

Abstract
C–N cross-coupling is one of the most valuable and widespread transformations in organic synthesis. Largely dominated by Pd- and Cu-based catalytic systems, it has proven to be a staple transformation for those in both academia and industry. The current study presents the development and mechanistic understanding of an electrochemically driven, Ni-catalyzed method for achieving this reaction of high strategic importance. Through a series of electrochemical, computational, kinetic, and empirical experiments the key mechanistic features of this reaction have been unraveled, leading to a second generation set of conditions that is applicable to a broad range of aryl halides and amine nucleophiles, including complex examples on oligopeptides, medicinally-relevant heterocycles, natural products, and sugars. Full disclosure of the current limitations as well as procedures for both batch and flow scale-ups (100 gram) are also described.
Graphical Abstract

Introduction
Given the ubiquitous nature of the aniline motif in drugs and natural products, there is a constant need for the development of methods for mild and selective C(sp2)–N bond formation.1 Currently, the stalwart methods to achieve cross-coupling of aryl halides and amines employ Pd- (Buchwald-Hartwig) and Cu-based (Ullmann) catalysts, which rank among one of the most employed transformations in modern pharmaceutical development.2 Indeed, there exists a myriad of reports investigating the classic limitations of these reactions such as the use of strong base and elevated temperature.1 Despite these reports, there is still documented and anecdotal evidence that several classes of amines or aryl halides remain particularly challenging substrates for C–N cross-coupling. As an example, the coupling of secondary alkyl amines and oligopeptides proceeds in low yields even under the most modern sets of conditions.3
Synthetic organic electrochemistry enables precise redox control to achieve either known transformations with less waste or to accomplish otherwise-intractable transformations.4 Examples from our own lab range from the anodic generation of alkyl radicals from sulfinate salts5 to the C–H oxidation of allylic6 and unactivated sites.7 Interactions with industrial collaborators in both process and medicinal chemistry inspired us to evaluate an electrochemical approach to C–N cross coupling. In 2017, we reported a Ni-catalyzed aryl amination—coined as e-amination—that exhibited a wide substrate scope for the coupling of aryl halides and triflates with amines, alcohols, and amides.8 The reaction uses commercial reticulated vitreous carbon (RVC) and Ni-foam electrodes in concert with an inexpensive and abundant Ni catalyst at room temperature. We speculated the success of this transformation relies on the curious co-existence of multiple different Ni-oxidation states in the same reaction vessel, a task perfectly suited to electrochemistry. Critical assessment of the original conditions revealed that aryl halide coupling partners were largely limited to electron-poor derivatives and attempts with historically challenging heteroaryl halides were deemed unsatisfactory. Additionally, the choice of amine was restricted to primary and secondary alkyl substrates; functionalized amines, including amino acids and oligopeptides, did not perform well.
The limitations in scope were attributed to a lack of understanding in the catalytic system. For instance, it was not clear what oxidation state of Ni was engaging in oxidative addition, the identity of the rate-determining step, or even the effect of simple experimental parameters such as ligand, electrolyte, or solvent. Thus, a comprehensive understanding of the reaction mechanism was needed in order to improve the previous set of conditions, with particular interest in identifying the rate-determining step, along with the ligation and oxidation states of catalytically active species. In this Article, a detailed reaction profile for e-amination is delineated, enabled by the strategic union of nuanced electrochemical analysis, DFT calculations, and empirical optimization. This collaborative endeavor facilitated the development of a robust, well-defined, catalytic system resulting in a greatly expanded substrate scope.
Background and Historical Context
Figure 1 graphically outlines the history of Ni-catalyzed C–N cross coupling.9 In 1950, Hughes and co-workers reported the first example of Ni mediated C–N cross-coupling between chlorobenzene and methylamine using NiCl2 at 200 °C (Figure 1A).10 Several decades later, Cramer11 and Cristau12 conducted comprehensive studies on the C–N bond formation between chlorobenzene and different amine coupling partners in the presence of a Ni° catalyst at elevated temperatures (100 °C to 230 °C). The limited substrate scope did not favor the uptake of this potentially valuable transformation. It was not until 1997, when Buchwald and co-workers explored Ni°-catalyzed amination in a synthetic context, that significant interest in Ni-catalyzed C–N cross coupling emerged.13 As recognized, Ni-based amination reactions might be more practical than related Pd-systems on large scale due to cost considerations. Moreover, owing to its high reactivity towards less reactive electrophiles such as aryl chlorides, Ni-catalysis provides an alternative approach to palladium and copper catalysis. However, the requirements of air-sensitive Ni° catalysts, high temperatures, and strong alkoxide bases remained important unsolved drawbacks that significantly curtailed the adoption of such systems.
Figure 1.

(A) Discovery of Ni-catalyzed amination; (B) Improvement of catalytic systems; (C) Accelerated reductive elimination via NiIII intermediate; (D) Ni-catalyzed amination with photo-induced electron transfer catalysis; (E) Electrochemical, Ni-catalyzed amination (e-amination).
Through the years, several groups tackled the challenges and limitations associated with the Ni-catalyzed C–N bond formation (Figure 1B). Nolan,14 Buchwald,15 Doyle,16 and Stewart17 independently addressed the air sensitivity issue of Ni° catalysts by developing innovative air stable Ni precatalysts. In 2010, Nicasio et al. reported the first room temperature amination reaction of heteroaromatic chlorides using a bulky allylnickel chloride/N-heterocyclic carbene complex as a well-defined precatalyst 1.18 Despite all these advances, the scope of the Ni-catalyzed amination of aryl halides has been limited to the coupling of secondary alkylamines and arylamines, while primary alkylamines, one of the most significant classes of amines for such cross coupling, have been shown to couple only with activated aryl chlorides. As a breakthrough for this problem, Stradiotto and Hartwig et al. independently reported the Ni-catalyzed amination for a broad range of unactivated aryl/heteroaryl chlorides and bromides with primary aliphatic amines.19 However, secondary aliphatic amines have proven to be poor coupling partners under their developed conditions, despite the use of a sophisticated ligand 2 or air-sensitive catalysts 3. These pioneering developments highlight the difficulty in finding an efficient, general and practical set of conditions for the Ni-catalyzed amination of aryl halides.
As a totally different approach to improve Ni-catalyzed amination, Hillhouse and co-workers demonstrated dramatic acceleration of C–N reductive elimination by using an oxidant in the context of aliphatic systems (Figure 1C).20 In 2012, Nakamura and co-workers suggested that facile C–N bond formation could be achieved under similar oxidative conditions for aryl systems as well.21 Therefore, the reductive elimination event can be promoted by the intermediacy of a high-valent Ni species accessible via oxidation. These results point to a critical challenge to achieve Ni-catalyzed amination of aryl halides; namely, the coexistence of a low-valent Ni catalyst to favor oxidative addition with a high-valent Ni catalyst to facilitate reductive elimination. In 2016, Buchwald, MacMillan and co-workers elegantly employed photoinduced electron transfer to allow mild generation of active Ni° species and oxidatively-induced reductive elimination in the same pot using an Ir photosensitizer (Figure 1D).22 Subsequently, our group reported an electrochemical approach to Ni-catalyzed amination (e-amination) that proceeds smoothly at room temperature or below without the aid of a strong inorganic base or co-catalyst, together with a promising substrate scope (Figure 1E).8
Mechanistic Studies of e-Amination
As alluded to above, e-amination likely involves accessing disparate Ni-oxidation states; the initial low-valent Ni (Ni° or NiI) for oxidative addition and the intermediate high-valent state (NiIII) required for efficient reductive elimination must harmoniously coexist in the same vessel. Realization of such contradictory redox events in a simultaneous manner is challenging under conventional chemical conditions but ideally suited to an electrochemical setup. In this section a careful study to demystify the underlying steps of the catalytic cycle is presented.
Extensive mechanistic studies involving detailed electrochemical analysis and DFT calculation have led to elucidation of the mechanistic underpinnings of the e-amination, which is now presented in Figure 2A. First, the ligation state of the NiII precatalyst was determined by UV-Vis spectroscopy (Figure 2B [A]). Concentration profiles for each NiLnBr2 ligation state (L = Mebpy) were constructed using reported λmax values and molar absorption coefficients.23 Interestingly, the data suggested a mixed and dynamic NiII ligation states, and predominant species varies depending on the ligand/NiII ratio. Importantly, the ligation profile suggests a substantial portion of NiII remains unligated when the ligand is present in only one equivalent relative to NiII. The presence of unligated NiII species is postulated to be undesirable because overreduction of such species could lead to excessive Ni° aggregation which would manifest as Ni-black deposition on the cathode. Consequently, the UV-Vis study suggested the possibility of more efficient catalysis with higher ligand loading.
Figure 2.

(A) Proposed mechanism; (B) Experimental and computational evidence to support the proposed mechanism; subpanels [A]-[G] represent evidence for each elementary step.
In parallel to these experiments, we performed density-functional theory (DFT) calculations were performed using the M06-L exchange-correlation functional, 6–31+G** basis set, and Stuttgart/Dresden effective core potentials, which took into account solvation effects with the SMD model using DMF as the implicit solvent to gain more insight about this equilibrium. The binding of the first Mebpy ligand (NiIIBr2 → NiIILBr2) is substantially exothermic with the calculated energy difference between unligated and mono-ligated species ΔE = −323 kJ/mol (See SI for details). Although much smaller energy gains are observed for the binding of the second and third ligands due to steric constraints, the energy differences indicate facile ligand association/dissociation is thermodynamically feasible. In reality the situation is likely competitive binding between the ligand, DMF, and amine nucleophile. Hence, the population of various NiII complexes is dependent on the molar fractions of all solution phase compounds, which creates a dynamic and complex ligation situation.
To shed light on the electrochemical reduction of NiII(Mebpy)nBr2, DMF solution of NiBr2•3H2O with various concentrations of Mebpy was analyzed by square wave voltammetry (SWV, Figure 2B [B]). There are two distinct sets of redox features grouped in the range of −0.8 to −1.2 V and −1.5 to −1.8 V (all potentials are reported vs Ag/AgNO3). Computational analysis of various NiII(Mebpy)nBr2 oxidation states (See SI for details) suggests that these experimentally observed redox potentials are similar to the calculated values for the Ni(II/I) and Ni(I/0) redox transitions. The large difference between the two redox couples suggest that the NiI species is likely formed predominantly under constant current conditions to initiate oxidative addition. Electrochemical analysis of NiII(Mebpy)nBr2 reduction using a microelectrode suggests that the nature of this step is complicated and the involvement of Ni° cannot be completely ruled out (See SI for details). However, the combination of computational evidence, SWV analysis, and the variety of known comproportionation pathways resulting in the rapid formation of NiI lead us to the conclusion that NiI is most likely the active species toward oxidative addition.24
Cyclic voltammograms (CVs) of NiII(Mebpy)Br2 in the absence and presence of 4-bromoanisole as an electrophile indicated the ability of NiI to undergo oxidative addition under the reaction conditions (Figure 2B [C][D]). A loss of electrochemical reversibility of the Ni(II/I) redox couple upon addition of 4-bromoanisole suggests oxidative addition is occurring rapidly relative to the time scale of the CV (100 mV/s), indicating this step is unlikely to be the rate-determining step. Reaction energy profiles determined by DFT (Figure 2B [C][D], bottom) further corroborate this conclusion; after endothermic ligand dissociation at ΔE = 99 kJ/mol, oxidative addition occurs with relatively low activation energy ΔE‡ = 73 kJ/mol. As indicated, this oxidative addition is quite exothermic (ΔE = −162 kJ/mol). This means that all NiII(Mebpy)nBr2 (n = 1–3) could undergo rapid oxidative addition upon reduction due to the relatively small activation barrier and large energy gain, even considering the unfavorable ligand dissociation to create open coordination sites. DFT results also suggest that the generated NiIII(Mebpy)(Ar)Br2 5a (Ar = 4-CF3C6H4) can be readily reduced to NiII(Mebpy)(Ar)Br 7a (Ered ~ −0.5 V vs Ag/AgNO3), which is experimentally supported by the observed increase in reductive current of the Ni(II/I) redox couple in the presence of aryl halide substrate.
The evidence for rapid oxidative addition suggests that either amine coordination/deprotonation or reductive elimination is rate-determining in the overall catalytic cycle. Experimentally, the reaction with a sterically demanding t-butyl amine became sluggish, indicating that slow coordination/deprotonation of the amine nucleophile is potentially rate-determining (Figure 2B [E][F], top). To further determine the energetic requirements of amine coordination versus deprotonation, we examined the coupling of morpholine with 4-trifluoromethylbromobenzene. The DFT results showed that in the absence of steric constraints, the dative amine coordination should be relatively facile, with energy release around ΔE = −46 kJ/mol for this step, while the subsequent deprotonation from the basic amine causes a large energy increase (ΔE = 193 kJ/mol) and similarly large values for the ensuing reductive elimination (ΔE ~ 204 kJ/mol, black line). However, when excess free amine molecules exist in solution, their action as a base in the deprotonation of the coordinated amine compensates for the large positive ΔE for this step (similarly accomplished by the addition of an exogenous base such as DBU). In addition, ΔE for reductive elimination drastically decreases if the NiII complex is oxidized to NiIII one. Overall, combination of additional base and oxidation of NiII to NiIII afford an energetically feasible reaction pathway.
To experimentally verify the oxidation of NiII to NiIII, intermediate NiII(tBubpy)(4-MeOC6H4)Br 12 was synthesized independently and cyclic voltammetry of 12 was studied in the presence or absence of hexyl amine. Neither 12 nor hexyl amine showed electrochemical oxidation within the electrochemical solvent/electrolyte window (Figure 2B [E][F] middle). However, the addition of a large excess of hexyl amine into the DMF solution of 12 (100 equivalents with respect to Ni) results in the formation of a complex that is oxidized irreversibly at ~0.8 V, which again indicates slow amine coordination/deprotonation on the NiIIL(Ar)Br intermediate 7.
Taken together, our results are consistent with the mechanism illustrated in Figure 2A involving [A] the dynamic ligation state of NiII precatalyst, [B] the electrochemical reduction of NiII to a NiI species 4 at the cathode, [C] the rapid oxidative addition of NiI to an aryl halide generating a transient NiIII species 5, [D] a second electrochemical reduction at the cathode resulting in a stable NiII aryl intermediate 7, [E] the coordination of the amine and rate-limiting deprotonation to the intermediate 8, [F] the electrochemical oxidation at the anode to generate a high-energy NiIII complex 6, followed by rapid reductive elimination to produce the arylamine product while regenerating 4 as the active catalyst. Basic kinetic analysis is in good agreement with this mechanistic picture (See SI for detail).
It should be noted the current mechanistic evidence does permit the possibility of a self-sustaining NiI/NiIII cycle (Figure 2A, step [G]) in which NiIIIL(Ar)Br2 5 is not immediately reduced at the cathode, but undergoes amine coordination/deprotonation to generate the intermediate 6. A similar NiI/NiIII cycle is also proposed by Miyake and co-workers in their photochemical amination work recently investigated.25 The feasibility of this NiI/NiIII pathway was tested by performing the reaction with a catalytic amount of electricity (Figure 2B [G]). While 5% yield of 10 was obtained after 15 minutes electrolysis/3 hours stirring, the current efficiency was merely 27%, which clearly suggests that continuous electrolysis is necessary in order to achieve appreciable yields. This apparent inefficiency of NiI/NiIII cycle could be explained by the comproportionation of 4 and 5 to generate stable NiII intermediate 7, considering redox potential of Ni(II/I) obtained by SWV (~ −1 V) and calculated reduction potential of 5a (−0.5 V). Therefore, the role of electricity is constant regeneration of the key catalytic species such as 4 and 6 throughout the duration of the reaction.
Collectively, the mechanistic results suggest the ligand/NiII ratio should be optimized to afford enhanced reactivity by avoiding deleterious Ni° deposit. Furthermore, the addition of appropriate amount of external base would appear to facilitate the rate-limiting deprotonation. With these mechanistic nuances in mind, we began our forays on expanding the scope of e-amination.
Optimization
Minimal arylation of glutamic acid di-tert-butyl ester (14) was observed under our previously reported e-amination conditions (Table 1A, conditions A, 0.1 mmol scale), and therefore it was selected as a model reaction to validate the insights obtained from the above mechanistic study. A slight improvement was observed by changing the electrolyte from LiBr to tetra-n-butylammonium bromide (conditions B). As suggested by the UV-Vis spectroscopic study, increasing the ligand/NiII ratio from 1/1 to 2/1 was found to be far superior with a greater than four-fold increase in yield obtained (conditions C). Although increasing the ligand loading further (ligand/NiII = 3/1, conditions D) seemed to have no effect on the yield, deposition of nickel black on the cathode was observed in some cases when 20 mol% ligand was employed; this phenomenon was almost completely suppressed at 30 mol%. Therefore, conditions D were chosen as the standard conditions for additional optimization with the impact of several reaction parameters summarized in Table 1B. As generally observed in transition-metal catalyzed coupling reactions, the choice of ligand was found to be crucial for the success of this reaction. After extensive screening of different ligand architectures (see SI for the complete list), the best result was obtained with simple, unsubstituted bipyridine (bpy, entry 1). This is fortuitous considering that the ligand is the most expensive component of this amination system. Similar results were also obtained with Mebpy (entry 2), whereas more sterically demanding 6,6’-Mebpy completely shut down the reaction (entry 3), indicating that this reaction is rather sensitive to the steric environment around the Ni catalyst. Pyridine-oxazoline ligands were found to be similarly effective for the of 14 (entry 4), but later were found not to be generally applicable. Reduced quantities of DBU were found to be deleterious to the reaction (entry 5), in good agreement with DFT calculations that the rate-determining step—displacement of the bromide on the intermediate 7 with the amine nucleophile—is facilitated by an external base (vide supra). Highly polar and aprotic solvents were found to be suitable for this reaction (entries 6–7), as these solvents have ideal characteristics for electrochemical synthesis: good solubility of the electrolyte and the Ni-catalyst, high dielectric constant required for efficient conductivity, and an appropriate electrochemical window.26 With regard to the counterion of the electrolyte, perchlorate and chloride were slightly less efficient than bromide (entries 8 and 9). Concentration of the electrolyte did not have a major impact on the reaction despite the apparent voltage difference (entry 10, terminal potential 3–4 V for 0.2 M vs 4–6 V for 0.05 M). Finally, it was found that Ni(bpy)3Br2 (17),27 a bench-stable and free-flowing solid with no apparent hygroscopicity, was an ideal precatalyst, which improves operational simplicity of this reaction by removing the need for the preparing stock solutions of hygroscopic NiBr2•3H2O (entry 11). The preparation of this known precatalyst is exceedingly simple as illustrated in Table 1C and is currently being commercialized (Sigma-Aldrich ALD00608).
Table 1.
(A) Initial re-optimization of amino acid arylation.; (B) Effect of various reaction parameters. These entries were performed in 0.1 mmol scale; (C) Simple preparation of the precatalyst Ni(bpy)3Br2 17.

|
Scope and Applications of e-Amination
Encouraged by the successful arylation of glutamic acid ester 14, the generality of the revised electrochemical C–N bond formation on various amino acid esters was explored (Table 2A). Using readily-available amino acid hydrochloride salts as starting materials, various amino acid esters (including non-canonical amino acids) were arylated efficiently with 4-trifluoromethylbromobenzene. Reactions (0.1 mmol scale) were conveniently carried out in an undivided cell with a readily-available RVC anode and a nickel foam cathode. Yields under the previous conditions are shown in parentheses, which highlight the drastic improvement afforded under the revised conditions. Although there have been multiple precedents for the arylation of amino acid esters employing palladium28, and copper catalysis,29 this is the first example of successful C–N bond formation on amino acid esters by Ni-catalysis. Racemization was minimized by short reaction times at ambient temperature.3 Of note, the sulfide in 24 and the indole ring in 26—both of which are commonly considered to be labile under oxidative conditions—were found to be compatible.
Table 2.
(A) Arylation of amino acid esters in 0.1 mmol scale. a () indicates yield under previous conditions (conditions A). (B) Optimization for the coupling of N-acetyl-4-bromoindole 29 with valine methyl ester 30.

|
As an extension of this scope, N-acetyl-4-bromoindole 29 was chosen (Table 2B) as an emblematic substrate for challenging C–N coupling.30 Similar systems were reported to require large amount of copper salt for traditional Ullmann coupling conditions,31 and applications of Buchwald-Hartwig coupling was found to be challenging.32 Not surprisingly, our first-generation conditions for e-amination failed to deliver the desired coupling product 31 (entry 1). The optimized conditions documented herein did in fact afforded 31 in 16% yield (entry 2), and ultimately a small screening of electrolyte, amount of base, ligands, nickel source (entries 3–7) was required to arrive at a workable solution (entry 7, 32%). Based on the conditions in entry 7, scale-up of the reaction was attempted with a slight change of substrate and electrolyte concentration. Fortunately, e-amination performed on a larger scale delivered superior yield, presumably due to the higher concentration of the substrate and more appropriate current density (entry 8, 51%). This small study demonstrates that like other C–N cross couplings, re-adjustment of reaction parameters such as precatalyst, ligand, and concentration of the substrate may be necessary for the success of certain substrate classes.
In our initial report, a limited substrate scope of heteroaryl amination was documented.8 Considering that heteroaryl amines are essential in medicinal chemistry, a more complete survey of this class of molecules was undertaken. N-Boc-piperazine was chosen as the amine coupling partner, since: (1) this motif frequently appears in pharmaceuticals, and (2) secondary amines are sometimes troublesome coupling partners even with state-of-the-art palladium catalysis at room temperature.3 As shown in Table 3, a wide range of heteroaryl halides deriving from quinoline (33, 34), indole (31, 35), benzofuran (36), pyridines (37-39), thiophene (40, 41), thiazole (42), pyrimidine (43) and azaindazole (44) were successfully coupled with N-Boc-piperazine in 0.2 mmol scale under essentially identical conditions optimized for amino acid esters. Both aryl bromides and chlorides were found to be competent in this reaction. Most of the heterocycles included in this table are privileged scaffolds in pharmaceuticals.33 The high functional group tolerance of this reaction deserves further comment. For instance, the free anilinic amine in 39 remained untouched. Sulfur-containing heterocycles afforded the desired coupling products 40-43, despite the fact that sulfur is commonly considered to be poisonous to transition-metal catalysts with the C–S bond being particularly labile in Ni-catalysis.34
Table 3.
Amination of heteroaryl halides with N-Boc-piperazine in 0.2 mmol scale. a () indicates yield under previous conditions (conditions A)

|
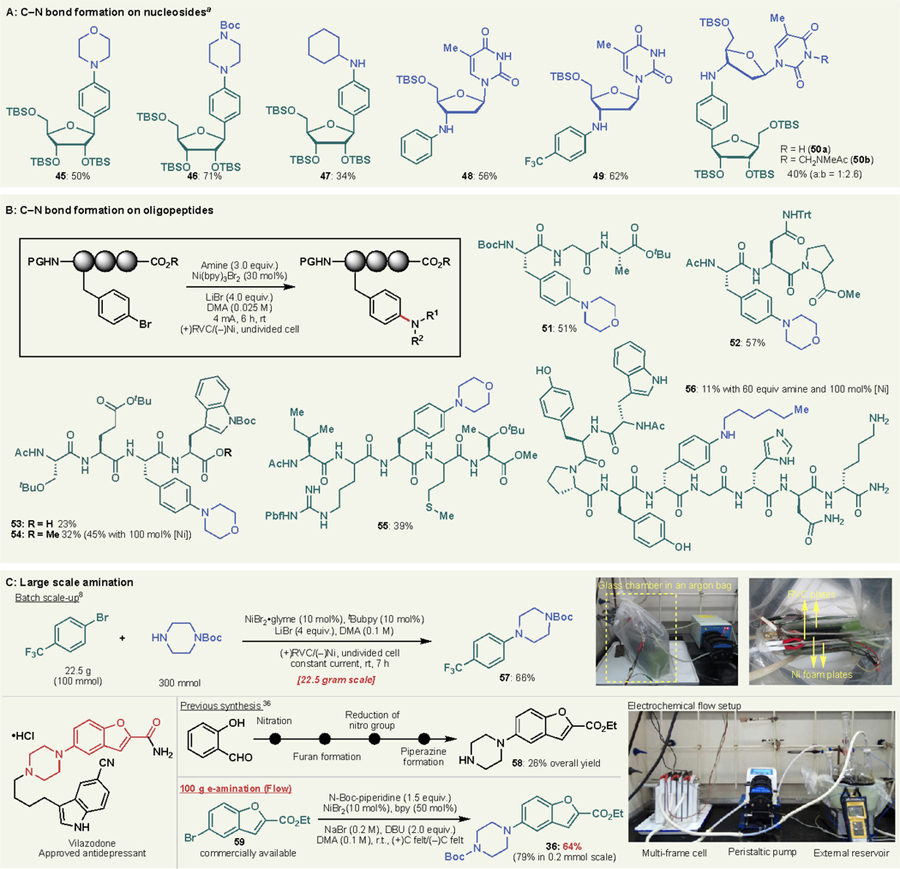
To further explore the capability of electrochemical C–N bond formation, the reaction was applied to more challenging substrates. Table 4A illustrates C–N bond formation on nucleoside analogs in 0.05–0.1 mmol scale. TBS-protected 1-(4-bromophenyl)ribose was successfully coupled with both primary and secondary amines. This method enables rapid access to various substituted 1-arylribose, which is potentially useful for the study of artificial base-pairs to expand the genetic alphabet.35 Likewise, arylation of 3-amino thymidine was also successful without protection on thymine. More interestingly, the powerful electrochemical C–N bond formation furnished a unique dinucleoside analog 5oa linked by an aniline moiety, which is a highly intriguing linker due to its non-hydrolyzable nature.
Table 4.
(A) C–N bond formation on nucleosides. a Reaction conditions: 10 mol% Ni(bpy)3Br2, nBu4N•Br (0.2 M), DBU (2.0 equiv.), DMA (0.025–0.05 M), (+)RVC/(−)Ni, 4 mA, r.t. in undivided cell. (B) C–N bond formation on oligopeptides. (C) Synthesis of arylomycin analogs. (D) Scale-up synthesis of compound 36.
|
Another interesting application of this method was found in oligopeptide modification (Table 4B). Slight modification to the established conditions were made to achieve synthetically useful efficiency in 0.05 mmol-scale reactions. Thus, the use of a LiBr electrolyte was found to give higher yield than tetra-n-butylammonium bromide, and DBU was omitted by using an excess amine coupling partner to avoid undesired racemization. With these modifications, e-amination on pendant 4-bromophenylalanine residues proceeded surprisingly well with only a catalytic amount of Ni precatalyst in several cases. Various functional groups in canonical amino acids were tolerated in protected form, as demonstrated in the products 51-55. More strikingly, the unprotected nona-peptide afforded the desired amination product 56 in appreciable yield. To the best of our knowledge, this is the first example of Ni-catalyzed C–N bond formation on oligopeptides, which holds promise for further application of such chemistry to biomolecule modifications. This work is a complementary addition to the growing area of transition-metal mediated oligopeptide functionalization.36
As we previously demonstrated in a batch decagram scale reaction (Table 4C, top), the e-amination is easily scalable. Compound 58 is an important intermediate for the synthesis of vilazodone, a drug recently approved by FDA for the treatment of major depressive disorder.37 Previously, 58 was prepared by four steps from salicylaldehyde in 26% overall yield,38 though a more straightforward synthesis is possible by using Pd-catalyzed amination to form the C–N bond between benzofuran fragment and piperidine.39 As an alternative approach, e-amination of 59 with N-Boc-piperidine was evaluated to prepare a closely-related compound 36. After a brief investigation, it was found that tetra-n-butylammonium bromide could be replaced with inexpensive sodium bromide to further reduce the cost of the production. Moreover, the RVC electrode was found to be replaceable with an even simpler carbon felt electrode. A 100 g scale reaction was successfully carried out by employing a flow system with only a small drop in yield (64% in 100 g vs 79% in 0.2 mmol). Notably, the multi-frame cell set up used in this scale-up study accommodates various reaction scales simply by changing the number of frames (See SI for details). This scale-up example clearly illustrates the simplicity and low-cost of scaling up e-amination.
Applicability of other nucleophiles and limitations of the e-amination
Finally, the efficiency of the modified conditions (higher ligand loading together with the use of DBU as an external base) over the first-generation conditions (Conditions A) as well as limitations of e-amination in a preparative scale are illustrated in Table 5. In the case of substrates previously described,8 the desired products 60-68 were obtained in similar or slightly higher yields under the modified conditions. With regard to the applicability of other types of nitrogen nucleophiles, a lactam and ammonia were both found to be competent (70, 71). However, e-amination with aniline and sulfonamide were not successful due to low nucleophilicity of these coupling partners (69, 72). Coupling with oxygen-based nucleophile was also found to be feasible as exemplified in the formation of 73-75. Phenol was not a suitable nucleophile likely due to its susceptibility toward oxidation (76). In contrast to relatively predictable reactivity of nucleophiles, the reactivity of the aryl bromides seems to be rather difficult to interpret. Some heteroaryl bromides gave complex mixtures, whereas others were unreactive. It implies that multiple mechanistic scenarios could exist in e-amination, depending on the substrate used. Oxidative addition, rather than amine coordination/deprotonation might be the possible rate-determining step in some of these cases. It also needs to be pointed out that electron-rich aryl halides are prone to give lower yields due to slow oxidative addition or oxidative degradation of the products, although e-amination smoothly took place on indole (35), benzofuran (36), thiophene (40) and electron-neutral substrates such as nucleoside analogs and oligopeptides.
Table 5.
Comparison to previously-successful substrates and limitations of e-amination in preparative scale. a Data obtained from ref 8. b 10 equiv. of nucleophile used. c 20 equiv. of water used.

|
Conclusion
The present study commenced with a deep interrogation of the mechanism of e-amination in order to demystify the nature of the catalytic cycle and aid in the elucidation of more general and robust conditions. By applying a range of techniques, from empirical optimizations to DFT calculations and kinetic studies, a second-generation set of conditions was invented using a simple Ni-precatalyst. The scope of e-amination was significantly expanded and supplemented with multiple applications. The use of e-amination is not limited to small scale applications and simple setups for both batch and flow scale up are delineated. Finally, this unique method for C–N bond formation has been field tested in multiple programs within the labs of our industrial collaborators. The mainstream adoption of electrochemistry in areas like medicinal chemistry has been stymied by a lack of standardized equipment and reaction classes that are relevant to drug discovery. It is anticipated that the current work will be an important step in accelerating the adoption of this useful means of controlling redox states in organic synthesis.
Supplementary Material
ACKNOWLEDGMENT
We thank Prof. Donna Blackmond for assistance in interpreting the kinetics data. We also thank Dr. D.-H. Huang and Dr. L. Pasternack for NMR spectroscopic assistance, Prof. A. L. Rheingold, Dr. C. E. Moore and Dr. Milan Gembicky for X- ray crystallographic analysis, Dr. Jason Chen and Brittany Sanchez (Scripps Automated Synthesis Facility) for assistance with HPLC, HRMS and LCMS. We gratefully acknowledge Dr. Jinshan Chen for helpful discussions during the early stage of this work. We also greatly appreciate insightful discussion with Dr. Martin D. Eastgate, Dr. Amy C. Hart and Dr. Jennifer X. Qiao.
Funding Sources
Financial support for this work was provided by NIH (GM-118176), CCI NSF Synthetic Organic Electrosynthesis Center (#1740656), George E. Hewitt foundation for postdoctoral fellowship (Y.K.), Universidad Autónoma de Madrid (A.F.G.-C.), NSF GRFP for graduate support (D.S.P. and J.N.D.) and JSPS for postdoctoral fellowship (H. N.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, additional electrochemical analysis data, the detail of DFT calculation and compound characterization data. (PDF)
X-ray crystallographic data for compound 17 (CIF)
REFERENCES
- (1).For selected reviews, see:; (a) Ruiz-Castillo P; Buchwald SL Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beletskaya IP; Cheprakov AV The Complementary Competitors: Palladium and Copper in C–N Cross-Coupling Reactions. Organometallics 2012, 31, 7753–7808. [Google Scholar]; (c) Fischer C; Koenig B Palladium- and Copper-Mediated N-Aryl Bond Formation Reactions for the Synthesis of Biological Active Compounds. Beilstein J. Org. Chem 2011, 7, 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Brown DG; Boström J Analysis of Past and Present Synthetic Methodologies on Medicinal Chemistry: Where Have All the New Reactions Gone? J. Med. Chem 2016, 59, 4443–4458. [DOI] [PubMed] [Google Scholar]
- (3).(a) Dennis JM; White NA; Liu RY; Buchwald SL Breaking the Base Barrier: an Electron-Deficient Palladium Catalyst Enables the Use of a Common Soluble Base in C–N Coupling. J. Am. Chem. Soc 2018, 140, 4721–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lee HG; Lautrette G; Pentelute BL; Buchwald SL Palladium-Mediated Arylation of Lysine in Unprotected Peptides. Angew. Chem. Int. Ed 2017, 56, 3177–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Balraju V; Iqbal J Synthesis of Cyclic Peptides Constrained with Biarylamine Linkers Using Buchwald–Hartwig C–N Coupling#. J. Org. Chem 2006, 71, 8954–8956. [DOI] [PubMed] [Google Scholar]
- (4).For selected reviews, see:; (a) Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemical Methods Since 2000: on the Verge of a Renaissance. Chem. Rev 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Horn EJ; Rosen BR; Baran PS Synthetic Organic Electrochemistry: an Enabling and Innately Sustainable Method. ACS Cent. Sci 2016, 2, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Francke R; Little RD Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]; (d) Yoshida J-I; Kataoka K; Horcajada R; Nagaki A Modern Strategies in Electroorganic Synthesis. Chem. Rev 2008, 108, 2265–2299. [DOI] [PubMed] [Google Scholar]; (e) Moeller KD Synthetic Applications of Anodic Electrochemistry. Tetrahedron 2000, 56, 9527. [Google Scholar]
- (5).O’Brien AG; Maruyama A; Inokuma Y; Fujita M; Baran PS; Blackmond DG Radical C–H Functionalization of Heteroarenes Under Electrochemical Control. Angew. Chem. Int. Ed 2014, 53, 11868–11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Horn EJ; Rosen BR; Chen Y; Tang J; Chen K; Eastgate MD; Baran PS Scalable and Sustainable Electrochemical Allylic C–H Oxidation. Nature 2016, 533, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kawamata Y; Yan M; Liu Z; Bao D-H; Chen J; Starr JT; Baran PS Scalable, Electrochemical Oxidation of Unactivated C–H Bonds. J. Am. Chem. Soc 2017, 139, 7448–7451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Li C; Kawamata Y; Nakamura H; Vantourout JC; Liu Z; Hou Q; Bao D; Starr JT; Chen J; Yan M; et al. Electrochemically Enabled, Nickel-Catalyzed Amination. Angew. Chem. Int. Ed 2017, 56, 13088–13093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Marín M; Rama RJ; Nicasio MC Ni-Catalyzed Amination Reactions: an Overview. Chem. Rec 2016, 16, 1819–1832. [DOI] [PubMed] [Google Scholar]
- (10).Hughes EC; Veatch F; Elersich V N-Methylaniline From Chlorobenzene and Methylamine. Ind. Eng. Chem 1950, 42,787–790. [Google Scholar]
- (11).Cramer R; Coulson DR Nickel-Catalyzed Displacement Reactions of Aryl Halides. J. Org. Chem 1975, 40, 2267–2273. [Google Scholar]
- (12).Cristau H-J; Desmurs J-R Arylation of Hard Heteroatomic Nucleophiles Using Bromoarenes Substrates and Cu, Ni, Pd-Catalysts; Industrial Chemistry Library; Elsevier, 1995; Vol. 7, pp 240–263. [Google Scholar]
- (13).Wolfe JP; Buchwald SL Nickel-Catalyzed Amination of Aryl Chlorides. J. Am. Chem. Soc 1997, 119, 6054–6058. [Google Scholar]
- (14).Kelly RA; Scott NM; Díez-González S; Stevens ED; Nolan SP Simple Synthesis of CpNi(NHC)Cl Complexes (Cp = Cyclopentadienyl; NHC = N-Heterocyclic Carbene). Organometallics 2005, 24, 3442–3447. [Google Scholar]
- (15).Park NH; Teverovskiy G; Buchwald SL Development of an Air-Stable Nickel Precatalyst for the Amination of Aryl Chlorides, Sulfamates, Mesylates, and Triflates. Org. Lett 2013, 16, 220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Shields JD; Gray EE; Doyle AG A Modular, Air-Stable Nickel Precatalyst. Org. Lett 2015, 17, 2166–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Kampmann SS; Skelton BW; Wild DA; Koutsantonis GA; Stewart SG An Air-Stable Nickel(0) Phosphite Precatalyst for Primary Alkylamine C-N Cross-Coupling Reactions. Eur. J. Org. Chem 2015, 2015, 5995–6004. [Google Scholar]
- (18).Iglesias MJ; Prieto A; Nicasio MC Well-Defined Allylnickel Chloride/N-Heterocyclic Carbene [(NHC)Ni(Allyl)Cl] Complexes as Highly Active Precatalysts for C–N and C–S Cross-Coupling Reactions. Adv. Synth. Catal 2010, 352, 1949–1954. [Google Scholar]
- (19).(a) Ge S; Green RA; Hartwig JF Controlling First-Row Catalysts: Amination of Aryl and Heteroaryl Chlorides and Bromides with Primary Aliphatic Amines Catalyzed by a BINAP-Ligated Single-Component Ni(0) Complex. J. Am. Chem. Soc 2014, 136, 1617–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lavoie CM; MacQueen PM; Rotta-Loria NL; Sawatzky RS; Borzenko A; Chisholm AJ; Hargreaves BKV; McDonald R; Ferguson MJ; Stradiotto M Challenging Nickel-Catalysed Amine Arylations Enabled by Tailored Ancillary Ligand Design. Nature Commun. 2016, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lavoie CM; MacQueen PM; Stradiotto M Nickel-Catalyzed N-Arylation of Primary Amides and Lactams with Activated (Hetero)Aryl Electrophiles. Chem. Eur. J 2016, 22, 18752–18755. [DOI] [PubMed] [Google Scholar]; (d) MacQueen PM; Tassone JP; Diaz C; Stradiotto M Exploiting Ancillary Ligation to Enable Nickel-Catalyzed C–O Cross-Couplings of Aryl Electrophiles with Aliphatic Alcohols. J. Am. Chem. Soc 2018, 140, 5023–5027. [DOI] [PubMed] [Google Scholar]; (e) Tassone JP; England EV; MacQueen PM; Ferguson MJ; Stradiotto M PhPAd-DalPhos: Ligand-Enabled, Nickel-Catalyzed Cross-Coupling of (Hetero)Aryl Electrophiles with Bulky Primary Alkylamines. Angew. Chem. Int. Ed 2019, 58, 2485–2489. [DOI] [PubMed] [Google Scholar]
- (20).(a) Koo K; Hillhouse GL Carbon-Nitrogen Bond Formation by Reductive Elimination From Nickel(II) Amido Alkyl Complexes. Organometallics 1995, 14, 4421–4423. [Google Scholar]; (b) Koo K; Hillhouse GL Indoline Synthesis via Coupling of Phenethyl Grignard Reagents with Organoazides Mediated by (Alkylphosphine)Nickel(II) Complexes. Organometallics 1996, 15, 2669–2671. [Google Scholar]; (c) Mindiola DJ; Hillhouse GL Terminal Amido and Imido Complexes of Three-Coordinate Nickel. J. Am. Chem. Soc 2001, 123, 4623–4624. [DOI] [PubMed] [Google Scholar]; (d) Lin BL; Clough CR; Hillhouse GL Interactions of Aziridines with Nickel Complexes: Oxidative-Addition and Reductive-Elimination Reactions That Break and Make C–N Bonds. J. Am. Chem. Soc 2002, 124, 2890–2891. [DOI] [PubMed] [Google Scholar]
- (21).Ilies L; Matsubara T; Nakamura E Nickel-Catalyzed Synthesis of Diarylamines via Oxidatively Induced C–N Bond Formation at Room Temperature. Org. Lett 2012, 14, 5570–5573. [DOI] [PubMed] [Google Scholar]
- (22).Corcoran EB; Pirnot MT; Lin S; Dreher SD; DiRocco DA; Davies IW; Buchwald SL; MacMillan DWC Aryl Amination Using Ligand-Free Ni(II) Salts and Photoredox Catalysis. Science 2016, 353, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Vander Griend DA; Bediako DK; DeVries MJ; DeJong NA; Heeringa LP Detailed Spectroscopic, Thermodynamic, and Kinetic Characterization of Nickel(II) Complexes with 2,2’-Bipyridine and 1,10-Phenanthroline Attained via Equilibrium-Restricted Factor Analysis. Inorg. Chem 2008, 47, 656–662. [DOI] [PubMed] [Google Scholar]
- (24).Lappin AG; McAuley A The Redox Chemistry of Nickel; Advances in Inorganic Chemistry; Elsevier, 1988; Vol. 32, pp 241–295. [Google Scholar]
- (25).Lim C-H; Kudisch M; Bin Liu; Miyake, G. M. C–N Cross-Coupling via Photoexcitation of Nickel–Amine Complexes. J. Am. Chem. Soc 2018, 140, 7667–7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).DMA seems to be oxidized under the reaction conditions as evident from the formation of aminal product 52b. However, this oxidation is likely to prevent undesirable consumption of amine nucleophile.
- (27).Dhar SK; Basolo F Thermal Decomposition of the Tris (2,2’-Bipyridine) Complexes of Some First Row Transition Group Elements in the Solid State. J. Inorg. Nucl. Chem 1963, 25, 37–44. [Google Scholar]
- (28).(a) Froese RDJ; Lombardi C; Pompeo M; Rucker RP; Organ MG Designing Pd–N-Heterocyclic Carbene Complexes for High Reactivity and Selectivity for Cross-Coupling Applications. Acc. Chem. Res 2017, 50, 2244–2253. [DOI] [PubMed] [Google Scholar]; (b) King SM; Buchwald SL Development of a Method for the N-Arylation of Amino Acid Esters with Aryl Triflates. Org. Lett 2016, 18, 4128–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sharif S; Mitchell D; Rodriguez MJ; Farmer JL; Organ MG N-Heteroarylation of Optically Pure α-Amino Esters Using the Pd-PEPPSI-IPentCl-o-picoline Precatalyst. Chem. Eur. J 2016, 22, 14860–14863. [DOI] [PubMed] [Google Scholar]; (d) Hammoud H; Schmitt M; Blaise E; Bihel F; Bourguignon J-J N-Heteroarylation of Chiral α-Aminoesters by Means of Palladium-Catalyzed Buchwald–Hartwig Reaction. J. Org. Chem 2013, 78, 7930–7937. [DOI] [PubMed] [Google Scholar]; (e) Surasani R; Kalita D; Rao AVD; Chandrasekhar KB Palladium-Catalyzed C–N and C–O Bond Formation of N-Substituted 4-Bromo-7-Azaindoles with Amides, Amines, Amino Acid Esters and Phenols. Beilstein J. Org. Chem 2012, 8, 2004–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Becica J; Dobereiner GE Acceleration of Pd-Catalyzed Amide N-Arylations Using Cocatalytic Metal Triflates: Substrate Scope and Mechanistic Study. ACS Catal. 2017, 7, 5862–5870. [Google Scholar]
- (29).(a) Sharma KK; Sharma S; Kudwal A; Jain R Room Temperature N-Arylation of Amino Acids and Peptides Using Copper(I) and α-Diketone. Org. Biomol. Chem 2015, 13, 4637–4641. [DOI] [PubMed] [Google Scholar]; (b) Ma D; Xia C CuI-Catalyzed Coupling Reaction of β-Amino Acids or Esters with Aryl Halides at Temperature Lower Than That Employed in the Normal Ullmann Reaction. Facile Synthesis of SB-214857. Org. Lett 2001, 3, 2583–2586. [DOI] [PubMed] [Google Scholar]
- (30).Nakamura H; Yasui K; Kanda Y; Baran PS 11-Step Total Synthesis of Teleocidins B-1–B-4. J. Am. Chem. Soc Articles ASAP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).(a) Noji T; Okano K; Tokuyama H A Concise Total Synthesis of (−)-Indolactam V. Tetrahedron 2015, 71, 3833–3837. [Google Scholar]; (b) Haynes-Smith J; Diaz I; Billingsley KL Modular Total Synthesis of Protein Kinase C Activator (−)-Indolactam V. Org. Lett 2016, 18, 2008–2011. [DOI] [PubMed] [Google Scholar]
- (32).Mari M; Bartoccini F; Piersanti G Synthesis of (−)-Epi-Indolactam V by an Intramolecular Buchwald-Hartwig C–N Coupling Cyclization Reaction. J. Org. Chem 2013, 78, 7727–7734. [DOI] [PubMed] [Google Scholar]
- (33).Welsch ME; Snyder SA; Stockwell BR Privileged Scaffolds for Library Design and Drug Discovery. Curr. Opin. Chem. Biol 2010, 14, 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).For recent reviews on C–S bond functionalization by nickel catalysis, see:; (a) Otsuka S; Nogi K; Yorimitsu H C–S Bond Activation. Top. Curr. Chem 2018, 376, 1–39. [DOI] [PubMed] [Google Scholar]; (b) Rentner J; Kljajic M; Offner L; Breinbauer R Recent Advances and Applications of Reductive Desulfurization in Organic Synthesis. Tetrahedron 2014, 70, 8983–9027. [Google Scholar]; (c) Wang L; He W; Yu Z Transition-Metal Mediated Carbon–Sulfur Bond Activation and Transformations. Chem. Soc. Rev 2013, 42, 599–621. [DOI] [PubMed] [Google Scholar]; (d) Pan F; Shi Z-J Recent Advances in Transition-Metal-Catalyzed C–S Activation: From Thioester to (Hetero)Aryl Thioether. ACS Catal. 2013, 4, 280–288. [Google Scholar]
- (35).(a) Feldman AW; Romesberg FE Expansion of the Genetic Alphabet: a Chemist’s Approach to Synthetic Biology. Acc. Chem. Res 2018, 51, 394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Joubert N; Urban M; Pohl R; Hocek M Modular Synthesis of 4-Aryl- and 4-Amino-Substituted Benzene C-2’-Deoxyribonucleosides. Synthesis 2008, 1918–1932. [Google Scholar]; (c) Wang; Schultz Expanding the Genetic Code. Chem. Commun 2001, 1–11. [DOI] [PubMed] [Google Scholar]
- (36).(a) Pentelute B; Zhang C; Vinogradova E; Spokoyny A; Buchwald S Arylation Chemistry for Bioconjugation. Angew. Chem. Int. Ed 2018, accepted article. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) deGruyter JN; Malins LR; Baran PS Residue-Specific Peptide Modification: a Chemist’s Guide. Biochemistry 2017, 56, 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hu B; Song Q; Xu Y Scale-Up Synthesis of Antidepressant Drug Vilazodone. Org. Process Res. Dev 2012, 16, 1552–1557. [Google Scholar]
- (38).Renuka J; Reddy KI; Srihari K; Jeankumar VU; Shravan M; Sridevi JP; Yogeeswari P; Babu KS; Sriram D Design, Synthesis, Biological Evaluation of Substituted Benzofurans as DNA gyraseB Inhibitors of Mycobacterium Tuberculosis. Bioorg. Med. Chem 2014, 22, 4924–4934. [DOI] [PubMed] [Google Scholar]
- (39).Shao Q-L; Jiang Z-J; Su W-K Solvent-Free Mechanochemical Buchwald-Hartwig Amination of Aryl Chlorides Without Inert Gas Protection. Tetrahedron Lett 2018, 59, 2277–2280. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.