling reaction of 2‐acyl imidazoles B with bromodifluoroacetamides A (ref. [116] – Section 7.1).
In a recent paper Wang et al.117 reported a copper‐catalyzed strategy to achieve simultaneous difluoroalkylation and γ‐lactonization by reacting amide‐containing difluoroalkylated bromides A with allylacetic acid B (Scheme 88).
Scheme 88.
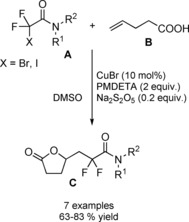
Copper‐mediated radical‐polar crossover reaction of amide‐containing difluoroalkylated bromides A with allylacetic acid B (ref. [117] – Section 7.1).
The double functionalization of the vinyl moiety occurred in the presence of CuBr and the ligand pentamethyldiethylenetriamine (PMDETA) in DMSO at 85 °C with Na2S2O5 as an additive reducing agent. Both tertiary and secondary amides are good partners in the copper catalyzed one‐pot reaction.
The initial regioselective addition of the fluorine‐containing radical (I) to the unsaturated carboxylic acid B generates the radical species (II) that undergos oxidation by Cu(II) to give a carbenium ion (III) (Scheme 89). The latter intercepts the tethered carboxyl group to generate the lactone moiety of the products C.
Scheme 89.
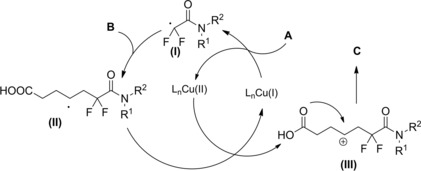
Mechanistic hypothesis for the copper‐catalyzed radical‐polar crossover reaction of amide‐containing difluoroalkylated bromides A with allylacetic acid B (ref. [117] – Section 7.1).
Thus, the copper‐mediated radical‐polar crossover mechanism entails two SET steps. In the first redox step, the Cu(I) complex increases its oxidation state by one abstracting a bromine atom from the amide substrate, whereas in a second SET‐step the Cu(II) is reduced to Cu(I) while the radical species (II) is oxidized to carbenium ion (III).
Removal of the halogen atom in α‐haloamides through a two‐electron transfer process allowed designing complementary syntheses of α‐alkylated amides where the C−C bond construction according to an “umpolung” approach was the featuring step.
Zincamide enolates reacted with electrophilic carbonyl compounds yielding β‐hydroxy amides according to the Reformatskii reaction course (Scheme 90). Cho and Kim118 reported the easy preparation of room‐temperature‐stable zinc enolates of amides (I) from chloroacetamides A. Their subsequent coupling reactions with various carbonyl derivatives B furnished β‐hydroxy amides C in moderate to good yields under mild conditions.
Scheme 90.
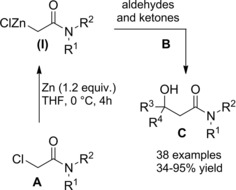
Zincamide coupling reactions with carbonyl derivatives (ref. [118] – Section 7.1).
Hashimoto, Saigo, and co‐workers119 disclosed that a combination of Ph3P and Sc(OTf)3 mediated Reformatsky‐type reaction of N,N‐diphenyl α‐bromopropanamide A with aldehydes B (Scheme 91). The reaction, performed in CH2Cl2 at room temperature, afforded synthetically useful β‐hydroxyamides C in good yields with syn‐selectivities. Thus, upon coordination of the Lewis acid to the carbonyl oxygen of the amide, the reducing agent triphenylphospine attacks the α‐bromine atom giving the Lewis acid enolate (I). The in situ formed nucleophilic species then intercepts the carbonyl group of aldehydes B affording the expected β‐hydroxyamides C through C−C bond formation (Scheme 91). Substituents on the nitrogen atom in α‐bromo amides strongly influenced the rate of the reduction, two phenyls being the best one both in term of isolated yields and selectivity. Other Lewis acids including BF3‚OEt2, TiCl4, and Yb(OTf)3 were less efficient, resulting in low reaction rate and/or poor selectivity.
Scheme 91.
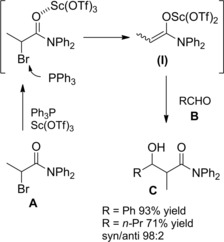
Ph3P/Sc(OTf)3 mediated Reformatsky‐type reaction of α‐bromopropanamide A with aldehydes B (ref. [119] – Section 7.1).
Karoyan et al.120 reported that the zincamide enolate prepared from the enantiopure α‐bromoamide A underwent stereospecific aminomethylation by reaction with a Mannich‐type iminium electrophile B (Scheme 92).
Scheme 92.
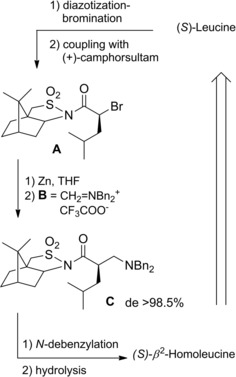
Stereospecific aminomethylation of zincamide enolates (ref. [120] – Section 7.1).
The Reformatskii‐like reaction proceeded with high diastereocontrol (de>98.5 %) providing the β2‐amino acid derivative C which was smoothly converted to β2‐homoleucine by nitrogen debenzylation and hydrolytic removal of the Oppolzer's camphorsultam. The stereochemistry of the resulting β‐amino acid could be deduced from comparison of the optical power of its N‐Boc derivative with an authentic sample. Notably, as the enantiopure α‐bromoamide A was prepared from (S)‐leucine (Leu), the overall process obtained the natural amino acid homologation.
Samarium‐based enolates derived by metalation of iodoacetamide or dichloroacetamide A with samarium diiodide smoothly added to the electrophilic carbon of aliphatic aldehydes as well as of aliphatic activated aldimines affording β‐hydroxy or β‐aminoamides B and C respectively (Scheme 93).121a–121d Noteworthy, the reaction with aldehydes afforded (E)‐α,β‐unsaturated primary amides provided that SmI2 was used in large excess (6 equiv.) and reaction time prolonged to 12 h.
Scheme 93.
Samarium‐based enolates addition to aldehydes and aldimines (ref. [121a–d]–Section 7.1).
7.2. From αCH(Hal) to αC(Alk)(Hal)
A diastereomeric mixture of monofluorinated amides EI−IV incorporating the (2R,5R)‐2,5‐dimethylpyrrolidine chiral framework has been obtained in a [3,3]‐sigmatropic Claisen rearrangement reaction (Scheme 94).122 In detail, the methyl triflate activation of the fluoroacetamide A generated the electrophilic keteniminium ion B which provided D, the suitable substrate for the Claisen rearrangement, upon reaction with trans‐2‐butenoxide C.
Scheme 94.
α‐Alkylation of fluoroacetamide A via [3,3]‐sigmatropic Claisen rearrangement (ref. [122] – Section 7.2).
The stereochemical outcome of the ensuing [3,3]‐sigmatropic reaction showed the chiral auxiliary directed the crotyl fragment to approach the Si face of the (E)‐N,O‐ketene acetal D.
Lithium enolates of 2‐chloro N,N‐diethylacetamides A reacted with the enoate B in which a vinylogous epoxysilane moiety is at the α‐position giving highly functionalized cyclopropane derivatives CI−V.123 The products formed exclusively with the internal (Z)‐olefin and predominant cis isomerism at the cyclopropane ring (Scheme 95).
Scheme 95.
Base‐promoted cascade process providing highly functionalized cyclopropane derivatives (ref. [123] – Section 7.2).
In the proposed reaction mechanism (Scheme 96), the anion (I) resulting from the Michael addition of A and B is involved in a base‐promoted cascade process entailing the epoxide ring opening to give a cyclic pentavalent silicon species (II). The latter is in turn involved in a Brook rearrangement producing the silyl enolether (III) from which the cyclopropane derivatives C are obtained via intramolecular diastereoselective akylation reaction.
Scheme 96.
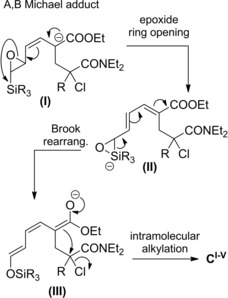
Mechanistic hypothesis for the cascade process giving cyclopropane derivatives C (ref. [123] – Section 7.2).
Arai and co‐workers124 utilized an easily prepared bis‐ammonium salt derived from BINOL as a chiral phase transfer catalyst (PTC) for the Darzens condensation of N,N‐diphenyl α‐haloamides A with both aromatic and aliphatic aldehydes B (Scheme 97). The catalytic asymmetric Darzens reaction proceeding in the presence of inorganic bases (RbOH or Cs2CO3) afforded anyway cis/trans mixtures of glycidic amide derivatives C. In addition to unsatisfactory diastereocontrol, the stereospecificity of the reaction was also scarce (up to 64 % ee for the major cis‐epoxides and up to 70 % ee for the minor trans‐epoxide). Three years later, results in step with the ones by Arai were reported by North and co‐workers which made use of a cobalt(salen) complex derived from (R,R)‐diaminocyclohexane to catalyze the Darzens condensation reaction (Scheme 97).125
Scheme 97.
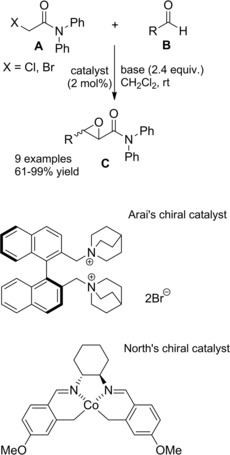
Catalytic asymmetric Darzens reactions using chiral catalysts (ref. [124], [125] – Section 7.2).
The reactions proceeding at room temperature in CH2Cl2 showed as a general trend that, when chloroacetamide/RbOH was the enolate source system, then tran‐epoxides were favoured, whilst cis‐epoxides were formed predominantly by employing the corresponding bromoamide in the presence of KOH. Moreover, the trans‐epoxides were generally produced with higher enantioselectivity than cis‐epoxides. However, for any aldehyde substrate, a higher enantiomeric excess of the epoxide was obtained employing the bromoamide. A chelated thermodynamically controlled aldol reaction was postulated as the critical step detemining the diastereomery of the Darzens condensations products.126
It was also demonstrated that the diastereoselectivity of epoxide formation was strongly influenced by the electronic nature of the aromatic rings in N,N‐diaryl α‐haloamides. In particular, electronwithdrawing nitro substituents favoured formation of the trans‐epoxides.
Shibasaki and co‐workers reported that 7‐azaindoline amides, including α‐halo derivatives, behaved as latent enolates under the action of a cooperative catalytic system consisting of a soft Lewis acid and a hard Brønsted base.11, 127a–127c Catalytic amounts of the Cu(I)/Barton's base binary system was widely effective for generating the corresponding Cu(I)‐enolates which were formidable nucleophiles to forge C−C connectivity through Mannich‐type reaction with both aromatic and aliphatic imine electrophiles (Scheme 98). Importantly, the method offered the potential of controlling the stereochemistry of the two adjacent stereogenic centers by using chiral phospine ligands to the Cu(I) center. Thus, the catalytic deprotonative enolization of α‐halo‐7‐azaindoline amides A afforded chiral halogen‐bearing Cu(I)‐enolates which reacting with imines B gave enantioenriched Mannich‐type adducts C. All α‐halo substituents, F, Cl, Br, and I, were successfully accommodated to provide the aimed adducts in a higly diastereo‐ and enantioselective manner.
Scheme 98.
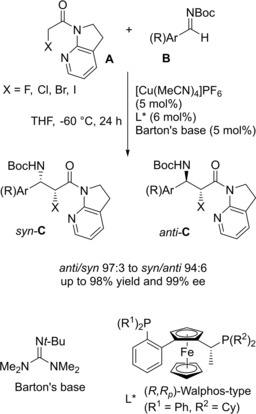
Catalytic asymmetric Mannich reactions using the Cu(I)/Barton's base binary system (ref.[11, 127a–b] – Section 7.2).
Intriguingly, it was found that the diastereoselectivity switched depending on the substitution pattern of the aromatic imines. Such a result was ascribed to stereochemical differentiation based on plausible open transition‐state models where a pentacoordinated Cu(I) complex is entrusted with the reagents assemblage.
Notably, the catalytic asymmetric Mannich protocol was scalable to a gram scale without any detrimental effects on conversion or stereoselectivity. Moreover, the obtained 7‐azaindoline amide derivatives, similarly to Weinreb amides, could take part to functional group interconversion reactions leading to high‐value compounds with halogens on a stereogenic carbon. As an example, 2,3,3,3‐tetrafluoro‐2‐methyl‐1‐arylpropan‐1‐amines D, to be used as fluorinated surrogates for α‐isopropylbenzylamines in SAR‐studies, were efficiently prepared reacting the 7‐azaindoline amide A with N‐Boc protected aryl imines B (Scheme 99).127c
Scheme 99.
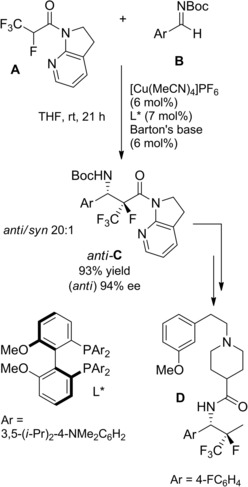
Catalytic asymmetric Mannich reaction using the Cu(I)/Barton's base binary system (ref. [127c] – Section 7.2).
8. α‐Alkenylation of α‐Haloamides
In the first eighties Mori and co‐workers128a–128b reported an attractive but low‐yielding approach to lactams where a Pd(0)‐catalyzed Heck cyclization of N‐benzyl‐iodoacetamides with internal double bond was the featuring step (Scheme 100).
Scheme 100.
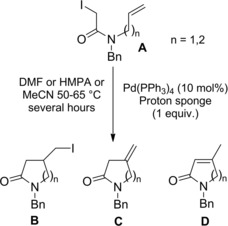
Pd(0)‐catalyzed Heck cyclization of N‐benzyl‐iodoacetamides with internal double bond (ref. [128a–b] – Section 8).
Thus, products with five and six‐membered rings, including pyrrolizidinone and indolizidinone derivatives, could be formed according to the basic steps of an intramolecular Heck reaction. Best results were obtained using a catalytic amount of Pd(PPh3)4 and the non‐nucleophilic base proton sponge. However, a mixture of lactams comprising the exo‐ and the endo‐cyclic unsaturated isomers C and D together with the saturated one B with the γ‐iodo group was invariably obtained.
The formation of compound B was explained through a reductive elimination process occurring on the Pd(II)‐intermediate (II). Usually, the endocyclic unsaturated lactams D were the dominant products, their formation resulting from the hydropalladation/dehydropalladation or base‐catalyzed double bond migration of the exocyclic isomers C (Scheme 101).
Scheme 101.
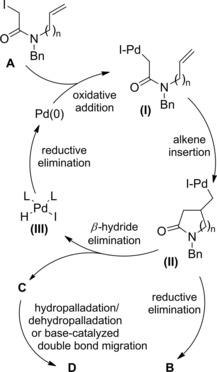
Mechanistic hypothesis for the Pd(0)‐catalyzed Heck cyclization (ref. [128a–b] – Section 8).
The strategy, was later129 successfully extended to N‐allyl‐N‐benzyl‐2‐bromopropanamide indicating that the corresponding σ‐alkyl Pd(II) intermediate did not underwent β‐hydride elimination to the undesired acyclic dehydrobrominated compound (Scheme 101).
Compound C, an advanced intermediate along the synthesis of the racemic alkaloid γ‐lycorane, has been prepared by way of two successive radical cyclizations (Scheme 102).130 In detail, the trichloroacetamido precursor A, in the presence of Ni(0) powder and acetic acid in refluxing 2‐propanol, generated the α‐amidyl radical (I) which cyclized in a 5‐endo‐trig mode. The ensuing radical (II) underwent oxidation to the corresponding cation (III) which afforded the lactam B through deprotonation, further reduction and a final elimination of HCl. Eventually, a Bu3SnH‐mediated 6‐exo‐trig cyclization of an aryl radical onto cyclohexene converted compound B into lactam C with the double bond at the ring junction
Scheme 102.
Tandem Ni‐catalyzed 5‐endo‐trig radical polar crossover reaction/Bu3SnH‐promoted 6‐exo‐trig cyclization (ref. [130] – Section 8).
The monochloro acetamido radical (I) from CuCl(bipyridine)‐mediated reduction of A takes part to an efficient 5‐endo cyclization process resulting in the formation of the octahydroindole radical intermediate (II) (Scheme 103).131 At this stage a Cu(II)‐mediated radical polar crossover reaction converts (II) into the corresponding carbenium ion (III) which following proton abstraction and dehydrochlorination gives B.
Scheme 103.
Copper catalyzed 5‐endo‐trig radical polar crossover reaction (ref. [131] – Section 8).
Efficient 5‐endo radical polar crossover reactions of N‐cyclohexenyl‐N‐benzyl‐2‐bromo‐2‐methyl propanamide have been reported to be mediated in solution by CuX/ Me6‐Tren‐complex132 (Me6‐Tren=[tris(N,N‐2‐dimethylamino)ethylamine]), CuX/PMDETA‐complex132 (PMDETA=N,N,N′,N′,N′′‐pentamethyldiethylenetriamine) and [RuCl2Cp*‐(PPh3)]/Mg(0)94 as well as under heterogeneous conditions using resin supported ligand/copper complexes such as the silica‐supported CuBr/Si‐PMI (PMI=pyridylmethanimine)133 and the JandaJel‐supported CuBr/ JJ‐TEDETA (TEDETA=N,N,N′,N′‐tetraethyldiethylenetriamine propanoate).132
The already discussed (Scheme 53, Section 5.) Clark's method2 for efficient synthesis of β‐lactams via 4‐exo‐trig ATRC served also the purpose of promoting 5‐endo radical‐polar crossover reaction. In detail, the α‐tetralone derivative A, when submitted to the copper wire mediated radical cyclization, afforded the five‐membered lactam B together with the corresponding oxyndole derivative C in turn formed by the in situ dehydrogenation of B (Scheme 104).
Scheme 104.
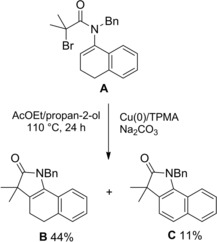
Copper wire mediated 5‐endo radical‐polar crossover reaction (ref. [2] – Section 8).
In 2004 Oshima and co‐workers[109],[134] established a protocol for the facile alkenylation of α‐halo carbonyl compounds, including α‐haloamides, that exploited divalent indium compounds as radical mediators (Scheme 105). The alkenylindium reagents were prepared in situ either via hydroindation of alkynes or via transmetalation of indium trichloride with β‐styryllithium derivatives. The latter compounds were in turn prepared by halogen‐metal exchange onto suitable Z and E vinyl bromides (or iodides) with t‐BuLi in ether. Treatment of the α‐haloamides A with a small excess of alkenylindium B and triethylborane afforded the aimed 3‐enamides C with considerable preservation of the alkene geometry.
Scheme 105.
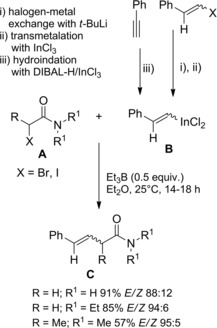
α‐Alkenylation of α‐haloamides using alkenyl indium compounds in the presence of Et3B and oxygen (ref. [109, 134] – Section 8).
The reaction resulted inhibited by the presence of radical scavengers thus, it was conjectured that an in situ formed carbamoylmethyl radical (I) adds to the carbon atom bearing the indium atom (Scheme 106). The resulting radical species (II) then evolves to the 3‐enamides C through elimination of dichloroindium radical that eventually abstracts iodine from the substrate A to generate a new carbamoylmethyl radical (I).
Scheme 106.
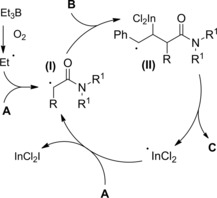
Mechanistic hypothesis for the α‐alkenylation of α‐haloamides using alkenyl indium compounds in the presence of Et3B and oxygen (ref. [109, 134] – Section 8).
A copper‐mediated formal cross‐electrophile coupling reaction of vinyl iodides B with various iododifluoroacetamides A was introduced by Hu and co‐workers in 2010 (Scheme 107).135
Scheme 107.
Cu‐mediated cross coupling reaction of vinyl iodides B with iododifluoroacetamides A (ref. [135] – Section 8).
Optimized reaction conditions giving the fluoroalkenylated products C entailed heating at 50–60 °C a mixture of the reactants in DMSO under an inert atmosphere and in the presence of Cu powder. Although no mechanism details were provided, it was hypothesized an electrophilic gem‐difluorinated radical species is involved. Interestingly, diastereomeric E/Z vinyl iodides cross‐coupled with iododifluoroacetamides providing products with retention of the original configuration.
Lei and co‐workers proposed the Ni(PPh3)4 and the bidentate ligand 1,3‐bis(diphenylphosphino)propane (dppp) as an efficient catalyst system to couple olefins with tertiary and secondary alkyl bromides alpha either to ester or amide functional groups (Scheme 108).136 The formation of the α‐alkenylated Heck‐type coupling products was explained as a radical‐polar crossover reaction involving Ni(I) and Ni(II) complexes as the active catalytic species (Scheme 109).
Scheme 108.
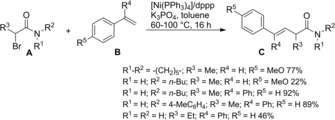
Ni‐catalyzed cross coupling reaction of olefins B with α‐bromoamides A (ref. [136] – Section 8).
Scheme 109.
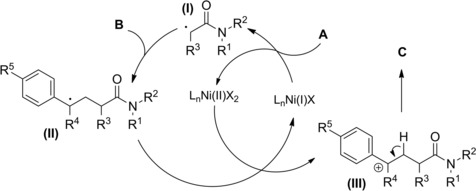
Mechanistic hypothesis for the Ni‐catalyzed radical‐polar crossover reaction (ref. [136] – Section 8).
3‐Enamides C could be obtained in good yields through formal cross‐electrophile coupling of alkenyl bromide with α‐chloro propanamides mediated by bis(cyclooctadiene)nickel(0) [Ni(COD)2] and Mn(0) (Scheme 110).137 The reductive Ni/Mn‐catalyzed process required pyridine as the sole labile ligand when styrenyl bromides B were the coupling partners of α‐chloroamides A, whereas alkyl‐substituted vinyl bromides required both pyridine and bipyridine as the co‐ligands.
Scheme 110.
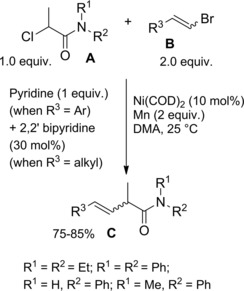
Reductive Ni/Mn‐catalyzed cross coupling of alkenyl bromides with α‐chloro propanamides (ref. [137] – Section 8).
With regard to the stereoselectivity, E‐vinyl bromides generated exclusively E‐products whereas Z‐vinyl bromides were moderately converted to the E‐products.
To note, both β‐ and α‐vinyl bromides were compatible, thus the latter partners easily provided α,α‐dialkyl substituted olefins, which remains a challenge under Heck coupling conditions. The cross‐electrophile coupling was advanced proceeding through a radical‐chain process wherein the Ni(0) oxidative addition to the vinyl halide B is the first step (Scheme 111).
Scheme 111.
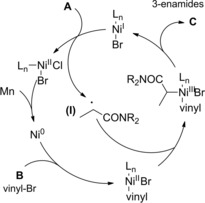
Mechanistic hypothesis for the Ni/Mn‐catalyzed cross coupling of alkenyl bromides B with α‐chloro propanamides A (ref. [137] – Section 8).
Afterward, a one‐electron oxidation step involving a carbamoyl radical (I) generates a Ni(III) complex which upon reductive elimination provides the coupling product C and a Ni(I) species. The latter through a one‐electron transfer to α‐chloroamide A generates the carbamoyl radical (I) and a Ni(II) species which is reduced to Ni(0) by the Mn.
Two examples of copper‐catalyzed coupling of dihydropyran derivative B and bromodifluoro tertiary acetamides A have been firstly described in 2014 (Scheme 112).138 Thus, the readily available α‐bromodifluoroacetamides became suitable reagents for the concomitant introduction of a gem‐difluoro group and an amide moiety into olefin, although modest yields of the 3‐enamides C were produced. Mechanistic experiments conducted with radical scavengers excluded the process involves radical intermediates, instead a highly electrophilic copper(III) species has been hypothesized (Scheme 113). Thus, Cu(I) oxidative addition to the C−Br bond of α‐bromoamide A gives the Cu(III) intermediate (I) which undergoes X‐ligand substitution with dihydropyran to generate an oxonium species (II). The base then release the corresponding enolether (III) eventually involved in the reductive elimination step leading to C through the C(sp 2)−C(sp 3) coupling while restoring the Cu(I) active catalyst.
Scheme 112.
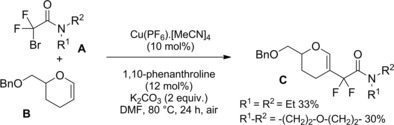
Cu‐catalyzed coupling of dihydropyran derivative B and bromodifluoro tertiary acetamides A (ref. [138] – Section 8).
Scheme 113.
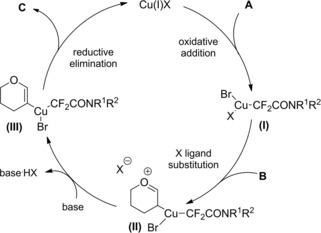
Mechanistic hypothesis for the Cu‐catalyzed coupling of dihydropyran B and bromodifluoro acetamides A (ref. [138] – Section 8).
Nishikata and co‐workers disclosed that styrene derivatives B and a range of secondary α‐bromoamides A cross‐coupled via a radical polar mechanism by heating in toluene in the presence of the [CuI/TPMA]‐catalyst system and benzyltributylammonium bromide (Scheme 114).139
Scheme 114.
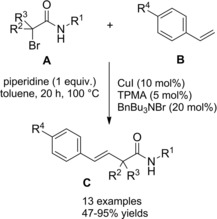
Cu‐catalyzed cross‐coupling of styrene derivatives B with secondary α‐bromoamides A (ref. [139] – Section 8).
The aimed 3‐enamides C were obtained in moderate to good yields provided that piperidine was the base. Interestingly, switching to the base triethylamine led to heterocyclic products as result of a competing carbooxygenation process we will discuss in sub‐Section 11.1. (Scheme 166).
Scheme 166.
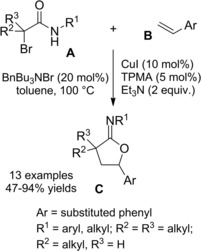
Cu‐catalyzed oxyalkylation reaction of styrenes B with α‐bromoamides A (ref. [139] – Section 11.1).
In 2017 a cobalt‐catalyzed cross‐coupling between α‐bromoamides and alkenyl Grignard reagents was disclosed by Cossy and co‐workers (Scheme 115).140
Scheme 115.
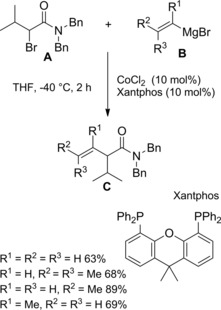
Co‐catalyzed cross‐coupling between α‐bromoamides A and alkenyl Grignard reagents B (ref. [140] – Section 8).
The alkenylation process providing β,γ‐unsaturated amides entailed reacting the nucleophilic and the elecrophilic partners in THF at −40 °C in the presence of CoCl2 and the bidentate diphosphine ligand Xantphos (Scheme 115). N,N‐Dibenzyl‐3‐methyl‐2‐bromo butanamide A cross‐coupled with alkenyl Grignard reagents B (2 equiv.) possessing methyl substituents variously allocated on the double bond giving the expected 3‐enamides C in good yields.
As discussed in Section 6.1. Ozaki et al. developed a method for effecting 5‐exo‐trig(and dig) ATRC of N‐propargyl α‐haloamides making use of electrogenerated Ni(I) species (Scheme 66).97a–97b
The electrochemical radical‐type cyclization converted the N‐propargyl bromoamide A into lactam B together with the reduced non cyclized product B’’, only trace of the brominated lactam B’ being formed (Scheme 116).
Scheme 116.
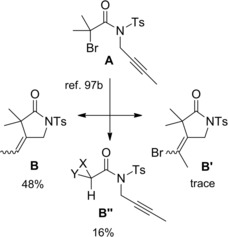
5‐exo‐dig ATRC of N‐propargyl α‐haloamides making use of electrogenerated Ni(I) (ref. [97b] – Section 8).
Good results in the 5‐exo‐dig ATRC of N‐propargyl α‐bromoisobutanamide derivatives A have been achieved by Clark and co‐workers using different CuBr/ligand combination both under homogeneous and heterogeneous catalyzed reaction conditions (Scheme 117).95, 132, 141
Scheme 117.
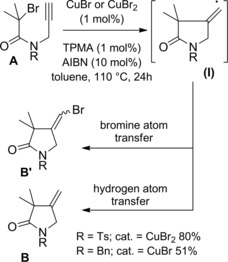
5‐exo‐dig radical cyclization of α‐bromoamides A using different CuBr/ligand combination (ref. [95] – Section 8).
Together with the expected mixture of the vinyl bromides B’ (E : Z=9 : 1) compounds B, probably derived by reduction of the intermediate vinyl radical (I), were also produced. Thus, intermediate (I) may evolve either to B’ via bromine atom transfer or to B via H abstraction from the ligand and/or the solvent. In particular, vinyl bromides B’ and the reduced products B were obtained in a 1 : 1 ratio either using CuBr‐TPMA or CuBr2‐TPMA in the presence of AIBN in toluene at reflux.95
Recently,142 a two‐component one‐pot reaction involving alkyne‐tethered α‐bromoamides A and commom alkynes B has been described to provide various heteropolycyclic frameworks C in the presence of K2CO3 as the base, CuSO4, the ligand TPMA, and KI (Scheme 118). The latter probably acting as an electron‐transfer reagent to improve the annulation cascade that starts with the carbamoyl radical (I) generation via Cu(I)‐mediated C−Br bond splitting of compounds A (Scheme 119).
Scheme 118.
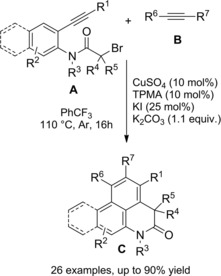
Copper‐catalyzed annulation cascade (ref. [142] – Section 8).
Scheme 119.
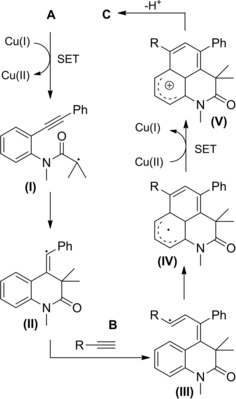
Mechanistic hypothesis for the Cu‐catalyzed annulation cascade (ref. [142] – Section 8).
The carbamoyl radical (I) is at first involved in a 5‐exo‐dig radical cyclization giving the vinyl radical (II). Through addition across the C≡C triple bond of an external alkyne B, the latter gives the intermediate radical (III) which is trapped by the aryl ring. The so formed aryl radical species (IV) is converted to the corresponding aryl cation (V) by Cu(II)‐mediated single electron oxidation. In the next step, a deprotonation concludes the domino process enabling the formation of three new C−C bond in a single step.
The method allowed preparing diverse heteropolycycles, including 1H‐benzo[de]quinolin‐2(3H)‐ones and 4H‐dibenzo[de,g]quinolin‐5(6H)‐one, which are valuable structural skeletons. Schemes 117, 118, 119 should be moved backward.
9. α‐Arylation of α‐Haloamides
The α‐aryl amide framework represents an important pharmacophore featuring various biologically active compounds and is therefore of high interest to the pharmaceutical industry.143 We have recognized three synthetic strategies giving access to α‐aryl amides, all of them adopting transition metal‐chemistry to mediate the cross‐coupling between the αC(sp3) carbon of the amide substrate and the αC(sp2) carbon belonging to an aryl ring of the reagent partner.
We have grouped the copious literature references according to the kind of pre‐functionalization presents on the aryl reagents.
Thus, in sub‐Section 9.1. we discuss methods where aryl halides are involved, whereas sub‐Section 9.2. deals with strategies exploiting aryl organometallic species, and sub‐Section 9.3. with technologies where unactivated arenes are the substrates for the cross‐coupling reactions.
9.1. Aryl Halides as Starting Materials
When α‐arylated amides are targeted for the synthesis according to a conventional Buchwald‐Hartwig reaction for carbon‐aryl bond formation, then alkali metal amide enolates are engaged in the slow transmetalation step with an aryl‐Pd(II) complex. Essentially due to the need of using strong bases (mandatory in order to produce amide enolates) and high temperatures, the intermolecular α‐arylation of amides presents several complications such as catalyst decomposition, quenching of the starting enolate by the α‐arylated product, and side reactions leading to diarylation products. To alleviate these drawbacks, Hartwig and co‐workers144 developed conditions that were more neutral than those for reactions of aryl halides with alkali metal enolates. Specifically, they succeeded in the α‐arylation by crosscoupling preformed zincamide enolates (Reformatsky's reagents) with aryl bromides at room temperature in the presence of the palladium catalyst formed from [Pd(dba)2] and pentaphenyl(di‐tert‐butylphosphino)ferrocene (Q‐Phos) as the optimal ligand. The Reformatsky's reagents could be prepared both from the α‐bromo amides or by quenching of an alkali metal amide enolate with zinc halide. Albeit the high functional group tolerance of zinc enolates gave a valuable contribution to the scope of the coupling processes, the reported reactions were limited to those of N,N‐diethylacetamide and N,N‐diethylpropionamide. The above encouraging results prompted Hartwig and co‐workers to develop an α‐arylation method where the Reformatsky reagents were generated in situ, thereby avoiding their problematic isolation (Scheme 120).
Scheme 120.
Hartwig's method for the α‐arylation of α‐bromoamides A with in situ generated Reformatsky reagents under Pd‐catalysis (ref. [144, 145] – Section 9.1).
Thus, the amide enolates prepared by adding activated zinc metal to a solution of the α‐bromoamides A at room temperature, could be converted into α‐arylated amides C following reaction with B under Pd‐catalysis.
The scope and limitations of the mild protocol were discussed in a full paper by Hartwig and co‐workers.145 Notably, acyclic acetamides, propionamides, isobutyramides, and morpholine amides A cross‐coupled with aryl halides B that were electron‐rich or electron‐poor including compounds containing protic and electrophilic functional groups incompatible with strongly basic or nucleophilic enolates.
Interestingly, a microwave accelerated reaction protocol for achieving α‐arylation of amides using zincamide enolates as nucleophilic partners of aryl bromides under palladium catalysis was also established.146
Molecules containing an α,α‐difluorobenzyl unit (ArCF2‐) are of great importance for use in medicinal chemistry as the two fluorine atoms are contained at a metabolically labile benzylic position. Therefore, the development of efficient protocols for the introduction of the CF2CONR1R2 moiety onto aryl/heteroaryl nuclei is a topical research interest.147
In 2014 Hartwig and co‐workers proposed an air‐ and moisture‐stable palladacyclic complex containing the sterically hindered P(t‐Bu)2Cy ligand to effect the crosscoupling between α,α‐difluoro‐α‐(trimethylsilyl)acetamides and aryl or heteroaryl bromides.148 The coupling reactions were conducted at 100 °C in dioxane/toluene solvent system using KF as activator of the nucleophilic enolates (Scheme 121).
Scheme 121.
Crosscoupling reaction between α,α‐difluoro‐α‐(trimethylsilyl)acetamides and aryl or heteroaryl bromides using a Palladacycle complex (ref. [148] – Section 9.1).
A broad range of aryl and heteroaryl bromides B, including bromopyridines, bromoquinolines, and bromoisoquinolines were efficiently coupled with α,α‐difluoro‐α‐(trimethylsilyl) tertiary acetamides A (2.0 equiv.). The latter compunds were easily prepared by reaction of TMSCl with the Grignard reagents derived from the corresponding α‐chloro amides. Interestingly, when aryl halides bearing both bromo and chloro substituents were the substrates, then the reaction occurred selectively at the bromide leaving the C−Cl bond intact. Importantly, the high electrophilic nature of the difluoroamide moiety allowed the facile conversion of the coupling products C into fluorinated alcohols, amines, ketones, aldehydes, and acids. In addition to alkenyl iodides, aryl iodides too were suitable partners (although to a lesser extent) of iododifluoroacetamides in the Hu's copper‐mediated cross‐electrophile coupling method (Scheme 107, Section 8.).135 Several α‐arylated difluoro acetamides C could be obtained in moderate to good yields reacting iododifluoroacetamides A and aryl iodides B in DMSO and in the presence of Cu powder (Scheme 122).
Scheme 122.
Cu(0)‐mediated cross‐electrophile coupling of iododifluoroacetamides A with aryl iodides B (ref. [135] – Section 9.1).
9.2. Aryl Organometallic Reagents as Starting Materials
Herein, complementary strategies to those grouped in sub‐Section 9.1. are discussed. In fact, aryl organometallic species (in the following arylboron, arylsilicon, arylmagnesium, and arylzinc) are the reagents partners of α‐halo amides, the use of electrophiles with C(sp 3)‐X bonds being a major synthetic challenge in transition metal catalyzed cross‐coupling reactions.
The reactions between α‐bromoacetic amides and arylboronic acids or esters in the presence of palladium catalysts had been firstly studied by Goossen and Deng.
Goossen149 obtained the N‐phenylacetylpiperidine in 81 % yield through palladium catalyzed cross‐coupling reaction between N‐bromoacetylpiperidine and phenylboronic acid in THF. The bulky, moderately electron‐donating tri(1‐naphthyl)phosphine [P(Nap)3] was the optimal ligand. Both Pd(OAc)2 and Pd2(dba)3 were used as Pd(0) precursors, while either K2CO3 or K3PO4 were equally efficient bases.
Interestingly, Duan and Deng150 found that adding Cu2O and PPh3 to palladium‐tetrakis(triphenylphosphine) [(Pd(PPh3)4] it was formed a catalyst‐system particularly active for the Suzuki cross‐coupling reaction between bromoacetamides A and phenylboronic acids B, establishing a mild and general approach to aryl acetamides C (Scheme 123). However, modest to good yields of the aimed products could be obtained, provided that tertiary or secondary amides were the substrates.
Scheme 123.
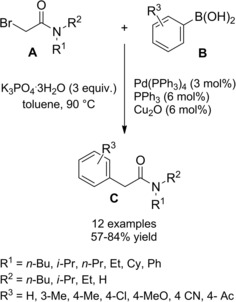
Suzuki cross‐coupling reaction between bromoacetamides A and phenylboronic acids B in the presence of Cu2O‐PPh3 (ref. [150] – Section 9.2)
Ni‐catalysts are less expensive than Pd‐catalysts, in addition, alkyl nickel complexes formed in the Suzuki catalytic cycle are less prone to β‐H elimination compared to the palladium ones. Thus, Ni‐based catalysis approaches for the α‐arylation of haloamides were explored with success.
Ni(PPh3)4 in the presence of K3PO4 served as a highly effective and versatile catalytic system for the arylations of α‐bromocarbonyl compounds with various arylboronic acids B in toluene (Scheme 124).151 Noteworthy, α‐bromoamides A bearing β‐hydrogens as well as free NH were anyway fairly compatible with the reaction conditions providing a variety of arylated derivatives C.
Scheme 124.
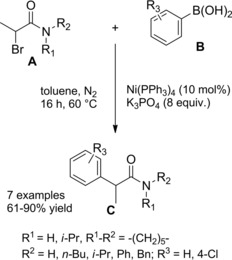
Cross‐coupling reaction between α‐bromoamides A and arylboronic acids B using a Ni‐catalyst (ref. [151] – Section 9.2).
In 2010, Lundin and Fu152 described a nickel‐catalyzed stereoconvergent method for the enantioselective Suzuki arylation of racemic α‐haloindoline amides A with Ar‐(9‐BBN) derivatives B (1.5 equiv.) (Scheme 125). The cross‐coupling reaction providing C in good yield and enantioselectivity required the additive i‐BuOH and the base KOt‐Bu together with NiBr2 ⋅ diglyme and the ligand (1 S,2 S)‐N,N′‐dimethyl‐1,2‐bis[3‐(trifluoromethyl)phenyl]ethanediamine. Both the pre‐catalyst and the chiral ligand L* were commercially available components. Notably, both enantiomers of the racemic starting material were preferentially converted into the same enantiomer of the product C. The coupling products could be transformed into useful enantio‐enriched α‐arylcarboxylic acids and primary alcohols without racemization.
Scheme 125.
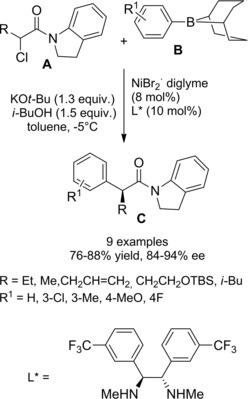
Nickel‐catalyzed stereoconvergent method for the enantioselective Suzuki arylation of racemic α‐haloindoline amides A with Ar‐(9‐BBN) derivatives B (ref. [152] – Section 9.2).
Lu and co‐workers153 developed a copper‐mediated cross‐coupling between N,N‐diethyl iododifluoroacetamide and various substituted phenylboronic acids. The reaction tolerated a wide range of functional groups on the aromatic ring, and could be easily scaled up (Scheme 126). Unlike most Suzuki‐Miyaura cross‐coupling reactions, the Lu's copper‐mediated protocol required neither bases nor ligand. Instead, the presence of air was very important for the coupling to occur. Thus, a mixture of arylboronic acids B (1.5 equiv.), iododifluoroacetamide A (1 equiv.), and Cu powder (1 equiv.) put in a solvent mixture of DMSO/DMF (5 : 1) under O2 atmosphere provided the aimed compounds C in moderate yields.
Scheme 126.
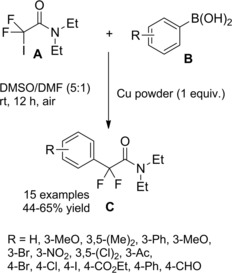
Copper‐mediated Suzuki‐Miyaura cross‐coupling reactions of iododifluoroacetamide A with arylboronic acids B (ref. [153] – Section 9.2).
In the proposed reaction mechanism, arylboronic acid B is first converted to arylcuprate [ArCu] in the presence of Cu powder and air (Scheme 127). In the following step the cuprate reacts with iododifluoroacetamide A by either an oxidative addition/reductive elimination mechanism or a nucleophilic substitution via a halogen “ate” intermediate to form the unstable organo‐cuprate [Et2NCOCF2Cu]. The latter is the active species introducing the CF2CONEt2 residue onto the aryl nucleus through oxidative arylation with arylboronic acid in the presence of air.
Scheme 127.
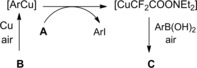
Mechanistic hypothesis for the Copper‐mediated Suzuki‐Miyaura cross‐coupling reactions (ref. [153] – Section 9.2).
In 2013, Molander and co‐workers154 developed an attractive Suzuki‐Miyaura cross‐coupling reaction exploiting the air and moisture stable potassium aryl(hetero)aryltrifluoroborate salts B as nucleophilic partners of a variety of α‐chloro tertiary amides A (Scheme 128). XPhos‐Pd−G2 was the Pd precatalyst used in the presence of Cs2CO3 in a 4 : 1 THF/H2O solvent system. It was reported that the method could also encompass secondary α‐chloroamides as suitable substrates by adding Cu2O (5 mol%) to the reaction mixture. Importantly, the reactions showed a wide substrate scope (a variety of functional groups and heterocyclic compounds were tolerated) leading to α‐arylated amides C in moderate to good yields using neither a large excess of organoboron reagents nor strong bases and inert atmosphere.
Scheme 128.
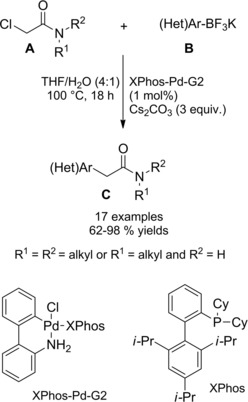
Suzuki‐Miyaura cross‐coupling reaction of α‐chloro tertiary amides A with potassium aryl(hetero)aryltrifluoroborate salts B (ref. [154] – Section 9.2).
Zhang and co‐workers155 first established in 2014 a palladium‐catalyzed cross‐coupling of aryl boronic acids B with bromodifluoroacetamides A providing aryldifluoroacetamides C (Scheme 129). The best catalyst system was [PdCl2(PPh3)2] together with the co‐promoter CuI and the bidentate diphosphine ligand Xantphos (10 mol%). The coupling reactions were performed in dioxane at 80 °C using K2CO3 as the base. Preliminary mechanistic studies suggested that a SET was probably involved in the reaction pathway.
Scheme 129.
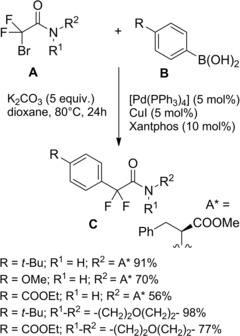
Palladium‐catalyzed difluoroalkylation of aryl boronic acids in the presence of CuI (ref. [155] – Section 9.2).
Meanwhile, the same research group156 disclosed that bromo and chlorodifluoroalkyl derivatives, including amides A, could cross‐couple to phenylboronic acid B with the intermediacy of the low costing nickel‐catalyst [Ni(NO3)2] in the presence of the ligand 2,2’‐bipyridine (bpy) (Scheme 130). The new protocol paved the way to highly cost‐efficient synthesis of a wide range of difluoroalkylated arenes C.
Scheme 130.
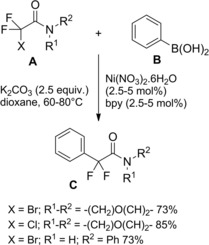
Nickel‐catalyzed difluoroalkylation of aryl boronic acids (ref. [156] – Section 9.2).
A reaction mechanism involving a Ni(I)/Ni(III) catalytic cycle where the transmetalation between [Ni(I)Ln] and aryl boronic acid B is the starting step was proposed (Scheme 131). The resulting [Ph−Ni(I)] complex through oxidative addition onto the fluoroalkyl halide A generates an aryl‐fluoroalkyl‐Ni(III) complex. Eventually, a reductive elimination releases the coupling product C while restoring the Ni(I) active catalytic species.
Scheme 131.
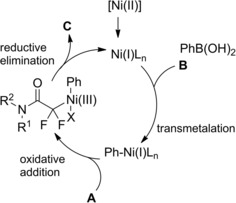
Mechanistic hypothesis for the Nickel‐catalyzed difluoroalkylation of aryl boronic acids (ref. [156] – Section 9.2).
The Hiyama cross‐coupling reaction between F3SiPh B and either unactivated or activated secondary alkyl halides including N‐2‐cloropropanoyl morpholine A provided the 2‐phenylpropanamide C in the presence of a nickel/norephedrine‐based catalyst (Scheme 132).157
Scheme 132.
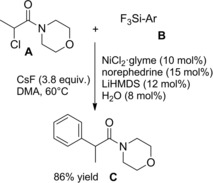
Cross‐coupling reaction between F3SiPh B and N‐2‐cloropropanoyl morpholine A in the presence of a nickel/norephedrine‐based catalyst (ref. [157] – Section 9.2).
The non‐toxic economical catalyst Fe(acac)3 was effective in the cross‐coupling reaction between aryl Grignard reagents and α‐bromocarboxylic acid derivatives including N,N‐diethylbromoacetamide (Scheme 133).158 The iron‐catalyzed reaction employing the p‐tolyl Grignard reagent B (1.5 equiv.) and the α‐bromo amide A in THF at −78 °C furnished the expected cross‐coupling product C in moderate yield (44 %) probably via bare ferrate species not bearing any auxiliary ligand.
Scheme 133.
Iron‐catalyzed cross‐coupling reaction between aryl Grignard reagents and N,N‐diethylbromoacetamide (ref. [158] – Section 9.2).
α‐Bromo tertiary amides A cross‐coupled with aryl Grignard reagents B following a protocol reaction developed by Cossy and co‐workers that used an equimolar mixture of CoCl2 and Xantphos as the optimal catalyst system in THF (Scheme 134).140
Scheme 134.
Cobalt‐catalyzed cross‐coupling reaction between aryl Grignard reagents B and α‐bromo tertiary amides A (ref. [140] – Section 9.2).
Optimized conditions for the cross‐coupling products C formation, effective also on a gram scale, entailed a B/A ratio of 2 : 1 and reaction temperatures in a range from 0 °C to rt; higher than the one in the related cobalt‐catalyzed α‐alkenylations (see Section 8.). Various substituents were tolerated at para and metha positions of the phenyl nucleus in the Grignard reagents, whereas an ortho methyl led the yield to drop. Moreover, a free NH of a secondary amide substrate led the expected coupling product to form in poor yield.
Excluding a radical mechanism, the oxidative addition of a reduced aryl‐cobalt species (I) to the C−Br bond of substrate A was hypothesized (Scheme 135).
Scheme 135.
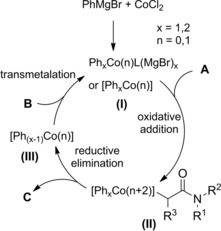
Mechanistic hypothesis for the Cobalt‐catalyzed cross‐coupling reaction between aryl Grignard reagents B and α‐bromo tertiary amides A (ref. [140] – Section 9.2).
The resulting cobalt‐complex (II) delivering the cross‐coupling product C via a reductive elimination is converted into complex (III). To close the catalytic cycle, transmetalation of (III) with the aryl Grignard reagent B regenerates the active catalyst (I).
Ando and co‐workers159 developed a nickel‐catalyzed α‐arylation of bromodifluoroacetamides using arylzinc reagents according to a Negishi coupling (Scheme 136). The reaction requiring both the bisoxazoline ligand and the additive tetramethylethylenediamine (TMEDA), smoothly proceeded in a short time in THF and at low temperature.
Scheme 136.
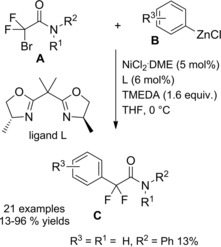
Nickel‐catalyzed cross‐coupling reaction between arylzinc reagents B and bromodifluoroacetamides A (ref. [159] – Section 9.2).
A wide variety of organozinc chlorides (3 equiv.) B reacted with structurally diverse bromodifluoroacetamides A giving rise to the corresponding products C in high yields. Both cyclic and acyclic tertiary amides, as well as N−H‐containing bromodifluoroacetamides were good partners of the arylzinc chlorides. However, some restrictions with respect to either the reagents were observed; thus, N‐aryl and primary amides were poor substrates, whereas alkyl and heteroarylzinc reagents were not tolerated.
9.3. Unactivated Arenes as Starting Materials
A classical approach to the oxindole systems is the Friedel‐Crafts cyclization of α‐haloacetanilides. As early as the 1930s, Stollè and co‐workers160a–160b used AlCl3 to promote cyclization from α‐halo anilides. Although meritorious, as a specifically functionalized precursor was not required, the strategy needed strong Lewis acids and high temperatures so limiting the range of tolerated functional groups.161
As mentioned in sub‐Section 7.1. (Scheme 77), silver‐promoted ionization of α‐bromo amides has been conveniently used as source of putative α‐carbonyl carbocation intermediates able to intercept carbon‐based nucleophiles. Thus, the unactivated arene B could act as nucleophile toward the α‐bromoamide A in the presence of AgOTf providing the Friedel‐Crafts reaction product C (Scheme 137).111
Scheme 137.
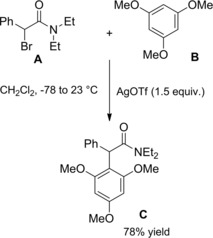
AgOTf‐promoted Friedel‐Crafts reaction of the α‐bromoamide A with unactivated arene B (ref. [111] – Section 9.3)
The intramolecular version of this scheme has been also investigated as a way to obtain heterocyclic compounds. Thus, oxindoles and dihydroisoquinolinones B with a range of substitution patterns were generated smoothly from the corresponding α‐bromoamides A under Ag(I)‐mediated conditions (Scheme 138).111 Unfortunately, attempts to develop diastereoselective variants of the intra‐ and intermolecular Friedel‐Crafts reactions using oxazolidinone‐based chiral auxiliaries have proved to be elusive.
Scheme 138.
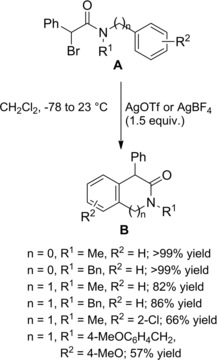
AgOTf‐promoted intramolecular Friedel‐Crafts reaction (ref. [111] – Section 9.3).
In 2003 Hennessy and Buchwald162 developed an important variant of the intramolecular Friedel‐Crafts procedure using palladium‐catalyzed aryl C−H functionalization, obviating the need for harsh reaction conditions (Scheme 139). Thus, readily available α‐chloroacetanilides A could be smoothly converted to oxindoles B in high yields in the presence of catalytic amounts of palladium acetate, the JohnPhos ligand, and triethylamine as a stoichiometric base. A variety of substituents on the aromatic nucleus were well tolerated including both EWG groups and functional groups incompatible with strong Lewis acids (−OMe, −CF3, −TMS). In such a way, the method was suitable for the preparation of substituted oxindoles, otherwise difficult to obtain using classical Friedel‐Crafts methodology. Moreover, substrates that could result in two distinct products cyclized with high regioselectivity (substitution to the 6‐position being preferred). However, the substrate scope was limited to N‐alkyl or N‐arylchloroacetanilides, N‐H or N‐acyl (amide, carbamate) containing substrates were unable to cyclize.
Scheme 139.
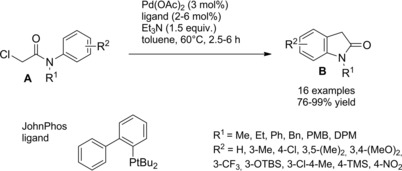
Intramolecular Friedel‐Crafts procedure using Pd‐catalyzed aryl C−H functionalization (ref. [162] – Section 9.3).
Three mechanistic hypothesis were advanced, all of them entailing an initial Pd(0) oxidative addition onto the C(sp 3)−Cl bond of the α‐chloroacetanilide A (Scheme 140). The resulting Pd(II) enolate (I) could be addressed to the oxindole system B via a carbopalladation/anti‐β‐hydride elimination sequence. An electrophilic palladation/reductive elimination or a C−H activation/reductive elimination, both the sequences entailing the intermediacy of a six‐membered palladacycle, were otherwise proposed.
Scheme 140.
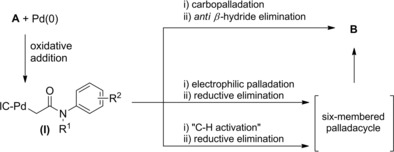
Mechanistic hypothesis for the intramolecular Friedel‐Crafts procedure using Pd‐catalyzed aryl C−H functionalization (ref. [162] – Section 9.3).
The Buchwald's palladium‐catalyzed aryl C−H functionalization served to establish the oxindole unit of the oxazolidinone Linezolid (Scheme 141).163
Scheme 141.
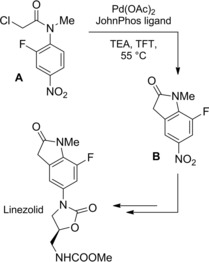
Pd‐catalyzed aryl C−H functionalization performed in trifluorotoluene (TFT) (ref. [163] – Section 9.3).
However, due to the base sensitivity of the oxindole product B, especially at elevated temperatures, the classical protocol led to its serious decomposition. Interestingly, switching the solvent from toluene to trifluorotoluene (TFT) gave complete reaction after 5 h at reduced temperature (55 °C). Moreover, decomposition of the oxindole intermediate B was minimized as it precipitated while forming. The modified Buchwald's conditions resulted in an improved yield and purity of the aimed compound while eliminating the need for an untenable purification. In such a way, the antibacterial Linezolid could be achieved in a kilogram‐scale starting from the chloroacetamide A.
A high‐yielding synthetic approach to a serine palmitoyl transferase inhibitor (SPT) took advantage of Buchwald's palladium‐catalyzed aryl C−H functionalization to convert the α‐chloroacetanilide A into kilogram quantities of oxindole B (Scheme 142).164 The latter compound was a key intermediate en route to the enzyme inhibitor with potential interest in the treatment of heart disease. To note, the use of 2‐methyltetrahydrofuran (MeTHF) and isopropyl alcohol (IPA) in 4 : 1 ratio as the solvent provided a more homogeneous reaction mixture, thus overcoming stirring issues associated to the use of toluene.
Scheme 142.
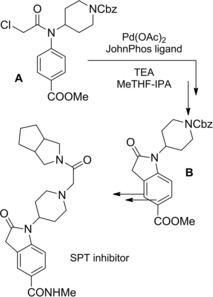
Pd‐catalyzed aryl C−H functionalization performed in MeTHF‐IPA solvent mixture (ref. [164] – Section 9.3).
The Buchwald intramolecular palladium‐catalyzed aryl C−H functionalization has been more recently applied to the preparation of a wide range of biologically relevant substituted 3,3‐difluoro‐oxindoles (Scheme 143).165
Scheme 143.
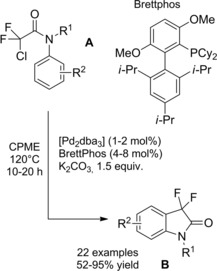
Preparation of substituted 3,3‐difluoro‐oxindoles via Pd‐catalyzed aryl C−H functionalization (ref. [165] – Section 9.3).
The reaction entailing the cyclization of chlorodifluoroacetanilides required the use of the BrettPhos as the exclusive bulky biarylphosphine ligand in the presence of the palladium dibenzylideneacetone in cyclopentyl methyl ether (CPME). A series of chlorodifluoroacetanilides A with electron‐rich, electron‐neutral, and electron‐deficient substituents on the aryl group were found to undergo the desired transformation; heterocycle substrates too encompassed the scope of the reaction affording the corresponding difluorooxindoles B in good yield. Unexpectedly, the cyclization of unsymmetrical indole and carbazole substrates occurred preferentially at the most sterically hindered position (Scheme 143). Studies based on kinetic isotope effects suggested that formation of the six‐membered palladacycle (III) could indeed proceed by an electrophilic aromatic substitution process occurring onto the Pd(II) enolate intermediate (I) (Scheme 144). Loss of a proton from the resulting carbenium (II) generates (III) which is converted to oxindole B with recycling of Pd(0) via reductive elimination.
Scheme 144.
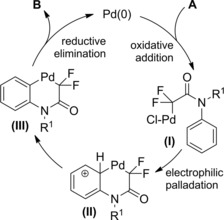
Mechanistic hypothesis for the Pd‐catalyzed difluorooxindoles B synthesis via Friedel‐Crafts cyclization of chlorodifluoroacetanilides A (ref. [165] – Section 9.3).
Visible light photoredox catalysis with transition metal complexes requiring neither complex ligands nor high temperature has been applied to the development of a wide range of new carbon‐carbon bond forming reactions including alkylation of arenes.166
Yu and co‐workers167 reported an efficient protocol for the synthesis of 3,3‐disubstituted oxindoles B from 2‐bromoanilides A based on fac‐(ppy)3Ir‐mediated visible light photoredox catalysis (Scheme 145). A prerequisite for the reaction to take place is the presence of an electron‐withdrawing group at the α‐position of the bromoanilides. The procedure results advantageous in terms of high yield, mildness of reaction conditions (irradiation with simple household lightbulbs) and tolerance of functional groups. Notably, a bromo atom on the phenyl ring, which is liable to loss with conventional free radical methods, is instead tolerated under visible light photoredox catalysis.
Scheme 145.
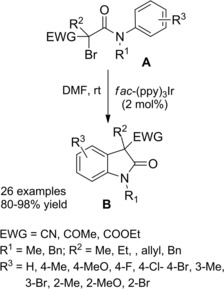
Synthesis of 3,3‐disubstituted oxindoles B from 2‐bromoanilides A based on fac‐(ppy)3Ir‐mediated visible light photoredox catalysis (ref. [167] – Section 9.3).
According to the proposed reaction mechanism, the bench stable Ir(III)‐complex, upon photoexcitation with visible light, is converted into fac‐(ppy)3Ir(III)* which is a very potent single‐electron‐transfer reagent (Scheme 146). In the next step the photoexcited complex is engaged in the reduction of the 2‐bromoanilide A. While generating the free amidoalkyl radical (I), the SET process produces fac‐(ppy)3Ir(IV). After intramolecular homolytic aromatic substitution reaction, intermediate (I) is converted into the cyclohexadiene radical intermediate (II) which interacts with fac‐(ppy)3Ir(IV). The SET process restores the active catalyst and generates a benzenonium ion from which the oxindole B is obtained by facile deprotonation.
Scheme 146.
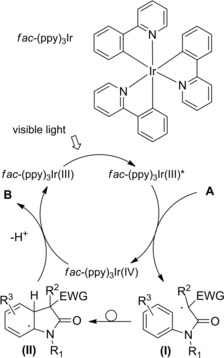
Mechanistic hypothesis for the fac‐(ppy)3Ir‐mediated visible light photoredox catalysis (ref. [167] – Section 9.3).
The Liu research group too turned to the photocatalyst fac‐(ppy)3‐Ir in order to make both inter‐ and intramolecular C(sp3)−C(aryl) bonds.168a–168b Thus, the introduction of a difluoroacetamide moiety onto unactivated arenes was reported in 2014 (Scheme 147). The photocatalytic process for the coupling of aryls(heteroaryls) B and tertiary as well as secondary bromo‐difluoroacetamides A proceeded smoothly at room temperature giving the expected products C in good to excellent yields. The visible‐light‐driven protocol displayed a broad scope toward aromatic and heteroaromatic systems with a wide range of functional group tolerance. Interestingly, due to the electrophilic nature of the key aminocarbonyldifluoromethyl radical intermediate, it was preferred its positioning onto the aryl ring at the site with greater electron density.
Scheme 147.
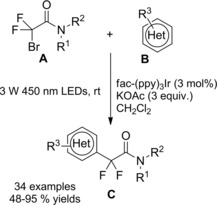
Visible‐light‐driven protocol for the introduction of a difluoroacetamide moiety onto unactivated arenes (ref. [168a] – Section 9.3).
After two years the fac‐Ir(ppy)3‐mediated visible light photoredox catalysis was successfully used in the synthesis of 3,3‐difluoro‐oxindoles B by intramolecular reaction of readily available 2‐bromo‐2,2‐difluoro‐N‐phenylacetamides A (Scheme 148).168b A broad array of substituents on the phenyl ring was tolerated albeit arenes bearing electron‐withdrawing substituents performed in a better way. The visible‐light‐driven protocol involved a Ir(III)/Ir(IV) catalyst cycle overlapping the one proposed by Yu and co‐workers (Scheme 146).167
Scheme 148.
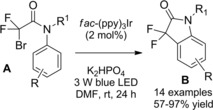
Synthesis of 3,3‐difluoro‐oxindoles B via visible‐light‐driven cyclization of A (ref. [168b] – Section 9.3).
The already discussed copper‐mediated methodology for the difluoroacetamidation of both alkenyl and aryl iodides, disclosed by Hu and co‐workers (Scheme 107, Section 8. and Scheme 122, Section 9.1.), was also effective onto unactivated arenes, provided that the reaction was intramolecular.135 Thus, oxindole derivatives B were obtained in moderate yield by heating DMSO solution of N‐disubstituted‐N‐aryl iododifluoroacetamides A in the presence of Cu powder (Scheme 149). As a plausible reaction mechanism, a gem‐difluoromethyl radical generated by the Cu‐catalyst is trapped intramolecularly at the ortho position of the phenyl ring. Interestingly, when secondary acetanilides A (R1=H) were the substrates, then homo‐coupling products were formed instead of oxindole derivatives. It has been proposed that N‐H containing substrates could permit the formation of the corresponding Cu(I) salt, thus modifying the reaction course.
Scheme 149.
Copper‐mediated intramolecular difluoroacetamidation and homo‐coupling reactions (ref. [135] – Section 9.3).
A variety of substituted oxindoles has been prepared via a nickel‐catalyzed aromatic C−H intramolecular alkylation of N‐disubstituted‐2‐bromo‐anilides (Scheme 150).169 The reaction proceeded both with tertiary and the more challenging secondary alkyl‐bromides A enabling the construction of 3‐mono‐ or 3,3‐disubstituted oxindoles B. Various functional groups were well tolerated and meta‐substituent‐containing substrates A gave the 6‐substituted together with the 4‐substituted oxindoles B in a 2 : 1 ratio.
Scheme 150.
Nickel‐catalyzed aromatic C.‐H intramolecular alkylation of N‐disubstituted‐2‐bromo‐anilides (ref. [169] – Section 9.3).
Addition of TEMPO, a known radical scavenger, to the reaction mixture under standard conditions only led to trace amount of the desired compound B. Thus, the nickel‐catalyzed aromatic C−H alkylation presumably involved radical species through a Ni(I)/Ni(II) catalytic cycle (Scheme 151).
Scheme 151.
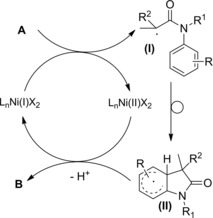
Mechanistic hypothesis for the Nickel‐catalyzed aromatic C−H intramolecular alkylation of N‐disubstituted‐2‐bromo‐anilides (ref. [169] – Section 9.3).
An unprecedented highly efficient electrosynthesis of C3‐fluorinated oxindoles B has been recently reported by Xu and co‐workers (Scheme 152).170 Very interestingly, the cyclization of the 2‐fluorinated malonate anilides A was accomplished electrochemically in an undivided cell where ferrocene (Fc) was the the redox catalyst. The reagent‐free cross‐dehydrogenative coupling (CDC) reaction proceeded smoothly to give oxindoles B in good to excellent yields with a broad substrate scope.
Scheme 152.
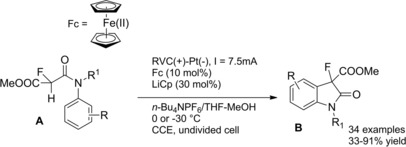
Electrochemically induced intramolecular cross‐dehydrogenative coupling reaction (ref. [170] – Section 9.3).
According to the proposed reaction mechanism, the anodic oxidation of Fc and the cathodic reduction of MeOH gives respectively Fc+ and the methoxide anion that abstracts a proton from the fluorinated carbon (Scheme 153).
Scheme 153.
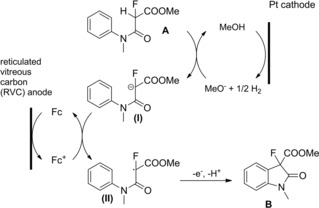
Mechanistic hypothesis for the electrochemical conversion of the anilides A into oxindoles B (ref. [170] – Section 9.3).
The resulting carbanion (I) is then converted to the corresponding radical (II) by SET to Fc+. At this stage, it is plausible the radical species is converted to the desired oxindoles through oxidation‐deprotonation steps.
10. α‐Alkynylation of α‐Haloamides
The Oshima research team active in developing protocols for the cross‐coupling of α‐iodo carbonyl compounds with both allyl‐ and alkenyl gallium (and indium) reagents (Scheme 75, Sections 7.1 and Scheme 105, Section 8 respectively)109 disclosed that alkynyl gallium reagents too could be successfully coupled with α‐haloamides under standard conditions (Scheme 154).171 Thus, terminal acetylenes prior metalated with butyllithium were added with gallium trichloride. The resulting alkynylgallium reagents B reacted in situ with N‐benzyl α‐iodo amides A in the presence of triethylborane and oxygen to give the acetylenic cross‐coupled products C.
Scheme 154.
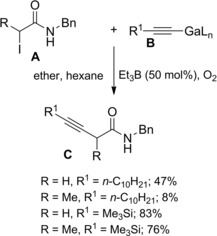
Cross‐coupling reactions of α‐iodoamides with alkynyl gallium reagents in the presence of Et3B and oxygen (ref. [171] – Section 10).
The reaction probably involved an iodine atom transfer reaction triggered by an ethyl radical in turn produced by the action of oxygen on Et3B.
Lei and co‐workers172 developed a Stille‐type protocol for the alkynylation of α‐bromo acetamides A with alkynylstannanes B in the presence of the pre‐catalyst PdCl2(PhCN)2 and the bidentate phosphine XantPhos as the ligand (Scheme 155). The method for C(sp)−C(sp 3) cross‐coupling provided tertiary as well as secondary 3‐alkynoamides C in moderate to high yields under neutral conditions. Notably, the competitive homo‐coupling reaction leading to diynes could be minimized by selecting XantPhos as the ligand for the palladium catalyst.
Scheme 155.
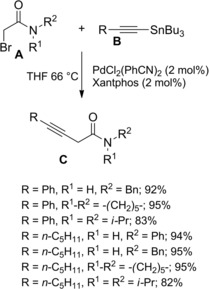
Pd‐catalyzed cross‐coupling reactions of α‐bromoacetamides with alkynyl stannanes (ref. [172] – Section 10).
Connell and Kang173 succeeded in the Stille‐type cross‐coupling encompassing secondary alkyl bromide carbamoyl derivatives A as suitable partners of alkynylstannanes B (Scheme 156). The resulting tertiary alkynyl carbonyl compounds C were obtained in moderate yields by heating a toluene solution of reactants with PdCl2(MeCN)2 as the pre‐catalyst and the bulky Xphos as the key ligand. However, the substrate scope was limited, as only tertiary α‐bromoamides and phenylethynylstannane were employed as reacting partners.
Scheme 156.
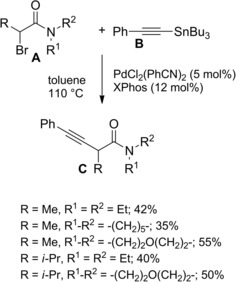
Pd‐catalyzed cross‐coupling reactions of secondary alkyl bromide amide derivatives with phenylethynylstannane (ref. [173] – Section 10).
In 2013, Molander and Traister174 reported the synthesis of β,γ‐alkynyl amides C through mild Suzuki‐Miyhaura cross‐coupling reaction conditions using low catalyst loading, air‐stable alkynyltrifluoroborates B and α‐chloroamides A (Scheme 157). Optimized conditions provided for the use of XPhos‐Pd−G2 precatalyst together with a second equivalent of XPhos (relative to Pd), K3PO4 as the base and a THF/H2O solvent mixture. With regard to the scope, both secondary and tertiary α‐chloro amides A were suited as well as a variety of alkynyltrifluoroborates B bearing aryl, straight‐chain alkyl, and branched alkyl substituents. To note, all the nucleophilic partners could be prepared from terminal alkynes in a simple and cost‐effective manner.
Scheme 157.
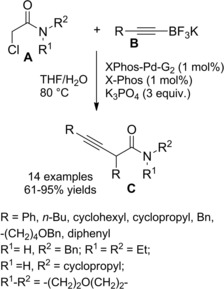
Pd‐catalyzed cross‐coupling reactions of α‐chloroacetamides A with alkynyltrifluoroborates B (ref. [174] – Section 10).
An intriguing method to synthesize alkynoates entailed on direct coupling of alkynes with α‐bromo carbonyl compounds, α‐bromo amides in this context (Scheme 158). The UV‐light‐driven reaction proceeded in aqueous medium without using any transition metal.175 Due to their low photo‐reactivity, bromoamides A were converted in situ into iodide by using the TBAI catalyst. Moreover, the addition of a mild reducing agent (Na2S2O3) helped to inhibit the competing reactions resulting from iodine radicals. The mild reaction conditions accounted for a broad functional group compatibility. Thus, various amides A, including aryl, primary aliphatic, and secondary aliphatic amides, reacted with phenylacetylene (1 : 3 molar ratio) to give the expected cross‐coupling products C with modest to good yields (40–76 %).
Scheme 158.
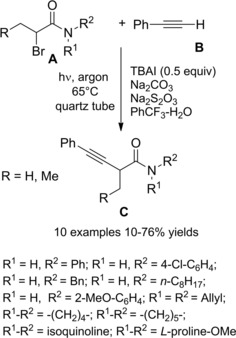
Direct coupling of phenylacetylene with α‐bromo amides (ref. [175] – Section 10).
Remarkably, the coupling compounds could be efficiently isomerized to allenoates under mild conditions (Et3N).
Recently, Nishikata and co‐workers176 developed a tert‐alkylative Sonogashira‐type coupling of α‐bromoamides with terminal alkynes requiring the sole presence of Cu(I) (Pd‐catalysts were absent) to give rise to the C(sp)−C(sp 3) bond forming process (Scheme 159).
Scheme 159.
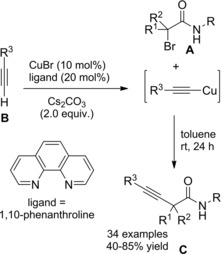
Sonogashira‐type coupling of α‐bromoamides with terminal alkynes without using Pd‐catalysts (ref. [176] – Section 10).
The cross‐coupling reaction proceeded at rt in toluene in the presence of two equivalents of Cs2CO3 by using CuBr/1,10‐phenanthroline as the optimal catalyst system. Importantly, the bidentate ligand inhibited the atom‐transfer radical addition (ATRA) onto the triple bond, that was the reaction competing with the Sonogashira coupling. The scope of substituents on the secondary amide group A was very broad, while terminal alkynes B possessing halogen, heterocycles, or a C−C double bond took part anyway to the room temperature cross‐coupling process with production of various congested alkynylated amide compounds C. The formation of a copper(I) acetylide species (I) from the reaction of CuBr and alkyne B in the presence of a base was hypothesized to start the reaction (Scheme 160). Then, the oxidative addition of (I) onto the C−Br bond of the haloamide A results in the formation of the copper(III)‐complex (II) which releases the coupling product C and re‐generates the active Cu(I)‐complex (III) in a reductive elimination step.
Scheme 160.
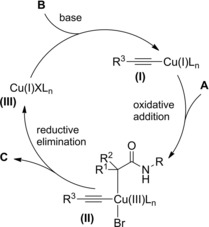
Mechanistic hypothesis for the Sonogashira‐type coupling of α‐bromoamides A with terminal alkynes B (ref. [176] – Section 10).
Lv and co‐workers established a methodology for the efficient and practical synthesis of trisubstituted 2,3‐allenamides C employing the CuI/Phen catalyst‐ligand system for the coupling reaction of 2‐bromopropanamides with terminal alkynes (Scheme 161).177 The radical‐free copper‐catalyzed process tolerated a variety of functionalized aromatic, heterocyclic, aliphatic acetylenes B and bromopropanamides A. The cross‐coupling proceeded at the best way by heating the reaction mixture in acetone in the presence of K2CO3 as the base. Interestingly, neither α‐halo‐ketones nor esters were suitable substrates. The authors proposed that alkynylated amides C’ were formed in the Sonogashira‐type coupling, with the C(sp)−C(sp 3) bond formation occurring along the steps outlined by Nishikata.176 Then, a base‐promoted rearrangement involving proton transfer from the α‐C to the γ‐C provided 2,3‐allenamides C.
Scheme 161.
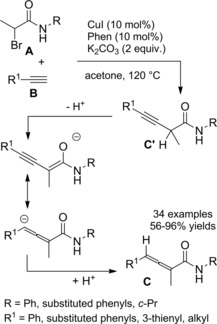
Coupling reaction of 2‐bromopropanamides with terminal alkynes yielding 2,3‐allenamides (ref. [177] – Section 10).
11. α‐Haloamides as a three‐Atom Unit for Concise Synthesis of Heterocycles
While in previous papers the αC of α‐haloamides is the only atom protagonist of the rich chemistry producing new carbon‐heteroatom (Sections 3–4) or carbon‐carbon bonds (Sections 5–10), in Section 11. we will deal with examples where both the αC atom and the adjacent carbamoyl group are involved together in the reaction process.
De facto, papers collected in sub Sections from 11.1. to 11.5. describe methods for the concise synthesis of heterocyclic systems featuring the α‐halogenated secondary amide substrates as three‐atom components in various annulation processes. As both radical and basic conditions result effective methodologies for achieving ring construction, we have arranged the copious literature in different sub‐Sections. Thus, in 11.1. and 11.2. we discuss methods involving radical species, whereas the remaining sub‐Sections 11.3.–5. deal with base‐promoted heterocyclization processes.
11.1. Carbooxygenation of Alkenes
The already discussed (Scheme 108, Section 8.)136 Ni‐catalyzed Heck‐type reaction for the cross‐coupling of olefins with tertiary and secondary alkyl bromides alpha to a carbamoyl group was also effective in cyclo‐coupling processes providing five‐membered iminolactones. Lei and co‐workers established a method for reacting 2‐bromo‐2‐methylpropanamides and alkenes with simultaneous new C−C/C−O bonds formation (Scheme 162).178
Scheme 162.
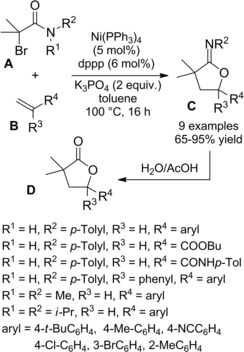
Ni‐catalyzed oxyalkylation reaction of olefins B with α‐bromoamides A (ref. [178] – Section 11.1).
Optimal conditions were the heating of reactants in toluene in the presence of the pre‐catalyst Ni(PPh3)4, the bidentate ligand dppp, and the base K3PO4. Thus, the Ni‐catalyzed oxyalkylation reaction of various olefins B, including electron‐deficient alkenes and various substituted styrenes, with NH‐containing α‐bromoamides A afforded γ‐iminolactones C in good to excellent yields. Compounds C could be converted into the corresponding γ‐lactones D following quenching of the reaction mixture by acid solution. Instead, γ‐lactones D were formed directly when tertiary α‐haloamides A were the substrates. The starting point in the catalytic cycle is the Ni(I)/Ni(II)‐controlled SET reduction of the α‐bromoamide leading to the α‐carbonyl alkyl radical (I) that ordinarily adds up to olefin B generating a γ‐carbonyl alkyl radical (II) (Scheme 163). The subsequent 5‐endo cyclization with formation of the C−O bond generates intermediate (III) where a carbon‐centered radical is bonded to two heteroatoms. DFT calculations supported this electron‐rich radical undergoes Ni(II)/Ni(I)‐mediated SET oxidation to cation (IV). Eventually, the carbooxygenation products C result by nitrogen deprotonation.
Scheme 163.
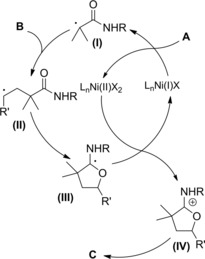
Mechanistic hypothesis for the Ni‐catalyzed oxyalkylation reaction of olefins B with α‐bromoamides A (ref. [178] – Section 11.1).
Lv and co‐workers179 developed a practical synthesis of iminolactones via a copper‐catalyzed cyclo‐coupling between styrenes B and readily available NHAr‐containing 2‐bromo‐2‐methyl propanamides A (Scheme 164). They smoothly achieved the carbooxygenated compounds C by conducting the reaction at 100 °C in acetone, under air, provided that 1,10‐phenanthroline (Phen) was the ligand and K2CO3 the base. The oxyalkylation radical process showed a wide range of functional group tolerance in styrenes and α‐bromoacetamides. Furthermore, the quenching of the reaction mixture with 2 M HCl resulted in a one‐pot two‐step tandem reaction for efficient synthesis of γ‐lactones.
Scheme 164.
Cu‐catalyzed oxyalkylation reaction of olefins B with α‐bromoamides A (ref. [179] – Section 11.1).
Investigations to probe the mechanism of the reaction supported a radical process triggered by an α‐carbonyl alkyl radical species in turn formed by SET reduction of the α‐bromoamide. The latter species evolves toward the oxyalkylated products C through a path congruent with the one proposed by Lei178 where a Cu(I)/Cu(II) catalytic cycle substitutes instead the Ni(I)/Ni(II) one.
At the same time, Song and Li180 succeeded in the oxyalkylation of p‐tolyl styrene with 2‐bromo‐2‐methyl‐N‐phenylpropanamide by using Cu(OTf)2 (10 mol%)/Phen (20 mol%)/Ag2CO3(1.0 equiv.) in MeCN/H2O (4.0 equiv.) at 120 °C. The catalyst system provided the expected five‐membered iminolactone in 58 % yield.
Nishikata and co‐workers181 succeded in the annulation reaction of acrylates B with α‐bromoamides A by using a catalyst consisting of Cu(I)‐TPMA in the presence of BnBu3NBr (probably increasing the catalyst solubility) (Scheme 165).
Scheme 165.
Cu‐catalyzed oxyalkylation reaction of acrylates B with α‐bromoamides A (ref. [181] – Section 11.1).
Iminolactones C were produced under slightly weak basic conditions in apolar solvent (tri‐ and dialkylamines in toluene). As detailed in the next Section 11.2. (Scheme 170), the above catalyst system was also employed to convert the reagent partners into γ‐lactams. Interestingly, the outcome of the copper‐catalyzed cyclo‐coupling process was decided by the strength of the base present in the reaction mixture. Various NHAr‐containing tertiary and secondary alkyl 2‐bromoamides A reacted with a broad range of acrylamides and esters B providing iminolactonization products C in good to excellent yields. The method, also effective on a gram scale, produced compounds which were easily converted into a wide range of derivatives using conventional organic reactions.
Scheme 170.
Cu‐catalyzed carboamidation reaction of acrylates B with α‐bromoamides A (ref. [181] – Section 11.2).
The divergent cyclo‐coupling process was strongly inhibited by the addition of TEMPO or BHT to the reaction mixture demonstrating that a radical process was involved. Thus, under weakly basic conditions and according to previous findings by Lv,179 the iminolactones C resulted through a multistep process commencing with the carbamoyl radical addition onto C=C double bond of the acrylate partners B. Afterward, a 5‐endo cyclization followed by an oxidation‐deprotonation step gave compound C.
The above copper‐catalyzed carbooxygenation (iminolactonization) encompassed styrene derivatives too as suitable substrates (Scheme 166).139
Intriguingly, while the carbooxygenated compound C was formed using triethylamine (2 equiv.), an olefination reaction took place in the presence of piperidine (1 equiv.) as the base (Scheme 114, Section 8.). The divergent process showed broad scope and proceeded by heating in toluene under a nitrogen atmosphere a mixture of α‐bromoamides A (1 equiv.), styrenes B (1.5 equiv.), CuI, TPMA, BnBu3NBr, and amine.
Recently, a variety of γ‐lactones has been obtained in good yields through an iron‐catalyzed radical annulation of alkenes with α‐halocarboxylic acids and derivatives, the N‐phenyl 2‐Br‐2‐methyl propanamide A being the sole amide tested in the cyclo‐coupling reaction with styrene B (Scheme 167).182
Scheme 167.
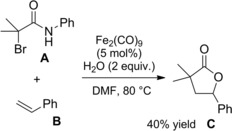
Iron‐catalyzed oxyalkylation reaction of styrene B with α‐bromoamides A (ref. [182] – Section 11.1).
Importantly, the iron‐catalyzed reaction went easily without any additional ligands, bases, or additives, the presence of deionized water significantly improving the yield. The reaction could also be conducted on gram scale employing a stoichiometric amount of the cheap iron powder albeit the nonacarbonyldiiron complex [Fe2(CO)9] (5 mol %) was found to be the best catalyst. Some mechanistic studies involving radical scavenger, radical clock, and isotope labeling experiments, suggested that radical species are involved in a reaction pathway that remained nevertheless unresolved.
11.2. Carboamidation of Alkenes
Chen and co‐workers envisioned an intramolecular nucleophilic substitution reaction taking place under basic conditions onto N‐phenyl‐γ‐iodo‐α,α‐difluoroamide derivatives as a convenient entry to γ‐lactams incorporating the biological rilevant difluoromethylene group (Scheme 168).183 The required γ‐iodoamides could in turn result from the regioselective radical addition of N‐phenyl‐iododifluoroacetamide A to terminal olefins B. Thus, the sodium hydrosulfite induced addition of A (1.0 equiv.) to monosubstituted alkenes B (2.0 equiv.) conducted in the presence of NaOH provided one‐pot the aimed 3,3‐difluoro‐5‐substituted γ‐lactams C in moderate to good yields.
Scheme 168.
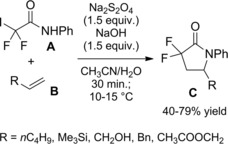
Carboamidation of alkenes B with iododifluoroacetamide A under sulfinatodehalogenation conditions (ref. [183] – Section 11.2).
As far as the mechanism for the carboamidation process it was conjectured tha sulfur dioxide radical anion from the Na2S2O4 decomposition is initially involved in the conversion of A into the radical species (I) which can add regioselectively to the alkene B (Scheme 169). The ensuing radical species (II) can abstract the iodine atom from the starting N‐phenyl‐iododifluoroacetamide A with production of radical (I) and γ‐iodoamide (II). The latter, is eventually transformed into the γ‐lactam C by deprotonation of the nitrogen atom and subsequent intramolecular alkylation occurring with expulsion of the γ‐iodine atom.
Scheme 169.
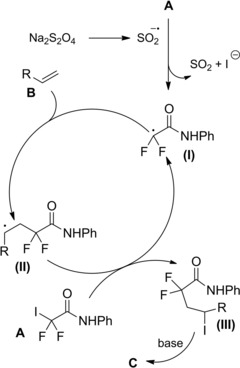
Mechanistic hypothesis for the carboamidation of alkenes B with iododifluoroacetamide A under sulfinatodehalogenation conditions (ref. [183] – Section 11.2).
As anticipated in sub‐Section 11.1. (Scheme 165),181 the Nishikata's copper‐catalyzed cyclo‐coupling of acrylates with α‐bromoamides could be addressed toward the synthesis of γ‐lactams just by switching from Et3N in toluene to K3PO4 in t‐BuNH2. Strongly basic conditions in a protic polar solvent were the conditions for controlling the ambident amide reactivity (N vs O nucleophilicity); thus, a carboamidation reaction took place (Scheme 170). The annulation reaction of acrylates B with α‐bromoamides A therefore provided the five‐membered lactams C, with almost perfect selectivities, through new C−C/C−N bonds formation.
As regard the mechanism for the lactamization reaction, it was proposed a Cu(I)/Cu(II)/Cu(III)‐catalytic cycle where a key [Cu(III)]‐metallacycle (IV) is generated by oxidative cyclization of a copper(II)‐amide complex (III) (Scheme 171). The formation of intermediate (III) occurs with the critical deprotonation of the amide NH; accordingly, secondary N‐alkyl amides are not good substrates for the transformation, presumably owing to the weaker acidity of alkyl N−H compared with aryl N−H bond. Eventually, a reductive elimination delivers lactam C while restores the Cu(I) catalyst.
Scheme 171.
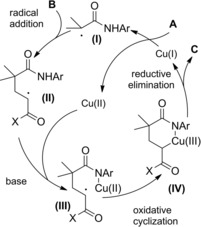
Mechanistic hypothesis for the Cu‐catalyzed carboamidation reaction of acrylates (ref. [181] – Section 11.2).
Zhu and co‐workers184 described the difluorocarboamidation of styrene derivatives B with N‐aryl bromodifluoroacetamides A by means of a dual catalyst system composed of the iridium photocatalyst Ir(III)(ppy)3 and the co‐catalyst NaI (Scheme 172). DMF was the best solvent for the cyclo‐coupling reaction that proceeded in the presence of AcONa as the base. Importantly, while the use of organic bases inhibited completely the reaction, the lack of NaI led the reaction to occur in a state of complete confusion. The method showed good substrate compatibility providing various α,α‐difluoro‐γ‐lactams C, compounds of interest for pharmacological applications.
Scheme 172.
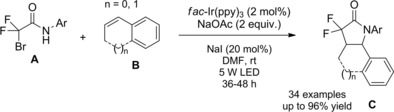
Light‐driven cyclo‐coupling reaction of styrenes B with N‐aryl bromodifluoroacetamides A (ref. [184] – Section 11.2).
It was proposed a mechanism where the SET from the photoexcited species fac‐*Ir(III)(ppy)3 to the activated C−Br bond of A generates the electrophilic radical (I) and fac‐Ir(IV)(ppy)3. The subsequent radical addition onto alkenes forms the intermediate (II) which is oxidized to the β‐difluoroalkylated carbocation intermediate (III) by SET with the Ir(IV)‐complex (Scheme 173). At this stage, the iodide ion trapping by (III) results in the formation of the electrophilic species (IV) which undergoes chemoselective nucleophilic attack by the nitrogen of the amide. Eventually, loss of a proton from (V) provides exclusively the carboamidation product C.
Scheme 173.
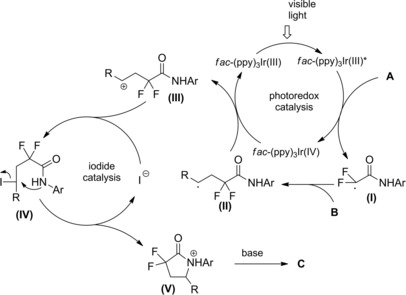
Mechanistic hypothesis for the light‐driven difluorocarboamidation of styrene derivatives (ref. [184] – Section 11.2).
The Lv research group in 2017185 reported a copper‐catalyzed external‐oxidant‐free regioselective difluorocarboamidation of alkenes (Scheme 174). Fluorinated nitrogen‐containing heterocycles C could be achieved reacting α‐bromodifluoroacetamides A and alkenes B using CuI/Phen as the catalyst system, and K2CO3 as a base. The method exhibited good functional group tolerance with respect to α‐bromodifluoroacetamides and alkenes, a variety of olefin ranging from styrenes, allylic hydrocarbons, N‐vinyl heterocyclic, alkyl vinyl ethers, aliphatic alkenes, trimethyl(vinyl)silane, and heterocyclic alkenes being suitable substrates.
Scheme 174.
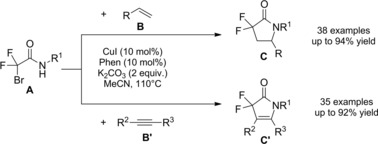
CuI/Phen‐catalyzed regioselective difluorocarboamidation of alkenes and alkynes (ref. [185, 186] – Section 11.2).
Interestingly, alkynes B’ too were suitable partners of α‐bromodifluoroacetamides A (Scheme 174).186 Thus, the protocol was also efficient for the preparation of 3,3‐difluoro‐1H‐pyrrol‐2(3H)‐ones C’ in a single step without using any extra oxidant. Remarkably, it was found that in the case of alkyne substrates not only N‐aryl amides but also N‐alkyl bromodifluoroamides worked well. The mechanism proposed for the radical cascade process follows the one by Nishikata et al. shown in Scheme 171.181
Concurrently, Wang and co‐workers found that pentamethyldiethylenetriamine (PMDETA) was an equal effective copper‐ligand in the difluorocarboamidation of alkenes (Scheme 175).187
Scheme 175.
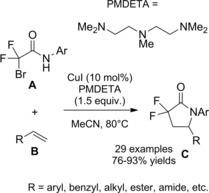
CuI/PMDETA‐catalyzed regioselective difluorocarboamidation of alkenes (ref. [187] – Section 11.2).
The optimal conditions featured reacting in CH3CN at 80 °C bromodifluoroacetamides A and alkenes B using CuI as the catalyst, and PMDETA as the base and tridentate ligand. The method showed a broad substrate scope respective to both alkenes and bromodifluoroacetamides, but was confined to N‐aryl amides, the N‐aliphatic ones being not accommodated; a result matching to the one found by Lv and co‐workers.185 Anyway, a number of α,α‐difluoro‐γ‐lactam derivatives C could be prepared in good to excellent yields.
11.3. Self and Cross Cyclo‐Condensations of α‐Haloamides
As shown in Section 3., base‐promoted conversion of α‐bromo amides into α‐lactams was made possible provided that the amide nitrogen bore a bulky group. On the contrary, substrates lacking the feature partake in self cyclo‐condensation reactions giving 2,5‐dioxopiperazines along with 2‐amino‐2‐bromoalkyl‐oxazolidinones (Scheme 176). Thus, dimerization of A under the action of NaH provided B and C, through double and single dehydrobromination respectively.[3a],[5a]
Scheme 176.
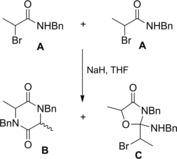
Base promoted self cyclo‐condensation reactions of 2‐bromopropanamide A (ref. [3a, 5a] – Section 11.3).
Instead, dehydrobromination of α‐bromo carboxamides A in the presence of DMF, propanamides, lactams, vinylogous amides (cyclic and acyclic enaminones), and thioamides, led to exclusive cross‐cyclo‐condensation reactions (Scheme 177).
Scheme 177.
Base promoted cross‐cyclo‐condensation reactions of α‐bromoamides A (ref. [5b–e] – Section 11.3).
Thus, various polysubstituted oxa‐ and thiazolidinones B–G could be prepared according to a formal [3+2] cycloaddition between A and the carbonyl moiety of the amide/enaminone partner reagents.5b–5e Among the cross‐cyclocondensation products, the intriguing spiro‐oxazolidin‐2‐ones F and G arose respectively from cyclic enaminones and 5‐, 6‐, and 7‐membered lactams.
The cross‐cyclo‐condensation process was also fulfilled by using an electrochemically generated base (EGB) in place of NaH. Thus, 2‐bromo amide anions (I) could be formed via electroreduction of the NH‐protic 2‐bromo amides A in DMF in the presence of a quaternary ammonium salt as the supporting electrolyte (bottom Scheme 178). A self‐protonation mechanism has been proposed. Accordingly, half of the starting bromoamide A undergoes two‐electron reduction to give the EGB carbanion and half acts as proton donor. Actually, 2‐dimethylamino oxazolidinone derivatives B could be prepared by electrochemical reduction of 2‐bromo‐carboxamides A in DMF or N,N‐dimethylacetamide (DMA) as dipolar aprotic amide solvents and reagents.188a–188c
Scheme 178.
bottom: EGB promoted cross‐cyclo‐condensation of A with DMF and DMA (ref. [188a‐c]); top: one‐pot electrosynthesis of the ester amide C (ref. [10] – Section 11.3).
The same authors disclosed a one‐pot electrosynthesis of the ester amide of 2,2‐dimethylmalonic acid C performing the electrochemical reduction in the presence of CO2 and a proper alkylating agent.10 The result could be explained by assuming a fast CO2 trapping by the EGB (top Scheme 178). The carboxylation having a rate constant 15‐fold the competing self‐protonation assures the formation of the carboxylate anion (II) efficiently alkylated in situ to provide the malonic acid derivative C in good yield.
Interestingly, Casadei and co‐workers189 established an expeditious high‐yielding electrosynthesis of oxazolidine‐2,4‐diones B performing the electrodic reduction of several secondary amides A in the presence of CO2 and suitable probases (PBs) (Scheme 179). Thus, the haloamide conjugated base (I), by capturing CO2 at the nitrogen atom, affords the carbamate intermediate (II). Eventually, the biologically relevant five‐membered heterocycles B are established through intramolecular O‐alkylation.
Scheme 179.
Electrosynthesis of oxazolidine‐2,4‐diones B through electrodic reduction of secondary amides A in the presence of CO2 and EGBs (ref.[189] – Section 11.3).
11.4. Ionic Domino Reactions
N‐substituted α‐iodoacetamides turned out useful starting material for building up erythrinan and homoerythrinan alkaloids (Scheme 180).190 The one‐pot establishment of the tetracyclic core of the natural substances entailed the alkylation of a cyclohexanone derivative with iodoacetamides A. The reaction prduced the hemiaminal (I) via simultaneous C−C and C−N bond formation. In the ensuing acidic step the intermediate compound (I) underwent a Mondon‐type cyclization191 delivering B via the N‐acyliminium ion intermediate (II).
Scheme 180.
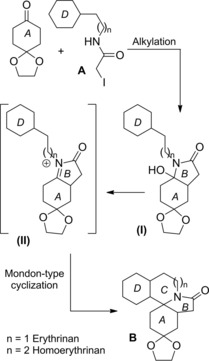
One‐pot establishment of the tetracyclic core of erythrinan and homoerythrinan alkaloids (ref. [190] – Section 11.4).
In Section 5. we introduced the diethyl malonate anion as the EGB suitable for the conversion of haloacetanilides into β‐lactams (Scheme 45).74 Intriguingly, the diazaspiro[4.4]nonane B was the product isolated conducting the electoreduction of diethyl bromomalonate in the presence of N‐benzyl‐α‐bromoacetamide A (Scheme 181). It was conjectured that the EGB could act as a nucleophile displacing bromine atom from A. The resulting α‐C‐alkylation product (I) underwent a base‐promoted cyclization‐alkylation‐cyclization domino process providing racemic bis‐succinimide product B via intermediate compounds (II) and (III). Indeed, compound B could be obtained (10 % yield) together with the 3‐ester succinimide (II) (40 % yield) in an independent synthesis entailing the reaction of diethyl malonate (1.0 mmol) and NaH (2.0 mmol) with N‐benzylbromoacetamide A (1.0 mmol) in DMF at room temperature.
Scheme 181.
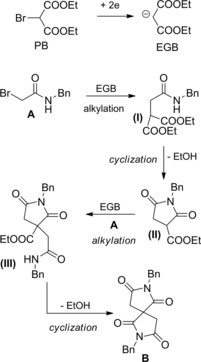
Electrosynthesis of diazaspiro[4.4]nonane B from bromoacetamide A and diethyl bromomalonate (ref. [74] – Section 11.4).
The reaction was reinvestigated 16 years later by Comesse and co‐workers with the aim of developing an efficient, easy access to symmetrical and unsymmetrical spiro‐bis‐imides, compounds with promising potential biological applications.192 In detail, K2CO3 in refluxing CH3CN transformed bromoacetamides A (2 equiv.) and dimethyl malonate (1 equiv.) into the bis‐spiro‐imides B via the one‐pot domino process (Scheme 182).
Scheme 182.
Synthesis of symmetrical and asymmetrical bis‐spiro‐imides C from α‐bromoacetamides A/A’ (ref. [192] – Section 11.4).
A two‐step sequence was also investigated in order to achieve unsymmetrical bis‐spiro‐imides. Thus, reacting bromoamides A (1 equiv.) with dimethyl malonate (2 equiv.) and NaH (1 equiv.) in anhydrous THF gave the 3‐ester succinimide B. Its reaction with various bromoacetamides A’ (1 equiv.) in the presence of K2CO3 (1.5 equiv.) in refluxing CH3CN gave the expected racemic diazaspiro[4.4]nonanes C in good to excellent yields (68‐98 %) (Scheme 182).
An aza‐Michael initiated ring closure (aza‐MIRC) was envisioned by Comesse and co‐workers as a straightforward way for achieving γ‐lactams C from N‐substituted α‐bromoacetamides A and Knoevenagel substrates B (Scheme 183).193
Scheme 183.
Synthesis of γ‐lactams C from α‐bromoacetamides A and Knoevenagel substrates B via aza‐MIRC (ref. [193] – Section 11.4).
Thus, the amidate anion addition onto substrates B followed by intramolecular alkylation afforded compounds C through a formal [3+2] cycloaddition reaction. The one‐pot tandem process provided diastereoselectively polysubstituted pyrrolidin‐2‐ones in moderate to good yields (49 to 69 %).
Even better results in the domino aza‐MIRC process were obtained by switching the Michael acceptor components to commercially available alkoxymethylene malonate derivatives B (Scheme 184).194 The formal [3+2] cycloaddition reactions provided 5‐alkoxy‐pyrrolidin‐2‐ones C which represented crucial reagents for alkaloids synthesis being direct precursors of N‐acyliminium ions.
Scheme 184.
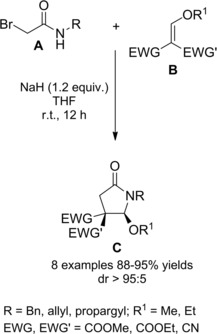
Synthesis of γ‐lactams C from α‐bromoacetamides A and alkoxymethylene malonate derivatives B via aza‐MIRC (ref. [194] – Section 11.4).
The 1,2‐dipolar properties displayed by ethoxymethylene malonate derivatives in the domino aza‐MIRC reaction could be efficiently exploited to achieve the one‐pot synthesis of Meyers bicyclic lactams (Scheme 185).195a–195c Precisely, polysubstituted oxazolo‐pyrrolidinones C were efficiently obtained allowing Michael components B to react with α‐bromoamido alcohols A in water‐free conditions and in the presence of NaH or K2CO3. Notably, tetrasubstuted Michael acceptors with unsymmetrical electron‐withdrawing groups led to the fully controlled formation of adjacent quaternary stereocenters in good yield. An Oxa‐Michael/Aza‐Michael/Cyclization domino sequence encompassing intermediates (I), (II), and (III) has been proposed for the one‐pot diastereoselective synthesis of densely functionalized oxazolo‐pyrrolidinone backbones.
Scheme 185.
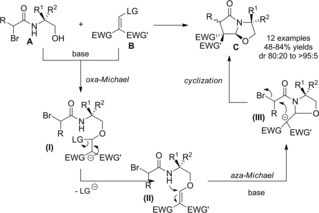
One‐pot synthesis of Meyers bicyclic lactam C from α‐bromoamides A and alkoxymethylene malonate derivatives B via Oxa‐Michael/Aza‐Michael/Cyclization domino sequence (ref. [195a–c] – Section 11.4).
More recently, in a contribute by the same research group, the enol ether moiety of B switched from Michael acceptor to a nucleophilic species simply by putting an excess of water in the medium.196 The change in reaction conditions opened an avenue to functionalized oxazolidin‐4‐one skeletons (Scheme 186). A domino process consisting of O‐alkylation/aza‐Michael/retro‐Claisen condensation was proposed as the plausible reaction pathway. Thus, the uncommon base KNaCO3 (more soluble than Na2CO3 and less basic than K2CO3) promoted the hydrolysis of the Michael acceptors B affording enolates (I) that were mandatory for the domino process. Indeed, their reaction with α‐bromoacetamide A gave enol ethers (II) which cyclized to (III) via an aza‐Michael addition. At this stage an unexpected intramolecular retro‐Claisen fragmentation with transfer of the (hetero)aroyl or alkanoyl group afforded 2,3‐disubstituted oxazolidin‐4‐ones C via the intermediate (IV). In a separate paper the same authors reported that by using Cs2CO3 as the base the polysubstituted oxazolo[3,2‐d][1,4]oxazepin‐5(3H)‐ones C/C′ were formed in good yields via an oxa‐Michael/aza‐Michael/Williamson cycloetherification domino reaction encompassing intermediates (I)‐(III) (Scheme 187).197 Thus, Cs2CO3 determined the 7‐membered ring formation in the last step of the domino process by switching nucleophilicity from carbon to oxygen in the ambident nucleophile enolate intermediate (III).
Scheme 186.
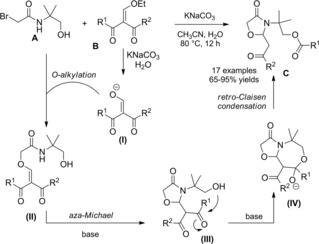
One‐pot synthesis of functionalized oxazolidin‐4‐one C from α‐bromoamide A and Michael acceptor B via O‐alkylation/aza‐Michael/retro‐Claisen domino sequence (ref. [196] – Section 11.4).
Scheme 187.
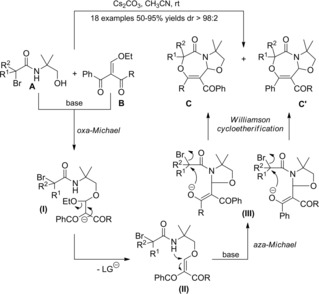
Synthesis of oxazolo[3,2‐d][1,4]oxazepin‐5(3H)‐ones C/C’ from α‐bromoamides A and Michael acceptors B via an oxa‐Michael/aza‐Michael/Williamson cycloetherification domino sequence (ref. [197] – Section 11.4).
The intriguing behavior turned out to be predominantly directed by steric effects. Thus, under mild conditions three different valuable heterocyclic skeletons, namely oxazolo‐pyrrolidinones (Scheme 185),195b oxazolidin‐4‐ones (Scheme 186),196 and oxazolo[3,2‐d][1,4]oxazepin‐5(3H)‐ones (Scheme 187)197 were easily accessible by simple tuning of the reaction conditions.
11.5. α‐Halohydroxamates as Precursors of Azaoxyallyl Cation Intermediates
Even though the existence of the aza‐oxyallylic cation was first suggested by Sheehan1 in the 1960s it was some years later that several other research groups envisaged that this zwitterion could be involved as an intermediate or at least as a transition state in transformations with α‐lactams, α‐haloamides, and related species.
As discussed in Section 3., Sheehan and Lengyel supposed that an aza‐oxyallyl cationic intermediate could be relevant to the regioselectivity trends of the nucleophilic ring‐opening reactions of α‐lactams (Scheme 28). However, experimental and theoretical studies ruled out its presence as an intermediate in these reactions.
Kikugawa and co‐workers198 demonstrated the importance of the alkoxy donor group on the nitrogen atom of N‐alkoxy‐N‐halo‐arylacetamides in order to attain their rearrangement into N‐alkoxy‐2‐ethoxyarylacetamides by ethanolysis. They suspected that the solvolysis reaction conducted in EtOH/Et3N entailed the intermediacy of an aza‐oxyallyl cation. Indeed, the existence of the aza‐oxyallylic cation remained a mere hypothesis until 2011 when Jeffrey and co‐workers199 captured it in a Diels‐Alder‐like cycloaddition reaction. Precisely, they found that N‐benzyloxy‐α‐haloamides when treated with a weak base could react with cyclic dienes giving bicyclic lactams. They anticipated that a hetero pericyclic reaction involving an aza‐oxyallyl cationic species as the pivotal dienophile was a plausible reaction mechanism. Thus, base‐mediated dehydrohalogenation of α‐halo‐hydroxamates produced in situ the putative zwitterion partaking in the aza‐[4+3] cycloaddition reaction with dienes. Computational data confirmed the Kikugawa's experimental findings pointing out that just the N‐alkoxy substituent was crucial in order to stabilize the aza‐oxyallyl cationic intermediate.
Cycloaddition reactions between N‐benzyloxy‐α‐haloamides A and either cyclopentadiene or furan B smoothly occurred in hexafluoroisopropanol (HFIP) at room temperature and in the presence of Et3N as the base (Scheme 188). The use of the fluorinated bulk solvent, with strong hydrogen‐bonding ability and excellent solvation properties, circumvented undesired solvolysis reactions providing the [4+3] cycloadducts C in good yields. The method proved to be quite general, a variety of α‐alkyl and α‐aryl substituted halohydroxamates being suitable substrates. Aryl substituents accelerated the overall rate of conversion, whereas the simple bromoacetamide was unreactive. Notably, much like the all‐carbon [4+3] cycloadditions with cyclic dienes, also the aza‐[4+3] cycloadditions involving aza‐oxyallyl cations demonstrated a kinetic preference for the endo‐cycloadducts C.
Scheme 188.
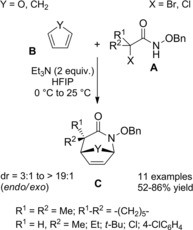
[4+3] Diels‐Alder‐like cycloaddition reactions (ref. [199] – Section 11.5).
A very nice review by Jeffrey and co‐workers,200 described the historical progress around the studies of aza‐oxyallylic cations as intermediates for the straightforward assembly of nitrogen heterocycles and polyheterocyclic scaffolds. Inter‐ and intramolecular aza‐[4+3] cycloaddition reactions with both furan and pyrrole derivatives provided fair to good yields of the cycloadducts with a bold array of functionality present in the resulting heterocyclic skeleton.
In 2018, a review by Xuan et al.201 highlighted recent advances in the rapidly growing area of heterocycloaddition reactions involving in situ generated azaoxyallyl cations. There is plenty of literature in recent years giving prominence to the 3‐atom unit intermediate as a formidable tool for concise and efficient synthesis of various heterocycles. Azaoxyallyl cations have been engaged in formal [4+3], [3+3], [3+2], and [3+1] cycloaddition processes originating seven‐membered to four‐membered heterocycloadducts respectively. Thus, six‐membered heterocycles were built up in [3+3] cycloadditions between in situ generated azaoxyallyl cations and nitrogen‐based 1,3‐dipoles (Scheme 189).
Scheme 189.
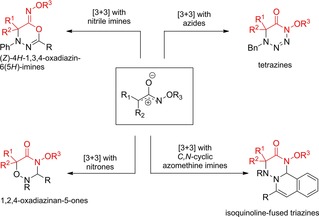
[3+3] cycloaddition reactions of in situ generated azaoxyallyl cations with nitrogen‐based 1,3‐dipoles (ref. [202–205] – Section 11.5).
Namely, nitrile imines gave 1,3,4‐oxadiazines,202 azides gave tetrazines,203 azomethine imines gave isoquinoline‐fused triazines,204 and nitrones provided 1,2,4‐oxadiazinan‐5‐ones.205a–205d
Furthermore, in a recent paper by Lin and co‐workers, N‐alkoxy‐α‐bromoamides A reacted with indoles B bearing Michael acceptors at C2 giving tetrahydro‐β‐carbolinones C through a formal [3+3] heterocycloaddition.206
Precisely, compounds A and B dissolved in HFIP reacted each other in the presence of Na2CO3 giving rise to tetrahydro‐β‐carbolinones C (Scheme 190).
Scheme 190.
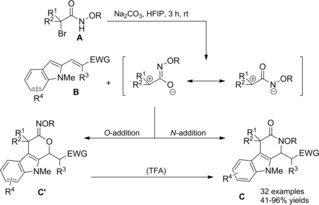
Formal [3+3] heterocycloaddition of N‐alkoxy‐α‐bromoamides A with indoles B (ref. [206] – Section 11.5).
Two possible reaction pathways were advanced for the δ‐lactam ring construction. Thus, both nitrogen and oxygen atoms of the in situ generated azaoxyallyl cation can add to the Michael acceptor moiety of B, while its indole nucleus is intercepting at C3 the electrophilic carbon center of the transient azaoxyallyl 1,3‐dipole.
Accordingly, both lactamization products C and iminolactonization products C’ can be formed with the latter ones undergoing spontaneous rearrangement (possibly accelerated by TFA) to tetrahydro‐β‐carbolinones C (Scheme 190).
Interestingly, computational studies by Gandon and co‐workers supported an oxirane‐2‐imine was more likely the intermediate involved in place of the azaoxyallyl 1,3‐dipole in a formal [3+3] heterocycloaddition with 4‐aminocyclopentenones B.207 Thus, a stepwise process via intermediate (I) was preferred over a concerted cycloaddition in forging the biological relevant skeleton of cyclopenta[b]piperazinones C (Scheme 191). In detail, the oxirane ring opening by the amine nucleophiles B afforded (I) from which cycloadducts C were formed following an intramolecular Michael reaction.
Scheme 191.
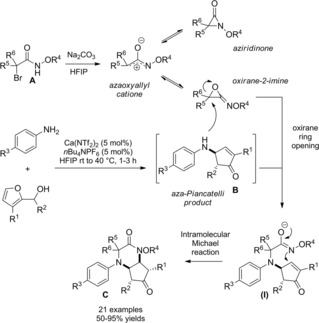
Formal [3+3] heterocycloaddition of N‐alkoxy‐α‐bromoamides A with 4‐aminocyclopentenones B (ref. [207] – Section 11.5).
The targeted highly functionalized compounds C could be achieved with complete diastereocontrol by combining the aza‐Piancatelli reaction affording intermediates B with the use of azaoxyallyl cations. The one‐pot sequence showing broad scope, converted a variety of anilines, 2‐furylcarbinols, and N‐alkoxy α‐bromoamides A into the desired cyclopenta[b]piperazinones in good to excellent yields (up to 95 %).
With regard to [3+2] cycloaddition processes, azaoxyallyl cation intermediates were key players in syntheses of γ‐lactams,208 pyrroloindolines,209a–209b 1,3‐dihydro‐2H‐pyrrol‐2‐ones,210a–210b 4‐imidazolones,211a–211b hydantoins,212 imidazolones,213 thiazolidin‐4‐ones,214 and oxazolidin‐4‐ones.215a–215e
On the other hand, azaoxyallyl cations were three‐atom components in [3+1] cycloadditions with sulfur ylides giving β‐lactams (Scheme 192).208
Scheme 192.
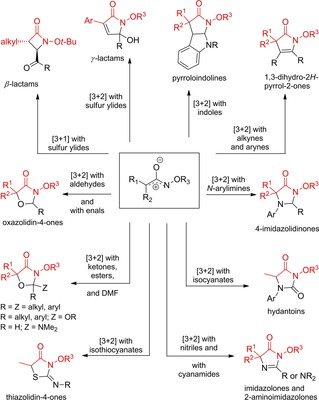
[3+2] and [3+1] cycloaddition reactions of in situ generated azaoxyallyl cations with various partners (ref. [208–215])–Section 11.5.
12. Miscellaneous
Dittmer and co‐workers established a telluride‐triggered Dieckmann cyclization of N‐protected‐N‐2‐bromoacyl chiral non‐racemic α‐aminoacid methylesters (or Weinreb amides) as an effective strategy for achieving medicinally important tetramic acid derivatives (Scheme 193).216
Scheme 193.
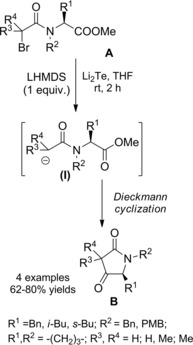
Synthesis of tetramic acid derivatives featuring a Telluride‐triggered Dieckmann cyclization of α‐bromoamides A (ref. [216] – Section 12).
The telluride protocol, not requiring strong basic conditions, could be applyed to synthesize chiral non‐racemic compounds preserving chirality of the starting aminoacid. In fact, NMR experiments supported that racemization at the C5 stereocenter of the emerging tetramic acid core was mainly avoided under reaction conditions. Thus, rapid and irreversible “nucleophilic reduction” of the α‐halocarbonyl group of A by the strongly nucleophilic telluride dianion provides the amide enolate (I) which undergoes Dieckmann cyclization to 2,4‐pyrrolidinediones B. LHMDS had to be used in order to prevent quenching of the pivotal amide enolate (I) by proton transfer from the tetramic acid or methanol products.
Pettus and co‐workers, en route towards the synthesis of Palau'imide, disclosed a general method to construct enantioselectively 3‐methyl‐4‐O‐methylated‐5‐substituted tetramic acid derivatives B (Scheme 194).6a
Scheme 194.
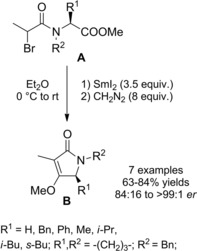
Synthesis of tetramic acid derivatives featuring a SmI2‐mediated Dieckmann cyclization of α‐bromoamides A (ref. [6a] – Section 12).
The aimed compounds could be prepared in good yields (63‐84 %) and high enantiomeric ratios by coupling a non‐basic SmI2‐mediated Dieckmann cyclization of N‐Cbz, N‐2‐bromopropionyl α‐amino acid methylesters A with the in situ enoletherification of the cyclized intermediates with diazomethane. To note, the enantiomeric integrity of compounds B could be restored to nearly >99 : 1 by a single recrystallization after cleavage of the protective Cbz group.
A practical route to the core structure of tetramic acid exploiting the advantageous of solid phase synthesis (SPS) has been recently developed by Krchňák and co‐workers (Scheme 195).6b
Scheme 195.
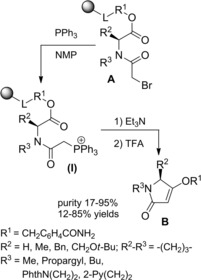
Synthesis of tetramic acid derivatives through unusual Wittig olefination of solid‐supported α‐bromoamides A (ref. [6b] – Section 12).
The method entailed assembling the acyclic resin‐bound precursors A in a modular fashion using simple building blocks and user‐friendly reaction conditions. In the key step, the resin‐bound N‐protected‐N‐bromoacetyl α‐aminoacid derivatives A are converted into the corresponding phosphonium salts (I) by treatment with PPh3 in anhydrous N‐methyl‐2‐pyrrolidone (NMP). Exposure of the resin‐bound phosphonium salts (I) to the action of Et3N in NMP promoted the unusual intramolecular Wittig olefination. Eventually, 4‐alkoxy‐1,5‐disubstituted‐1,5‐dihydro‐2H‐pyrrol‐2‐ones B were isolated by dethaching them from the resin by treatment with TFA in dichloromethane.
An efficient method for preparing biologically and medicinally interesting N‐substituted 2H‐1,4‐benzoxazin‐3‐(4H)‐ones C entailed a one‐pot two‐step reaction of 2‐chloroacetamides A with 2‐halophenols B (Scheme 196).217
Scheme 196.
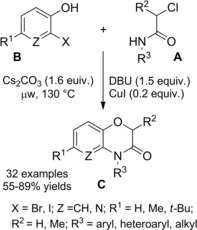
Synthesis of benzoxazinones by reaction of 2‐chloroacetamides A with 2‐halophenols B (ref. [217] – Section 12).
Thus, to a base‐promoted aryl etherification reaction, a copper(I)‐catalyzed coupling cyclization was connected in order to produce C. The transformation was complete within minutes under microwave heating (130 °C) in DMSO. Both N‐aryl‐ and N‐alkyl‐2H‐1,4‐benzoxazin‐3‐(4H)‐ones, as well as N‐heterocyclic substituted ones were obtained in moderate to good yields.
In 2017 Liu and co‐workers disclosed a valid process for the preparation of various amides with a quaternary α‐carbon stereocenter.218 In detail, N‐(arylsulfonyl)acrylamides B underwent α,β‐difunctionalization by reaction with α,α‐difluoro‐α‐(TMS)‐acetamides A in the presence of the CuBr‐mediator (Scheme 197). Featuring steps in the one‐pot process were the β‐difluoromethylation of the acrylic moiety of B, a 1,4 sulfur to carbon aryl migration, and the SO2 elimination.
Scheme 197.
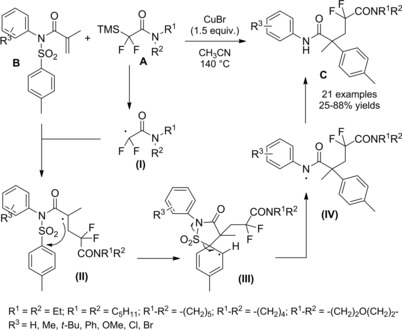
Copper‐mediated reaction providing amides C with a quaternary α‐carbon stereocenter (ref. [218] – Section 12)
The intriguing copper‐mediated reaction showed a good substrate scope providing a range of coupling products C featuring both a secondary N‐aryl amide functional group and a tertiary difluorinated one.
To note, the chemoselective manipulation of the latter functional group provided synthetically useful compounds such as difluorofunctionalized esters and alcohols in excellent yields. Mechanistic investigations suggested the radical cascade process started with a copper‐mediated conversion of A into the difluoroacetamido radical (I). The latter, following addition to the β‐carbon atom of the acrylamide moiety of B afforded the carbon‐centered radical (II) which was intramolecularly intercepted by the aryl nucleus. The 5‐exo‐dig cyclization generated the spirocyclic radical intermediate (III) from which SO2 could be eliminated. Eventually, the resulting amidyl radical intermediate (IV) gave compounds C through a hydrogen atom abstraction.
Two independent research group engaged in developing palladium‐catalyzed carbonylative fluoroalkylation reactions disclosed their results almost at the same time in 2016 (Scheme 198).219, 220 Exactly, a diverse selection of α,α‐difluoro‐β‐aryl‐β‐ketoamides C/C′ were prepared by carbonylative cross‐coupling between bromodifluoroacetamides A and either (hetero)arylboronic acids B (Zhang et al.220) or boronate esters B′ (Skrydstrup et al.219). Both strategies, proceeding under one atmosphere pressure of CO, made use of the Pd/Xantphos catalyst‐ligand system together with a copper salt as a co‐catalyst.
Scheme 198.
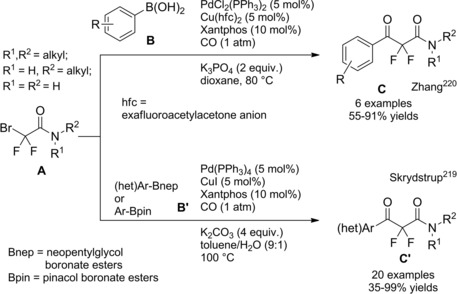
Carbonylative cross‐coupling between bromodifluoroacetamides A and either boronate esters B’ or (hetero)arylboronic acids B (ref. [219, 220] – Section 12).
Very recently, Skrydstrup et al. have successfully employed alkyl‐9‐BBN B as suitable partners of both tertiary and secondary bromodifluoroacetamides A in their Pd‐mediated carbonylative Suzuki coupling method (Scheme 199).221 Thus, a variety of α,α‐difluoro‐β‐alkyl‐β‐ketoamides C were produced in moderate to good yields by using a wide range of boron reagents B. The latter were in turn easily prepared by hydroboration of terminal alkenes with the 9‐BBN dimer. The resulting α,α‐difluoro‐β‐alkyl‐β‐ketoamides could be either transformed into different reduction products applying different reduction conditions or used to form fluorine‐containing heterocycles. Notably, 13C‐labeled α,α‐difluoro‐β‐alkyl‐β‐ketoamides, diols and heterocycles could be achieved by employing 13C‐COgen.
Scheme 199.
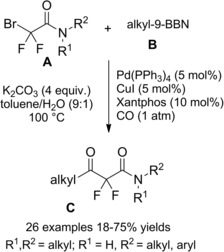
Synthesis of α,α‐difluoro‐β‐alkyl‐β‐ketoamides C through Pd‐mediated carbonylative Suzuki coupling method (ref. [221] – Section 12).
A tentative mechanism explaining the carbonylative Suzuki coupling reaction features an initial Pd(0)‐mediated SET step converting A into the carbon‐centered radical (I) while generating a Pd(I) complex (Scheme 200). The two species then combines together and with CO forming the CO ligated Pd(II) complex (II). The catalytic cycle then proceeds with migratory insertion giving the acyl Pd(II) complex (III) and trans‐metalation with B (probably facilitated by the copper co‐catalyst) giving complex (IV). Eventually, a reductive elimination delivers compound C while the Pd(0) catalyst is restored.
Scheme 200.
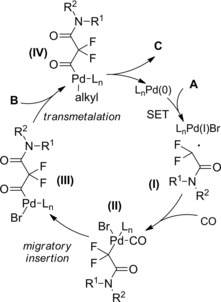
Mechanistic hypothesis for the Pd‐mediated carbonylative Suzuki coupling method (ref. [221] – Section 12).
The conjugative catalytic amination of readily available α‐fluoroenamides A with both primary and secondary amines B led to synthesize branched γ‐amino‐α,β‐unsaturated amides C which were useful intermediates toward hybrid peptides and indolizidines (Scheme 201).222 Both Pd(0) and Pt(0) were effective catalysts promoting both the inter‐ and intramolecular allylic substitution reaction.
Scheme 201.
Conjugative catalytic amination of α‐fluoroenamides A (ref. [222] – Section 12).
α‐Bromoamides A reacted with p‐toluenesulfonylmethyl isocyanide B (TosMIC) in the presence of catalytic Cu(OTf)2 giving α‐sulfonyl substituted products C (Scheme 202).223 The unusual transformation occurring in toluene required Cs2CO3 and water as additives.
Scheme 202.
Cu‐catalyzed reaction of α‐bromoamides A with TosMIC (ref. [223] – Section 12).
Mechanistic studies suggested that the Cu(OTf)2‐catalyzed hydration of TosMIC gave the formamide intermediate (I) which underwent base‐promoted elimination of N‐methyleneformamide (II) via facile C−S bond cleavage. Finally, a nucleophilic substitution reaction between the in situ generated sulfinate cesium salt (III) and α‐bromoamides A provided α‐sulfonated compounds C (Scheme 203).
Scheme 203.
Mechanistic hypothesis for the Cu‐catalyzed reaction of α‐bromoamides A with TosMIC (ref. [223] – Section 12).
Tertiary alkyl α‐bromoamides A were converted into the corresponding tertiary fluorinated compounds B by means of a copper‐catalyzed halogen substitution reaction occurring in the presence of CsF (Scheme 204).224 Optimized conditions selected PMDETA and the cheap CsF reagent as the nitrogen ligand and the fluoride source providing highest yields of the desirable tertiary alkyl‐F bonds. Radical scavengers suppressed the reaction, whereas bromoamides A with an amidic NH−Ar group were the only suitable substrates. Importantly, the radical process showed to be site‐selective, other primary or secondary alkyl‐halogen bonds being resistant under reaction conditions.
Scheme 204.
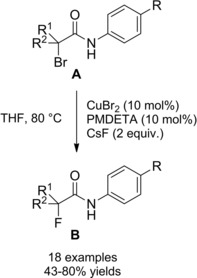
Copper‐catalyzed halogen substitution reaction for the conversion of alkyl α‐bromoamides A into the corresponding tertiary fluorinated compounds B (ref. [224] – Section 12).
Accordingly, the first step of the proposed reaction mechanism entails a SET from Cu(I) to A generating the tertiary alkyl radical (I) together with a Cu(II) species (Scheme 205). By reacting with CsF the latter provides the active fluorinating reagent CuF2. At this stage, the intermediate (I), with the amide moiety acting as a directing group, reacts with CuF2 via the copper complex (II). The latter step allows for the concomitant formation of B and a Cu(I) species to complete the catalytic cycle.
Scheme 205.
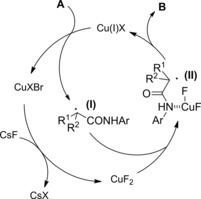
Mechanistic hypothesis for the copper‐catalyzed halogen substitution reaction (ref. [224] – Section 12).
13. Summary and Outlook
Compared to the richly‐developed chemistry of halocompounds, the chemistry of α‐haloamides has been largely underexplored. α‐Haloamides have traditionally been exploited as alkyl electrophiles in nucleophilic substitution providing C−N, C−O and C−S bonds. Despite the success in this field, there is still a number of challenges. For example, development of chiral catalysts to carry out enantioconvergent nucleophilic substitution reactions is highly desirable.
More recently the α‐functionalization of α‐haloamides has shifted towards cross‐coupling reactions, the installation of new C−C bonds being of utmost importance in synthesis. Reactions between α‐haloamides with C(sp3), C(sp2) and C(sp) coupling partners is a growing field, in particular, metal‐ and non‐metal‐catalyzed radical‐mediated transformations that feature mild and efficient conditions. Future advancements in the field will lead to protocols for the delivery of fine and useful chemicals otherwise difficult to prepare by other methods.
The scope of amide arylation by reaction of aryl halides has been greatly expanded using zinc enolates generated from α‐bromoamides instead of alkali metal enolates.225 On the other hand, a polarity reversal by using a metal‐catalyzed cross‐coupling between α‐haloamides and aryl organometallics showed to be a valuable synthetic alternative. Within this context, efforts to individuate asymmetric cross‐couplings requiring low catalyst loading is a worthwhile goal for making valuable building blocks for medicinal chemistry.
In recent years, there has been increasing interest in the use of visible light to drive organic reactions because of its infinite availability, ease of handling, and promising application in industry (leading reviews ref. [226a–e]). Consequently, widespread attention has received photoredox catalysis with transition metal complexes as a tool for the development of waste‐reducing processes involving α‐haloamides as suitable reagent partners for a wide range of new carbon‐carbon bond forming reactions.
α‐Halogenated 7‐azaindoline amides are particularly useful in direct enolization chemistry due to their facilitated enolization in soft Lewis acid/Brønsted base cooperative catalysis allowing for direct aldol and Mannich‐type reactions with chiral Cu(I) catalysts in a highly stereoselective manner.227
Eventually, the discovery of practical methods for the introduction of difluoroalkyl groups into target molecules continue to be a challenging topic.228 Protocols working under metal free conditions and showing wide substrate scope are still highly desirable.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Anna Fantinati was born in Rovigo in 1990. She is a PhD student in organic chemistry and her main research interests are focused in the synthesis of bio‐active spiro‐compounds mitochondria target.

Biographical Information
Paolo Marchetti was born in Mantua on 13 May 1959. After graduation in Chemistry and Pharmaceutical Technology he obtained the PhD with a thesis entitled “Transformation of a natural aminoacid in homochiral N‐alkyl derivatives via 2‐bromoamides. Preliminary applications to peptides synthesis and biological studies”. He is currently an aggregate professor at the Department of Chemical and Pharmaceutical Sciences of the University of Ferrara.

Biographical Information
Claudio Trapella was born in Ferrara in 1972, completed his PhD in pharmaceutical sciences in 2002 and is now assistant professor of organic chemistry. The main research interests are focused in the organotransition metal chemistry for the synthesis of biological active compounds especially in the pain field.

Biographical Information
Vinicio Zanirato was appointed Researcher at the University of Ferrara in 1990 and Associate Professor of Organic Chemistry at the University of Siena in 1998. Today he holds Organic Chemistry and Advanced Organic Chemistry courses at the Department of Chemical and Pharmaceutical Sciences of the University of Ferrara. His research interests include developmnet of new reaction methodologies, design and synthesis of natural products, light‐driven artificial molecular switches, and biologically active small molecules.

Acknowledgements
This work was supported by funds from the Ferrara University grant FAR 2018 (C.T. V.Z, P.M.) We address a special word of gratitude to Prof. G. P. Pollini for his continuous support and encouragement.
A. Fantinati, V. Zanirato, P. Marchetti, C. Trapella, ChemistryOpen 2020, 9, 100.
In memory of the recently deceased Prof. Ferruccio D'Angeli
References
- 1. Lengyel I., Sheehan J. C., Angew. Chem. Int. Ed. 1968, 7, 25–36; [Google Scholar]; Angew. Chem. 1968, 80, 27–37. [Google Scholar]
- 2. Clarck A. J., Duckmanton J. N., Felluga F., Gennaro A., Ghelfi F., Hardiman J. R. D., Isse A. A., Manferdini C., Spinelli D., Eur. J. Org. Chem. 2016, 2479–2491. [Google Scholar]
- 3.
- 3a. D'Angeli F., Rondanin R., Marchetti P., Bertolasi V., J. Org. Chem. 1996, 61, 1252–1255; [Google Scholar]
- 3b. Lee M-s., Kim Y., Youk E., Park Y. S., Bull. Korean Chem. Soc. 2016, 37, 981–984. [Google Scholar]
- 4. Marchetti P., Tetrahedron Lett. 2003, 44, 4121–4123. [Google Scholar]
- 5.
- 5a. Zanotti G., Filira F., Del Pra A., Cavicchioni G., Veronese A. C., D'Angeli F., J. Chem. Soc. Perkin Trans. 1 1980, 2249–2253; [Google Scholar]
- 5b. Cavicchioni G., Scrimin P., Veronese A. C., D'Angeli F., J. Chem. Commun. 1981, 416–417; [Google Scholar]
- 5c. Scrimin P., D'Angeli F., Veonese A. C., Baioni V., Tetrahedron Lett., 1983, 24, 4473–4476; [Google Scholar]
- 5d. Cavicchioni G., Scrimin P., Veronese A. C., Balboni G., D'Angeli F., J. Chem. Soc. Perkin Trans. 1 1982, 2969–2972; [Google Scholar]
- 5e. Scrimin P., Cavicchioni G., D'Angeli F., Goldblum A., Maran F., J. Chem. Soc. Perkin Trans. 1 1988, 43–47; [Google Scholar]
- 5f. Veronese A. C., Scrimin P., Vecchiati G., Sferra S., D'Angeli F., J. Chem. Soc. Perkin Trans. 1 1984, 781–784. [Google Scholar]
- 6.
- 6a. Bai W.-J., Jackson S. K., Pettus T. R. R., Org. Lett. 2012, 14, 3862–3865; [DOI] [PubMed] [Google Scholar]
- 6b. Schütznerová E., Oliver A. G., Pribylka A., Krchnák V., Adv. Synth. Catal. 2018, 360, 3693–3699. [Google Scholar]
- 7. Catelani G., Nejad F. M. K., D'Angeli F., Cavicchioni G., Marchetti P., Gazz. Chim. It. 1992, 122, 51–54. [Google Scholar]
- 8.
- 8a. D'Angeli F., Marchetti P., Cavicchioni G., Catelani G., Nejad F. M. K., Tetrahedron: Asymmetry 1990, 1, 155–158; [Google Scholar]
- 8b. Cavicchioni G., Tetrahedron Lett. 1987, 28, 2427–2430; [Google Scholar]
- 8c. Cavicchioni G., D'Angeli F., Casolari A., Orlandini P., Synthesis 1988, 947–950; [Google Scholar]
- 8d. D'Angeli F., Marchetti P., Cavicchioni G., Bertolasi V., Maran F., Tetrahedron: Asymmetry 1991, 2, 1111–1121; [Google Scholar]
- 8e. Nam J., Chang J.-Y., Shin E-k., Kim H. J., Kim Y., Jang S., Park Y. S., Tetrahedron 2004, 60, 6311–6318; [Google Scholar]
- 8f. Kubo A., Kubota H., Takahashi M., Nunami K-i., J. Org. Chem. 1997, 62, 5830–5837; [Google Scholar]
- 8g. Nam J., Chang J.-Y., Hahm K.-S., Park Y. S., Tetrahedron Lett. 2003, 44, 7727–7730. [Google Scholar]
- 9. Cavicchioni G., Synth. Commun. 1994, 24, 2223–2227. [Google Scholar]
- 10. Maran F., Fabrizio M., D'Angeli F., Vianello E., Tetrahedron 1988, 44, 2351–2358. [Google Scholar]
- 11. Sun B., Balaji P. V., Kumagai N., Shibasaki M., J. Am. Chem. Soc. 2017, 139, 8295–8301. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Bowman W. R., Bridge C. F., Brookes P., J. Chem. Soc. Perkin Trans. 1 2000, 1–14; [Google Scholar]
- 12b. Ishibashi H., Haruki S., Uchiyama M., Tamura O., Matsuo J−I., Tetrahedron Lett. 2006, 47, 6263–6266. [Google Scholar]
- 13. Clark A. J., Chem. Soc. Rev. 2002, 31, 1–11. [DOI] [PubMed] [Google Scholar]
- 14. Fava E., Nakajima M., Tabaka M. B., Rueping M., Green Chem. 2016, 18, 4531–4535. [Google Scholar]
- 15. Fischer C., Fu G. C., J. Am. Chem. Soc. 2005, 127, 4594–4595. [DOI] [PubMed] [Google Scholar]
- 16. El Bouakher A., Martel A., Comesse S., Org. Biomol. Chem. 2019, 17, 8467–8485. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Speziale A. J., Hamm P. C., J. Am. Chem. Soc. 1956, 78, 2256–2259; [Google Scholar]
- 17b. Pazcal A., Bényei A. C., Kotschy A., J. Org. Chem. 2006, 71, 5969–5979; [DOI] [PubMed] [Google Scholar]
- 17c. Kumar M. R., Park K., Lee S., Adv. Synth. Catal. 2010, 352, 3255–3266. [Google Scholar]
- 18. Abd El-Aal H. K., Khalaf A. A., Arkivoc 2013, iv, 306–322. [Google Scholar]
- 19. Harte A. J., Gunnlaugsson T., Tetrahedron Lett. 2006, 47, 6321–6324. [Google Scholar]
- 20. Duran F., Ghosez L., Tetrahedron Lett. 1970, 3, 245–248. [Google Scholar]
- 21.
- 21a. Casadei M. A., Micheletti Moracci F., J. Chem. Soc. Perkin Trans. 2 1986, 419–423; [Google Scholar]
- 21b. Casadei M. A., Inesi A., Micheletti Moracci F., Occhialini D., Tetrahedron 1989, 45, 6885–6890. [Google Scholar]
- 22.J. A. Dubland, MSc Thesis 2010.
- 23. Morimoto H., Fujiwara R., Shimizu Y., Morisaki K., Ohshima T., Org. Lett. 2014, 16, 2018–2021. [DOI] [PubMed] [Google Scholar]
- 24. Le Pironnec M.-G., Guinamant J.-L., Robert A., Baudy-Floc'h M., Synthesis 1997, 229–232. [Google Scholar]
- 25.
- 25a. Vora H. U., Rovis T., J. Am. Chem. Soc. 2007, 129, 13796–13797; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Vora H. U., Wheeler P., Rovis T., Adv. Synth. Catal. 2012, 354, 1617–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kirihara M., Ogawa S., Noguchi T., Okubo K., Monma Y., Shimizu I., Shimosaki R., Hatano A., Hirai Y., Synlett 2006, 14, 2287–2289. [Google Scholar]
- 27. Ilayaraja N., Noel M., J. Electroanal. Chem. 2009, 632, 45–54. [Google Scholar]
- 28.
- 28a. Pace V., Castoldi L., Mamuye A. D., Holzer W., Synthesis 2014, 46, 2897–2909; [Google Scholar]
- 28b. Pace V., Castoldi L., Holzer W., Chem. Commun. 2013, 49, 8383–8385. [DOI] [PubMed] [Google Scholar]
- 29. Ng F.-N., Lau Y.-F., Zhou Z., Yu W.-Y., Org. Lett. 2015, 17, 1676–1679. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Pan F., Li X.-L., Chen X.-M., Shu C., Ruan P.-P., Shen C.-H., Lu X., Ye L.-W., ACS Catal. 2016, 6, 6055–6062; [Google Scholar]
- 30b. Li L., Zhou B., Wang Y.-H., Shu C., Pan Y.-F., Lu X., Ye L.-W., Angew. Chem. Int. Ed. 2015, 54, 8245–8249. [DOI] [PubMed] [Google Scholar]
- 31. Gless R. D. Jr., Synth. Commun. 1986, 16, 633–638. [Google Scholar]
- 32. Souers A. J., Schürer S., Kwack H., Virgilio A. A., Ellman J. A., Synthesis 1999, 4, 583–585. [Google Scholar]
- 33. Ward R. S., Pelter A., Goubet D., Pritchard M. C., Tetrahedron: Asymmetry 1995, 6, 469–498. [Google Scholar]
- 34. Thom C., Kocienski P., Synthesis 1992, 582–586. [Google Scholar]
- 35. Caddick S., Jenkins K., Tetrahedron Lett. 1996, 37, 1301–1304. [Google Scholar]
- 36. Lee S.-K., Lee S. Y., Park Y. S., Synlett 2001, 12, 1941–1943. [Google Scholar]
- 37. Azim A., Sharma S. K., Olsen C. E., Parmar V. S., Bioorg. Med. Chem. 2001, 9, 1345–1348. [DOI] [PubMed] [Google Scholar]
- 38. Westerbeek A., Szymanski W., Wijma H. J., Marrink S. J., Feringa B. L., Janssen D. B., Adv. Synth. Catal. 2011, 353, 931–944. [Google Scholar]
- 39. Westerbeek A., Szymanski W., Feringa B. L., Janssen D. B., ACS Catal. 2011, 1, 1654–1660. [Google Scholar]
- 40. Wheeler P., Vora H. U., Rovis T., Chem. Sci. 2013, 4, 1674–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dong X., Yang W., Hu W., Sun J., Angew. Chem. Int. Ed. 2015, 54, 660–663. [DOI] [PubMed] [Google Scholar]
- 42.
- 42a. Baumgarten H. E., J. Am. Chem. Soc. 1962, 84, 4975–4976; [Google Scholar]
- 42b. Baumgarten H. E., Fuerholzer J. F., Clark R. D., Thompson R. D., J. Am. Chem. Soc. 1963, 85, 3303–3305. [Google Scholar]
- 43. Sheehan J. C., Lengyel I., J. Am. Chem. Soc. 1964, 86, 1356–1359. [Google Scholar]
- 44. Scrimin P., D'Angeli F., Veronese A. C., Synthesis 1982, 586–587. [Google Scholar]
- 45. Cesare V., Lyons T. M., Lengyel I., Synthesis 2002, 12, 1716–1720. [Google Scholar]
- 46. Hoffman R. V. in The Amide Linkage: Selected Structural Aspects in Chemistry Biochemistry and Material Science, (Eds.: A. Greenberg, C. M. Breneman, J. F. Liebman), John Wiley & Sons: New York, 2000, p 137. [Google Scholar]
- 47. Lengyel I., Cesare V., Chen S., Taldone T., Heterocycles 2002, 57, 677–695. [Google Scholar]
- 48. Sarel S., D'Angeli F., Klug J. T., Taube A., Isr. J. Chem. 1964, 2, 167–170. [Google Scholar]
- 49. Fitzpatrick D. E., Maujean T., Evans A. C., Ley S. V., Angew. Chem. Int. Ed. 2018, 57, 15128–15132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dieter R. K., Deo N., Lagu B., Dieter J. W., J. Org. Chem. 1992, 57, 1663–1671. [Google Scholar]
- 51. Mancilla T., Zamudio-Rivera L. S., Carrillo L., Beltran H. I., Farfan N., Arkivoc 2003, 37–47. [Google Scholar]
- 52. Kelly N. M., Wellejus A., Elbrond-Bek H., Weidner M. S., Jorgensen S. H., Synth. Commun. 2013, 43, 1243–1249. [Google Scholar]
- 53.
- 53a. Sanchez J. P., Parcell R. F., J. Heterocycl. Chem. 1990, 27, 1601–1607; [Google Scholar]
- 53b. Du W., Jewell J. P., Lin L. S., Colandrea V. J., Xiao J. C., Lao J., Shen C. P., Bateman T. J., Reddy V. B. G., Ha S. N. Bioorg. Med. Chem. Lett. 2009, 19, 5195–5199. [DOI] [PubMed] [Google Scholar]
- 54. Guziec F. S., Torres F. F., J. Org. Chem. 1993, 58, 1604–1606. [Google Scholar]
- 55. Lai J. T., Tetrahedron Lett. 1982, 23, 595–598. [Google Scholar]
- 56. Scrimin P., Dangeli F., Cavicchioni G., Synthesis 1982, 1092–1094. [Google Scholar]
- 57. D'angeli F., Marchetti P., Bertolasi V., J. Org. Chem. 1995, 60, 4013–4016. [Google Scholar]
- 58. D′Angeli F., Marchetti P., Ind. Chem. Libr. 1995, 7, 160–170. [Google Scholar]
- 59. Vachal P., Fletcher J. M., Hagmann W. K., Tetrahedron Lett. 2007, 48, 5761–5765. [Google Scholar]
- 60. Park Y. S., Tetrahedron: Asymmetry 2009, 20, 2421–2427. [Google Scholar]
- 61. Nunami K. I., Kubota H., Kubo A., Tetrahedron Lett. 1994, 35, 8639–8642. [Google Scholar]
- 62.
- 62a. Kubota H., Kubo A., Takahashi M., Shimizu R., Date T., Okamura K., Nunami K., J. Org. Chem. 1995, 60, 6776–6784; [Google Scholar]
- 62b. Kubo A., Kubota H., Takahashi M., Nunami K., Tetrahedron Lett. 1996, 37, 4957–4960. [Google Scholar]
- 63. Caddick S., Jenkins K., Treweeke N., Candeias S. X., Afonso C. A. M., Tetrahedron Lett. 1998, 39, 2203–2206. [Google Scholar]
- 64. Santos A. G., Pereira J., Afonso C. A. M., Frenking G., Chem. Eur. J. 2005, 11, 330–343. [DOI] [PubMed] [Google Scholar]
- 65. Ward R. S., Pelter A., Goubet D., Pritchard M. C., Tetrahedron: Asymmetry 1995, 6, 93–96. [Google Scholar]
- 66. Fisher D. J., Burnett G. L., Velasco R., de Alaniz J. R., J. Am. Chem. Soc. 2015, 137, 11614–11617. [DOI] [PubMed] [Google Scholar]
- 67. Kainz Q. M., Matier C. D., Bartoszewicz A., Zultanski S. L., Peters J. C., Fu G. C., Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ishida S., Takeuchi K., Taniyama N., Sunada Y., Nishikata T., Angew. Chem. Int. Ed. 2017, 56, 11610–11614; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11768–11772. [Google Scholar]
- 69.
- 69a. Gerona-Navarro G., Bonache M. A., Herranz R., García-López M. T., González-Muñiz R., Synlett 2000, 9, 1249–1252; [DOI] [PubMed] [Google Scholar]
- 69b. Gerona-Navarro G., Bonache M. A., Herranz R., García-López M. T., González-Muñiz R., J. Org. Chem. 2001, 66, 3538–3547; [DOI] [PubMed] [Google Scholar]
- 69c. Gerona-Navarro G., García-López M. T., González-Muñiz R., J. Org. Chem. 2002, 67, 3953–3956; [DOI] [PubMed] [Google Scholar]
- 69d. Bonache M. A., Gerona-Navarro G., Martín-Martínez M., García-López M. T., López P., Cativiela C., González-Muñiz R., Synlett 2003, 7, 1007–1011; [Google Scholar]
- 69e. Bonache M. A., Gerona-Navarro G., García-Aparicio C., Alías M., Martín-Martínez M., García-López M. T., López P., Cativiela C., González-Muñiz R., Tetrahedron: Asymmetry 2003, 14, 2161–2169; [Google Scholar]
- 70.
- 70a. Fuji K., Kawabata T., Chem. Eur. J. 1998, 4, 373–376; [Google Scholar]
- 70b. Kawabata T., Suzuki H., Nagae Y., Fuji K., Angew. Chem. Int. Ed. 2000, 39, 2155–2157; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2239–2241; [Google Scholar]
- 70c. Kawabata T., Chen J. Y., Suzuki H., Nagae Y., Kinoshita T., Chancharunee S., Fuji K., Org. Lett. 2000, 2, 3883–3885; [DOI] [PubMed] [Google Scholar]
- 70d. Kawabata T., Fuji K., Top. Stereochem. 2003, 23, 175–205. [Google Scholar]
- 71. Bonache M. A., López P., Martín-Martínez M., García-López M. T., Cativiela C., González-Muñiz R., Tetrahedron 2006, 62, 130–138. [Google Scholar]
- 72. Pérez-Faginas P., O′Reilly F., O′Byrne A., García-Aparicio C., Martín-Martínez M., Pérez. de Vega M. J., García-López M. T., González-Muñiz R., Org. Lett. 2007, 9, 1593–1596. [DOI] [PubMed] [Google Scholar]
- 73. Pérez-Faginas P., Alkorta I., García-López M. T., González-Muñiz R., Tetrahedron Lett. 2008, 49, 215–218. [Google Scholar]
- 74.
- 74a. Carelli I., Inesi A., Carelli V., Casadei M. A., Liberatore F., Micheletti Moracci F., Synthesis 1986, 591–593; [Google Scholar]
- 74b. Casadei M. A., Dirienzo B., Inesi A., Micheletti Moracci F., J. Chem. Soc. Perkin Trans. 1 1992, 3, 379–382. [Google Scholar]
- 75.
- 75a. Feroci A., Orsini M., Rossi L., Sotgiu G., Inesi A., Electrochim. Acta 2006, 51, 5540–5547; [Google Scholar]
- 75b. Feroci M., Lessard J., Orsini M., Inesi A., Tetrahedron Lett. 2005, 46, 8517–8519. [Google Scholar]
- 76. Feroci M., Adv. Synth. Catal. 2007, 349, 2177–2181. [Google Scholar]
- 77. Sotgiu G., Chiarotto I., Feroci M., Orsini M., Rossi L., Inesi A., Electrochim. Acta 2008, 53, 7852–7858. [Google Scholar]
- 78. Feroci M., Chiarotto I., Orsini M., Sotgiu G., Inesi A., Adv. Synth. Catal. 2008, 350, 1355–1359. [Google Scholar]
- 79. Pedroni J., Boghi M., Saget T., Cramer N., Angew. Chem. Int. Ed. 2014, 53, 9064–9067; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 9210–9213. [Google Scholar]
- 80. Pedroni J., Cramer N., Angew. Chem. Int. Ed. 2015, 54, 11826–11829; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 11992–11995. [Google Scholar]
- 81. Chatgilialoglu C., Ferreri C., Guerra M., Timokhin V., Froudakis G., Gimisis T., J. Am. Chem. Soc. 2002, 124, 10765–10772. [DOI] [PubMed] [Google Scholar]
- 82. Ishibashi H., So T. S., Okochi K., Sato T., Nakamura N., Nakatani H., Ikeda M., J. Org. Chem. 1991, 56, 95–102. [Google Scholar]
- 83. Jeffs P. W., Cortese N. A., Wolfram J., J. Org. Chem. 1982, 47, 3881–3886. [Google Scholar]
- 84. Sanchez I. H., Lopez F. J., Soria J. J., Larraza M. I., Flores H. J., J. Am. Chem. Soc. 1983, 105, 7640–7643. [Google Scholar]
- 85.
- 85a. Bryans J. S., Large J. M., Parsons A. F., Tetrahedron Lett. 1999, 40, 3487–3490; [Google Scholar]
- 85b. Bryans J. S., Large J. M., Parsons A. F., J. Chem. Soc. Perkin Trans. 1 1999, 20, 2897–2904. [Google Scholar]
- 86. Gilbert B. C., Kalz W., Lindsay C. I., McGrail P. T., Parsons A. F., Whittaker D. T. E., Tetrahedron Lett. 1999, 40, 6095–6098. [Google Scholar]
- 87.
- 87a. Isse A. A., Visona G., Ghelfi F., Roncaglia F., Gennaro A., Adv. Synth. Catal. 2015, 357, 782–792; [Google Scholar]
- 87b. Clark A. J., Cornia A., Felluga F., Gennaro A., Ghelfi F., Isse A. A., Menziani C., Muniz-Miranda F., Roncaglia F., Spinelli D., Eur. J. Org. Chem. 2014, 6734–6745; [Google Scholar]
- 87c. Bellesia F., Clark A. J., Felluga F., Gennaro A., Isse A. A., Roncaglia F., Ghelfi F., Adv. Synth. Catal. 2013, 355, 1649–1660; [Google Scholar]
- 87d. Clark A. J., Collis A. E. C., Fox D. J., Halliwell L. L., James N., O′Reilly R. K., Parekh H., Ross A., Sellars A. B., Willcock H., Wilson P., J. Org. Chem. 2012, 77, 6778–6788; [DOI] [PubMed] [Google Scholar]
- 87e. Casolari R., Felluga F., Frenna V., Ghelfi F., Pagnoni U. M., Parsons A. F., Spinelli D., Tetrahedron 2011, 67, 408–416; [Google Scholar]
- 87f. Clark A. J., Wilson P., Tetrahedron Lett. 2008, 49, 4848–4850; [Google Scholar]
- 87g. Bregoli M., Felluga F., Frenna V., Ghelfi F., Pagnoni U. M., Parsons A. F., Petrillo G., Spinelli D., Synthesis 2011, 1267–1278; [Google Scholar]
- 87h. Ghelfi F., Pattarozzi M., Roncaglia F., Giangiordano V., Parsons A. F., Synth. Commun. 2010, 40, 1040–1051; [Google Scholar]
- 87i. Pattarozzi M., Roncaglia F., Giangiordano V., Davoli P., Prati F., Ghelfi F., Synthesis 2010, 694–700; [Google Scholar]
- 87j. Pattarozzi M., Roncaglia F., Accorsi L., Parsons A. F., Ghelfi F., Tetrahedron 2010, 66, 1357–1364; [Google Scholar]
- 87k. Motoyama Y., Kamo K., Yuasa A., Nagashima H., Chem. Commun. 2010, 46, 2256–2258. [DOI] [PubMed] [Google Scholar]
- 88.
- 88a. Fernandez-Zumel M. A., Kiefer G., Thommes K., Scoppelliti R., Severin K., Eur. J. Inorg. Chem. 2010, 3596–3601; [Google Scholar]
- 88b. Thommes K., Kiefer G., Scopelliti R., Severin K., Angew. Chem. Int. Ed. 2009, 48, 8115–8119; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 8259–8263; [Google Scholar]
- 88c. Wolf J., Thommes K., Briel O., Scopelliti R., Severin K., Organometallics 2008, 27, 4464–4474; [Google Scholar]
- 88d. Thommes K., Icli B., Scopelliti R., Severin K., Chem. Eur. J. 2007, 13, 6899–6907; [DOI] [PubMed] [Google Scholar]
- 88e. Kanbayashi N., Takenaka K., Okamura T., Onitsuka K., Angew. Chem. Int. Ed. 2013, 52, 4897–4901; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4997–5001; [Google Scholar]
- 88f. McGonagle F. I., Brown L., Cooke A., Sutherland A., Tetrahedron Lett. 2011, 52, 2330–2332; [Google Scholar]
- 88g. Dutta B., Scoppelliti R., Severin K., Organometallics 2008, 27, 423–429; [Google Scholar]
- 88h. Edlin C. D., Faulkner J., Fengas D., Helliwell M., Knight C. K., House D., Parker J., Preece I., Quayle P., Raftery J., Richards S. N., J. Organomet. Chem. 2006, 691, 5375–5382; [Google Scholar]
- 88i. Motoyama Y., Hanada S., Shimamoto N., Nagashima H., Tetrahedron 2006, 62, 2779–2788; [Google Scholar]
- 88j. Benedetti M., Forti L., Ghelfi F., Pagnoni U. M., Ronzoni R., Tetrahedron 1997, 53, 14031–14042; [Google Scholar]
- 88k. Liu Q., Chen C., Tong X., Tetrahedron Lett. 2015, 56, 4483–4485; [Google Scholar]
- 88l. Tseng C. K., Teach E. G., Simons R. W., Synth. Commun. 1984, 14, 1027–1031; [Google Scholar]
- 88m. Rachita M. A., Slough G. A., Tetrahedron Lett. 1993, 34, 6821–6824. [Google Scholar]
- 89. Nagashima H., Wakamatsu H., Itoh K., J. Chem. Soc. Chem. Commun. 1984, 10, 652–653. [Google Scholar]
- 90. Nagashima H., Wakamatsu H., Ozaki N., Ishii T., Watanabe M., Tajima T., Itoh K., J. Org. Chem. 1992, 57, 1682–1689. [Google Scholar]
- 91.
- 91a. Nagashima H., Ozaki N., Ishii M., Seki K., Washiyama M., Itoh K., J. Org. Chem. 1993, 58, 464–470; [Google Scholar]
- 91b. Iwamatsu S., Matsubara K., Nagashima H., J. Org. Chem. 1999, 64, 9625–9631. [Google Scholar]
- 92. Nagashima H., Isono Y., Iwamatsu S., J. Org. Chem. 2001, 66, 315–319. [DOI] [PubMed] [Google Scholar]
- 93. Bregoli M., Felluga F., Frenna V., Ghelfi F., Pagnoni U. M., Parsons A. F., Petrillo G., Spinelli D., Synthesis 2011, 8, 1267–1278. [Google Scholar]
- 94. Thommes K., Icli B., Scopelliti R., Severin K., Chem. Eur. J. 2007, 13, 6899–6907. [DOI] [PubMed] [Google Scholar]
- 95. Clark A. J., Wilson P., Tetrahedron Lett. 2008, 49, 4848–4850. [Google Scholar]
- 96. Kanbayashi N., Takenaka K., Okamura T., Onitsuka K., Angew. Chem. Int. Ed. 2013, 52, 4897–4901; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4997–5001. [Google Scholar]
- 97.
- 97a. Ozaki S., Matsushita H., Ohmori H., J. Chem. Soc. Perkin Trans. 1 1993, 19, 2339–2344; [Google Scholar]
- 97b. Ozaki S., Matsushita H., Emoto M., Ohmori H., Chem. Pharm. Bull. 1995, 43, 32–36. [Google Scholar]
- 98. Chaminade X., Dunach E., Esteves A. P., Medeiros M. J., Neves C. S., Olivero S., Electrochim. Acta 2009, 54, 5120–5126. [Google Scholar]
- 99. Isse A. A., Visona G., Ghelfi F., Roncaglia F., Gennaro A., Adv. Synth. Catal. 2015, 357, 782–792. [Google Scholar]
- 100.
- 100a. Cornia A., Felluga F., Frenna V., Ghelfi F., Parsons A. F., Pattarozzi M., Roncaglia F., Spinelli D., Tetrahedron 2012, 68, 5863–5881; [Google Scholar]
- 100b. Clark A. J., Geden J. V., Thom S., Wilson P., J. Org. Chem. 2007, 72, 5923–5926; [DOI] [PubMed] [Google Scholar]
- 100c. Bryans J. S., Chessum N. E. A., Hunther N., Parsons A. F., Ghelfi F., Tetrahedron 2003, 59, 6221–6231; [Google Scholar]
- 100d. Parsons A. F., C. R. Acad. Sci. Paris, Chimie/Chemistry 2001, 4, 391–400; [Google Scholar]
- 100e. Clark A. J., Dell C. P., McDonagh J. P., C. R. Acad. Sci. Paris, Chimie/Chemistry 2001, 4, 575–579. [Google Scholar]
- 101. Parsons A. F., C. R. Acad. Sci. 2001, 4, 391–400. [Google Scholar]
- 102. Coussanes G., Vila X., Diaba F., Bonjoch J., Synthesis 2017, 49, 1481–1499. [Google Scholar]
- 103.
- 103a. Ishibashi H., Nakamura N., Sato T., Takeuchi M., Ikeda M., Tetrahedron Lett. 1991, 32, 1725–1728; [Google Scholar]
- 103b. Ishibashi H., Sato T., Ikeda M., Synthesis 2002, 6, 695–713. [Google Scholar]
- 104.
- 104a. Ishibashi H., Kodama K., Kameoka C., Kawanami H., Ikeda M., Tetrahedron 1996, 52, 13867–13880; [Google Scholar]
- 104b. Ishibashi H., Higuchi M., Ohba M., Ikeda M., Tetrahedron Lett. 1998, 39, 75–78. [Google Scholar]
- 105. Sato T., Nakamura N., Ikeda K., Okada M., Ishibashi H., Ikeda M., J. Chem. Soc. Perkin Trans. 1 1992, 18, 2399–2407. [Google Scholar]
- 106.
- 106a. Baker S. R., Parsons A. F., Pons J. F., Wilson M., Tetrahedron Lett. 1998, 39, 7197–7200; [Google Scholar]
- 106b. Baker S. R., Burton K. I., Parsons A. F., Pons J. F., Wilson M., J. Chem. Soc. Perkin Trans. 1 1999, 4, 427–436. [Google Scholar]
- 107. Tamura O., Matsukida H., Toyao A., Takeda Y., Ishibashi H., J. Org. Chem. 2002, 67, 5537–5545. [DOI] [PubMed] [Google Scholar]
- 108. Curran D. P., Guthrie D. B., Geib S. J., J. Am. Chem. Soc. 2008, 130, 8437–8445. [DOI] [PubMed] [Google Scholar]
- 109. Takami K., Usugi S. I., Yorimitsu H., Oshima K., Synthesis 2005, 5, 824–839. [Google Scholar]
- 110. Lou S., Fu G. C., Bryan C. S., Lautens M., Org. Synth. 2010, 87, 330–338.21533010 [Google Scholar]
- 111. Lai P. S., Dubland J. A., Sarwar M. G., Chudzinski M. G., Taylor M. S., Tetrahedron 2011, 67, 7586–7592. [Google Scholar]
- 112. Nakajima M., Lefebvre Q., Rueping M., Chem. Commun. 2014, 50, 3619–3622. [DOI] [PubMed] [Google Scholar]
- 113. Esumi N., Suzuki K., Nishimoto Y., Yasuda M., Org. Lett. 2016, 18, 5704–5707. [DOI] [PubMed] [Google Scholar]
- 114. Huang W. H., Chen W. X., Wang G. Q., Li J., Cheng X., Li G. G., ACS Catal. 2016, 6, 7471–7474. [Google Scholar]
- 115. Zhou F., Zhang Y., Xu X. F., Zhu S. L., Angew. Chem. Int. Ed. 2019, 58, 1754–1758. [DOI] [PubMed] [Google Scholar]
- 116. Liang H., Xu G. Q., Feng Z. T., Wang Z. Y., Xu P. F., J. Org. Chem. 2019, 84, 60–72. [DOI] [PubMed] [Google Scholar]
- 117. Yuan F. Y., Zhou S., Yang Y. Y., Guo M. J., Tang X. Y., Wang G. W., Org. Chem. Front. 2018, 5, 3306–3309. [Google Scholar]
- 118. Cho H. H., Kim S. H., Bull. Korean Chem. Soc. 2015, 36, 1274–1277. [Google Scholar]
- 119. Kagoshima H., Hashimoto Y., Saigo K., Tetrahedron Lett. 1998, 39, 8465–8466. [Google Scholar]
- 120. Moumne R., Lavielle S., Karoyan P., J. Org. Chem. 2006, 71, 3332–3334. [DOI] [PubMed] [Google Scholar]
- 121.
- 121a. Concellón J. M., Rodríguez-Solla H., Simal C., Adv. Synth. Catal. 2009, 351, 1238–1242; [Google Scholar]
- 121b. Concellón J. M., Rodríguez-Solla H., Simal C., del Amo V., García-Granda S., Díaz M. R., Adv. Synth. Catal. 2009, 351, 2991–3000; [Google Scholar]
- 121c. Concellón J. M., Rodríguez-Solla H., Concellón C., Simal C., Alvaredo N. J. Org. Chem. 2010, 75, 3451–3453; [DOI] [PubMed] [Google Scholar]
- 121d. Concellón J. M., Rodríguez-Solla H., Concellón C., Simal C., Alvaredo N., Synlett 2010, 14, 2119–2121. [Google Scholar]
- 122. Yamazaki T., Welch J. T., Plummer J. S., Gimi R. H., Tetrahedron Lett. 1991, 32, 4267–4270. [Google Scholar]
- 123. Okamoto N., Sasaki M., Kawahata M., Yamaguchi K., Takeda K., Org. Lett. 2006, 8, 1889–1891. [DOI] [PubMed] [Google Scholar]
- 124. Arai S., Tokumaru K., Aoyama T., Tetrahedron Lett. 2004, 45, 1845–1848. [Google Scholar]
- 125. Achard T. J. R., Belokon′ Y. N., Ilyin M., Moskalenko M., North M., Pizzato F., Tetrahedron Lett. 2007, 48, 2965–2969. [Google Scholar]
- 126. North M., Pizzato F., Lett. Org. Chem. 2009, 6, 552–556. [Google Scholar]
- 127.
- 127a. Brewitz L., Arteaga F. A., Yin L., Alagiri K., Kumagai N., Shibasaki M., J. Am. Chem. Soc. 2015, 137, 15929–15939; [DOI] [PubMed] [Google Scholar]
- 127b. Li Z., Noda H., Kumagai N., Shibasaki M., Tetrahedron 2018, 74, 3301–3305; [Google Scholar]
- 127c. Brewitz L., Kumagai N., Shibasaki M., J. Fluorine Chem. 2017, 194, 1–7. [Google Scholar]
- 128.
- 128a. Mori M., Oda I., Ban Y., Tetrahedron Lett. 1982, 23, 5315–5318; [Google Scholar]
- 128b. Mori M., Kanda N., Oda I., Ban Y., Tetrahedron 1985, 41, 5465–5474. [Google Scholar]
- 129. Yang S. C., Shea F. R., J. Chin. Chem. Soc. 1995, 42, 969–972. [Google Scholar]
- 130. Cassayre J., Zard S. Z., Synlett 1999, 4, 501–503. [Google Scholar]
- 131. Davies D. T., Kapur N., Parsons A. F., Tetrahedron 2000, 56, 3941–3949. [Google Scholar]
- 132. Clark A. J., Geden J. V., Thom S., J. Org. Chem. 2006, 71, 1471–1479. [DOI] [PubMed] [Google Scholar]
- 133. Clark A. J., Filik R. P., Haddleton D. M., Radigue A., Sanders C. J., Thomas G. H., Smith M. E., J. Org. Chem. 1999, 64, 8954–8957. [DOI] [PubMed] [Google Scholar]
- 134. Takami K., Yorimitsu H., Oshima K., Org. Lett. 2004, 6, 4555–4558. [DOI] [PubMed] [Google Scholar]
- 135. Zhu J. M., Zhang W., Zhang L. J., Liu J., Zheng J., Hu J. B., J. Org. Chem. 2010, 75, 5505–5512. [DOI] [PubMed] [Google Scholar]
- 136. Liu C., Tang S., Liu D., Yuan J., Zheng L., Meng L., Lei A., Angew. Chem. Int. Ed. 2012, 51, 3638–3641; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3698–3701. [Google Scholar]
- 137. Qiu C. B., Yao K., Zhang X. H., Gong H. G., Org. Biomol. Chem. 2016, 14, 11332–11335. [DOI] [PubMed] [Google Scholar]
- 138. Belhomme M. C., Dru D., Xiong H. Y., Cahard D., Besset T., Poisson T., Pannecoucke X., Synthesis 2014, 46, 1859–1870. [Google Scholar]
- 139. Nishikata T., Itonaga K., Yamaguchi N., Sumimoto M., Org. Lett. 2017, 19, 2686–2689. [DOI] [PubMed] [Google Scholar]
- 140. Barde E., Guerinot A., Cossy J., Org. Lett. 2017, 19, 6068–6071. [DOI] [PubMed] [Google Scholar]
- 141. Clark A. J., Battle G. M., Bridge A., Tetrahedron Lett. 2001, 42, 1999–2001. [Google Scholar]
- 142. Liu B., Yu J. X., Li Y., Li J. H., He D. L., Org. Lett. 2018, 20, 2129–2132. [DOI] [PubMed] [Google Scholar]
- 143. Johansson C. C. C., Colacot T. J., Angew. Chem. Int. Ed. 2010, 49, 676–707; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 686–718. [Google Scholar]
- 144. Hama T., Liu X. X., Culkin D. A., Hartwig J. F., J. Am. Chem. Soc. 2003, 125, 11176–11177. [DOI] [PubMed] [Google Scholar]
- 145. Hama T., Culkin D. A., Hartwig J. F., J. Am. Chem. Soc. 2006, 128, 4976–4985. [DOI] [PubMed] [Google Scholar]
- 146. Bentz E., Moloney M. G., Westaway S. M., Tetrahedron Lett. 2004, 45, 7395–7397. [Google Scholar]
- 147. Belhomme M. C., Besset T., Poisson T., Pannecoucke X., Chem. Eur. J. 2015, 21, 12836–12865. [DOI] [PubMed] [Google Scholar]
- 148. Ge S. Z., Arlow S. I., Mormino M. G., Hartwig J. F., J. Am. Chem. Soc. 2014, 136, 14401–14404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Goossen L. J., Chem. Commun. 2001, 7, 669–670. [Google Scholar]
- 150. Duan Y. Z., Deng M. Z., Tetrahedron Lett. 2003, 44, 3423–3426. [Google Scholar]
- 151. Liu C., He C., Shi W., Chen M., Lei A., Org. Lett. 2007, 9, 5601–5604. [DOI] [PubMed] [Google Scholar]
- 152. Lundin P. M., Fu G. C., J. Am. Chem. Soc. 2010, 132, 11027–11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Qi Q., Shen Q., Lu L., J. Am. Chem. Soc. 2012, 134, 6548–6551. [DOI] [PubMed] [Google Scholar]
- 154. Molander G. A., Traister K. M., Barcellos T., J. Org. Chem. 2013, 78, 4123–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Feng Z., Min Q. Q., Xiao Y. L., Zhang B., Zhang X. G., Angew. Chem. Int. Ed. 2014, 53, 1669–1673; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 1695–1699. [Google Scholar]
- 156. Xiao Y. L., Guo W. H., He G. Z., Pan Q., Zhang X. G., Angew. Chem. Int. Ed. 2014, 53, 9909–9913; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10067–10071. [Google Scholar]
- 157. Strotman N. A., Sommer S., Fu G. C., Angew. Chem. Int. Ed. 2007, 46, 3556–3558; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 3626–3628. [Google Scholar]
- 158. Jin M., Nakamura M., Chem. Lett. 2011, 40, 1012–1014. [Google Scholar]
- 159. Tarui A., Shinohara S., Sato K., Omote M., Ando A., Org. Lett. 2016, 18, 1128–1131. [DOI] [PubMed] [Google Scholar]
- 160.
- 160a. Stollè R., Bergdoll R., Luther M., Auerhahn A., Wcker W., J. Prakt. Chem. 1922, 105, 137–148; [Google Scholar]
- 160b. Stollè R., Bergdoll R., Luther M., Auerhahn A., Wcker W., J. Prakt. Chem. 1930, 128, 1–43. [Google Scholar]
- 161. Beckett A. H., Daisley R. W., Walker J., Tetrahedron 1968, 24, 6093–6109. [Google Scholar]
- 162. Hennessy E. J., Buchwald S. L., J. Am. Chem. Soc. 2003, 125, 12084–12085. [DOI] [PubMed] [Google Scholar]
- 163. Choy A., Colbry N., Huber C., Pamment M., Van Duine J., Org. Process Res. Dev. 2008, 12, 884–887. [Google Scholar]
- 164. Kiser E. J., Magano J., Shine R. J., Chen M. H., Org. Process Res. Dev. 2012, 16, 255–259. [Google Scholar]
- 165. Shi S. L., Buchwald S. L., Angew. Chem. Int. Ed. 2015, 54, 1646–1650; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1666–1670. [Google Scholar]
- 166. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ju X. H., Liang Y., Jia P. J., Li W. F., Yu W., Org. Biomol. Chem. 2012, 10, 498–501. [DOI] [PubMed] [Google Scholar]
- 168.
- 168a. Wang L., Wei X. J., Jia W. L., Zhong J. J., Wu L. Z., Liu Q., Org. Lett. 2014, 16, 5842–5845; [DOI] [PubMed] [Google Scholar]
- 168b. Wei X. J., Wang L., Du S. F., Wu L. Z., Liu Q., Org. Biomol. Chem. 2016, 14, 2195–2199. [DOI] [PubMed] [Google Scholar]
- 169. Liu C., Liu D., Zhang W., Zhou L. L., Lei A. W., Org. Lett. 2013, 15, 6166–6169. [DOI] [PubMed] [Google Scholar]
- 170. Wu Z. J., Xu H. C., Angew. Chem. Int. Ed. 2017, 56, 4734–4738; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 4812–4816. [Google Scholar]
- 171. Usugi S., Yorimitsu H., Shinokubo H., Oshima K., Bull. Chem. Soc. Jpn. 2002, 75, 2687–2690. [Google Scholar]
- 172. Shi W., Liu C., Yu Z., Lei A. W., Chem. Commun. 2007, 23, 2342–2344. [DOI] [PubMed] [Google Scholar]
- 173. Kang J. Y., Connell B. T., J. Org. Chem. 2011, 76, 6856–6859. [DOI] [PubMed] [Google Scholar]
- 174. Molander G. A., Traister K. M., Org. Lett. 2013, 15, 5052–5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Liu W. B., Chen Z. W., Li L., Wang H. N., Li C. J., Chem. Eur. J. 2016, 22, 5888–5893. [DOI] [PubMed] [Google Scholar]
- 176. Yamane Y., Miwa N., Nishikata T., ACS Catal. 2017, 7, 6872–6876. [Google Scholar]
- 177. Lv Y. H., Pu W. Y., Zhu X. L., Zhao T. T., Lin F. F., Adv. Synth. Catal. 2018, 360, 1397–1401. [Google Scholar]
- 178. Liu D., Tang S., Yi H., Liu C., Qi X. T., Lan Y., Lei A. W., Chem. Eur. J. 2014, 20, 15605–15610. [DOI] [PubMed] [Google Scholar]
- 179. Lv Y. H., Pu W. Y., Mao S. K., Ren X. R., Wu Y. T., Cui H., RSC Adv. 2017, 7, 41723–41726. [Google Scholar]
- 180. Pan G. H., Song R. J., Li J. H., Org. Chem. Front. 2018, 5, 179–183. [Google Scholar]
- 181. Yamane Y., Miyazaki K., Nishikata T., ACS Catal. 2016, 6, 7418–7425. [Google Scholar]
- 182. Iwasaki M., Miki N., Ikemoto Y., Ura Y., Nishihara Y., Org. Lett. 2018, 20, 3848–3852. [DOI] [PubMed] [Google Scholar]
- 183. Li B.-H., Li K.-L., Chen Q.-Y., J. Fluorine Chem. 2012, 133, 163–166. [Google Scholar]
- 184. Zhang M. L., Li W. P., Duan Y. Q., Xu P., Zhang S. L., Zhu C. J., Org. Lett. 2016, 18, 3266–3269. [DOI] [PubMed] [Google Scholar]
- 185. Lv Y. H., Pu Y. W. Y., Wang Q. Q., Chen Q., Niu J. J., Zhang Q., Adv. Synth. Catal. 2017, 359, 3114–3119. [Google Scholar]
- 186. Lv Y. H., Pu W. Y., Chen Q., Wang Q. Q., Niu J. J., Zhang Q., J. Org. Chem. 2017, 82, 8282–8289. [DOI] [PubMed] [Google Scholar]
- 187. Chen H. T., Wang X. Y., Guo M. J., Zhao W. T., Tang X. Y., Wang G. W., Org. Chem. Front. 2017, 4, 2403–2407. [Google Scholar]
- 188.
- 188a. Maran F., Vianello E., Cavicchioni G., Dangeli F., J. Chem. Soc. Chem. Commun. 1985, 10, 660; [Google Scholar]
- 188b. Maran F., Vianello E., Dangeli F., Cavicchioni G., Vecchiati G., J. Chem. Soc. Perkin Trans. 2 1987, 1, 33–38; [Google Scholar]
- 188c. Maran F., J. Am. Chem. Soc. 1993, 115, 6557–6563. [Google Scholar]
- 189. Casadei M. A., Cesa S., Inesi A., Tetrahedron 1995, 51, 5891–5900. [Google Scholar]
- 190. Gao S. H., Tu Y. Q., Hu X. D., Wang S. H., Hua R. B., Jiang Y. J., Zhao Y. M., Fan X. H., Zhang S. Y., Org. Lett. 2006, 8, 2373–2376. [DOI] [PubMed] [Google Scholar]
- 191. Maryanoff B. E., Zhang H. C., Cohen J. H., Turchi I. J., Maryanoff C. A., Chem. Rev. 2004, 104, 1431–1628. [DOI] [PubMed] [Google Scholar]
- 192. Allous I., Comesse S., Daich A., Lett. Org. Chem. 2008, 5, 73–78. [Google Scholar]
- 193. Comesse S., Sanselme M., Daich A., J. Org. Chem. 2008, 73, 5566–5569. [DOI] [PubMed] [Google Scholar]
- 194. Saber M., Comesse S., Dalla V., Daich A., Sanselme M., Netchitailo P., Synlett 2010, 14, 2197–2201. [Google Scholar]
- 195.
- 195a. Comesse S., Martel A., Daich A., Org. Lett. 2011, 13, 4004–4007; [DOI] [PubMed] [Google Scholar]
- 195b. Le Goff R., Sanselme M., Lawson A. M., Daich A., Comesse S., Eur. J. Org. Chem. 2015, 33, 7244–7248; [Google Scholar]
- 195c. Le Goff R., Martel A., Sanselme M., Lawson A. M., Daich A., Comesse S., Chem. Eur. J. 2015, 21, 2966–2979. [DOI] [PubMed] [Google Scholar]
- 196. El Bouakher A., Le Goff R., Tasserie J., Lhoste J., Martel A., Comesse S., Org. Lett. 2016, 18, 2383–2386. [DOI] [PubMed] [Google Scholar]
- 197. El Bouakher A., Tasserie J., Le Goff R., Lhoste J., Martel A., Comesse S., J. Org. Chem. 2017, 82, 5798–5809. [DOI] [PubMed] [Google Scholar]
- 198. Kikugawa Y., Shimada M., Kato M., Sakamoto T., Chem. Pharm. Bull. 1993, 41, 2192–2194. [Google Scholar]
- 199. Jeffrey C. S., Barnes K. L., Eickhoff J. A., Carson C. R., J. Am. Chem. Soc. 2011, 133, 7688–7691. [DOI] [PubMed] [Google Scholar]
- 200. Barnes K. L., Koster A. K., Jeffrey C. S., Tetrahedron Lett. 2014, 55, 4690–4696. [Google Scholar]
- 201. Xuan J., Cao X., Cheng X., Chem. Commun. 2018, 54, 5154–5163. [DOI] [PubMed] [Google Scholar]
- 202. Zhao H. W., Zhao Y. D., Liu Y. Y., Zhao L. J., Song X. Q., Chen X. Q., Pang H. L., Du J., Feng N. N., RSC Adv. 2017, 7, 55106–55109. [Google Scholar]
- 203. Xu X. Y., Zhang K. F., Li P. P., Yao H. Q., Lin A. J., Org. Lett. 2018, 20, 1781–1784. [DOI] [PubMed] [Google Scholar]
- 204. Cheng X., Cao X., Xuan J., Xiao W. J., Org. Lett. 2018, 20, 52–55. [DOI] [PubMed] [Google Scholar]
- 205.
- 205a. Zhao H. W., Zhao Y. D., Liu Y. Y., Zhao L. J., Feng N. N., Pang H. L., Chen X. Q., Song X. Q., Du J., RSC Adv. 2017, 7, 12916–12922; [Google Scholar]
- 205b. Xuan J., Cheng X., Cao X., ChemistrySelect 2017, 2, 4364–4367; [Google Scholar]
- 205c. Jia Q. F., Li D. L., Lang M., Zhang K., Wang J., Adv. Synth. Catal. 2017, 359, 3837–3842; [Google Scholar]
- 205d. Chen R. X., Sun S. F., Wang G. Q., Guo H. B., Tetrahedron Lett. 2018, 59, 1916–1920. [Google Scholar]
- 206. Zhang K. F., Xu X. Y., Zheng J., Yao H. Q., Huang Y., Lin A. J., Org. Lett. 2017, 19, 2596–2599. [DOI] [PubMed] [Google Scholar]
- 207. Balde B., Force G., Marin L., Guillot R., Schulz E., Gandon V., Leboeuf D., Org. Lett. 2018, 20, 7405–7409. [DOI] [PubMed] [Google Scholar]
- 208. Li C., Jiang K., Ouyang Q., Liu T. Y., Chen Y. C., Org. Lett. 2016, 18, 2738–2741. [DOI] [PubMed] [Google Scholar]
- 209.
- 209a. DiPoto M. C., Hughes R. P., Wu J., J. Am. Chem. Soc. 2015, 137, 14861–14864; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209b. Acharya A., Anumandla D., Jeffrey C. S., J. Am. Chem. Soc. 2015, 137, 14858–14860. [DOI] [PubMed] [Google Scholar]
- 210.
- 210a. Wang G. Q., Chen R. X., Wu M. H., Sun S. F., Luo X., Chen Z., Guo H. B., Chong C., Xing Y. L., Tetrahedron Lett. 2017, 58, 847–850; [Google Scholar]
- 210b. Singh R., Nagesh K., Yugandhar D., Prasanth A. V. G., Org. Lett. 2018, 20, 4848–4853. [DOI] [PubMed] [Google Scholar]
- 211.
- 211a. Eyilcim O., Issever S., Ocal N., Gronert S., Erden I., Tetrahedron Lett. 2018, 59, 3674–3677; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211b. Ji D. Q., Sun J. T., Org. Lett. 2018, 20, 2745–2748. [DOI] [PubMed] [Google Scholar]
- 212. Sun S. F., Chen R. X., Wang G. Q., Wang J., Org. Biomol. Chem. 2018, 16, 8011–8014. [DOI] [PubMed] [Google Scholar]
- 213. DiPoto M. C., Wu J., Org. Lett. 2018, 20, 499–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Wang G. Q., Zhao S., Chen R. X., Yang L. F., Wang J., Guo H. B., Wu M. H., Domena J., Xing Y. L., Sun S. F., Tetrahedron Lett. 2017, 58, 4308–4311. [Google Scholar]
- 215.
- 215a. Zhang K. F., Yang C., Yao H. Q., Lin A. J., Org. Lett. 2016, 18, 4618–4621; [DOI] [PubMed] [Google Scholar]
- 215b. Jia Q. F., Du Z. Y., Zhang K., Wang J., Org. Chem. Front. 2017, 4, 91–94; [Google Scholar]
- 215c. Acharya A., Montes K., Jeffrey C. S., Org. Lett. 2016, 18, 6082–6085; [DOI] [PubMed] [Google Scholar]
- 215d. Jiang S. S., Li K., Yan J., Shi K. X., Zhao C. T., Yang L. M., Zhong G. F., J. Org. Chem. 2017, 82, 9779–9785; [DOI] [PubMed] [Google Scholar]
- 215e. Shao P. L., Li Z. R., Wang Z. P., Zhou M. H., Wu Q., Hu P., He Y., J. Org. Chem. 2017, 82, 10680–10686. [DOI] [PubMed] [Google Scholar]
- 216. Dittmer D. C., Avilov D. V., Kandula V. S., Purzycki M. T., Martens Z. J., Hohn E. B., Bacler M. W., Arkivoc 2010, 61–83. [Google Scholar]
- 217. Feng E. G., Huang H., Zhou Y., Ye D. J., Jiang H. L., Liu H., J. Org. Chem. 2009, 74, 2846–2849. [DOI] [PubMed] [Google Scholar]
- 218. Liu K., Sui L. C., Jin Q., Li D. Y., Liu P. N., Org. Chem. Front. 2017, 4, 1606–1610. [Google Scholar]
- 219. Andersen T. L., Frederiksen M. W., Domino K., Skrydstrup T., Angew. Chem. Int. Ed. 2016, 55, 10396–10400; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10552–10556. [Google Scholar]
- 220. Zhao H. Y., Feng Z., Luo Z. J., Zhang X. G., Angew. Chem. Int. Ed. 2016, 55, 10401–10405; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10557–10561. [Google Scholar]
- 221. Yin H. F., Kumke J. J., Domino K., Skrydstrup T., ACS Catal. 2018, 8, 3853–3858. [Google Scholar]
- 222. Reddy K. H. V., Bedier M., Bouzbouz S., Eur. J. Org. Chem. 2018, 12, 1455–1459. [Google Scholar]
- 223. Chen J. J., Guo W., Wang Z. R., Hu L., Chen F., Xia Y. Z., J. Org. Chem. 2016, 81, 5504–5512. [DOI] [PubMed] [Google Scholar]
- 224. Nishikata T., Ishida S., Fujimoto R., Angew. Chem. Int. Ed. 2016, 55, 10008–10012; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 10162–10166. [Google Scholar]
- 225. Barde E., Guérinot A., Cossy J., Synthesis 2019, 51, 178–184. [Google Scholar]
- 226.
- 226a. Prier C. K., Rankic D. A., MacMillan D. W. C., Chem. Rev. 2013, 113, 5322–5363; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226b. Romero N. A., Nicewicz D. A., Chem. Rev. 2016, 116, 10075–10166; [DOI] [PubMed] [Google Scholar]
- 226c. Marzo L., Pagire S. K., Reiser O., König B., Angew. Chem. Int. Ed. 2018, 57, 10034–10072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10188–10228; [Google Scholar]
- 226d. Bogdos M. K., Pinard E., Murphy J. A., Beilstein J. Org. Chem. 2018, 14, 2035–2064; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226e. Lemos A., Lemaire C., Luxen A., Adv. Synth. Catal. 2019, 361, 1500–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Kumagai N., Shibasaki M., Synthesis 2019, 51, 185–193. [Google Scholar]
- 228. Yerien D. E., Barata-Vallejo S., Postigo A., Chem. Eur. J. 2017, 23, 14676–14701. [DOI] [PubMed] [Google Scholar]