

Abstract
β-N-methylamino-l-alanine (BMAA) is a nonproteinogenic amino acid that has been associated with neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) and Alzheimer's disease (AD). BMAA has been found in human protein extracts; however, the mechanism by which it enters the proteome is still unclear. It has been suggested that BMAA is misincorporated at serine codons during protein synthesis, but direct evidence of its cotranslational incorporation is currently lacking. Here, using LC-MS–purified BMAA and several biochemical assays, we sought to determine whether any aminoacyl-tRNA synthetase (aaRS) utilizes BMAA as a substrate for aminoacylation. Despite BMAA's previously predicted misincorporation at serine codons, following a screen for amino acid activation in ATP/PPi exchange assays, we observed that BMAA is not a substrate for human seryl-tRNA synthetase (SerRS). Instead, we observed that BMAA is a substrate for human alanyl-tRNA synthetase (AlaRS) and can form BMAA-tRNAAla by escaping from the intrinsic AlaRS proofreading activity. Furthermore, we found that BMAA inhibits both the cognate amino acid activation and the editing functions of AlaRS. Our results reveal that, in addition to being misincorporated during translation, BMAA may be able to disrupt the integrity of protein synthesis through multiple different mechanisms.
Keywords: aminoacyl tRNA synthetase, protein synthesis, translation, transfer RNA (tRNA), amino acid, β-N-methylamino-l-alanine (BMAA), nonproteinogenic amino acid (NPA), alanyl-tRNA synthetase (AlaRS), amyotrophic lateral sclerosis (ALS), neurodegeneration and cyanotoxin
Introduction
β-N-methylamino-l-alanine (BMAA)2 is a nonproteinogenic amino acid that was first isolated from the seeds of the cycad species Cycas circinalis (1). BMAA acts as a glutamate agonist when bicarbonated under physiological conditions, causing excitotoxicity in neurons (2). The correlation between BMAA and neurodegeneration was initially observed in Guam, where the rate of ALS-Parkinson's (ALS-parkinsonism-dementia complex) symptoms was 50- to 100-fold higher than the average within the continental United States and other developed countries (3). Because the symptoms were geographically restricted, the etiology of disease was considered to be related to the unique lifestyle of the indigenous Chamorro people. It was noticed that the Chamorros regularly consumed tortillas made from cycad seeds; therefore, BMAA was proposed to be the cause of the Guam symptom (4). However, it was later found that the concentrations of BMAA in the tortillas were not sufficient to achieve the observed toxicity (5). The hypothesis of BMAA being the cause of the Guam ALS-parkinsonism-dementia complex cases was not further investigated until 2003, when bioaccumulation of BMAA was identified in the Guam dietary chain by Cox et al. (6). Accumulated BMAA was found not only in the free amino acid pool, but also in the protein-associated fraction of the foods (7). Since then, studies have explored the question of how BMAA enters or associates with the proteome. In 2013, Dunlop et al. (8) revealed that BMAA competes with serine for incorporation and suggested that it enters the proteome during protein synthesis at serine codons. However, this hypothesis is still debated, and the mechanism of BMAA misincorporation remains unclear.
Although the number of ALS-parkinsonism-dementia complex cases has significantly declined in Guam, BMAA is still a widespread problem. Environmental factors such as BMAA accumulation are believed to play important roles in the development of sporadic ALS and AD (9–11). BMAA is synthesized by cyanobacteria and algae, and it has been found in many bodies of water all over the world (12–14). Both ocean and freshwater animals have been shown to contain BMAA, including commonly consumed seafood (15, 16). As BMAA has previously been shown to bioaccumulate through an isolated dietary chain, elevated incidence of eutrophication in domestic and international waters makes BMAA contamination a worldwide health concern. Therefore, there is an urgent need to understand the mechanism of BMAA protein incorporation.
For a substrate to be incorporated during de novo protein synthesis, it must be aminoacylated on a tRNA by an aminoacyl-tRNA synthetase (aaRS). aaRSs are essential enzymes catalyzing the aminoacylation reaction through a two-step process: first, the amino acid is activated to form an aminoacyl-adenylate (AA-AMP); then the AA-AMP is transferred to the 3′ end of the corresponding tRNA. Accurate aminoacylation is required for faithful translation of the encoded genetic information. However, because of the structural similarities of many amino acids, errors in amino acid activation can occur (17). For example, AlaRS misacylates both Ser and Gly onto tRNAAla. Fortunately, these enzymes have evolved a proofreading (editing) function to hydrolyze misactivated amino acids or misaminoacylated tRNAs (18). The loss of proofreading function can be detrimental to cells because of the accumulation of misfolded and malfunctional proteins (19).
In this study, we found that BMAA is a substrate for human AlaRS (HsAlaRS), and is not subject to the AlaRS intrinsic proofreading activity. Furthermore, we showed that BMAA can inhibit both the activation and editing catalytic efficiency of HsAlaRS, revealing new mechanisms by which BMAA can disrupt the integrity of protein synthesis.
Results
BMAA is a substrate of human AlaRS
To identify the aaRS catalyzing BMAA aminoacylation, an initial screen to examine BMAA activation was performed. Because it was previously suggested that SerRS is responsible for aminoacylating BMAA (8), we monitored amino acid activation using ATP/PPi exchange assays with recombinant human SerRS, AlaRS, and prolyl-tRNA synthetase (ProRS), respectively. ATP/PPi exchange is an assay estimating the level of substrate activation by measuring the production of radiolabeled ATP in the presence of excess [32P]PPi in the reaction. AlaRS and ProRS were chosen because they misacylate noncognate amino acids that are either serine or share a structural similarity with serine (Fig. 1A): AlaRS can misacylate Gly and Ser to tRNAAla (20), whereas ProRS misacylates Ala and Cys to tRNAPro (21). Chromatographically purified BMAA was used throughout the study to eliminate any artificial effects because of amino acid contamination. Among the aaRSs examined, only HsAlaRS was able to activate BMAA (Fig. 1B). To further elucidate the kinetics of BMAA misactivation, the steady-state activation kinetics of HsAlaRS for Ala, Ser, and BMAA were determined (Table 1). The results showed that BMAA shares a similar Km to the known AlaRS noncognate amino acid, Ser, but the kcat was 40-fold lower than Ser, revealing that the rate of BMAA-AMP formation was comparatively slow. Because aminoacylation is a two-step process, we further investigated whether BMAA can be transferred to tRNAAla to form BMAA-tRNAAla. BMAA proved to be a substrate for AlaRS aminoacylation, resulting in ∼3.5% of active tRNAAla being charged with BMAA (Fig. 1, C and D). Besides the no amino acid negative control as a background, we also tested phenylalanine as a noncognate amino acid and no aminoacylation was observed (Fig. S2).
Figure 1.

BMAA is a substrate of human AlaRS. A, the chemical structure of Ser, Ala, and BMAA. B, ATP/PPi exchange assays were carried out using three human aaRSs, respectively: ProRS, SerRS, and AlaRS to screen for BMAA activation. C, aminoacylation assays were carried out using [32P]-labeled human tRNAAla and WT HsAlaRS in the presence of 50 mm BMAA and 0.1 mm Ala. A no amino acid control was also conducted to determine the background level. D, relative level of charged tRNA at 30 min was determined by normalizing the results to Ala-tRNAAla after subtracting the background value of the no amino acid control. All of the experiments were done at least three times, and the error bars indicate the standard deviations of the means.
Table 1.
Steady-state activation kinetics of HsAlaRS and C723A
ATP/PPi exchange assays were carried out to determine the activation kinetics. The specificity is defined by the ratio of kcat/Km of cognate over noncognate amino acids. C723A activation kinetics is also compared with the WT AlaRS to examine if differences exist between these two enzymes.
| Km (mm) | kcat (s−1) | kcat/Km (s−1 mm−1) | Specificity | ||
|---|---|---|---|---|---|
| AlaRS | Ala | 0.04 ± 0.01 | 4 ± 0.3 | 100 | 1 |
| Ser | 25 ± 3 | 6 ± 1.6 | 0.2 | 500 | |
| BMAA | 38 ± 16 | 0.15 ± 0.06 | 0.004 | 25,000 | |
| C723A | Ala | 0.06 ± 0.01 | 8 ± 2 | 131 | 0.8 |
| Ser | 31 ± 10 | 6 ± 0.6 | 0.2 | 500 | |
| BMAA | 19 ± 12 | 0.07 ± 0.02 | 0.004 | 25,000 | |
To understand if codon-specific misincorporation of BMAA can be observed in vivo, we used MS-READ, an MS-based misincorporation reporter technique, to investigate BMAA misincorporation at Ala codons in yeast (22). However, no BMAA misincorporation was identified in our reporter (Fig. S3). We also examined if BMAA is excluded from the cyanobacterial translational machinery, as it is a cyanotoxin. We performed the activation screen using AlaRS from Anabaena pcc.7120, a model nitrogen-fixing cyanobacterial species, and found that similar to HsAlaRS, AnaAlaRS is able to activate BMAA (Fig. S4).
BMAA is not subject to AlaRS intrinsic editing
The consequence of AlaRS misaminoacylation is evolutionarily problematic; hence, cells have developed redundant mechanisms to solve this problem (18). Two checkpoints exist to prevent the accumulation of Ser-tRNAAla. One is the intrinsic editing domain in AlaRS, which is able to deacylate Ser-tRNAAla both in cis and in trans (20); another is AlaXP, the genome encoded free-standing AlaRS editing domain homolog that is able to hydrolyze mischarged tRNAAla in trans (23). An editing-deficient AlaRS variant (HsAlaRS C723A) was used to determine the role of proofreading on BMAA activation and aminoacylation. HsAlaRS is highly similar to mouse AlaRS, and the human Cys-723 residue corresponds to the previously characterized mouse Cys-723 (24) and the Escherichia coli Cys-666 (20) residues in the editing domain, both of which are critical for editing (Fig. S1). Our results showed no significant differences in BMAA activation and aminoacylation between the WT AlaRS and the C723A editing-deficient variant (Fig. 2, A and B). This was further supported by the activation kinetics performed with C723A, whose kinetic parameters toward BMAA activation showed no significant differences from the WT enzyme's (Table 1). To exclude any possible artifacts of using the C723A variant, we carried out an ATP-consumption assay using the WT AlaRS enzyme as reported previously (25). Measuring the consumption of ATP is an indirect measure of pre- and post-transfer editing because of ATP futile cycling as a consequence of iterative rounds of substrate activation and product hydrolysis. If a substrate is subject to proofreading, higher levels of ATP consumption should be observed. We tested ATP consumption using Ala, Ser, and BMAA, with or without the presence of tRNAAla to monitor both pre- and post-transfer editing. The futile cycling of ATP was only observed with Ser, indicating that it is subject to both pre- and post-transfer editing as reported previously (26). The level of ATP consumption with BMAA was similar to Ala, supporting the observation with C723A that BMAA is not subject to HsAlaRS proofreading (Fig. 2C).
Figure 2.

BMAA is not subject to AlaRS proofreading. A, pyrophosphate exchange assay using human AlaRS and the AlaRS editing mutant C723A in the presence of 50 mm BMAA. B, BMAA aminoacylation is carried out using 500 nm C723A. C, an ATP consumption assay is used to examine ATP futile cycling with and without the presence of tRNAAla. All of the experiments were done at least three times, and the error bars indicate the standard deviations of the means.
BMAA is a competitive inhibitor of AlaRS activation of Ala
As presented earlier in the activation kinetics data with AlaRS and C723A (Table 1), we noticed that the Km of BMAA activation is similar to Ser, but the rate of product formation is extremely low. This led us to investigate whether BMAA could act as an activation inhibitor of HsAlaRS by sequestering enzyme from its cognate substrate. In general, there are three modes of reversible enzyme inhibition: Competitive, uncompetitive and noncompetitive. The mode of inhibition can be characterized by the change of Km and Vmax in the presence of the inhibitor (27). To establish BMAA's mode of inhibition, the inhibition kinetics were measured to estimate the Km and Vmax of AlaRS cognate amino acid activation in the presence of BMAA. Upon BMAA treatment, the Km of AlaRS activation increased whereas the Vmax was unaffected, suggesting that BMAA is a competitive inhibitor of HsAlaRS (Fig. 3A). Inhibition assays were then carried out using two different concentrations of BMAA and the data were analyzed on a Lineweaver-Burk plot. The results demonstrated that with increasing concentration of the inhibitor, the slopes increased, and the y-intercept remained unchanged, confirming that BMAA is indeed a competitive inhibitor (Fig. 3B). The inhibition constant KI is defined as the concentration required to produce half of the maximum inhibition, and hence is used to represent how potent an inhibitor is. Here, we determined the KI of BMMA in inhibiting amino acid activation by AlaRS to be 25.8 mm using the equation KI = Km [I]/(Km, apparent − Km).
Figure 3.

BMAA is a competitive inhibitor to AlaRS activation. A, steady-state activation kinetics is performed with or without 25 mm BMAA, and the results were used to calculate the kinetics of inhibition. B, aminoacylation assays were carried out with 100 nm human AlaRS in the presence of 12.5 mm (filled circles), 25 mm (filled squares), or no BMAA (filled triangles), and plotted on a Lineweaver-Burk plot to identify the mode of inhibition.
BMAA perturbs the proofreading function of AlaRS
The role of BMAA as an enzyme inhibitor has recently been shown by van Onselen et al. (28), where multiple enzymes were exposed to BMAA and the activity was evaluated via commercially available kits. The authors found that BMAA tends to inhibit enzymes with exposed amino acids sharing terminal hydroxyl groups. The editing domain of AlaRS is composed of three critical residues, Ser-587, Thr-567 and Cys-666 (E. coli numbering) (29). As serine and threonine both have terminal hydroxyl groups, we were curious if the editing domain is also sensitive to BMAA treatment. To explore this hypothesis, we performed deacylation assays using misacylated Ser-tRNAAla and estimated the efficiency of deacylation by HsAlaRS following BMAA treatment. A 2.4-fold increase in the half-life of Ser-tRNAAla following BMAA treatment was observed (Fig. 4 and Table S1), showing that BMAA perturbs the intrinsic AlaRS editing activity.
Figure 4.

BMAA perturbs AlaRS editing. A deacylation assay using [3H]Ser-tRNAAla is carried out with 1 mm HsAlaRS, 1 mm HsAlaRS with 50 mm BMAA, 1 mm C723A, and a no enzyme control. The percentage of misacylated tRNA remaining in the reaction is calculated at each time point. The counts of time point zero was set as 100% for the deacylation analysis. All of the experiments were done at least three times, and the error bars indicate the standard deviations of the means.
Discussion
BMAA misincorporation is facilitated through AlaRS
Mistranslation generates aberrant or misfolded protein and can eventually result in protein aggregation, which is a feature of many neurodegenerative diseases including Alzheimer's and Parkinson's (30). Neurons are notably sensitive to erroneous translation; for example, a minor defect in AlaRS editing can lead to neurodegeneration in mice (19). Thus, many neurodegenerative diseases have been associated with mutations in the protein synthesis machinery (31). We attempted to observe codon-specific misincorporation of BMAA at Ala codons in yeast using MS-READ (22), but failed to identify any BMAA misincorporation at the resolution at which our reporter was capable of detecting (Fig. S3). A previous report was also unable to detect BMAA misincorporation in human cell lines when examining SDS-PAGE-purified proteins (32). Because these experiments were conducted in growth media supplemented with 4 mm and 2 mm BMAA respectively, the frequency of BMAA misincorporation may be too low to be detected, which is consistent with our biochemical assays which only monitored a low fraction (3.5%) of BMAA-tRNAAla formation using 50 mm BMAA. Although we found that BMAA is a rather weak substrate for HsAlaRS aminoacylation, it is able to escape the intrinsic proofreading mechanism of AlaRS. Thus, low level misincorporation may still accumulate throughout the lifespan of the individual and eventually become problematic, as the capability for cells to maintain proteostasis declines over the course of aging (33).
Because it was reported in 2013 that BMAA competes with Ser for cellular incorporation, others have tried to observe Ser codon misincorporation by in vitro translation, but the outcome remains controversial (8). Glover et al. (34) used the PURE system to investigate the efficiency of BMAA misincorporation to replace nine different amino acids by quantifying total protein in the reaction and found that the highest substitution occurred with Ala and Ser. However, a recent work by Beri et al. (35) had failed to identify protein synthesis in the reaction lacking Ser (−Ser + BMAA). These contradicting results have raised doubts on the hypothesis of BMAA misincorporation at Ser codons. We showed here that BMAA is not a substrate for HsSerRS but instead for HsAlaRS. The competition observed previously may be because of other reasons, such as BMAA competing with Ser for aminoacylation by AlaRS (1); TCA precipitated protein was used in the previous study, so the interaction of BMAA and peptides may not be covalent (2); other amino acid contaminants existing in commercial BMAA products may interfere with the experiments (3).
Although considered a cyanotoxin, the biosynthesis and functions of BMAA in cyanobacteria are still unclear. It was reported that BMAA is produced under nitrogen-limiting conditions in Synechocystis, whereas exogenous BMAA inhibits nitrogenase activity and the formation of heterocysts (specialized cells that perform nitrogen fixation) in Anabaena (36–38). Using the AlaRS from Anabaena pcc.7120, we found that AnaAlaRS also activates BMAA (Fig. S4). It is unknown if mistranslation or inhibition of aaRS activity contributes to the failure of heterocyst development.
BMAA perturbs HsAlaRS function through multiple pathways
Nonproteinogenic amino acids (NPAs) are amino acids that are not naturally encoded in the genome. Many of them share structural similarities with cognate amino acids, hence are misrecognized by aaRS, leading to misincorporation. NPAs are often more toxic than naturally encoded noncognate amino acids, because they are not evolutionarily involved in the translational machinery and have a higher chance to elude intrinsic quality control mechanisms. For example, azetidine-2-carboxylic acid (Aze), an NPA found in sugar beets, is reported to misincorporate at Pro codons by evading ProRS quality control (26). Many NPAs generate cellular toxicity in vivo, which is believed to be a consequence of mistranslation (39). Here we reported that the toxicity of BMAA to AlaRS is achieved through multiple pathways (Fig. 5). We showed that in addition to misincorporation, BMAA acts as a competitive inhibitor to HsAlaRS activation and perturbs the editing function of the enzyme. By targeting the activation pocket, cognate amino acid activation is inhibited, potentially resulting in the activation of multiple stress response pathways. For example, halofuginone, a ProRS active site competitive inhibitor, is reported to induce the GCN2 pathway because of the accumulation of deacylated tRNA in the cells (40). When GCN2 is activated by post-translational phosphorylation, it inhibits the eukaryotic translation initiation factor 2α (eIF2α), leading to the reduction of global translation. It is unclear if a similar effect may occur in BMAA-treated cells.
Figure 5.
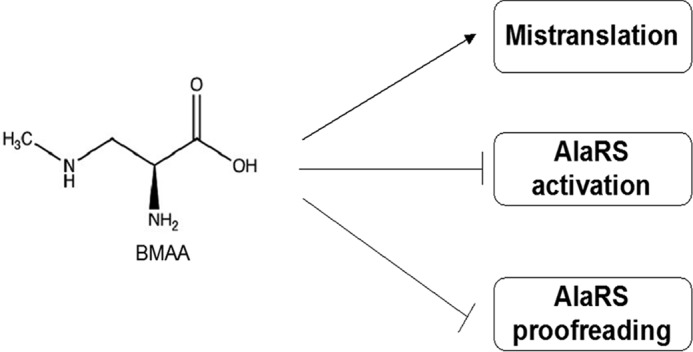
A model for BMAA's impact on protein synthesis. We showed that BMAA affects the function of AlaRS via multiple mechanisms, which could potentially lead to disrupting the stability of protein synthesis. Besides acting as a noncognate substrate for misincorporation, it also perturbs the activation and editing function of AlaRS.
In addition to inhibiting AlaRS amino acid activation, BMAA also perturbs the deacylation activity of HsAlaRS on Ser-tRNAAla. It is well-documented that AlaRS editing is crucial in mammals. As it was recently reported, a vertebrate-specific protein ANKRD16 was identified to facilitate the pre-transfer editing of AlaRS, suggesting that mistranslation by AlaRS is evolutionarily problematic (41, 42). In eukaryotes, the endoplasmic reticulum (ER) is responsible for facilitating appropriate protein folding, and the loss of aaRS quality control can lead to ER stress (43, 44). To alleviate the stress, the unfolded protein response is triggered, where protein synthesis is halted, and the expression of chaperones is increased. If the ER stress is still unable to be resolved, apoptosis will be activated. Up-regulation of ER stress and unfolded protein response gene expression have both been reported in cells treated with BMAA (35), supporting the finding in our study that BMAA possesses the potential to perturb faithful protein synthesis.
Materials and methods
BMAA purification
BMAA (Sigma, B107–50 mg) was dissolved in water (1.3 ml) and aliquots (250 μl) were injected onto a reversed phase HPLC column system (Keystone Scientific Aquasil C18, 250 × 10 mm connected in series to a Keystone Scientific C18 250 × 4.6 mm column) equilibrated in water/TFA (100/0.1) and eluted isocratically (0.5 ml/min) with the same solution. UV absorption of the eluate was monitored (215 and 280 nm) and collected fractions (0.5 min) were screened off-line for BMAA, Ala, and Ser content. This was done by first drying aliquots of the collected fractions (typically 10 μl) in a vacuum centrifuge and treating them first with methanolic HCl to make the methyl esters and then with acetic anhydride to make the N-acetyl derivatives. The fully derivatized samples, along with authentic Ser and Ala standards, were analyzed by combined GC/MS. Neither Ser nor Ala (as the N-acetylmethyl esters) was detected in the peak BMAA-containing fractions, which were then pooled. Aliquots were taken of the final pooled BMAA sample and screened again by GC/MS for verification of the absence of Ser and Ala contamination. Again, no contamination of Ser or Ala was detected in the final pooled BMAA sample that was then dried in a vacuum centrifuge. The BMAA content of the final sample was determined by weighing.
Strains, plasmids, and genetic techniques
Anabaena PCC 7120 was grown on BG11 plates until a visible lawn formed. Genomic DNA was extracted as previously reported with modification of the lysis method (45) in which the cells were frozen in liquid nitrogen and then crushed and ground using a mortar and pestle while still immersed in liquid nitrogen. The resulting powder was resuspended in 400 μl TE buffer with 0.5% SDS, followed by the purification process reported by Singh et al. (45). The human AlaRS-pET21b (Rosetta (DE3)) and human ProRS-pET21b (BL21(DE3)) constructs are gifts from Dr. Karin Musier-Forsyth at The Ohio State University. The EcAlaRS, and AnaAlaRS expression construct were made by Gibson, assembling the PCR product of AlaRS-encoding genes from the genomic DNA to NdeI and Xho1 digested pET21b. The fusion proteins were overexpressed in BL21(DE3). The human SerRS-pQE80 expression construct was a gift from Dr. Miljan Simonović from The University of Illinois at Chicago (46). Site-directed mutagenesis was used to generate the EcC666A and HsC723A overexpression construct.
Protein expression and purification
All the fusion protein expression constructs were grown overnight in lysogeny broth with the corresponding antibiotics based on the selection markers. The overnight cultures were back-diluted in the same media at 1:100 and induced when the optical density reached 0.4–0.6 with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside (IPTG) at 16 °C overnight except C666A, which was induced at 37 °C for 4 h with 1 mm IPTG. Cells were then harvested by centrifugation (5000 × g, 20 min) and resuspended in 10 ml lysis buffer containing 50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10 mm imidazole, and half a tablet of the cOmpleteTM EDTA-free protease inhibitor mixture (Roche) before being subjected to sonication. The homogenate was centrifuged (75,000 × g, 20 min, twice), and the supernatant was first filtered through a 0.22-μm filter disk then subjected to affinity column purification using TALON® metal affinity resin (Takara) for His-tag protein purification. The column was pre-equilibrated with 20 resin bed volumes of wash buffer (50 mm Tris-HCl, pH 8.0, 300 mm NaCl, 10 mm imidazole) before loading the sample, followed by another wash with 20 resin bed volumes of wash buffer. The fusion protein was eluted with 2 resin bed volumes of elution buffer (50 mm Tris-HCl, pH 8.0, 300 mm NaCl and 250 mm imidazole), and the collected fractions were screened by SDS-PAGE. Fractions of interest were concentrated; dialyzed against 20 mm Tris-HCl, pH 8.0, 300 mm NaCl, 5 mm 2-mercaptoethanol, and 10% glycerol overnight; and followed by a second dialysis for 4 h using the same buffer with 20% glycerol. The concentrations of active enzymes were determined by active site titration as described previously (47).
tRNA preparation and radiolabeling
tRNAAla was transcribed by T7 polymerase in 40 mm Tris, pH 8.0, 2 mm spermidine, 22 mm MgCl2, 5 mm DTT, 50 μg/ml BSA, 20 mm 5′GMP, pyrophosphatase, 4 mm NTP, RNase inhibitor, and 50 μg template DNA at 42 °C for 12–16 h. DEAE-cellulose anionic exchange resin was used for tRNA purification and was pre-equilibrated with 20 mm Tris, pH 8.0, and 5 mm MgCl2. After the in vitro transcribed sample was loaded, the column was washed in the same buffer with 250 mm NaCl and eluted with 1 m NaCl. The eluted fractions were pooled and incubated at −20 °C overnight in 3 volumes of ice-cold ethanol and 0.1 volume of 3 m sodium acetate (pH 5.5). After centrifugation (4500 × g, 10 min) the supernatant was decanted, and the pellet was air-dried. Finally, the purified tRNA pellet was resuspended in water, and the active concentration was determined through aminoacylation assays as described below. The [3′-32P]-labeled tRNAAla was prepared as described previously with slight modification (48). In this study, the Ala-76 was removed through in vitro transcription, and this tRNA lacking the 3′ terminal adenine was used for the radiolabel reaction. The [3′-32P]-labeled tRNA was purified by acid phenol chloroform extraction, resuspended in water and further purified with G25 size-exclusion chromatography to remove free [α-32P]ATP. The labeled tRNA was refolded at 70 °C for 2 min in 5 mm MgCl2, and slow-cooled to room temperature. Cognate amino acid aminoacylation was examined using the TLC method described below (Fig. S2).
Aminoacylation assay
In general, aminoacylation assays were carried out at 37 °C in aminoacylation buffer (100 mm Na-HEPES, 30 mm KCl, 10 mm MgCl2), 4 mm ATP, 100 μm radiolabeled amino acid, tRNA, and aaRS. Time points were taken by spotting aliquots of the reaction onto 3MM Whatman filter disks presoaked with 5% TCA for quenching. The disks were washed with 5% TCA three times, ethanol once, and dried in an oven before measuring the radioactivity by scintillation counting (Beckman Coulter LS6500). Because not all in vitro transcripts are active, we used 5 μl stock tRNA and 150 nm HsAlaRS to determine the concentration of active human tRNAAla. Longer time points were taken to ensure that the tRNAs were saturated with amino acids, and the highest count was selected for calculating the active concentration. [32P] tRNAAla was used for the BMAA aminoacylation assay. 500 nm AlaRS/C723A was added to initiate the reaction in aminoacylation buffer containing [32P]tRNA, 10 mm ATP, 10 mMDTT, and the designated amino acids (100 μm Ala/50 mm BMAA/no aa). Aliquots were quenched at chosen time points by an equal volume of S1 mix (1× S1 buffer, 0.5 m sodium acetate, pH 4.5, and S1 nuclease). Reactions were incubated at room temperature for 30 min and aliquots were spotted on a PEI cellulose TLC plate (Sigma-Aldrich). Free [α-32P]AMP and aminoacyl [α-32P]AMP were separated by developing the TLC with 100 mm ammonium acetate and 5% acetic acid. The TLC plate was imaged by Typhoon FLA9000 phosphorimager (GE Healthcare), and the intensity was analyzed using ImageJ. The fraction of charged tRNA was determined by the ratio of charged/uncharged tRNA as described previously (49) after subtracting the background values of the no amino acid control. The results were normalized to Ala-tRNAAla to determine the relative level of aminoacylation.
Pyrophosphate exchange
Pyrophosphate exchange assays were carried out at 37 °C in HKM buffer (100 mm Na-Hepes, pH 7.2, 30 mm KCl, 10 mm MgCl2, 2 mm NaF) containing 2 mm ATP, 2 mm [32P]PPi (2–4 cpm/pmol) and 500 nm aaRS as described previously (50). Briefly, the reactions were quenched at designated time points by aliquoting 25 μl into 970 μl charcoal solution (1% charcoal, 5.6% HClO4, and 75 mm PPi). The quenched reactions were deposited on 3 MM Whatman filter discs by vacuum filtration, followed by washing three times with 5 ml water, once with 5 ml EtOH, and dried in the oven. Finally, the radioactivity was determined by liquid scintillation counting. Results were fit to a Michaelis-Menten curve using KaleidaGraph to determine the steady state activation kinetics.
ATP consumption assay
ATP consumption assays were performed as described previously with slight modification (25). In this study, the assay was carried out at 37 °C in aminoacylation buffer containing 100 mm amino acids, 3 mm [γ-32P]ATP, 2 units/ml of yeast pyrophosphatase (Roche), and1 μm human AlaRS with or without 2 μm tRNA. Reactions were quenched at each time point by aliquoting 2-μl samples into 2-μl glacial acetic acid. Quenched reaction was spotted on a PEI cellulose plate prewashed with water, and the separation of [γ-32P]ATP and [γ-32P]Pi was accomplished by developing in 0.7 m potassium phosphate (pH 3.5). The TLC plate was imaged by Typhoon FLA9000 phosphorimager (GE Healthcare), and the intensity was analyzed using ImageJ. The concentration of Pi formation was determined by multiplying the Pi/ATP ratio with the initial ATP concentration (3 mm).
Enzyme inhibition assay
To determine the inhibition kinetics, aminoacylation was carried out in the same condition with 50 mm BMAA and 400, 250, 200, 125,100, 50, 25, or 12.5 μm [14C]Ala. The radioactivity was determined by liquid scintillation counting, and the results were fitted into a Michaelis-Menten curve using KaleidaGraph as mentioned previously. For the Lineweaver-Burk plot, aminoacylation was carried out at 37 °C with 50 mm, 25 mm, or no BMAA in aminoacylation buffer, including 4 mm ATP, 5 μm tRNAAla, 100 nm HsAlaRS, and different concentrations of [14C]Ala (5, 10, 20, or 40 μm).
Misaminoacylation and deacylation
The misaminoacylation reaction was carried out at 37 °C for 1 h in aminoacylation buffer (100 mm Na-HEPES, 30 mm KCl, 10 mm MgCl2) containing 50 μm tRNAAla, pyrophosphatase, 800 μm [3H]Ser, and 2.5 μm C666A. Ser-tRNAAla was extracted by adding 1 volume of acid phenol chloroform, mixing thoroughly, and centrifuging at 11,000 × g for 5 min. The aqueous phase was collected and the tRNA was precipitated overnight at −80 °C in 2.7 volumes of cold ethanol and 0.1 volumes of sodium acetate (pH 4.5). The sample was centrifuged at 11,000 × g for 30 min, the supernatant was decanted, and the pellet was air-dried for 10 min. Finally, the pellet was resuspended in 100 mm sodium acetate (pH 4.5) and was used for the deacylation assay. Deacylation reactions were carried out at 37 °C in aminoacylation buffer containing [3H]Ser-tRNAAla and100 nm AlaRS. C723A was also tested as a negative control. 50 mm BMAA was treated to determine the effect of BMAA on AlaRS editing. A no enzyme control was also included to estimate spontaneous deacylation. The reactions were quenched at different time points followed by washing as described previously in the aminoacylation section above. Time point zero was taken immediately prior to addition of the enzyme to initiate the reaction, and was set as 100% misaminoacylation for downstream analysis.
MS-READ sample preparation
Saccharomyces cerevisiae MS-READ reporter samples were prepared as previously described with slight modification (22). Briefly, S. cerevisiae strain KMO3 (51) was transformed with pXRH3 containing the Ala codon MS-READ reporter. The Ala codon reporter (GCU) was constructed through site-directed mutagenesis from the previously described Phe codon MS-READ reporter (UUU) plasmid (22). The cells were grown with or without 4 mm BMAA until the outer diameter reached 1, and was lysed by sonication. The MS-READ reporter peptides were purified as described previously (22).
Protein digestion and MS
Affinity-purified, buffer-exchanged protein was digested and analyzed by MS as described previously with some modifications (22). Briefly, the concentration of protein was determined by UV280 spectroscopy and 5 μg ELP-GFP (MS-READ) reporter from S. cerevisiae was dissolved in 12.5 μl solubilization buffer consisting of 10 mm Tris-HCl, pH 8.5 (23 °C), 10 mm DTT, 1 mm EDTA, and 0.5% acid labile surfactant (ALS-101, Protea). Samples were heat denatured for 20 min at 55 °C in a heat block. Alkylation of cysteines was performed with iodoacetamide (IAA) using a final IAA concentration of 24 mm. The alkylation reaction proceeded for 30 min at room temperature in the dark. Excess IAA was quenched with DTT and the buffer concentration was adjusted using 1 m Tris-HCl, pH 8.5, resulting in a final Tris-HCl concentration of 150 mm. The reaction was then diluted with water and 1 m CaCl2 solution to obtain an ALS-101 concentration of 0.045% and 2 mm CaCl2, respectively. Finally, sequencing grade porcine trypsin (Promega) was added to obtain an enyzme/protein ratio of 1/5.3 and the digest was incubated for 15 h at 37 °C without shaking. The digest was quenched with 20% TFA solution resulting in a sample pH of 2. Cleavage of the acid-cleavable detergent proceeded for 15 min at room temperature. Digests were frozen at −80 °C until further processing. Peptides were desalted on C18 UltraMicroSpin columns (The Nest Group Inc.) essentially following the instructions provided by the manufacturer but using 300 μl elution solvent consisting of 80% ACN, 0.1% TFA for peptide elution. Peptides were dried in a vacuum centrifuge at room temperature. Dried peptides were reconstituted and analyzed by LC-MS/MS. LC-MS/MS was performed on an Orbitrap Velos using a Top10 HCD method. The trapping column consisted of a 3 cm × 150 μm Kasil frit-terminated fused silica capillary packed with 3 μm particle size Reprosil-Pur 120 C18-AQ (Dr. Maisch GmBH). The analytical column was a 20 cm × 75 μm ID picofrit (New Objective) column packed with 1.9 μm particle size Reprosil-Pur 120 C18-AQ (Dr. Maisch GmBH) to a length of 20 cm. Trapping column and analytical column were connected using a vented split setup using a low dead volume T metal connector where the spray voltage was applied. Eluent A was 0.1% formic acid in water and eluent B was 0.1% formic acid in acetonitrile. Trapping of peptides was performed for 5 min at a flow rate of 2.5 μl/min with an eluent composition of 2% eluent B. Gradient separation of peptides proceeded at a flow rate of 300 nl/min using the following linear gradient program (min/% eluent B): 0.0/2.0, 0.1/2.0, 60.00/25.0, 70.0/40, 72.0/95.0, 78.0/95.0, 80.0/2.0, 90.0/2.0. An estimated 150 ng of the digest was injected for each experiment. A database search was performed with MaxQuant v. 1.5.3.30 against a custom database of GFP X16 containing all natural AAs at position X16, and a S. cerevisiae protein database (UniProt, strain AWRI1631, 5450 sequences). Forward and decoy database searches were carried out using full trypsin specificity with up to three missed cleavages and using a mass tolerance of 30 ppm for the precursor and 0.1 Da for fragment ions, respectively. Peptides were analyzed and quantified in Skyline.
Author contributions
N.-C. H., T. J. B., and M. I. conceptualization; N.-C. H. and M. I. data curation; N.-C. H. and M. I. formal analysis; N.-C. H., K. F. F., K. M., and J. R. validation; N.-C. H., K. F. L., K. F. F., K. M., and J. R. investigation; N.-C. H. visualization; N.-C. H., K. F. F., K. M., J. R., and M. I. methodology; N.-C. H. writing-original draft; N.-C. H., T. J. B., and M. I. writing-review and editing; M. I. supervision; M. I. funding acquisition; M. I. project administration.
Supplementary Material
Acknowledgments
We thank Dr. Musier-Forsyth and Dr. Simonović for the fusion protein expression constructs and P. Kelly for the valuable discussions. The Pasarow Mass Spectrometry Laboratory (KFF) gratefully acknowledges support from Hidden Wings, Solvang, CA.
This work was supported by National Science Foundation Grant MCB-1715840 (to M. I.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4 and Table S1.
- BMAA
- β-N-methylamino-l-alanine
- aaRS
- aminoacyl-tRNA synthetase
- AlaRS
- alanyl-tRNA synthetase
- SerRS
- seryl-tRNA synthetase
- ProRS
- prolyl-tRNA synthetase
- ER
- endoplasmic reticulum
- MS-READ
- mass spectrometry reporter for exact amino acid decoding
- NPA
- nonproteinogenic amino acids.
References
- 1. Vega A., and Bell E. A. (1967) α-Amino-β-methylaminopropionic acid, a new amino acid from seeds of Cycas circinalis. Phytochemistry 6, 759–762 10.1016/S0031-9422(00)86018-5 [DOI] [Google Scholar]
- 2. Weiss J. H., Christine C. W., and Choi D. W. (1989) Bicarbonate dependence of glutamate receptor activation by β-N-methylamino-l-alanine: Channel recording and study with related compounds. Neuron 3, 321–326 10.1016/0896-6273(89)90256-0 [DOI] [PubMed] [Google Scholar]
- 3. Mulder D. W., Kurland L. T., and Iriarte L. L. (1954) Neurologic diseases on the island of Guam. U. S. Armed Forces Med. J. 5, 1724–1739 [PubMed] [Google Scholar]
- 4. Spencer P. S., Nunn P. B., Hugon J., Ludolph A. C., Ross S. M., Roy D. N., and Robertson R. C. (1987) Guam amyotrophic lateral sclerosis-parkinsonism dementia linked to a plant excitant neurotoxin. Science 237, 517–522 10.1126/science.3603037 [DOI] [PubMed] [Google Scholar]
- 5. Duncan M. W., Steele J. C., Kopin I. J., and Markey S. P. (1990) 2-Amino-3-(methylamino)-propanoic acid (BMAA) in cycad flour: An unlikely cause of amyotrophic lateral sclerosis and parkinsonism-dementia of Guam. Neurology 40, 767–772 10.1212/WNL.40.5.767 [DOI] [PubMed] [Google Scholar]
- 6. Cox P. A., Banack S. A., and Murch S. J. (2003) Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc. Natl. Acad. Sci. U.S.A. 100, 13380–13383 10.1073/pnas.2235808100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Murch S. J., Cox P. A., and Banack S. A. (2004) A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc. Natl. Acad. Sci. U.S.A. 101, 12228–12231 10.1073/pnas.0404926101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dunlop R. A., Cox P. A., Banack S. A., and Rodgers K. J. (2013) The non-protein amino acid BMAA is misincorporated into human proteins in place of l-serine causing protein misfolding and aggregation. PLoS One 8, e75376 10.1371/journal.pone.0075376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Caller T. A., Field N. C., Chipman J. W., Shi X., Harris B. T., and Stommel E. W. (2012) Spatial clustering of amyotrophic lateral sclerosis and the potential role of BMAA. Amyotroph. Lateral Scler. 13, 25–32 10.3109/17482968.2011.621436 [DOI] [PubMed] [Google Scholar]
- 10. Masseret E., Banack S., Boumédiène F., Abadie E., Brient L., Pernet F., Juntas-Morales R., Pageot N., Metcalf J., Cox P., Camu W., and French Network on ALS Clusters Detection and Investigation (2013) Dietary BMAA exposure in an amyotrophic lateral sclerosis cluster from southern France. PLoS One 8, e83406 10.1371/journal.pone.0083406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Torbick N., Ziniti B., Stommel E., Linder E., Andrew A., Caller T., Haney J., Bradley W., Henegan P. L., and Shi X. (2018) Assessing cyanobacterial harmful algal blooms as risk factors for amyotrophic lateral sclerosis. Neurotox. Res. 33, 199–212 10.1007/s12640-017-9740-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cox P. A., Banack S. A., Murch S. J., Rasmussen U., Tien G., Bidigare R. R., Metcalf J. S., Morrison L. F., Codd G. A., and Bergman B. (2005) Diverse taxa of cyanobacteria produce β-N-methylamino-l-alanine, a neurotoxic amino acid. Proc. Natl. Acad. Sci. U.S.A. 102, 5074–5078 10.1073/pnas.0501526102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Al-Sammak M. A., Hoagland K. D., Cassada D., and Snow D. D. (2014) Co-occurrence of the cyanotoxins BMAA, DABA and anatoxin-a in Nebraska reservoirs, fish, and aquatic plants. Toxins (Basel). 6, 488–508 10.3390/toxins6020488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Esterhuizen M., and Downing T. G. (2008) β-N-methylamino-l-alanine (BMAA) in novel South African cyanobacterial isolates. Ecotoxicol. Environ. Saf. 71, 309–313 10.1016/j.ecoenv.2008.04.010 [DOI] [PubMed] [Google Scholar]
- 15. Davis D. A., Mondo K., Stern E., Annor A. K., Murch S. J., Coyne T. M., Brand L. E., Niemeyer M. E., Sharp S., Bradley W. G., Cox P. A., and Mash D. C. (2019) Cyanobacterial neurotoxin BMAA and brain pathology in stranded dolphins. PLoS One 14, e0213346 10.1371/journal.pone.0213346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang L., Kiselova N., Rosén J., and Ilag L. L. (2014) Quantification of neurotoxin BMAA (β-N-methylamino-L-alanine) in seafood from Swedish markets. Sci. Rep. 4, 6931 10.1038/srep06931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mohler K., and Ibba M. (2017) Translational fidelity and mistranslation in the cellular response to stress. Nat. Microbiol. 2, 17117 10.1038/nmicrobiol.2017.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Guo M., Chong Y. E., Shapiro R., Beebe K., Yang X. L., and Schimmel P. (2009) Paradox of mistranslation of serine for alanine caused by AlaRS recognition dilemma. Nature 462, 808–812 10.1038/nature08612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee J. W., Beebe K., Nangle L. A., Jang J., Longo-Guess C. M., Cook S. A., Davisson M. T., Sundberg J. P., Schimmel P., and Ackerman S. L. (2006) Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443, 50–55 10.1038/nature05096 [DOI] [PubMed] [Google Scholar]
- 20. Beebe K., Ribas De Pouplana L., and Schimmel P. (2003) Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J. 22, 668–675 10.1093/emboj/cdg065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wong F.-C., Beuning P. J., Nagan M., Shiba K., and Musier-Forsyth K. (2002) Functional role of the prokaryotic proline-tRNA synthetase insertion domain in amino acid editing. Biochemistry 41, 7108–7115 10.1021/bi012178j [DOI] [PubMed] [Google Scholar]
- 22. Mohler K., Aerni H. R., Gassaway B., Ling J., Ibba M., and Rinehart J. (2017) MS-READ: Quantitative measurement of amino acid incorporation. Biochim. Biophys. Acta Gen. Subj. 1861, 3081–3088 10.1016/j.bbagen.2017.01.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chong Y. E., Yang X. L., and Schimmel P. (2008) Natural homolog of tRNA synthetase editing domain rescues conditional lethality caused by mistranslation. J. Biol. Chem. 283, 30073–30078 10.1074/jbc.M805943200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu Y., Satz J. S., Vo M.-N., Nangle L. A., Schimmel P., and Ackerman S. L. (2014) Deficiencies in tRNA synthetase editing activity cause cardioproteinopathy. Proc. Natl. Acad. Sci. 111, 17570–17575 10.1073/pnas.1420196111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roy H., Ling J., Irnov M., and Ibba M. (2004) Post-transfer editing in vitro and in vivo by the β subunit of phenylalanyl-tRNA synthetase. EMBO J. 23, 4639–4648 10.1038/sj.emboj.7600474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Song Y., Zhou H., Vo M.-N., Shi Y., Hussain Nawaz M., Vargas-Rodriguez O., Diedrich J. K., Yates J. R., Kishi S., Musier-Forsyth K., and Schimmel P. (2017) Double mimicry evades tRNA synthetase editing by toxic vegetable-sourced non-proteinogenic amino acid. Nat. Commun. 8, 2281 10.1038/s41467-017-02201-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dixon M., and Webb E. C. (1979) Enzymes, 3rd Ed., 332–352, Academic Press, New York [Google Scholar]
- 28. van Onselen R., and Downing T. G. (2018) BMAA-protein interactions: A possible new mechanism of toxicity. Toxicon 143, 74–80 10.1016/j.toxicon.2018.01.011 [DOI] [PubMed] [Google Scholar]
- 29. Pawar K. I., Suma K., Seenivasan A., Kuncha S. K., Routh S. B., Kruparani S. P., and Sankaranarayanan R. (2017) Role of D-aminoacyl-tRNA deacylase beyond chiral proofreading as a cellular defense against glycine mischarging by AlaRS. Elife 6, e24001 10.7554/eLife.24001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Selkoe D. J. (2004) Cell biology of protein misfolding: The examples of Alzheimer's and Parkinson's diseases. Nat. Cell Biol. 6, 1054–1061 10.1038/ncb1104-1054 [DOI] [PubMed] [Google Scholar]
- 31. Kapur M., and Ackerman S. L. (2018) MRNA translation gone awry: Translation fidelity and neurological disease. Trends Genet. 34, 218–231 10.1016/j.tig.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Onselen R., Downing S., Kemp G., and Downing T. (2017) Investigating β-N-methylamino-l-alanine misincorporation in human cell cultures: A comparative study with known amino acid analogues. Toxins 9, E400 10.3390/toxins9120400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saez I., and Vilchez D. (2014) The mechanistic links between proteasome activity, aging and age-related diseases. Curr. Genomics. 15, 38–51 10.2174/138920291501140306113344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Glover W. B., Mash D. C., and Murch S. J. (2014) The natural non-protein amino acid N-β-methylamino-l-alanine (BMAA) is incorporated into protein during synthesis. Amino Acids 46, 2553–2559 10.1007/s00726-014-1812-1 [DOI] [PubMed] [Google Scholar]
- 35. Beri J., Nash T., Martin R. M., and Bereman M. S. (2017) Exposure to BMAA mirrors molecular processes linked to neurodegenerative disease. Proteomics 17, 170061 10.1002/pmic.201700161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Downing S., Banack S. A., Metcalf J. S., Cox P. A., and Downing T. G. (2011) Nitrogen starvation of cyanobacteria results in the production of β-N-methylamino-l-alanine. Toxicon 58, 187–194 10.1016/j.toxicon.2011.05.017 [DOI] [PubMed] [Google Scholar]
- 37. Berntzon L., Erasmie S., Celepli N., Eriksson J., Rasmussen U., and Bergman B. (2013) BMAA inhibits nitrogen fixation in the cyanobacterium Nostoc sp. PCC 7120. Mar. Drugs 11, 3091–3108 10.3390/md11083091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Popova A. A., Rasmussen U., Semashko T. A., Govorun V. M., and Koksharova O. A. (2018) Stress effects of cyanotoxin β-methylamino-l-alanine (BMAA) on cyanobacterial heterocyst formation and functionality. Environ. Microbiol. Rep. 10, 369–377 10.1111/1758-2229.12647 [DOI] [PubMed] [Google Scholar]
- 39. Bullwinkle T., Lazazzera B., and Ibba M. (2014) quality control and infiltration of translation by amino acids outside of the genetic code. Annu. Rev. Genet. 48, 149–166 10.1146/annurev-genet-120213-092101 [DOI] [PubMed] [Google Scholar]
- 40. Keller T. L., Zocco D., Sundrud M. S., Hendrick M., Edenius M., Yum J., Kim Y.-J., Lee H.-K., Cortese J. F., Wirth D. F., Dignam J. D., Rao A., Yeo C.-Y., Mazitschek R., and Whitman M. (2012) Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat. Chem. Biol. 8, 311–317 10.1038/nchembio.790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ahel I., Korencic D., Ibba M., and Söll D. (2003) Trans-editing of mischarged tRNAs. Proc. Natl. Acad. Sci. U.S.A. 100, 15422–15427 10.1073/pnas.2136934100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vo M. N., Terrey M., Lee J. W., Roy B., Moresco J. J., Sun L., Fu H., Liu Q., Weber T. G., Yates J. R., Fredrick K., Schimmel P., and Ackerman S. L. (2018) ANKRD16 prevents neuron loss caused by an editing-defective tRNA synthetase. Nature 557, 510–515 10.1038/s41586-018-0137-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu J., Bergert M., Walther A., and Suter B. (2014) Double-sieving-defective aminoacyl-tRNA synthetase causes protein mistranslation and affects cellular physiology and development. Nat. Commun. 5, 5650 10.1038/ncomms6650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nangle L. A., Motta C. M., and Schimmel P. (2006) Global effects of mistranslation from an editing defect in mammalian cells. Chem. Biol. 13, 1091–1100 10.1016/j.chembiol.2006.08.011 [DOI] [PubMed] [Google Scholar]
- 45. Singh S. P., Rastogi R. P., Häder D.-P., and Sinha R. P. (2011) An improved method for genomic DNA extraction from cyanobacteria. World J. Microbiol. Biotechnol. 27, 1225–1230 10.1007/s11274-010-0571-8 [DOI] [Google Scholar]
- 46. Holman K. M., Puppala A. K., Lee J. W., Lee H., Simonović M., and Simonović S. (2017) Insights into substrate promiscuity of human seryl-tRNA synthetase. RNA 23, 1685–1699 10.1261/rna.061069.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wilkinson A. J., Fersht A. R., Blow D. M., and Winter G. (1983) Site-directed mutagenesis as a probe of enzyme structure and catalysis: Tyrosyl-tRNA synthetase cysteine-35 to glycine-35 mutation. Biochemistry 22, 3581–3586 10.1021/bi00284a007 [DOI] [PubMed] [Google Scholar]
- 48. Ledoux S., and Uhlenbeck O. C. (2008) [3′-32P]-labeling tRNA with nucleotidyltransferase for assaying aminoacylation and peptide bond formation. Methods 44, 74–80 10.1016/j.ymeth.2007.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wolfson A. D., and Uhlenbeck O. C. (2002) Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc. Natl. Acad. Sci. U.S.A. 99, 5965–5970 10.1073/pnas.092152799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Roy H., Ling J., Alfonzo J., and Ibba M. (2005) Loss of editing activity during the evolution of mitochondrial phenylalanyl-tRNA synthetase. J. Biol. Chem. 280, 38186–38192 10.1074/jbc.M508281200 [DOI] [PubMed] [Google Scholar]
- 51. Mohler K., Mann R., Bullwinkle T. J., Hopkins K., Hwang L., Reynolds N. M., Gassaway B., Aerni H. R., Rinehart J., Polymenis M., Faull K., and Ibba M. (2017) Editing of misaminoacylated tRNA controls the sensitivity of amino acid stress responses in Saccharomyces cerevisiae. Nucleic Acids Res. 45, 3985–3996 10.1093/nar/gkx077 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.