

Summary
Post-translational substrate modification with ubiquitin is essential for eukaryotic cellular signaling. Polymeric ubiquitin chains are assembled with specific architectures, which convey distinct signaling outcomes depending on the linkages involved. Recently, branched K11/K48-linked polyubiquitins were shown to enhance proteasomal degradation during mitosis. To better understand the underlying structural mechanisms, we determined the crystal and NMR structures of branched K11/K48-linked tri-ubiquitin and discovered a previously unobserved interdomain interface between the distal ubiquitins. Small-angle neutron scattering and site-directed mutagenesis corroborated the presence of this interface, which we hypothesized to be influential in the physiological role of branched K11/K48-linked chains. Yet, experiments probing polyubiquitin interactions – deubiquitination assays; binding to proteasomal shuttle hHR23A – showed negligible differences between branched K11/K48-linked tri-ubiquitin and related di-ubiquitins. However, significantly stronger binding affinity for branched K11/K48-linked tri-ubiquitin was observed with proteasomal subunit Rpn1, thereby suggesting a functional impact of this interdomain interface and pinpointing the mechanistic site of enhanced degradation.
Keywords: branched mixed-linkage polyubiquitin, K11-linked ubiquitin, K48-linked ubiquitin, deubiquitinase, Rad23 UBA domain, proteasome
Graphical Abstract
Blurb
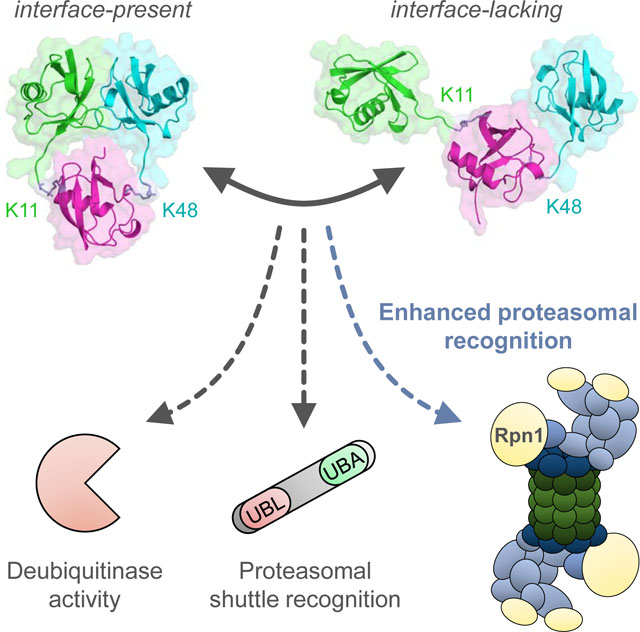
Boughton et al. characterize branched K11/K48-linked tri-ubiquitin structurally (X-ray crystallography, NMR, SANS) and biochemically and show that it possesses a unique, previously unobserved hydrophobic interface between the distal ubiquitins. This branching enhances binding affinity for proteasomal receptor Rpn1, allowing branched K11/K48-linked polyubiquitins to act as high-priority signals for proteasomal degradation.
Introduction
Post-translational modification of cellular proteins with ubiquitin (Ub) is one of the most critical and diverse signaling pathways in eukaryotes (Fushman and Wilkinson, 2011). Conjugation of Ub to specific substrates is responsible for regulating cellular division, differentiation, immune response, DNA damage repair, and functions as the primary signal for controlled protein turnover (Hershko and Ciechanover, 1998; Wang and Maldonado, 2006). This diversity of signaling outcomes reflects Ub’s ability to form polyubiquitin (polyUb) chains through a covalent linkage between the C-terminus of one Ub (‘distal’) and any of seven lysines or the N-terminus of another Ub (‘proximal’). All eight Ub-Ub linkages have been detected in cells, with varying abundance (Xu et al., 2009). Each linkage bestows unique structural and dynamical features that enable distinct recognition and signaling patterns for polyUb of every linkage type, promoting the concept of the ‘ubiquitin code’ (Komander and Rape, 2012; Pickart and Fushman, 2004). PolyUbs linked through K48 or K63, termed canonical Ub chains, have been extensively studied. K48-linked chains target substrates for proteasomal degradation, while K63-linked chains regulate NF-κB signaling, DNA damage response, and endocytosis (Haglund and Dikic, 2005).
Considerably less information is available regarding non-canonical Ub chains, which are linked through any of the other six attachment sites. Of the non-canonical linkages, K11-linkages are the most common, and might even be as abundant as K48-linkages in yeast (Peng et al., 2003; Xu et al., 2009). Numerous functions for K11-linked polyUb have been proposed, such as functioning in cellular division, membrane trafficking, and DNA repair pathways (Bremm and Komander, 2011; Matsumoto et al., 2010; Wickliffe et al., 2011).
Attention has recently shifted to mixed-linkage polyUb chains (Nakasone et al., 2013), for which novel physiological roles are rapidly being identified (Yau and Rape, 2016). Such chains have either unbranched (each proximal Ub is modified with only one distal Ub) or branched (at least one Ub is modified with at least two distal Ubs) linkage architecture. NF-κB signaling is activated by M1/K63-linked chains (Emmerich et al., 2013) and enhanced by branched K48/K63-linked chains (Ohtake et al., 2016), which also signal for proteasomal degradation (Ohtake et al., 2018). Chains consisting of K11/K63-linkages initiate endocytosis (Boname et al., 2010), while branched K29/K48-linked chains participate in the ER-associated degradation pathway (Liu et al., 2017). Most noteworthy to this study, branched K11/K48-linked polyUb unexpectedly enhances proteasomal degradation of cell-cycle regulators in mitotic cells (Meyer and Rape, 2014), even though homogenous K48-linked chains are considered the primary degradation signal.
The complexity of Ub signaling remains poorly understood, even in solely the context of the Ub-proteasome system (UPS). We aimed to clarify the role of K11/K48-linked polyUb in this carefully orchestrated pathway in order to understand how ‘spiking’ K48-linked chains with branched K11-linked Ub enhances proteasomal degradation. Here, we investigated the structure of branched K11/K48-linked Ub3 using a combination of solution nuclear magnetic resonance (NMR) spectroscopy, X-ray crystallography, and small-angle neutron scattering (SANS) complemented with ensemble modeling. These methods consistently indicated the presence of a distinct interface between the distal Ubs, even though these Ubs are not directly connected to each other; to our knowledge, this is the first observation of such an interaction. Based on similarities between this interface and the well-established binding surface in K48-linked Ub2, we speculated that it may constitute a novel recognition surface. Thus, we considered three components of the UPS in which the unique interface in branched K11/K48-linked Ub3 could improve signal recognition and, ultimately, enhance proteasomal degradation: nascent deubiquitinase activity, proteasomal shuttling factors, and proteasomal receptors. Our results identified the third component – proteasomal subunit Rpn1 – as the focal point of this phenomenon, thereby pinpointing the specific location in the UPS at which branched K11/K48-linked polyUb enhances substrate recognition and further emphasizing the significant signaling diversity of mixed-linkage polyUb.
Results
To convey the architecture of branched polyUb chains, we will follow the nomenclature (Nakasone et al., 2013) in which branched K11/K48-linked Ub3 is written as [Ub]2–11,48Ub (Table S1). The same Ub3 with a 15N-enriched distal K11-linked Ub or distal K48-linked Ub is Ub(15N)[Ub]–11,48Ub or Ub[Ub(15N)]–11,48Ub, respectively. For consistency, this notation is used for all Ub chains (e.g., K48-linked Ub2 is Ub–48Ub).
Our objective was to identify salient structural features of branched K11/K48-linked polyUb to understand its apparent physiology. We focused on the shortest form of branched K11/K48-linked polyUb, [Ub]2–11,48Ub, which was assembled using an established approach (Methods) to control chain length, architecture, and selective isotopic labeling of any Ub.
NMR reveals the presence of a unique hydrophobic interface in [Ub]2–11,48Ub
Structural characteristics of [Ub]2–11,48Ub were first investigated by comparing its NMR spectra with those of related dimers and monomers. Two chains were assembled, each with a specific distal Ub 15N-enriched: Ub(15N)[Ub]–11,48Ub and Ub[Ub(15N)]–11,48Ub.
NMR spectra revealed significant differences in amide signal positions of Ub(15N)[Ub]–11,48Ub compared to Ub(15N)–11Ub and 15N-monoUb (Fig. S1A). The NMR signal shifts, quantified as chemical shift perturbations (CSPs), are shown in Fig. 1A. The majority of signal shifts were clustered around the hydrophobic surface-patch residues L8, I44, H68, and V70 (Beal et al., 1996). These perturbations were surprising, as the published NMR data (Castaneda et al., 2016a; Castañeda et al., 2013) and crystal structures of K11-linked polyUb (Bremm et al., 2010; Matsumoto et al., 2010) (Fig. 1D) do not exhibit extensive inter-Ub contacts that would produce such signal shifts. Since the only structural difference between [Ub]2–11,48Ub and Ub–11Ub is an extra distal Ub connected to the proximal Ub through K48, we speculated that these unexpected signal perturbations were caused by direct interactions between the distal Ubs.
Figure 1: Structural characterization of [Ub]2–11,48Ub.

Overlaid CSP (Δδ) plots for (A) Ub(15N)[Ub]–11,48Ub versus 15N-monoUb (green) and Ub(15N)–11Ub versus 15N-monoUb (light grey); (B) Ub[Ub(15N)]–11,48Ub versus 15N-monoUb (cyan) and Ub(15N)–48Ub versus 15N-monoUb (light grey); (C) Ub(15N)–11Ub–48Ub versus 15N-monoUb (black) and Ub(15N)–11Ub versus 15N-monoUb (light grey). Insets in (A-C) show a schematic of Ub3 with the 15N-enriched moiety colored and an example of one NMR signal shifting between respective Ub (black), Ub2 (red), and Ub3 (blue) species. (D) Crystal structure of Ub–11Ub (PDB: 2XEW). (E) Crystal structure of Ub–48Ub (PDB: 1AAR). (F) Crystal structure of [Ub]2–11,48Ub, with a semi-transparent surface representation in grey. In (D-F), the distal K11-linked Ub is green, the distal K48-linked Ub is cyan, the proximal Ub is magenta, and hydrophobic patch residues are shown as sticks (red), and the isopeptide linkages are periwinkle. (G) Electrostatic surface potential (positive is blue, negative is red) of [Ub]2–11,48Ub, with a ±3 kT/e coloring range. (H) Residues with CSPs between respective Ub3 and Ub2 species larger than one standard deviation above the average are shown as orange sticks on the crystal structure of [Ub]2–11,48Ub. Ub coloring scheme is as in (D-F). See also Figures S1–4.
Therefore, we next investigated the distal K48-linked Ub. Distinct differences were observed between the spectra of Ub[Ub(15N)]–11,48Ub and 15N-monoUb (Fig. 1B). Such perturbations could reflect the formation of a hydrophobic interface between the distal K48-linked Ub and the proximal Ub, akin to the characteristic interface in isolated Ub–48Ub (Varadan et al., 2002) (Fig. 1E). However, substantial differences between the NMR spectra of Ub[Ub(15N)]–11,48Ub and Ub(15N)–48Ub indicated that the interactions involving the distal K48-linked Ub in [Ub]2–11,48Ub were not the same as in Ub–48Ub (Fig. 1B). Notably, signals from Ub[Ub(15N)]–11,48Ub continued shifting further (Fig. S1B); thus, it appears that residues in the distal K48-linked Ub of [Ub]2–11,48Ub experience stronger interdomain contacts than in Ub–48Ub.
Since both distal Ubs of [Ub]2–11,48Ub exhibited significant CSPs compared to respective dimers, and the CSPs were predominantly observed around the hydrophobic patch residues, we hypothesized that the distal K11-linked Ub and distal K48-linked Ub form a hydrophobic interface with each other. The two distal Ubs cannot simultaneously contact the hydrophobic patch on the proximal Ub as these interactions are mutually exclusive; therefore, the proximal Ub is not significantly involved.
Interestingly, the observed interface is unique to the branched form of K11/K48-linked Ub3. Indeed, the NMR spectra of unbranched K11/K48-linked Ub3 (Ub(15N)–11Ub–48Ub) showed inconsequentially small differences from Ub(15N)–11Ub (Fig. 1C), indicating that the distal K11-linked Ub in Ub–11Ub–48Ub behaves similarly to that of Ub–11Ub. Thus, adding a K48-linked Ub to Ub–11Ub by unbranched extension does not affect the overall Ub3 structure; i.e., the distal K11-linked Ub and K48-linked Ub behave as they normally do in respective Ub2 species. However, adding a branched K48-linked Ub to Ub–11Ub produces a unique structural change through interactions between the distal Ubs. This behavior is in stark contrast to other mixed-linkage Ub3 species, such as K48/K63-linked Ub3 (Nakasone et al., 2013), where both unbranched and branched chains show no direct interactions between non-linked Ubs. To our knowledge, this is the first observation of Ubs that are not directly linked to each other forming an interface in mixed-linkage polyUb. Therefore, we speculated that this unique structural feature of [Ub]2–11,48Ub may be responsible for enhancing proteolytic degradation (Meyer and Rape, 2014).
Crystal structure of [Ub]2–11,48Ub identifies unique interdomain interface
We used X-ray crystallography to test our hypothesis about the unique interface in [Ub]2–11,48Ub. We obtained [Ub]2–11,48Ub crystals that diffracted at 2.2 Å resolution (Fig. S2 and Table 1).
The crystal structure of [Ub]2–11,48Ub exhibits a distinct interface between the distal Ubs (Fig. 1F). This interface is primarily mediated by interactions between the hydrophobic patches of both distal Ubs, which – to a cursory degree – agrees with NMR data. Further examination of this structure revealed that the interface between K48-linked Ubs, which is present in K48-linked Ub2 (Fig. 1E) and Ub4 (Cook et al., 1992; Eddins et al., 2007; Varadan et al., 2002) and considered a hallmark of this linkage, is completely broken. Instead, the distal K48-linked Ub is shifted and rotated ~180° to form a new interface, now with the distal K11-linked Ub (Fig. 1F). Since the majority of Ub-receptor binding occurs through the hydrophobic patch (Hurley et al., 2006), we hypothesized that this interdomain interface may constitute a novel binding surface exclusive to branched K11/K48-linked polyUb.
Interestingly, the outer rim of [Ub]2–11,48Ub carries a negative charge, while positive charges are grouped at the core, resulting in a bimodal electrostatic surface potential distribution (Fig. 1G). We speculated that this segregation of charges may be important for ligand recognition and binding that is partially mediated by electrostatic interactions, such as with ubiquitin-associated (UBA) domains (Walinda et al., 2014) and proteasomal subunit Rpn1 (Shi et al., 2016).
To ascertain the agreement between the crystal structure of [Ub]2–11,48Ub and our solution NMR data, residues with significant CSPs were mapped onto the structure. Contrary to the expectation that large CSPs should be exhibited by residues directly at the interface between distal Ubs, the most-affected residues for the distal K48-linked Ub appeared adjacent to the interface in the crystal structure (Fig. 1H); in fact, many of these residues are solvent-exposed. This discrepancy suggests that although the crystal structure is generally correct in that the distal Ubs form a hydrophobic interface, their relative orientation is not representative of [Ub]2–11,48Ub structure in solution. We therefore resorted to comprehensive structural characterization in solution to further refine the structural ‘portrait’ of [Ub]2–11,48Ub.
Characterization of [Ub]2–11,48Ub in solution corroborates the interdomain contact
To independently characterize the structure of [Ub]2–11,48Ub in solution, a paramagnetic spin label (MTSL) was attached to specific sites in selectively 15N-labeled [Ub]2–11,48Ub. This allowed determination of interdomain distances via paramagnetic relaxation enhancement (PRE) effects induced by MTSL (Varadan et al., 2005a).
Attachment of MTSL to C48 in the distal K11-linked Ub of UbK48C[Ub(15N)]–11,48Ub resulted in significant region-specific signal attenuations in the distal K48-linked Ub, especially pronounced in and around the hydrophobic patch and C-terminal tail residues (Fig. 2A), indicating that they are located within ~25 Å of MTSL. Likewise, a reverse experiment showed strong signal attenuations in similar residues of the distal K11-linked Ub when MTSL was attached to C48 in the distal K48-linked Ub of Ub(15N)[UbK48C]–11,48Ub (Fig. 2B). In this case, the attenuations were stronger, suggesting a closer distance to MTSL. The signal attenuations and crystal structure of [Ub]2–11,48Ub were used to reconstruct the location of MTSL’s unpaired electron relative to each Ub using program SLfit (Ryabov and Fushman, 2006).
Figure 2: Solution structure of [Ub]2–11,48Ub.

PREs (I/I0) for (A) UbK48C~MTSL[Ub(15N)]–11,48Ub, (B) Ub(15N)[UbK48C~MTSL]–11,48Ub, and (C) 15N-monoUbK48C~MTSL. Colored bars depict experimental PREs, while red circles represent PREs back-calculated from the reconstructed MTSL position. Residues denoted with an asterisk were not observed in the NMR spectra and not included in analysis. (D) Top-down view of the crystal structure of [Ub]2–11,48Ub, where the actual positions of MTSL attached to each distal Ub are represented by grey spheres, while the reconstructed MTSL positions are shown as red spheres. (E) Superposition of the ten top-scoring [Ub]2–11,48Ub NMR structures. (F) Alignment, via the proximal Ubs, between the crystal (grey) and NMR (colored) structures of [Ub]2–11,48Ub. (G) NMR structure of [Ub]2–11,48Ub, with hydrophobic patch residues shown as red sticks. (H) Residues with significant CSPs shown as orange sticks on the NMR structure of [Ub]2–11,48Ub. CSPs were between respective Ub3 and Ub2 species; only residues with CSPs larger than one standard deviation above the average were selected. (I) Superposition of the ten best [Ub]2–11,48Ub NMR structures, where the actual positions of MTSL attached to each distal Ub are represented by grey spheres, while the reconstructed MTSL positions are shown as red spheres. For (D-I), the Ub coloring scheme is as in Fig. 1. See also Figures S2–3 and Tables S3–4.
A control experiment was also performed, whereby MTSL was attached to C48 in 15N-monoUbK48C (Fig. 2C). The coordinates of MTSL’s unpaired electron determined from the observed PREs were in excellent agreement with its expected location, confirming the actual position of the Ub-attached MTSL. Since MTSL was attached to C48 in all of our PRE experiments, this location could be applied to both distal Ubs in [Ub]2–11,48Ub.
The actual location of MTSL attached to one distal Ub and its reconstructed position based on PREs detected in the other distal Ub were mapped onto the crystal structure of [Ub]2–11,48Ub (Fig. 2D). These MTSL positions were in stark disagreement: the distances (ΔMTSL) between the two were 24.7 Å for MTSL attached to the distal K11-linked Ub and 25.0 Å for MTSL attached to the distal K48-linked Ub. These data – along with CSP mapping – were a clear indication that the crystal structure does not completely reflect the structure of [Ub]2–11,48Ub in solution.
Structural modeling of [Ub]2–11,48Ub improves agreement with solution NMR data
Since, in the crystal structure, both reconstructed spin label positions were misplaced from the actual position by equivalent distances, we speculated that a rigid-body rotation and/or translation of the distal Ubs may fix this discrepancy. To test this, we first tested if it is physically possible to reposition the distal Ubs in [Ub]2–11,48Ub such that the reconstructed and actual MTSL locations align simultaneously. We generated an ensemble of ~38,000 structures using Torsion Angle Monte Carlo (TAMC) simulation in SASSIE (Curtis et al., 2012), starting with the crystal structure of [Ub]2–11,48Ub and allowing flexibility of each Ub’s C-terminal tail. The reconstructed MTSL position for each distal Ub was compared with the actual MTSL position for every structure; the best match was obtained with ΔMTSL of 3.2 Å and 5.9 Å for the distal K11-linked Ub and distal K48-linked Ub (Fig. S2E). This supported the feasibility of rearranging the crystal structure to agree with solution NMR data and also set the lower limits for expected ΔMTSL values.
Encouraged by these results, we used HADDOCK (van Zundert et al., 2016) to generate a structural model for [Ub]2–11,48Ub that reflected the solution NMR data more appropriately. We began with the crystal structure of [Ub]2–11,48Ub and utilized CSPs, which identified residues at the interdomain interface, and PREs, which provided distance restraints between the distal Ubs. The ten best structures from the highest scoring HADDOCK cluster (hereafter ‘NMR structure’) are shown in Fig. 2E. Superimposing the NMR and crystal structures of [Ub]2–11,48Ub by the proximal Ub shows that the distal K11-linked Ub has undergone a ~90° rotation in the NMR structure, while the distal K48-linked Ub has undergone a ~180° rotation and a ~25 Å translation (Fig. 2F). Even though these structures do not superimpose well, the alterations primarily reflect movement in the flexible linkers between Ubs and local rearrangement of the distal Ubs. Most notably, the hydrophobic Ub:Ub interface is preserved in the NMR structure (Fig. 2G).
The NMR structure of [Ub]2–11,48Ub agreed well with both CSP data (Fig. 2H) and PRE data (Fig. 2I). The ΔMTSL distances decreased to 3.6 Å for the distal K11-linked Ub and 6.2 Å for the distal K48-linked Ub. Both distances were adequately small and within the realm of MTSL and side-chain flexibility in solution. The NMR structure was also consistent with the best structure from TAMC modeling (Fig. S2F).
Small-angle neutron scattering reveals existence of multiple conformations in solution
Finally, we compared our structures to solution SANS data. Neither the crystal structure nor the NMR structure provided an appropriate match to the observed scattering profile of [Ub]2–11,48Ub (Fig. 3A–B). Indeed, the apparent radius of gyration (Rg) extracted from SANS data, 24.3 ± 0.2 Å, was significantly larger than predicted Rg values of 18.3 Å and 19.2 Å for the crystal and NMR structures. Furthermore, none of the ~38,000 TAMC-generated structures fully agreed with the SANS data by itself (see Fig. 3C, ‘one conformer’), suggesting that [Ub]2–11,48Ub adopts multiple conformational states in solution. The comparison of atom pair distributions, P(r) (Fig. 3B), indicates that in addition to the aforementioned compact structure, [Ub]2–11,48Ub also exists in extended conformations; this is evident from the well-defined second P(r) maximum at 31 Å and a shoulder at ~50 Å, reflecting larger interdomain distances. This is not surprising given the flexibility of Ub-Ub linkages and the dynamic nature of polyUb (Castaneda et al., 2016a; Castaneda et al., 2016b; Castañeda et al., 2013; Datta et al., 2009; Ryabov and Fushman, 2006; Varadan et al., 2002).
Figure 3: SANS-based conformational ensemble modeling of [Ub]2–11,48Ub.

(A) Comparison of the experimental SANS profile, I(q), for [Ub]2–11,48Ub (grey circles) with predicted I(q) for: the crystal structure (blue line), the NMR structure (red line), and a two-conformer ensemble comprising the NMR structure and the best-match second conformer (orange line). Errors on the experimental data are standard errors of the mean based on the number of pixels used in data averaging. Error-normalized residuals of fit between the experimental data and the two-conformer ensemble are shown (bottom). (B) Comparison of the experimental P(r) profile for [Ub]2–11,48Ub (grey circles) with predicted P(r) for the crystal structure (blue line) and the NMR structure (red line) of [Ub]2–11,48Ub. Errors on the experimental P(r) curve are standard deviations based on multiple fits to the data using a series of Monte Carlo simulations. (C) Comparison of χ2 values (per data point) quantifying the agreement between the experimental and predicted I(q) for individual structures (crystal, NMR, or best-fit single conformer from the TAMC-generated structures) and/or best-fit multi-conformer ensembles. (D) Structures of [Ub]2–11,48Ub from the two-conformer ensemble comprising the NMR structure (left, 40% populated) and the best-match second conformer (right, 60% populated). (E) Structures of [Ub]2–11,48Ub from the three-conformer ensemble comprising the NMR structure (left, 36% populated) and the two best-match additional conformers (middle, 45% populated; right, 19% populated). For (D-E), the Ub coloring scheme is as in Fig. 1. See also Figure S6.
Therefore, we investigated if the agreement with experimental SANS data can be improved by considering contributions from multiple conformers. To test this, we used our TAMC-generated structures supplemented with the NMR or crystal structure of [Ub]2–11,48Ub. Remarkably, a two-conformer ensemble that included the NMR structure (Fig. 3D) provided a substantially improved agreement with scattering data (Fig. 3A, C), resulting in χ2 per data point (χ2/N) of 1.3, which is similar to the χ2/N value of 1.1 for the best-fit combination of two conformers (Fig. 3C). A three-conformer ensemble containing the NMR structure (Fig. 3E) reduced the χ2/N to 0.96, indicating a good agreement with experimental data. Similar results were obtained for ensembles that included the crystal structure (Fig. 3C). This analysis suggested that in solution [Ub]2–11,48Ub exists in equilibrium between the aforementioned compact conformation (~40% populated) and one or more extended conformations (~60% populated), which do not exhibit a close noncovalent contact between distal Ubs.
The crystal structure, NMR structure, and SANS ensembles of [Ub]2–11,48Ub all displayed a compact conformation with a Ub:Ub interface involving the hydrophobic patch residues in each distal Ub; CSP and PRE data also consistently exhibited significant effects in those residues. To provide further evidence of this interaction, we introduced mutations (L8A/I44A) in the distal K48-linked Ub to disrupt contacts involving the hydrophobic patch (Beal et al., 1996; Varadan et al., 2005b). Indeed, the NMR spectrum of Ub(15N)[UbL8A/I44A/K48C]–11,48Ub was nearly identical to that of Ub(15N)–11Ub, producing negligible CSPs (Fig. S3A), thereby suggesting that the distal K11-linked Ub could no longer form a stable interface with the mutated distal K48-linked Ub. Furthermore, attachment of MTSL to Ub(15N)[UbL8A/I44A/K48C]–11,48Ub resulted in drastically diminished signal attenuations compared to those in Ub(15N)[UbK48C]–11,48Ub (Fig. S3B); the average distance between backbone amides and the derived MTSL position increased from 16 Å to 26 Å, indicating that the distal Ubs were farther apart from each other.
Irrespective of the precise orientation of the distal Ubs, all structural and mutagenesis data indicate the presence of a direct hydrophobic contact between these Ubs, which is a unique feature of branched K11/K48-linked Ub3.
Deubiquitination assays show that K48-linkages primarily dictate cleavage rate
With the structure of [Ub]2–11,48Ub characterized, we wanted to determine what components of the UPS pathway are responsible for branched K11/K48-linked polyUb functioning as an enhanced degradation signal. A surprising feature of [Ub]2–11,48Ub is the resemblance of its interface (formed by the distal Ubs) to that of Ub–48Ub, seen by comparing buried surface areas in each species (Fraczkiewicz and Braun, 1998). For Ub–48Ub, 34.2% of residues are buried, and the total buried interfacial surface area is 1515 Å2; for the NMR structure of [Ub]2–11,48Ub, 34.7% of residues are buried, resulting in a buried interfacial surface area of 1539 Å2 (Fig. S4). Notably, the buried interfacial residues correspond to the hydrophobic patch of Ub in both cases. Not only do these data emphasize the similarities between the interfaces, they suggest that the interface in [Ub]2–11,48Ub is at least as tight as that of Ub–48Ub. Thus, we speculated that the interface in [Ub]2–11,48Ub may exhibit similar recognition properties to those of the Ub–48Ub interface; moreover, it may constitute a novel binding surface, thereby contributing to the distinctive functionality of branched K11/K48-linked polyUb.
We considered three ways (Scheme S1) in which the unique hydrophobic interface of branched K11/K48-linked polyUb could enhance degradation efficiency via the UPS: (1) deubiquitinating enzyme (DUB) activity could be affected, either while ubiquitinated substrates are being transported to the proteasome, or once they have reached the proteasome; (2) binding to proteasomal shuttles could be augmented, allowing substrates tagged with branched K11/K48-linked chains to be recognized and directed to the proteasome more efficiently; and/or (3) direct binding to the proteasome could be enhanced.
The first scenario was tested using five DUBs (Castaneda et al., 2016b). Two of the DUBs are linkage-specific: OTUB1 exclusively cleaves K48-linkages, while Cezanne preferentially cleaves K11-linkages. The other three DUBs act non-specifically: USP2, IsoT/USP5, and proteasome-associated UBP6. DUB assays were performed with [Ub]2–11,48Ub, Ub–11Ub, and Ub–48Ub, which enabled us to determine – both qualitatively (Fig. 4A–E) and quantitatively (Fig. 4F; Table S2) – if the addition of K11-linkages to K48-linked chains, or vice versa, affects DUB activity.
Figure 4: Disassembly of Ub–11Ub, Ub–48Ub, and [Ub]2–11,48Ub by deubiquitinases.

SDS-PAGE gels showing the results of treating Ub–11Ub (top), Ub–48Ub (middle), and [Ub]2–11,48Ub (bottom) at indicated timepoints with (A) OTUB1; (B) IsoT/USP5; (C) Cezanne; (D) USP2; and (E) UBP6. (F) Band intensities of initial Ub–11Ub (blue), Ub–48Ub (red), and [Ub]2–11,48Ub (purple) remaining after each timepoint were quantified, and disassembly rate constants were calculated by simultaneously fitting the intensities to respective differential equations (Equations 1–4). See also Scheme S2 and Table S2.
The cleavage rate for OTUB1 was similar between Ub–48Ub and [Ub]2–11,48Ub (Fig. 4A), indicating that the extra K11-linked Ub in [Ub]2–11,48Ub does not significantly affect OTUB1 activity. A similar observation was made for UBP6 (Fig. 4E) and IsoT/USP5 (Fig. 4B), where the cleavage rate of [Ub]2–11,48Ub mirrored that of Ub–48Ub. Therefore, we concluded that the K48-linkage dictates cleavage rate for OTUB1, UBP6, and IsoT/USP5, while the extra K11-linkage in [Ub]2–11,48Ub does not impact DUB activity.
Unexpectedly, Cezanne cleaved [Ub]2–11,48Ub remarkably faster than Ub–11Ub (Fig. 4C), indicating that the presence of a K48-linkage is influential for Cezanne activity. This further suggests that the K48-linkage, not the K11-linkage, primarily dictates DUB activity for [Ub]2–11,48Ub. This may be explained by the observation that Cezanne engages three Ubs simultaneously, thereby facilitating more efficient destruction of the isopeptide linkage in [Ub]2–11,48Ub (Mevissen et al., 2013). Note that although Cezanne preferentially recognizes K11-linkages, it weakly cleaves K48-linkages as well (Bremm et al., 2010; Mevissen et al., 2013).
For USP2, the cleavage rate was the slowest for Ub–48Ub, followed by Ub–11Ub and then [Ub]2–11,48Ub (Fig. 4D). The enhanced activity in [Ub]2–11,48Ub appears to primarily be an additive effect, and the ratio of relative rates for cleaving K11- and K48-linkages in the trimer is similar to that of respective dimers. Thus, for four of the five DUBs (OTUB1, UBP6, IsoT/USP5, and Cezanne), activity was predominately controlled by K48-linkages and not affected by the presence of an additional K11-linkage, indicating that DUBs are likely not responsible for the degradation enhancement exhibited with branched K11/K48-linked polyUb.
[Ub]2–11,48Ub binds to hHR23A-UBA2 across the K48-linkage and with typical affinity
To test the hypothesis that the unique hydrophobic interface in [Ub]2–11,48Ub might represent a new binding surface, we utilized the UBA2 domain of proteasomal shuttle protein hHR23A. UBA2 binds Ub–48Ub in a ‘sandwich’-like complex by intercalating into the hydrophobic interface between two Ubs (Varadan et al., 2005b). UBA2 binds significantly more tightly to Ub–48Ub than to Ub–11Ub, with Kd values of ~18 μM and ~155 μM, respectively (Castañeda et al., 2013; Varadan et al., 2005b). The structure of Ub–48Ub positions the hydrophobic patches of both Ubs in close proximity, which facilitates their simultaneous binding to UBA2. This structural feature is not readily available in Ub–11Ub, hence weaker UBA2 binding. Since [Ub]2–11,48Ub contains a similar hydrophobic surface to Ub–48Ub, albeit across the distal Ubs, we hypothesized that it may bind to UBA2 with strong affinity.
UBA2 could bind to [Ub]2–11,48Ub across the K48-linkage (high-affinity), across the K11-linkage (low-affinity), or across the novel distal Ub:Ub interface (Fig. 5A). The binding mode was examined by NMR as UBA2 was titrated into Ub(15N)[Ub]–11,48Ub. Small shifts were immediately apparent in a few signals and started to saturate once the molar ratio of both proteins approached one, after which several signals began to shift significantly and did not approach saturation until a six-fold excess of UBA2 (Fig. 5B). This suggests two binding events: one at a site far from the NMR-visible distal K11-linked Ub, and a second directly involving the distal K11-linked Ub.
Figure 5: hHR23A-UBA2 binding to [Ub]2–11,48Ub.

(A) Schematic representing the three possible UBA2 binding modes to [Ub]2–11,48Ub, where UBA2 (orange) could bind across the K48-linkage (left), across the K11-linkage (middle), or across the distal Ub:Ub interface (right). (B) NMR titration curves for select residues in Ub(15N)[Ub]–11,48Ub as a function of UBA2:Ub3 molar ratio. Signals from some residues did not shift until reaching an equimolar ratio (top), while other signals shifted slightly initially, then more substantially after reaching an equimolar ratio (bottom). The curves represent the fit to a binding model assuming two independent nonequivalent binding sites (Varadan et al., 2005b). Residue numbers are shown to the right of titration curves. (C) NMR titration curves for select residues in Ub(15N)[Ub]–11,48Ub for the UBA2:Ub3 molar ratio range from one to six (phase II). The curves represent the fit to a single-site binding model; residue numbers are shown (right). (D) CSPs (Δδ) in Ub(15N)[Ub]–11,48Ub caused by UBA2 binding, where total CSPs at the titration endpoint are grey, while CSPs from a UBA2:Ub3 molar ratio of zero to one (phase I) are red. (E) CSPs corresponding to phase I were scaled to the same average intensity as in phase II. The difference (ΔΔδ) between phase II CSPs and scaled phase I CSPs is plotted as a function of residue number. Negative (red) ΔΔδ values indicate residues with a greater contribution during phase I, while positive (orange) ΔΔδ values correspond to residues that have a greater impact on phase II. (F) Mapping the binding results onto the NMR structure of [Ub]2–11,48Ub (PDB: 6OQ2), where the Ub coloring scheme is as in Fig. 1. Residues with ΔΔδ values more negative than −0.025 ppm or more positive than 0.025 ppm are shown as sticks, indicating their respective presence in either phase I (red sticks) or phase II (orange sticks). (G) Proposed mechanism for UBA2 binding to [Ub]2–11,48Ub, where UBA2 (orange) initially binds across the K48-linkage, thereby breaking the distal Ub:Ub interface and releasing the distal K11-linked Ub from its contact with the distal K48-linked Ub. Addition of excess UBA2 results in weak binding to the single distal K11-linked Ub. See also Figures S5–6.
Thus, analysis was performed assuming two binding events: phase I, corresponding to a ligand-to-protein ratio range from zero to one; and phase II, corresponding to a ligand-to-protein ratio range from one to six. The two phases were first analyzed independently as consecutive single-site binding events using program Kdfit (Varadan et al., 2004). The Kd for phase I could not be determined as signal shifts did not saturate, while the Kd for phase II was 505 ± 67 μM (Fig. 5C). The data were also analyzed simultaneously using a model assuming two independent nonequivalent binding sites (Varadan et al., 2005b), resulting in Kd values of 26 ± 13 μM for the first site and 248 ± 23 μM for the second (Fig. 5B, S5). Thus, the first binding event exhibited a Kd close to the reported value of ~18 μM for UBA2 binding to Ub–48Ub, while the Kd for the second event fell in the range of 300–500 μM reported for UBA2 binding to monoUb (Mueller et al., 2004; Ryu et al., 2003; Varadan et al., 2005b).
Quantifying (Fig. 5D–E) and mapping (Fig. 5F) UBA2-induced CSPs onto the structure of [Ub]2–11,48Ub revealed that the initial pattern of signal shifts involved residues in the interdomain interface, while phase II exhibited a signal shifts profile indicative of UBA2 binding directly to monoUb (Mueller et al., 2004; Varadan et al., 2005b). Therefore, we concluded that UBA2 first bound across the K48-linkage of [Ub]2–11,48Ub in the traditional ‘sandwich’ mode (Varadan et al., 2005b), thereby destroying the interface between the distal Ubs. Once UBA2 and [Ub]2–11,48Ub were present in equimolar ratios, excess UBA2 bound weakly to the distal K11-linked Ub, which effectively behaved as a tethered monoUb moiety (Fig. 5G).
To verify this binding mode structurally, we performed SANS measurements with an equimolar mixture of [Ub]2–11,48Ub and UBA2 in D2O-based buffer, using either protonated or 82%-deuterated UBA2 (Fig. S6A). The striking change in the scattering profile – combined with the well-pronounced P(r) dip and extended second maximum (Fig. S6B) – under contrast matching conditions (when deuterated UBA2 was transparent to neutrons) was consistent with the model where UBA2 intercalates between Ubs, thereby pushing them away from each other (Varadan et al., 2005b). Furthermore, we were able to replicate the observed SANS scattering profile using a model of the [Ub]2–11,48Ub:UBA2 complex in which UBA2 was bound across the two K48-linked Ubs in the same binding mode as in Ub–48Ub (Fig. S6C–D), thus breaking the interface between the distal Ubs.
We therefore concluded that the novel hydrophobic surface in [Ub]2–11,48Ub is not directly relevant in binding to the UBA2 domain of hHR23A. Most importantly, these data suggest that branched K11/K48-linked chains do not enhance proteolytic degradation through augmented binding to shuttling factors.
[Ub]2–11,48Ub binds to Rpn1 with enhanced affinity
We then investigated the relationship between [Ub]2–11,48Ub and the third component of the UPS: proteasomal receptors. Three proteasomal subunits can recognize polyUb: Rpn1 (Chojnacki et al., 2017; Shi et al., 2016), Rpn10 (Deveraux et al., 1994; Elsasser et al., 2004), and Rpn13 (Husnjak et al., 2008). Rpn10 and Rpn13 contain a single Ub-binding entity (UIM and Pru, respectively), while Rpn1 is the largest subunit and contains multiple Ub-binding sites. We postulated that if branched K11/K48-linked polyUb were to directly interact with the proteasome at higher affinity, it would likely be via Rpn1, since its multiple Ub-binding sites could accommodate a more complex binding model. This hypothesis was indirectly supported by the recent finding that S5a, the mammalian form of Rpn10 containing two UIMs, does not have greater affinity for branched K11/K48-linked polyUb compared to K48-linked polyUb (Liang et al., 2018). To test our hypothesis, we utilized an Rpn1 construct encompassing residues 391–642 (Rpn1391–642), which contains at least two Ub binding sites spread out over three α-helices (Shi et al., 2016).
Binding was first examined via NMR titration experiments between Rpn1391–642 and three controls: 15N-monoUb, Ub(15N)–48Ub, and Ub(15N)–11Ub. In each case, the addition of Rpn1391–642 caused significant perturbations in NMR spectra of the 15N-labeled Ub. CSPs and strong signal attenuations showed nearly identical profiles among all three species (Fig. 6A–C), suggesting that they employ a similar mode of binding to Rpn1391–642. We found that Rpn1391–642 binds to 15N-monoUb with a Kd of 219 ± 60 μM; Ub(15N)–48Ub with a Kd of 123 ± 34 μM; and Ub(15N)–11Ub with a Kd of 28 ± 6 μM. The Kd values for monoUb and Ub–48Ub agree well with published values of 214 ± 68 μM and 116 ± 30 μM, respectively (Chojnacki et al., 2017). Surprisingly, Rpn1391–642 exhibited roughly four-fold stronger affinity for Ub–11Ub than for Ub–48Ub, even though K48-linked polyUb is considered the canonical degradation signal.
Figure 6: [Ub]2–11,48Ub binds Rpn1391–642 with enhanced affinity.

NMR titration data for Rpn1391–642 binding to (A) 15N-monoUb; (B) Ub(15N)–48Ub; (C) Ub(15N)–11Ub; (D) Ub(15N)–48Ub–48Ub; (E) Ub(15N)–11Ub–48Ub; and (F) Ub(15N)[Ub]–11,48Ub. The left column shows residue-specific CSPs (Δδ, black bars) in the respective proteins at the endpoint of titration with Rpn1391–642; residues exhibiting complete signal attenuation are indicated with light grey bars. The middle column shows titration curves as a function of total Rpn1391–642 concentration; the curves represent data fit to a single-site binding model. Residue numbers are shown to the right of titration curves. Insets show a polyUb schematic with the 15N-enriched moiety colored grey. In the right column, residues affected by Rpn1391–642 binding are mapped onto Ub structure as follows: complete signal attenuations are colored red; CSPs greater than one standard deviation above the average are orange.
We next investigated binding between Rpn1391–642 and Ub(15N)[Ub]–11,48Ub. Significant signal shifts were immediately seen upon addition of Rpn1391–642, and substantial signal attenuations were apparent, an indicator of strong binding resulting in intermediate or slow exchange on the NMR timescale (Fig. 6F). Spectral perturbations were evident in and around the hydrophobic patch residues, as with monoUb, Ub–11Ub, and Ub–48Ub. Surprisingly, however, strong signal attenuations were also observed in the α-helix (residues 23–34) and I36, suggesting that the binding mode of Rpn1391–642 with [Ub]2–11,48Ub differs from those of the respective dimers. The Kd value for this interaction was 8 ± 4 μM, fifteen times and four times tighter than Rpn1391–642 binding to Ub–48Ub and Ub–11Ub, respectively. These data suggest a synergistic relationship involving K11- and K48-linkages: combining both linkages together yields higher affinity than either linkage individually.
To confirm that the increase in binding affinity was not simply a result of adding a third Ub to the chain, binding between Rpn1391–642 and Ub(15N)–48Ub–48Ub was investigated. As above, the addition of Rpn1391–642 caused significant spectral perturbations indicative of binding to the hydrophobic patch region (Fig. 6D). There were some perturbations in the α-helix, but not to the extent as for [Ub]2–11,48Ub. The Kd of Rpn1391–642 binding to Ub(15N)–48Ub–48Ub was 89 ± 10 μM, roughly eleven times weaker than that of Ub(15N)[Ub]–11,48Ub.
A final comparison was performed with the unbranched mixed-linkage trimer, Ub(15N)–11Ub–48Ub. Significant spectral perturbations were observed in the hydrophobic patch region and in some residues of the α-helix (Fig. 6E). The Kd was 24 ± 5 μM, comparable to that for Rpn1391–642 binding to Ub–11Ub. This binding affinity is three times weaker than that of Ub(15N)[Ub]–11,48Ub, even though both Ub3 species contain the same number of Ubs connected through the same linkages; the only difference between them is the unbranched versus branched linkage architecture.
We conclude from these data that incorporation of a K11-linked Ub into Ub–48Ub dramatically enhances binding to Rpn1391–642, whereas elongation of Ub–48Ub by the addition of another K48-linked Ub does not substantially affect binding affinity. Furthermore, the addition of a branched K11-linkage increases binding affinity by three times compared to an unbranched addition. Therefore, it is evident that the integration of a branched K11-linkage into Ub3 is the primary factor responsible for imparting enhanced affinity for Rpn1.
Discussion
Characterization of [Ub]2–11,48Ub by NMR, X-ray crystallography, and SANS revealed a unique interface between the distal Ubs that has not been observed in other mixed-linkage polyUb species, and site-directed mutagenesis provided further evidence that the interface involves hydrophobic patch residues. Ensemble analysis corroborated the presence of the ‘closed’ conformation featuring this interface, but also suggested structural heterogeneity that includes ‘open’ (interface-lacking) conformers. Although these methods showed minor discrepancies regarding the exact domain positioning and orientation for the closed structure, they uniformly indicated the prevalence of a novel hydrophobic interface in [Ub]2–11,48Ub. Notably, the interface was consistently observed across a broad range (0–200 mM salt, pH 6.5–7.6) of typical physiological conditions.
The presence of multiple [Ub]2–11,48Ub conformations was not evident in our 1H-15N NMR spectra, where only one signal corresponding to each backbone amide was detected. This indicates that conformational interconversion occurs on the fast-exchange (sub-millisecond) timescale, whereby signal positions reflect a weighted average of multiple states. The open conformers do not contain any direct contact between distal Ubs; thus, they do not contribute directly to the observed CSPs. Similarly, the minimum distance between the distal Ubs in the open conformer is ~30 Å, outside the ~25 Å range for PRE detection with MTSL. Therefore, the observed CSPs and PREs primarily correspond to the closed conformation of [Ub]2–11,48Ub, indicating that our NMR structure is a valid model of this conformer.
The dynamic nature of polyUb chains, reflecting the flexibility of Ub-Ub linkages and relatively weak noncovalent Ub:Ub interactions, is well-documented (Castaneda et al., 2016a; Castaneda et al., 2016b; Castañeda et al., 2013; Datta et al., 2009; Ryabov and Fushman, 2006; Varadan et al., 2002). Given the multiple reported structural snapshots of Ub–11Ub and Ub–48Ub, it is not unexpected for [Ub]2–11,48Ub to exhibit conformational heterogeneity. We retrospectively modeled the structure of [Ub]2–11,48Ub by superimposing the known structures of the respective dimers (Fig. S7). Aligning the interface-containing structure of Ub–48Ub (PDB: 1AAR) with one crystal structure of Ub–11Ub (PDB: 2XEW) by their proximal Ubs produced an extended structure analogous to the open conformer of [Ub]2–11,48Ub (Fig. S7F). Using a different Ub–11Ub crystal structure (PDB: 3NOB) generated a structure resembling the shape of the closed conformer of [Ub]2–11,48Ub (Fig. S7G), where the distal Ubs are in close contact with each other, albeit with drastically different interfacial residues. These modeled structures generally agree with our analysis of the SANS data, suggesting the existence of compact and extended conformations of [Ub]2–11,48Ub (Fig. 3D–E). Meanwhile, modeling with the NMR structures of Ub–11Ub (PDB: 2MBO and 2MBQ) generated ‘intermediate’ conformers of [Ub]2–11,48Ub (Fig. S7H).
These structural models of [Ub]2–11,48Ub reflect transitions of the distal K11-linked Ub between multiple positions. It is known that the distal K48-linked Ub can also adopt different positions and orientations (Ryabov and Fushman, 2006). As seen in the multi-conformer ensembles of [Ub]2–11,48Ub (Fig. 3D–E), the distal K11-linked Ub and the proximal Ub compete with each other for interface formation with the distal K48-linked Ub. The distal Ubs contact each other in the closed conformation, while the interface across the K48-linkage is nearly restored in the open state. Thus, it is natural to expect that the distal K48-linked Ub may alternate between forming an interface with the distal K11-linked Ub and the proximal Ub. Clearly, the structural ensemble of [Ub]2–11,48Ub is more complex than just the few discrete conformers discussed here; however, they illustrate the existence of both compact and extended states.
The branched triUb presents an example of a system where an integrated structural biology approach is needed because any single technique has difficulties in explaining the composite of the data. Crystallography is biased towards states that pack well to form crystals, and NMR provides population-averaged information over relatively short distances. These techniques thus characterize a subset of the full conformational ensemble. It is therefore not surprising that the presence of open [Ub]2–11,48Ub conformers was only detected by SANS, a low-resolution technique that senses molecular shape. Our data demonstrate that ubiquitin chains have a larger conformational space than previously thought based on data from short-range methods, which may explain why polyUb chains typically exhibit relatively low binding affinities.
DUB assays demonstrated that adding a branched K11-linkage to a K48-linked chain had minimal effect on cleavage rates for four of the five DUBs investigated. These results are consistent with previous observations (Mevissen et al., 2016; Meyer and Rape, 2014). The one exception to this trend was USP2, which exhibited an additive rate increase for [Ub]2–11,48Ub, although the in vivo relevance of this observation is unclear. Most significantly, proteasome-associated UBP6, which facilitates cleavage of substrate-attached polyUb, exhibited no difference in cleavage rate between [Ub]2–11,48Ub and Ub–48Ub. It is important to emphasize that our experiments were only performed on a trimer, the minimal branched signal; chains found in cells are likely more extended and complex, which could conceivably affect their disassembly by DUBs.
For reasons not yet fully understood, certain polyUb signals interact with UBL-UBA shuttle proteins before reaching the proteasome. The UBA domain binds to polyUb conjugated to a substrate, while the UBL domain is recognized by the proteasome. We speculated that branched K11/K48-linked polyUb could support a stronger, novel binding mechanism with the UBA domain of shuttles, thereby enabling more efficient delivery of substrates to the proteasome. However, we found that binding of hHR23A-UBA2 to K48-linked polyUb is essentially unaffected by the presence of an additional K11-linkage or the unique interface in [Ub]2–11,48Ub.
We did not examine [Ub]2–11,48Ub binding to the UBA domain of the other proteasomal shuttle, Dsk2, because this UBA does not exhibit linkage specificity (Raasi et al., 2005; Zhang et al., 2008). Recent studies (Meyer and Rape, 2014; Yau et al., 2017) identified p97/VCP segregase as a prominent effector of branched K11/K48-linked polyUb. p97 is involved in recruitment of ubiquitinated conjugates from large cellular complexes, so it is possible that [Ub]2–11,48Ub is also implicated in this process.
Our binding studies with proteasomal subunit Rpn1 revealed significantly enhanced binding affinity for [Ub]2–11,48Ub compared to related chains, thus identifying Rpn1 as the mechanistic location of enhanced proteolytic activity. Experiments with Ub2 species provided a rationale for this observation: unexpectedly, Rpn1391–642 exhibited substantially stronger binding affinity for Ub–11Ub compared with Ub–48Ub, even though K48-linked Ub chains are considered the primary degradation signal. In line with this observation, Rpn1391–642 binds [Ub]2–11,48Ub more than ten times stronger than Ub–48Ub–48Ub.
Ub recognition by Rpn1391–642 is modulated by three α-helices, which simultaneously bind multiple Ubs (Shi et al., 2016). We propose two possible reasons for enhanced [Ub]2–11,48Ub affinity to Rpn1: the hydrophobic interface between distal Ubs in [Ub]2–11,48Ub may facilitate tighter interactions with the α-helices in Rpn1; and/or the structure of [Ub]2–11,48Ub may enable more favorable positioning of the Ubs for Rpn1 association. The affinity of [Ub]2–11,48Ub for Rpn1 is three times stronger than that of Ub–11Ub–48Ub, suggesting that the unique distal Ub:Ub interface may be involved in Rpn1 recognition. Furthermore, Ub–48Ub, which contains a hydrophobic interface, has a weaker affinity for Rpn1 than does Ub–11Ub, which has no defined interface. Significant [Ub]2–11,48Ub spectral perturbations in the α-helix of the distal K11-linked Ub are indicative of Rpn1 binding through a non-canonical surface, a feature that is not present with relevant dimers, Ub–48Ub–48Ub, or even Ub–11Ub–48Ub. This is suggestive of an altered recognition mode for [Ub]2–11,48Ub, perhaps involving α-helices from both Ub and Rpn1. It cannot be distinguished from the current data whether the unique interface of [Ub]2–11,48Ub is directly involved in enhanced recognition of Rpn1, or if the branched arrangement allows multiple Ubs to optimally interact with Rpn1.
In line with the second possibility, physiological branched chains likely include longer stretches of homogenously linked Ubs, rather than a ‘minimal’ branched trimer moiety. This architecture may allow polyUb chains to simultaneously interact with multiple proteasomal receptors – either inside Rpn1 or across proteasomal subunits (Zhang et al., 2009) – which could synergistically increase proteasomal affinity for branched conjugates beyond what we observed for the trimer. Subsequent structural studies are required to further elucidate this binding mechanism.
Irrespective of a detailed binding picture, [Ub]2–11,48Ub clearly exhibits a novel interface-containing structure and enhanced affinity for Rpn1. Surprisingly, Rpn1 preferentially recognizes K11-linkages over K48-linkages, even though K48-linked polyUb is considered the quintessential degradation signal. Thus, spiking K48-linked polyUb with K11-linkages results in enhanced proteasomal recognition; this is especially true for branched K11-linkages, indicating that branching is a critical mechanistic feature. These observations signify the expanding relevance of mixed-linkage polyUb and suggest additional signaling diversity. This does not diminish the role of homogenous polyUb chains, nor does it imply that linkage connectivity is unimportant; rather, the ubiquitin code may be more elaborate than previously thought.
Table 1:
Statistics of [Ub]2–11,48Ub X-ray diffraction and structure refinement.
| Data Collection | Refinement | ||
|---|---|---|---|
| Space group | P 1 21 1 | Resolution (Å) | 57.69 – 2.20 (2.26 – 2.20) |
| No. of reflections | 20984 | ||
| Rwork / Rfree | 0.213 / 0.230 | ||
| α, β, γ (°) | 90.00, 90.21, 90.00 | ||
| Resolution (Å) | 57.69 – 2.20 (2.27 – 2.20) | ||
| Rmerge | 0.180 | Water | 33 |
| I/σI | 5.3 | ||
| Completeness (%) | 100.0 (99.9) | ||
| Redundancy | 4.3 (4.4) | Water | 35.33 |
| CC1/2 | 0.987 (0.265) | ||
| Wavelength (Å) | 0.97921 | ||
| Bond angles (°) | 1.652 | ||
Statistics for [Ub]2–11,48Ub (PDB: 6OQ1) X-ray diffraction data collection and structure refinement; values in parentheses are for the highest-resolution shell. See also Figure S2.
STAR METHODS
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: David Fushman (fushman@umd.edu).
Experimental Model and Subject Details
All Ub monomers (Varadan et al., 2004; Varadan et al., 2005b; Varadan et al., 2002), hHR23A-UBA2 (Varadan et al., 2004), and Rpn1391–642 (Chojnacki et al., 2017) were expressed in E. coli BL21(DE3) pJY2 or Codon Plus competent cells and purified as described previously.
Method Details
Certain commercial equipment, instruments, materials, suppliers, or software are identified in this paper to foster understanding. Such identification does not imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the materials or equipment identified are necessarily the best available for the purpose.
In addition to the structures of [Ub]2–11,48Ub presented in this work, previously deposited structures were also used for interpretation and discussion of results. These structures are listed as follows: crystal structure of Ub–48Ub (PDB: 1AAR) (Cook et al., 1992); crystal structure of Ub–11Ub (PDB: 2XEW) (Bremm et al., 2010); crystal structure of Ub–11Ub (PDB: 3NOB) (Matsumoto et al., 2010); NMR structure of Ub–11Ub (PDB: 2MBO) (Castañeda et al., 2013); NMR structure of Ub–11Ub (PDB: 2MBQ) (Castañeda et al., 2013); crystal structure of Ub (PDB: 1UBQ) (Vijay-Kumar et al., 1987); NMR structure of Ub (PDB: 1D3Z) (Cornilescu et al., 1998); NMR structure of Ub–48Ub:UBA2 complex (PDB: 1ZO6) (Varadan et al., 2005b).
PolyUb chain assembly
PolyUb chains were generated by using controlled-length enzymatic assembly (Pickart and Raasi, 2005; Varadan et al., 2005a) combined with domain-specific isotopic enrichment (Varadan et al., 2002). The linkage-specific conjugating enzymes Ube2S (Bremm et al., 2010) and E2–25K (Ube2K) (Varadan et al., 2002) were used to form K11- and K48-linkages, respectively. Specific Ub mutants controlled the length and linkage architecture of [Ub]2–11,48Ub: K11R/K48R/K63R Ub (distal) and K63R/D77 Ub (proximal). K11R, K48R, and D77 prevented chain elongation, while K63R blocked Ube2S from forming a small amount of K63-linkages (Bremm et al., 2010). Ub–11Ub and Ub–48Ub were assembled using the same Ub variants as for [Ub]2–11,48Ub. Assembly reactions were carried out overnight at 37°C with equimolar amounts of respective Ub species, catalytic amounts of E1 and E2 enzymes, and 2 mM ATP. Reaction products were separated by cation chromatography using 50 mM ammonium acetate, pH 4.5 as the equilibration buffer and 50 mM ammonium acetate, 1 M sodium chloride, pH 4.5 as the elution buffer. Mass spectrometry was used to verify each product.
X-ray crystallography data collection
The initial [Ub]2–11,48Ub concentration was 5.1 mg/mL in 50 mM HEPES, pH 7.4. Screening of conditions was performed in sitting drop vapor diffusion 96-well plates at 22°C. Crystallization was observed after one week in 100 mM Bis-Tris, 200 mM NaCl, 25% polyethylene glycol (PEG) 3350, pH 6.5. Crystals were briefly soaked in 25% PEG 3350 and submerged in liquid nitrogen prior to data collection. Diffraction data were collected at the APS on beam line NE-CAT 24-ID-E with a wavelength of 0.97921 Å and rotation angle of 0.8°.
X-ray crystallography data analysis
Diffraction images were indexed and integrated with iMOSFLM (Battye et al., 2011) and scaled with AIMLESS (Evans and Murshudov, 2013). Molecular replacement with the crystal structure of Ub (PDB: 1UBQ) as the search model was carried out in Phaser (McCoy et al., 2007). Six copies of Ub were present in the asymmetric unit, corresponding to two trimers. Refinement was performed with Coot (Emsley et al., 2010), CCP4i (Winn et al., 2011), and Phenix (Adams et al., 2011). Electron density was not visible for residues 72–77 in the proximal Ub. Although analysis with the assigned space group P 1 21 1 produced a β-angle disconcertingly close to 90°, analysis with other space groups yielded poor agreement with the data, suggesting that our space group assignment is indeed correct. The final structure was validated by MolProbity (Chen et al., 2010) and the PDB Validation Service (Read et al., 2011). Complete collection and refinement statistics are shown in Table 1.
Torsion Angle Monte Carlo ensemble modeling
Structure ensembles of [Ub]2–11,48Ub were generated with the TAMC module (Zhang et al., 2017) in the SASSIE software suite (Curtis et al., 2012). Monte Carlo simulations were performed for the isopeptide bond regions using Markov chain Monte Carlo sampling of protein backbone torsion angles. Energetics of the torsion angles were determined using CHARMM force field parameters. Flexible regions were defined as the amino acids comprising the isopeptide bonds (residues 72–76 of the distal Ubs; residues K11 and K48 of the proximal Ub) and the C-terminal residues 72–77 of the proximal Ub. The maximum torsion angle step was initially set to 30°. Initial TAMC runs started wit h the crystal structure of [Ub]2–11,48Ub and generated 11569 accepted structures; i.e., those with no overlap of the heavy (non-hydrogen) atoms. Hydrogens and the missing C-terminal residues 72–77 of the proximal Ub were added, and the structures were energy-minimized and equilibrated. The structures were compared with PRE data; the best-matching structure was used as the starting structure for additional runs with a smaller maximum torsion angle step of 10° (18390 structures generated) and also with fewer trial steps (5) before returning to the previous structure (19527 structures generated) to provide fine sampling of the conformational space. Theoretical SANS curves were calculated using the SasCalc module in SASSIE.
TAMC structure ensembles of [Ub]2–11,48Ub in complex with UBA2 were generated using a similar approach as above. The starting structure was created by superimposing (by the proximal Ub) the structure of Ub–48Ub:UBA2 complex (PDB: 1ZO6) with the crystal structure of Ub–11Ub (PDB: 2XEW). The missing flexible N-terminal residues of the UBA2 construct were added, and the structure was energy-minimized and equilibrated before the production run. The final ensemble contained 23560 structures.
HADDOCK structural modeling
The HADDOCK2.2 webserver (van Zundert et al., 2016) was used to generate a structural model for [Ub]2–11,48Ub based on solution NMR data. Starting structures were taken from the atomic coordinates of each Ub in the crystal structure of [Ub]2–11,48Ub. Unambiguous restraints were determined from PRE-calculated distances: only residues with an I/I0 value less than 0.8 were included. CSPs were used to generate ambiguous interdomain restraints (AIR). Active residues were determined as residues with greater than 40% solvent accessibility and a CSP value larger than the average for that Ub. Passive residues were defined as those within 6.5 Å of an active residue. In total, four sets of experimental constraints were used: ambiguous constraints based on CSPs in (1) Ub(15N)[Ub]–11,48Ub and (2) Ub[Ub(15N)]–11,48Ub; unambiguous long-distance constraints determined from PREs in (3) Ub(15N)[UbK48C~MTSL]–11,48Ub and (4) UbK48C~MTSL[Ub(15N)]–11,48Ub. Additional unambiguous restraints preserved the isopeptide linkages between K11 and K48 of the proximal Ub and G76 of respective distal Ubs (Fushman and Walker, 2010; van Dijk et al., 2005). Residues 72–76 of each Ub, as well as residues 11 and 48 in the proximal Ub, were defined as fully flexible. A summary of all restraints is listed in Tables S3 and S4.
Docking was performed by following standard protocols. Energy minimization generated 5000 rigid-body docking solutions, of which the 400 best solutions according to AIR energy (Dominguez et al., 2003) were selected for semi-flexible refinement. A final refinement was carried out in water. The resulting 400 structures were clustered according to a Fraction of Common Contacts cutoff of 0.6 and a minimum of 4 structures per cluster.
Deubiquitination assays
Deubiquitination assays were performed as described (Castaneda et al., 2016b) using 35 μM of each Ub2 or Ub3 species and the appropriate enzyme concentration in PBS, pH 7.4 at 30°C. Enzyme concentrations were: 3 μM OTUB1, 1 μM GST-Cezanne, 3 μM IsoT/USP5, 4 μM USP2, and 8 μM UBP6. Reaction samples were taken at various timepoints (see Fig. 4) and quenched with SDS-PAGE loading dye, then frozen at −20°C until being resolved on a 15% polyacrylamide gel by SDS-PAGE and visualized by Coomassie staining.
Quantification of deubiquitinase activity
Band intensities from Figure 4 were quantified using ImageJ (Schneider et al., 2012), and numerical fitting was performed using the in-house Matlab program DUBfit (Mansour et al., 2015). Disassembly of [Ub]2–11,48Ub by DUBs proceeds via breakage of respective isopeptide linkages, and therefore involves intermediate Ub2 products. This schematic (Scheme S2) is described by the following differential equations:
| (1) |
where [11,48], [11], [48], and [U] are respective molar concentrations of [Ub]2–11,48Ub, Ub–11Ub, Ub–48Ub, and monoUb; k11(48), k48(11), k11, and k48 are cleavage rate constants corresponding to specific disassembly steps shown in Scheme S2. These equations consider that [Ub]2–11,48Ub can be cleaved into two distinct Ub2 + Ub products. Equation 1 can be re-written in matrix form as:
| (2) |
and the general solution can be written as follows:
| (3) |
where t is time and t=0 corresponds to the initial time point. Since the starting concentrations of monomer and dimers are zero, Equation 3 can be simplified further like this:
| (4) |
which can be fit to experimental intensities to determine respective cleavage rate constants.
NMR studies
NMR experiments were performed at 23°C or 25°C on B ruker Avance III 600 MHz and 800 MHz spectrometers equipped with cryoprobes. NMR samples containing Rpn1391–642 were prepared in 50 mM HEPES, 50 mM KCl, 1 mM TCEP, 0.02% NaN3, and 5–10% D2O at pH 7.6. All other samples were prepared in 20 mM sodium phosphate, 1 mM TCEP, 0.02% NaN3, and 5–10% D2O at pH 6.8. NMR data were processed using TopSpin 3.0 (Bruker) and analyzed with Sparky (Goddard and Kneller).
CSPs (Δδ) for each residue were calculated as follows:
| (5) |
where ΔδH and ΔδN correspond to chemical shifts differences for the 1H and 15N resonances.
Site-directed spin labeling was performed by attaching a nitroxide paramagnetic spin label, (1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl) methanesulfonate (MTSL), via disulfide bond to a cysteine residue introduced at position 48 in the distal K11-linked or distal K48-linked Ub of [Ub]2–11,48Ub. 1H-15N HSQC spectra were recorded with MTSL in the oxidized (paramagnetic) and reduced (diamagnetic) states. MTSL was reduced with ascorbate. It is important to mention here that the NMR spectra of samples reduced with ascorbate agreed well with the spectra that were collected prior to attaching MTSL, thereby verifying that attachment of MTSL did not adversely affect the structure of respective Ub species or their contacts with each other.
PRE effects were quantified by measuring the ratio of signal intensities (I/I0) between the oxidized (I) and reduced (I0) states (Varadan et al., 2005a). The experimentally-determined position of MTSL’s unpaired electron was reconstructed using the in-house Matlab program SLfit (Ryabov and Fushman, 2006). The distance information from PRE experiments was used to generate constraints for modeling the solution structure of [Ub]2–11,48Ub in HADDOCK (van Zundert et al., 2016). These distance constraints were created between HN of the affected residue on one distal Ub and Nε of R48 on the other distal Ub, since our PRE experiments with monoUb showed that Nε of this arginine is positioned at roughly the same location as the unpaired electron of MTSL attached to a cysteine.
Binding studies were conducted with starting concentrations of 15N-labeled protein ranging from 50–150 μM. Stock concentrations of ligand ranged from 500 μM to 2 mM. Ligand was titrated into 15N-labeled protein, and binding was monitored by recording 1H-15N SOFAST-HMQC spectra at each titration point. The dissociation constant, Kd, was calculated by fitting experimental CSP values for various titration points to a single-site binding model:
| (6) |
where Δδmax is the CSP at saturation and [Pt] and [Lt] are molar concentrations of the total amount of protein and ligand, respectively. Kd and Δδmax were treated as global fitting parameters using the in-house Matlab program Kdfit (Varadan et al., 2004).
Titration data for hHR23A-UBA2 were analyzed using a model assuming that [Ub]2–11,48Ub contains two independent nonequivalent UBA2-binding sites with different affinities, and the bound fraction of UBA2 is partitioned between these sites at each titration point according to the respective Kd values. This model has been described in detail previously (Varadan et al., 2005b). The Kd values for both binding sites were treated as fitting parameters, and analysis was performed using the in-house Matlab program Kdfit_2s (Varadan et al., 2005b).
SANS data collection and analysis
Scattering measurements were performed on the CHRNS NGB 30m SANS instrument (Glinka et al., 1998) at the NIST Center for Neutron Research. Samples were prepared at a total protein concentration of 4.0 mg/mL in D2O buffer containing 20 mM sodium phosphate, 3 mM TCEP at pD 6.8. The neutron wavelength (λ) was 6 Å, with a wavelength spread (Δλ/λ) of 0.15. Scattered neutrons were detected with a 64 × 64 cm two-dimensional position-sensitive detector with 128 × 128 pixels at a resolution of 0.5 cm/pixel. A sample-to-detector distance of 1.5 m was used to cover the range 0.03 Å−1 ≤ q ≤ 0.4 Å−1; q = 4πsinθ/λ and the scattering angle is 2θ.
SANS data were reduced using the IGOR program and routines developed at the NIST Center for Neutron Research (Kline, 2006) to produce scattering intensity, I(q). Guinier analysis was carried out using the equation I(q)/I(0) ≈ exp(−q2Rg2/3) to estimate the radius of gyration (Rg) and the forward scattering intensity, I(0), of the samples. The atom pair distribution, P(r), was calculated using GNOM (Semenyuk and Svergun, 1991).
The comparison of experimental SANS profile, I(q)exp, with prediction, I(q)pred, for each tested ensemble of N conformers (N = 1, 2, 3) was performed by minimizing the target function:
| (7) |
where σ(q) is the experimental error in I(q)exp,
| (8) |
is a weighted sum of the theoretical (calculated using SasCalc) SANS profiles Ik(q)calc for each conformer k, and wk is the corresponding weight of the conformer (treated as adjustable parameter). For the analysis, the conformers were taken from the TAMC-generated ensemble of structures and included, when specified, the NMR or crystal structure.
Quantification and Statistical Analysis
Information on statistical analysis and corresponding analysis software is described in relevant sections of the methods and figure legends.
Data and Code Availability
Coordinates for the crystal structure (PDB: 6OQ1) and the NMR structure (PDB: 6OQ2) of [Ub]2–11,48Ub have been deposited with the PDB. Chemical shifts for [Ub]2–11,48Ub have been deposited with the BMRB (BMRB: 30602). Matlab software (SLfit, Kdfit, Kdfit_2s, DUBfit) and relevant data are available upon reasonable request.
Supplementary Material
Highlights (up to 85 characters each, including spaces).
Branched K11/K48-triUb possesses a unique hydrophobic interface between distal Ubs
Branched K11/K48-triUb also exists in extended, interface-lacking conformations
Branching via K11 and K48 does not affect deubiquitination or shuttle recognition
Branched K11/K48-triUb binds proteasomal receptor Rpn1 with enhanced affinity
Acknowledgements
We thank Christina M. Camara for help during ensemble analysis, Betsegaw Lemma for providing Ub–48Ub–48Ub, Paul J. Paukstelis for crystallography advice, and Rajesh K. Singh for help with X-ray diffraction data collection. NMR instrumentation was supported in part by NSF grant DBI-1040158. NE-CAT beamline 24-ID-E was supported by NIH grant GM124165 and equipped with an Eiger detector supported by NIH grant OD021527 at the APS (Argonne National Laboratory) under DOE Contract No. DE-AC02–06CH11357. Access to the NGB 30m SANS instrument was provided by the Center for High Resolution Neutron Scattering (CHRNS), a joint NIST/NSF partnership under Agreement No. DMR-1508249. This work was supported by NIH grant GM065334 to D.F. and benefitted from CCP-SAS software developed through a joint EPSRC (EP/K039121/1) and NSF (CHE-1265821) grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Echols N, Headd JJ, Hung LW, Jain S, Kapral GJ, Grosse Kunstleve RW, et al. (2011). The Phenix software for automated determination of macromolecular structures. Methods 55, 94–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TG, Kontogiannis L, Johnson O, Powell HR, and Leslie AG (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal R, Deveraux Q, Xia G, Rechsteiner M, and Pickart C (1996). Surface hydrophobic residues of multiubiquitin chains essential for proteolytic targeting. Proc Natl Acad Sci U S A 93, 861–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boname JM, Thomas M, Stagg HR, Xu P, Peng J, and Lehner PJ (2010). Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic 11, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremm A, Freund SM, and Komander D (2010). Lys11-linked ubiquitin chains adopt compact conformations and are preferentially hydrolyzed by the deubiquitinase Cezanne. Nat Struct Mol Biol 17, 939–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremm A, and Komander D (2011). Emerging roles for Lys11-linked polyubiquitin in cellular regulation. Trends Biochem Sci 36, 355–363. [DOI] [PubMed] [Google Scholar]
- Castaneda CA, Chaturvedi A, Camara CM, Curtis JE, Krueger S, and Fushman D (2016a). Linkage-specific conformational ensembles of non-canonical polyubiquitin chains. Phys Chem Chem Phys 18, 5771–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda CA, Dixon EK, Walker O, Chaturvedi A, Nakasone MA, Curtis JE, Reed MR, Krueger S, Cropp TA, and Fushman D (2016b). Linkage via K27 Bestows Ubiquitin Chains with Unique Properties among Polyubiquitins. Structure 24, 423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda CA, Kashyap TR, Nakasone MA, Krueger S, and Fushman D (2013). Unique structural, dynamical, and functional properties of k11-linked polyubiquitin chains. Structure 21, 1168–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chojnacki M, Mansour W, Hameed DS, Singh RK, El Oualid F, Rosenzweig R, Nakasone MA, Yu Z, Glaser F, Kay LE, et al. (2017). Polyubiquitin-Photoactivatable Crosslinking Reagents for Mapping Ubiquitin Interactome Identify Rpn1 as a Proteasome Ubiquitin-Associating Subunit. Cell Chem Biol 24, 443–457.e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook WJ, Jeffrey LC, Carson M, Chen Z, and Pickart CM (1992). Structure of a diubiquitin conjugate and a model for interaction with ubiquitin conjugating enzyme (E2). J Biol Chem 267, 16467–16471. [DOI] [PubMed] [Google Scholar]
- Cornilescu G, Marquardt J, Ottiger M, and Bax A (1998). Validation of Protein Structure from Anisotropic Carbonyl Chemical Shifts in a Dilute Liquid Crystalline Phase. J. Am. Chem. Soc 120, 6836–6837. [Google Scholar]
- Curtis JE, Raghunandan S, Nanda H, and Krueger S (2012). SASSIE: A program to study intrinsically disordered biological molecules and macromolecular ensembles using experimental scattering restraints. Computer Physics Communications 183, 382–389. [Google Scholar]
- Datta AB, Hura GL, and Wolberger C (2009). The structure and conformation of Lys63-linked tetraubiquitin. J Mol Biol 392, 1117–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveraux Q, Ustrell V, Pickart C, and Rechsteiner M (1994). A 26 S protease subunit that binds ubiquitin conjugates. J Biol Chem 269, 7059–7061. [PubMed] [Google Scholar]
- Dominguez C, Boelens R, and Bonvin AM (2003). HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J Am Chem Soc 125, 1731–1737. [DOI] [PubMed] [Google Scholar]
- Eddins MJ, Varadan R, Fushman D, Pickart CM, and Wolberger C (2007). Crystal Structure and Solution NMR Studies of Lys48-linked Tetraubiquitin at Neutral pH. J Mol Biol 367, 204–211. [DOI] [PubMed] [Google Scholar]
- Elsasser S, Chandler-Militello D, Muller B, Hanna J, and Finley D (2004). Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J Biol Chem 279, 26817–26822. [DOI] [PubMed] [Google Scholar]
- Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, and Cohen P (2013). Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A 110, 15247–15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraczkiewicz R, and Braun W (1998). Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. Journal of Computational Chemistry 19, 319–333. [Google Scholar]
- Fushman D, and Walker O (2010). Exploring the linkage dependence of polyubiquitin conformations using molecular modeling. J Mol Biol 395, 803–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fushman D, and Wilkinson KD (2011). Structure and recognition of polyubiquitin chains of different lengths and linkage. F1000 Biol Rep 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glinka CJ, Barker JG, Hammouda B, Krueger S, Moyer JJ, and Orts WJ (1998). The 30 m Small-Angle Neutron Scattering Instruments at the National Institute of Standards and Technology. Journal of Applied Crystallography 31, 430–445. [Google Scholar]
- Goddard TD, and Kneller DG SPARKY 3. University of California, San Francisco. [Google Scholar]
- Haglund K, and Dikic I (2005). Ubiquitylation and cell signaling. EMBO J 24, 3353–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, and Ciechanover A (1998). The ubiquitin system. Annu Rev Biochem 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Lee S, and Prag G (2006). Ubiquitin-binding domains. Biochem J 399, 361–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K, Elsasser S, Zhang N, Chen X, Randles L, Shi Y, Hofmann K, Walters KJ, Finley D, and Dikic I (2008). Proteasome subunit Rpn13 is a novel ubiquitin receptor. Nature 453, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline SR (2006). Reduction and analysis of SANS and USANS data using IGOR Pro. Journal of Applied Crystallography 39, 895–900. [Google Scholar]
- Komander D, and Rape M (2012). The ubiquitin code. Annu Rev Biochem 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Liang L-J, Si Y, Tang S, Huang D, Wang ZA, Tian C, and Zheng J-S (2018). Biochemical properties of K11,48-branched ubiquitin chains. Chinese Chemical Letters. [Google Scholar]
- Liu C, Liu W, Ye Y, and Li W (2017). Ufd2p synthesizes branched ubiquitin chains to promote the degradation of substrates modified with atypical chains. Nat Commun 8, 14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour W, Nakasone MA, von Delbruck M, Yu Z, Krutauz D, Reis N, Kleifeld O, Sommer T, Fushman D, and Glickman MH (2015). Disassembly of Lys11 and mixed linkage polyubiquitin conjugates provides insights into function of proteasomal deubiquitinases Rpn11 and Ubp6. J Biol Chem 290, 4688–4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D, Phu L, Kirkpatrick DS, Hymowitz SG, Rape M, et al. (2010). K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell 39, 477–484. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, et al. (2013). OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TET, Kulathu Y, Mulder MPC, Geurink PP, Maslen SL, Gersch M, Elliott PR, Burke JE, van Tol BDM, Akutsu M, et al. (2016). Molecular basis of Lys11-polyubiquitin specificity in the deubiquitinase Cezanne. Nature 538, 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HJ, and Rape M (2014). Enhanced protein degradation by branched ubiquitin chains. Cell 157, 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller TD, Kamionka M, and Feigon J (2004). Specificity of the interaction between ubiquitin-associated domains and ubiquitin. J Biol Chem 279, 11926–11936. [DOI] [PubMed] [Google Scholar]
- Nakasone MA, Livnat-Levanon N, Glickman MH, Cohen RE, and Fushman D (2013). Mixed-linkage ubiquitin chains send mixed messages. Structure 21, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtake F, Saeki Y, Ishido S, Kanno J, and Tanaka K (2016). The K48-K63 Branched Ubiquitin Chain Regulates NF-κB Signaling. Mol Cell 64, 251–266. [DOI] [PubMed] [Google Scholar]
- Ohtake F, Tsuchiya H, Saeki Y, and Tanaka K (2018). K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, Schwartz D, Elias JE, Thoreen CC, Cheng D, Marsischky G, Roelofs J, Finley D, and Gygi SP (2003). A proteomics approach to understanding protein ubiquitination. Nat Biotechnol 21, 921–926. [DOI] [PubMed] [Google Scholar]
- Pickart CM, and Fushman D (2004). Polyubiquitin chains: polymeric protein signals. Curr Opin Chem Biol 8, 610–616. [DOI] [PubMed] [Google Scholar]
- Pickart CM, and Raasi S (2005). Controlled synthesis of polyubiquitin chains. Methods Enzymol 399, 21–36. [DOI] [PubMed] [Google Scholar]
- Raasi S, Varadan R, Fushman D, and Pickart CM (2005). Diverse polyubiquitin interaction properties of ubiquitin-associated domains. Nat Struct Mol Biol 12, 708–714. [DOI] [PubMed] [Google Scholar]
- Read RJ, Adams PD, Arendall WB, Brunger AT, Emsley P, Joosten RP, Kleywegt GJ, Krissinel EB, Lütteke T, Otwinowski Z, et al. (2011). A new generation of crystallographic validation tools for the protein data bank. Structure 19, 1395–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryabov Y, and Fushman D (2006). Interdomain Mobility in Di-Ubiquitin Revealed by NMR. Proteins 63, 787–796. [DOI] [PubMed] [Google Scholar]
- Ryu KS, Lee KJ, Bae SH, Kim BK, Kim KA, and Choi BS (2003). Binding surface mapping of intra- and interdomain interactions among hHR23B, ubiquitin, and polyubiquitin binding site 2 of S5a. J Biol Chem 278, 36621–36627. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenyuk AV, and Svergun DI (1991). GNOM - a program package for small-angle scattering data processing. J Appl Cryst 24, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Chen X, Elsasser S, Stocks BB, Tian G, Lee BH, Zhang N, de Poot SA, Tuebing F, Sun S, et al. (2016). Rpn1 provides adjacent receptor sites for substrate binding and deubiquitination by the proteasome. Science 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk AD, Fushman D, and Bonvin AM (2005). Various strategies of using residual dipolar couplings in NMR-driven protein docking: application to Lys48-linked di-ubiquitin and validation against 15N-relaxation data. Proteins 60, 367–381. [DOI] [PubMed] [Google Scholar]
- van Zundert GCP, Rodrigues JPGL, Trellet M, Schmitz C, Kastritis PL, Karaca E, Melquiond ASJ, van Dijk M, de Vries SJ, and Bonvin AMJJ (2016). The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J Mol Biol 428, 720–725. [DOI] [PubMed] [Google Scholar]
- Varadan R, Assfalg M, and Fushman D (2005a). Using NMR spectroscopy to monitor ubiquitin chain conformation and interactions with ubiquitin-binding domains. Methods Enzymol 399, 177–192. [DOI] [PubMed] [Google Scholar]
- Varadan R, Assfalg M, Haririnia A, Raasi S, Pickart C, and Fushman D (2004). Solution conformation of Lys63-linked di-ubiquitin chain provides clues to functional diversity of polyubiquitin signaling. J Biol Chem 279, 7055–7063. [DOI] [PubMed] [Google Scholar]
- Varadan R, Assfalg M, Raasi S, Pickart C, and Fushman D (2005b). Structural determinants for selective recognition of a Lys48-linked polyubiquitin chain by a UBA domain. Mol Cell 18, 687–698. [DOI] [PubMed] [Google Scholar]
- Varadan R, Walker O, Pickart C, and Fushman D (2002). Structural properties of polyubiquitin chains in solution. J Mol Biol 324, 637–647. [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar S, Bugg CE, and Cook WJ (1987). Structure of ubiquitin refined at 1.8 A resolution. J Mol Biol 194, 531–544. [DOI] [PubMed] [Google Scholar]
- Walinda E, Morimoto D, Sugase K, Konuma T, Tochio H, and Shirakawa M (2014). Solution structure of the ubiquitin-associated (UBA) domain of human autophagy receptor NBR1 and its interaction with ubiquitin and polyubiquitin. J Biol Chem 289, 13890–13902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, and Maldonado MA (2006). The ubiquitin-proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol 3, 255–261. [PubMed] [Google Scholar]
- Wickliffe KE, Williamson A, Meyer HJ, Kelly A, and Rape M (2011). K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol 21, 656–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, and Peng J (2009). Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau R, and Rape M (2016). The increasing complexity of the ubiquitin code. Nat Cell Biol 18, 579–586. [DOI] [PubMed] [Google Scholar]
- Yau RG, Doerner K, Castellanos ER, Haakonsen DL, Werner A, Wang N, Yang XW, Martinez-Martin N, Matsumoto ML, Dixit VM, and Rape M (2017). Assembly and Function of Heterotypic Ubiquitin Chains in Cell-Cycle and Protein Quality Control. Cell 171, 918–933.e920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Raasi S, and Fushman D (2008). Affinity makes the difference: nonselective interaction of the UBA domain of Ubiquilin-1 with monomeric ubiquitin and polyubiquitin chains. J Mol Biol 377, 162–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Wang Q, Ehlinger A, Randles L, Lary JW, Kang Y, Haririnia A, Storaska AJ, Cole JL, Fushman D, and Walters KJ (2009). Structure of the s5a:k48-linked diubiquitin complex and its interactions with rpn13. Mol Cell 35, 280–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Howell SC, Wright DW, Heindel A, Qiu X, Chen J, and Curtis JE (2017). Combined Monte Carlo/torsion-angle molecular dynamics for ensemble modeling of proteins, nucleic acids and carbohydrates. J Mol Graph Model 73, 179–190. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates for the crystal structure (PDB: 6OQ1) and the NMR structure (PDB: 6OQ2) of [Ub]2–11,48Ub have been deposited with the PDB. Chemical shifts for [Ub]2–11,48Ub have been deposited with the BMRB (BMRB: 30602). Matlab software (SLfit, Kdfit, Kdfit_2s, DUBfit) and relevant data are available upon reasonable request.