

Abstract
Background
Genetic risk factors can provide insight into susceptibility for acute pancreatitis (AP) and disease progression towards (infected) necrotizing pancreatitis and persistent organ failure. The aim of the study was to undertake a systematic review of the genetic evidence for AP.
Methods
Online databases (MEDLINE, Embase, BIOSIS, Web of Science, Cochrane Library) were searched to 8 February 2018. Studies that reported on genetic associations with AP susceptibility, severity and/or complications were eligible for inclusion. Meta‐analyses were performed of variants that were reported by at least two data sources. Venice criteria and Bayesian false‐discovery probability were applied to assess credibility.
Results
Ninety‐six studies reporting on 181 variants in 79 genes were identified. In agreement with previous meta‐analyses, credible associations were established for SPINK1 (odds ratio (OR) 2·87, 95 per cent c.i. 1·89 to 4·34), IL1B (OR 1·23, 1·06 to 1·42) and IL6 (OR 1·64, 1·15 to 2·32) and disease risk. In addition, two novel credible single‐nucleotide polymorphisms were identified in Asian populations: ALDH2 (OR 0·48, 0·36 to 0·64) and IL18 (OR 1·47, 1·18 to 1·82). Associations of variants in TNF, GSTP1 and CXCL8 genes with disease severity were identified, but were of low credibility.
Conclusion
Genetic risk factors in genes related to trypsin activation and innate immunity appear to be associated with susceptibility to and severity of AP.
A comprehensive systematic review and meta‐analysis of the genetic evidence for acute pancreatitis was conducted, and identified credible associations of SPINK1, ALDH2, IL1B, IL6 and IL18 with the risk of acute pancreatitis. A lack of replication studies precludes definite conclusions to be drawn regarding the role of genetics in severity and severe clinical phenotypes of acute pancreatitis.
Genes affecting trypsin activation and innate immunity important
Antecedentes
Los factores de riesgo genético pueden contribuir a determinar la susceptibilidad para desarrollar una pancreatitis aguda (acute pancreatitis, AP) y de su progresión a pancreatitis necrotizante (infectada) e insuficiencia orgánica crónica. Nuestro objetivo fue revisar de forma sistemática la evidencia genética de la pancreatitis aguda.
Métodos
Se revisaron las bases de datos electrónicas (MEDLINE, Embase, BIOSIS, Web of Science, Cochrane Library) hasta febrero de 2018. Se incluyeron estudios que presentaban información de las asociaciones genéticas y la susceptibilidad de AP, gravedad y/o complicaciones. Se realizó un metaanálisis de las variantes genéticas descritas en al menos dos fuentes. Se aplicaron los criterios de Venecia y la probabilidad bayesiana de falsa alarma para la valoración de la credibilidad.
Resultados
Se identificaron 96 estudios que analizaron 181 variantes en 79 genes. De acuerdo con un metaanálisis previo, se establecieron asociaciones creibles con el riesgo de enfermedad para SPINK1 (razón de oportunidades, odds ratio, OR 2,87, i.c. del 95% 1,89‐4,34), IL1B (OR 1,23, i.c. del 95% 1,06‐1,42) e IL6 (OR 1,64, i.c. del 95% 1,15‐2,32). Además, en poblaciones asiáticas, se identificaron dos nuevos polimorfismos de nucleótico único (SNP) creibles en ALDH2 (OR 0,48, i.c. del 95% 0,36‐0,64) e IL18 (OR 1,47, i.c. del 95% 1,18‐1,82). En cuanto a la gravedad de la enfermedad se identificaron variantes en los genes TNF, GSTP1 y CXCL8, pero de baja credibilidad en función de nuestra evaluación.
Conclusión
Los factores de riesgo genéticos en genes relacionados con la activación de la tripsina y la inmunidad innata parecen ser estar asociados con la susceptibilidad y gravedad de la pancreatitis aguda.
Introduction
Acute pancreatitis (AP) is a common inflammatory disease of the pancreas with an incidence of 13–45 per 100 000 population in Western countries1. Most patients experience a mild disease course with hospital discharge within 1 week, but around 20 per cent develop severe AP. Pancreatic necrosis occurs in about 30 per cent of patients, and severe complications such as secondary bacterial infection of (peripancreatic) necrosis and (persistent) organ failure lead to mortality rates of up to 30 per cent2.
Biliary disease and alcohol are the leading causes of AP, and account for 70 per cent of cases; other causes are hypertriglyceridaemia, hypercalcaemia, iatrogenic (mostly related to endoscopic retrograde cholangiopancreatography and drugs) and autoimmune disease, and some are classified as idiopathic3. The risk of developing AP in patients with asymptomatic gallstone disease or heavy alcohol consumers is 2–3 per cent, indicating a complex multifactorial pathogenesis4, 5. As well as host factors such as smoking and diabetes, genetic variants are suspected to play an important role in disease susceptibility, outcome and progression6. Improved understanding of genetic risk factors may help to identify patients at risk of AP, severe complications and progression to chronic pancreatitis.
Since the discovery of the causal variant of hereditary pancreatitis in the PRSS1 gene by Whitcomb and colleagues7, two decades of mostly candidate gene genetic association studies have been initiated. Other disease‐causing variants (such as SPINK1, CTRC, CASR and CFTR 8) were discovered, and stimulated the initiation of genetic association studies in patients with AP. However, the majority of identified associations could not be replicated in similar or other populations, probably because most studies lacked statistical power owing to insufficient sample sizes. Summarization and analysis of current knowledge is needed9.
Field synopsis and meta‐analysis are powerful tools to integrate genetic data from a large field and identify credible genetic associations10, 11, 12. In the present review, all genetic data associated with AP were systematically collected and summarized. In addition, credibility of the evidence was assessed by applying the Venice criteria9 and the Bayesian false‐discovery probability (BFDP) method13. The aim was to provide a framework for clinicians and researchers to guide genetic research, and the development of clinical diagnostic tools and personalized therapeutic interventions.
Methods
This systematic study was conducted by following a protocol, and is concordant with the Updated Guidance on Human Genome Epidemiology Reviews and Meta‐Analysis of Genetic Associations14, and the PRISMA guidelines15 for systematic reviews and meta‐analysis (Appendix S1 , supporting information).
Data sources and searches
A systematic search of five online databases (MEDLINE, Embase, BIOSIS, Web of Science, Cochrane Library) was performed, using the keywords and index terms ‘pancreatitis’ combined with ‘mutation*’, ‘polymorphism*’ and ‘variant*’, and applying the limit ‘human’. After removal of duplicates, identified articles were screened for eligibility in two steps: first by title and/or abstract independently by two authors, and second by reading of the full text, and selected based on predefined inclusion and exclusion criteria. The references of identified studies and reviews were cross‐checked for additional studies. In case of disagreement, consensus was resolved by discussion.
Study selection
Genetic association studies (case–control and cohort) associated with AP and recurrent AP in humans published in a peer‐reviewed journal before 8 February 2018 that met the criteria were included with no restriction on language. Genome‐wide association studies were considered, but not identified. Studies reporting on other than bi‐allelic markers were excluded from quantitative analyses. Studies reporting on tropical, hereditary or autoimmune pancreatitis are considered as chronic pancreatitis, and were excluded. Abstracts, case reports, economic evaluations, in vitro and animal studies were excluded. Studies reporting exclusively on paediatric patients (age below 18 years) were also excluded, because of differences in aetiology16.
Published meta‐analyses of identified variants were considered for comparison. Studies with identical, or largely overlapping, sets of data were compared, and the largest data set was included in the meta‐analysis; when data were identical, the first published study was included.
Data extraction and quality assessment
Data extraction was performed separately by two authors, and differences were resolved by discussion. As well as study identifiers such as first author, year of publishing, journal and language, the following demographic data were extracted: study location and design, inclusion and exclusion criteria, sample size, reported outcomes, studied aetiology, ethnic background of patients and control subjects, source of controls, age and sex.
The following data points for each variant were extracted: Single Nucleotide Polymorphism database (dbSNP) identifier number, Human Gene Nomenclature Committee symbol and name, type of genetic variation, position and genotype distribution. Genotype counts were calculated when genotype frequencies and numbers of cases and controls were reported. If only allele frequencies were available, genotype counts were calculated assuming distribution according to the Hardy–Weinberg equilibrium (HWE).
The corresponding author was contacted when genotype counts were not presented in the article or could not be calculated, or if an inconsistency was noticed. They were asked to supply raw data (genotype distribution) or clarification. The data points were excluded for quantitative analysis because of insufficient data if no, or only a partial, response was received after two attempts.
The credibility of significant associations was assessed according the Venice criteria, based on the published recommendations of the HuGENet Working Group9. The Venice criteria involve a three‐point grading system, assessing the amount of evidence (power), replication and protection from bias. To correct for multiple testing, the BFDP method was applied. The overall assessment of credibility was, consistent with the recommendations, based on both the BFDP and Venice grade, and was rated strong (A), moderate (B) or weak (C).
Statistical analysis
All analyses were performed using R studio version 3.3.217. The packages meta and metafor18 were used for meta‐analysis and funnel plotting. Because mode of inheritance is mostly unknown in complex traits, the primary meta‐analysis was conducted using the allelic contrast model, in which allele frequencies of the major and minor allele are compared (A versus a). In addition, meta‐analysis were performed using dominant (AA + Aa versus aa) and recessive (AA versus Aa + aa) genetic models. Unless stated otherwise, the effect of the minor allele (based on the Ensemble database) was investigated19. For extracted variants that had been reported on by multiple studies, summary odds ratios (ORs) and 95 per cent c.i. were calculated using a random‐effects model (DerSimonian and Laird). Between‐study heterogeneity was calculated with the Q test and I 2 statistic. Deviation from HWE was calculated systematically, and defined as P < 0·050 using a χ2 test with 1 degree of freedom. Funnel plot analysis and the Harbord statistical test20 was performed for significant associations to detect possible publication bias and small‐study effects. Sensitivity analysis was performed for significant associations excluding studies that deviated from HWE. Where possible, subgroup meta‐analysis for severity and ethnicity was done.
Results
The search resulted in 841 studies potentially eligible for inclusion, of which 96 articles met the inclusion and exclusion criteria (Fig. 1; Table S1 , supporting information). Eighty‐seven studies reported on AP, six studies on recurrent disease and three on both. This resulted in a total of 181 studied variants in 79 genes for AP and 24 variants in 12 genes for recurrent AP. Eight published meta‐analyses of genetic variants associated with AP were identified, reporting on nine single‐nucleotide polymorphisms (SNPs) in nine different genes (Table 1). The remaining SNPs in the present review had not previously been subjected to meta‐analysis.
Figure 1.
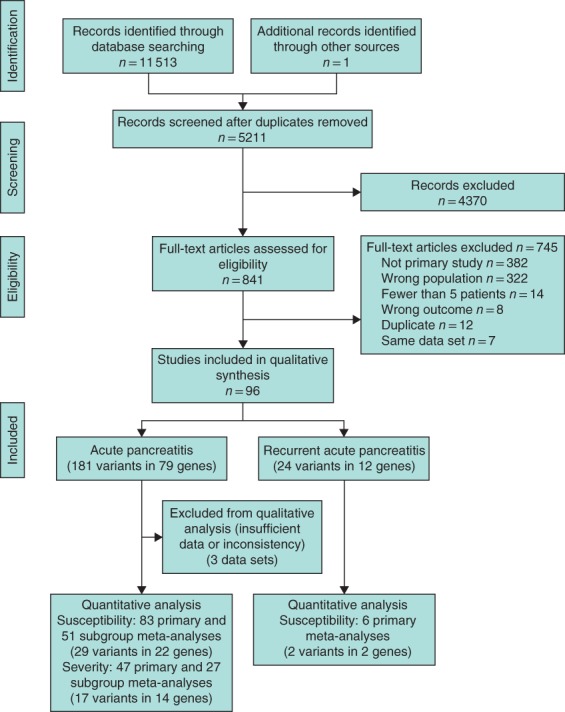
Flow diagram of study selection and quantitative analysis
Table 1.
Genes and variants associated with susceptibility for acute pancreatitis compared with previously published meta‐analyses
| Present meta‐analysis | Previous meta‐analyses | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variant | No. of cases | No. of controls | No. of studies | Outcome | Credibility* | Reference | No. of cases | No. of controls | No. of studies | Outcome† |
| SPINK1 | rs17107315 | 1493 | 2595 | 9 | + | A | Joergensen et al.21 | 1135 | 2822 | 8 | + |
| IL1B | rs1143634 | 1301 | 1171 | 6 | + | C | Yin et al.22 | 519 | 388 | 3 | + |
| CXCL8 (IL8) | rs4073 | 1770 | 1990 | 9 | + | C | Yin et al.22 | 503 | 758 | 5 | + |
| ACE | rs4646994 | 770 | 4878 | 4 | − | Fang et al.23 | 245 | 1455 | 3 | + | |
| CCL2 | rs1024611 | 470 | 562 | 4 | − | Fang et al.24 | 567 | 562 | 4 | + | |
| CD14 | rs2569190 | 1328 | 1195 | 6 | − | Yuan and Wang25 | 1211 | 932 | 5 | − | |
| IL10 | rs1800896 | 1738 | 1691 | 8 | − | Yin et al.22 | 339 | 243 | 2 | − | |
| TLR4 | rs4986790 | 994 | 794 | 4 | − | Zhou et al.26 | 1255 | 998 | 6 | − | |
| TNF | rs1800629 | 1335 | 1076 | 6 | − | Yin et al.27 | 1006 | 782 | 6 | − | |
Assessed by Venice criteria, based on sample size, heterogeneity among studies, and risk of bias.
Reported by original author.
+, positive association; −, no association.
To provide a comprehensive overview, the variants extracted from the studies are presented per gene category (Table S2 , supporting information). Together, the studies included 18 138 patients and 32 227 control subjects. Two‐thirds of the studies were performed in a Caucasian population; the remaining populations were Asian (30·5 per cent), Hispanic/Latin American (1·7 per cent) or of mixed origin (0·7 per cent). Further study characteristics are presented in Appendix S1 (supporting information).
Subgroup analysis based on severity of the disease course in AP was done in 45 studies. Other clinical outcome measures such as pancreatic necrosis, infectious and systemic complications, mortality and need for surgery were reported for four, 12, 41, four and three variants respectively (Table S3 , supporting information).
Thirty‐one of the 205 extracted variants were reported by at least two articles and were subsequently selected for meta‐analysis. Three data sets were excluded due to insufficient or inconsistent data. A mean of 750 (median 512 (i.q.r. 325)) patients and 940 (median 652 (i.q.r. 418)) controls per variant were included in the primary meta‐analyses. Results of the primary and subgroup meta‐analyses, sensitivity analyses and credibility assessments are presented in Tables S3 – S8 (supporting information). Nine nominal significant associations in six variants (genes SPINK1, ALDH2, IL1B, IL6, GSTT1 and IL18) were identified using the allelic contrast genetic model (Table 2). Additional meta‐analyses using dominant and recessive genetic models revealed six more significant associations (Table 3) in five additional variants (genes IL18, IL1B, CCL2, CXCL8 and IL10). After credibility assessment, five and three associations were rated as having strong and moderate evidence respectively. The significant associations are discussed in more detail below.
Table 2.
Nominal significant associations for variants identified for meta‐analysis using the allelic contrast genetic model with credibility factors in (recurrent) acute pancreatitis
| Effect size | Heterogeneity | Credibility | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variant | Group | Minor allele | Model | Ethnicity | No. of cases | No. of controls | No. of studies | OR | P | I 2 (%) | P | BFDP | Venice | Overall |
| SPINK1 | rs17107315 | AP | C | Allelic | Mixed | 1493 | 2595 | 9 | 2·87 (1·89, 4·34) | 6·3 × 10−7 | 0·0 | 0·493 | 0·055 | AAA | A |
| SPINK1 | rs17107315 | AP | C | Allelic | White | 1085 | 1687 | 5 | 2·49 (1·55, 3·98) | 1·5 × 10−4 | 0·0 | 0·244 | 0·529 | AAA | A |
| SPINK1 | rs17107315 | RAP | C | Allelic | Mixed | 233 | 1300 | 3 | 7·51 (3·20, 17·64) | 3·7 × 10−6 | 15 | 0·389 | 0·75 | AAA | A |
| SPINK1 | rs17107315 | RAP | C | Allelic | White | 136 | 758 | 2 | 5·65 (2·69, 11·88) | 4·9 × 10−6 | 0·0 | 0·847 | 0·664 | BAA | B |
| ALDH2 | rs671 | AP | A | Allelic | Asian | 350 | 272 | 2 | 0·48 (0·36, 0·64) | 3·3 × 10−7 | 0·0 | 0·415 | 0·008 | BAA | B |
| IL1B | rs1143634 | AP | A | Allelic | Asian | 997 | 899 | 4 | 1·23 (1·03, 1·45) | 0·018 | 0·0 | 0·318 | 0·788 | AAC | C |
| IL6 | rs1800795 | AP | C | Allelic | Asian | 607 | 607 | 2 | 1·22 (1·00, 1·47) | 0·045 | 0·0 | 0·726 | 0·882 | AAA | C |
| GSTT1 | Null | AP | Null | Allelic | White | 575 | 804 | 4 | 0·66 (0·44, 0·99) | 0·045 | 41·8 | 0·138 | 0·907 | AAA | C |
| IL18 | rs1946518 | AP | T | Allelic | Asian | 325 | 418 | 2 | 1·25 (1·00, 1·57) | 0·049 | 0·0 | 0·781 | 0·905 | BAA | C |
Values in parentheses are 95 per cent confidence intervals. OR, odds ratio; BFDP, Bayesian false‐discovery probability; AP, acute pancreatitis; RAP, recurrent acute pancreatitis.
Table 3.
Nominal significant associations for variants identified for meta‐analysis using dominant or recessive genetic models with credibility factors in (recurrent) acute pancreatitis
| Effect size | Heterogeneity | Credibility | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variant | Group | Minor allele | Model | Ethnicity | No. of cases | No. of controls | No. of studies | OR | P | I 2 (%) | P | BFDP | Venice | Overall |
| IL1B | rs16944 | AP | A | Dominant | Asian | 857 | 861 | 4 | 1·23 (1·06, 1·42) | 0·005 | 0·0 | 0·519 | 0·622 | AAA | A |
| IL6 | rs1800795 | AP | C | Recessive | Asian | 607 | 607 | 2 | 1·64 (1·15, 2·32) | 0·006 | 0·0 | 0·685 | 0·754 | AAA | A |
| IL18 | rs187238 | AP | G | Dominant | Asian | 325 | 418 | 2 | 1·47 (1·18, 1·82) | 4·9 × 10−4 | 0·0 | 0·328 | 0·234 | BAA | B |
| CCL2 | rs1024611 | RAP | G | Dominant | Mixed | 302 | 652 | 2 | 2·19 (1·23, 3·89) | 0·008 | 58·6 | 0·120 | 0·876 | BCA | C |
| CXCL8 (IL8) | rs4073 | AP | T | Recessive | Mixed | 1770 | 1990 | 9 | 1·29 (1·06, 1·57) | 0·010 | 57·7 | 0·015 | 0·760 | ACC | C |
| CXCL8 (IL8) | rs4073 | AP | T | Recessive | White | 368 | 400 | 4 | 1·19 (1·01, 1·40) | 0·034 | 0·0 | 0·300 | 0·885 | BAC | C |
| IL10 | rs1800872 | AP | C | Dominant | Mixed | 830 | 802 | 4 | 1·17 (1·01, 1·35) | 0·040 | 0·0 | 0·992 | 0·880 | AAA | C |
| IL1B | rs1143634 | AP | T | Recessive | Mixed | 1301 | 1171 | 6 | 1·41 (1·00, 1·99) | 0·048 | 26·9 | 0·295 | 0·906 | ABC | C |
Values in parentheses are 95 per cent confidence intervals. OR, odds ratio; BFDP, Bayesian false‐discovery probability; AP, acute pancreatitis; RAP, recurrent acute pancreatitis.
Variants associated with disease risk
Serine protease inhibitor Kazal type 1 (SPINK1)
Premature intrapancreatic trypsinogen activation leads to autodigestion and is believed to be a key initiating process in the pathogenesis of pancreatitis3, 28. Transcriptions of the SPINK1 gene in the pancreas function as a trypsin inhibitor, and loss‐of‐function variants are believed to increase autoactivation of trypsinogen29. Twenty variants in the SPINK1 gene were described in ten different studies. Only two of the ten studies reported an association of AP susceptibility with the N34S variant (c.101A>G, rs17107315). Functionality of the variant remains unclear30. The present meta‐analysis of nine studies (1493 patients, 2595 controls) identified an association in the allelic model with susceptibility for AP (OR 2·87, 95 per cent c.i. 1·89 to 4·34; P = 6·3 × 10−7) (Table 2). This association remained robust following subgroup analysis of five studies of Caucasians (OR 2·49, 1·55 to 3·98; P = 1·5 × 10−4), but not of Asians. The association was rated as strong, indicating a highly credible finding.
A Caucasian population of 72 patients with recurrent disease and 670 controls showed a significant association with the N34S variant31, and has been confirmed in a Japanese study32 but not in an Italian population33. Meta‐analysis of these studies confirmed this association in the overall population (3 studies) (OR 7·51, 95 per cent c.i. 3·20 to 17·64; P = 3·7 × 10−6) and the Caucasian population (2 studies) (OR 5·65, 2·69 to 11·88; P = 4·9 × 10−6) (Table 2). Two articles34, 35 reported on the functional c.194+2T>C (IVS3+T>C, rs148954387) variant, but found no association. The present meta‐analysis of 210 patients and 707 control subjects confirmed this result.
Meta‐analysis of variants of other members of the trypsin family of serine proteases and peptidases, such as PRSS1 and PRSS2, showed no correlation with disease occurrence or phenotype (Tables S3–S14 , supporting information).
Cytokines: interleukins , 6 and 18, and C‐C motif chemokine ligand 2
Interleukins are produced mainly by macrophages and lymphocytes, and act as proinflammatory or anti‐inflammatory communication molecules between cells of the immune system. Cytokines are likely causal candidates and have therefore been investigated extensively. Positive associations with AP susceptibility were reported in genes IL1B, IL1RA, CXCL8 (IL8), IL10 and IL18 (Table S2 , supporting information).
IL1B codes for interleukin (IL) 1β, one of the proinflammatory cytokines that is increased during an episode of AP36. The functional 511 C/T (g.4490 T>C, rs16944) promoter variant enhances gene expression of IL1B 37, and has been associated with the severity of the episode38, 39. Meta‐analysis of four studies (all Asian populations) including 857 patients and 861 controls showed a correlation with AP risk in the dominant genetic model (OR 1·23, 95 per cent c.i. 1·06 to 1·42), but not in allelic contrasts. Between‐study heterogeneity was low, and the association was rated as strong with a BFDP of 0·622 (Table 3). There was no association of the synonymous variant rs1143634 in allelic contrasts in six studies of patients with mixed ethnicity (4 Asian, 2 Caucasian). However, when performing ethnic subgroup analysis, a correlation was found for the Asian populations (OR 1·23, 1·03 to 1·45; P = 0·018) (Table 2). Meta‐analysis of the recessive genetic model found positive associations in both Asian and mixed populations. Significance was lost in all associations when excluding one study in which the control group deviated from HWE.
IL‐6 is another proinflammatory cytokine that is correlated with disease severity36. The −174G>C intronic variant (g.4880C>G, rs1800795) demonstrated lower expression of IL6 in vitro, and is associated with susceptibility to juvenile rheumatoid arthritis40. Pooled analyses of two studies (both Asian populations) in 607 patients and controls were associated with susceptibility to AP in both the allelic contrast (OR 1·22, 95 per cent c.i. 1·00 to 1·47; P = 0·045) (Table 2) and the recessive genetic model (OR 1·64, 1·15 to 2·32; P = 0·006) (Table 3). Credibility of the associations was weak and strong respectively, based on BFDP values of 0·882 and 0·754.
Two functional intronic variants of IL18 have been studied in AP. The −607C/A (g.4383A>C, rs1946518) and − 137G>C (g.4853G>C, rs187238) variants have been shown to increase IL‐18 mRNA levels in expression analyses assays41. Meta‐analyses of the rs1946518 variant in two studies reached nominal significance (OR 1·25, 95 per cent c.i. 1·00 to 1·57; P = 0·049), but was deemed weak evidence after BFDP correction (Table 2). The rs187238 variant was associated with AP risk in the dominant genetic model only (OR 1·47, 1·18 to 1·82; P = 4·9 × 10−4), and was rated as moderate evidence because of the limited sample size (Table 3).
C‐C motif chemokine ligand 2 (CCL2), also referred to as monocyte chemoattractant protein 1 (MCP‐1), is involved in the migration of inflammatory cells, and is expressed during pancreatic inflammation42. Papachristou et al.43 found an association of the functional rs1024611 variant with severe AP. This was not replicated in patients with AP in another Caucasian population; however, an over‐representation of the G allele was found in patients with recurrent disease33. Meta‐analysis did not confirm an association with AP. However, this was established in two studies of patients with recurrent disease in the dominant genetic model (OR 2·19, 95 per cent c.i. 1·23 to 3·89; P = 0·008). Owing to heterogeneity, the credibility was rated as weak.
Meta‐analyses of other interleukin variants resulted in weak associations, owing to failure to pass correction for multiple testing (IL1B rs1143634), between‐study heterogeneity (IL10 rs1800872), or both (CXCL8 rs4073).
Mitochondrial aldehyde dehydrogenase (ALDH2)
Four variants in genes related to the ethanol metabolism pathway have been investigated in Asian populations. Variants in these genes are common in Asian populations and associated with susceptibility to alcoholism, alcohol sensitivity and oesophageal cancer.
The *2 allele of ALDH2 (g.42421G>A, rs671) was found significantly less often in patients with alcoholic pancreatitis, but not in those with a biliary aetiology44, 45. When pooling patients with both aetiologies from two studies, a protective association was found for AP risk with the minor *2 allele (OR 0·48, 95 per cent c.i. 0·36 to 0·64; P = 3·3 × 10−7) in allelic contrasts (Table 2). The credibility score was moderate (BFDP 0·008, Venice grade BAA) owing to the relatively small sample size. Meta‐analysis of variants in other genes related to ethanol metabolism, such as ADH2, ADH3 and CYP2E1, did not show significant associations with disease risk or severity.
Antioxidant enzymes: glutathione S‐transferase
The antioxidant enzyme glutathione S‐transferase (GTS) protects tissues from free radical injury and has four classes: alpha (GTSA), mu (GSTM), pi (GSTP) and theta (GSTT)46. The effect of ten variants of these genes on AP susceptibility was investigated. The effect of four functional alleles in three studies was studied, but only one SNP in the GSTM1 gene showed an association with AP susceptibility47. Meta‐analysis could not confirm this, but found instead an overrepresentation of the null allele of GSTT1 in controls (OR 0·66, 95 per cent c.i. 0·44 to 0·99; P = 0·045); however, this was rated as weak evidence owing to the likelihood of being a false‐positive association based on a BFDP value of 0·907 (Table 2).
Variants associated with severity of acute pancreatitis
Reported associations with disease severity and complications were assessed systematically; 17 variants reported on by more than two articles were identified (Tables S9 – S14 , supporting information). Meta‐analysis showed three significant associations in the genes CXCL8, GSTP1 and TNF (tumour necrosis factor (TNF) α) (Table 4). However, the BFDP value for the CXCL8 and GSTP1 variants (both greater than 0·8) indicated a high likelihood of a false‐positive outcome. The BFDP value for the TNF variant was very low, indicating a highly significant association. However, significance was lost when sensitivity analysis excluded one study that deviated from HWE, indicating the presence of genotyping error or population stratification47. The credibility of all three associations was subsequently rated as weak. Nine positive associations with disease severity in the genes TLR3, TLR4, TLR6, CD14, NFKBIA, PKA2G7, PPARG and SERPINE1 were found that were not replicated in another study (Table 5). Although the limited data on other disease phenotypes did not allow for pooled analyses, positive associations were identified for infectious complications (TLR4, CD14, DEFB1, IL10, REN), systemic complications (TNF, TNFAIP3, PLA2G7), pancreatic necrosis (HMOX1), mortality (REN) and surgery (TLR2). These are viable candidate genes, and replication is needed (Table 5).
Table 4.
Nominal significant associations for variants identified for meta‐analysis with credibility factors comparing patients with mild and severe acute pancreatitis
| Effect size | Heterogeneity | Credibility | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Variant | Group | Minor allele | Model | Ethnicity | No. of cases | No. of controls | No. of studies | OR | P | I 2 (%) | P | BFDP | Venice | Overall |
| TNF | rs1800629 | AP | A | Recessive | Mixed | 703 | 702 | 6 | 0·54 (0·41, 0·71) | 1·3 × 10−5 | 0 | 0·707 | 0·041 | AAC | C |
| GSTP1 | rs1695 | AP | G | Recessive | White | 282 | 125 | 2 | 1·43 (1·05, 1·93) | 0·022 | 0 | 0·950 | 0·845 | BAA | C |
| CXCL8 (IL8) | rs4073 | AP | T | Dominant | Mixed | 387 | 200 | 6 | 0·74 (0·56, 0·98) | 0·039 | 29 | 0·159 | 0·950 | BBC | C |
Values in parentheses are 95 per cent confidence intervals. OR, odds ratio; BFDP, Bayesian false‐discovery probability; AP, acute pancreatitis; RAP, recurrent acute pancreatitis.
Table 5.
Genes and variants associated with disease severity or complications of acute pancreatitis that were not replicated in another study*
| Gene | Variant | Reference | Outcome | Incidence (%) | No. of cases | No. of controls | Risk allele |
|---|---|---|---|---|---|---|---|
| Severity | |||||||
| CD14 | rs5744455 | Masamune et al.34 | SAP | 31 | 107 | 238 | C |
| TLR3 | rs3775291 | Matas‐Cobos et al.48 | SAP | 13 | 36 | 233 | C |
| TLR4 | G11367C | Zhang et al.49 | SAP | 33 | 150 | 300 | C |
| TLR6 | rs7543795 | Matas‐Cobos et al.48 | SAP | 13 | 36 | 233 | A |
| NFKBIA | rs696 | Zhang et al.49 | SAP | 33 | 150 | 300 | T |
| PLA2G7 | rs16874954 | Ma et al.50 | SAP | 51 | 486 | 46 | A |
| PLA2G7 | rs1805017 | Ma et al.50 | SAP | 51 | 48 | 46 | A |
| PPARG | rs1801282 | Zhang et al.51 | SAP | 33 | 150 | 300 | G |
| SERPINE1 | rs1799889 | Tukiainen et al.52 | SAP | 33 | 150 | 300 | 5G |
| Pancreatic necrosis | |||||||
| HMOX1 | S/L | Gulla et al.53 | Pancreatic necrosis | 50 | 63 | 64 | L |
| Infectious complications | |||||||
| CD14 | rs5744455 | Masamune et al.34 | Infected necrosis | 9 | 32 | 314 | C |
| TLR4 | rs4986790 | Gao et al.54 | Infected necrosis | 24 | 30 | 95 | G |
| DEFB1 | rs11362 | Tiszlavicz et al.55 | Infected necrosis | 23 | 29 | 95 | A |
| DEFB1 | rs1799946 | Tiszlavicz et al.55 | Infected necrosis | 23 | 29 | 95 | C |
| IL10 | rs1800896 | Zhang et al.56 | Septic shock | 30 | 33 | 76 | G |
| REN | rs5707 | Skipworth et al.57 | Infected necrosis | 14 | 52 | 317 | G |
| Systemic complications | |||||||
| TNF | rs1799964 | Bishehsari et al.58 | MODS | 11 | 23 | 188 | C |
| TNF | rs1800630 | Bishehsari et al.58 | MODS | 11 | 23 | 188 | A |
| TNF | rs361525 | de‐Madaria et al.59 | Systemic complications | 11 | 9 | 75 | A |
| TNFAIP3 | rs5029924 | Liu et al.60 | SIRS | 35 | 47 | 88 | T |
| PLA2G7 | rs16874954 | Ma et al.50 | MODS | 6 | 6 | 88 | A |
| Mortality | |||||||
| REN | rs5707 | Skipworth et al 57 | Mortality | 7 | 36 | 477 | C |
| Surgery | |||||||
| TLR2 | Microsatellite | Takagi et al.61 | Surgery | 15 | 30 | 172 | S |
Reported by original author.
SAP, severe acute pancreatitis; MODS, multiple organ dysfunction syndrome; SIRS, systemic inflammatory response syndrome.
Discussion
This systematic review and meta‐analysis of genetic association studies in AP found two genetic variants in SPINK1 and ALDH2 genes that showed moderate or strong credible associations with disease risk in the allelic contrasts model, and three credible associations using dominant and recessive models in IL1B, IL6 and IL18. However, except for the SPINK1 variants, credible associations were found only with pooled analysis of Asian populations. In addition, three weak associations with disease severity were found in CXCL8, GSTP1 and TNF (TNFα). The application of credibility criteria ensures a high likelihood of identifying only true and robust associations.
The loss‐of‐function N34S variant (c.101A>G, rs17107315) in the SPINK1 gene is presumed to be a disease modifier for recurrent and chronic, but not for acute, pancreatitis6, 62. However, a meta‐analysis by Joergensen and colleagues21 showed an association with AP risk, which remained robust in the present updated meta‐analyses of both AP and recurrent AP. The trypsin inhibitory function of SPINK1, the relatively large sample size of approximately 1500 patients, and the absence of significant heterogeneity indicates a reliable role in susceptibility to AP and progression to recurrent or chronic disease. Pathogenic variants in the trypsinogen gene PRSS1 are causative for hereditary pancreatitis with a penetrance of 80 per cent7. Because of its known functional consequence, PRSS1 would also be expected to be associated with AP or recurrent AP. Surprisingly, such an association was not found. However, as PRSS1 variants are associated with early‐onset pancreatitis, they are possibly underrepresented in the present data set owing to the exclusion of studies performed exclusively in paediatric populations. Other possibilities are data paucity, lack of statistical power, and the fact that different variants were studied.
A protective association for the ALDH2 rs671 variant was found after pooled analyses of two studies by the same authors containing 350 patients and 272 controls of Chinese origin. The functional consequence of this non‐synonymous variant in ALDH2 is a defective enzyme involved in ethanol breakdown, and is common in Asian, but rare in non‐Asian, populations. The A allele is related to alcohol sensitivity, and shown to be protective against alcoholism and alcohol‐associated diseases, such as alcoholic pancreatitis63. Due to lack of appropriate control groups (alcoholics without alcoholic pancreatitis or other alcohol‐associated disease) in the original studies, it remains uncertain whether this variant has a true association with alcoholic pancreatitis, or rather is associated with a reduced alcohol intake.
The role of cytokines in the pathogenesis of AP has long been recognized31. Previous meta‐analyses of variants have identified associations with AP in CXCL8 (IL8) and CCL2, but not in other cytokine genes (IL1B, IL10 or TNF)22, 24, 27. Meta‐analysis showed significant associations of only low credibility with disease risk in TNF, CXCL8 (IL8) and GSTP1 genes. A previous meta‐analysis24 reported significant associations between AP risk and the G allele of the CCL2 rs1024611 variant. The present meta‐analysis of the same studies contradicted these results. The present authors explain this discrepancy by an inconsistency in the data of one study64, which was verified by the original author during data assessment. In addition, the meta‐analyses performed by Fang et al.24 included patients with recurrent disease.
A variant in the ACE gene was previously reported as having a positive association with AP, but this was not confirmed in the present study; meta‐analysis of the ACE insertion–deletion (rs4646994) in 245 patients and 1455 controls showed a positive association with the insertion23. This meta‐analysis was updated with a large study, and the association was lost in both genetic models. In general, discrepancies between the present and former meta‐analyses can be explained by the fact that, in some cases, additional studies were included, credibility criteria were applied, and there was bias in the earlier studies based on the choice of genetic model (dominant or recessive). Here, the allelic contrasts model, which is independent of mode of inheritance, was used for the primary analysis.
No credible associations with disease severity (defined by the Atlanta criteria65) could be established in the pooled analyses. A paucity of data relating variants to disease outcome and complications is the main explanation for the lack of credible associations. Reported unreplicated genetic variants associated with infected (peri)pancreatic necrosis34, 54, 55, 56, 57 and multiple organ failure syndrome50, 58, 59 are involved mainly in pathways of innate immunity (Fig. 2). Lipopolysaccharide (endotoxin), produced by Gram‐negative bacteria, is an important activator of the Toll‐like receptor 4 (TLR4) pathway that leads to innate immune system activation, the production of proinflammatory cytokines (TNFα, IL‐1β, IL‐6) and antimicrobial peptides (β‐defensins). A role has been suggested for cytokine polymorphisms in the development of severe complications28, 66; however, redundancy in these downstream inflammatory responses makes it less likely that these variants alone can account for progression to severe AP. Associated variants in these genes, together with (unidentified) variants in more upstream genes of the pathway, need replication to investigate causality in the pathogenesis of disease progression.
Figure 2.
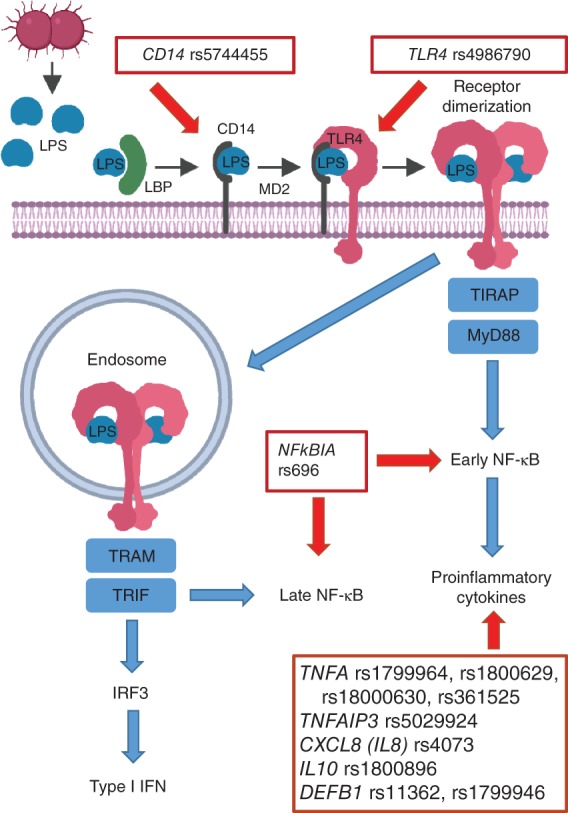
TLR4/CD14 pathway and genetic variants significantly associated with disease progression (severity or complications), identified by unreplicated primary studies or meta‐analysis CD, cluster of differentiation; LPS, lipopolysaccharide; TLR, Toll‐like receptor; LBP, lipopolysaccharide binding protein; MD2, lymphocyte antigen 96; TIRAP, Toll–interleukin 1 receptor (TIR) domain‐containing adaptor protein; MyD, myeloid differentiation primary response 88; NF, nuclear factor; TRAM, translocating chain‐associated membrane protein; TRIF, TIR‐domain‐containing adapter‐inducing interferon β; IRF, interferon regulatory transcription factor; IFN, interferon.
This review has some limitations, so the results of the present meta‐analyses should be interpreted with caution. A general limitation of synthesis of genetic association studies in complex diseases is the lack of replication and small sample sizes. This is supported by a study67 showing that only 3·6 per cent of associations can be replicated consistently. Usually, large sample sizes are needed to establish a causal effect. Therefore, most small‐scale studies are underpowered and at risk of not detecting true associations (type II error). This means that a non‐significant association does not necessarily mean that there is no causal relationship with the phenotype of interest. To correct for multiple testing of the variants, the authors used the BFDP method. Although this method is independent of the number of associations being evaluated, and therefore less conservative than, for example, the Bonferroni method, there is a risk of losing true associations (false‐negatives). Second, there is significant heterogeneity in the definition of severity and complications of AP. Some studies use clinical scoring systems that are frequently used by ICUs, such as Acute Physiology And Chronic Health Evaluation (APACHE) II and Ranson scores. These are, however, limited by poor sensitivity and specificity68, 69, leading to underestimation of the true effect size. Third, due to lack of data reporting in the included studies, it was often difficult to make a complete risk of bias assessment. For example, most studies did not report whether the studied population included patients with recurrent disease. Some 17 and 8 per cent of patients with a first episode of AP progress within 5 years to recurrent AP and chronic pancreatitis respectively68. Owing to this limitation, it is not possible to draw definitive conclusions regarding the effect of genetic variation on sentinel AP. Furthermore, only a few studies reported on methodologically important factors such as blinding of laboratory personnel, genotyping quality controls, missing data and ethnicity. In addition, details of statistical analyses were often missing; only around half of the studies mentioned whether they calculated deviation from HWE of the control group. Correction for multiple testing was used in less than half of the studies reporting on three or more variants. Finally, the source of control subjects was heterogeneous and often not specified. Control subjects are ideally sex‐ and age‐matched, and selected from the same population as the patients70.
Although the clinical relevance of identifying single associated genetic variants remains debatable, it can potentially provide clues about pathophysiological mechanisms and lead to the discovery of novel therapeutic targets. Furthermore, the accumulation of genetic risk factors can have a significant impact on diagnostic and prognostic strategies, as shown for other complex diseases such as Parkinson's disease71.
Future genetic studies need to be well designed, adequately powered, and include an extensively phenotyped patient cohort with clear definitions of aetiology. International collaboration and joint analyses will increase the power and methodological quality of genetic research. For standardization of definitions, the authors recommend using the revised Atlanta criteria65. Currently, there is no consensus regarding when to classify patients as having recurrent AP. Follow‐up of 5 years allows for the exclusion of patients who progress to recurrent or chronic pancreatitis, and for the identification of true causal variants for sentinel AP. High‐throughput sequencing, such as exome sequencing, has been successful for the identification of novel genetic risk factors in complex diseases, but generally requires a large sample size and a control group drawn from the same population. Variants associated with specific phenotypes (infected necrotizing pancreatitis or multiple organ dysfunction syndrome) can be identified by applying strategies that reduce the need for large sample sizes, such as selection of patients from opposing phenotypic extremes72, 73, 74.
Genetic risk factors in genes related to trypsin activation and innate immunity appear to be associated with AP susceptibility and severity. However, robust replication studies among genetic associations with severe clinical phenotypes are needed to push forward clinical applications.
Although open for debate, there are indications that multiple variants with a modest effect size contribute to the occurrence of disease progression processes such as pancreatic necrosis and secondary systemic or infectious complications.
Supporting information
Appendix S1. Supplementary methods and results
Table S1. Characteristics of included studies
Table S2. Comprehensive overview of extracted genetic variants associated with (recurrent) acute pancreatitis, categorized by gene function
Table S3. Primary meta‐analysis of variants associated with susceptibility for (recurrent) acute pancreatitis
Table S4. Subgroup meta‐analysis of variants associated with susceptibility for (recurrent) acute pancreatitis in patients of Caucasian ethnicity
Table S5. Subgroup analysis of variants associated with susceptibility for (recurrent) acute pancreatitis in patients of Asian ethnicity
Table S6. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with susceptibility for (recurrent) acute pancreatitis
Table S7. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with susceptibility for (recurrent) acute pancreatitis in patients of Caucasian ethnicity
Table S8. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with susceptibility for (recurrent) acute pancreatitis in patients of Asian ethnicity
Table S9. Primary meta‐analysis of variants associated with disease severity
Table S10. Subgroup meta‐analysis of variants associated with disease severity in patients of Caucasian ethnicity
Table S11. Subgroup analysis of variants associated with disease severity in patients of Asian ethnicity
Table S12. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with disease severity
Table S13. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with disease severity in patients of Caucasian ethnicity
Table S14. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with disease severity in patients of Asian ethnicity
Acknowledgements
The authors thank all original authors who provided raw data and/or clarification upon request.
Disclosure: The authors declare no conflict of interest.
Funding information
No funding
References
- 1. Spanier B, Bruno MJ, Dijkgraaf MG. Incidence and mortality of acute and chronic pancreatitis in the Netherlands: a nationwide record‐linked cohort study for the years 1995‐2005. World J Gastroenterol 2013; 19: 3018–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Santvoort HC, Bakker OJ, Bollen TL, Besselink MG, Ahmed Ali U, Schrijver AM et al; Dutch Pancreatitis Study Group . A conservative and minimally invasive approach to necrotizing pancreatitis improves outcome. Gastroenterology 2011; 141: 1254–1263. [DOI] [PubMed] [Google Scholar]
- 3. Abela JE, Carter CR. Acute pancreatitis – a review. Surgery 2010; 28: 205–211. [Google Scholar]
- 4. Lankisch PG, Lowenfels AB, Maisonneuve P. What is the risk of alcoholic pancreatitis in heavy drinkers? Pancreas 2002; 25: 411–412. [DOI] [PubMed] [Google Scholar]
- 5. Lowenfels AB, Lankisch PG, Maisonneuve P. What is the risk of biliary pancreatitis in patients with gallstones? Gastroenterology 2000; 119: 879–880. [DOI] [PubMed] [Google Scholar]
- 6. Whitcomb DC. Genetic risk factors for pancreatic disorders. Gastroenterology 2013; 144: 1292–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Whitcomb DC, Gorry MC, Preston RA, Furey W, Sossenheimer MJ, Ulrich CD et al Hereditary pancreatitis is caused by a mutation in the cationic trypsinogen gene. Nat Genet 1996; 14: 141–145. [DOI] [PubMed] [Google Scholar]
- 8. Issa Y, Bruno MJ, Bakker OJ, Besselink MG, Schepers NJ, van Santvoort HC et al Treatment options for chronic pancreatitis. Nat Rev Gastroenterol Hepatol 2014; 11: 556–564. [DOI] [PubMed] [Google Scholar]
- 9. Ioannidis JP, Boffetta P, Little J, O'Brien TR, Uitterlinden AG, Vineis P et al Assessment of cumulative evidence on genetic associations: interim guidelines. Int J Epidemiol 2008; 37: 120–132. [DOI] [PubMed] [Google Scholar]
- 10. Allen NC, Bagade S, McQueen MB, Ioannidis JP, Kavvoura FK, Khoury MJ et al Systematic meta‐analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet 2008; 40: 827–834. [DOI] [PubMed] [Google Scholar]
- 11. Montazeri Z, Theodoratou E, Nyiraneza C, Timofeeva M, Chen W, Svinti V et al Systematic meta‐analyses and field synopsis of genetic association studies in colorectal adenomas. Int J Epidemiol 2016; 45: 186–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brouwer MC, de Gans J, Heckenberg SG, Zwinderman AH, van der Poll T, van de Beek D. Host genetic susceptibility to pneumococcal and meningococcal disease: a systematic review and meta‐analysis. Lancet Infect Dis 2009; 9: 31–44. [DOI] [PubMed] [Google Scholar]
- 13. Wakefield J. A Bayesian measure of the probability of false discovery in genetic epidemiology studies. Am J Hum Genet 2007; 81: 208–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gwinn M, Ioannidis JP, Little J, Khoury MJ. Editorial: updated guidance on human genome epidemiology (HuGE) reviews and meta‐analyses of genetic associations. Am J Epidemiol 2014; 180: 559–561. [DOI] [PubMed] [Google Scholar]
- 15. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group . Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Med 2009; 6: e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bai HX, Lowe ME, Husain SZ. What have we learned about acute pancreatitis in children? J Pediatr Gastroenterol Nutr 2011; 52: 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. R Core Team . R: a Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, 2016. [Google Scholar]
- 18. Viechtbauer W. Conducting meta‐analyses in R with the metafor package. J Stat Softw 2010; 36: 1–48. [Google Scholar]
- 19. Yu W, Ned R, Wulf A, Liu T, Khoury MJ, Gwinn M. The need for genetic variant naming standards in published abstracts of human genetic association studies. BMC Res Notes 2009; 2: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Harbord RM, Egger M, Sterne JA. A modified test for small‐study effects in meta‐analyses of controlled trials with binary endpoints. Stat Med 2006; 25: 3443–3457. [DOI] [PubMed] [Google Scholar]
- 21. Joergensen MT, Brusgaard K, Novovic S, Andersen AM, Hansen MB, Gerdes AM et al Is the SPINK1 variant p.N34S overrepresented in patients with acute pancreatitis? Eur J Gastroenterol Hepatol 2012; 24: 309–315. [DOI] [PubMed] [Google Scholar]
- 22. Yin YW, Sun QQ, Feng JQ, Hu AM, Liu HL, Wang Q. Influence of interleukin gene polymorphisms on development of acute pancreatitis: a systematic review and meta‐analysis. Mol Biol Rep 2013; 40: 5931–5941. [DOI] [PubMed] [Google Scholar]
- 23. Fang F, Pan J, Xu L, Su G, Li G, Wang J. Association between angiotensin‐converting enzyme gene insertion/deletion polymorphism and pancreatitis risk: a meta‐analysis. J Renin Angiotensin Aldosterone Syst 2015; 16: 820–826. [DOI] [PubMed] [Google Scholar]
- 24. Fang F, Pan J, Xu LX, Su GG, Li G, Wang J. Association between chemokine (C‐C motif) ligand 2 gene‐2518 A/G polymorphism and pancreatitis risk: a meta‐analysis. Pancreatology 2015; 15: 53–58. [DOI] [PubMed] [Google Scholar]
- 25. Yuan X, Wang H. Lack of association between CD14‐159 C/T polymorphism and acute pancreatitis: a meta‐analysis. Int J Clin Exp Med 2015; 8: 4134–4139. [PMC free article] [PubMed] [Google Scholar]
- 26. Zhou XJ, Cui Y, Cai LY, Xiang JY, Zhang Y. Toll‐like receptor 4 polymorphisms to determine acute pancreatitis susceptibility and severity: a meta‐analysis. World J Gastroenterol 2014; 20: 6666–6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yin YW, Hu AM, Sun QQ, Liu HG, Wang Q, Zeng YH et al Association between tumor necrosis factor‐alpha gene −308A/G polymorphism and the risk of acute pancreatitis: a meta‐analysis. J Surg Res 2012; 178: 409–414. [DOI] [PubMed] [Google Scholar]
- 28. Sah RP, Dawra RK, Saluja AK. New insights into the pathogenesis of pancreatitis. Curr Opin Gastroenterol 2013; 29: 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Whitcomb DC. How to think about SPINK and pancreatitis. Am J Gastroenterol 2002; 97: 1085–1088. [DOI] [PubMed] [Google Scholar]
- 30. Kuwata K, Hirota M, Shimizu H, Nakae M, Nishihara S, Takimoto A et al Functional analysis of recombinant pancreatic secretory trypsin inhibitor protein with amino‐acid substitution. J Gastroenterol 2002; 37: 928–934. [DOI] [PubMed] [Google Scholar]
- 31. Aoun E, Muddana V, Papachristou GI, Whitcomb DC. SPINK1 N34S is strongly associated with recurrent acute pancreatitis but is not a risk factor for the first or sentinel acute pancreatitis event. Am J Gastroenterol 2010; 105: 446–451. [DOI] [PubMed] [Google Scholar]
- 32. Masamune A, Ariga H, Kume K, Kakuta Y, Satoh K, Satoh A et al Genetic background is different between sentinel and recurrent acute pancreatitis. J Gastroenterol Hepatol 2011; 26: 974–978. [DOI] [PubMed] [Google Scholar]
- 33. Cavestro GM, Zuppardo RA, Bertolini S, Sereni G, Frulloni L, Okolicsanyi S et al Connections between genetics and clinical data: role of MCP‐1, CFTR, and SPINK‐1 in the setting of acute, acute recurrent, and chronic pancreatitis. Am J Gastroenterol 2010; 105: 199–206. [DOI] [PubMed] [Google Scholar]
- 34. Masamune A, Kume K, Kikuta K, Watanabe T, Hirota M, Satoh K et al −651C/T promoter polymorphism in the CD14 gene is associated with severity of acute pancreatitis in Japan. J Gastroenterol 2010; 45: 225–233. [DOI] [PubMed] [Google Scholar]
- 35. Kume K, Masamune A, Mizutamari H, Kaneko K, Kikuta K, Satoh M et al Mutations in the serine protease inhibitor Kazal Type 1 (SPINK1) gene in Japanese patients with pancreatitis. Pancreatology 2005; 5: 354–360. [DOI] [PubMed] [Google Scholar]
- 36. Chen CC, Wang SS, Lee FY, Chang FY, Lee SD. Proinflammatory cytokines in early assessment of the prognosis of acute pancreatitis. Am J Gastroenterol 1999; 94: 213–218. [DOI] [PubMed] [Google Scholar]
- 37. Chen H, Wilkins LM, Aziz N, Cannings C, Wyllie DH, Bingle C et al Single nucleotide polymorphisms in the human interleukin‐1B gene affect transcription according to haplotype context. Hum Mol Genet 2006; 15: 519–529. [DOI] [PubMed] [Google Scholar]
- 38. Brivet FG, Emilie D, Galanaud P. Pro‐ and anti‐inflammatory cytokines during acute severe pancreatitis: an early and sustained response, although unpredictable of death. Parisian Study Group on Acute Pancreatitis. Crit Care Med 1999; 27: 749–755. [DOI] [PubMed] [Google Scholar]
- 39. Mayer J, Rau B, Gansauge F, Beger HG. Inflammatory mediators in human acute pancreatitis: clinical and pathophysiological implications. Gut 2000; 47: 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fishman D, Faulds G, Jeffery R, Mohamed‐Ali V, Yudkin JS, Humphries S et al The effect of novel polymorphisms in the interleukin‐6 (IL‐6) gene on IL‐6 transcription and plasma IL‐6 levels, and an association with systemic‐onset juvenile chronic arthritis. J Clin Invest 1998; 102: 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Giedraitis V, He B, Huang WX, Hillert J. Cloning and mutation analysis of the human IL‐18 promoter: a possible role of polymorphisms in expression regulation. J Neuroimmunol 2001; 112: 146–152. [DOI] [PubMed] [Google Scholar]
- 42. Norman JG, Fink GW, Franz MG. Acute pancreatitis induces intrapancreatic tumor necrosis factor gene expression. Arch Surg 1995; 130: 966–970. [DOI] [PubMed] [Google Scholar]
- 43. Papachristou GI, Sass DA, Avula H, Lamb J, Lokshin A, Barmada MM et al Is the monocyte chemotactic protein‐1 −2518 G allele a risk factor for severe acute pancreatitis? Clin Gastroenterol Hepatol 2005; 3: 475–481. [DOI] [PubMed] [Google Scholar]
- 44. Chao YC, Wang LS, Hsieh TY, Chu CW, Chang FY, Chu HC. Chinese alcoholic patients with esophageal cancer are genetically different from alcoholics with acute pancreatitis and liver cirrhosis. Am J Gastroenterol 2000; 95: 2958–2964. [DOI] [PubMed] [Google Scholar]
- 45. Chao YC, Wang SJ, Chu HC, Chang WK, Hsieh TY. Investigation of alcohol metabolizing enzyme genes in Chinese alcoholics with avascular necrosis of hip joint, pancreatitis and cirrhosis of the liver. Alcohol Alcohol 2003; 38: 431–436. [DOI] [PubMed] [Google Scholar]
- 46. Mannervik B, Board PG, Hayes JD, Listowsky I, Pearson WR. Nomenclature for mammalian soluble glutathione transferases. Methods Enzymol 2005; 401: 1–8. [DOI] [PubMed] [Google Scholar]
- 47. Markova EV, Zotova NV, Savchenko AA, Titova NM, Slepov EV, Cherdantsev DV et al [Lymphocyte metabolism in patients with acute pancreatitis and different genotypes of GSTM1 and GSTT1 genes.] Biomed Khim 2006; 52: 317–326. [PubMed] [Google Scholar]
- 48. Matas‐Cobos AM, Redondo‐Cerezo E, Alegria‐Motte C, Martinez‐Chamorro A, Saenz‐López P, Jiménez P et al The role of Toll‐like receptor polymorphisms in acute pancreatitis occurrence and severity. Pancreas 2015; 44: 429–433. [DOI] [PubMed] [Google Scholar]
- 49. Zhang C, Guo L, Qin Y, Li G. [Interaction between polymorphisms of TLR4 gene G11367C in 3′ untranslated region and IκB‐α Hae III in acute pancreatitis and the degree of severity.] Zhong Nan Da Xue Xue Bao Yi Xue Ban 2016; 41: 272–281. [DOI] [PubMed] [Google Scholar]
- 50. Ma M, Zhai CX, Sun CX. Correlations between LP‐PLA2 gene polymorphisms and susceptibility and severity of acute pancreatitis in a Chinese population. Gastroenterol Res Pract 2017; 21: 206–212. [DOI] [PubMed] [Google Scholar]
- 51. Zhang CX, Guo LK, Zhang LL, Qin YM, Chang TM. [Interaction of polymorphisms of TNF‐α gene promoter −308G/A and PPAR‐γ2 gene −C34G with acute pancreatitis and its severity degree.] Journal of Xi'an Jiaotong University (Medical Sciences) 2017; 38: 76–82. [Google Scholar]
- 52. Tukiainen E, Kylänpää ML, Repo H, Orpana A, Methuen T, Salaspuro M et al Hemostatic gene polymorphisms in severe acute pancreatitis. Pancreas 2009; 38: e43–e46. [DOI] [PubMed] [Google Scholar]
- 53. Gulla A, Evans BJ, Navenot JM, Pundzius J, Barauskas G, Gulbinas A et al Heme oxygenase‐1 gene promoter polymorphism is associated with the development of necrotizing acute pancreatitis. Pancreas 2014; 43: 1271–1276. [DOI] [PubMed] [Google Scholar]
- 54. Gao HK, Zhou ZG, Li Y, Chen YQ. Toll‐like receptor 4 Asp299Gly polymorphism is associated with an increased risk of pancreatic necrotic infection in acute pancreatitis: a study in the Chinese population. Pancreas 2007; 34: 295–298. [DOI] [PubMed] [Google Scholar]
- 55. Tiszlavicz Z, Szabolcs A, Takacs T, Farkas G, Kovacs‐Nagy R, Szantai E et al Polymorphisms of beta defensins are associated with the risk of severe acute pancreatitis. Pancreatology 2010; 10: 483–490. [DOI] [PubMed] [Google Scholar]
- 56. Zhang DL, Zheng HM, Yu BJ, Jiang ZW, Li JS. Association of polymorphisms of IL and CD14 genes with acute severe pancreatitis and septic shock. World J Gastroenterol 2005; 11: 4409–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Skipworth JR, Nijmeijer RM, van Santvoort HC, Besselink MG, Schulz HU, Kivimaki M et al The effect of renin angiotensin system genetic variants in acute pancreatitis. Ann Surg 2015; 261: 180–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bishehsari F, Sharma A, Stello K, Toth C, O'Connell MR, Evans AC et al TNF‐alpha gene (TNFA) variants increase risk for multi‐organ dysfunction syndrome (MODS) in acute pancreatitis. Pancreatology 2012; 12: 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. de‐Madaria E, Martinez J, Sempere L, Lozano B, Sanchez‐Paya J, Uceda F et al Cytokine genotypes in acute pancreatitis: association with etiology, severity, and cytokine levels in blood. Pancreas 2008; 37: 295–301. [DOI] [PubMed] [Google Scholar]
- 60. Liu Y, Dan G, Wu LJ, Chen GY, Wu AL, Zeng P et al Functional effect of polymorphisms in the promoter of TNFAIP3 (A20) in acute pancreatitis in the Han Chinese population. PLoS One 2014; 9: e103104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Takagi Y, Masamune A, Kume K, Satoh A, Kikuta K, Watanabe T et al Microsatellite polymorphism in intron 2 of human Toll‐like receptor 2 gene is associated with susceptibility to acute pancreatitis in Japan. Hum Immunol 2009; 70: 200–204. [DOI] [PubMed] [Google Scholar]
- 62. Aoun E, Chang CC, Greer JB, Papachristou GI, Barmada MM, Whitcomb DC. Pathways to injury in chronic pancreatitis: decoding the role of the high‐risk SPINK1 N34S haplotype using meta‐analysis. PLoS One 2008; 3: e2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li D, Zhao H, Gelernter J. Strong protective effect of the aldehyde dehydrogenase gene (ALDH2) 504lys (*2) allele against alcoholism and alcohol‐induced medical diseases in Asians. Hum Genet 2012; 131: 725–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chen WC, Nie JS. Genetic polymorphism of MCP‐1‐2518, IL‐8‐251 and susceptibility to acute pancreatitis: a pilot study in population of Suzhou, China. World J Gastroenterol 2008; 14: 5744–5748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG et al; Acute Pancreatitis Classification Working Group . Classification of acute pancreatitis – 2012: revision of the Atlanta classification and definitions by international consensus. Gut 2013; 62: 102–111. [DOI] [PubMed] [Google Scholar]
- 66. Mounzer R, Whitcomb DC. Genetics of acute and chronic pancreatitis. Curr Opin Gastroenterol 2013; 29: 544–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hirschhorn JN, Altshuler D. Once and again – issues surrounding replication in genetic association studies. J Clin Endocrinol Metab 2002; 87: 4438–4441. [DOI] [PubMed] [Google Scholar]
- 68. Ahmed Ali U, Issa Y, Hagenaars JC, Bakker OJ, van Goor H, Nieuwenhuijs VB et al; Dutch Pancreatitis Study Group . Risk of recurrent pancreatitis and progression to chronic pancreatitis after a first episode of acute pancreatitis. Clin Gastroenterol Hepatol 2016; 14: 738–746. [DOI] [PubMed] [Google Scholar]
- 69. Bollen TL, Singh VK, Maurer R, Repas K, van Es HW, Banks PA et al A comparative evaluation of radiologic and clinical scoring systems in the early prediction of severity in acute pancreatitis. Am J Gastroenterol 2012; 107: 612–619. [DOI] [PubMed] [Google Scholar]
- 70. Little J, Bradley L, Bray MS, Clyne M, Dorman J, Ellsworth DL et al Reporting, appraising, and integrating data on genotype prevalence and gene‐disease associations. Am J Epidemiol 2002; 156: 300–310. [DOI] [PubMed] [Google Scholar]
- 71. Latourelle JC, Beste MT, Hadzi TC, Miller RE, Oppenheim JN, Valko MP et al Large‐scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson's disease: a longitudinal cohort study and validation. Lancet Neurol 2017; 16: 908–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sham PC, Purcell SM. Statistical power and significance testing in large‐scale genetic studies. Nat Rev Genet 2014; 15: 335–346. [DOI] [PubMed] [Google Scholar]
- 73. Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, Knowles MR et al Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet 2012; 44: 886–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Li D, Lewinger JP, Gauderman WJ, Murcray CE, Conti D. Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies. Genet Epidemiol 2011; 35: 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supplementary methods and results
Table S1. Characteristics of included studies
Table S2. Comprehensive overview of extracted genetic variants associated with (recurrent) acute pancreatitis, categorized by gene function
Table S3. Primary meta‐analysis of variants associated with susceptibility for (recurrent) acute pancreatitis
Table S4. Subgroup meta‐analysis of variants associated with susceptibility for (recurrent) acute pancreatitis in patients of Caucasian ethnicity
Table S5. Subgroup analysis of variants associated with susceptibility for (recurrent) acute pancreatitis in patients of Asian ethnicity
Table S6. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with susceptibility for (recurrent) acute pancreatitis
Table S7. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with susceptibility for (recurrent) acute pancreatitis in patients of Caucasian ethnicity
Table S8. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with susceptibility for (recurrent) acute pancreatitis in patients of Asian ethnicity
Table S9. Primary meta‐analysis of variants associated with disease severity
Table S10. Subgroup meta‐analysis of variants associated with disease severity in patients of Caucasian ethnicity
Table S11. Subgroup analysis of variants associated with disease severity in patients of Asian ethnicity
Table S12. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with disease severity
Table S13. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with disease severity in patients of Caucasian ethnicity
Table S14. Sensitivity analysis (excluding studies that deviate from HWE) of variants associated with disease severity in patients of Asian ethnicity