

Abstract
Tissue immunostaining provides highly specific and reliable detection of proteins of interest within a given tissue. Here we describe a complete and simple protocol to detect protein expression during craniofacial morphogenesis/pathogenesis using mouse craniofacial tissues as examples. The protocol consists of preparation and cryosectioning of tissues, indirect immunofluorescence, image acquisition, and quantification. In addition, a method for preparation and cryosectioning of undecalcified hard tissues for immunostaining is described, using craniofacial tissues and long bones as examples. Those methods are key to determine the protein expression and morphological/anatomical changes in various tissues during craniofacial morphogenesis/pathogenesis. They are also applicable to other tissues with appropriate modifications. Knowledge of the histology and high quality of sections are critical to draw scientific conclusions from experimental outcomes. Potential limitations of this methodology include but are not limited to specificity of antibodies and difficulties of quantification, which are also discussed here.
Keywords: Developmental Biology, Issue 147, developmental biology, transgenic mice, BMP signaling, immunostaining, craniofacial morphogenesis, undecalcified hard tissues, quantification
Introduction
The face is a key part of human identity, and is composed of several different types of tissues, such as epithelium, muscle, bone, cartilage, tooth. Those tissues are derived from all three germ layers: ectoderm, endoderm, and mesoderm1,2. For proper patterning and development of craniofacial tissues, cell proliferation, death and differentiation need to be highly coordinated and regulated by specific signaling pathways, such as Wnt, Fgf, Hh and Bmp pathways3,4,5. Defects in proliferation, survival or differentiation of cells will lead to craniofacial malformations, which are among the most frequently occurring congenital birth defects. Transgenic mice are useful tools to study mechanisms of craniofacial morphogenesis and pathogenesis1,2,3,4,5. Understanding the changes in craniofacial structures during development and pathogenesis will help to clarify key developmental principles as well as the mechanisms of craniofacial malformations1,2,3,4,5.
The staining of whole mount or sectioned tissues with specific antibodies is an invaluable technique for determining spatial distribution of proteins of interest6. Formally, tissue immunostaining can rely either upon immunohistochemistry (IHC) or immunofluorescence (IF). Compared with the opaque reaction product generated with a chromogenic substrate such as 3,3’-Diaminobenzidine (DAB) by IHC, IF involves the use of fluorescent conjugates visible by fluorescence microscopy. Therefore, IF may clearly differentiate positive cells from background noise, and allows images to be quantitatively analyzed and enhanced in a straightforward fashion by software such as ImageJ and Adobe Photoshop7,8. The whole mount staining approach works on small blocks of tissue (less than 5 mm thick), which can provide three-dimensional information about the location of proteins/antigens without the need for reconstruction from sections9,10. However, compared with tissue sections, whole mount immunostaining is time consuming and requires large volumes of antibody solutions. Not all antibodies are compatible with the basic whole mount approach. In addition, the incomplete penetration of antibodies will result in uneven staining or false negative staining. Here we will focus on the immunofluorescence detection of proteins/antigens on sectioned tissues. For hard tissues (eg, head, tooth, long bone), calcium deposition during development/pathogenesis makes the sample difficult to section and easily rinsed off during immunostaining treatment11,12. Most of the currently available protocols decalcify hard tissues before embedding to make sectioning easier, which is time consuming and can destroy morphology and antigens of samples if handled improperly11,12. To overcome the issues, we optimized an approach for cryosectioning of hard tissues without decalcification, leading to improved visualization of their morphology and distribution of signaling proteins.
The protocol described here is being used to determine morphometric and histological changes in the craniofacial tissues of BMP transgenic mice. Specifically, the protocol includes (1) harvesting and dissecting head tissues, (2) section and immunostaining of experimental markers (Ki67, pSmad1/5/9) along with TUNEL staining, (3) imaging the sections using fluorescence microscope, and finally (4) analyzing and quantifying the results. The protocol to prepare and cryosection hard tissues without decalcification is also described13. Those methods are optimized for craniofacial tissues. They are also applicable to other tissues from various ages of samples with appropriate modifications.
Protocol
All mouse experiments were carried out in accordance with University of Michigan guidelines covering the humane care and use of animals in research. All animal procedures used in this study were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan (Protocol #PRO00007715).
1. Tissue Preparation
- Preparation of embryonic tissues
-
Prepare one 10 cm dish and several 3.5 cm dishes containing phosphate buffered saline (PBS), and one 12-well culture plate containing 2 mL 4% paraformaldehyde (PFA) in PBS in each well for each pregnant mouse. Place all Petri dishes and the plate on ice.NOTE: Handle 4% PFA in a fume hood.
- Dissect embryos from pregnant mice in ice-cold PBS with forceps and scissors as previously described14.
- Briefly, euthanize a pregnant mouse with CO2, grab the skin below the center of the belly with forceps and cut through the skin only, then gently pull at the skin to separate it from the underlying the abdominal muscle wall.
- Next, cut into the abdominal cavity following the same line of the skin incision. Remove the uterus containing a string of embryos and remove the embryos by gently cutting away the uterine wall. The extraembryonic tissues such as the yolk sac and amnion will be removed.
- Cut and isolate head from each embryo.
-
Transfer each head into each well of a 12-well plate containing 4% PFA with a plastic transfer pipette or forceps. Fix samples in 4% PFA at 4 °C for 4 h. Rinse samples in PBS at 4 °C with gentle shaking for 12 h.NOTE: For embryos younger than embryonic day 16.5 (E16.5), fix embryo heads with 4% PFA directly after isolation. For embryos at E16.5 or later, remove to discard skin and adipose tissue from the heads and rinse several times in ice cold PBS before fixation.
- Cryoprotect heads.
- Transfer each head into a new 12-well plate containing 2 mL of 30% sucrose in PBS using a plastic transfer pipette or forceps. Agitate gently at 4 °C until the head sinks to the bottom of the dish.
- Embed heads.
- Transfer the cryoprotected head into a mold containing Optimal Cutting Temperature (OCT) compound. Equilibrate samples in OCT for several minutes. Adjust the location and direction of samples with forceps.
-
Place the mold on dry ice to freeze. Store resulting cryomolds in a plastic bag at −80 °C until ready for cryosectioning.NOTE: The trimmed side of the samples must face the bottom of the embedding mold.
-
- Preparation of postnatal undecalcified hard tissues
- Euthanize at 3 week or 3 month old mouse with CO2. Remove the skin and adipose tissues. Cut and isolate the head or long bones from the mouse.
- Fix and cryoprotect the head or long bone of mice as described in steps 1.1.3–1.1.4.
-
Embed in 8% gelatin in a similar manner as step 1.1.5. Keep the cryomolds in a plastic bag at −80 °C until cryosectioning.NOTE: Decalcification is not necessary here. To prepare 8% gelatin, mix 8 g of gelatin with 100 mL of PBS and boil using a microwave. Be aware that the mixture boils over easily.
2. Cryosectioning
Set cryostat temperature to −18 °C for soft tissues embedded in OCT or −25 °C and lower for undecalcified hard tissues embedded in gelatin. Keep samples in the cryostat chamber for about 30 min to equilibrate to the cryostat temperature.
Expel the block from the cryomold. Freeze the block onto the specimen chuck (tissue holder) via mounting with an OCT drop. Keep the trimmed side of the sample furthest from the chuck (facing the operator).
Load the block-mounted chuck onto the cryostat object holder. Adjust the blade holder to make the angle of the blade 3°–5° relative to the sample.
Collect 10 μm sections onto coated microscope slides. Dry sections completely at RT, then store them at −80 °C.
3. Histological Staining and Microscopic Imaging
- Immunofluorescence staining
- Take out slides from −80 °C. Keep slides at RT for 1 h to airdry sections. Rinse slides in 0.1% PBST (0.1 % Polyethylene glycol tert-octylphenyl ether in PBS; see Table of Materials) three times for 5 min each to wash out OCT and permeabilize sections.
- Optionally, perform antigen retrieval (optional).
- Preheat citrate buffer (10 mM sodium citrate pH 6) in the staining dish with steamer or water bath to 95–100 °C. Immerse slides in the citrate buffer, incubate for 10 min.
-
Take the staining dish out from steamer or water bath to RT. Cool the slides at RT for 20 min or longer15.NOTE: As alternatives, use Tris-EDTA buffer (10 mM Tris base, 1 mM EDTA, 0.05% Tween 20, pH 9.0) or EDTA buffer (1 mM EDTA, 0.05% Tween 20, pH 8.0) for heat-induced antigen retrieval. Use a pressure cooker, microwave, or water bath for heat-induced antigen retrieval, in addition to the hot steamer. An enzyme-induced antigen retrieval using trypsin or pepsin is another alternative. Optimize the concentration and treatment time of enzymatic retrieval to avoid damaging sections. Optimize the antigen retrieval method for each antibody/antigen combination.
- Incubate each slide with 200 μL of blocking solution (5% donkey serum diluted in 0.1 % PBST) at RT for 30 min, then remove the blocking solution without rinse.
- Incubate each slide with 100 μL of primary antibody or antibodies diluted in blocking solution for 1 h at RT or O/N at 4 °C. Rinse slides with PBS three times for 10 min each at RT.
- Incubate each slide with 100 μL of secondary antibody diluted in blocking solution for 1 h at RT. Rinse slides in PBS three times for 10 min each at RT. Protect slides from light.
- Mount slides.
- Add two drops of anti-fade medium with DAPI (4’, 6-diamidino-2-phenylindole) on the slide. Then cover with a coverslip.
-
Store at 4 °C in dark until ready to image.NOTE: As an alternative, label the nuclei with DAPI or Hoechst 33324 dye diluted 1:2,000 in PBS at RT first, then mount with glycerol.
-
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining.
NOTE: Double-stranded DNA with 3’-hydroxyl termini (3’OH DNA termini) will form during apoptosis in the cell. Here, we provide a protocol that label the free 3’OH DNA termini in situ via labeling DNA fragments with the digoxigenin-nucleotide utilizing terminal deoxynucleotidyl transferase (TdT) by specific staining using a commercial kit (see Table of Materials).-
Optionally, stain sections with primary and Alexa Fluor-488 labeled secondary antibodies prior to the TUNEL staining. Rinse the slides in PBS three times for 10 min each.NOTE: This step is optional for a double staining of a protein and TUNEL in the same slide.
-
Incubate each slide with 100 μL Proteinase K (10 μg/mL in 10 mM Tris pH 7.5 and 5 mM EDTA) for 5 min at RT. Rinse slides with PBS three times for 10 min each at RT.NOTE: Adjust the incubation time and temperature of Proteinase K for each tissue type. For 10 μm sections of embryo heads fixed in 4% PFA, incubate for 5 min at RT. In addition to the method using Proteinase K, use alternative treatments as needed, including (1) freshly prepared 0.1% Polyethylene glycol tert-octylphenyl ether, 0.1% sodium citrate, 10 min at 37 °C; (2) 0.25%–0.5% Pepsin in HCl (pH 2) or 0.25% trypsin, 10 min at 37 °C; and (3) microwave irradiation with 0.1 M citrate buffer (pH 6).
- Apply 200 μL of blocking solution (5% donkey serum diluted in 0.1 % PBST) to each slide, incubate at RT for 30 min, tap off the blocking solution without rinse.
- Apply 50 μL of the equilibration buffer supplied by the kit to each slide at RT for at least 10 s. Tap off the buffer without rinse.
- Prepare reaction mixture (working strength TdT enzyme) by mixing TdT Enzyme with the reaction buffer supplied by the kit at the ratio of 3:7. Apply 50 μL of the reaction mixture to each slide, and incubate at 37 °C for 1 h. Tap off the buffer without rinsing.
- Apply 200 μL of the stop buffer (1:30 diluted in ddH2O) supplied by the kit to each slide, then incubate at RT for 10 min. Rinse slides with PBS three times for 10 min each.
- Label with Rhodamine antibody.
- Apply 50 μL of pre-warmed (RT) anti-digoxigenin conjugate (rhodamine) (1:1 diluted in blocking solution) to each slide. Incubate at RT for 30 min in dark.
- Rinse slides with PBS three times for 10 min each. Mount slides as step 3.1.6.
-
Table of Materials
| Name of Material/ Equipment | Company | Catalog Number | Comments/Description |
|---|---|---|---|
| Adhesive tape | Leica | #39475214 | |
| Alexa fluor 488-goat anti-Rabbit secondary antibody | Invitrogen | A-11034 | |
| Antifade Mountant with DAPI | Invitrogen | P36931 | |
| bovine serum albumin | Sigma | A2153 | |
| Coverslips | Fisher Brand | 12–545-E | |
| Cryostat | Leica | CM1850 | |
| EDTA | Sigma | E6758 | |
| Fluorescence microscope | Olympus | BX51 | |
| Gelatin | Sigma | G1890 | |
| In Situ Cell Death Detection Kit | Millipore | S7165 | |
| Microscope slides | Fisher Brand | 12-550-15 | |
| OCT Compound | Fisher Healthcare | 23-730-571 | |
| Paraformaldehyde (PFA) | Sigma | P6148 | |
| Phosphate buffered saline (PBS) | Sigma | P4417 | |
| Polyethylene glycol tert-octylphenyl ether | Sigma | T9284 | Triton™ X-100 |
| Proteinase K | Invitrogen | AM2542 | |
| Rabbit anti-Ki67 antibody | Cell Signaling Technology | 9129 | Lot#:3; RRID:AB_2687446 |
| Rabbit anti-pSmad1/5/9 antibody | Cell Signaling Technology | 13820 | Lot#:3; RRID:AB_2493181 |
| Sodium citrate | Sigma | 1613859 | |
| Sucrose | Sigma | S9378 | |
| Tris | Sigma | 10708976001 |
4. Imaging Acquisition
Use positive controls (tissues positive for the target antigen) to check the signal labeling and negative controls (omit the primary antibody, isotype control, or tissues negative for the target antigen) to evaluate the background of images under the fluorescent microscope.
-
Set the equipment and camera conditions (exposures and other general settings) for imaging based on the signal intensity of negative and positive controls.
NOTE: These conditions vary by (1) cameras and microscopes used for imaging, (2) antibodies, and (3) tissues for each experiment. Common conditions used for craniofacial tissues are ISO 200 with an exposure time ranging from 1/100 s to 1 s depend on the quality and specificity of antibodies. Appropriate magnifications vary depending on the size of the samples and purpose of experiments.
Acquire images with conventional epifluorescence microscope or confocal microscope. Acquire images (including those of corresponding controls) in the same conditions for each color channel. Save images with the same format (tiff is best to preserve information).
5. Fluorescence Quantification
NOTE: Statistically comparing the staining between different groups will be more informative in many cases. With the immunofluorescence images, quantify the relative level of the protein by measuring signal density, counting positive cells, or calculating positive areas. For statistical analysis, the minimum number of biologically independent samples is 3. A typical method is to generate at least three sections from each sample and take images for at least three representative areas in each section.
- Quantification of fluorescence intensity using ImageJ
- Open the software, and use Analyze > Set Measurements to check that only Area and Integrated Density are selected. Use File > Open to open images to be analyzed.
- Use Toolbar to select either the square or circle icon on the far left. Select the area to be analyzed on the image using the selection tool. Use Analyze > Measure to get the readout of the selected area and integrated density in the Results window. Select a region next to a positive cell that has no fluorescence to read out the background.
- Repeat step 5.1.2 to analyze other images. Adjusted the area to be analyzed to match with that of the first image.
- Copy all the data in the Results window and paste into a spreadsheet when finished analyzing.
- Calculate the corrected fluorescence intensity (CTCF) as Integrated Density — (Area of selected cell × Mean fluorescence of background readings). Compare the difference of the corrected total cell fluorescence between samples and make a graph.
- Quantification of the positive cell number of fluorescent images using ImageJ
- Manual cell counting.
- Use ImageJ > Plugins > Analysis to install the Cell Counter plugin.
-
Use File > Open to open images to be analyzed. Use Plugins > Analysis > Cell counter to open the Counter window and the Results window.NOTE: Cell counter does not work on stacks. For counting stacks, plugin Plot Z Axis Profile, then use Image > Stacks > Plot Z Axis Profile to monitor the intensity of a moving ROI using a particle tracking tool. This tool can be either manual or automatic.
- Clicking one of buttons at the bottom of the Counter window to initiate counting. Click directly on a cell/object to be count until finishing.
- Click the Results button in the Count window. The total number of cells counted will be shown in the Results window. Save the result log as spreadsheet and analyze.
- Automated cell counting.
- Use File > Open to open images to be analyzed. Convert the RGB image into a grey scale image before proceeding.
- Use Image > Adjust > Threshold to select all the areas that need to be counted.
-
Use Analyze > Analyze Particles to get the number of cells/particles. Set a range of the valid particle size (e.g., 100-Infinity) instead of the default of 0-Infinity to count cells/particles within a specific range. Save the result log as spreadsheet and analyze.NOTE: To get other information from the image, besides area, go to Analyze > Set Measurements and select the box next to the information needed.
Representative Results
Embryonic craniofacial tissue sections
Following the above steps, heads were dissected from control (P0-Cre) or mutant (constitutively activated Bmpr1a in neural crest cells, P0-Cre; caBmpr1a) embryos at embryonic day (E) 16.5 or 18.5. After fixing in 4% PFA for 4 h, samples were embedded in OCT and cryosectioned coronally. Resulted sections were immunostained with antibodies against pSmad1/5/9 (downstream BMP signaling factors) or Ki67 (a cell proliferation marker) without antigen retrieval according to the protocol. As shown, pSmad1/5/9 (Figure 1A) and Ki67 (Figure 1C) were positive in the frontal bones of control embryos. In mutant embryos, the levels of pSmad1/5/9 was increased (Figure 1B), while those of Ki67 was decreased (Figure 1D) in the frontal bones. Cell death in those samples were also checked according to the protocol. As shown, more apoptotic cells were observed in the frontal bones of mutant embryos than those of control embryos (Figure 1E,F).
Figure 1: Examples of IF results of pSmad1/5/9, Ki67 or TUNEL in control embryos and mutant embryos with enhanced BMP activity.
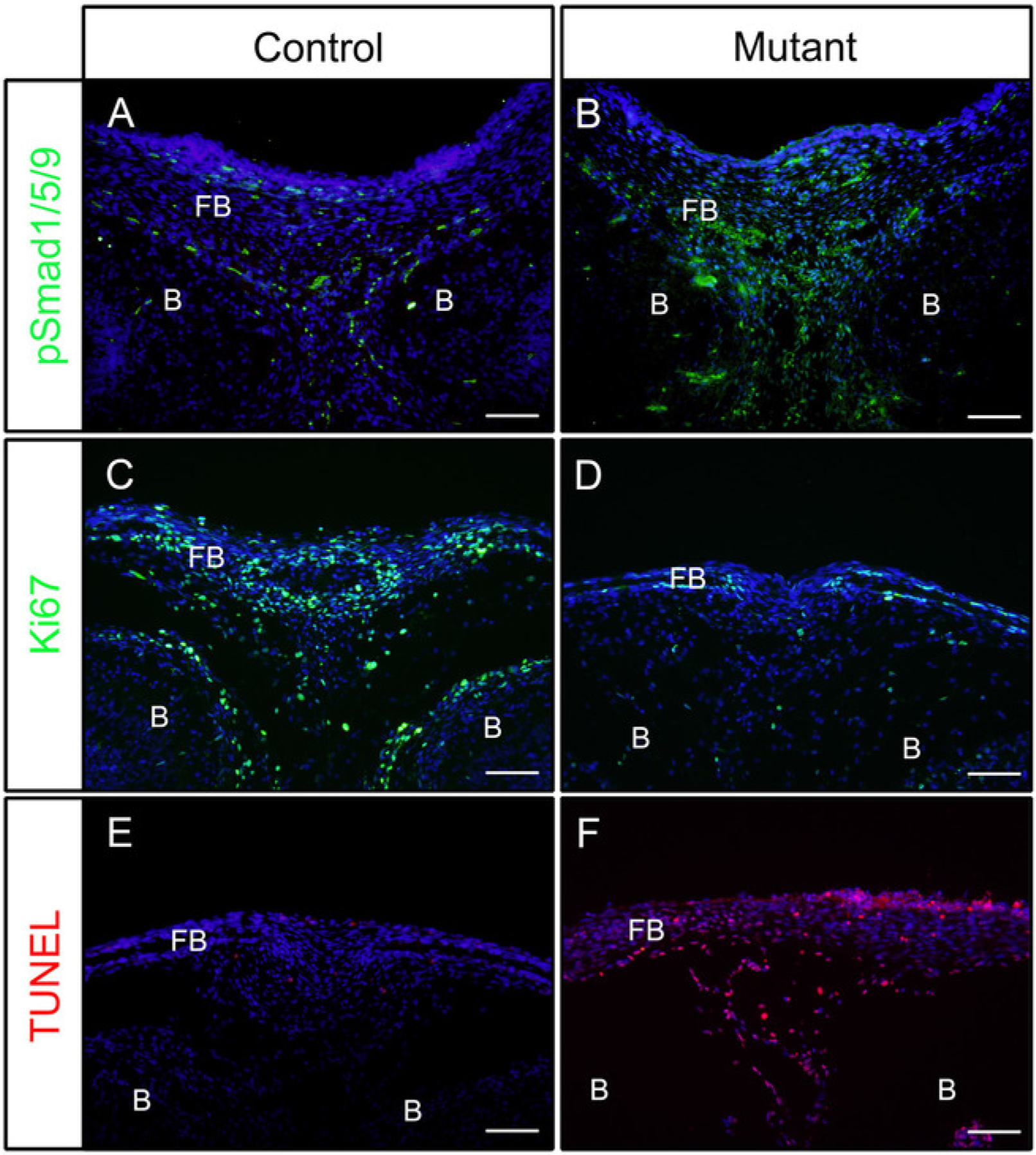
Constitutively activated Bmpr1a (caBmpr1a) mice were crossed with P0-Cre mice to increase BMP signaling activity in neural crest cells (NCCs). Heads of control (P0-Cre; caBmpr1a+/+) and mutant (P0-Cre; caBmpr1afx/+) embryos were dissected at E16.5 or E18.5, fixed with 4% PFA for 4h, cryoprotected with 30% sucrose for 1 day, embedded in OCT, and cryosectioned at −18 °C. Sections of the frontal bone (similar level with the eye) were used for immunodetection against pSmad1/5/9, Ki67, or TUNEL staining. (A, B) pSmad1/5/9 (green) staining patterns in the frontal bones of control (A) or mutant (B) embryos at E16.5. (C, D) Ki67 (green) staining patterns in the frontal bones of control (C) or mutant (D) embryos at E18.5. (E, F) TUNEL (red) staining patterns in the frontal bones of control (E) or mutant (F) embryos at E18.5. Nuclei were stained with DAPI (blue). FB = frontal bone, B = brain. Scale bars = 100 μm.
Undecalcified craniofacial tissues or long bone sections
Following the above steps for undecalcified hard tissues, heads from 3 week old mice (P0-Cre; mTmG (membrane-tomato and membrane GFP)) were fixed with 4% PFA and embedded in 8% gelatin. Coronal cryosections were washed with PBST and mounted with anti-fade medium with DAPI. Figure 2A,B demonstrate that gelatin does not interfere with fluorescent signals from sectioned tissues.
Figure 2: Examples of mTmG reporter signal results of undecalcified tissues in the head.
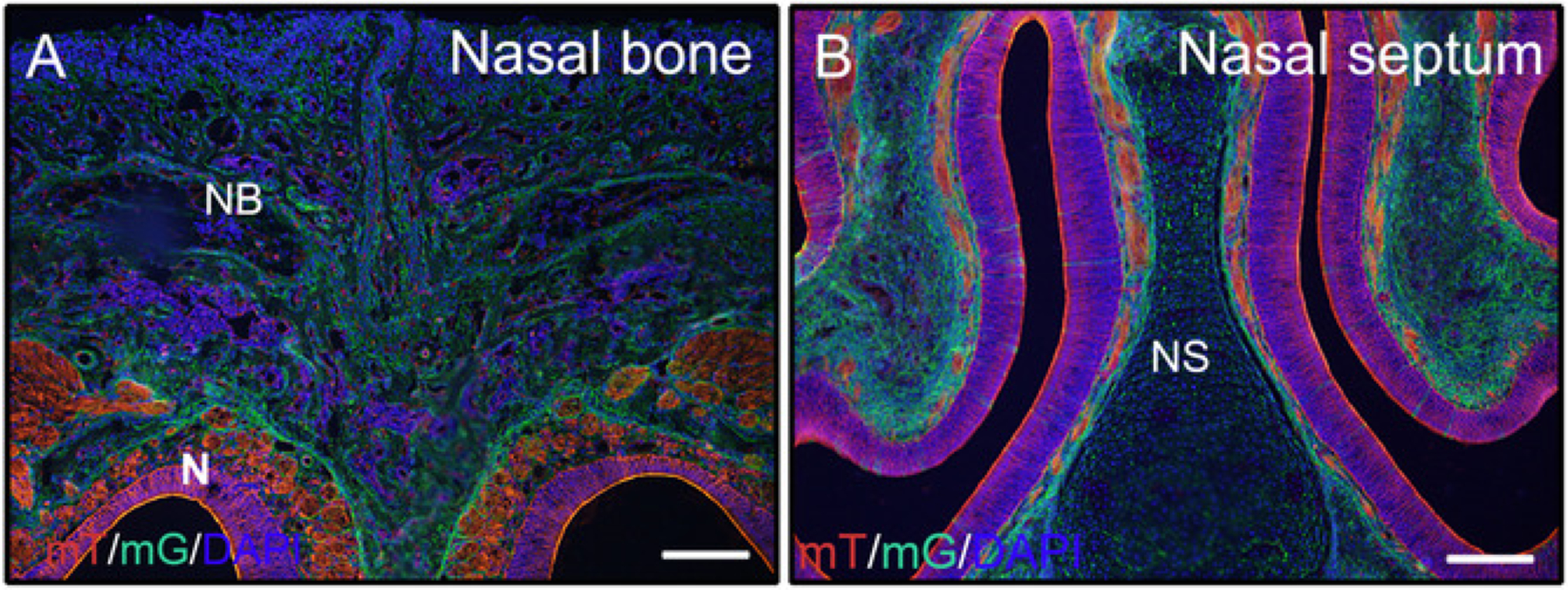
Heads from 3 week old P0-Cre mice with membrane-tomato and membrane GFP (mTmG) reporter were dissected, fixed with 4% PFA for 4h, cryoprotected with 30% sucrose for 2 days, embedded in 8% gelatin, and cryosectioned at −25 °C. Head sections clearly show GFP (green, Cre recombination positive) and Tomato (red, Cre recombination negative) signal in the nasal bone and nasal tissues (A, B). Nuclei were stained with DAPI (blue). NB = nasal bone, N = nasal tissues, NS = nasal septum. Scale bars = 250 μm.
Heads and femora from 3 week-old or 3 month-old mice were employed to check whether gelatin embedded undecalcified tissues are good for IF. The whole heads and femora were processed and sectioned according to the protocol. The resulted sections were used for SOX9 immunostaining (Figure 3) or OSX and E11/Podoplanin double immunostaining (Figure 4). As shown, good quality sections were obtained from most of the 3 week hard tissues, including the trabecular and the cortical compartments of the femur (Figure 3A,B, Figure 4A–D), the frontal bones (Figure 4E,F), the incisor (Figure 3E,F, Figure 4I,J), nasal tissues (Figure 3C,D), and the skull including the nasal-premaxilla suture and surrounding bones (Figure 4G,H) of the head. While, with 3-month-old samples, good quality sections were only obtained in some of the hard tissues, including the trabecular compartments of the femur (Figure 3G,H, Figure 4K,L), nasal tissues (Figure 3I,J), and the skull including the nasal-premaxilla suture and surrounding bones (Figure 4M,N) of the head. As shown in Figure 3, SOX9 positive cells were detected specifically in the chondrocytes of the growth plate (Figure 3B) and the joint (Figure 3H) from the femur, and the nasal septum (Figure 3D,J). In the 3 week-old incisor, SOX9 was detected in the mesenchymal cells (Figure 3F). OSX and E11 double staining results showed that OSX was detected in osteoblasts, while E11 was detected in osteocytes of bones from the femur and the head (Figure 4B,D,H,L,N). In the 3 week incisor, OSX was positive in odontoblasts, while E11 was positive in follicle mesenchymal cells (Figure 4J). Those results indicate that undecalcified hard tissues embedded with gelatin well-preserve antigen functions.
Figure 3: Examples of SOX9 immunostaining results of undecalcified tissues in the head and the femora.
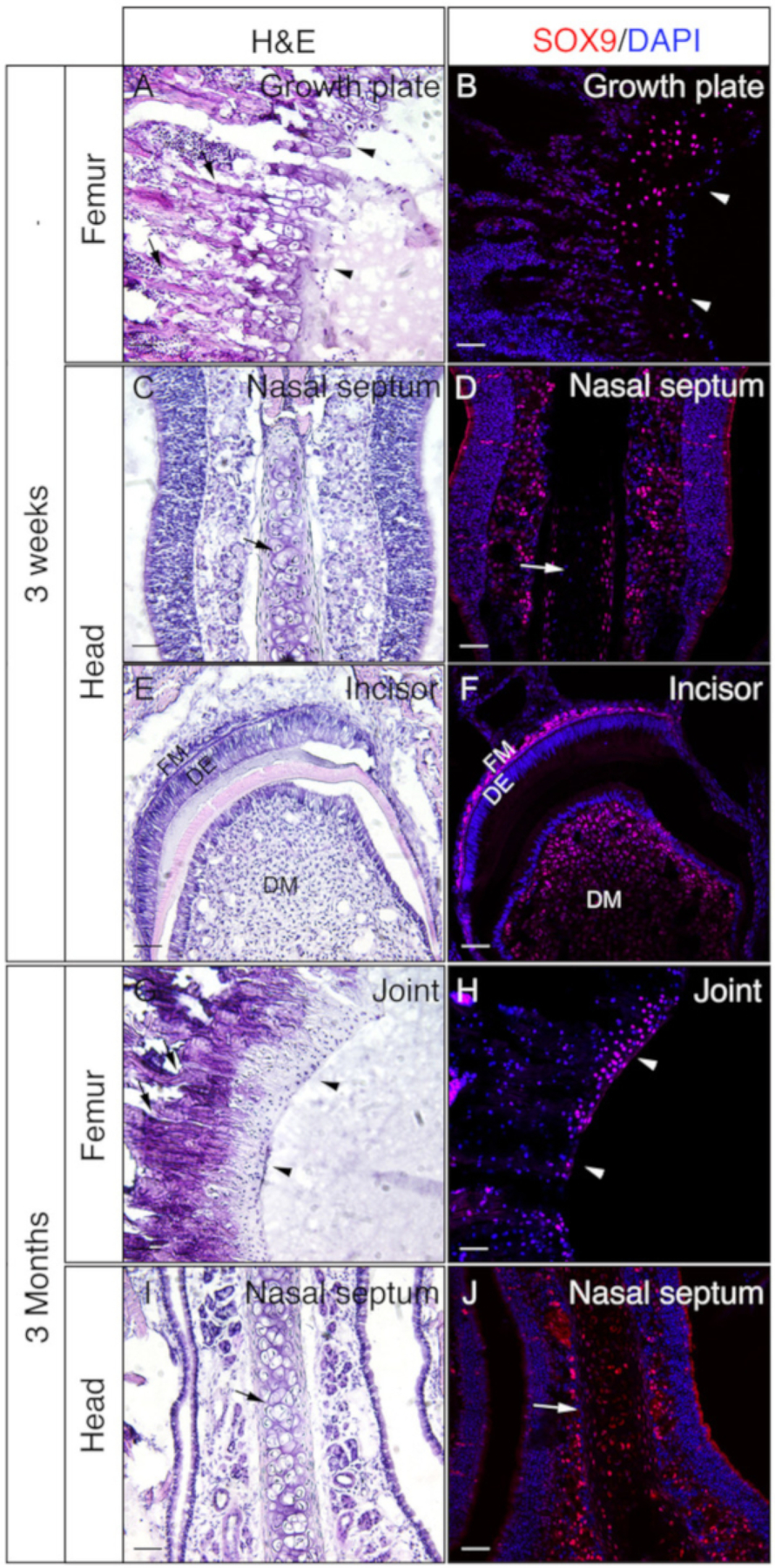
Heads and femora were dissected from 3 week or 3 month old mice, fixed with 4% PFA for 4h, cryoprotected with 30% sucrose for 2 days, embedded in 8% gelatin, and cryosectioned at −25 °C. Slides were used for immunodetection against SOX9 (red). Nuclei were stained with DAPI (blue) (B, D, F, H, J). Adjacent sections of those tissues were used for Hematoxylin & Eosin (H&E) staining (A, C, E, G, I). Arrow heads in A and B indicate growth plate and in G and H, articular cartilage. Arrows in A and G indicate trabecular bones and in C, D, I, and J, nasal septum. DM = dental mesenchyme, DE = dental epithelium, FM = follicle mesenchyme. Scale bars = 50 μm.
Figure 4: Examples of OSX and E11 double immunostaining results of undecalcified tissues in the head and the femora.
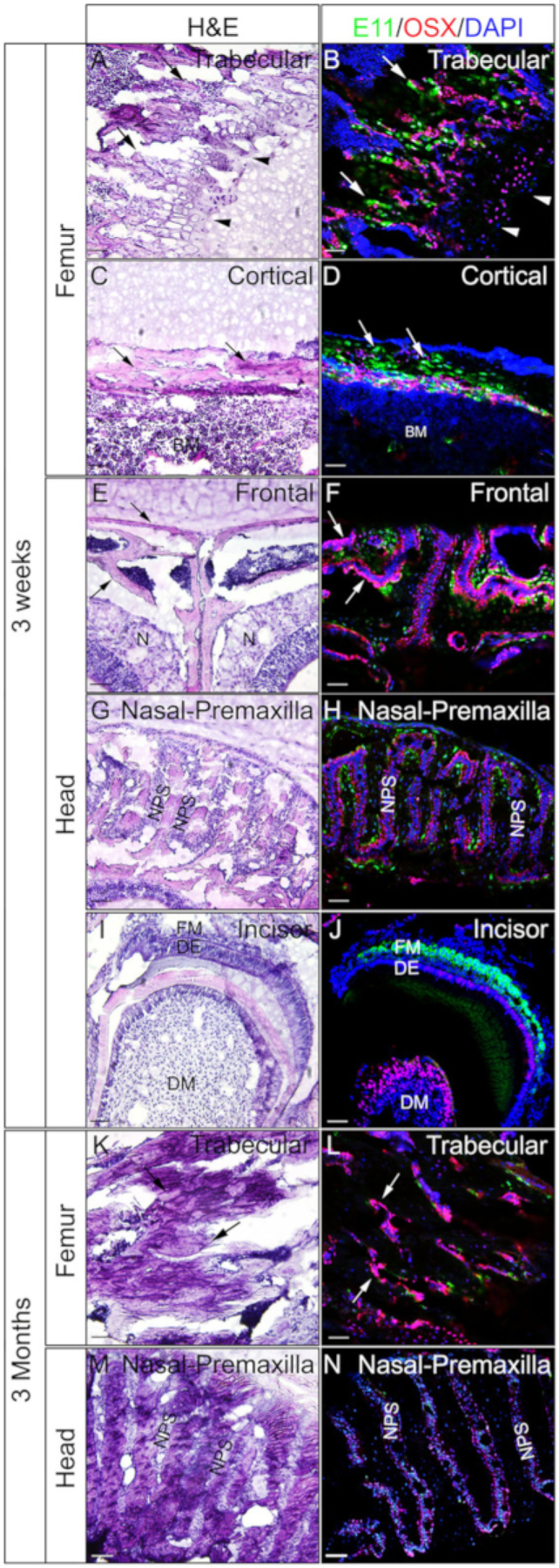
Heads and femora were dissected from 3 week-old or 3 month-old mice, fixed with 4% PFA for 4h, cryoprotected with 30% sucrose for 2 days, embedded in 8% gelatin, and cryosectioned at −25 °C. Sections were used for double immunostaining with antibodies against OSX (Red) and E11/Podoplanin (Green). Nuclei were stained with DAPI (blue) (B, D, F, H, J, L, N). Adjacent sections of those tissues were used for H&E staining (A, C, E, G, I, K, M). Arrows in A, B, K, and L indicate trabecular compartments of the femur; C and D, cortical compartments of the femur; and in E and F, the frontal bones. Arrowheads in A and B indicate growth plate. BM = bone marrow, N = nasal tissues, DM = dental mesenchyme, DE = dental epithelium, FM = follicle mesenchyme, NPS = nasal premaxilla suture. The frontal bones (E, F) and the nasal-premaxilla suture and surrounding bones (G, H, M, N) are also shown. Scale bars = 50 μm.
Discussion
Here we provide a detailed protocol for preparation of mouse head and undecalcified bone tissues, and cryosectioning for immunostaining of cell proliferation, cell death, and BMP signaling markers. We also detail the strategy for obtaining quantitative data from immunofluorescent images. Those methods can also be applicable to other tissues with appropriate modifications.
Conditions for tissue preparation vary by the size and type of tissues. The fixation and cryoprotection time usually need several hours to overnight. After fixation, the tissue can also be embedded in paraffin and sectioned with a microtome16. Though both paraffin and OCT work well for immunostaining, there are some differences between them. Paraffin blocks can be kept for multiple years at RT, while OCT blocks are for 1 year at −80 °C. Paraffin preserves tissue morphology, while ice crystal formed during OCT embedding may negatively affect tissue structures. Paraffin sometimes masks epitopes of antigens, while OCT preserves enzyme activities and antigen epitopes. Therefore, there is no need of antigen retrieval for most of the antibodies if fixed in 4% PFA for only 4 h or less and embedded in OCT. It is, however, still possible to get better results by antigen retrieval, if positive controls did not show good staining results in cryosections.
Both Hoechst dye and DAPI can be used for nuclear counter-staining. They have similarities, as both (1) are UV-excited, minor groove-binding chemicals to emit signals proportional to total DNA content, and (2) are subjected to photo-bleaching after a long exposure. However, Hoechst dyes are typically used for staining DNA content in live cells due to their high permeability. DAPI is typically used for staining DNA in fixed cells due to its low membrane permeability. In addition, DAPI generates a stronger and more stable signal than Hoechst.
Proper controls are essential for IF. The specificity of every new antibody should be confirmed by Western blot analysis, if applicable. The optimal working concentration of a particular primary antibody should be determined through the use of serial dilutions. A positive control (tissue or cell which is proved to express the protein/antigen) should be included to check the IF process and specificity of antibody. A negative control should also be included, e.g., absence of the primary antibody, or the substitution of normal IgG from the same species for the primary antibody, or tissues negative for the target antigen. When taking pictures, the sample without secondary antibodies (background control) should be examined independently with each channel to set the limits of signal gain and offset to be adapted for the final imaging. For detection of multiple labels, background control and single-labeled controls need to be prepared to avoid spectral overlap artifacts. All channels that will be used to obtain an image of a multiple-label sample must be subjected to independent background correction, because the level of autofluorescence in each channel varies substantially.
We also provide the protocol for preparation and cryosectioning of undecalcified hard tissues embedded in gelatin. For the OCT embedded decalcified hard tissues, most of the hard tissue sections will be detached from slide glasses during immunostaining procedures, because of their low adhesion character on the slide. The adhesive tape designed to facilitate cryosectioning helps to generate good quality sections. But, those sections are easily damaged when the tape is peeling off. For gelatin embedded tissues, there is no need for a tape-transfer system to generate good quality sections. As an embedding medium, gelatin can infiltrate the sample well, although it has a lower viscosity compared with OCT. Gelatin has been used in other histological applications, such as brain tissue17,18 and ultrathin sections of cells for immunocytochemistry19. Here, gelatin was used to embed undecalcified bone, which generates blocks easier to cryosection than OCT. There are several small tips to get good sections of gelatin embedded undecalcified hard tissues. The critical step is to embed with gelatin instead of OCT. To get better penetration, keep samples in 30% sucrose one more day after samples sink to the bottom. It is equally important to set the temperature lower than usual at about −25 °C. An ultra-sharp blade is not necessary. Although a lower cryo temperature (−25 °C) makes some improvements for cryosectioning of OCT embedded undecalcified hard tissues, it is still difficult to get a good integrity of tissue structures. As shown in Figure 2, Figure 3, and Figure 4, good quality sections applicable for immunostaining were obtained from gelatin embedded hard tissues (e.g., trabecular bones, cortical bones, skull bones, nasal tissues, and incisor). Those results proved that gelatin embedding significantly improves the specimen integrity of hard tissue sections, but also enhances adhesion of the sections to slide glasses. In addition, gelatin preserves antigen functions, and exhibits compatibility with fluorescent signals and immunostaining. However, this technique only works well for up to 3 month-old samples. Potential improvements of this method are (1) to dissect the samples further to separate the target tissue from other parts to make the structure of the tissue simple (in the case of teeth, the mandible or maxilla should be dissected and fixed instead of the whole head) and (2) to use 10% EDTA to decalcify tissues for only 2–3 days before cryoprotection. This short time of decalcification will not compromise the immunostaining results. Another concern is that, as a non-aqueous embedding media, gelatin cannot be easily removed from slides, which may lead to higher background depending on staining methods (e.g., H&E staining).
Immunostaining results are not easy to quantify, so they are usually used semi-quantitatively. Difficulties and limitations of the quantification of immunostaining of craniofacial tissues include but are not limited to the following: (1) it is difficult to define the area to be counted due to the complexity of the structure of craniofacial tissues; (2) it is difficult to define the labeled area or labeled cells due to the non-linear nature of immunostaining; (3) there is limited information for the dynamic range of the signal; (4) it is hard to compare the intensity of signals between images or groups due to the fading of fluorescent signal during image acquisition; and (5) the signal background may change significantly among antibodies, slides, and samples. To increase the reliability of the quantification results, the experiment should be carefully and strictly performed. All samples should be processed under the same conditions. During immunostaining, various controls are required to evaluate signal background and define the positive area or cells for the signal. Take convincing and representative images to clearly show the area to be counted and labeled cells with good contrast. In addition, during image acquisition, the camera setting and equipment setting must be kept consistent.
Taken together, we present a simple standard protocol for immunofluorescence on mouse craniofacial tissues, especially for undecalcified hard tissues. Immunostaining analysis of craniofacial tissues will not only help to understand the mechanism of morphogenesis during development, but also illustrate the changes during pathogenesis. In addition, immunostaining can also be used to study the expression pattern of other signaling pathway ligands, receptors, or other phenotypic markers, besides cell proliferation, cell death, and the BMP signaling pathway. However, the critical steps of an immunostaining experiment must be modified appropriately for each antigen/antibody or tissue to get specific staining and minimized non-specific background signals.
Acknowledgments
This work was supported by the National Institutes of Health (R01DE020843 to Y.M.), the International FOP Association (Y.M.), and a grant-in-aid from the National Natural Science Foundation of China (31500788 to J.Y.).
Footnotes
Video Link
The video component of this article can be found at https://www.jove.com/video/59113/
Disclosures
The authors have nothing to disclose.
References
- 1.Trinh Le A., Fraser SE Imaging the cell and molecular dynamics of craniofacial development: challenges and new opportunities in imaging developmental tissue patterning. Current Topics in Developmental Biology. 115, 599–629 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Marcucio R et al. Facial morphogenesis: physical and molecular interactions between the brain and the face. Current Topics in Developmental Biology. 115, 299–320 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graf D et al. Common mechanisms in development and disease: BMP signaling in craniofacial development. Cytokine & Growth Factor Reviews. 27, 129–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Snider TN, Mishina Y Cranial neural crest cell contribution to craniofacial formation, pathology, and future directions in tissue engineering. Birth Defects Research Part C: Embryo Today. 102 (3), 324–332 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mishina Y, Snider TN Neural crest cell signaling pathways critical to cranial bone development and pathology. Experimental Cell Research. 325 (2), 138–147 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Hecke D Routine Immunohistochemical Staining Today: Choices to Make, Challenges to Take. Journal of Histotechnology. 1, 45–54 (2002). [Google Scholar]
- 7.Xiao C, Dan-Bi C Double staining immunohistochemistry. North American Journal of Medical Sciences. 2, (5), 241–245 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zongli Q et al. Comparison of immunofluorescence and immunohistochemical staining with anti-insulin antibodies on formalin-fixed paraffin-embedded human pancreatic tissue microarray sections. International Journal of Clinical and Experimental Pathology. 10, (3), 3671–3676 (2017). [Google Scholar]
- 9.Montgomery SC, Cox BC Whole mount dissection and immunofluorescence of the adult mouse cochlea. Journal of Visualized Experiments. (107), e53561 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dun XP, Parkinson DB Whole mount immunostaining on mouse sciatic nerves to visualize events of peripheral nerve regeneration. Methods in Molecular Biology. 1739, 339–348 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Akkiraju H et al. An Improved Immunostaining and Imaging Methodology to Determine Cell and Protein Distributions within the Bone Environment. Journal of Histochemistry & Cytochemistry. 64, (3), 168–178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gonzalez-Chavez SA et al. Assessment of different decalcifying protocols on Osteopontin and Osteocalcin immunostaining in whole bone specimens of arthritis rat model by confocal immunofluorescence. International Journal of Clinical and Experimental Pathology. 6 (10), 1972–1983 (2013). [PMC free article] [PubMed] [Google Scholar]
- 13.Kapelsohn K Improved Methods for Cutting, Mounting, and Staining Tissue for Neural Histology. Protocol Exchange. (2015). [Google Scholar]
- 14.Kalaskar VK, Lauderdale JD Mouse embryonic development in a serum-free whole embryo culture system. Journal of Visualized Experiments. (85), e50803 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi SR et al. Antigen retrieval techniques: current perspectives. Journal of Histochemistry & Cytochemistry. 49, (8), 931–937 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Adell T et al. Immunohistochemistry on paraffin-embedded planarian tissue sections. Methods in Molecular Biology. 1774, 367–378 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Griffioen HA et al. Gelatin embedding to preserve lesion-damaged hypothalami and intracerebroventricular grafts for vibratome slicing and immunocytochemistry. Journal of Neuroscience Methods. 43, 43–47 (1992). [DOI] [PubMed] [Google Scholar]
- 18.Sarkar S et al. In situ demonstration of Fluoro-Turquoise conjugated gelatin for visualizing brain vasculature and endothelial cells and their characterization in normal and kainic acid exposed animals. Journal of Neuroscience Methods. 219 (2), 276–284 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Oorschot V et al. A novel flat#embedding method to prepare ultrathin cryosections from cultured cells in their in situ orientation. Journal of Histochemistry & Cytochemistry. 50, 1067–1080 (2002). [DOI] [PubMed] [Google Scholar]