

UV irradiation is becoming common for disinfection in water treatment plants, but little is known about the effectiveness of this treatment for enteric RNA viruses. Here, we observed that 220-nm UV irradiation was efficacious against rotavirus (RV) and Tulane virus (TV). UV irradiation at 254 nm inactivated TV to a greater extent than RV. Additional assays showed that UV irradiation compromised different portions of the RV and TV life cycles. UV irradiation decreased the binding of TV to its host receptor and mutagenized the TV genome. UV irradiation at 220 nm appeared to allow RV–host receptor interaction but halted RV genome replication. These findings provide knowledge about the disinfection of waterborne viruses, information that is important for the safe reuse or release of treated wastewater.
KEYWORDS: UV disinfection, noroviruses, rotavirus
ABSTRACT
Enteric viruses are shed in fecal material by humans and other animals and are common contaminants in wastewater and surface water. Wastewater treatment plants often disinfect this effluent with low-pressure and medium-pressure UV lamps, which emit 254-nm and 220- to 280-nm irradiation, respectively. It is not known whether this treatment is efficacious against enteric viruses or how such treatments may inactivate these enteric viruses. This study examined UV disinfection for two enteric viruses: rotavirus (RV) (strain OSU with double-stranded RNA and a three-layer capsid) and Tulane virus (TV) (a cultivable surrogate for human norovirus with single-stranded RNA and a single-layer capsid). Viruses were treated with UV irradiation at 220 or 254 nm under conditions relevant to wastewater stabilization ponds, whose water is often used for irrigation. TV was susceptible to 220- or 254-nm UV at similar levels. It appears that UV irradiation inactivated TV by mutagenizing both its genome and capsid binding proteins. RV was more susceptible to UV at 220 nm than to UV at 254 nm. UV irradiation of RV at either 220 or 254 nm resulted in a virus that retained its ability to bind to its host cell receptor. After 220-nm treatment, the VP7 segment of the RV genome could not be amplified by PCR, suggesting that this treatment mutagenized the viral genome. However, this correlation was not observed when UV at 254 nm was used. Thus, RV and TV, with different genome and capsid contents, are targeted by UV irradiation in different ways.
IMPORTANCE UV irradiation is becoming common for disinfection in water treatment plants, but little is known about the effectiveness of this treatment for enteric RNA viruses. Here, we observed that 220-nm UV irradiation was efficacious against rotavirus (RV) and Tulane virus (TV). UV irradiation at 254 nm inactivated TV to a greater extent than RV. Additional assays showed that UV irradiation compromised different portions of the RV and TV life cycles. UV irradiation decreased the binding of TV to its host receptor and mutagenized the TV genome. UV irradiation at 220 nm appeared to allow RV–host receptor interaction but halted RV genome replication. These findings provide knowledge about the disinfection of waterborne viruses, information that is important for the safe reuse or release of treated wastewater.
INTRODUCTION
Enteric RNA viruses, such as rotaviruses (RVs) and human norovirus (HuNoV), account for the majority of gastrointestinal outbreaks worldwide (1–3). RVs caused an estimated 200,000 deaths in children under the age of 5 years worldwide in 2013 (4). HuNoV is the leading cause of gastrointestinal outbreaks in the United States (5, 6). These viruses were detected in wastewater effluent and surface water contaminated with wastewater effluent (7). HuNoV concentrations increased in surface water after norovirus outbreaks occurred (7, 8). From a public health perspective, there is a concern that untreated wastewater can spread these viruses to other humans. In support of this concern is the finding that the RV isolated from sewage water is genetically similar to the RV isolated from the stools of RV-infected children (8). Infection and diseases can also occur when humans directly contact virus-contaminated surface water through recreational activities or vegetables irrigated by contaminated water (9–11).
Wastewater treatment plants in the United States use either chlorine or UV irradiation as a method of disinfecting water (12, 13). While chlorine is the most widely used disinfectant for water, UV irradiation is gaining popularity because it inactivates a wide range of pathogens, including those that are resistant to chlorine disinfection (14–16). There are trade-offs to consider when one is deciding whether to use low-pressure (254-nm) or medium-pressure (220- to 280-nm) UV irradiation for wastewater disinfection. Medium-pressure UV disinfection uses more energy than low-pressure UV disinfection, but medium-pressure UV irradiation has shown more-promising outcomes in the inactivation of adenovirus and bacteriophage MS2 (16–18). Most studies have focused on adenovirus, a double-stranded DNA virus that infects humans, or MS2, a single-stranded RNA bacteriophage that is used as a surrogate for enteric RNA viruses (14–20). The general knowledge obtained from these studies is that the 220-nm wavelength in medium-pressure UV is more effective at viral inactivation than the 254-nm wavelength in low-pressure UV (16–18) and that double-stranded DNA or RNA bacteriophages are more resistant than single-stranded DNA or RNA bacteriophages (20). It should be noted that the bacteriophages and viruses of eukaryotic cells differ greatly in terms of their mechanisms for introducing viral genetic material into cells. Thus, the mechanisms described for bacteriophage inactivation cannot necessarily be assumed to be identical to those for enteric viruses of humans. UV irradiation can cause several types of damage to single- or double-stranded DNA or RNA: photochemical modification leading to pyrimidine dimerization, cross-linking of nucleotides, and oxidative damage (21, 22). However, a recent study conducted at a full-scale Canadian wastewater treatment plant showed that RVs and HuNoV were resistant to 254-nm UV irradiation (23). These studies demonstrate the limited information available about the inactivation of enteric RNA viruses. It is crucial to determine the inactivation mechanisms of RVs and HuNoV or a surrogate for HuNoV in order to ensure that UV-treated wastewater does not spread these viruses to recycled wastewater or surface water receiving wastewater effluent.
To fill in the gaps in our knowledge about UV inactivation of enteric RNA viruses, we performed an initial characterization of virion sensitivity to UV irradiation, and if the virions were susceptible to such disinfection, we sought to understand the portion(s) of the virion that was targeted by UV irradiation. We chose to examine two enteric RNA viruses, RV strain OSU and Tulane virus (TV) (a HuNoV surrogate). Although recent studies have shown that HuNoV can be grown in B cells and human enteroids (24, 25), in vitro propagation of HuNoV still poses a technical challenge. TV has been proposed as a suitable surrogate for HuNoV because TV causes enteric infection in rhesus monkeys, and this infection is similar to the disease caused by HuNoV (19, 26). Both TV and HuNoV recognize histo-blood group antigens, the cellular receptors for TV and HuNoV (27–30). The findings on RV and TV inactivation mechanisms and the efficacies of UV irradiation will provide insights enabling the design of effective UV treatment systems that take into consideration the characteristics of enteric RNA viruses so as to reduce the risk that these viruses pose to public health.
RESULTS AND DISCUSSION
Inactivation kinetics of RV and TV in waste stabilization pond water by 220- and 254-nm irradiation.
We first measured the efficacies of UV irradiation against TV and RV. Figure 1 shows the reduction in the infectivity of TV and RV as measured by plaque assays when purified viruses were treated with increasing doses of UV irradiation. Linear correlations were used to describe both TV and RV inactivation as a function of a UV dose of either 220 nm or 254 nm. The slopes of these regression lines indicate the susceptibilities of these viruses to UV irradiation. Figure 1a shows that the inactivation kinetics of TV by UV irradiation at 220 nm and at 254 nm were statistically similar (P = 0.6), and the data can be fitted with one regression line with an R2 value of 0.95. In contrast, 220-nm irradiation was more effective at inactivating RV than 254-nm irradiation (P < 0.05) (Fig. 1b). Specifically, a dose of 22.5 mJ/cm2 resulted in a 2-log10 inactivation by 254-nm irradiation but caused a 5-log10 inactivation by 220-nm irradiation (Fig. 1b). Based on the slopes of these regression lines, the most effective inactivation condition was RV treatment with 220-nm UV (slope = 0.22); the next most effective was TV treatment with either 220 or 254 nm (slope = 0.18), and the least effective was RV treatment with 254 nm (slope = 0.09).
FIG 1.
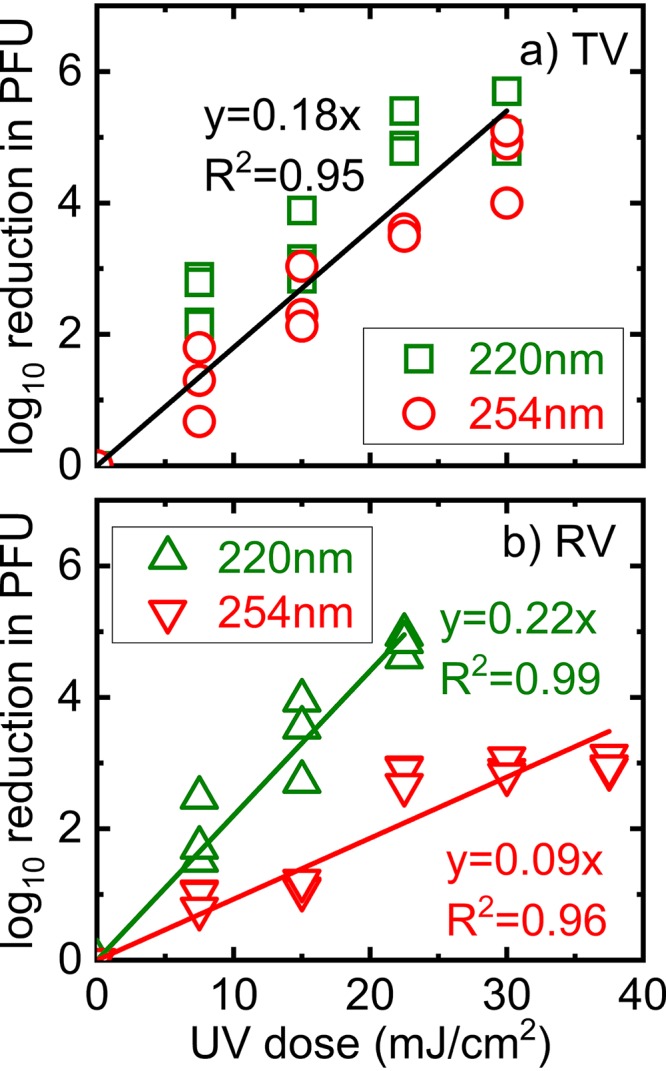
Inactivation kinetics of TV and RV in waste stabilization pond water by 220- and 254-nm irradiation. The log10 reduction values were calculated by dividing the titer at time zero by the titers of solutions collected at different irradiation times. All data collected were plotted. For each experimental condition, experiments were performed at least three times independently and in triplicate. The y axis was calculated as log10 (PFU0/PFU), where PFU0 and PFU are the infectivities detected before and after UV irradiation, respectively.
Influence of UV irradiation on virus binding to an in vitro substrate (PMG-MBs) as an indicator of host cell receptor binding.
The results in Fig. 1 suggest that UV acts on different portions of the RV and TV virions for disinfection. To understand how UV irradiation damages RV and TV virions, we asked if UV irradiation altered the ability of a virus to bind to an artificial substrate that is similar to its host cell receptors (sialic acid for RV OSU; sialic acid and histo-blood group antigens for TV). The cell-free assay we used provides a robust means of quantitatively measuring the abilities of RV and TV to bind to artificial cellular receptors present in porcine gastric mucin (PGM) (31, 32). Nonspecific binding to host cell receptors is unavoidable but minimal, as we showed in a previous study (33). We vigorously washed magnetic beads coated with PGM (PGM-MBs) to remove nonspecifically bound virions. We used this assay to compare the abilities of RV OSU and TV to interact with sialic acid or histo-blood group antigens, respectively. Virus solutions were first treated with UV and then incubated with RNase before incubation with PGM-MBs. With this strategy, RNase is expected to destroy viral RNA that is no longer encapsulated or is capsulated in a partially damaged capsid. Only RNA from presumably infectious and intact virions would be amplified by reverse transcription-quantitative PCR (RT-qPCR). For TV, RNA copies decreased significantly when virions were treated with 220-nm irradiation at doses between 22.5 and 37.5 mJ/cm2 or with 254-nm irradiation at doses of 30 or 37.5 mJ/cm2 (P < 0.05) (Fig. 2a). However, irradiation at lower doses did not significantly influence TV binding (P > 0.05) (Fig. 2a). A different pattern was observed when RV was studied. UV irradiation did not diminish the number of RV virions bound to PGM-MBs (P > 0.05) (Fig. 2b), suggesting that UV irradiation did not alter RV–host receptor interactions to a level that could be quantified by the binding assay.
FIG 2.
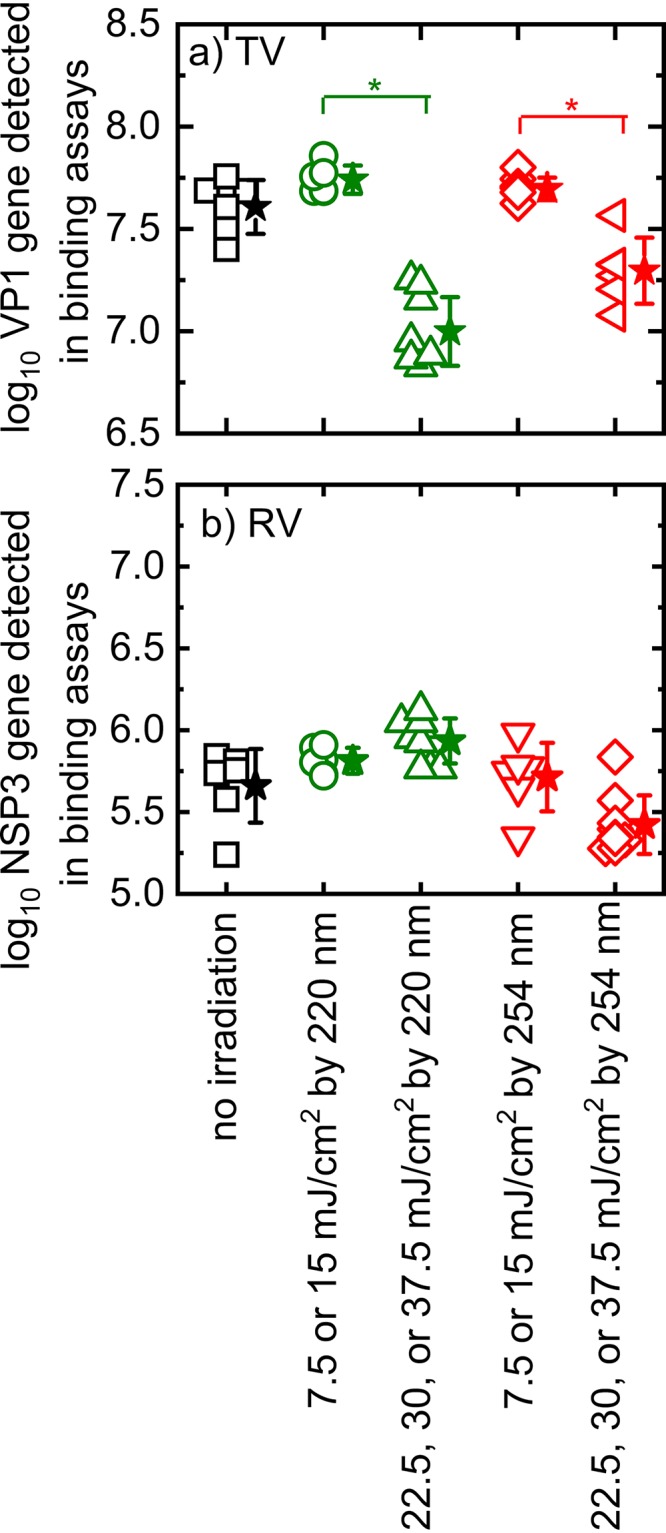
Detected TV (a) and RV OSU (b) virions bound to PGM-MBs coated with host cell receptors. Squares, circles, triangles, and diamonds all represent collected data. Stars represent average results for conditions, and error bars show standard deviations. An asterisk indicates statistically significant data. Because the results for some irradiation conditions overlapped, we combined these data for statistical analysis.
Influence of 220- and 254-nm irradiation on the TV and RV RNA genomes located inside the virion.
To determine if UV irradiation damaged the TV and RV RNA genomes, we determined the copy numbers of the TV NSP gene and the RV VP7 and VP1 genes before and after the virions were exposed to UV irradiation. As shown in Fig. 3a, 220-nm UV irradiation was more potent than 254-nm UV irradiation at damaging the TV NSP segment. For a given UV dose, there was a significantly greater reduction in the copy number of the NSP segment of TV after 220-nm irradiation than after 254-nm irradiation (P < 0.05). The linear correlation between the reduction in the copy number of the TV NSP segment and the reduction in TV titers after irradiation by 220- or 254-nm UV (Fig. 3b) suggests that the inactivation of TV was due to genome damage.
FIG 3.
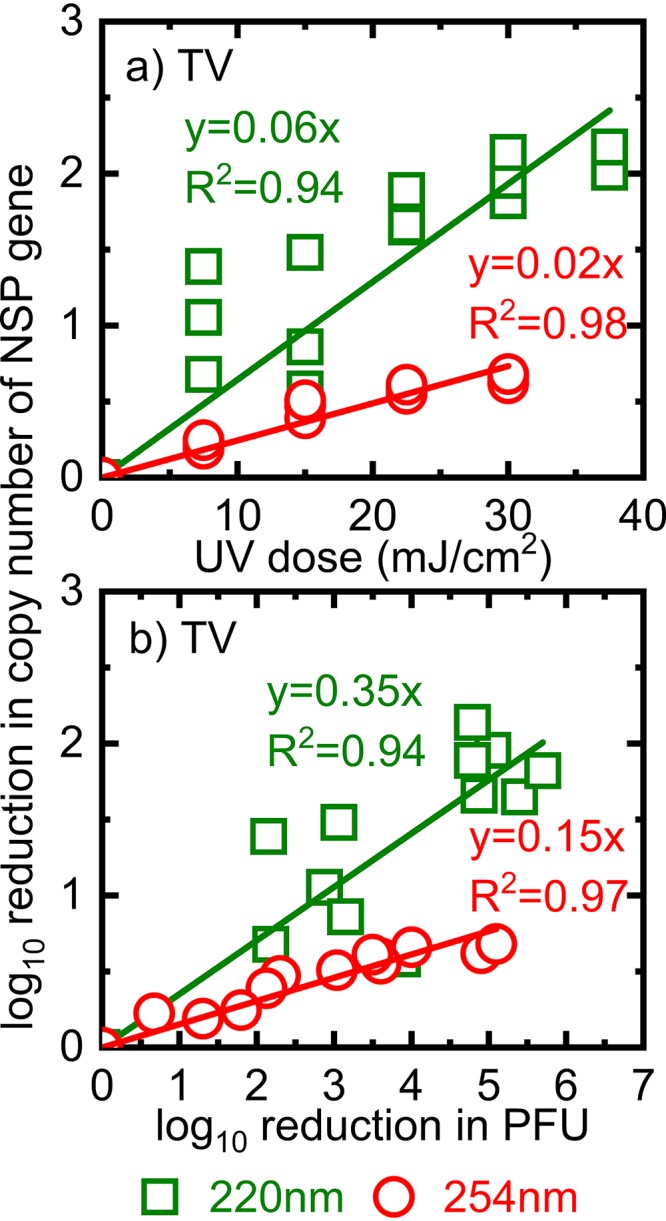
Influence of 220- and 254-nm irradiation on the TV genome. The log10 reduction in copy numbers of TV RNA is plotted as a function of the UV dose (a) and as a function of the log10 reduction in PFU (b). The NSP segment with a size of 1,305 bp was quantified.
We observed a linear correlation between the log10 reduction in the copy number of the VP7 gene of RV and 220-nm UV doses (Fig. 4a). In addition, we found insignificant differences between the reductions in TV NSP and RV VP7 gene copy numbers (P > 0.05). The regression lines that represented the reductions in the copy numbers of TV NSP and RV VP7 due to 220-nm UV irradiation had the same slope of 0.06 (Fig. 3a and 4a). Similarly, the log10 reduction in the copy number of RV OSU VP7 correlated linearly with the log10 reduction in PFU (Fig. 4b). Thus, similar trends for TV and RV OSU inactivation and genome damage due to 220-nm irradiation suggested that 220-nm irradiation caused damage to both the TV and RV genomes.
FIG 4.
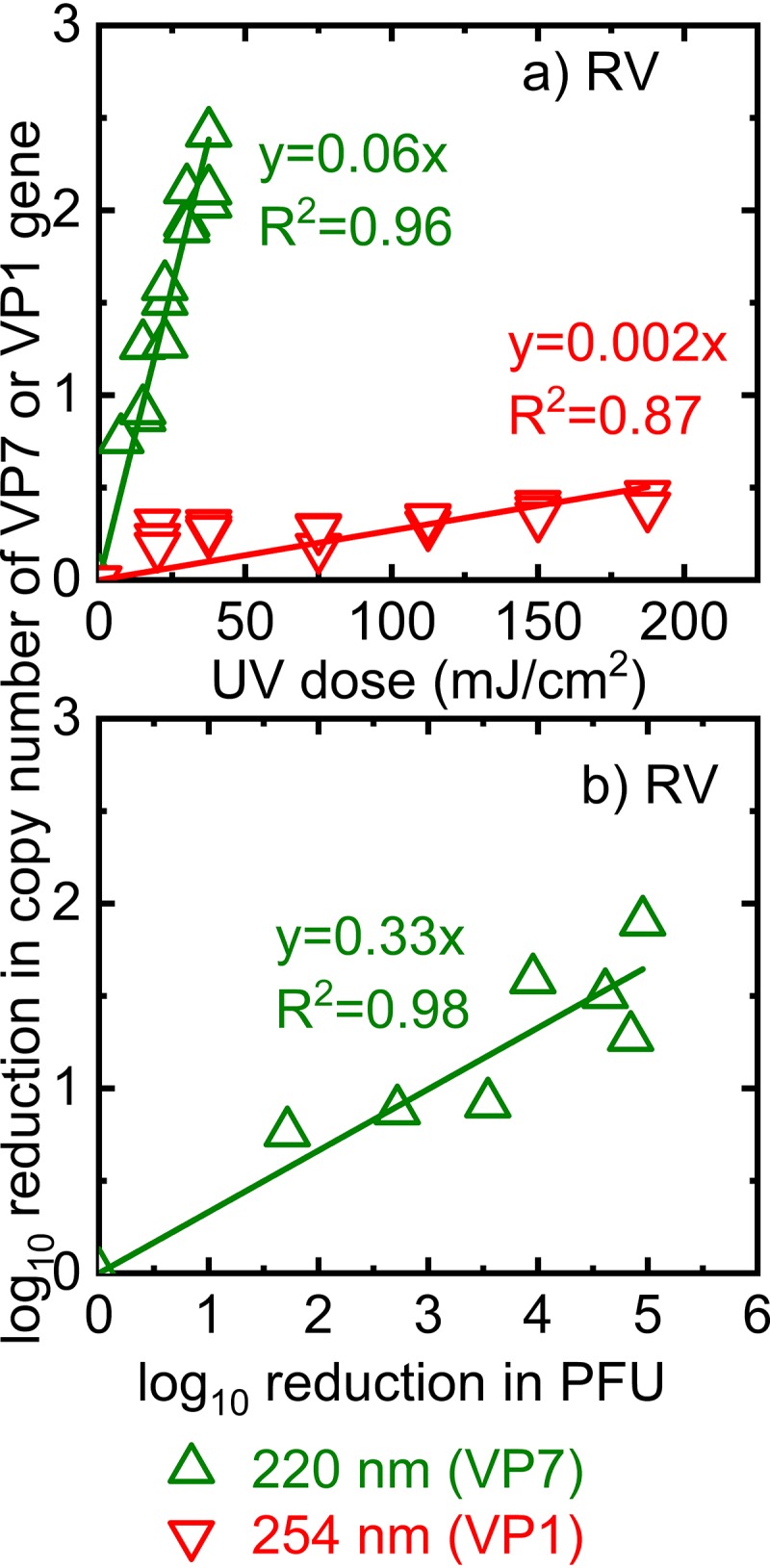
Log10 reduction in copy numbers of RV OSU RNA as a function of RV virions treated with 220-nm or 254-nm irradiation (a) and as a function of the log10 reduction in PFU of RV OSU irradiated at 220 nm (b). The y axis was calculated as the log10 of the amplicon copy number detected after UV irradiation of the RV solutions normalized by the copy number detected in these solutions right before UV irradiation.
We did not detect significant differences in the reduction in the VP7 gene copy number between the control (unirradiated) and irradiated RV (P > 0.05), indicating that the VP7 gene was not mutagenized by treatment with 254-nm UV (data not shown). When we increased the UV dose of 254-nm irradiation to 180 mJ/cm2, we still did not detect significant genome damage in the VP7 gene of RV (data not shown). Because the VP7 gene is only about 1,000 bp, and the damage may occur in other parts of the RV genome, we also examined the VP1 gene, which is 3,302 bp, in the same manner. Under these conditions, there was a reduction in RV VP1 gene amplification when virions were treated with 254-nm UV (Fig. 4a), suggesting that irradiation at high doses can indeed damage RV genomes. However, the highest UV dose of 180 mJ/cm2 corresponded to only a 70%, or 0.5-log10, reduction in the VP1 gene copy number. Although 254-nm irradiation can damage the RNA genome of RV, the results of the assays targeting the VP1 and VP7 genes of RV did not correlate with decreased RV titers.
Influence of 220- and 254-nm irradiation on the genomes of TV and RV RNA independent of the virions.
To remove the influence of the protein encapsulating the RNAs of TV and RV on the degradation of these RNAs, we exposed the RNA genomes extracted from TV and RV to irradiation at 220 nm and 254 nm. The copy numbers of the NSP segment of TV and the VP7 and VP1 genes of RV were determined after these genomes were exposed to different doses of UV irradiation. As shown in Fig. 5, an increase in UV exposure resulted in a linear increase in the log10 reduction in the copy number of the NSP segment for TV or in those of the VP7 and VP1 genes for RV. Notably, irradiation by 220-nm UV caused significantly larger reductions in the targeted genes than 254-nm irradiation for both TV and RV (P < 0.05). We also observed significantly larger reductions in the TV NSP segment than in the RV VP1 or VP7 gene for both 220-nm and 254-nm irradiation. The trends observed in Fig. 5 were consistent with the trends described above for the reductions in gene copy numbers when the whole virions were exposed to UV irradiation, suggesting that UV irradiation damaged the RNA genomes of both TV and RV.
FIG 5.
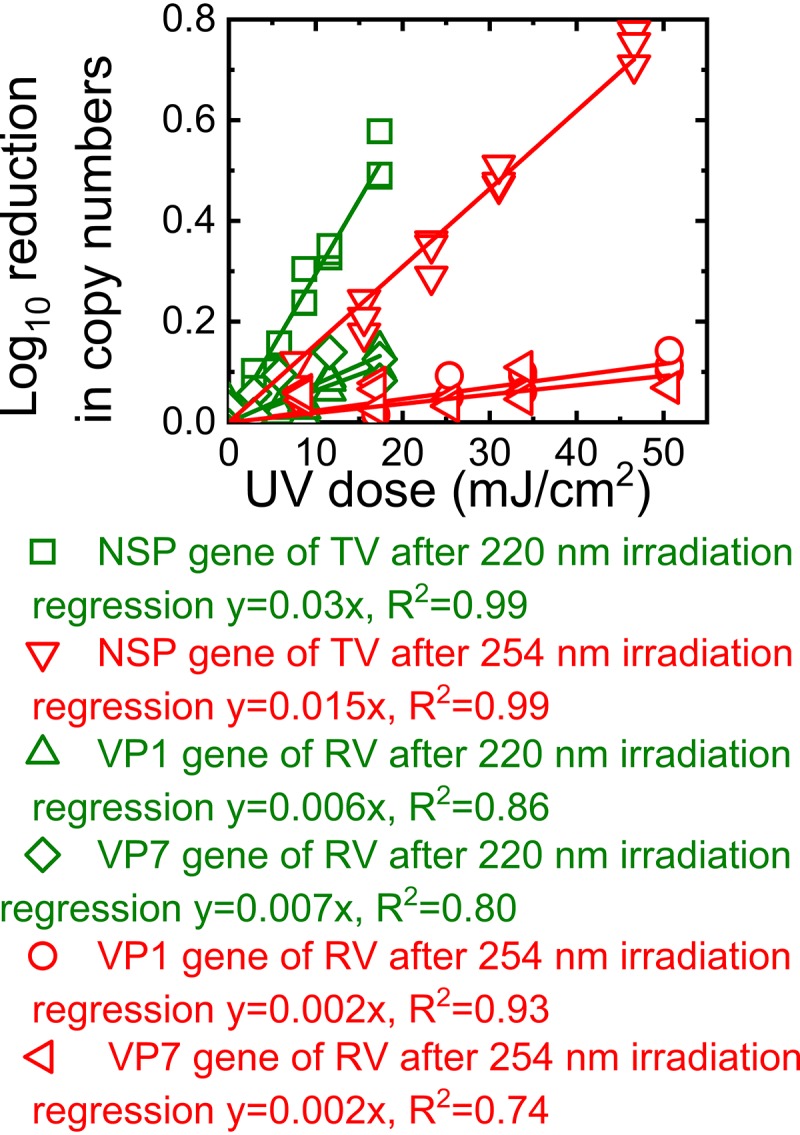
Log10 reductions in copy numbers of the NSP gene of TV and the VP7 and VP1 genes of RV after 220-nm or 254-nm irradiation.
Proposed inactivation mechanisms.
The goal of this study was to understand how different amounts of UV irradiation may alter enteric viruses that have different properties. RV is a nonenveloped virus containing a segmented, double-stranded RNA genome, while TV is a nonenveloped virus containing a single-stranded RNA genome. A dose of 22.5 mJ/cm2 of 220-nm irradiation achieved approximately 4-log10 inactivation of both TV and RV. In contrast, a dose of 22.5 mJ/cm2 of 254-nm irradiation produced only 2-log10 inactivation of RV. Thus, 220-nm UV irradiation is equally effective for inactivating RV and TV, while 254-nm UV irradiation is much less effective for inactivating RV than TV. Previous studies have shown that 220-nm UV is also very effective at inactivating adenovirus 2 and bacteriophage MS2 (16–18, 34). Perhaps 220-nm UV is more effective at inactivating viruses because of the stronger photon energy of 220-nm irradiation than of 254-nm irradiation. These data are important for considering the best practices for wastewater treatment: 220-nm UV may be more effective at eliminating TV, RV, and adenovirus, but medium-pressure UV disinfection uses more energy than low-pressure UV (16–18). It is known that 220-nm UV targets proteins/amino acid residues (18). Thus, 220-nm UV irradiation may be a good disinfection strategy for viruses, because it can nonspecifically mutagenize viral capsid proteins, including those that bind to host cell receptors.
Several reports show that viruses possessing double-stranded DNA or RNA genomes are at least 10 times more resistant to UV disinfection than viruses with single-stranded nucleic acid genomes (16, 17, 20). Double-stranded RNA or DNA is more resistant, probably because the additional strand can be used as a template for replication inside host cells (35). When comparing results from RV (double-stranded RNA genome) and TV (single-stranded RNA genome), we observed a similar trend: 254-nm UV irradiation inactivated TV to a greater extent than RV. We asked why RV was relatively more resistant to 254-nm UV inactivation than TV. We observed that the double-stranded RNA genome of RV was more resistant to 254-nm irradiation than the single-stranded RNA genome of TV when we were studying these naked (capsid-independent) viral genomes, and this may be the molecular basis for the difference in resistance. For TV, we observed a decrease in TV-receptor interactions and a decrease in NSP gene amplification when viruses were subjected to UV treatment. This finding implies that 254-nm UV and 220-nm UV are likely affecting both the virus capsid and viral genomes to inactivate viruses. UV at 254 nm chemically modifies individual nucleotide bases of RNA, and these modifications include pyrimidine doublets. It would be expected that a viral RNA polymerase would stall on this pyrimidine doublet, preventing the replication or translation of viral RNA genomes. Alternatively, such mutagenized viral RNA may be more unstable than normal viral RNA genomes, such that viral genomes are degraded (22). Ariza-Mateos et al. (21) found that the 5′ internal ribosome entry sites (IRES) of some viral RNA genomes are cleaved by 254-nm UV irradiation. Thus, another possibility is that viral proteins are no longer translated after UV irradiation. We have not examined viral protein synthesis levels in our experiments, so we cannot formally rule out this possibility.
We observed that 220-nm or 254-nm irradiation inactivates TV. We also observed that irradiation mutagenizes the TV genome such that the genome cannot replicate or be used to synthesize proteins once inside the host cells. Irradiation also mutagenizes the TV capsid such that the virus can no longer bind to the host cell, thus aborting virus infection. Thus, our data suggested that UV irradiation compromised both TV-host interactions and TV genome replication. A study by Wang et al. in 2014 examined the effect of UV (60 and 600 mJ/cm2) on the TV life cycle (35). They reported that UV irradiation did not affect virus-host interactions but did decrease viral genome replication. Note that the binding assay used by Wang et al. detected TV–anti-TV interactions via enzyme-linked immunosorbent assays (ELISAs) (35), whereas our assays detected TV–sialic acid interactions. Thus, these differences in outcomes may be due to the different types of assays used.
For UV inactivation of RV, a different mechanism appears to be in effect. In this case, RV replication was hampered to a greater degree when virions were treated with 220-nm than with 254-nm UV irradiation. Interaction with the host receptor was not affected when the virus was treated with either 220-nm or 254-nm UV. This finding implied that 220-nm- or 254-nm-treated RV could still bind to its host cell but that a postentry event was affected by UV treatment. Treatment of viruses with 220-nm UV either (i) may compromise the structure of the capsid, such that some or all of the small segments of the RV genome escape from capsids that are no longer intact, or (ii) may not mutagenize the VP8* protein of RV (the protein important for binding to the sialic acid host receptor) but instead may mutagenize other capsid proteins such that RV cannot be endocytosed into the host cell or such that an internalized capsid may not release the viral genome. Our findings presented here suggest that 254-nm UV irradiation had the same effects on the virions as 220-nm UV energy but at a lower level.
One caveat for the interpretation of the RT-qPCR assays is that we examined only portions of the RV and TV genomes for genome stability. For RV, we targeted two regions, VP7 (a 1,062-bp amplicon) and VP1 (a 3,302-bp amplicon), which cover about 5% and 15% of the total 18-kbp RV genome consisting of 11 segments (36). For TV, we PCR amplified a 1,305-bp fragment, which is about 20% of the TV genome of 6,714 bp (https://www.ncbi.nlm.nih.gov/genomes/GenomesGroup.cgi?taxid=11974). Thus, we cannot rule out the possibility that other parts of the viral genomes may be damaged, and this damage may be an additional cause of inactivation. If this were indeed the case, then it would be reflected by a reduction in the titers in plaque assays, but not in the RT-qPCR assays that quantify the RV and TV genome portions we examined here.
The findings presented here show that different types of UV irradiation target different portions of RV versus TV capsids. Thus, one cannot assume that all viruses will respond to UV treatment in the same manner. Effective disinfection treatment requires knowledge of how viruses with potential public health impacts are affected by different kinds of UV irradiation.
MATERIALS AND METHODS
Cell culture, virus propagation, and confirmation of viral stocks.
Rotavirus (RV) porcine strain OSU was obtained from the ATCC (Manassas, VA) and was cultivated as described previously (31, 37). Tulane virus (TV) was from Xi Jiang and has been used in previous studies (32, 37). Because TV recognizes the sialic acid present on MA-104 cell surfaces (38), TV was also propagated in MA-104 cells cultured in Eagle’s minimum essential medium (EMEM) supplemented with 2% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA). Both viruses were purified by using 40% sucrose cushion centrifugation, as described previously (31). Concentrated viruses were aliquoted and stored at –80°C until use.
The identity and purity of RV OSU and TV stocks were confirmed by reverse transcription (RT)-PCR, amplifying and sequencing the RV OSU VP4 gene and a portion (899 bp) of the TV NSP ORF1 gene. The protocol for confirming that the sequenced VP4 gene indeed belongs to RV OSU has been reported in our previous study (31). To sequence the TV NSP segment, total TV RNA was extracted from virus stocks by using the QIAamp viral RNA minikit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The extracted RNA was subjected to RT using reverse primer 5′-TGAGGTCTTCTTCAACGCCC-3′ to amplify 899 bp of the NSP ORF1 region of the TV genome. The RT parameters were as follows: 48°C for 30 min for the RT step, followed by 94°C for 2 min to inactivate the RT enzyme. Next, cDNA was PCR amplified using the SuperScript III PCR kit with Platinum Taq High Fidelity DNA polymerase (Invitrogen). PCR conditions were as follows: 40 cycles of 94°C for 15 s, 57°C for 1 min, and 68°C for 1 min, and one cycle at 68°C for 10 min for the final elongation. A portion of the PCR product was visualized by Sybr green staining of the agarose gel to confirm that the PCR product was the predicted size. The remaining PCR product was purified using the ExoSAP-IT PCR product cleanup reagent (Thermo Fisher Scientific, USA). The purified TV DNA was sequenced at the High-Throughput Sequencing and Genotyping Unit at the University of Illinois at Urbana–Champaign. Sequence conversion and alignment based on the published TV sequence (GenBank accession no. EU391643.1) were conducted using the BLAST tool of NCBI. The comparison results confirmed the identity of the TV propagated through MA-104 cells.
Sampling and characterization of wastewater.
Water from a reservoir, which is the last step in a series of waste stabilization ponds at a wastewater treatment facility in Illinois, was collected as described previously (39). At this facility, water from this reservoir is regularly disinfected by UV irradiation at 254 nm before being used for irrigation. This water was characterized by a total organic carbon (TOC) content of 18.7 mg/liter, 0.65 mg/liter ammonia, a total chemical oxygen demand (COD) of 59 mg/liter, and 18 mg/liter volatile suspended solids (VSS) (39). All disinfection experiments were conducted with this water, and the pH of this water was 8.57, which is typical for the waste stabilization pond (40). The absorbance of the water used for the irradiation experiments is shown in Fig. S1 in the supplemental material.
UV disinfection experiments.
The UV irradiation conditions and calculations have been described previously (34). Briefly, all experiments were conducted in a 1-kW Rayox collimated UV system equipped with a polychromatic MPUV (medium-pressure UV) lamp (Calgon Carbon Corp., Pittsburgh, PA). To isolate the 220-nm or 254-nm wavelength of UV irradiation from the UV spectrum generated by the MPUV lamp, a bandpass optical filter (Andover Corporation, Salem, NH) with a full width at half maximum (FWHM) bandwidth of about 10 nm, and at half of the maximum peak transmittance at 220 or 254 nm, was placed at the end of the collimator. The desired wavelength was confirmed by a BLK-C-50 spectrometer (StellarNet Inc., Tampa, FL). The average photon irradiance rate was calculated according to the method of Bolton et al. (41). The UV dose was calculated based on the lamp emission spectrum, the absorbance spectra of the solution used for disinfection experiments, and correcting factors, including the Petri factor, reflection factor, water factor, and divergence factor (34). Before UV irradiation, the water was collected and passed through a 0.45-μm sterilized nitrocellulose membrane filter to remove coarse particles and bacteria, which would interfere with the infectivity assays. UV disinfection experiments were conducted in reactors made of 100-ml glass beakers, with both the sides and edges covered with dark tape to prevent UV scattering. The volume of the UV-irradiated virus solution in each reactor was 21 ml, and the depth of the wastewater solution in the reactors was 1 cm. The volume and the depth of the wastewater solution were selected to reduce the attenuation of UV light within the wastewater solutions and to minimize the change in the solution depth when samples were taken at 0, 2.5, 5, 7.5, 10, and 15 min. The irradiation experiments were conducted at room temperature with continuous stirring at 300 rpm by a magnetic stir bar. The TV or RV OSU titer in each reactor before exposure to irradiation was 105 PFU/ml. We collected 250 μl of the virus-containing wastewater sample at each sampling point (0, 2.5, 5, 7.5, 10, and 15 min; the corresponding UV doses were 0, 7.5, 15, 22.5, 30, and 37.5 mJ/cm2, respectively). The samples were kept at –80°C until use for plaque assays or RT-qPCR within about a week. All wastewater samples were tested in triplicate. Disinfection data were expressed as the log10 reduction in PFU, or log10(PFU0/PFU), where PFU is the infectivity determined for the solution after exposure to a specific UV dose, and PFU0 is the infectivity of the sample taken just before exposure to UV (a UV dose of zero). The monodispersivity of the viral suspension for the UV experiments was confirmed by a single peak in the dynamic light-scattering measurement (Zetasizer Nano; Malvern Panalytical). Interactions preventing RV particle aggregation have been studied previously (42, 43). These studies show that RV aggregation occurs when at least 20 mM Ca2+ is present (42, 43). Thus, to avoid aggregation, we stored RV OSU stocks in a solution containing about 1.8 mM Ca2+.
Plaque assays to quantify infectious viral particles.
A standard plaque assay was conducted using monolayers of MA-104 cells in 6-well plates (33). For RV OSU, viral solutions were serially diluted with MEM without FBS before being added to cellular monolayers. Viruses were incubated with the monolayers for 1 h at 37°C in a 5% CO2 incubator with hand agitation every 10 to 15 min. Next, MEM with 2.5 μm/ml trypsin and agarose was added to the infected cells for 72 h at 37°C for RV OSU. For TV, incubation was carried out for 48 h in the absence of trypsin. Plaques were visualized after the cells were fixed in 10% formaldehyde and stained with a 0.05% crystal violet solution in 10% ethanol.
Quantification of TV and RV RNA damage after UV irradiation by real-time RT-PCR.
To determine if UV irradiation damaged the viral genome, two-step RT-qPCR was used to amplify an extended portion of the TV or RV genome. RNA was extracted by use of a QIAamp viral RNA minikit (Qiagen, Germantown, MD) from 70 μl of an RV or TV solution collected from the irradiation experiments. Alternatively, 70 μl of a RV or TV solution receiving no UV irradiation was used as a control. Viral RNA was then subjected to RT using the primers listed in Table 1. The RT step was carried out at 48°C for 60 min and 65°C for 25 min using an M-MuLV (Moloney murine leukemia virus) reverse transcriptase kit (New England Biolabs, Ipswich, MA). For RV, we determined the copy number of VP7 for samples after irradiation at 220 nm and those of VP7 and VP1 after irradiation at 254 nm. The reverse primer VP7OSU-R was used to synthesize the cDNA of the VP7 gene, starting at the position of bp 1062 to cover 1,051 bp. The reverse primer VP1OSU-R was used to synthesize cDNA with a length of 3,002 bp, starting at bp 3256 of the VP1 gene. For TV, the reverse primer TV NSP-R was used to synthesize cDNA with a length of 1,305 bp, starting at position 4097. Next, cDNA from each RT reaction was quantified by quantitative PCR (qPCR) using primers specific for a small region (approximately 100 bp) at the 3′ end of each synthesized cDNA. For RV, qPCR was conducted using forward primer VP7OSU-qPCR-F and reverse primer VP7OSU-qPCR-R or forward primer VP1OSU-qPCR-F and reverse primer VP1OSU-qPCR-R to quantify VP7 or VP1 cDNA, respectively. For TV, forward primer TVNSP-qPCR-F and reverse primer TVNSP-qPCR-R were used to amplify the 3′ region of NSP cDNA. All the primer sequences used in this study are given in Table 1. The iTaq Universal SYBR green iScript reverse transcriptase (Bio-Rad) reagents were used according to the manufacturer’s directions. The qPCRs comprised 40 cycles (94°C for 10 s, followed by 57°C for 30 s). Standard curves for the RV OSU VP1 gene were performed for each qPCR assay using synthetic cDNA purchased from Integrated DNA Technologies (IDT, Coralville, IA), as reported previously (31, 44). The standard sequence is given in Table 1.
TABLE 1.
Primers and standards used for two-step RT-qPCR to detect genome damage
| Step | Primer or standard | Sequencea | Position in the genome (bp) | Fragment size (bp) |
|---|---|---|---|---|
| Reverse transcription for RV OSU VP7 | Reverse primer VP7OSU-R | 5′-GGTCACATCATACAGTTCTAAC-3′ | VP7 (1041–1062) | 1,062 |
| Quantitative PCR for RV OSU VP7 | Forward primer VP7OSU-qPCR-F | 5′-GAGAGAATTTCCGACTGG-3′ | VP7 (11–28) | 130 |
| Reverse primer VP7OSU-qPCR-R | 5′-GTCCATTGTTCTAGTAACTGA-3′ | VP7 (121–141) | ||
| Standard curve for RV OSU VP7 | Standard sequence for RV OSU VP7 | GGCTTTAAAA GAGAGAATTT CCGACTGGCT ATCGGATAGC TCTTTTTAAT GTATGGTATT GAATATACCA CAGTTCTAAC TTTTTTGATA TCGCTTGTAT TTGTCAATTA TATACTGAAA TCAGTTACTA GAACAATGGA CTTTATCATT TATAGATTCT TATTGGTTAT AGTCGTACTT GCACCGCTCA | VP7 | 1,062 |
| Reverse transcription for RV OSU VP1 | Reverse primer VP1OSU-R | 5′-AGGGCGATCGTAATGCTGTT-3′ | VP1 (3235–3256) | 3,002 |
| Quantitative PCR for RV OSU VP1 | Reverse primer VP1OSU-qPCR-R | 5′-CAGCTGATTGGAATAATTCAGAAGT-3′ | VP1 (285–361) | 108 |
| Forward primer VP1OSU-qPCR-F | 5′-ACGATAAGTATAATGCTGTAGAACGG-3′ | VP1 (253–279) | ||
| Standard curve | Standard sequence for RV OSU VP1 | GCATCCATA TTATCGTATT CCTACGATAA GTATAATGCT GTAGAACGGA AGTTAGTTAA TTATGCTAA AGGTAAACCA TTAGAAGCAG ATTTGACGGCG AATGAACTCG ATTATGAAAA TAATAAAATA ACTTCTGAAT TATTCCAATC AGCTGAAGAA TATACTGATT CATTAATGGA TCCTGCTATA CTGACTTCAT TATCATCTAA TTTAAATGCA GTTATGTTTT GGTTGGAACG | VP1 | 3,302 |
| Reverse transcription for TV NSP | Reverse primer TV NSP-R | 5′-GGTCACATCATACAGTTCTAAC-3′ | NSP (4076–4097) | 1,305 |
| Quantitative PCR for TV NSP | Forward primer TVNSP-qPCR-F | 5′-GGCAGCTGGGAAGAAATCTG-3′ | VP1 (2774–2793) | 110 |
| Reverse primer TVNSP-qPCR-R | 5′-CCTGCTGTGTGAATGCCTAC-3′ | NSP (2865–2884) |
Underlined sequence, the sequence to which primers attach; boldface sequence, sequence not included in the amplicon but included in the standard to conserve the DNA from degradation.
For TV, cDNA formed after the RT reaction using the TV NSP-R primer was used as the standard. For both RV and TV, each cDNA concentration was determined by Qubit (Thermo Fisher), and the copy numbers were calculated using an online tool (https://cels.uri.edu/gsc/cndna.html). The qPCR efficiency ranged between 90 and 99%, with R2 values of 0.97 to 0.99. These values were based on the standard curves generated with serially diluted solutions of cDNAs, which were then quantified by qPCR in parallel with experimental samples. Genome damage data were expressed as the log10 reduction in the copy numbers of either the NSP gene for TV or the VP1 gene for RV, or log10(C0/C), where C0 is the genome copy number of the sample taken at a UV dose of zero (i.e., before UV irradiation) and C is the number of genome copies determined for the solution after exposure to a specific UV dose. We used the complete genome sequences of RV OSU and TV available from NCBI to design the OSU VP7, OSU VP1, and TV NSP primers that were used for this work. The primers were chosen based on specificity to a genomic region, annealing temperatures, and a lack of obvious primer-dimer formations. In addition to analyzing the melting curve after each qPCR, we analyzed a small portion of each PCR product by agarose gel electrophoresis to ensure that an amplicon of the correct size was present.
Effects of UV irradiation on TV and RV genomes when genomes are not encapsidated.
To determine the effect of UV irradiation on the viral genome, we extracted total RNA from RV OSU (∼105 PFU/ml for VP1 or VP7 gene detection) or TV (105 PFU/ml for NSP gene detection). A 100-μl volume of the extracted RNA of RV OSU or TV was added to 1,600 μl phosphate-buffered saline (PBS) containing 50 μl SUPERase·In RNase inhibitor (at a 1:10 dilution) (Invitrogen, Grand Island, NY). A 350-μl volume of this RNA suspension was added to 1 of 9 wells of a black 96-well plate. This plate was irradiated using the same setup as that for the disinfection experiments (described above) for 0, 2.5, 5, 7.5, 10, or 15 min with UV at 220 or 254 nm. A 50-μl sample from each well was taken and was examined for RNA integrity using two-step RT-qPCR as described above.
Preparation of PGM-MBs.
We applied a cell-free assay using porcine gastric mucin-conjugated magnetic beads (PGM-MBs) to determine whether UV irradiation compromised the binding proteins of TV and RV. The PGM-MBs were prepared as described previously (31, 32). Briefly, 1 ml MagnaBind carboxyl-derivatized beads (Thermo Scientific, Rockford, IL) was placed in a 1.5-ml low-adhesion microcentrifuge tube (USA Scientific Inc., Orlando, FL). A bead attractor was used to separate the beads from the suspension buffer. The beads were washed three times with sterile PBS, and the bead attractor was used to concentrate the beads after each wash. Ten milligrams of PGM type III, containing 1 to 1.5% sialic acid (Sigma, St. Louis, MO), was dissolved in 1 ml of conjugation buffer [0.1 M 2-(N-morpholino)ethanesulfonic acid (MES), 0.9% NaCl (pH 4.7)] in a 15-ml plastic tube. One milliliter of PGM suspension was added to wash the beads, and 0.1 ml of a 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC)-based cross-linking solution was added to each bead suspension immediately. The cross-linking solution was prepared by dissolving 0.05 g of EDC per 500 μl of conjugation buffer. The beads were shaken for 30 min at room temperature on a benchtop rotator at 8 rpm. Next, the beads were washed three times with PBS and were then resuspended in 1 ml PBS solution containing 0.05% sodium azide. Bead suspensions were stored at 4°C until use.
Virus binding assays using PGM-MBs.
In previous studies (31, 37), we calibrated the binding assays to determine the ranges of RV and TV virion concentrations at which there are linear relationships between the gene copies quantified by RT-qPCR and the infectivity quantified by the plaque assay. These linear relationships are established for RV titers of 102 to 107.3 PFU/ml and 104.4 to 107.9 copies of the OSU NSP3 gene/ml and for TV concentrations of 10 to 107 PFU/ml and 103 to 109 RNA copies of the VP1 gene/ml (31, 37). These linear relationships showed that untreated virions could bind to the PGM-MBs under the conditions of the binding assay, including the incubation time and the ratios between the virions added and the PGM-MBs. The detection limits of the plaque assay were 100 infectious virions/ml for RV OSU and 10 infectious virions/ml for TV. For RT-qPCR, the detection limits were 104 genome copies/ml for RV OSU and 103 genome copies/ml for TV. Based on these findings, these ranges for RV and TV were used for all binding experiments with PGM-MBs as follows. A 70-μl volume of purified TV or RV was either left untreated or UV irradiated and was added to a 1.5-ml low-adhesion microcentrifuge tube (USA Scientific Inc., Orlando, FL). Next, 2 μl of RNase A and RNase T1 (40 μg/ml of RNase A and 100 U/ml of RNase T) (Thermo Scientific, Rockford, IL) was added to each tube. The mixture was incubated in a water bath at 37°C for 30 min. To remove the RNase that was left after the reaction, we added 2 μl of SUPERase·In RNase inhibitor (Invitrogen) to the samples and incubated them for another 20 min at room temperature. Next, 80 μl of PGM-MBs was added to each tube. Sterilized PBS was added to each tube to a final volume of 1 ml. The tubes were gently shaken on a bench shaker at room temperature for 30 min at 8 rpm to allow virus attachment to PGM-MBs. Next, the beads were separated from the suspension by use of a magnetic bead attractor. The beads were washed three times with 1 ml sterilized PBS. Finally, the beads were collected by magnets a final time and were then resuspended in 140 μl DNase- and RNase-free water. Factors that can influence the results of binding assays may include the adsorption of wastewater components to virions, which would inhibit virion–-PGM-MB interactions. However, this is not likely, because the numbers of virions bound to the PGM-MBs in the wastewater used here were comparable to those bound to the PGM-MBs in PBS.
Quantification of the virions bound to PGM-MBs.
After UV treatment, the virus solution was subjected to RNase treatment before being incubated with the PGM-MBs. This RNase treatment was required to degrade viral RNA that either was no longer encapsidated or was present in a partially damaged capsid. This strategy allowed us to quantify only the RNA from presumably infectious and intact virions. RNA was extracted from beads by use of the QIAamp viral RNA minikit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. The extracted RNA was quantified by one-step RT-qPCR using primers targeting either the TV VP1 or the RV NSP3 gene (Table 2) using iTaq Universal SYBR green iScript reverse transcriptase (Bio-Rad). The RT parameters were as follows: 48°C for 10 min for the RT step, followed by 95°C for 1 min. For qPCR, there were 40 cycles of 94°C for 10 s, followed by 57°C for 30 s. Standard curves for RV were generated for each assay by including serial dilutions of synthetic DNA carrying the RV OSU NSP3 gene (31). For TV, RNA extracted from TV was diluted sequentially and used as standards. The RNA concentration of each dilution was quantified by Qubit, and the copy number of the VP1 gene was calculated accordingly. The targeted amplicon sizes were 130 bp for RV and 110 bp for TV. These short amplicons were used to avoid PCR targeting of the damaged RNA after UV irradiation. We have confirmed that this one-step RT-qPCR assay did not detect RNA damage for RV and TV.
TABLE 2.
Primers and standard used for amplification in one-step RT-qPCR to detect RV and TV bound to PMG-MBs
|
Step |
Primer or standard | Sequence | Position in the genome | Fragment size (bp) |
|---|---|---|---|---|
| Quantitative PCR for RV OSU NSP3 for the binding assay | Forward primer JVKF | 5′-CAGTGGTTGAT CT AAAT-3′ | NSP3 (17–36) | 130 |
| Reverse primer JVKR | 5′-TCATTGTAATCATATTGAATACCCA-3′ | NSP3 (123–147) | ||
| Standard curve for RV OSU NSP3 | Standard sequence for RV OSU NSP3 | 5′-GGCTTTTAATGCTTTTCAGTGGTTGATGCTCAAGATGGAGTCTACTCAACAGATGGCATCTTCTATTATTAACTCTTCTTTTGAAGCTGCAGTTGTCGCTGCAACTTCTACATTAGAATTAATGGGTATTCAATATGATTATAATGAAGTATACACTAGAGTTAAAAGTAAGTTTGATTTTGTAATGGATGATTCTGGTG-3′ | NSP3 | 1,305 |
| Quantitative PCR for TV VP1 for the binding assay | Forward primer TVVP1-qPCR-F | 5′-TTGCAGGAGGGTTTCAAGATG-3′ | VP1 (5380–5400) | 110 |
| Reverse primer TVVP1-qPCR-R | 5′-CACGGTTTCATTGTCCCCCATA-3′ | VP1 (5468–5490) |
Statistical analysis.
Linear correlations of the log10 reduction in infectivity (measured by the infectivity assay) or the log10 reduction in gene copy numbers (measured by two-step RT-qPCR) with the UV dose were conducted. For each regression, the coefficients of determination (R2) and Pearson’s coefficient were calculated to assess the goodness of fit and correlation strength, respectively. Analysis of covariance (ANOCOVA) using OriginPro was applied to compare inactivation kinetics data, as described previously (34). For the binding assays, we first confirmed that the number of gene copies followed a normal distribution; then we conducted one-factor analysis of variance (ANOVA) using OriginPro. In all cases, 95% confidence intervals were used. For each experimental condition and each type of measurement, experiments were performed at least three times independently, and three technical replicates were conducted for each sample in the PCRs and plaque assays.
Supplementary Material
ACKNOWLEDGMENTS
This project was partially supported by grant RD83582201-0 from the U.S. Environmental Protection Agency (EPA). The contents of this paper are solely the responsibility of the grantee and do not necessarily represent the official views of the EPA. Further, the EPA does not endorse the purchase of any commercial products or services mentioned in this publication.
We acknowledge B. J. Mariñas and B. Vazquez-Bravo for guidance on the UV experimental setup. We also thank Yulin Wang and Grant Lesak for assistance. We thank Xi Jiang for providing the Tulane virus for this study.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Karst SM. 2010. Pathogenesis of noroviruses, emerging RNA viruses. Viruses 2:748–781. doi: 10.3390/v2030748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oude Munnink BB, van der Hoek L. 2016. Viruses causing gastroenteritis: the known, the new and those beyond. Viruses 8:E42. doi: 10.3390/v8020042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wilhelmi I, Roman E, Sanchez-Fauquier A. 2003. Viruses causing gastroenteritis. Clin Microbiol Infect 9:247–262. doi: 10.1046/j.1469-0691.2003.00560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.WHO. 2013. Estimated rotavirus deaths for children under 5 years of age: 2013, 215 000. https://www.who.int/immunization/monitoring_surveillance/burden/estimates/rotavirus/en/.
- 5.Grytdal SP, DeBess E, Lee LE, Blythe D, Ryan P, Biggs C, Cameron M, Schmidt M, Parashar UD, Hall AJ. 2016. Incidence of norovirus and other viral pathogens that cause acute gastroenteritis (AGE) among Kaiser Permanente member populations in the United States, 2012–2013. PLoS One 11:e0148395. doi: 10.1371/journal.pone.0148395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verstraeten T, Cattaert T, Harris J, Lopman B, Tam CC, Ferreira G. 2017. Estimating the burden of medically attended norovirus gastroenteritis: modeling linked primary care and hospitalization datasets. J Infect Dis 216:957–965. doi: 10.1093/infdis/jix410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hata A, Hanamoto S, Ihara M, Shirasaka Y, Yamashita N, Tanaka H. 2018. Comprehensive study on enteric viruses and indicators in surface water in Kyoto, Japan, during 2014–2015 season. Food Environ Virol 10:353–364. doi: 10.1007/s12560-018-9355-3. [DOI] [PubMed] [Google Scholar]
- 8.Ruggeri FM, Bonomo P, Ianiro G, Battistone A, Delogu R, Germinario C, Chironna M, Triassi M, Campagnuolo R, Cicala A, Giammanco GM, Castiglia P, Serra C, Gaggioli A, Fiore L. 2015. Rotavirus genotypes in sewage treatment plants and in children hospitalized with acute diarrhea in Italy in 2010 and 2011. Appl Environ Microbiol 81:241–249. doi: 10.1128/AEM.02695-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobayashi N, Oshiki M, Ito T, Segawa T, Hatamoto M, Kato T, Yamaguchi T, Kubota K, Takahashi M, Iguchi A, Tagawa T, Okubo T, Uemura S, Harada H, Motoyama T, Araki N, Sano D. 2017. Removal of human pathogenic viruses in a down-flow hanging sponge (DHS) reactor treating municipal wastewater and health risks associated with utilization of the effluent for agricultural irrigation. Water Res 110:389–398. doi: 10.1016/j.watres.2016.10.054. [DOI] [PubMed] [Google Scholar]
- 10.Dalahmeh SS, Lalander C, Pell M, Vinnerås B, Jönsson H. 2016. Quality of greywater treated in biochar filter and risk assessment of gastroenteritis due to household exposure during maintenance and irrigation. J Appl Microbiol 121:1427–1443. doi: 10.1111/jam.13273. [DOI] [PubMed] [Google Scholar]
- 11.Gao T, Chen R, Wang X, Ngo HH, Li Y-Y, Zhou J, Zhang L. 2016. Application of disease burden to quantitative assessment of health hazards for a decentralized water reuse system. Sci Total Environ 551–552:83–91. doi: 10.1016/j.scitotenv.2016.01.210. [DOI] [PubMed] [Google Scholar]
- 12.Jackson MR, Meschke JS, Simmons J, Isaksen TB. 2019. Fecal coliform concentrations in effluent from ultraviolet disinfection units installed in onsite wastewater treatment systems. J Water Health 17:113–123. doi: 10.2166/wh.2018.256. [DOI] [PubMed] [Google Scholar]
- 13.Metcalf & Eddy, Inc, Tchobanoglous G, Stensel HD, Tsuchihashi R, Burton F, Abu-Orf M, Bowden G, Pfrang W. 2013. Wastewater engineering: treatment and resource recovery, 5th ed McGraw-Hill Education, New York, NY. [Google Scholar]
- 14.Weng S, Dunkin N, Schwab KJ, McQuarrie J, Bell K, Jacangelo JG. 2018. Infectivity reduction efficacy of UV irradiation and peracetic acid-UV combined treatment on MS2 bacteriophage and murine norovirus in secondary wastewater effluent. J Environ Manage 221:1–9. doi: 10.1016/j.jenvman.2018.04.064. [DOI] [PubMed] [Google Scholar]
- 15.Rattanakul S, Oguma K, Takizawa S. 2015. Sequential and simultaneous applications of UV and chlorine for adenovirus inactivation. Food Environ Virol 7:295–304. doi: 10.1007/s12560-015-9202-8. [DOI] [PubMed] [Google Scholar]
- 16.Beck SE, Hull NM, Poepping C, Linden KG. 2018. Wavelength-dependent damage to adenoviral proteins across the germicidal UV spectrum. Environ Sci Technol 52:223–229. doi: 10.1021/acs.est.7b04602. [DOI] [PubMed] [Google Scholar]
- 17.Beck SE, Rodriguez RA, Hawkins MA, Hargy TM, Larason TC, Linden KG. 2015. Comparison of UV-induced inactivation and RNA damage in MS2 phage across the germicidal UV spectrum. Appl Environ Microbiol 82:1468–1474. doi: 10.1128/AEM.02773-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez-Bravo B, Gonçalves K, Shisler JL, Mariñas BJ. 2018. Adenovirus replication cycle disruption from exposure to polychromatic ultraviolet irradiation. Environ Sci Technol 52:3652–3659. doi: 10.1021/acs.est.7b06082. [DOI] [PubMed] [Google Scholar]
- 19.Rattanakul S, Oguma K. 2017. Analysis of hydroxyl radicals and inactivation mechanisms of bacteriophage MS2 in response to a simultaneous application of UV and chlorine. Environ Sci Technol 51:455–462. doi: 10.1021/acs.est.6b03394. [DOI] [PubMed] [Google Scholar]
- 20.Qiao Z, Ye Y, Chang PH, Thirunarayanan D, Wigginton KR. 2018. Nucleic acid photolysis by UV254 and the impact of virus encapsidation. Environ Sci Technol 52:10408–10415. doi: 10.1021/acs.est.8b02308. [DOI] [PubMed] [Google Scholar]
- 21.Ariza-Mateos A, Prieto-Vega S, Díaz-Toledano R, Birk A, Szeto H, Mena I, Berzal-Herranz A, Gómez J. 2011. RNA self-cleavage activated by ultraviolet light-induced oxidation. Nucleic Acids Res 40:1748–1766. doi: 10.1093/nar/gkr822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simonet J, Gantzer C. 2006. Inactivation of poliovirus 1 and F-specific RNA phages and degradation of their genomes by UV irradiation at 254 nanometers. Appl Environ Microbiol 72:7671–7677. doi: 10.1128/AEM.01106-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pang X, Qiu Y, Gao T, Zurawell R, Neumann NF, Craik S, Lee BE. 2019. Prevalence, levels and seasonal variations of human enteric viruses in six major rivers in Alberta, Canada. Water Res 153:349–356. doi: 10.1016/j.watres.2019.01.034. [DOI] [PubMed] [Google Scholar]
- 24.Ettayebi K, Crawford SE, Murakami K, Broughman JR, Karandikar U, Tenge VR, Neill FH, Blutt SE, Zeng X-L, Qu L, Kou B, Opekun AR, Burrin D, Graham DY, Ramani S, Atmar RL, Estes MK. 2016. Replication of human noroviruses in stem cell–derived human enteroids. Science 353:1387–1393. doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones MK, Watanabe M, Zhu S, Graves CL, Keyes LR, Grau KR, Gonzalez-Hernandez MB, Iovine NM, Wobus CE, Vinjé J, Tibbetts SA, Wallet SM, Karst SM. 2014. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346:755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lacombe A, Niemira BA, Gurtler JB, Kingsley DH, Li XH, Chen HQ. 2018. Surfactant-enhanced organic acid inactivation of Tulane virus, a human norovirus surrogate. J Food Prot 81:279–283. doi: 10.4315/0362-028X.JFP-17-330. [DOI] [PubMed] [Google Scholar]
- 27.Farkas T, Cross RW, Hargitt E III, Lerche NW, Morrow AL, Sestak K. 2010. Genetic diversity and histo-blood group antigen interactions of rhesus enteric caliciviruses. J Virol 84:8617–8625. doi: 10.1128/JVI.00630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kniel KE. 2014. The makings of a good human norovirus surrogate. Curr Opin Virol 4:85–90. doi: 10.1016/j.coviro.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Shao LX, Chen HQ, Hicks D, Wu CQ. 2018. Thermal inactivation of human norovirus surrogates in oyster homogenate. Int J Food Microbiol 281:47–53. doi: 10.1016/j.ijfoodmicro.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 30.Yang ZH, Chambers H, DiCaprio E, Gao G, Li JR. 2018. Internalization and dissemination of human norovirus and Tulane virus in fresh produce is plant dependent. Food Microbiol 69:25–32. doi: 10.1016/j.fm.2017.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Araud E, Shisler JL, Nguyen TH. 2018. Inactivation mechanisms of human and animal rotaviruses by solar UVA and visible light. Environ Sci Technol 52:5682–5690. doi: 10.1021/acs.est.7b06562. [DOI] [PubMed] [Google Scholar]
- 32.Araud E, DiCaprio E, Ma Y, Lou F, Gao Y, Kingsley D, Hughes JH, Li J. 2016. Thermal inactivation of enteric viruses and bioaccumulation of enteric foodborne viruses in live oysters (Crassostrea virginica). Appl Environ Microbiol 82:2086–2099. doi: 10.1128/AEM.03573-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuzawa M, Ku KM, Palma-Salgado SP, Nagasaka K, Feng H, Juvik JA, Sano D, Shisler JL, Nguyen TH. 2016. Effect of leaf surface chemical properties on efficacy of sanitizer for rotavirus inactivation. Appl Environ Microbiol 82:6214–6222. doi: 10.1128/AEM.01778-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Y, Araud E, Shisler JL, Nguyen TH, Yuan B. 2019. Influence of algal organic matter on MS2 bacteriophage inactivation by ultraviolet irradiation at 220 nm and 254 nm. Chemosphere 214:195–202. doi: 10.1016/j.chemosphere.2018.09.065. [DOI] [PubMed] [Google Scholar]
- 35.Wang D, Xu S, Yang D, Young GM, Tian P. 2014. New in situ capture quantitative (real-time) reverse transcription-PCR method as an alternative approach for determining inactivation of Tulane virus. Appl Environ Microbiol 80:2120–2124. doi: 10.1128/AEM.04036-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Desselberger U. 2014. Rotaviruses. Virus Res 190:75–96. doi: 10.1016/j.virusres.2014.06.016. [DOI] [PubMed] [Google Scholar]
- 37.Fuzawa M, Araud E, Li J, Shisler JL, Nguyen TH. 2019. Free chlorine disinfection mechanisms of rotaviruses and human norovirus surrogate Tulane virus attached to fresh produce surfaces. Environ Sci Technol 53:11999–12006. doi: 10.1021/acs.est.9b03461. [DOI] [PubMed] [Google Scholar]
- 38.Tan M, Wei C, Huang P, Fan Q, Quigley C, Xia M, Fang H, Zhang X, Zhong W, Klassen JS, Jiang X. 2015. Tulane virus recognizes sialic acids as cellular receptors. Sci Rep 5:11784. doi: 10.1038/srep11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Massalha N, Dong S, Plewa MJ, Borisover M, Nguyen TH. 2018. Spectroscopic indicators for cytotoxicity of chlorinated and ozonated effluents from wastewater stabilization ponds and activated sludge. Environ Sci Technol 52:3167–3174. doi: 10.1021/acs.est.7b05510. [DOI] [PubMed] [Google Scholar]
- 40.Davies-Colley RJ, Donnison AM, Speed DJ, Ross CM, Nagels JW. 1999. Inactivation of faecal indicator micro-organisms in waste stabilisation ponds: interactions of environmental factors with sunlight. Water Res 33:1220–1230. doi: 10.1016/S0043-1354(98)00321-2. [DOI] [Google Scholar]
- 41.Bolton JR, Mayor-Smith I, Linden KG. 2015. Rethinking the concepts of fluence (UV dose) and fluence rate: the importance of photon-based units—a systemic review. Photochem Photobiol 91:1252–1262. doi: 10.1111/php.12512. [DOI] [PubMed] [Google Scholar]
- 42.Gutierrez L, Mylon SE, Nash B, Nguyen TH. 2010. Deposition and aggregation kinetics of rotavirus in divalent cation solutions. Environ Sci Technol 44:4552–4557. doi: 10.1021/es100120k. [DOI] [PubMed] [Google Scholar]
- 43.Gutierrez L, Nguyen TH. 2012. Interactions between rotavirus and Suwannee River organic matter: aggregation, deposition, and adhesion force measurement. Environ Sci Technol 46:8705–8713. doi: 10.1021/es301336u. [DOI] [PubMed] [Google Scholar]
- 44.Romero-Maraccini OC, Shisler JL, Nguyen TH. 2015. Solar and temperature treatments affect the ability of human rotavirus Wa to bind to host cells and synthesize viral RNA. Appl Environ Microbiol 81:4090–4097. doi: 10.1128/AEM.00027-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.