

Abstract
Site-selective bioconjugation to native protein residues is a powerful tool for protein functionalization, with cysteine and lysine side chains being the most common points for attachment owing to their high nucleophilicity. We now report a strategy for histidine modification using thiophosphorodichloridate reagents that mimic post-translational histidine phosphorylation, enabling fast and selective labeling of protein histidines under mild conditions where various payloads can be introduced via copper-assisted alkyne–azide cycloaddition (CuAAC) chemistry. We establish that these reagents are particularly effective at covalent modification of His-tags, which are common motifs to facilitate protein purification, as illustrated by selective attachment of polyarginine cargoes to enhance the uptake of proteins into living cells. This work provides a starting point for probing and enhancing protein function using histidine-directed chemistry.
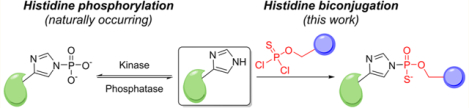
Graphical Abstract
INTRODUCTION
Site-selective bioconjugation chemistry offers a versatile strategy to probe and expand the function of proteins.1–5 The most common and robust chemoselective and regiose-lective protein bioconjugation strategies have focused on functionalization of cysteine thiol6–9 and lysine amine10–12 sites and related nucleophilic hydroxyl13,14 and carboxyl15 side chains. More recent advances in protein bioconjugation technologies have targeted less nucleophlic amino acids,16 including tyrosine,17–19 tryptophan,20,21 and methionine.2,23 In contrast, selective modification of histidine, which is commonly found in enzyme active sites and metal-binding sites,24,25 remains underexplored. Because the imidazole side chain of histidine is a good metal ligand, metal coordination can enable protein modification through metal-directed covalent labeling proximal to the histidine group26,27 or direct noncovalent metal-histidine complexation,28–32 the latter of which can be labile under biological contexts or mass spectrometry conditions. However, histidine is a useful catalytic component owing to its ability to serve as both a good nucleophile and leaving group, but this character also makes it difficult to form stable bonds with the imidazole side chain through electrophilic functionalization. Indeed, selected nucleophiles including epoxides are histidine-reactive but typically require harsh reaction conditions such as high temperatures and/or strong bases,33,34 as well as sequence-dependent motifs35,36 and/or an affinity-directed ligand.37–40 As such, selective and direct covalent labeling of histidine remains a challenge.
RESULTS AND DISCUSSION
Design and Synthesis.
Inspired by observations of reversible histidine phosphorylation as an emerging post-translational modification in prokaryotes and eukaryotes and elegant studies to probe its biological functions,41–45 we sought to develop a histidine-selective bioconjugation method that mimics this type of chemistry (Figure 1). We turned our attention to phosphorus-based electrophiles, based on precedent that potassium phosphoramidate can selectively phosphorylate histidine46,47 and that thiophosphoryl chloride and potassium thiophosphoramidate can generate thiophosphohistidine analogs with improved aqueous stability.48–50 To this end, we synthesized a series of phosphorus electrophiles with varying reactivity as suggested by their different synthesis and handling methods (Scheme 1). We first tested thiophosphoramidate 1 combining a phosphosulfide moiety for histidine labeling and an alkyne group for further functionalization, but this compound did not show an observable reaction with Fmoc-His-OH in buffered aqueous solution (Scheme 2). We then tested compound 2, where we hypothesized that installation of chloride as a better leaving group might enhance reactivity. Indeed, thiophosphochloride 2 did show appreciable labeling of Fmoc-His-OH, but the resulting product underwent significant hydrolysis back to the unmodified histidine over prolonged incubation at neutral pH (Scheme 2), presumably due to the high acidity of the thiophosphoric acid diester as a leaving group.51 To prevent this observed hydrolysis, we further replaced the methoxy group with another chloride that eventually hydrolyzes to the corresponding hydroxy congener to introduce a negative charge, akin to stable phosphate diester linkers found in nucleotides. The resulting compound 4, termed thiophosphoro alkyne dichloridate (TPAC), gives higher conversion to product that is resistant to hydrolysis (Scheme 2, Table S1 of the Supporting Information, SI). The thiophosphorus electrophiles 2 and TPAC are superior histidine labeling reagents compared to their oxygen counterparts 3 and 5, presumably owing to the greater stabilization provided by the less electronegative sulfur. Alternatively, phosphorus electrophiles with nitrogen (6) or sulfur (7) as the bridging atom between phosphorus an alkyne show reduced reactivity toward histidine compared to TPAC. The thiophosphodichloridate can be functionalized with other clickable handles such as azide 8 and sterically hindered alkyne 9 that show comparable reactivity. Further characterization using TPAC as a representative thiophosphodichloridate reagents shows optimal histidine labeling at more basic pH values, affording a 60% yield of bioconjugate within 20 min at pH 8.5 (Figure 2a). The short observed reaction time for TPAC-mediated histidine labeling is primarily controlled by the fast hydrolysis of TPAC itself (Figure S1), which is advantageous since the reaction does not require quenching of residual TPAC to halt its reactivity and the reagent can thus be used in excess. Moreover, the thiophospho-histidine product shows reasonable stability under various conditions including high temperature, acidic, basic, reducing, and alkylating environments (Table S1). The conjugate also shows robust stability in serum at 37 °C, whereas in cell lysate at 37 °C the modification undergoes efficient hydrolysis (Figure S2). Interestingly, the adduct is stable in cell lysate at 4 °C, suggesting that cleavage at higher temperatures is possibly due to some catalyzed activity (Figure S2). Indeed, such behavior can be potentially useful for traceless delivery of proteins or drugs into cells. Most importantly, TPAC exhibits high selectivity for histidine, showing negligible reactivity on other nucleophilic amino acids (Figures 2b and S3). Reaction on model peptides also supports its histidine selectivity, where peptide with nucleophilic amino acids residues other than histidine shows insignificant reaction, and conversion yield increases with increasing number of histidine residues on peptide (Figure S4). The results collectively identify TPAC as a promising candidate for chemoselective bioconjugation to histidine.
Figure 1.

Histidine labeling and the following click reaction for bioconjugation. X, Y = Cl, OH, OMe or NH2, Z = O, NH, or S.
Scheme 1.

Different Routes for Synthesis of Phosphorus Electrophiles
Scheme 2.

Reaction Yields of Phosphorus Electrophiles on Fmoc-His-OH.a
aConditions: 0.5 mM Fmoc-His-OH, 5 mM electrophile, 25 mM HEPES pH 8.5, 20% MeCN, 1h; yields were determined by HPLC. Q = S or O, Y = Cl, OH, OMe or NH2, Z = O, NH, or S. bReactant readily degenerates and forms precipitate.
Figure 2.

TPAC labeling on small molecules. (a) Kinetics of reaction between TPAC and Fmoc-His-OH. Triangle: pH 8.5. Circle: pH 7.5. (n = 3, average ± s.d.). (b) HPLC chromatograph showing the reaction between TPAC and nucleophilic Fmoc-amino acid mixtures. Bottom: before TPAC treatment. Top: after TPAC treatment. Left to right: Fmoc-Lys, Fmoc-Arg, Fmoc-His, Fmoc-Ser, Fmoc-Glu, Fmoc-Thr, and Fmoc-Tyr. Arrows point to Fmoc-His-OH (left) and TPAC-labeled Fmoc-His-OH (right, ESI-MS m/z expected 512.1, found 512.2 for [M + H]+).
TPAC Bioconjugation on Model Proteins.
With these results in hand, we moved on to test TPAC labeling of histidine on intact protein substrates (Figure 3a). We used ribonuclease A as a model protein and analyzed bioconjugation reactions by mass spectrometry. The TPAC labeling is dose-dependent, generating ca. 45% singly modified protein and 11% doubly modified protein at pH 8.5 (Figure 3b). Similar to what was observed in small-molecule amino acid models, the TPAC-ribonuclease A coupling reaction is more effective at slightly basic conditions compared to neutral pH (Figure 3c). We further performed LC-MS/MS of the digested protein to analyze the site of modification. Similar to other phosphopeptides, the TPAC modification undergoes significant neutral loss in collision-induce dissociation (CID), but the fragment peaks are sufficient for identifying the site of modification (Figures S5a and S6b,c). We also used electron-transfer dissociation (ETD) on peptides with ambiguous modification sites for supplementary information (Figure S6a). Interestingly, the LC-MS/MS of the digested protein indicates that the reaction occurs primarily on H48, a surface-exposed histidine rather than histidines at the catalytic center (H12 and H119). In addition, two other histidine sites, one at the active site (H119) and one that is not (H105), are also modified to lesser extent, as shown qualitatively by extracted precursor ion chromatogram (Figure S5a). The data are also in line with the negligible loss of activity of ribonuclease A after TPAC treatment (Figure S5b), and site-directed mutagenesis also support that H48 is the major modified residue on this protein (Figure S5c).
Figure 3.

Model protein tagging with TPAC. (a) Structure and mass difference of the histidine modification by TPAC. (b) Deconvoluted mass spectra showing ribonuclease A labeled by TPAC with concentration ranging from 0 to 2 mM. Single and double TPAC modified RNaseA: expected mass 13 817, 13 951 Da, found 13 816, 13 950 Da. Its crystal structure (PDB 1bel) is shown in the inset highlighting its major modified histidine. (c) Deconvoluted mass spectra showing ribonuclease A labeled with 1.5 mM of TPAC with different buffered pH. (d) Calmodulin, (e) myoglobin and (f) lysozyme are also labeled by TPAC as shown by their deconvoluted mass spectra. TPAC-modified calmodulin: expected mass 16 924 Da, found 16 924 Da; single, double and triple TPAC modified myoglobin: expected mass 17 085, 17 219, and 17 353 Da, found 17 085, 17 218, and 17 352 Da; single and double TPAC modified lysozyme: expected mass 14 439, 14 573 Da, found 14438, 14 572 Da. Their crystal structures (PDB 2o60, 1bje, and 193l) are shown as insets highlighting the major modified histidines. Conditions: 20 μM protein, 2 mM TPAC in 25 mM HEPES with pH 8.5, room temperature, 1 h unless otherwise noted.
We then demonstrated that the TPAC bioconjugation method is amenable to labeling histidine residues on other proteins, including calmodulin, myoglobin and lysozyme. The yields vary for these proteins (Figure 3d–f), and the reaction shows excellent histidine selectivity on calmodulin and myoglobin with only small amount of side reaction on lysine and threonine (Figures S7 and S8). Modest side-reactivity toward lysine and tyrosine is observed on lysozyme, but lysine labeling can be suppressed by lowering the pH to 7.5 to further block the nucleophilicity of the lysine residues by protonation, while increasing TPAC concentrations to compensate the decreased histidine reactivity (Figure S9). TPAC labeling in HeLa lysates also proceeds smoothly as shown by in-gel fluorescence (Figure S10a), and more importantly, pretreatments of lysates with competing electrophiles that block cysteine, lysine or serine residues do not strongly affect the observed TPAC signal, suggesting that the reagent does not appreciably react with these competing amino acid residues (Figure S10b–e). Taken together, the results show that TPAC is effective at selectively labeling native histidine residues on proteins.
We then sought to apply this histidine bioconjugation method to install clickable payloads onto proteins. Considering that TPAC converts the slightly basic histidine residue into a rather acidic thiophosphoric acid derivative, we reasoned that the labeled protein can be separated readily from the unreacted protein by its charge difference. Indeed, the separation of the reaction mixture of ribonuclease A and TPAC is effective on a strong cation exchange column buffered at pH 4.2, giving rise to three fractions (Figure 4a). The mass spectra of these fractions confirm our hypothesis: unreacted ribonuclease A carries more positive charge and elutes last, the doubly TPAC-modified ribonuclease A protein is less positively charged and elutes first, and the singly TPAC-modified protein elutes between these two fractions (Figure 4b). After the facile isolation of TPAC-modified reaction products, we further performed copper(I)-catalyzed alkyne–azide cycloaddition (CuAAC) between monolabeled ribonuclease A and either Cy3-N3 or desthiobiotin-N3 payloads for detection and enrichment, respectively (Figure S11). As expected, the obtained protein is uniform with only one payload per protein molecule (Figure 4b). Collectively, TPAC labeling with subsequent ion exchange chromatography and CuAAC chemistry provides a easy and straightforward workflow to create homogeneous proteins modified on histidine residues.
Figure 4.

Protein bioconjugation with TPAC. (a) Chromatogram at 280 nm showing the separation of the reaction mixture between ribonuclease A and TPAC by strong cation exchange column. Three fractions, F1–3, were collected for analysis. (b) Deconvoluted mass spectra of ribonuclease A in F1–3, and TPAC-modified ribonuclease A in F2 reacted with the model azide compounds by CuAAC. Found masses are labeled next to the peak. Expected masses for non-, single-, and double-labeled ribonuclease A are 13 683, 13 817, and 13 951 Da, respectively. Expected masses for single TPAC/desthiobiotin-N3 and TPAC/Cy3-N3 labeled ribonuclease A are 14 231 and 14 623 Da, respectively.
TPAC Bioconjugation on His-Tag for Protein Delivery.
To showcase the potential merits of TPAC bioconjugation chemistry at the cellular level, we turned our attention to modification of polyhistidine-tagged proteins (i.e., His-tag). His-tag is a widely used method for purification of proteins by introducing a short polyhistidine peptide fused to the surface exposed portion of the protein of interest for subsequent resin capture and separation. We envisioned that this polyhistidine motif would greatly enhance the labeling efficiency of TPAC in this region, and we demonstrated this possibility by combining TPAC labeling with further bioconjugation by CuAAC with polyarginine, a cell-penetrating peptide, to create a general method for enabling protein delivery into living cells (Figure 5a). We utilized GFP bearing an N-terminal 10 × His-tag as a proof-of-concept protein, where TPAC-labeling greatly reduces binding and recognition of His-tag GFP by a His-tag antibody, consistent with efficient labeling of TPAC in this His-tag region (Figure 5b). We further performed the cleavage of the His-tag from the protein by enterokinase treatment to verify the labeling sites. Indeed, TPAC labeling followed by CuAAC with desthiobiotin-N3 on His-tag GFP produces a significantly, stronger signal compared to GFP without the His-tag on a streptavidin blot (Figure 5c), further indicating that on this protein TPAC selectively reacts with histidine residues on the His-tag rather than other 10 histidine residues with 5 sites on the surface. Similarly, LC-MS analysis of the digested protein after TPAC treatment only identifies one to three modifications, all of which appear on the His-tag region (Figure S12). With this information in hand we performed CuAAC between TPAC-labeled His-tag GFP and N3–(Arg)9–OH to introduce cell-penetrating capabilities onto the protein. Indeed, the (Arg)9-functionalized GFP is now capable of being delivered into HeLa cells as shown by increases in intracellular green fluorescence as observed by confocal microscopy, whereas His-tagged GFP without TPAC or N3–(Arg)9–OH treatment results in low fluorescence signal over dark background (Figure 4d). Similar results are also observed with an mCherry construct carrying an N-terminus 6 × His-tag, although on this protein histidine residues other than the His-tag are also labeled by TPAC to a smaller extent (Figure S13). For both cases the internalization of the modified proteins were confirmed by Z-stack images of the cells (Figure S15). Taken together, this line of experiments shows that TPAC can provide covalent, histidine-selective bioconjugation for protein functionalization.
Figure 5.

Functionalization of His-tag on GFP to enable protein delivery. (a) Scheme of bioconjugation of polyarginine onto His-tag of a fluorescent protein using TPAC. (b) Streptavidin-HRP blot and coomassie stain showing that removal of the His-tag significantly reduces the labeling by TPAC. His-tagged GFP and native GFP prepared by enterokinase cleavage of His-tag were treated with TPAC, followed by CuAAC with desthiobiotin-N3 and gel-analysis. Cleavage of His-tag leads to less coomassie staining but with similar migration on SDS-PAGE (Figure S14a). (c) Western blot and coomassie stain showing that labeling of TPAC with His-tagged GFP significantly reduces its detection by His-tag antibody. (d) Transduction of functionalized GFP into live HeLa cells. Cells were incubated with GFP (0.1 μg/μL), stained with Hoechst 33 342 and imaged; quantification is shown on the bottom (n = 3, average ± s.d. ***P ≤ 0.001; two-tailed Student’s t test.). Scale-bars: 20 μm.
CONCLUSIONS
To close, we have reported thiophosphorodichloridates inspired by native histidine phosphorylation processes as reagents for histidine-selective covalent modification of proteins. With TPAC, an alkyne-tagged version of this family of probes, we demonstrated efficient bioconjugation on model proteins with excellent selectivity for histidine over other potentially reactive amino acids. TPAC-labeled small model proteins are readily purified and can undergo further coupling with bioorthogonal click chemistry to introduce a variety of payloads. In one example to illustrate the utility of this approach, TPAC labeling was applied to introduce polyarginine motifs onto His-tag proteins to endow membrane permeability and enable delivery into living cells. Owing to the central importance of histidine residues in enzyme chemistry, this work provides a starting point for probing histidine function in native contexts. Likewise, the efficient labeling of polyhistidine tags can enable versatile protein functionalization for biochemical and protein engineering studies in both fundamental and applied settings. Coupled with recent work from our laboratory on the development of selective methionine bioconjugation reagents that operate by redoxmediated nitrogen group transfer processes inspired by native oxygen atom-transfer oxidations that transform methionine to methionine sulfoxide,22 mimicking histidine phosphorylation can provide a starting point for developing histidine-selective bioconjugation chemistry. We are currently expanding and applying the toolbox of activity-based sensing reagents in this direction to develop chemoselective probes for other reactive amino acid sites in proteins and proteomes.
EXPERIMENTAL SECTION
Chemical and Protein Materials.
All commercial reagents were used without further purification. Fmoc-Cys-OH was purchased from Chem-Impex (Wood Dale IL). All other Fmoc-protected amino acids were purchased from Ark Pharm (Arlington Heights IL). 2-(4-((bis((1-(terf-butyl)-1H-1,2,3-triazol-4-yl)methyl)amino)methyl)-1H-1,2,3-triazol-1-yl)acetic acid (BTTAA) was purchased from Click Chemistry Tools (Scottsdale, AZ). Custom peptides for model reactions were purchased from GenScript (Piscataway, NJ). Ribonuclease A from bovine pancreas, lyzome from chicken egg white, myoglobin from equine heart and all other chemicals were purchased from Sigma-Aldrich (St. Louis MO). Calmodulin, porcine was purchased from rPeptide (Watkinsville GA). His-tagged GFP was purchased from Sino Biological (Beijing, China). His-tagged mCherry was purchased from Origene (Rockville MD). Enterokinase was purchased from New England Biolabs (Ipswich MA). TEV protease was purchased from QB3 (San Francisco CA). Enzymes were used following the protocols provided by their manufacturer.
Reaction between Phosphorus Electrophiles and Amino Acids.
Fmoc-protected amino acid was dissolved in DMSO (100 mM) and diluted to a final concentration of 0.5 mM in 25 mM HEPES buffer, pH 8.5 containing 20% MeCN unless otherwise noted. To this solution was added 5 mM of phosphorus electrophile (50× stock in MeCN). The reaction was performed at room temperature for 1 h unless otherwise noted, filtered and immediately subject to LC/MS analysis. For reaction with Fmoc-protected amino acid mixtures, 0.3 mM of each protected amino acid in 50 mM HEPES buffer, pH 8.5 containing 20% MeCN was reacted with 3 mM of TPAC.
Protein Labeling with TPAC.
Proteins were diluted into 25 mM HEPES, pH 8.5 to a concentration of 20 μM unless otherwise noted. Samples were labeled with 2 mM of TPAC (50× stock in MeCN). The reaction was performed at room temperature for 1 h. Samples were then subject to LC-MS analysis or further click reaction.
Purification of TPAC-Labeled Ribonuclease A.
The reaction mixture of RNase A with TPAC was diluted in 20 mM sodium succinate, pH 4.2 and was loaded onto HiTrap SP HP cation exchange chromatography column (1 mL, GE Healthcare, Chicago IL). The protein was then separated with a linear gradient from 20 mM sodium succinate, pH 4.2 to 25 mM succinate with 1 M NaCl, pH 4.2 at a flow rate of 1.0 mL/min over 40 min. The collected fractions were concentrated and buffer-exchanged into desired buffer for click reaction or LC/MS analysis.
Click Reactions on TPAC-Labeled Proteins.
We followed the protocol recommended by Finn et al. for click reactions.52 Namely, protein solution in HEPES or PBS buffer was treated with 1 mM aminoguanidine hydrochloride (100× stock in water), 100 μM CuSO4 (100× stock in water), 500 μM THPTA or BTTAA (100× stock in water), 100 μM organic azide (100× stock in DMSO or water), and 5 mM sodium ascorbate (100× stock in water). For reaction on His-tagged protein 100 μM NiCl2(100× stock in water) was also added. The reaction was mixed thoroughly and placed in the dark at room temperature. After 1 h the reaction was quenched by adding 500 μM EDTA.
For click reaction with Cy3-N3, proteins were precipitated with acetone prior to click reaction to remove excess TPAC. The pellet was dissolved in PBS containing 0.1% SDS for click reaction, and precipitated again to remove unreacted dye. The pellet was then dissolved in running buffer and analyzed by SDS-PAGE.
For click reaction on TPAC-labeled, His-tagged proteins, proteins were buffer-exchanged into 25 mM HEPES, pH 7.5 by extensive ultrafiltration (Amnicon 10K, EMD Millipore, Hayward CA) prior to click reaction. After the click reaction, proteins were buffer-exchanged into PBS by extensive ultrafiltration to remove unwanted chemicals.
Cell Culture and Imaging.
Cells were grown in the Cell Culture Facility at the University of California, Berkeley. HeLa cells were cultured in DMEM supplemented with 10% FBS and glutamine (2 mM). One day before imaging, cells were passed and plated on eight-well chamber slides (Lab-Tek, Thermo Fisher).
For imaging, cells were grown on 8-well chamber slides (LabTek, Thermo Fisher) to desired confluency, washed with PBS and incubated with 0.1 mg/mL (Arg)9-labeled GFP or mCherry in PBS for 30 min at 37 °C. The cells were then washed with PBS and stained with 1 μM Hoechst 33342 for 15 min, washed again and imaged on a Zeiss LSM710 laser-scanning microscope with a 20× objective lens for quantification and a 63× oil-immersion objective lens for images. Excitation was provided at 405 for Hoechst 33 342, 488 nm for GFP and 543 nm for mCherry.
Synthesis of Methyl Propargyl Thiophosphorochloridate (2).
To a flask containing PSCl3 (2 mL, 20 mmol) cooled in ice/water bath was added dry methanol (2.0 mL, 49 mmol) dropwise. The mixture was stirred for 15 min on ice and excess methanol was distilled under vacuum at the same temperature to give crude methyl thiophosphorodichloridate. Sodium (0.453 g, 19.7 mmol) was dissolved in cooled propargyl alcohol (6.0 mL, 104 mmol) to form an orange, thick solution, which was added dropwise to methyl thiophosphorodichloridate cooled in ice/water bath. The suspension was further stirred for 2 h at room temperature, diluted with CH2Cl2, filtered, and purified by column chromatography (30:1 hexanes/EtOAc) to give product 2 as a colorless oil (2.4 g, 67%). 1H NMR (400 MHz, CDCl3) δ 4.85−4.80 (m, 2H), 3.91 (d, J = 16.1 Hz, 3H), 2.63 (t, J = 2.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 77.16 (s), 76.39 (d, J = 10.5 Hz), 56.90 (d, J = 4.4 Hz), 55.98 (d, J = 7.1 Hz). 31P NMR (162 MHz, CDCl3) δ 71.91. HRMS (APCI+) m/z calcd 184.9587, found 184.9586 for C4H7ClO2PS+ (M+H+).
Synthesis of Methyl Propargyl Thiophosphoramidate (1).
Crude 2 in propargyl alcohol and CH2Cl2 was prepared as described above. Excess ammonia was led through this mixture to form NH4O as a precipitate. The mixture was filtered, concentrated, and purified by column chromatography (2:1 hexanes/EtOAc) to give product 1 as a light yellow oil (2.1 g, 65% overall yield). 1H NMR (400 MHz, MeOD) δ 4.62 (dd, J = 10.5, 2.5 Hz, 2H), 3.68 (d, J = 13.8 Hz, 3H), 2.94 (t, J = 2.5 Hz, 1H). 13C NMR (101 MHz, MeOD) δ 79.47 (d, J = 10.9 Hz), 76.35, 55.04 (d, J = 2.8 Hz), 53.72 (d, J = 5.3 Hz). 31P NMR (162 MHz, MeOD) δ 78.25. HRMS (APCI+) m/z calcd 166.0086, found 166.0097 for C4H9NO2PS+ (M+H+).
Synthesis of Propargyl Thiophosphorodichloridate (4, TPAC).
To a solution of PSCl3 (1.0 mL, 9.8 mmol) in CH2Cl2 (10 mL) were added propargyl alcohol (0.57 mL, 9.8 mmol) and K2CO3 (1.36 g, 9.8 mmol). After overnight stirring at room temperature, the mixture was filtered and purified by column chromatography (50:1 hexanes/EtOAc) to give product TPAC as a colorless oil with a pungent smell (1.1 g, 55%). 1H NMR (400 MHz, CDCl3) δ 4.94 (dd, J = 15.9, 2.5 Hz, 2h), 2.72 (t, J = 2.5 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 78.37, 75.48 (d, J = 11.1 Hz), 58.52 (d, J = 7.4 Hz). 31P NMR (162 MHz, CDCl3) δ 59.68. HRMS (APCI+) m/z calcd 188.9092, found 188.9115 for C3H4Cl2OPS+ (M+H+).
Synthesis of Propargyl Phosphorodichloridate (5).
POCl3 (1.0 mL, 11 mmol) and propargyl alcohol (0.62 mL, 11 mmol) were dissolved in Et2O (20 mL) and cooled in dry ice/acetone bath under N2. Triethylamine (1.5 mL, 11 mmol) was dissolved in Et2O (20 mL) and added dropwise via addition funnel to form a white suspension. The reaction mixture was warmed to room temperature and was furthered stirred at room temperature for 2 h. Trimethylamine hydrochloride was removed by filtration, and the solution was concentrated to give product 5 as a light yellow oil with a pungent smell (1.8 g, 99%). The product was used without further purification. 1H NMR (400 MHz, CDCl3) δ 4.92 (dd, J = 14.5, 1.0 Hz, 2H), 2.76 (t, J = 1.0 Hz 1H). 13C NMR (101 MHz, CDCl3) δ 78.93, 74.91 (d, J = 9.6 Hz), 58.53 (d, J = 7.2 Hz). 31P NMR (162 MHz, CDCl3) δ 8.70. LRMS (EI+) m/z calcd 137.0, found 136.9 for C3H3ClO2P+ (M−Cl−).
Synthesis of Methyl Propargyl Phosphorochloridate (3).
Crude 5 with trimethylamine hydrochloride in Et2O was prepared as described above. The mixture was cooled again in dry ice/acetone bath. Dry methanol (0.43 mL, 11 mmol) and trimethylamine (1.5 mL, 11 mmol) in Et2O (20 mL) was added dropwise via addition funnel. The slurry was further stirred at room temperature for 3 h, filtered and concentrated to give product 3 as light yellow oil with a pungent smell (1.7 g, 93%). 1H NMR (400 MHz, CDCl3) δ 4.79 (dd, J = 11.5, 2.5 Hz, 2H), 3.92 (d, J = 13.8 Hz, 3H), 2.66 (t, J = 2.5 Hz, 1H). 13C NMR (101 MHz, CDO3) δ 78.92, 76.04 (d, J = 9.0 Hz), 56.66 (d, J = 5.3 Hz), 55.97 (d, J = 7.0 Hz). 31P NMR (162 MHz, CDCl3) δ 6.74. HRMS (APCI+) m/z calcd 168.9816, found 168.9855 for C4H7ClO3P+ (M+H+).
Synthesis of N-Propargyl Thiophosphoramidic Dichloride (6).
To a solution of PSCl3 (0.30 mL, 2.9 mmol) in CH2Cl2 (5 mL) was added propargyl amine (0.19 mL, 2.9 mmol) and K2CO3 (0.41 g, 2.9 mmol). After 2 h stirring at room temperature, the mixture was filtered and purified by column chromatography (30:1 hexanes/EtOAc) to give product 6 as a yellow oil with a pungent smell (0.25 g, 49%). 1H NMR (400 MHz, Acetone) δ 4.09 (dd, J = 20.7, 2.5 Hz, 2H), 2.89 (t, J = 2.5 Hz, 1H). 13C NMR (101 MHz, Acetone) δ79.90 (d, J = 8.8 Hz), 74.13, 33.42. 31P NMR (162 MHz, Acetone) δ 57.59. LRMS (EI+) m/z calcd 151.9, found 152.0 for C3H4ClNPS+ (M−Cl−).
Synthesis of Propargyl Dithiophosphorodichloridate (7).
S- Propargyl thioacetate (0.35 mg, 3.1 mmol) was stirred with NaOH (0.16 g, 4.0 mmol) in MeOH (10 mL) under N2 for 30 min. The mixture was then diluted with CH2Cl2 (30 mL), washed with H2O (×4) and dried (Na2SO4). To this solution was added PSCl3 (0.37 mL, 3.7 mmol) and K2CO3 (0.51g, 3.7 mmol) and the mixture was stirred overnight. The mixture was filtered and purified by column chromatography (100:1 hexanes/EtOAc) to give product 7 as a light yellow oil, which degenerates into a solid mixture within minutes after concentration. We were able to obtain a crude 1H NMR before degeneration and also confirmed its negligible reactivity in aqueous buffer with histidine when generated in situ. 1H NMR (600 MHz, CDCl3) δ 3.75 (dd, J = 15.6, 2.7 Hz, 2H), 2.35 (t, J = 2.7 Hz, 1H).
Synthesis of 2-Azidoethyl Thiophosphorodichloridate (8).
2-Azidoethanol (0.43 g, 4.9 mmol) was dissolved in dry THF (15 mL) under N2 and cooled in dry ice/acetone bath. To this solution was added dropwise nBuLi (2.0 mL, 2.5 M in hexanes) and stirred for 20 min at room temperature to form the lithium salt. To another flask cooled in dry ice/acetone bath was added THF (15 mL) and PSCl3 (1.0 mL, 4.9 mmol) under N2. The lithium salt solution was then added dropwise at this temperature and the mixture was then stirred for 1 h at room temperature. The mixture was concentrated, diluted in CH2Cl2, filtered to remove lithium chloride and purified by column chromatography (50:1 hexanes/EtOAc) to give product 8 as a light yellow oil with a pungent smell (0.60 g, 56%). 1H NMR (400 MHz, CDCl3) δ 4.45 (dt, J = 11.0, 5.1 Hz, 2H), 3.63 (t, J = 4.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 69.57 (d, J = 9.9 Hz), 50.11 (d, J = 10.6 Hz). 31P NMR (162 MHz, CDCl3) δ 59.05. HRMS (APCI+) m/z calcd 191.9202, found 191.9219 for C2H5Cl2NOPS+ (M−N2+H+).
Synthesis of (±)-3-Butyn-2-yl Thiophosphorodichloridate (9).
(±)-3-Butyn-2-ol (0.77 mL, 9.8 mmol) was dissolved in dry THF (10 mL) under N2 and cooled in dry ice/acetone bath. To this solution was added dropwise nBuLi (3.9 mL, 2.5 M in hexanes) with stirring for 20 min at room temperature to form the lithium salt. To another flask cooled in dry ice/acetone bath were added THF (20 mL) and PSCl3 (1.0 mL, 9.8 mmol) under N2. The lithium salt solution was then added dropwise at this temperature, and the mixture was then stirred for 3 h at room temperature. The mixture was concentrated, diluted in CH2Cl2, filtered to remove lithium chloride and purified by column chromatography (100:1 hexanes/EtOAc) to give product 9 as a colorless oil with a pungent smell (0.53 g, 26%). 1H NMR (400 MHz, CDCl3) δ 5.46 (dqd, J = 13.3, 6.6, 2.1 Hz, 1H), 2.71 (d, J = 2.2 Hz, 1H), 1.71 (dd, J = 6.6, 0.8 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 79.95 (d, J = 6.4 Hz), 76.64, 69.20 (d, J = 8.6 Hz), 23.18 (d, J = 7.6 Hz). 31P NMR (162 MHz, CDCl3) δ 58.98. HRMS (APCI+) m/z calcd 202.9249, found 202.9261 for C4H6Cl2OPS+ (M+H+).
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (ES 028096) and Novartis Institutes for BioMedical Research and the Novartis-Berkeley Center for Proteomics and Chemistry Technologies (NB-CPACT) for supporting this work. C.J.C. is an investigator of the Howard Hughes Medical Institute and a CIFAR Senior Fellow. D.H. is supported by Tang Distinguished Scholarship (UC Berkeley, QB3). The QB3/Chemistry Mass Spectrometry Facility at the University of California, Berkeley received support from National Institutes of Health (grant 1S10OD020062-01). The Catalysis Facility of Lawrence Berkeley National Laboratory is supported by the Office of Science of the US Department of Energy under contract no. DE-AC02-05CH11231.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b11912.
Supplementary experimental materials and analytical methods, supplementary reaction characterization, summary of LC-MS/MS results, analysis of bioconjugation on cell lysate and mCherry protein, and supplementary synthesis and characterization (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Stephanopoulos N; Francis MB Choosing an Effective Protein Bioconjugation Strategy. Nat. Chem. Biol 2011, 7 (12), 876–884. [DOI] [PubMed] [Google Scholar]
- (2).Spicer CD; Davis BG Selective Chemical Protein Modification. Nat. Commun 2014, 5, 4740. [DOI] [PubMed] [Google Scholar]
- (3).McKay CS; Finn MG Click Chemistry in Complex Mixtures: Bioorthogonal Bioconjugation. Chem. Biol 2014, 21 (9), 1075–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Koniev O; Wagner A Developments and Recent Advancements in the Field of Endogenous Amino Acid Selective Bond Forming Reactions for Bioconjugation. Chem. Soc. Rev 2015, 44 (15), 5495–5551. [DOI] [PubMed] [Google Scholar]
- (5).Boutureira O; Bernardes GJL Advances in Chemical Protein Modification. Chem. Rev 2015, 115 (5), 2174–2195. [DOI] [PubMed] [Google Scholar]
- (6).Vinogradova EV; Zhang C; Spokoyny AM; Pentelute BL; Buchwald SL Organometallic Palladium Reagents for Cysteine Bioconjugation. Nature 2015, 526 (7575), 687–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Zhang C; Welborn M; Zhu T; Yang NJ; Santos MS; Van Voorhis T; Pentelute BL π-Clamp-Mediated Cysteine Conjugation. Nat. Chem 2016, 8 (2), 120–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Willwacher J; Raj R; Mohammed S; Davis BG Selective Metal-Site-Guided Arylation of Proteins. J. Am. Chem. Soc 2016, 138 (28), 8678–8681. [DOI] [PubMed] [Google Scholar]
- (9).Messina MS; Stauber JM; Waddington MA; Rheingold AL; Maynard HD; Spokoyny AM Organometallic Gold(III) Reagents for Cysteine Arylation. J. Am. Chem. Soc 2018, 140 (23), 7065–7069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Matos MJ; Oliveira BL; Martfnez-Saez N; Guerreiro A; Cal PMSD; Bertoldo J; Maneiro M; Perkins E; Howard J; Deery MJ; et al. Chemo- and Regioselective Lysine Modification on Native Proteins. J. Am. Chem. Soc 2018, 140 (11), 4004–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Ward CC; Kleinman JI; Nomura DK NHS-Esters As Versatile Reactivity-Based Probes for Mapping Proteome-Wide Ligandable Hotspots. ACS Chem. Biol 2017, 12 (6), 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hacker SM; Backus KM; Lazear MR; Forli S; Correia BE; Cravatt BF Global Profiling of Lysine Reactivity and Ligandability in the Human Proteome. Nat. Chem 2017, 9 (12), 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Liu Y; Patricelli MP; Cravatt BF Activity-Based Protein Profiling: The Serine Hydrolases. Proc. Natl. Acad. Sci. U. S. A 1999, 96 (26), 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kidd D; Liu Y; Cravatt BF Profiling Serine Hydrolase Activities in Complex Proteomes. Biochemistry 2001, 40 (13), 4005–4015. [DOI] [PubMed] [Google Scholar]
- (15).McGrath NA; Andersen KA; Davis AKF; Lomax JE; Raines RT Diazo Compounds for the Bioreversible Esterification of Proteins. Chem. Sci 2015, 6 (1), 752–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).deGruyter JN; Malins LR; Baran PS Residue-Specific Peptide Modification: A Chemist’s Guide. Biochemistry 2017, 56 (30), 3863–3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Schlick TL; Ding Z; Kovacs EW; Francis MB DualSurface Modification of the Tobacco Mosaic Virus. J. Am. Chem. Soc 2005, 127 (11), 3718–3723. [DOI] [PubMed] [Google Scholar]
- (18).Romanini DW; Francis MB Attachment of Peptide Building Blocks to Proteins Through Tyrosine Bioconjugation. Bioconjugate Chem. 2008, 19 (1), 153–157. [DOI] [PubMed] [Google Scholar]
- (19).Ban H; Gavrilyuk J; Barbas CF Tyrosine Bioconjugation through Aqueous Ene-Type Reactions: A Click-Like Reaction for Tyrosine. J. Am. Chem. Soc 2010, 132 (5), 1523–1525. [DOI] [PubMed] [Google Scholar]
- (20).Antos JM; Francis MB Selective Tryptophan Modification with Rhodium Carbenoids in Aqueous Solution. J. Am. Chem. Soc 2004, 126 (33), 10256–10257. [DOI] [PubMed] [Google Scholar]
- (21).Antos JM; McFarland JM; Iavarone AT; Francis MB Chemoselective Tryptophan Labeling with Rhodium Carbenoids at Mild PH. J. Am. Chem. Soc 2009, 131 (17), 6301–6308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Lin S; Yang X; Jia S; Weeks AM; Hornsby M; Lee PS; Nichiporuk RV; Iavarone AT; Wells JA; Toste FD; et al. Redox-Based Reagents for Chemoselective Methionine Bioconjugation. Science 2017, 355 (6325), 597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Taylor MT; Nelson JE; Suero MG; Gaunt MJ A Protein Functionalization Platform Based on Selective Reactions at Methionine Residues. Nature 2018, 562 (7728), 563–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Gutteridge A; Thornton JM Understanding Nature’s Catalytic Toolkit. Trends Biochem. Sci 2005, 30 (11), 622–629. [DOI] [PubMed] [Google Scholar]
- (25).Dokmanić I; šikić M; Tomić S Metals in Proteins: Correlation between the Metal-Ion Type, Coordination Number and the Amino-Acid Residues Involved in the Coordination. Acta Crystallogr., Sect. D: Biol. Crystallogr 2008, 64 (3), 257–263. [DOI] [PubMed] [Google Scholar]
- (26).Ohata J; Minus MB; Abernathy ME; Ball ZT Histidine-Directed Arylation/Alkenylation of Backbone N-H Bonds Mediated by Copper(II). J. Am. Chem. Soc 2016, 138 (24), 7472–7475. [DOI] [PubMed] [Google Scholar]
- (27).Ohata J; Zeng Y; Segatori L; Ball ZT A Naturally Encoded Dipeptide Handle for Bioorthogonal Chan-Lam Coupling. Angew. Chem., Int. Ed 2018, 57 (15), 4015–4019. [DOI] [PubMed] [Google Scholar]
- (28).Waibel R; Alberto R; Willuda J; Finnern R; Schibli R; Stichelberger A; Egli A; Abram U; Mach J-P; Pluckthun A; et al. Stable One-Step Technetium-99m Labeling of His-Tagged Recombinant Proteins with a Novel Tc(I)-Carbonyl Complex. Nat. Biotechnol 1999, 17 (9), 897–901. [DOI] [PubMed] [Google Scholar]
- (29).Kapanidis AN; Ebright YW; Ebright RH Site-Specific Incorporation of Fluorescent Probes into Protein: Hexahistidine-Tag-Mediated Fluorescent Labeling with (Ni2+:Nitrilotriacetic Acid)N-Fluorochrome Conjugates. J. Am. Chem. Soc 2001, 123 (48), 12123–12125. [DOI] [PubMed] [Google Scholar]
- (30).Lata S; Gavutis M; Tampé R; Piehler J Specific and Stable Fluorescence Labeling of Histidine-Tagged Proteins for Dissecting Multi-Protein Complex Formation. J. Am. Chem. Soc 2006, 128 (7), 2365–2372. [DOI] [PubMed] [Google Scholar]
- (31).Wang X; Jia J; Huang Z; Zhou M; Fei H Luminescent Peptide Labeling Based on a Histidine-Binding Iridium(III) Complex for Cell Penetration and Intracellular Targeting Studies. Chem. - Eur. J 2011, 17 (29), 8028–8032. [DOI] [PubMed] [Google Scholar]
- (32).Lai Y-T; Chang Y-Y; Hu L; Yang Y; Chao A; Du Z-Y; Tanner JA; Chye M-L; Qian C; Ng K-M; et al. Rapid Labeling of Intracellular His-Tagged Proteins in Living Cells. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (10), 2948–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li X; Ma H; Dong S; Duan X; Liang S Selective Labeling of Histidine by a Designed Fluorescein-Based Probe. Talanta 2004, 62 (2), 367–371. [DOI] [PubMed] [Google Scholar]
- (34).Li X; Ma H; Nie L; Sun M; Xiong S A Novel Fluorescent Probe for Selective Labeling of Histidine. Anal. Chim. Acta 2004, 515 (2), 255–260. [Google Scholar]
- (35).Cong Y; Pawlisz E; Bryant P; Balan S; Laurine E; Tommasi R; Singh R; Dubey S; Peciak K; Bird M; et al. Site-Specific PEGylation at Histidine Tags. Bioconjugate Chem. 2012, 23 (2), 248–263. [DOI] [PubMed] [Google Scholar]
- (36).Peciak K; Laurine E; Tommasi R; Choi J; Brocchini S Site-Selective Protein Conjugation at Histidine. Chem. Sci 2019, 10 (2), 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Takaoka Y; Tsutsumi H; Kasagi N; Nakata E; Hamachi I One-Pot and Sequential Organic Chemistry on an Enzyme Surface to Tether a Fluorescent Probe at the Proximity of the Active Site with Restoring Enzyme Activity. J. Am. Chem. Soc 2006, 128 (10), 3273–3280. [DOI] [PubMed] [Google Scholar]
- (38).Wakabayashi H; Miyagawa M; Koshi Y; Takaoka Y; Tsukiji S; Hamachi I Affinity-Labeling-Based Introduction of a Reactive Handle for Natural Protein Modification Chem.—Asian J. 2008, 3 (7), 1134–1139. [DOI] [PubMed] [Google Scholar]
- (39).Olszewska A; Pohl R; Brázdová M; Fojta M; Hocek M Chloroacetamide-Linked Nucleotides and DNA for Cross-Linking with Peptides and Proteins. Bioconjugate Chem. 2016, 27 (9), 2089–2094. [DOI] [PubMed] [Google Scholar]
- (40).Adusumalli SR; Rawale DG; Singh U; Tripathi P; Paul R; Kalra N; Mishra RK; Shukla S; Rai V Single-Site Labeling of Native Proteins Enabled by a Chemoselective and Site-Selective Chemical Technology. J. Am. Chem. Soc 2018, 140 (44), 15114–15123. [DOI] [PubMed] [Google Scholar]
- (41).Puttick J; Baker EN; Delbaere LTJ Histidine Phosphorylation in Biological Systems. Biochim. Biophys. Acta, Proteins Proteomics 2008, 1784 (1), 100–105. [DOI] [PubMed] [Google Scholar]
- (42).Besant PG; Attwood PV Detection and Analysis of Protein Histidine Phosphorylation. Mol. Cell. Biochem 2009, 329 (1–2), 93–106. [DOI] [PubMed] [Google Scholar]
- (43).Kee J-M; Muir TW Chasing Phosphohistidine, an Elusive Sibling in the Phosphoamino Acid Family. ACS Chem. Biol 2012, 7 (1), 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wilke KE; Francis S; Carlson EE Activity-Based Probe for Histidine Kinase Signaling. J. Am. Chem. Soc 2012, 134 (22), 9150–9153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Potel CM; Lin M-H; Heck AJR; Lemeer S Widespread Bacterial Protein Histidine Phosphorylation Revealed by Mass Spectrometry-Based Proteomics. Nat. Methods 2018, 15 (3), 187–190. [DOI] [PubMed] [Google Scholar]
- (46).Medzihradszky KF; Phillipps NJ; Senderowicz L; Wang P; Turck CW Synthesis and Characterization of Histidine-Phosphorylated Peptides. Protein Sci. 1997, 6 (7), 1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hohenester UM; Ludwig K; Konig S Chemical Phosphorylation of Histidine Residues in Proteins Using Potassium Phosphoramidate - a Tool for the Analysis of Acid-Labile Phosphorylation. Curr. Drug Delivery 2013, 10 (1), 58–63. [DOI] [PubMed] [Google Scholar]
- (48).Lasker M; Bui CD; Besant PG; Sugawara K; Thai P; Medzihradszky G; Turck CW Protein Histidine Phosphorylation: Increased Stability of Thiophosphohistidine. Protein Sci. 1999, 8 (10), 2177–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pirrung MC; James KD; Rana VS Thiophosphorylation of Histidine. J. Org. Chem 2000, 65 (25), 8448–8453. [DOI] [PubMed] [Google Scholar]
- (50).Ruman T; Dlugopolska K; Jurkiewicz A; Rut D; Fraczyk T; Cieśla J; Leś A; Szewczuk Z; Rode W Thiophosphorylation of Free Amino Acids and Enzyme Protein by Thiophosphoramidate Ions. Bioorg. Chem 2010, 38 (2), 74–80. [DOI] [PubMed] [Google Scholar]
- (51).Westheimer FH Why Nature Chose Phosphates. Science 1987, 235 (4793), 1173–1178. [DOI] [PubMed] [Google Scholar]
- (52).Hong V; Presolski SI; Ma C; Finn MG Analysis and Optimization of Copper-Catalyzed Azide-Alkyne Cycloaddition for Bioconjugation. Angew. Chem., Int. Ed 2009, 48 (52), 9879–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.