

Abstract
Objectives
Enhanced inactivated influenza vaccines (eIIV) aim to increase immunogenicity and protection compared with the widely used standard IIV (S‐IIV).
Methods
We tested four vaccines in parallel, FluZone high dose, FluBlok and FluAd versus S‐IIV in a randomised controlled trial of older adults and in a mouse infection model to assess immunogenicity, protection from lethal challenge and mechanisms of action.
Results
In older adults, FluAd vaccination stimulated a superior antibody profile, including H3‐HA antibodies that were elevated for up to 1 year after vaccination, higher avidity H3HA IgG and larger HA stem IgG responses. In a mouse model, FluAd also elicited an earlier and larger induction of HA stem antibodies with increased germinal centre responses and upregulation and long‐term expression of B‐cell switch transcription factors. Long‐term cross‐reactive memory responses were sustained by FluAd following lethal heterosubtypic influenza challenge, with reduced lung damage and viral loads, coinciding with increased T‐ and B‐cell recall. Advantages were also noted for the high‐dose FluZone vaccine in both humans and mice.
Conclusion
The early, broadly reactive and long‐lived antibody response of FluAd indicates a potential advantage of this vaccine, particularly in years when there is a mismatch between the vaccine strain and the circulating strain of influenza viruses.
Keywords: adjuvant, antibody, B cell, HA stem, infection, influenza vaccine
In this randomised clinical trial, we showed that enhanced influenza vaccines boost antibody quality in older adults, and showed a particular advantage of adjuvanted vaccines for long‐term high‐avidity and haemagglutinin‐stalk antibodies. This action translated to an increased protection by the same adjuvanted vaccines in mice for reduced viral loads and inflammation as a result of early and enhanced B‐cell recruitment and activation for antibody production.

Introduction
Vaccination against influenza viruses aims to mitigate morbidity and mortality associated with infection. Adults over 65 years of age are the most susceptible group to complications of influenza infection, account for the majority of influenza‐associated mortality and are widely recommended for annual vaccination.1 Influenza vaccine effectiveness varies from year to year and can be impacted by antigenic drift,2 mismatch,3, 4 pandemic emergence,5 egg adaptations6 and age.7 Standard‐dose inactivated influenza vaccines (S‐IIV) contain 15 µg of haemagglutinin (HA) per strain and are the most widely used vaccine formulation to reduce the burden of seasonal influenza epidemics.8 Prioritising the use of newly available enhanced inactivated influenza vaccines (eIIVs) may increase the longevity, breadth and quality of vaccine‐mediated protection against influenza virus infections and associated morbidity and mortality in older adults.9, 10
Available eIIVs include vaccines that have a higher antigen content than S‐IIV, such as recombinant FluBlok (recombinant HA protein, three times of HA content) or FluZone high dose (subunit vaccine, four times of HA content), or contain adjuvants, such as FluAd (MF59). The adjuvanted eIIV (A‐eIIV), FluAd, contains MF59, an oil‐in‐water emulsion acting as both adjuvant and carrier for the antigens, previously increasing the breadth, diversity and avidity of HA antibodies.11 A recombinant HA‐only high‐dose IIV (R‐eIIV), FluBlok, has a reported higher vaccine efficacy than S‐IIV.12 However, focusing the FluBlok response to a single HA protein target, whilst a critical protein for viral entry, may diminish the breadth of vaccine‐mediated protection, neglect NA‐mediated protection and affect CD4+ T‐cell‐mediated help directed at internal proteins.13 The high‐dose inactivated IIV (H‐eIIV), FluZone, induces significantly higher antibody responses and provides better protection against laboratory‐confirmed influenza than S‐IIV among the elderly.14 H‐eIIV has also shown improved immunogenicity by HAI measures in HIV‐immunocompromised patients, despite their low overall CD4+ T‐cell counts. This indicates that increasing the antigenic dose may result in a correspondingly increased immune response,15 which is particularly important to older adults who experience immunosenescence.
Structural and post‐translational modifications of the HA as a result of vaccine production methods, antigen dose and the inclusion of adjuvants may impact serological and cellular immunity.16, 17 Initial results of HAI and T‐cell responses from our ongoing trial have shown R‐eIIV elicits higher titre microneutralisation antibody titres against influenza A (H3N2) and CD4+ T‐cell boosting, followed by H‐eIIV, then A‐eIIV.18 Parallel comparison of eIIVs through experimental immunogenicity studies may help elucidate their mechanism of action, as large‐scale efficacy studies are rare for already‐licensed vaccines. In this study, we extended serological results from our ongoing randomised trial of eIIVs in older adults to a vaccination‐challenge mouse model to assess eIIV‐mediated protection.
Results
A‐eIIV increases the avidity, stem and long‐term H3‐HA‐specific IgG responses in vaccinated human subjects
A randomised trial of community‐dwelling older adults between 65 and 82 years old was initiated in 2017/2018 in Hong Kong to evaluate the immunogenicity of eIIVs (http://ClinicalTrials.gov NCT03330132).18 Older adults (Supplementary table 1) were randomly allocated to receive the 2017/2018 Northern Hemisphere formulation of either standard FluQuadri (S‐IIV, Sanofi Pasteur, Lyon, France) or one of three eIIVs, including adjuvanted (FluAd, A‐eIIV, Seqirus, Maidenhead, UK), high dose (FluZone high dose, H‐eIIV, Sanofi Pasteur) and recombinant HA (FluBlok, R‐eIIV, Sanofi Pasteur) (Table 1).18 Sera were collected for baseline responses (day 0) and then after vaccination at days 7 (acute), 30 (short‐term memory) and 365 (long‐term memory) (Figure 1a). Standard HAI serological responses and T‐cell responses are reported elsewhere.18
Table 1.
IIV composition and dosage for human and mouse vaccination
| Abbreviation | Type | Vaccine name | Valence | Strains included | HA content per vial (μg) | HA content per mouse dose (μg) | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H1 | H3 | B/Vic | B/Yam | Per strain | Total | Per strain | Total | ||||
| S | Standard | FluQuadri | QIV | ● | ● | ● | ● | 15 | 60 | 5 | 20 |
| A | Adjuvant | FluAd | TIV | ● | ● | ● | – | 15 | 45 | 5 | 15 |
| H | High dose | FluZone | TIV | ● | ● | ● | – | 60 | 180 | 20 | 60 |
| R | Recombinant HA protein | FluBlok | QIV | ● | ● | ● | ● | 45 | 180 | 15 | 60 |
Figure 1.
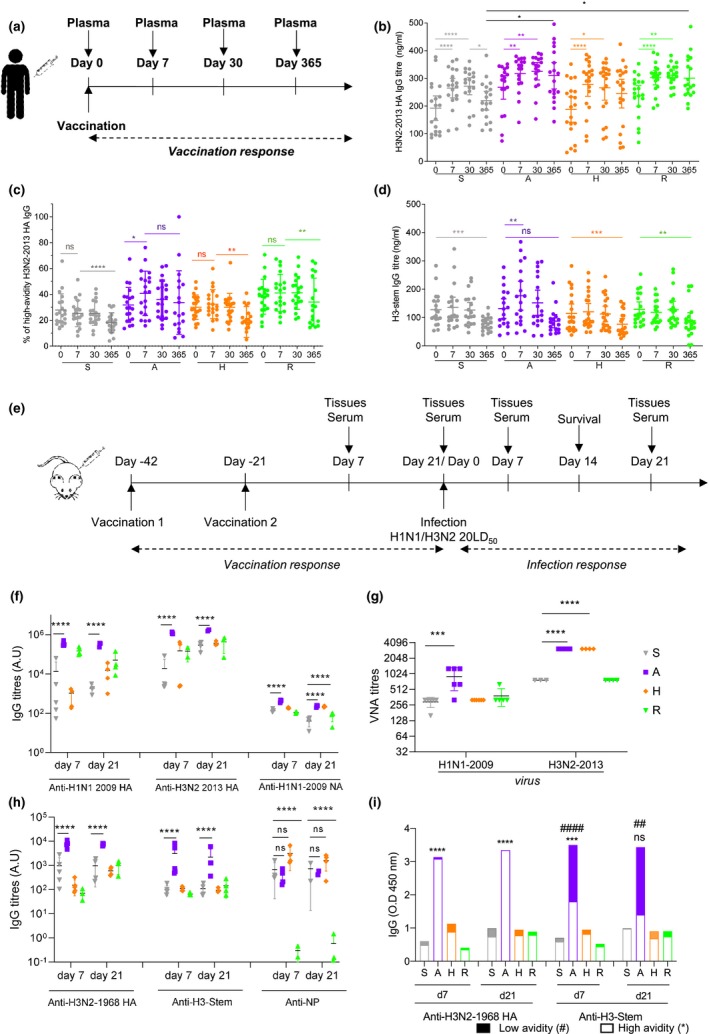
A‐eIIV induces high‐avidity H3‐2013 and H3‐stem IgG in humans and mice. A randomised clinical trial to assess eIIV immunogenicity compared with S‐IIV, sampled blood plasma at days 0 (baseline), 7, 30 and 365 after vaccination (a). H3N2‐2013 HA titres (b), high‐avidity H3N2‐2013 HA (c) and H3‐stem‐specific (d) IgG responses were measured by ELISA. The same vaccines were assessed in a mouse model of vaccination and infection (e). Vaccine strains, H1N1‐2009 and H3N2‐2013 HA‐specific and H1N1‐2009 NA‐specific, day 7 and 21 post‐vaccination IgG titres by ELISA (f) and day 21 post‐vaccination by VNA (g). Cross‐reactive, H3N2‐1968, H3‐stem HA‐specific and NP‐specific, day 7 and 21 post‐vaccination IgG responses by ELISA (h). Proportion of low‐ and high‐avidity H3N2‐1968 HA and H3‐stem‐specific IgG (i). Data represent the mean, SEM and individual responses, (b–d) n = 20 per group and (f–i) n = 4 or 5 per group. For b–d, *shows statistical significance by one‐way ANOVA with the Friedman test in each vaccine group day 0 versus days 7, 30 and 365. For (f–i), *shows statistical significance by one‐way ANOVA (mixed model) for eIIV versus S‐IIV by Tukey's multiple comparison test. For (i), #shows statistical significance for low‐avidity antibodies and *for high‐avidity antibodies for eIIV versus S‐IIV by one‐way ANOVA (mixed model) and Tukey's multiple comparison test. # or *P < 0.05, ## or **P < 0.01, ### or ***P < 0.005, #### or ****P < 0.001, *P < 0.05, **P < 0.01 and ***P < 0.005, experiments were repeated twice. Human serological experiments were performed with n = 20 individuals/group, experiments were repeated twice. Mouse experiments were performed with individual mouse serum from 4‐5 mice, and experiments were repeated twice.
We assessed anti‐HA IgG titres (Figure 1b) and avidity (Figure 1c) against a representative vaccine strain H3N2‐2013. Antibody responses were equivalent between all vaccine groups at days 7 and 30, but importantly, A‐eIIV and R‐eIIV H3N2‐2013 HA IgG responses were elevated to long‐term memory 1 year after vaccination compared with S‐IIV. The A‐eIIV vaccine also elicited a significant selective increase in high‐avidity H3N2‐2013 HA IgG at day 7, which was maintained 1 year after vaccination (ns, day 7 versus day 365; Figure 1c). A‐eIIV also elicited an increase in HA stem‐specific antibodies at day 7 from both H3‐stem (Figure 1d, P = 0.0015), which were unchanged at 1 year later (ns, day 7 versus day 365; Figure 1d), and H1‐stem (Supplementary figure 1a). Furthermore, H‐eIIV also mounted a short‐lived H1‐stem response at day 7 post‐vaccination (Supplementary figure 1a). Therefore, A‐eIIV induced the most superior quality IgG responses in terms of long‐term memory, high‐avidity H3‐IgG and H3‐stem‐specific responses.
Subsequent experiments focused on H3‐HA‐specific responses as a result of the challenge of antigenic matching for current vaccines and reduced vaccine efficacy.3, 6 Our initial study also found improved responses to enhanced vaccines for H3N2 viruses,18 and we also observed similar trends for H1‐HA‐specific responses (Supplementary figures 1 and 5) and influenza B responses with limited reagents (data not shown).
A‐eIIV in mice induces superior influenza antibody responses for vaccine antigens, cross‐reactive proteins and neutralisation
To understand the protective potential of high‐avidity H3‐HA and H3‐stem IgG by eIIV in older adults, especially A‐eIIV, we evaluated the same vaccines in mice (Figure 1e). Mice were vaccinated twice, and post‐vaccination immune responses were assessed at days 7 and 21. The A‐eIIV group had significantly increased day 7 and 21 post‐vaccination magnitude of HA IgG responses to vaccine strains, H1N1‐2009 and H3N2‐2013 HA (Figure 1f), whilst other eIIVs were equivalent to S‐IIV for these HA IgG responses. In addition, the H1N1‐2009 NA IgG response was elevated for A‐eIIV at days 7 and 21 post‐vaccination, whilst H‐eIIV was delayed to until day 21 post‐vaccination compared with S‐IIV (Figure 1f). As R‐eIIV also does not contain NA, no substantial NA‐specific IgG responses were detected. Furthermore, virus neutralisation antibodies (VNAs) at day 7 post‐vaccination (Figure 1g) were significantly increased for A‐eIIV against H1N1‐2009 virus (907 ± 174 versus 293 ± 27 S‐IIV) and H‐eIIV and A‐eIIV against H3N2‐2013 virus compared with S‐IIV.
Cross‐reactive antibodies for an antigenically older H3N2 virus (H3‐1968), conserved proteins (NP) and functionally conserved targets (H3‐stem and H1‐stem) were also assessed (Figure 1h and Supplementary figure 1b). Again, only A‐eIIV compared with S‐IIV had significantly elevated IgG for H3N2‐1968 HA, H3‐stem (Figure 1h) and H1‐stem (Supplementary figure 1b). Cross‐reactive NP‐specific IgG responses were comparable for S‐IIV, A‐eIIV and H‐eIIV (Figure 1h). As the recombinant HA protein vaccine, R‐eIIV, also does not contain NP, no substantial NP‐specific IgG responses were detected. Cross‐reactive H3N2‐1968 HA IgG responses by A‐eIIV were predominantly high‐avidity and significantly increased in magnitude (Figure 1i), whilst both high‐ and low‐avidity H3‐stem‐specific IgG were increased for A‐eIIV (P = 0.003; Figure 1i).
A‐eIIV increases memory B‐cell and long‐term antibody responses in mice
We next investigated the effects of eIIV on the generation of isotype‐switched memory B‐cell responses (B220+ CD95/Faslow IgD− IgG/IgM+) (Figure 2a), which are important for long‐term antibody production. Shortly after vaccination (day 7), there was no significant increase in total IgM+ or IgG+ B memory cells in the vaccination site draining inguinal lymph node (iLN) for S‐IIV compared with unvaccinated PBS mock‐treated mice (Figure 2b and c). Meanwhile, there was a significant induction of both IgG+ and IgM+ memory B cells in by A‐eIIV compared with S‐IIV at day 7, which was maintained at 21 days post‐vaccination.
Figure 2.
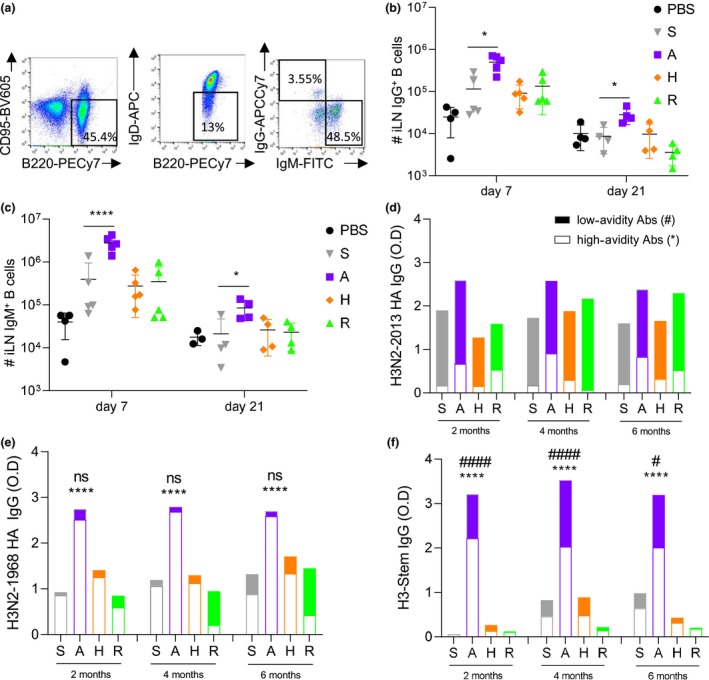
Memory B‐cell responses and long‐term antibody responses are increased in A‐eIIV‐vaccinated mice. BALB/c mice were vaccinated twice i.m. with either PBS, S‐IIV, A‐eIIV, H‐eIIV or R‐eIIV vaccines 3 weeks apart. iLN was collected at days 7 and 21 post‐vaccination to quantify by FACS (a), IgG+ (b) and IgM+ (c) B memory cells. Longitudinal serum samples were serially sampled from individual mice at 2, 4 and 6 months post‐vaccination for H3N2‐2013 (d), H3N2‐1968 (e) and H3‐stem (f) IgG by ELISA for avidity. Data represent the mean, SEM and individual responses, n = 4–8 mice per group. For (b, c), *shows statistical significance for eIIV versus S‐IIV by one‐way ANOVA (mixed model) and Tukey's multiple comparison test. For d–f, #shows statistical significance for low‐avidity antibodies and *for high‐avidity antibodies for eIIV versus S‐IIV by one‐way ANOVA (mixed model) and Tukey's multiple comparison test. # or *P < 0.05, ## or **P < 0.01, ### or ***P < 0.005, and #### or ****P < 0.001. Experiments were repeated twice. Representative FACS plots were gated on lymphocytes for FSC/SSC and live/dead (a).
To asses post‐vaccination waning, the kinetics of antibody responses at 2, 4 and 6 months were assessed longitudinally (Figure 2d–f). All vaccines induced stable long‐term H3‐specific IgG until 6 months post‐vaccination; however, A‐eIIV was consistently superior for increased magnitude and avidity of cross‐reactive H3N2‐1968 HA (Figure 2e) and H3‐stem responses (Figure 2f).
Increased class switch and hypermutation‐associated transcripts in A‐eIIV B cells
A‐eIIV elicited stable long‐lived high‐avidity antibodies to vaccine strains and cross‐reactive HA proteins, H3N2‐1968 and H3‐stem. Therefore, we investigated further the germinal centre (GC) B‐cell reaction and associated CD4+ T follicular helper (Tfh)‐cell responses in the iLN induced by the eIIVs (Figure 3a). A‐eIIV elicited a significantly increased Tfh response, which was more than double that of S‐IIV at day 7 and still ongoing at day 21 (Figure 3b). Concurrently, the GC B‐cell response was also doubled at day 7 in the A‐eIIV group compared with S‐IIV responses (Figure 3c). At day 21, Tfh and GC cell counts decreased by 30‐fold in most vaccine groups, but the A‐eIIV group still maintained significantly higher GC responses than S‐IIV (P = 0.02; Figure 3c). In comparison with S‐IIV, H‐eIIV and R‐eIIV vaccination did not display any significant increase in GC or Tfh cells (Figure 3b and c).
Figure 3.
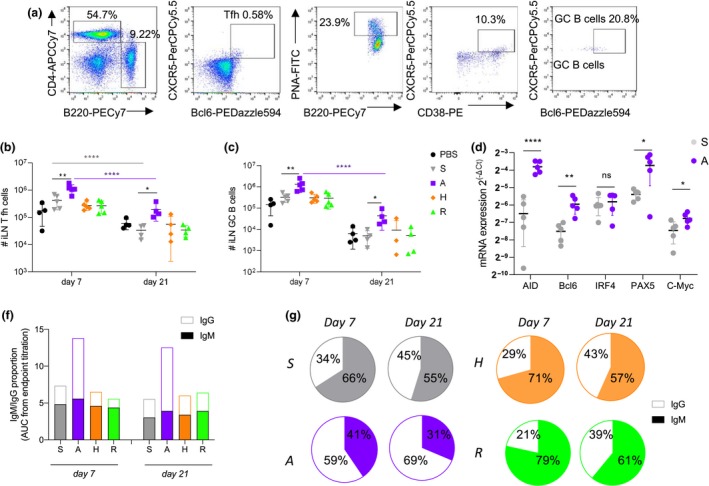
Elevated CD4+ Tfh and GC B cells by increased expression of SHM and CSR genes lead to early IgG class switch for A‐eIIV. Vaccine responses were assessed by FACS (a) for CD4+ Tfh cells (b) and GC B cells (c) at days 7 and 21 post‐vaccination in the iLN. Expression of AID, Bcl6, IRF4, Pax5 and c‐Myc in B cells from iLN (relative expression to GAPDH expression, expressed in ) at day 7 post‐vaccination (d). Quantity (f) and proportion (g) of H3N2‐1968 HA‐specific IgM and IgG at days 7 and 21 post‐vaccination, measured by ELISA AUC endpoint titres (Supplementary figure 2). Data represent the mean, SEM and individual responses, n = 4 or 5 mice per group. *shows statistical significance by one‐way ANOVA (mixed model) for eIIV versus S‐IIV, *P < 0.05, **P < 0.01 and ***P < 0.005, experiments were repeated twice. Representative FACS plots were gated on lymphocytes for FSC/SSC and live/dead (a).
A‐eIIV increased the avidity and magnitude of cross‐reactive antibody responses, which was accompanied by increased Tfh and GC cellular responses in the iLN, suggesting an early and enhanced somatic hypermutation (SHM) and class switch recombination (CSR) response. Therefore, we assessed the transcription levels of Aicda for activation‐induced cytidine deaminase (AID) enzyme, which is responsible for the B‐cell receptor maturation through the SHM process in the light zone of LN.19 Expression of AID was increased in iLN B cells at 7 days after A‐eIIV compared with S‐IIV (P < 0.0001; Figure 3d). Additional transcription factors controlling GC formation and AID activation, namely Bcl6, Pax5 and c‐Myc,20 were also significantly increased in A‐eIIV B‐cell responses compared with S‐IIV (Figure 3d).
As AID is also responsible for the CSR that takes place in the light zone of LN, we determined the level of class switching (from IgM to IgG responses) at days 7 and 21 post‐vaccination using ELISA endpoint titrations of H3‐2013 HA‐specific IgG and IgM responses in mouse serum (Figure 3f and g and Supplementary figure 2). At day 7, the S‐IIV H3N2‐2013 HA response was predominantly IgM (66%) rather than IgG (34%), whereas the A‐eIIV response was predominantly IgG (59%) rather than IgM (41%) (Figure 3h). This indicates an earlier switch from the default IgM isotype towards the IgG isotype by A‐eIIV. By day 21, the S‐IIV H3N2‐2013 HA IgG response increased (55% IgM, 45% IgG) but was still significantly lower than the A‐eIIV response (31% IgM, 69% IgG) (Figure 3h and Supplementary figure 2). Similar results were obtained for the cross‐reactive H3N2‐1968 HA‐specific antibodies, consolidating an earlier class switch induced by the A‐eIIV for cross‐reactive responses as well (Supplementary figure 2). The H‐eIIV and R‐eIIV, H3N2‐2013 HA IgM and IgG switch ratios were comparable to S‐IIV at both time points (Figure 3h). Also, the H3N2‐1968 HA IgG subclass response observed in the A‐eIIV group of mice was increased for both IgG1 and IgG2a compared with the S‐IIV, and for H3‐stem for IgG1, IgG2a and IgG2b, which are associated with greater effector functions and maturation of B‐cell selection,21 suggesting an increased IgG subclass maturation in response to A‐eIIV (Supplementary figure 3).
Increased HA stem neutralisation by A‐eIIV but comparable HA landscape to S‐IIV
The A‐eIIV vaccine in mice and humans elicited significantly higher magnitude of antibodies binding to the H3‐stem (Figure 1d, h, and i) and H1‐stem (Supplementary figure 1b). We determined whether these antibodies were also neutralising using a chimeric H9/1N3 virus that contains a H9 HA head domain (A/guinea fowl/Hong Kong/WF10/99) and a H1‐stem domain (A/Puerto Rico/8/1934) for H1‐stem‐mediated neutralisation.22 The day 7 VNA titres against cH9/1N3 by A‐eIIV were significantly increased compared with S‐IIV response (Figure 4a); therefore, HA stem binding antibodies stimulated by A‐eIIV are also neutralising.
Figure 4.
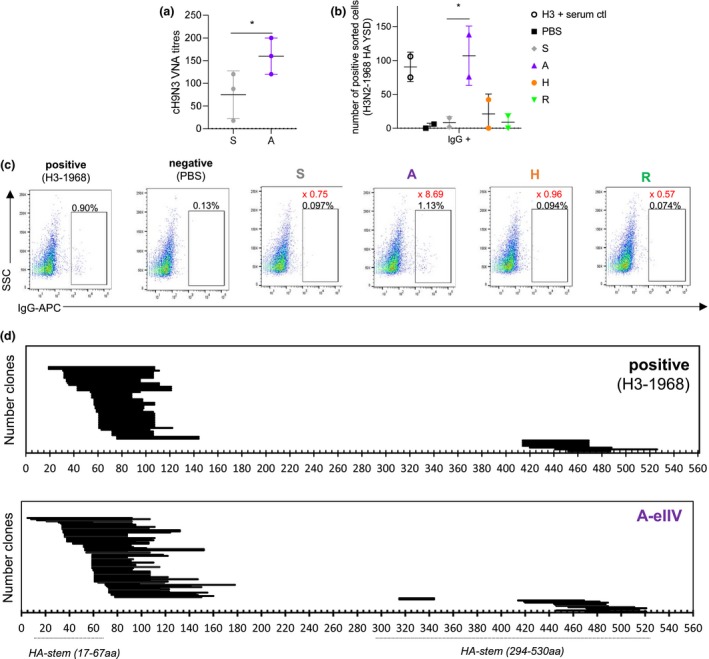
HA stem antibodies are neutralising and map to the HA stem for A‐eIIV responses. VNA titres for cH9/1N3 virus measuring HA stem‐mediated neutralisation of A‐eIIV and S‐IIV serum from day 7 post‐vaccination (a) (n = 4 pooled serum in triplicate). YSD H3N2‐1968 library binding to eIIV and control pooled H3N2‐1968 convalescent mouse serum (b) for FACS sorting. Mapping of the yeast clone HA amino acid sequence by serum binding for A‐eIIV (n = 85 clones) and recovered H3N2‐1968 (n = 48 clones) serums (c). YSD binding (b, c), pooled serum from n = 5 mice per group. In (d), the percentage of the population of interest is shown in black on the FACS plots, and the fold change of the percentage versus PBS is shown in red. Experiments were repeated twice, and HA stem amino acid positions are as previously described.29
A H3N2‐1968 HA yeast surface display (YSD) library in combination with mouse serum from day 7 post‐vaccination was used to confirm and map the presence of cross‐reactive anti‐H3N2‐1968 HA antibodies from the 2017/2018 vaccination. Assay controls showed specific binding by H3N2‐1968 HA convalescent serum (0.82% HA‐YSD+) and minimal background from PBS‐vaccinated mice (0.05% HA‐YSD+) (Figure 4b). However, we did not detect enough HA‐YSD binding in the serum from S‐IIV, H‐eIIV or R‐eIIV (Figure 4b) for further HA‐YSD mapping, whilst A‐eIIV serum had a reliable population for HA‐YSD mapping (1.4% HA‐YSD+; Figure 4bc). HA‐YSD sequence analysis showed similar H3N2‐1968 HA binding of the A‐eIIV serum compared with the H3N2‐1968‐positive serum (Figure 4d), without an enrichment of HA stem antibodies despite cH9/1N3 neutralisation and H3‐stem binding.
In vitro protection by mouse and human serum by A‐eIIV against heterologous viruses
Because of the presence of HA stem antibodies and cross‐reactivity against H3N2‐1968, we assessed further cross‐reactivity against an avian influenza virus, H7N7, another group 2 influenza virus. Firstly, A‐eIIV elicited significantly increased H7‐HA‐specific IgG response compared with S‐IIV (Figure 5a), which correlated with the group 2 HA stem responses (Figure 5b, R 2 = 0.6246). An in vitro serum protection assay by FACS (Figure 5c) showed that the increased A‐eIIV cross‐reactive antibody responses also lead to a significant reduction of H7N7‐infected cells relative to naïve mouse serum, whilst S‐IIV had no in vitro serum protection against H7N7 and was comparable to naïve mouse serum (Figure 5d). In the same in vitro protection FACS assay, the human antibody responses after S‐IIV and A‐eIIV vaccination showed equivalent serum protection against homologous H3N2‐2013 virus; however, A‐eIIV responses significantly reduced cells infected with heterologous viruses, H3N2‐1968 and H7N7, reflecting the high level of antibody cross‐reactivity induced by A‐eIIV (Figure 5e and f).
Figure 5.
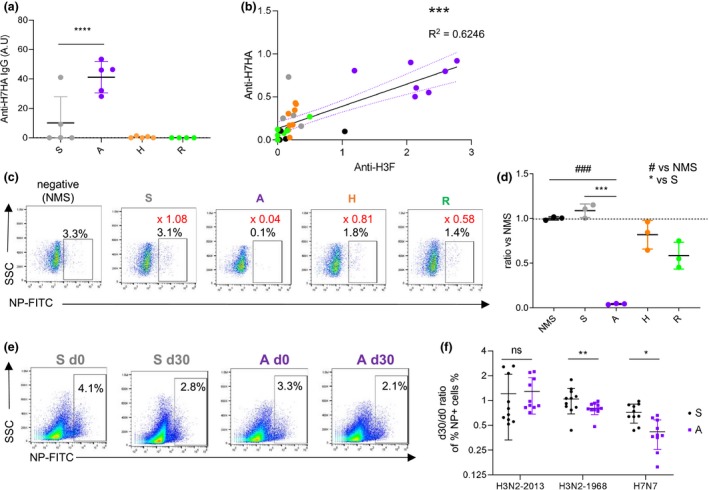
H7N7 cross‐reactive A‐eIIV antibodies in mice and humans. H7N9‐HA‐specific IgG titres measured by ELISA in mice at 7 days post‐vaccination (n = 5 mice per group) (a). Correlation of H7N9‐HA and H3‐stem IgG titres in mice serum by the Pearson correlation, R 2 = 0.624, P < 0.0001 (b). In vitro protection assay of H7N7‐infected Raji cells with mouse serum from 21 days post‐vaccination (pooled serum in triplicate, n = 3 mice per group) assessed by FACS (c), and responses were normalised to naïve mouse serum (d). In vitro protection assay of homologous H3N2‐2013, heterologous H3N2‐1968 or H7N7‐infected Raji cells with human pre‐ and 30‐day post‐vaccination serums (e), individual responses (n = 10 per group) for S‐IIV and A‐eIIV, and fold change post‐vaccination (f). Experiments were repeated twice. Data represent the mean, SEM and individual responses. In c, the percentage of the population of interest is shown in black on the FACS plots, and the ratio of the mean percentage versus naïve mouse serum (NMS) is shown in red. #shows statistical significance versus NMS, and *shows statistical significance versus S‐IIV. *P < 0.05, **P < 0.01 and ***/### P < 0.005.
Recall of H3‐specific T and B cells by A‐eIIV during heterologous influenza challenge in mice
Human and mouse data for A‐eIIV have shown increased H3‐HA‐specific antibodies that are high‐avidity and cross‐reactive, but the protective potential of these responses is not known. Therefore, to determine vaccine‐mediated protection we challenged eIIV mice with a lethal dose of H3N2‐1968. We selected the H3N2‐1968 virus for mouse challenge as a well‐established model of heterosubtypic protection in T cell23 and HA stem‐based vaccine models.24 The H3N2‐1968 virus has 85.3% amino acid similarity to the HA of the H3N2‐2014 virus in the 2017/2018 vaccines used. Challenge experiments were performed at 6 months post‐vaccination (long‐term memory) to study the potential waning of vaccine responses (Figure 6), and at 21 days after vaccination (short term; Supplementary figure 4a–c).
Figure 6.
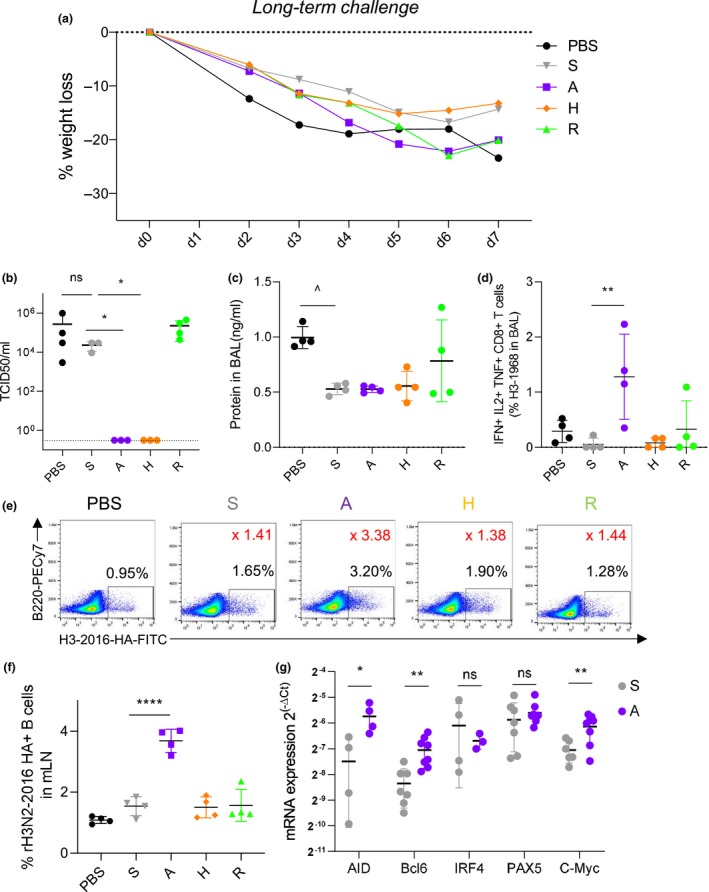
Influenza‐specific T‐ and B‐cell recall responses increased by A‐eIIV after H3N2‐1968 challenge. Vaccinated mice were challenged i.n. with 20LD50 of H3N2‐1968 virus at 6 months (long‐term memory) post‐vaccination, and at day 7 post‐infection, lungs and BALF were collected. Long‐term memory H3N2‐1968 challenge weight loss (a), lung viral load (b) and protein concentration in BALF (c). By FACS, polyfunctional H3N2‐1968‐specific CD8+ T cells (IFN‐γ+ TNF‐α+ IL‐2+) were measured in the BAL (d). By FACS (e), fluorescent‐labelled recombinant HA probes for H3‐2016‐specific B cells were measured in the mLN (f). Expression of AID, Bcl6, IRF4, Pax5 and c‐Myc in B cells from spleen (relative expression to GAPDH expression, expressed in ) (g). Data represent the mean, SEM and individual responses of n = 4 mice per group. In e, the percentage of the population of interest is shown in black on the FACS plots, and the ratio of the mean percentage versus PBS is shown in red. ^shows statistical significance of S‐IIV versus PBS. *shows statistical significance of eIIV versus S‐IIV. *P < 0.05, **P < 0.01 and ***P < 0.005, experiments were repeated twice.
Challenge of mice with H3N2‐1968 virus at 6 months after vaccination induced a significant weight loss in unvaccinated mice, whilst all vaccinated mice were still protected with no lethality and no differences between weight loss between S‐IIV and eIIV (Figure 6a). Therefore, our definition of protection is based on lung viral loads and damage to the lung by total protein concentration. Importantly, A‐eIIV and H‐eIIV exhibited earlier viral clearance in the lungs by day 7 than S‐IIV (Figure 6b). Meanwhile, S‐IIV‐mediated protection had waned and viral loads were comparable to unvaccinated PBS mice (Figure 6b); however, there was a trend for less damage to the lung (by total protein concentration) for S‐IIV mice compared with the PBS groups (P = 0.009; Figure 6c).
Similar observations were made at short‐term challenge with H3N2‐1968 and H1N1‐2009. In the short‐term challenge, no difference was observed between S‐IIV and eIIV in terms of weight loss, but the local lung damage (by total protein concentration) was reduced in A‐eIIV and H‐eIIV compared with S‐IIV, whereas R‐eIIV was elevated compared with S‐IIV (Supplementary figure 4). Homologous short‐term challenge with H1N1‐2009 virus also showed protection for all vaccines and a strong superiority for the A‐eIIV in terms of VNA titres and H1N1‐2009 HA‐specific antibodies but comparable serum antibody ADCC activity (Supplementary figure 5).
Differences in inflammation and viral clearance after influenza challenge at long term may indicate changes in cellular responses; therefore, we assessed recall of influenza‐specific T cells (Figure 6d) and B cells (Figure 6e and f). Following influenza virus challenge, A‐eIIV mice showed increased H3N2‐1968‐specific polyfunctional CD8+ T cells (IFN‐γ+ TNF‐α+ IL‐2+) in the bronchoalveolar lavage (BAL), whilst no other vaccination group had any substantial T‐cell response (Figure 6d, A‐eIIV versus S‐IIV, P = 0.005), whereas H3N2‐1968‐specific CD4+ T‐cell responses were comparable between the vaccination groups in the lung draining mediastinal lymph node (mLN) (Supplementary figure 4d and e). Furthermore, in the mLN, only A‐eIIV elicited a substantial recall of the H3N2‐2016 HA‐specific B‐cell response (Figure 6e and f), which was twice the magnitude of S‐IIV (Figure 6f, P < 0.0001). The A‐eIIV B‐cell recall responses at 6 months post‐vaccination also maintained elevated expression of B‐cell CSR‐ and SHM‐promoting responses for AID, Bcl6 and c‐Myc versus S‐IIV (Figure 6g).
Recall of high‐quality antibody responses of A‐eIIV short‐ and long‐term memory
To investigate the local inflammatory response, we performed a 12‐panel cytokine bead array for Th1, Th2 cytokines and chemokines (Figure 7a) in BAL after challenge. S‐IIV had a significant fold‐change increase in pro‐inflammatory IFN‐γ, IL‐6, TNF‐α and IL‐2 and anti‐inflammatory IL‐4 compared with unvaccinated PBS mice (Figure 6d), whilst A‐eIIV compared with S‐IIV induced a significant increase in pro‐inflammatory IFN‐γ, TNF‐α, IL‐9 and IL‐2, which may drive viral clearance in the lung, and IL‐4 driving a Th2 response. Further, H‐eIIV had an opposite effect compared with S‐IIV, with a significant reduction of both types of Th1 and Th2 cytokines, and also of the CCL2 and CCL4 chemokines, which may lead to reduced recruitment of immune cells to the lung. Finally, R‐eIIV had comparable responses to S‐IIV.
Figure 7.
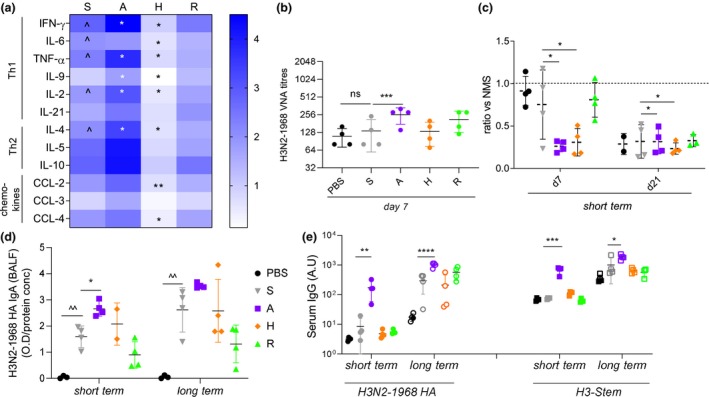
Lung elevated cytokine production and secretory‐specific IgA, along with high serum H3‐stem antibodies in A‐eIIV mice after challenge. Mice were challenged with 20LD50 H3N2‐1968 virus 21 days or 6 months post‐vaccination, and inflammatory responses were sampled at day 7 post‐infection. Inflammatory cytokine and chemokine concentrations in individual lung homogenates (n = 4) by CBA were normalised to fold change from PBS response (a). H3N2‐1968 VNA endpoint titres from serum from short‐term challenge at day 7 post‐infection (b). In vitro protection assay of H3N2‐1968‐infected Raji cells with mouse serums from 7 and 21 days post‐infection (n = 3 or 4), responses were normalised to naïve mouse serum (c). Day 7 post‐challenge, H3N2‐1968 HA‐specific secretory IgA in BALF for short‐ and long‐term memory recall measured by ELISA (d), normalised to total protein concentration by BCA. H3N2‐1968 HA and H3‐stem‐specific IgG responses by ELISA for short‐ and long‐term memory recall at day 21 post‐challenge (e). Data represent the mean, SEM and individual responses, n = 4 mice per group. *shows statistical significance of eIIV versus S‐IIV. *P < 0.05, **P < 0.01 and ***P < 0.005, experiments were repeated twice.
As antibody quality is a consequence of AID activity, the properties of immune sera at influenza challenge time points were further assessed. The H3N2‐1968 VNA titre at day 7 post‐infection was comparable between unvaccinated PBS mice and S‐IIV (Figure 7b), suggesting no recall of existing vaccine antibodies by heterologous H3N2‐1968 challenge; however, A‐eIIV had significantly increased early VNA responses during infection. Our FACS‐based in vitro serum protection assay also showed an early increased day 7 neutralisation response for A‐eIIV and H‐eIIV (Figure 7c), whilst other vaccine groups only developed in vitro neutralisation activity later by day 21 post‐challenge when virus is well cleared. Importantly, the local mucosal IgA H3N2‐1968 HA response from the BAL of the lung of A‐eIIV mice showed significant early recall of vaccine antibody responses, with a significant H3N2‐1968 HA IgA response at the site of infection at short‐term post‐vaccination challenge, which was not observed in other vaccination groups, and a trend for increased IgA responses at long‐term challenge compared with S‐IIV (Figure 7d). Challenge at both short‐ and long‐term post‐vaccination time points showed A‐eIIV had significantly increased IgG titres and avidity of cross‐reactive antibody responses against H3N2‐1968 HA (Figure 7e and Supplementary figure 4g), H3‐stem (Figure 7e) and NP (Supplementary figure 4h) compared with S‐IIV.
Discussion
Influenza virus is the only pathogen that is recommended for yearly vaccination, with over 500 million doses used globally every year.25 IIV has limited protection against infecting strains that are antigenically distinct from those contained in the vaccine. This can create a public health problem, such as recent H3N2 viruses, which have been more difficult to match and control with S‐IIV.26 Newly available vaccine approaches, including eIIVs, could be utilised to provide longer duration and breadth of protection, as broadly protecting vaccines are needed, but no universal influenza vaccine is licensed yet.27, 28 We therefore compared the immunogenicity, mechanism of action and protective potential of three commercially available enhanced vaccines, FluAd (A‐eIIV), FluZone‐HD (H‐eIIV) and FluBlok (R‐eIIV), with a standard seasonal influenza vaccine, FluQuadri (S‐IIV), in humans and mice. We found evidence to indicate that A‐eIIV and H‐eIIV may provide longer‐lasting and broader cross‐protection against influenza viruses than S‐eIIV and R‐eIIV.
Results from our randomised trial of eIIVs in older adults showed that A‐eIIV increased the long‐term avidity of H3‐2013‐specific HA IgG early after vaccination in conjunction with an increased IgG response towards H1‐stem and H3‐stem and, to a lesser extent, H‐eIIV also increased H1‐stem IgG responses. The HA stem of the HA protein harbours functionally conserved neutralising epitopes, which may enable broader protection against different influenza strains,29 rendering the induction of anti‐HA stem antibodies by A‐eIIV and partly H‐eIIV of particular interest for increasing the breadth of protection by influenza vaccination strategies.
Our randomised clinical trial was designed to assess immunogenicity, not vaccine efficacy against clinical outcomes. Therefore, we assessed S‐IIV versus three different eIIVs in parallel in mice followed by challenge with a lethal heterologous influenza strain, enabling access to infected tissues and immune sites that are constrained in human studies. In addition, the naïve mouse status enables direct comparison of direct vaccine immunogenicity without interference of prior immunity. Whilst IgG subclass function, age, exposure history, vaccine dosage and duration of responses may differ between mice and humans, the mouse model is an important but limited resource to benchmark vaccine responses.
Our mouse vaccination results showed an increase in the avidity and magnitude of antibodies targeting the vaccine H3N2‐2013 HA, H1N1‐2009 HA and NA, and more importantly an induction of cross‐reactive H3N2‐1968 HA and H3‐stem antibodies by A‐eIIV. We further confirmed the induction of antibody cross‐reactivity by the induction of neutralising anti‐HA stem antibodies using a chimeric H9/1N3 virus.22 Furthermore, challenge of mice with the heterologous H3N2‐1968 virus showed substantial early recall of IgA antibodies to the site of infection. Altogether, these data reinforce the findings on the effect of the MF59 adjuvant of A‐eIIV increasing the breadth and cross‐reactivity of vaccine responses.11 MF59 is an oil‐in‐water emulsion that enhances the persistence of the vaccine antigens at the injection site and subsequently increases the activation of antigen‐presenting cells.30 We found that MF59 induces an earlier and higher expression of AID, the enzyme responsible for antibody SHM and CSR in the GC, together with Bcl6, Pax5 and c‐Myc, leading to an elevated GC reaction and magnitude of Tfh‐ and GC B‐cell responses. Because of the enhanced expression of AID and related transcription factors involved in GC development, we observed an enhancement of the avidity of cross‐reactive H3N2‐1968 HA and H3‐stem IgG by A‐eIIV, and an earlier class switch from the default IgM towards the IgG isotype, which did not occur in other vaccine groups. Expression of AID and other genes of the CSR process plays a vital role for protection from influenza infections, as AID−/− mice showed severe morbidity with delayed virus clearance compared with AID+/− mice.31 As these important B‐cell transcription factors are induced in the local LN by A‐eIIV and not S‐IIV, the adjuvant promotes the GC reaction, SHM and CSR to generate high‐quality antibody responses.
The earlier and more intense induction of GC B cells by A‐eIIV and not H‐eIIV or R‐eIIV may also be as a result of the persistent deposition of the antigen in the LN by follicular dendritic cells facilitated by the MF59 adjuvant, as sustained antigen availability has previously been shown to increase the number of GC and Tfh in the LN.32, 33 A‐eIIV also induced larger memory B‐cell responses than the high‐dose vaccines, H‐eIIV and R‐eIIV. Previous vaccine studies for hepatitis B and pneumococcal vaccines have also reported that low doses of antigen tend to favor immune memory34, 35, 36 over higher antigen doses, whilst we did not see this for S‐IIV, the lower dose and use of adjuvant of A‐eIIV may have favored the establishment of long‐term immune memory responses. The formation of long‐term memory is an advantage for extended seasonal protection against influenza infection.
Vaccine‐mediated protection was further assessed by lethal H3N2‐1968 challenge at long‐term (6 months) and short‐term (21 days) memory time points post‐vaccination. Consistent with our vaccine immunogenicity results, A‐eIIV showed superiority during infection by reducing lung inflammation and increasing Th1 and Th2 cytokines that are key for viral clearance.37, 38 The H3N2‐1968 HA‐specific antibody response from A‐eIIV, most importantly lung IgA, was recalled at a higher magnitude and avidity compared with other vaccine groups. Influenza‐specific polyfunctional CD8+ T cells and B cells were also selectively recalled by the A‐eIIV long‐term memory response, unlike other vaccine groups.
HA stem‐specific antibodies have shown remarkable cross‐reactivity in some cases for different strains, subtypes and even pan‐influenza reactivity,39 and clinical trials are underway for HA stem vaccines to induce broadly reactive antibodies.40 A‐eIIV induced HA stem antibodies that may contribute to H3N2‐1968 cross‐reactivity and also bring an additional level of protection against other IAV subtypes such as avian H7N7.41 From our in vitro protection assay, we saw that A‐eIIV dramatically reduced H7N7‐infected cells after vaccination in mice and humans, whilst other vaccine responses did not. These in vitro antiviral responses could possibly be contributed to by antibodies specific to the H3‐stem and NP proteins. Antibodies targeting the highly conserved internal NP were also highly induced after vaccination in most vaccines, excluding R‐eIIV that does not contain NP, and A‐eIIV‐treated mice had greater recall of NP antibodies during influenza challenge. The NP protein has an even higher level of sequence conservation than the HA stem (95% versus 70–40%), and is expressed on the surface of infected cells and internally of virions,42 and could therefore be an additional important universal vaccine target.
Whilst A‐eIIV stimulated the strongest cross‐reactive responses, H‐eIIV also had some promising results. H‐eIIV displayed interesting protective features in the challenge experiments with a reduction of the lung damage and all inflammatory cytokines, particularly the chemokine responses, CCL2 (MCP‐1) and CCL4 (MIP‐1β), thus potentially reducing the local recruitment of inflammatory cells and deleterious inflammation, along with efficient viral clearance. H‐eIIV had an equivalent protective effect to A‐eIIV for reduction of the viral loads after long‐term challenge, which illustrates sustained vaccine memory responses. Also, H‐eIIV compared with S‐IIV elicited significant day 21 post‐vaccination antibody responses against the NA, NP proteins and H3‐2013 virus by VNA in mice and H1‐stem in humans.
Our study focused on H3N2 responses stimulated by eIIV because of the recent lower vaccine efficacy against this virus subtype.26 Some limitations of our study include the use of mismatched antigens to characterise vaccine immune responses. Because of the lack of commercial full‐length HA proteins for the H3N2‐2014 virus (A/Hong Kong/4801/2014), we used the H3N2‐2013 HA protein from a recent virus (A/Switzerland/9715293/2013), which has over 98% amino acid similarity and high level of cross‐reactivity. Furthermore, H3N2‐2016 HA probes from B‐cell staining at long‐term challenge were from a different H3N2 strain again, H3‐2016 (A/Singapore/INFIMH‐16‐0019/2016) – as these are precious non‐commercial trimeric‐HA fluorescent probes, we used that which was available to us. Challenge of mice with a more closely related H3N2 virus may have yielded different results, but the use of H3N2‐1968 virus is warranted to illustrate vaccine mismatch and heterologous protection. Despite a number of antigenic differences between the H3N2 vaccine and antigens within our immunological characterisation, A‐eIIV showed continued superiority. Another limitation is the use of young mice (6 weeks old) and older adults (> 65 years) for the randomised clinical trial. The use of the mouse model was to study vaccine immunogenicity and protective potential directly rather than effect of age on vaccine usage and immunosenescence.
In Australia since 2018, H‐eIIV and A‐eIIV have been preferentially given to older adults,43 whilst the UK switched to the A‐eIIV for older adults in 2018/19. In the United States, the H‐eIIV is now given to 40% of vaccinated older adults.44 Therefore, no current consensus on vaccine utilisation has been achieved. In our initial report,18 we identified an improved microneutralisation response against cell‐grown H3N2 by R‐eIIV compared with the A‐eIIV and H‐eIIV. Therefore, combining those earlier findings with the results of this study, we have found that each of the eIIVs has an advantage over S‐IIV, with indications that the R‐eIIV could provide better protection against vaccine‐matched H3N2, whilst A‐eIIV and H‐eIIV provide better cross‐protection against a broader array of influenza viruses. Future studies could examine whether combination strategies, for example alternating use of different eIIVs from year to year, could take advantage of the distinct strengths of the different eIIVs.
Methods
Human subjects and randomised clinical trial of eIIV
We conducted a randomised trial to determine the vaccine immunogenicity of standard versus enhanced IIV in community‐dwelling older adults, 65–82 years old, which was initiated before the 2017/2018 winter influenza season in Hong Kong (http://ClinicalTrials.gov NCT03330132). Older adults (Supplementary table 1) were randomly allocated to receive the 2017/2018 WHO‐recommended IIV formulation of either standard FluQuadri IIV (S‐IIV) or one of three enhanced vaccines adjuvanted (FluAd, A‐eIIV), recombinant HA (FluBlok, R‐eIIV) and high dose (FluZone high dose, H‐eIIV) (Table 1).18 Plasma was isolated from blood samples at baseline (day 0), and after vaccination at days 7 (acute) and 30 (short‐term memory) (Figure 1a). Standard HAI serological responses and T‐cell responses are reported elsewhere.18 All participants provided written informed consent. The study protocol was approved by the Institutional Review Board of the University of Hong Kong (UW:16‐2014).
Vaccination and infection of mice
To assess eIIV immunogenicity and vaccine‐mediated protection, female 6‐week‐old BALB/c mice were vaccinated twice 21 days apart via the intramuscular (i.m.) route with either the S‐IIV, eIIV or PBS (Figure 1e), with a third of a human dose in 100 μL (Table 1). Mice were vaccinated with the Northern Hemisphere 2017/2018 containing A/Michigan/45/2015 (H1N1)pdm09‐like virus, A/Hong Kong/4801/2014 (H3N2)‐like virus and B/Brisbane/60/2008‐like virus (Victoria lineage). S‐IIV and R‐eIIV were quadrivalent and contained a second influenza B virus, B/Phuket/3073/2013 (Yamagata lineage). For assessment of vaccination responses, mice were culled at day 7 or 21 after the second booster vaccination dose.
Vaccinated mice were challenged 21 days or 6 months after the booster dose, and anaesthetised mice were infected intranasally with a lethal dose (20LD50) of either H1N1‐2009 (A/California/07/2009, 1.375 × 106 pfu 25 μL−1) or H3N2‐1968 (A/Hong Kong/1/1968 MAC20, 1 × 104 pfu 25 μL−1), and culled at day 7 or 21 after challenge (Figure 1e). H1N1 (A/California/04/2009) virus was used for ‘homologous’ challenge representing the A/Michigan/45/2015 H1N1 vaccine component (HA amino acid conservation of 97%). Alternatively, to assess the breadth of vaccine‐mediated protection from eIIV versus S‐IIV, mice were challenged with a ‘drifted’ H3N2 strain, from 46 years earlier of the vaccine strain (A/Hong Kong/4801/2014) with A/Hong Kong/1/1968 (HK/68) for ‘heterologous’ challenge (HA amino acid conservation of 85.3%).
Blood was collected by cardiac puncture and clotted (MiniCollect, Greiner Bio‐one, Kremsmünster, Austria), and serum was harvested after centrifugation and aliquoted and heat‐inactivated, for 30 min at 56°C, before all in vitro experiments. To quantify virus replication, antibody and cellular responses, the lungs, BAL and lymphoid organs (spleen, mLN for challenged mice or iLN for vaccinated mice) were processed as previously described.23 Lung viral titres were determined from homogenates by standard TCID50 assay on MDCK cells, and viral titres were calculated by the Reed–Muench method, as previously described.23 All experimental procedures were conducted in accordance with the standards and approved by the Committee on the Use of Live Animals in Teaching and Research, The University of Hong Kong.
Antibody quantification by enzyme‐linked immunosorbent assay (ELISA)
To assess influenza‐specific antibodies from vaccination or infection, serum was probed in ELISA. Commercial proteins (Sinobiological, Beijing, China) to represent challenge viruses and vaccine strains included the following: H1N1‐2009 HA (A/California/07/2009), H3N2‐2013 HA (A/Switzerland/9715293/2013), H3N2‐1968 HA (A/Hong Kong/1/1968), H7N7‐2013 HA (A/Anhui/01/2013), and NP (A/Switzerland/9715293/2013). Because of commercial availability, the H3N2‐2013 protein (A/Switzerland/9715293/2013(H3N2)) was used to represent the H3N2 vaccine strain (A/Hong Kong/4801/2014 (H3N2)), which shared 98% HA amino acid homology and showed a high level of cross‐reactivity.
The H1‐stem (A/California/07/2009) and H3‐stem (A/Aichi/2/1968) proteins (from Raghavan Varadarajan, Indian Institute of Science) were made as previously described.24, 45 Recombinant HA, NP proteins (at 80 ng mL−1) and HA stem protein (at 800 ng mL−1) were coated on 96‐well flat‐bottom immunosorbent plates (Nunc Immuno MaxiSorp, Roskilde, Denmark), in 100 μL coating buffer (PBS with 53% Na2CO3 and 42% NaHCO3, pH 9.6) at 4°C overnight. An additional plate coated with a non‐specific protein (blocking buffer, PBS with 5% FBS) was used to measure the background binding of each individual mouse serum and used as a background subtraction from protein‐specific responses. Following FBS blocking and thorough washing, diluted serum samples (starting at 1:100) were bound for 2 h, further washed and then detected by an anti‐mouse or anti‐human Ig secondary antibody labelled with HRP specific for selected antibody classes or subclasses as indicated (IgG, or anti‐mouse IgG1, IgG2a, IgG2b, IgA or IgM; Invitrogen, Carlsbad, CA, USA).
High‐avidity antibodies were measured by an incubation 15 min with 8 m urea, after serum binding to remove low‐avidity antibodies, and then, the remaining IgG response represents high‐avidity antibodies.46 TMB/peroxide was used as substrate, and the reaction was stopped by addition of sulphuric acid (R&D systems, Minneapolis, MN, USA), and optical density (O.D.) absorbance was read at 450 nm. A standard range was performed for all ELISAs as an interexperimental control, using pooled recovered post‐infection mouse serum for H1N1‐2009, H3N2‐1968 or H7N7 viruses, and enabling the expression of results in arbitrary units (A.U.) relative to either H1, H3 or H7 proteins in A.U. Finally, to allow a quantitative comparison of the diverse IgG subclasses, the levels of the IgG1, IgG2a and IgG2b in these positive control serums were measured in ng mL−1 with total IgG1, IgG2a and IgG2b ELISA kits (Chondrex, Redmond, WA, USA). IgA quantification in the BAL fluid (BALF) was normalised per protein concentration as measured by BCA assay.
Haemagglutinin yeast surface display (HA‐YSD) epitope mapping
To map HA‐specific antibodies to the length of the HA protein, we used a fragmented YSD HA library to bind immune mouse serum. A H3N2‐1968 HA‐YSD library was constructed as previously described.23 Yeast surface expressing HA (length 100–500 bp) was constructed from random digestion of the HA fragments derived from the H3N2‐1968 HA gene. Antibody‐positive yeast were isolated by cell sorting (FACSAria I cell sorter, BD Bioscience, Franklin Lakes, NJ, USA) and grown overnight in selective medium. Plasmids were then extracted by Zymoprep yeast plasmid miniprep II kit (Zymo Research, Irvine, CA, USA) and transformed in E. coli. A representative of 120 individual clones was picked for sequencing analysis from A‐eIIV and H3N2‐1968 convalescent serums. Sequences were then aligned to the original H3N2‐1968 HA sequence.
Virus neutralisation antibody (VNA) assays
Influenza‐specific VNA was measured by standard microneutralisation (MN) assay. Immune serum was pre‐treated with receptor‐destroying enzyme (RDE) (Denka Seiken, Tokyo, Japan). Twofold serial dilutions from 1:5 to 1:3200 of mouse serum samples were prepared in virus medium (MEM, 1% Pen/Strep). An equal volume of 200TCID50 35 μL−1 of influenza viruses, H1N1‐2009, H3N2‐2013, H3N2‐1968 and H7N7 (low pathogenic avian influenza (LPAI); A/Northern Shoveller/MPF518/2008), was added to the sample dilutions (final serum dilution 1:10 to 1:1280) and incubated for 2 h. Then, 35 μL of antibody/virus was added to MDCK cells and incubated for 72 h. Standard haemagglutination assay and visualisation of cytopathic effect were used to measure virus inhibition.
Immune cell profiling by flow cytometry
To quantify and profile adaptive immune cells recruited during vaccination and infection, we used multi‐parameter flow cytometry using a panel of phenotypic immune cell markers. Single‐cell suspensions were prepared from lymph nodes (mLN from infection and iLN from vaccination), spleen or BAL, as previously described.47 Cells were then stained with Zombie live/dead (Biolegend, San Diego, CA, USA), FcR‐blocked (anti‐CD16/CD32; BD Bioscience), and stained with one of the three cocktails containing a panel of monoclonal antibodies (all from Biolegend unless otherwise indicated) for 30 min on ice in FACS buffer (PBS, 1% FBS and 0.5% NaN3). Cocktail 1 for GC B and Tfh cells contains the following: anti‐mouse CD3‐BV605, B220‐PECy7, CD4‐APCCy7, PNA‐FITC (Vector Labs, Burlingame, CA, USA), CXCR5‐PerCPCy5.5, CD38‐PE and intracellular Bcl6‐PEDazzle594. For intracellular stains, cells were fixed in fixation/permeabilisation buffer (eBioscience, San Diego, CA, USA) for 30 min on ice and stained with anti‐Bcl6‐PEDazzle594 in Perm/Wash buffer (BD Bioscience). GC B cells were defined as B220+ CD3− PNA+ CXCR5+ CD38low Bcl6+, and Tfh cells were defined as CD3+ B220− CD4+ CXCR5+ Bcl6+. Cocktail 2 for B memory cells contains the following: anti‐mouse B220‐PECy7, CD38‐PE, IgD‐APC, CD95 (Fas)‐BV605, IgM‐FITC and IgG‐APCCy7. B memory cells were defined as B220+ CD38+/− Faslow IgD− IgM+ or IgG+. Cocktail 3 for influenza‐specific B cells contains the following: B220‐PECy7 and rHA‐probe‐SA‐APC (H3N2 A/Singapore/INFIMH‐16‐0019/2016) (from Christopher Mok, HKU and Nicholas Wu, Scripps). Cells were finally fixed with 100 μL−1 of 4% PFA for 20 min on ice. Samples were acquired by flow cytometry on a FACS Attune (Invitrogen) and analysed with FlowJo software.
Inflammation and damage by total protein in BAL and cytokine bead array (CBA)
Bronchoalveolar lavage samples were harvested in total 3 mL MEM, and cells were pelted for immunological staining and supernatant for measuring total protein and cytokine concentrations. Total protein was measured on BAL fluids with the BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) with a twofold standard range using BSA, according to the manufacturer's instructions. Optical densities were read on a plate reader at 562 nm. For the measurement of cytokines in the BAL, a custom 12‐cytokine LegendPlex (Biolegend) panel included the following: IFN‐γ, IL‐6, TNF‐α, IL‐9, IL‐2, IL‐21, IL‐4, IL‐5, IL‐10, CCL2, CCL3 and CCL4. Standard wells were run in duplicates from individual lung homogenate supernatants (n = 4 per group). Samples were acquired by flow cytometry on a FACS Attune (Invitrogen) and analysed with LegendPlex software (Biolegend) as per the manufacturer's instructions.
Intracellular cytokine staining (ICS) of influenza‐specific T cells
To quantify influenza‐specific T cells producing polyfunctional antiviral cytokines, single‐cell suspensions of BAL and mLN were infected with a MOI of 4 of H3N2‐1968 virus plus 10 U mL−1 rhIL‐2 (Roche, Basel, Switzerland), purified CD49d and CD28 (Biolegend) for 6 h before the addition of BFA Golgi‐Plug, and then incubated for further 12 h at 37°C. At 18 h post‐infection, cells were washed with FACS buffer, stained with anti‐mouse CD8‐PerCPCy5.5 and CD4‐APCCy7 (Biolegend) for 30 min on ice, and then fixed (BD Cytoperm/Cytofix) for 20 min on ice. Cells were then stained intracellularly with anti‐mouse IFN‐γ‐FITC, TNF‐α‐APC and IL‐2‐PE for 30 min on ice. Samples were acquired by flow cytometry on a FACS Attune (Invitrogen) and analysed with FlowJo software. Total cytokine production was calculated by subtracting background fluorescence using the no‐virus controls.
In vitro FACS protection assay
To determine whether vaccination elicited antibodies that reduced or increased infection, we used an in vitro FACS protection assay. Raji cells (1 × 105 per well) were infected with a MOI of 4 of H3N2‐2013, H3N2‐1968 or H7N7 virus in the presence of mice or human sera (neat, 9 μL) for 16 h in MEM (1% Pen/Strep). Cells were then fixed (BD Cytofix/Cytoperm) and stained with anti‐NP‐FITC (Abcam, Cambridge, UK) in BD permeabilisation buffer for 30 min on ice. Samples were acquired by flow cytometry on a FACS Attune (Invitrogen) and analysed with FlowJo software. H3N2‐1968‐recovered mouse serum and naïve mouse serum were used as positive and negative controls, respectively. The % NP+ cells were normalised to naïve mouse serum, and a ratio above 1 indicates increased infection and below 1 indicates reduced infection. For human samples, the fold change versus day 0 responses was calculated.
Quantitative RT‐PCR (qRT‐PCR) of genes associated with SHM and CSR
Quantitative RT‐PCR was performed for the murine Aicda (AID), Bcl6, Pax5, IRF4 and C‐Myc genes on B cells isolated from mouse iLNs using magnetic B‐cell isolation kit (Miltenyi Biotec, Bergisch Gladbach, Germany). RNA was extracted using RNeasy mini kit (Qiagen, Hilden, Germany), and a reverse transcription using oligo‐dT primers with PrimeScript RT reagent kit (TAKARA, Kusatsu, Japan) was performed. The cDNA was then used in the qPCR (Fast SYBR Green, ABI, St. Louis, MO, USA) with gene‐specific primers (Supplementary table 2), then run and analysed in the LightCycler 480 (Roche). To quantify the gene expression, relative qRT‐PCR was performed by normalising the data to the GAPDH gene expression.
Statistical analysis
Results represent the individual result, mean ± SEM of 4–6 mice per group, unless indicated otherwise. Statistical significance was compared between the S‐IIV group versus PBS (indicated as ^ P < 0.05, ^^ P < 0.01 and ^^^ P < 0.0001) and eIIV versus S‐IIV group (indicated as *P < 0.05, **P < 0.01 and ***P < 0.0001) using a one‐way ANOVA (unless indicated) on GraphPad Prism software v8. When only two groups were present in an experiment, a Student– t‐test was performed on GraphPad Prism software v8. The Pearson correlations were also performed on GraphPad Prism software v8. To compare the distributions of the AUC IgM and IgG between groups, a binomial test of the percentage values was performed for each observed value of eIIV groups versus the S‐IIV group on GraphPad Prism software v8 with a Wilson/Brown calculation of the confidence intervals.
Funding
Human serum samples were derived from a study supported by the Centers for Disease Control and Prevention (Cooperative Agreement Number, IP001064‐02). Mouse studies were supported by NIH/NIAID CEIRS grant (contract HHSN272201400006C) and the General Research Fund of the University Grants Committee of Hong Kong (17113718). This paper content is solely the responsibility of the authors and does not necessarily represent the official views of the Centers for Disease Control and Prevention or the Department of Health and Human Services.
Conflict of interest
BJC has received honoraria from Sanofi and Roche for advisory committees. The authors report no other potential competing interests.
Author contributions
NK and SAV designed the study and wrote the first draft of the manuscript. NK, AH, AL, CC, SAV and AWC performed the experiments. NK, SAV, BJC, NHL, VJF and LLMP provided analysis and interpretation of experiments. All authors critically revised the paper. VJF and BJC were the consulting statisticians.
Supporting information
Acknowledgments
The authors thank Echo Chen (HKU) for sample collection and coordination, Jodi Chan (HKU) for technical assistance, Mahen Perera (HKU) and Florian Krammer (Mt Sinai) for preparing the cH9/1N3 virus, Christopher Mok (HKU) and Nicholas Wu (Scripps Institute) for preparing the H3‐HA B‐cell probe, Scarlett Limeng Yan (HKU) for preparing the H3N2 challenge virus, and Raghavan Varadarajan (Indian Institute of Science) for providing HA stem proteins.
References
- 1. Iuliano AD, Roguski KM, Chang HH et al Estimates of global seasonal influenza‐associated respiratory mortality: a modelling study. Lancet 2018; 391: 1285–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Appiah GD, Blanton L, D'Mello T et al Influenza activity ‐ United States, 2014–15 season and composition of the 2015–16 influenza vaccine. MMWR Morb Mortal Wkly Rep 2015; 64: 583–590. [PMC free article] [PubMed] [Google Scholar]
- 3. Xie H, Wan XF, Ye Z et al H3N2 mismatch of 2014–15 Northern hemisphere influenza vaccines and head‐to‐head comparison between human and ferret antisera derived antigenic maps. Sci Rep 2015; 5: 15279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ambrose CS, Levin MJ. The rationale for quadrivalent influenza vaccines. Hum Vaccin Immunother 2012; 8: 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vijaykrishna D, Poon LL, Zhu HC et al Reassortment of pandemic H1N1/2009 influenza A virus in swine. Science 2010; 328: 1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wu NC, Zost SJ, Thompson AJ et al A structural explanation for the low effectiveness of the seasonal influenza H3N2 vaccine. PLoS Pathog 2017; 13: e1006682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Russell K, Chung JR, Monto AS et al Influenza vaccine effectiveness in older adults compared with younger adults over five seasons. Vaccine 2018; 36: 1272–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barberis I, Martini M, Iavarone F et al Available influenza vaccines: immunization strategies, history and new tools for fighting the disease. J Prev Med Hyg 2016; 57: e41–e46. [PMC free article] [PubMed] [Google Scholar]
- 9. Wong SS, Webby RJ. Traditional and new influenza vaccines. Clin Microbiol Rev 2013; 26: 476–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Xu C, Thompson MG, Cowling BJ. Influenza vaccination in tropical and subtropical areas. Lancet Respir Med 2017; 5: 920–922. [DOI] [PubMed] [Google Scholar]
- 11. Khurana S, Verma N, Yewdell JW et al MF59 adjuvant enhances diversity and affinity of antibody‐mediated immune response to pandemic influenza vaccines. Sci Transl Med 2011; 3: 85ra48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cox MM, Izikson R, Post P et al Safety, efficacy, and immunogenicity of Flublok in the prevention of seasonal influenza in adults. Ther Adv Vaccines 2015; 3: 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nayak JL, Richards KA, Yang H et al Effect of influenza A(H5N1) vaccine prepandemic priming on CD4+ T‐cell responses. J Infect Dis 2015; 211: 1408–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. DiazGranados CA, Dunning AJ, Kimmel M et al Efficacy of high‐dose versus standard‐dose influenza vaccine in older adults. N Engl J Med 2014; 371: 635–645. [DOI] [PubMed] [Google Scholar]
- 15. El Sahly HM, Davis C, Kotloff K et al Higher antigen content improves the immune response to 2009 H1N1 influenza vaccine in HIV‐infected adults: a randomized clinical trial. J Infect Dis 2012; 205: 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zost SJ, Parkhouse K, Gumina ME et al Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg‐adapted vaccine strains. Proc Natl Acad Sci USA 2017; 114: 12578–12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Francis ME, King ML, Kelvin AA. Back to the future for influenza preimmunity‐looking back at influenza virus history to infer the outcome of future infections. Viruses 2019; 11: 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ranjeva S, Subramanian R, Fang VJ et al Age‐specific differences in the dynamics of protective immunity to influenza. Nat Commun 2019; 10: 1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Muramatsu M, Kinoshita K, Fagarasan S et al Class switch recombination and hypermutation require activation‐induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000; 102: 553–563. [DOI] [PubMed] [Google Scholar]
- 20. Basso K, Schneider C, Shen Q et al BCL6 positively regulates AID and germinal center gene expression via repression of miR‐155. J Exp Med 2012; 209: 2455–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cooper LJ, Shikhman AR, Glass DD et al Role of heavy chain constant domains in antibody‐antigen interaction. Apparent specificity differences among streptococcal IgG antibodies expressing identical variable domains. J Immunol 1993; 150: 2231–2242. [PubMed] [Google Scholar]
- 22. Pica N, Hai R, Krammer F et al Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc Natl Acad Sci USA 2012; 109: 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valkenburg SA, Li OT, Mak PW et al IL‐15 adjuvanted multivalent vaccinia‐based universal influenza vaccine requires CD4+ T cells for heterosubtypic protection. Proc Natl Acad Sci USA 2014; 111: 5676–5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Valkenburg SA, Mallajosyula VV, Li OT et al Stalking influenza by vaccination with pre‐fusion headless HA mini‐stem. Sci Rep 2016; 6: 22666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Palache A, Abelin A, Hollingsworth R et al Survey of distribution of seasonal influenza vaccine doses in 201 countries (2004–2015): the 2003 World Health Assembly resolution on seasonal influenza vaccination coverage and the 2009 influenza pandemic have had very little impact on improving influenza control and pandemic preparedness. Vaccine 2017; 35: 4681–4686. [DOI] [PubMed] [Google Scholar]
- 26. Rondy M, Gherasim A, Casado I et al Low 2016/17 season vaccine effectiveness against hospitalised influenza A(H3N2) among elderly: awareness warranted for 2017/18 season. Euro Surveill 2017; 22: pii=17-00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Erbelding EJ, Post DJ, Stemmy EJ et al A universal influenza vaccine: the strategic plan for the National Institute of Allergy and Infectious Diseases. J Infect Dis 2018; 218: 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kumar A, Meldgaard TS, Bertholet S. Novel platforms for the development of a universal influenza vaccine. Front Immunol 2018; 9: 600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kirkpatrick E, Qiu X, Wilson PC et al The influenza virus hemagglutinin head evolves faster than the stalk domain. Sci Rep 2018; 8: 10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ott G, Barchfeld GL, Chernoff D et al Design and evaluation of a safe and potent adjuvant for human vaccines. Pharm Biotechnol 1995; 6: 277–296. [DOI] [PubMed] [Google Scholar]
- 31. Harada Y, Muramatsu M, Shibata T et al Unmutated immunoglobulin M can protect mice from death by influenza virus infection. J Exp Med 2003; 197: 1779–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tam HH, Melo MB, Kang M et al Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc Natl Acad Sci USA 2016; 113: e6639–e6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baumjohann D, Preite S, Reboldi A et al Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 2013; 38: 596–605. [DOI] [PubMed] [Google Scholar]
- 34. Ahman H, Kayhty H, Vuorela A et al Dose dependency of antibody response in infants and children to pneumococcal polysaccharides conjugated to tetanus toxoid. Vaccine 1999; 17: 2726–2732. [DOI] [PubMed] [Google Scholar]
- 35. Lambert PH, Liu M, Siegrist CA. Can successful vaccines teach us how to induce efficient protective immune responses? Nat Med 2005; 11: S54–S62. [DOI] [PubMed] [Google Scholar]
- 36. Cassidy WM, Watson B, Ioli VA et al A randomized trial of alternative two‐ and three‐dose hepatitis B vaccination regimens in adolescents: antibody responses, safety, and immunologic memory. Pediatrics 2001; 107: 626–631. [DOI] [PubMed] [Google Scholar]
- 37. Beshara R, Sencio V, Soulard D et al Alteration of Flt3‐Ligand‐dependent de novo generation of conventional dendritic cells during influenza infection contributes to respiratory bacterial superinfection. PLoS Pathog 2018; 14: e1007360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oshansky CM, Gartland AJ, Wong SS et al Mucosal immune responses predict clinical outcomes during influenza infection independently of age and viral load. Am J Respir Crit Care Med 2014; 189: 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dreyfus C, Laursen NS, Kwaks T et al Highly conserved protective epitopes on influenza B viruses. Science 2012; 337: 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paules CI, Marston HD, Eisinger RW et al The pathway to a universal influenza vaccine. Immunity 2017; 47: 599–603. [DOI] [PubMed] [Google Scholar]
- 41. LaMere MW, Lam HT, Moquin A et al Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J Immunol 2011; 186: 4331–4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yewdell JW, Frank E, Gerhard W. Expression of influenza A virus internal antigens on the surface of infected P815 cells. J Immunol 1981; 126: 1814–1819. [PubMed] [Google Scholar]
- 43. Therapeutic Goods Administration . 2019 seasonal influenza vaccines; https://www.tga.gov.au/alert/2019-seasonal-influenza-vaccines. Therapeutic Goods Administration Department of Health Australian Government; 2019. [e‐pub ahead of print 31 October 2019].
- 44. National Health Service . Flu vaccine overview; https://www.nhs.uk/conditions/vaccinations/flu-influenza-vaccine/. 2019. [e‐pub ahead of print 31 October 2019].
- 45. Mallajosyula VV, Citron M, Ferrara F et al Hemagglutinin sequence conservation guided stem immunogen design from influenza A H3 subtype. Front Immunol 2015; 6: 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Herrera‐Ortiz A, Conde‐Glez CJ, Vergara‐Ortega DN et al Avidity of antibodies against HSV‐2 and risk to neonatal transmission among Mexican pregnant women. Infect Dis Obstet Gynecol 2013; 2013: 140142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Valkenburg SA, Li OTW, Li A et al Protection by universal influenza vaccine is mediated by memory CD4 T cells. Vaccine 2018; 36: 4198–4206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials