

Broad-spectrum antibiotics can indiscriminately kill most bacteria, including commensal species that are a part of the normal human flora. This can potentially lead to the proliferation of drug-resistant bacteria upon elimination of competing species and to unwanted autoimmune effects in patients. Bacteriophage-derived lysin proteins are an alternative to conventional antibiotics that have coevolved alongside specific bacterial hosts. Lysins are capable of targeting conserved substrates in the bacterial cell wall essential for its viability. To engineer these proteins to exhibit improved therapeutically relevant properties, homology-guided statistical approaches can be used to identify compelling sites for mutation and to quantify the functional constraints acting on these sites to direct mutagenic library creation. The platform described herein couples this informed approach with a visual plate assay that can be used to simultaneously screen hundreds of mutants for catalytic activity, allowing for the streamlined identification of improved lysin variants.
KEYWORDS: Potts model, covariation, epistasis, homology, lysin, vancomycin-resistant Enterococcus
ABSTRACT
Bacteriophage-derived lysin proteins are potentially effective antimicrobials that would benefit from engineered improvements to their bioavailability and specific activity. Here, the catalytic domain of LysEFm5, a lysin with activity against vancomycin-resistant Enterococcus faecium (VRE), was subjected to site-saturation mutagenesis at positions whose selection was guided by sequence and structural information from homologous proteins. A second-order Potts model with parameters inferred from large sets of homologous sequence information was used to predict the average change in the statistical fitness for mutant libraries with diversity at pairs of sites within the secondary catalytic shell. Guided by the statistical fitness, nine double mutant saturation libraries were created and plated on agar containing autoclaved VRE to quickly identify and segregate catalytically active (halo-forming) and inactive (non-halo-forming) variants. High-throughput DNA sequencing of 873 unique variants showed that the statistical fitness was predictive of the retention or loss of catalytic activity (area under the curve [AUC], 0.840 to 0.894), with the inclusion of more diverse sequences in the starting multiple-sequence alignment improving the classification accuracy when pairwise amino acid couplings (epistasis) were considered. Of eight random halo-forming variants selected for more sensitive testing, one showed a 1.8 (±0.4)-fold improvement in specific activity and an 11.5 ± 0.8°C increase in melting temperature compared to those of the wild type. Our results demonstrate that a computationally informed approach employing homologous protein information coupled with a mid-throughput screening assay allows for the expedited discovery of lysin variants with improved properties.
IMPORTANCE Broad-spectrum antibiotics can indiscriminately kill most bacteria, including commensal species that are a part of the normal human flora. This can potentially lead to the proliferation of drug-resistant bacteria upon elimination of competing species and to unwanted autoimmune effects in patients. Bacteriophage-derived lysin proteins are an alternative to conventional antibiotics that have coevolved alongside specific bacterial hosts. Lysins are capable of targeting conserved substrates in the bacterial cell wall essential for its viability. To engineer these proteins to exhibit improved therapeutically relevant properties, homology-guided statistical approaches can be used to identify compelling sites for mutation and to quantify the functional constraints acting on these sites to direct mutagenic library creation. The platform described herein couples this informed approach with a visual plate assay that can be used to simultaneously screen hundreds of mutants for catalytic activity, allowing for the streamlined identification of improved lysin variants.
INTRODUCTION
The misuse of antibiotics is a growing problem in the twenty-first century (1). In addition to the development of antibacterial resistance and subsequent loss of treatment efficacy, the use of broad-spectrum antibiotics can reduce the diversity of a patient’s commensal flora (2). This reduction in diversity has been correlated with the onset of multiple health issues, including several inflammatory and autoimmune diseases (3). The development of alternative antimicrobial strategies that offer improved specificity could help mitigate both of these issues.
Native bacteriophage-derived lysin proteins are released during the last stage of the virus’s lytic cycle to degrade the cell wall of the Gram-positive bacterial host (4). These antimicrobial proteins have the potential to be used as effective alternatives that ameliorate many of the negative side effects of conventional antibiotics. The mechanism of action of lysins generally involves the cleavage of an essential and highly conserved peptidoglycan bond in the bacterial cell wall; as such, the development of resistance to these antimicrobial proteins is expected to occur less easily (5–8). Additionally, many lysins are specific, enabling them to kill pathogens without exhibiting significant activity against commensal bacterial species (9, 10). However, engineering lysins to have more desirable properties that contribute to improvements in functional bioavailability, such as higher rates of catalytic activity, heightened solubility, or increased thermal stability, is almost always necessary before a sufficient therapeutic response in the infected host can be achieved (5, 11). Improvements to catalytic activity in particular can reduce the necessary concentration, both in formulation and physiologically, thus decreasing the required solubility.
Lysins generally possess both a catalytic domain and cell wall-binding domain (CWBD) connected together via a flexible linker (12). Catalytic domains can be categorized into five groups depending on their substrate: N-acetyl-β-d-muramidases (lysozymes), lytic transglycosylases, N-acetyl-β-d-glucosaminidases, N-acetylmuramoyl-l-alanine amidases, and endopeptidases (8). N-Acetylmuramoyl-l-alanine amidases hydrolyze the amide bond between N-acetylmuramic acid, a constituent in the repeating disaccharide of the glycan chain in the cell walls of Gram-positive bacteria, and l-alanine, the first amino acid residue of the stem peptide responsible for cross-linking neighboring glycan chains (13). The structure of each major type of catalytic domain is well conserved between lysins derived from different phage species (14), as is generally observed for functional sites in enzymes (15). The CWBD of a lysin, in contrast, is responsible for colocalizing the catalytic domain with its substrate and usually possesses specific affinity for a particular species or subgroup of bacteria (5). The two domains, although connected, are often thought of as capable of carrying out mechanistically distinct functions. This has allowed for the creation of chimeric lysins with altered activity and specificity via domain swapping (16, 17).
LysEFm5 is a lysin with an N-acetylmuramoyl-l-alanine amidase as its catalytic domain. LysEFm5 was previously isolated and described as having killing activity against vancomycin-resistant Enterococcus faecium (VRE) (18). E. faecium is found in the gastrointestinal tracts of healthy individuals but can pose a serious threat if it spreads to the bloodstream, urinary tract, or wound of an immunocompromised patient, most often from a nosocomial infection. Vancomycin is typically only used as a “last resort” to treat infections of Gram-positive bacteria that are unresponsive to other antibiotics. As such, vancomycin resistance in patient-derived E. faecium isolates has been correlated with poor patient outcome and even death (19–21).
LysEFm5 was shown to have a broader antibacterial range than IME-EFm5, its parent phage. LysEFm5 was able to lyse 19 of 23 strains of E. faecium, 7 of them VRE (compared to 1 of 23 strains of E. faecium lysed by IME-EFm5) but possessed no apparent killing activity against the other Gram-positive or Gram-negative bacteria tested. The homology-based structure of the catalytic domain of LysEFm5 has also been reported (18). E90 and T138 have been identified as putative catalytic residues, and H27, H132, and C140 were identified as putative zinc-coordinating residues. These two sets of residues are generally well conserved in the ligand-binding grooves of zinc-dependent peptidoglycan hydrolases (22, 23).
LysEFm5 was chosen for further study based on the clinical relevance of its target, availability of homology-based structural information, and specificity toward E. faecium (in contrast to other broadly active anti-E. faecium lysins [24]).
Nine site-saturation mutagenic libraries were created to study the effectiveness of using structure and sequence information to direct lysin engineering efforts. To determine which residue positions in LysEFm5 to diversify, it was desired to find sites in the catalytic domain that were not critical for the catalytic activity of the protein but played a role in stabilizing other key functional residues. In addition to identifying these residues using the crystal structure of a close homolog to LysEFm5, the choice of positions used in double mutant libraries was refined further using a computationally informed approach. The overall methodology is given in Fig. 1.
FIG 1.

Research methodology. (1) LysEFm5 catalytic domain is used in an iterative homology search. (2) Resulting homologous sequences are subject to length cutoffs. (3 to 4) A structure-based MSA is created for each group of sequences. PLMC is used to infer site-dependent and pairwise coupling parameters and create a generative model for predicting the change in statistical fitness, ΔE, of mutants. (5) Residues in the putative secondary interaction shell of LysEFm5 are identified using the ligand-binding crystal structure of a homologous protein. (6) A matrix of predicted double mutation outcomes is created using PLMC. This is used to guide position selection for combinatorial library design. (7) Halo-forming and non-halo-forming variants from each library are observed, binned, and deep sequenced. (8) The experimental retention of function is compared to the predicted statistical fitness for mutants.
Homologous protein sequences contain information about the structural and functional constraints imposed on a protein over the course of its evolution, which can be of value when directing engineering efforts (14, 25, 26). The natural sequence record is assumed to contain mutations that allow for the retention of a protein’s biological function. Sequences of protein homologs are often highly variable despite marked similarities in their structure and function. This suggests that the site-specific, or independent, trends in amino acid conservation alone may be insufficient to model sequence constraints experienced by proteins over evolutionary time (27). Recently, statistical methods that consider the interactions between pairs of residues in an attempt to capture the nature of nonindependent, or epistatic, mutations have emerged (27–32). Models such as these that take epistatic interactions into account have been shown to more accurately predict the effects of mutations on a protein’s function than independent models that neglect pair couplings (28, 32).
It has been shown that if the mutation of a protein is assumed to be a reversible Markov process, the resulting maximum-entropy ensemble that represents the distribution of natural sequences at equilibrium (functionally, long evolutionary times from the shared ancestral protein) obeys a Boltzmann distribution (33). Thus, the probability of observing any full-length amino acid sequence, σ, in the system can be computed.
| (1) |
It is further assumed that the energy function in equation 1 takes the form of a second-order Potts model with parameters that are fitted to reproduce the empirically observed sitewise and pairwise statistics in the multiple sequence alignment (MSA) (34):
| (2) |
where is the statistical fitness, hi are the site-dependent constraints, and Ji,j are the pairwise coupling constraints at positions i and j in the full-length amino acid sequence, σ.
The exact calculation of the parameters in the Potts model requires determination of the partition function, Z, in the Boltzmann distribution equation—a sum over all possible 20L protein sequences. The pseudolikelihood maximization inference method can be used to simplify this generally intractable calculation, requiring instead the calculation of L individual sums over 20 amino acids (27). A Potts model with parameters inferred using pseudolikelihood maximization has been shown to accurately identify strongly coupled pairs of amino acids, making pseudolikelihood maximization a useful inference method that is less computationally intensive than alternative more precise methods (27, 29, 34, 35).
Although not linked to any one molecular phenotype, the statistical fitness is more likely to correlate with a phenotype directly related to an organism’s survival that would be selected for throughout its evolutionary history (28). In this manner, the effect of a mutation(s) on a protein can be predicted by calculating ΔE = E(σmutant) − E(σwild type), insofar as predicting whether the mutation(s) increases (ΔE > 0) or decreases (ΔE < 0) the probability of observing the new sequence in the protein family described originally by the MSA.
The framework of this methodology was previously developed and released as an open-source code by the Marks lab at Harvard under the name pseudolikelihood maximization coupling inference (PLMC) (28). PLMC was used to build a predictive model of mutational outcomes in the LysEFm5 catalytic domain and direct the selection of amino acid sites for site-saturation mutagenesis.
RESULTS
Statistically guided design and construction of a mutant lysin library.
Only the catalytic domain of LysEFm5 was chosen for alteration; the CWBD was not edited in order to maintain the desired specificity of the protein. Within the catalytic domain of LysEFm5, a network of residues interact with the peptidoglycan substrate to hydrolyze the amide bond between N-acetylmuramic acid and l-alanine at the first position of the stem peptide. Within this network, there are residues that directly interact with the substrate (primary shell) and residues which position and stabilize primary residues without directly interacting with the substrate (secondary shell). We hypothesized that mutating these so-called secondary residues could optimize the catalytic performance of the enzyme, as has been seen before in other enzymes (36), and possibly improve antimicrobial activity.
The catalytic domain of the major pneumococcal autolysin LytA, initially evaluated due to the availability of the solved crystal structure of the domain bound to a synthetic peptidoglycan ligand (37), was identified as a homolog of the catalytic domain of LysEFm5 via sequence alignment (with a sequence similarity of 0.23). SWISS-Model provided a QMEAN score of −7.58, sequence identity of 8.33, and coverage of 1.00 when the catalytic domain of LytA (PDB code 5CTV) was used as a template to model the first 180 amino acids in the catalytic domain of LysEFm5. Thirteen primary residues and ten putative secondary residues were identified in the structure of the LytA amidase (Fig. 2A and B). Eleven structurally analogous secondary residues (N32, S33, T34, A35, E38, T40, M45, N47, A74, I87, and V91) were selected as candidates for mutation (Fig. 2C). One primary residue (N83) that occupied the same space in the structural homolog model of the LysEFm5 molecule as in the aligned structure of the LytA molecule was also included for comparison.
FIG 2.

Primary and secondary amino acids in LytA and their putative structural analogs in LysEFm5. Molecules are aligned using the “align” command in PyMOL. (A) Surface representation of the LytA molecule, showing primary residues (yellow) interacting with the synthetic peptidoglycan ligand (red). (B) Putative secondary residues (green) that interact with primary residues. (C) Structural analogs of the eleven secondary residues and single primary residue in LysEFm5. (The ligand was superimposed following the structural alignment of LytA and LysEFm5 in PyMOL and is not part of the reported structure of LysEFm5.) (D) Map of the location of the relevant putative secondary and primary residues in the amino acid sequences of LytA and LysEFm5. Note that although the hydroxyl group, -OH, of E38 in LytA was predicted to bind to the O in the CH2OH group of N-acetylmuramic acid in the peptidoglycan ligand, the analog E38 in LysEFm5 was still selected as a secondary residue (most primary residues were found to bind the ligand twice or more).
A computational model of sitewise and pairwise interactions, based on the sequence alignment of homologous sequences, was used to determine which pairs of sites to simultaneously mutate in the experimental libraries. To identify homologs to the LysEFm5 amidase domain sequence, a search of the UniProtKB protein database was performed via Jackhmmer (38). Sequence searches were constrained to three levels of evolutionary depth by restricting the acceptable taxonomy of the host organism to all organisms (no restrictions), bacteria only, or Firmicutes only. Sequences that were either extremely short or long were excluded from further consideration by applying either a lax or stringent cutoff criterion for outlier detection (see Materials and Methods). This generated a total of six sets of starting homologous sequences (Table 1). Each set was independently input into PROMALS3D, an alignment tool that incorporates both sequence and structure information (39), to create an MSA.
TABLE 1.
Predictive performance of the statistical fitness when different groups of homologous sequences are used in the starting MSA
| Designation | Length (no. of aa)a | No. of sequences | Effective no. of sequencesb (±SD) | AUC (±SE)c |
|
|---|---|---|---|---|---|
| Epistatic model | Independent model | ||||
| Alllax-29k | 141–199 | 29,498 | 6,352 | 0.894 | 0.807 |
| Allstringent-23k | 155–186 | 23,176 | 4,809 | 0.856 | 0.857 |
| Alllax-3k | 3,037 | 1,420 ± 33 | 0.815 ± 0.013 | 0.744 ± 0.026 | |
| Bacterialax-27k | 137–200 | 26,950 | 5,565 | 0.868 | 0.888 |
| Bacteriastringent-23k | 149–188 | 23,194 | 4,595 | 0.851 | 0.871 |
| Bacterialax-3k | 3,037 | 1,309 ± 39 | 0.839 ± 0.006 | 0.782 ± 0.013 | |
| Firmicuteslax-3k | 133–201 | 3,037 | 940 | 0.852 | 0.795 |
| Firmicutesstringent-2k | 163–192 | 2,007 | 600 | 0.840 | 0.830 |
| Firmicutes+alllax-3k | 3,037 | 1,344 ± 34 | 0.856 ± 0.005 | 0.818 ± 0.014 | |
| Firmicutes+Bacterialax-3k | 3,037 | 1,317 ± 30 | 0.851 ± 0.004 | 0.814 ± 0.014 | |
| Firmicutes+nonbacterialax-2k | 5,585 | 0.865 | 0.799 | ||
The acceptable amino acid lengths across the six initial groupings.
The effective number is the sum of the inverse of the neighborhood size of each sequence, where the neighborhood is defined as the number of sequences within 80% identity. SD, standard deviation.
Results are presented as mean values from 20 subsamplings from the parent group(s). SE, standard error.
PLMC was then used to infer the parameters of a second-order Potts model for each MSA. Without knowledge a priori regarding the effect of the sequence diversity in the starting MSA on prediction accuracy, it was hypothesized that the most constrained and least diverse set of data would enable the most accurate prediction of activity. Thus, the MSA containing the least diverse set of sequences (Firmicutesstringent-2k) was used to predict the change in statistical fitness, ΔE, compared to that of the wild type (WT) for all possible double mutants across the twelve sites of interest (Fig. 3). Simultaneous mutation of S33 and T40 yielded the highest ΔE values; as such, these sites were randomized in library 1. To evaluate the predictive performance of the statistical fitness parameter, seven additional combinations of positions were selected with a range of average ΔE values upon mutation, from the highest value of 4 ± 2 observed for library 1 to the lowest value of −12 ± 3 observed for library 8 (Fig. 3 and 4). Library 8 contained the putative primary residue, N83.
FIG 3.

The predicted changes in statistical fitness for double mutants. The change in statistical fitness compared to that of the WT, ΔE, for all double mutants with diversity at positions 32, 33, 34, 35, 38, 40, 45, 47, 74, 83, 87, and/or 91 was computed using PLMC with inputs from MSA group Firmicutesstringent-2k (Table 1). (Left) The eight libraries chosen for creation are boxed. (Right) A closer look at the predicted ΔE values for libraries 1 (the library with the highest average ΔE value, −4 ± 2) and 8 (the library with the lowest average ΔE value, −12 ± 3). The dotted squares represent WT residues.
FIG 4.
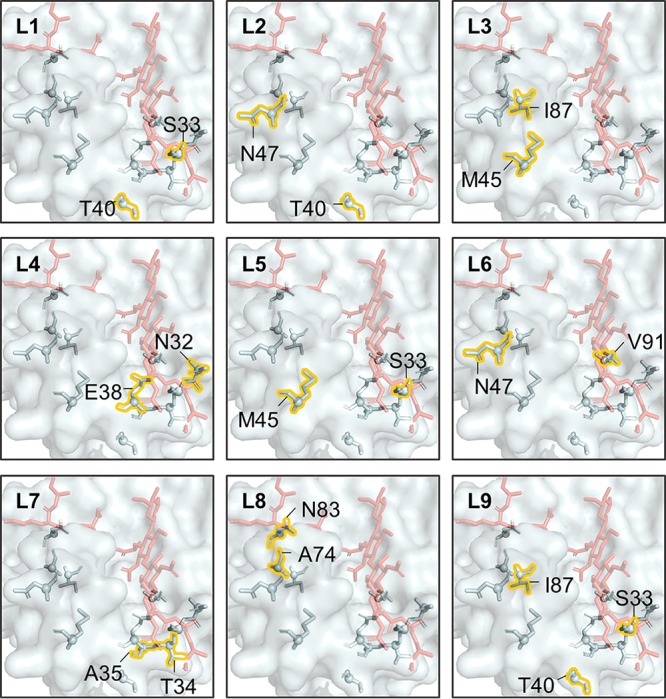
Location of residues in each library (L) relative to superimposed ligand in red.
One additional library was designed with amino acid diversity constrained based on the predicted statistical fitness. A matrix of the predicted ΔE values from PLMC for single mutants occurring at each of the twelve sites was discretized and input into SwiftLib, an algorithm that specifies a degenerate codon library to yield the desired amino acids at several positions based on a user-defined array of integers describing a favoring or disfavoring of all amino acids (here, based on ΔE) (40). The resulting optimal library (library 9) diversified the same two positions as library 1 (33 and 40) but also included the single mutation I87L. This mutation had a positive predicted ΔE value (+0.21) and was thus highly favored; only three positive ΔE values were observed across the single mutants in general, the other two of which occurred at site 40 (T40N and T40D).
To generate the libraries, gene fragments of the WT were amplified via PCR with mutation-encoding primers. Overlapping fragments were combined via Gibson assembly to yield a collection of plasmids encoding the entire LysEFm5 gene with two randomized codons at the desired sites for each library (41). Upon assembly, clones from each library were transformed into high-efficiency electrocompetent cells. Sequencing of random colonies confirmed that the libraries encoded the entire LysEFm5 gene with diversity at the expected sites.
VRE halo assay to screen LysEFm5 variants for catalytic activity.
Recombinant plasmids encoding LysEFm5 mutants were transformed into Escherichia coli for protein expression, and individual clones were assayed for their ability to digest autoclaved 8-E9 VRE by plating the transformed E. coli on top of agar plates containing autoclaved VRE and isopropyl-β-d-1-thiogalactopyranoside (IPTG) used to induce lysin expression. The plating density was such that the majority of individual colonies were easily distinguishable and separate from their neighbors. Upon incubation, colonies expressing an active lysin variant formed a visible halo due to degradation of the surrounding VRE, leading to a localized decrease in optical density (Fig. 5). Halo formation did not occur when E. coli transformed with a plasmid encoding a phage lysin with specific activity against Clostridium perfringens (42) was plated in an identical manner (see Fig. S2 in the supplemental material). This observation supports the hypothesis that autoclaving the VRE prior to use did not increase its susceptibility to native lysozymes produced by E. coli or to the activity of a lysin with an alternative specificity. The size of the observed halo is the result of a number of physical properties of the expressed lysins, including stability, expression and degradation rates, per-molecule activity, diffusivity, etc. (43). Therefore, this format does not result in equal amounts of protein produced and subsequently released from each colony, and halo size cannot be directly correlated to specific activity. This assay was instead used in a binary sense to designate a variant as either halo forming or non-halo forming, with halo-forming variants assumed to be a subset of all active variants. Assays similar to the one described here were previously used to screen expression libraries and identify endolysin-producing clones (44) as well as to confirm the production in E. coli of two phage-derived lysins with broad activity against multiple strains of E. faecium and Enterococcus faecalis (45). Although the E. coli in these previous studies was chemically permeabilized to release expressed proteins, the method presented herein relied only on the intrinsic leakage of the host. The mechanism of this release is not known but hypothesized to be the result of cell lysis upon death or increased permeabilization as a result of the overexpression of lysY.
FIG 5.
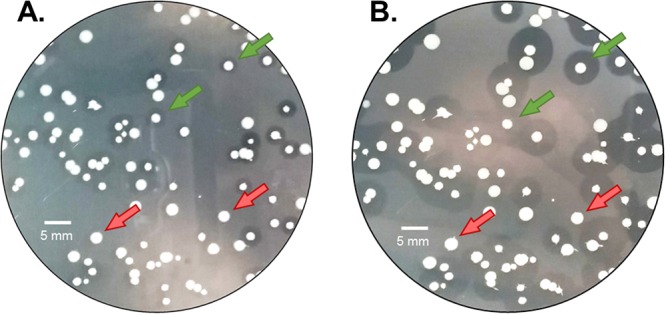
Halo formation over time. This image is representative of the appearance of halo-forming and non-halo-forming variants in general. Libraries were plated on top of LB plus kan-VRE-IPTG plates. (A) At approximately 16 to 18 h following incubation at 37°C, discernible halos appeared around catalytically active variants (green arrows) and not around catalytically inactive variants (red arrows). (B) At longer times (>18 h), the halo radii continued to grow.
Three parallel runs were performed for each library, in which each of approximately 375 colonies was classified as halo forming or non-halo forming. This resulted in a total of six bins, corresponding to the replicate number and halo formation designation, per library. DNA isolated from these six bins was sequenced using Illumina MiSeq. Sequences with fewer than 100 reads were deemed to be erroneous and excluded. Protein sequences translated from the remaining DNA reads were excluded if present in only a single replicate or if they lacked a majority of either halo-forming or non-halo-forming designations. Applying the latter constraint reduced the number of usable data points from 1,731 unique sequences to 873, while greatly improving the classification accuracy of the statistical fitness associated with this method (see Fig. S5).
Secondary site restriction allows for focused library design resulting in a high retention of catalytic activity.
Sequencing results showed a rate of activity of between 84% and 100% per library for all classified variants in libraries 1 to 7 and 9 (Table 2). There was no general trend in the experimental retention of catalytic activity and the average statistical fitness of these libraries (Fig. 6). However, library 8, which had the lowest predicted statistical fitness and diversity at positions N83 and A74, demonstrated a low activity retention of 30%. N83 in LysEFm5, the only putative primary residue considered in this analysis, is structurally analogous to N79 in LytA. N79 is a ligand-binding residue that is highly conserved across multiple prokaryote- and eukaryote-derived peptidoglycan recognition proteins (PGRPs), both with and without amidase activity (37). In AmiE (the amidase domain of the major autolysin of Staphylococcus epidermidis) and in human PGRP-Iα, this conserved asparagine residue was shown to hydrogen bond with the carbonyl groups in the second and third amino acids in the peptide stem of N-acetylmuramic acid (MurNAc)-l-Ala-d-isoGln-l-Lys, a peptidoglycan analog (23, 46). A74, in contrast, is not predicted to be a primary residue from direct structural comparison, but the average changes in statistical fitness associated with independently mutating A74 and N83 were similar (ΔE = −6 ± 2 for both, compared to ΔE = −3 ± 2 on average for the other ten residues). Even if not directly bound to the ligand and/or playing a pivotal role in stabilizing the transitional state between substrate and product, A74 may stabilize one or more neighboring primary residues in an essential way. The low rate of activity retention observed for library 8 provides evidence that the statistical fitness parameter was able to predict highly detrimental mutations at a key conserved site critical for the function of the protein.
TABLE 2.
Numbers of active and inactive classified mutants in each library
| Library no. | Diversified positions | No. of double mutants |
Total no. |
||
|---|---|---|---|---|---|
| Active (% of total) | Inactive | Active (% of total) | Inactive | ||
| 1 | 33, 40 | 184 (94) | 11 | 211 (95) | 11 |
| 2 | 40, 47 | 83 (98) | 2 | 114 (98) | 2 |
| 3 | 45, 87 | 53 (82) | 12 | 67 (84) | 13 |
| 4 | 32, 38 | 96 (96) | 4 | 109 (96) | 4 |
| 5 | 33, 45 | 92 (100) | 0 | 114 (100) | 0 |
| 6 | 47, 91 | 24 (100) | 0 | 45 (98) | 1 |
| 7 | 34, 35 | 18 (90) | 2 | 36 (95) | 2 |
| 8 | 74, 83 | 12 (21) | 46 | 22 (30) | 51 |
| 9 | 33, 40, 87a | 74b (96) | 3 | 313 (95) | 15 |
Specific mutation I87L, not implementation of a randomized codon, at this site.
Triple mutants.
FIG 6.
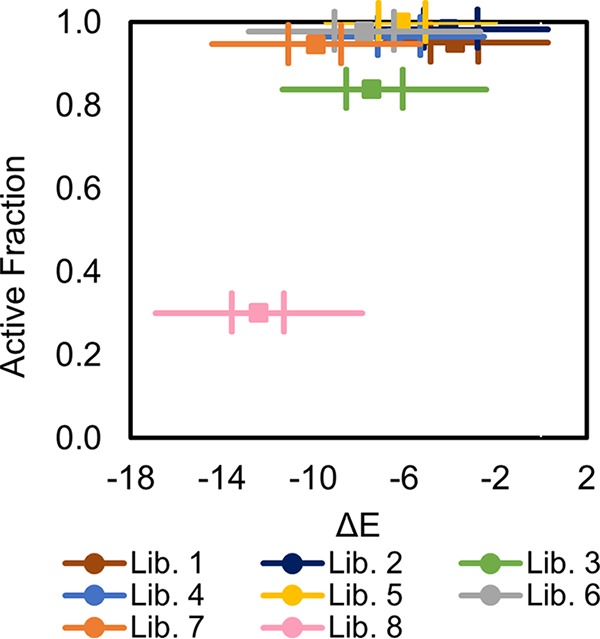
Active fraction of variants in library as a function of average ΔE. The difference between the statistical fitness of each variant (using MSA group Firmicutesstringent-2k) and the WT, ΔE, is plotted against the active fraction of classified mutants in a library. The central square is the median ΔE value and the left- and rightmost vertical lines represent the 25th (Q1) and 75th (Q3) percentiles. Whiskers extend to the most extreme points not considered outliers; outliers are defined as values less than Q1 − WM(Q3 − Q1) or greater than Q3 + WM(Q3 − Q1), where WM is the maximum whisker length.
The rates of activity retention for library 9 (with constrained diversity at sites 33 and 40 and the mutation I87L) and library 1 (with full diversity at sites 33 and 40) were nearly identical at 95%. Further analysis revealed that there were 73 triple mutants present in library 9 with analogous sequences in library 1 (sequences with the same diversity at sites 33 and 40, but with the WT residue at site 87). Of these, 73/73 were active in library 1 (100%) compared to 72/73 in library 9 (99%). Taken together, these results highlight the flexibility in amino acid identity of the residue at site 87, from the WT I to L.
A similar ex post facto analysis was performed for all remaining libraries by comparing the active fraction of classified double mutant sequences to the active fraction of sequences in a hypothetical constrained library (built using a discretized matrix of predicted independent mutation outcomes at each of the two library-specific sites) (Table 3). For libraries 6, 7, and 8, SwiftLib predictions were so constrained that none of the allowable sequences were among those that were experimentally observed. Among the remaining five libraries, only the constrained subset of library 3 showed any substantial improvement in activity retention (from 82% to 100%).
TABLE 3.
Active fractions for all double mutants in a library compared to active fractions of the subset predicted by SwiftLib
| Library no. | Diversified positions | SwiftLib codona |
SwiftLib theoretical aa diversity | Overall active fraction | SwiftLib active fraction (no. of observations)b | |
|---|---|---|---|---|---|---|
| Position 1 | Position 2 | |||||
| 1 | 33, 40 | VNC | NNS | 252 | 0.94 | 0.97 (173) |
| 2 | 40, 47 | NNS | NDC | 252 | 0.98 | 0.97 (68) |
| 3 | 45, 87 | RNS | NWC | 96 | 0.82 | 1.00 (17) |
| 4 | 32, 38 | NNM | BRS | 190 | 0.96 | 0.92 (38) |
| 5 | 33, 45 | NNS | RBG | 126 | 1.00 | 1.00 (48) |
| 6 | 47, 91 | NNM | GTA | 19 | 1.00 | NA (0) |
| 7 | 34, 35 | AVC | GCA | 3 | 0.90 | NA (0) |
| 8 | 74, 83 | GCA | AAC | 1 | 0.21 | NA (0) |
N = G, T, A, or C; V = G, T, or A; B = G, T, or C; M = A or C; R = A or G; W = A or T; S = G or C; K = G or T.
NA, not applicable.
Figure 7 shows the fraction of halo-forming sequences of all classified single, double, and triple mutants based on the identity of the amino acid at a specified position (note that the same positions were mutated in multiple libraries). A deeper analysis of library 8, which is the only library that included mutations at sites A74 and N83, revealed that the rate of activity retention of single mutants (67% [10/15]) was higher than that of double mutants (21% [12/58]). When the WT residue was retained at A74 and only N83 was mutated, the active fraction was 78% (7/9); conversely, when the WT residue was retained at N83 and only A74 was mutated, the active fraction was 50% (3/6). The intolerance to simultaneous mutations occurring at both sites but moderate tolerance to single mutations at either site suggests that the retention of one of these two WT residues (or of a close analog with similar polarity and size characteristics) is critical for catalytic activity. In general, the polar uncharged residues serine, cysteine, and glutamine were the best tolerated at site N83 across all mutants (63% [5/8], 50% [1/2], and 50% [2/4], respectively). Mutation to the alanine analog valine was additionally well tolerated at site A74 (100% [4/4]). In contrast, mutation to the positively charged hydrophilic residues arginine and lysine, or to proline and tryptophan, was not tolerated at either A74 or N83. Mutation to arginine was also not tolerated at sites A35, I87, or V91 (0/2, 0/5, and 0/1, respectively).
FIG 7.

Average active fractions for a sequence based on the amino acid at the specified position. For each position of interest, the amino acid at that site is related to the active fraction of sequences having that particular mutation. The total number of sequences considered in the calculation of the active fraction value is given in the top-right corner of each cell. Note that this is based on sequence data for single, double, and triple mutants across libraries mutating redundant positions and is therefore not a canonical heat map of independent mutation outcomes.
Increasing the diversity of protein sequences used in the MSA improves the binary classification ability of the statistical fitness property.
The halo-forming or non-halo-forming designation of each sequence was compared to the predicted statistical fitness calculated using different sets of sequence inputs in the starting MSA and assessed via a receiver operating characteristic (ROC) curve.
All six ROC curves for the initial homology sequence sets yielded area under the ROC curve (AUC) values between 0.840 (Firmicutesstringent-2k) and 0.894 (alllax-29k), demonstrating the ability of the statistical fitness to discriminate between active and inactive variants (Fig. 8). Moreover, relaxing the restrictions placed on the protein sequences in the starting MSA, in terms of both acceptable sequence length (stringent→lax cutoff) and acceptable taxonomy (Firmicutes→Bacteria→all), was shown to consistently improve the AUC and thus increase the reliability of the predictive model (Fig. 9A). This agrees with previous findings by Hopf et al., who found that progressively excluding evolutionarily distant sequences led to poorer PLMC predictive performance across 34 sets of data, 21 of which involved proteins (28).
FIG 8.
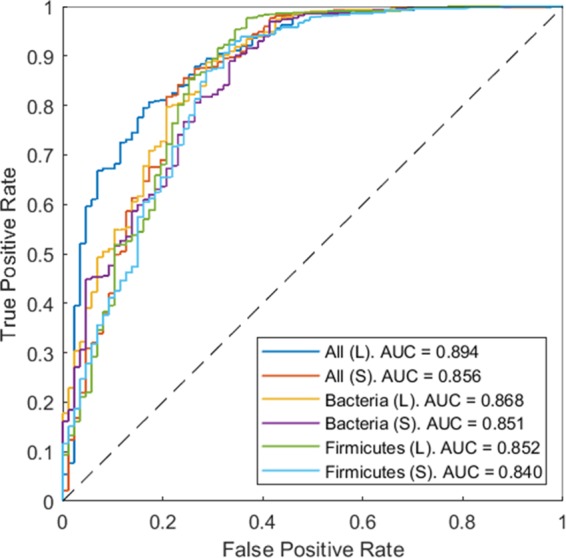
ROC curves demonstrating that the statistical fitness is indicative of variant activity. The experimentally observed outcome (retention or ablation of catalytic activity) for each qualifying variant was evaluated against the variant’s statistical fitness, calculated using one of the six sets of protein sequences in the starting MSA (Table 1). “(L)” denotes that the lax cutoff threshold for outlier detection was used, and “(S)” denotes that the stringent cutoff threshold was used. The AUC consistently improved when restrictions placed on the protein sequences used in the MSA, in terms of both allotted sequence length and taxonomy, were relaxed. The best predictive method employed the least constrained MSA containing the most sequence information.
FIG 9.
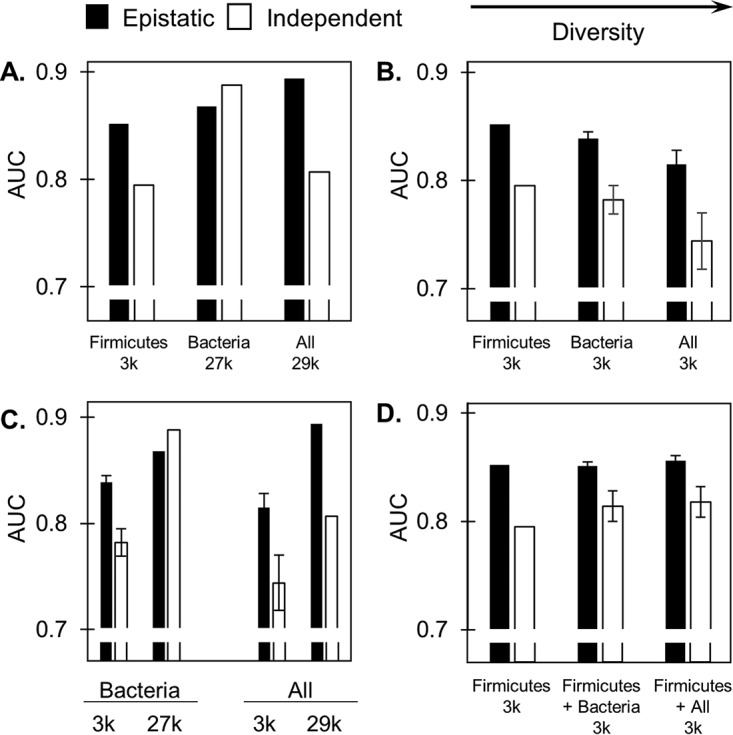
Effects of varying sequence diversity and depth on the predictive performance of the statistical fitness. Predictive performance of the statistical fitness when different groups of homologous sequences are used in the starting MSA (summarizing key results from Table 1, with the average AUC given for designations consisting of 20 subsampled groups containing 3,000 sequences and error bars representing the standard errors). Comparison of different diversity sources including every available sequence (A) or only 3,037 sequences (B) within each category. (C) Comparison of performance at different sequence depths for Bacteria and “all.” (D) Comparison of performance for 1,500 Firmicute sequences plus 1,500 additional sequences from Firmicutes, Bacteria, or “all.” All data are for the lax length threshold.
Including increasingly evolutionarily distant sequences in the starting MSA also increased the number of sequences under consideration. To decouple the individual effects that the evolutionary distance and sequencing depth have on predictive performance, the groups alllax-29k and Bacterialax-27k were randomly sampled 20 times each to create subgroups containing the same number of sequences as in Firmicuteslax-3k (3,037). All subgroups were independently aligned with PROMALS3D, and PLMC was used to generate a Potts model for each. Additionally, epistatic coupling was considered in the model as before or toggled off by omitting pairwise contributions during model inference. An AUC value for the ROC curve relating the statistical fitness to the experimental results was calculated for each of these subgroups (Table 1).
When the sequencing depth was fixed, the inclusion of sequences phylogenetically closest to that of the WT, i.e., those that were less diverse, led to the best predictive performance. This was true of both the epistatic and independent models (Fig. 9B). Conversely, when the acceptable diversity was fixed, the inclusion of more sequences in the starting MSA improved the predictive performance of both models (Fig. 9C).
Notably, 91% of the 2.9 × 104 sequences in the alllax-29k group are bacteria, and 89% of these bacterial sequences are nonfirmicutes. For the epistatic model, supplementing the 3,037 firmicute-only sequences with 2.4 × 104 additional nonfirmicute bacterial sequences (to yield the group Bacterialax-27k) led to a +0.016 improvement in AUC. Supplementing further with only 2,548 nonbacterial sequences (to yield the group alllax-29k) led to a further +0.026 improvement in AUC. The same was not true in the independent model: supplementing the group Firmicuteslax-3k to yield Bacterialax-27k improved the AUC from 0.795 to 0.888 (+0.093), but further supplementing the group Bacterialax-27k to yield alllax-29k led to a decrease in the AUC from 0.888 to 0.807 (−0.081). These results suggest that epistasis must be considered in order for highly diverse sequences to be beneficial and improve predictive performance; otherwise, they can have a negative impact if incorporated into the starting MSA. The group Firmicuteslax-3k was additionally supplemented with 2,548 nonbacterial sequences, and the inclusion of the nonfirmicute bacterial sequences was circumvented entirely. Compared to that for the group Bacterialax-27k, this improved the AUC slightly, from 0.852 to 0.865 (+0.013) in the epistatic case and from 0.795 to 0.799 (+0.004) in the independent case. This suggests that divergent sequences outside those of bacteria offer predominantly sparse information, and their contribution is significant only when pairwise information is considered.
Thus, although using a small set of sequences that were phylogenetically similar to the WT led to a superior predictive performance compared to that using a small set of diverse sequences (AUC Fimicuteslax-3k > AUC Bacterialax-3k > AUC alllax-3k), once the MSA was seeded with a collection of close homologs, the inclusion of more diverse sequences further improved the predictive performance of the epistatic model (AUC alllax-29k > AUC Bacterialax-27k). To further evaluate the relative benefits of appending sequences with different diversities, a set of 1.5 × 103 firmicute sequences was supplemented with 1.5 × 103 sequences subsampled from either alllax-29k (to yield the subgroup Firmicutes+alllax-3k) or Bacterialax-27k (to yield Firmicutes+Bacterialax-3k), once again resulting in a total of 3,037 sequences per subgroup. The performance of both subgroups was compared to that of Firmicuteslax-3k. For the epistatic model, the predictive performances of each group were similar. For the independent model, the inclusion of more diverse sequences was slightly more advantageous than only including more firmicute sequences (Fig. 9D).
A mid-throughput binary screen, coupled with a computationally informed library design, resulted in the efficient isolation of lysin mutants with improved specific activity and/or thermal stability.
Ten halo-forming variants from libraries 1 to 9 were randomly selected during the halo plate assay to more sensitively quantify their specific activity. Variant 2 (R17L/T40L) had an off-target mutation that was not found in any libraries and was therefore not pursued further. When expressed, sufficient amounts of protein were able to be recovered for eight of the nine remaining variants, with final purities ranging from 90% to 99% (median, 97%) (see Fig. S4). Variant 3 (M45E/I87D, library 3) was unable to be produced in sufficient quantities and excluded from further testing. Activity was assayed using the turbidity reduction method, in which 0.1 μg, 0.3 μg, or 0.5 μg of each variant or the WT was coincubated with crude VRE cell wall material. The optical density at 600 nm (OD600) of each sample was monitored over the course of 4 h (Fig. 10A to C). The largest slope over a 10-min time frame was calculated for each replicate as a proxy for specific catalytic activity (Fig. 10D). Under all starting conditions, the WT exhibited a normalized change in OD600 of 0.048 ± 0.007 −ΔOD600/min/μg (n = 32). Five variants (4, 5, 6, 9, and 10) exhibited activities that were moderately diminished compared to that of the WT. Two variants (1 and 8) exhibited activity that was statistically indistinguishable from that of the WT. Variant 7 exhibited a markedly improved activity of 0.09 ± 0.01 −ΔOD600/min/μg (P < 0.001). To further validate the halo assay, we also tested eight random non-halo-forming variants (Table 4). Six of the eight yielded negligible recombinant production (four of which were revealed via sequencing to encode undigested pET-24 vector), which validates their inability to generate halos. Variants 13 and 14, which were non-halo forming, had activities of 0.005 ± 0.004 and 0.008 ± 0.004 −ΔOD600/min/μg, respectively, which were statistically indistinguishable from the performance of the buffer negative control, 0.004 ± 0.004 −ΔOD600/min/μg (n = 32), further supporting the validity of the halo assay. See Table 4 for a summary of variant mutations, libraries of origin, and −ΔOD600 per minute per microgram values.
FIG 10.
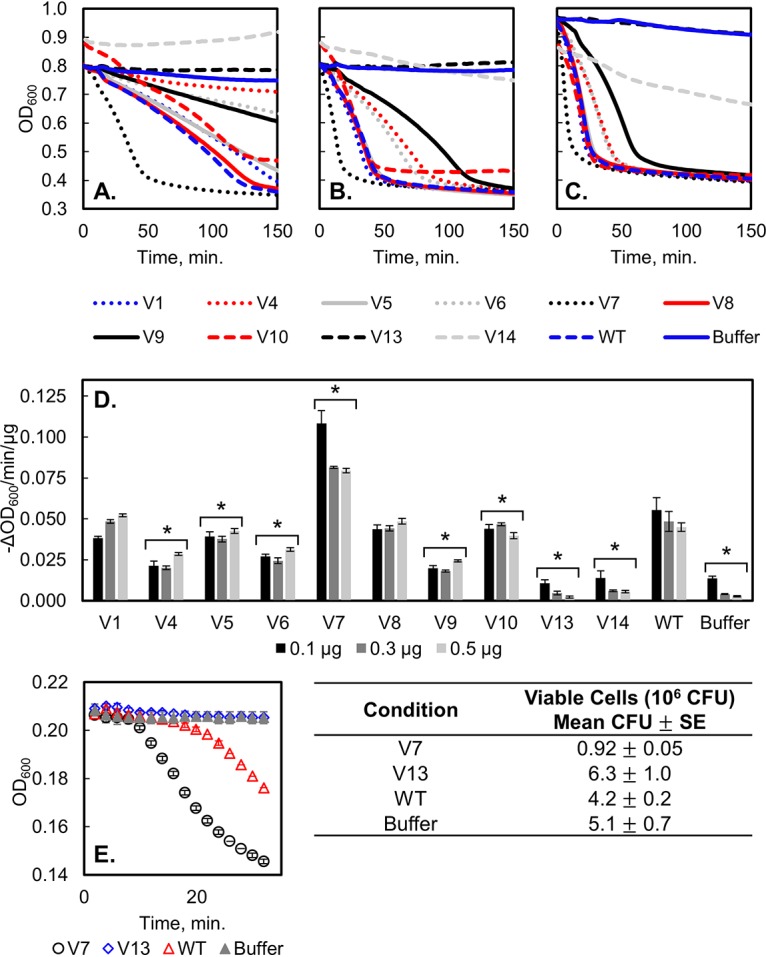
LysEFm5 variant and WT activity against VRE. Turbidity reduction assay results where (A) 0.1 μg (n = 4 for variants, 8 for the WT), (B) 0.3 μg (n = 4 for variants, 8 for the WT), or (C) 0.5 μg (n = 8 for variants, 16 for the WT) of each lysin was combined with VRE cell wall fragments in 200 μl of PBS (total). Additionally, a buffer negative control was included where no lysin was added (n = 32). OD600 was monitored over time, with data collected every 2 min. Error bars are not shown for visual clarity; a measure of the uncertainty between replicates is given in Fig. 10D as the standard deviations in the slope of the linear regions. (D) Quantified activity for variants, WT, and the buffer negative control. The maximum change in OD600 over a 10-min period was calculated from the turbidity assay results for each replicate. *, P < 0.004 (compared to the WT) for a two-tailed, two-sample heteroscedastic Student's t test with a Bonferroni’s correction applied (n = 12). The activity of variants 13 and 14 was additionally statistically insignificant from that of the buffer negative control (P = 0.54 and P = 0.009, respectively). (E) Bacteriolytic activity of variants 7 and 13, the WT, and buffer against live 8-E9 VRE; 0.5 μg of lysin was applied to mid-exponential phase Enterococcus faecium 8-E9 resuspended in PBS. Cell lysis was monitored dynamically via OD600 reduction (left). Killing activity was assessed by plating serially diluted cell suspensions after approximately 30 min (right). Data are presented as means ± standard errors.
TABLE 4.
Information for purified variants and the WT
| Variant | Halo forming? | Library | Mutation(s) | −ΔOD600/min/μg |
|---|---|---|---|---|
| WT | Yes | NAa | NA | 0.048 ± 0.007 |
| 1 | Yes | 2 | T40P, N47V | 0.048 ± 0.006 |
| 2 | Yes | NA | R17L, T40L | NA |
| 3 | Yes | 3 | M45E, I87D | NA |
| 4 | Yes | 4 | N32G, E38T | 0.025 ± 0.004 |
| 5 | Yes | 5 | S33P, M45W | 0.041 ± 0.003 |
| 6 | Yes | 6 | N47A, V91P | 0.028 ± 0.003 |
| 7 | Yes | 7 | T34S, A35V | 0.087 ± 0.013 |
| 8 | Yes | 1 | T40A | 0.046 ± 0.003 |
| 9 | Yes | 1 | S33G, T40S | 0.022 ± 0.003 |
| 10 | Yes | 9 | S33G, T40E, I87L | 0.043 ± 0.003 |
| No | NA | Uncut vector | NA | |
| 11 | No | 3 | M45P, I87G | NA |
| No | NA | Uncut vector | NA | |
| No | NA | Uncut vector | NA | |
| 12 | No | 7 | A35G, T34T | NA |
| No | NA | Uncut vector | NA | |
| 13 | No | 8 | A74R, N83M | 0.005 ± 0.004 |
| 14 | No | 8 | A74T, N83Y | 0.008 ± 0.004 |
NA, not applicable.
Additionally, variant 7 (most active), variant 13 (least active), WT, and buffer were tested against live 8-E9 VRE in exponential phase to evaluate if the turbidity reduction assay, which used purified cell wall material, correlated to the killing of live cells. The reduction in OD600 of cultured VRE cells was found to agree well with the results of the turbidity reduction assay (Fig. 10E).
To determine whether the perceived change in catalytic activity of any of the variants could be attributed in part to a change in the marginal thermal stability of the WT under assay conditions, the stability of variants 4 to 9, 13, and the WT was assessed by Sypro orange thermal denaturation assay (Fig. 11). Variant 1 was unable to be produced in sufficient quantities to use in this assay and was not tested. The melting temperature (Tm) of the WT was 43.4 ± 0.5°C. Variant 4 did not exhibit a signal consistent with unfolding, perhaps resulting from a low Tm or increased disorder in the molecule. Variants 8 and 9 exhibited Tms that were statistically indistinguishable from that of the WT. Variants 5, 6, 7, and 13 exhibited improved Tms at or above 46.5 ± 0.4°C, with variant 7 exhibiting the highest value, 54.9 ± 0.6°C, an 11.5 ± 0.8°C improvement over the WT. This variant comprises chemically homologous mutations—T34S and A35V—at adjacent sites (Fig. 4). The retention of amino acid characteristics at these two sites may be key to the observed improvements in both activity and stability.
FIG 11.
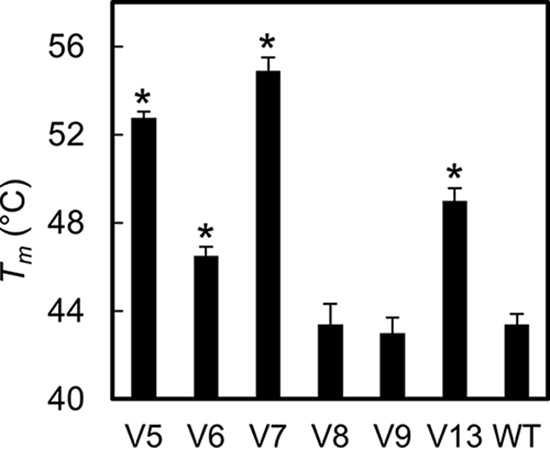
Lysin thermal stability. The midpoint of thermal denaturation was measured for lysin variants 5 to 9 and 13 and the WT by Sypro orange assay. *, P < 0.001 for a two-tailed, two-sample heteroscedastic Student's t test.
DISCUSSION
Improvements to the specific activity of a lysin allow for a lower required dose to achieve the same therapeutic effect, potentially reducing dose-related toxicity and mitigating the immune response to lysin-specific antibodies produced upon administration of the lysin in vivo (47). Improvements in lysin stability allow for more flexibility in the protein production process, a longer shelf life, and a reduction in the tendency to unfold, thereby reducing aggregate formation (48). Improvements in one or both characteristics can contribute to heightened bioavailability in an infected host, which can increase treatment efficacy. Of the eight randomly selected halo-forming lysin variants that were assayed for catalytic activity, three exhibited activity that was indistinguishable from that of the WT (0.048 ± 0.007 −ΔOD600/min/μg) or was improved. Six of the seven variants assayed for thermal stability exhibited a Tm that was indistinguishable from that of the WT (43.4 ± 0.5°C) or was improved. Variant 7 in particular demonstrated both a considerably higher catalytic activity of 0.09 ± 0.01 −ΔOD600/min/μg and Tm of 54.9 ± 0.6°C. The improved characteristics of this variant suggest that it could be used as a starting point for future LysEFm5 engineering efforts.
The notion that one of the eight randomly selected halo-forming variants tested was able to be produced in sufficient quantities and exhibited improvements in catalytic activity and stability, and that two others tested exhibited improvements in stability and retained a fraction of the catalytic activity, is a promising result given the limited sampling. Extending the study presented herein, additional clones could be sampled for more sensitive testing to improve the characterization of the PLMC-informed libraries. This extension may reveal variants with physical properties that are further improved in comparison with those described here. Ultimately, this platform may be able to expedite the discovery process by requiring sensitive testing of a focused set of prescreened variants rather than uninformed libraries orders of magnitude larger in size.
Of the eight double mutant protein libraries that were studied, library 8, with diversity at the putative primary residue N83 and secondary residue A74, was predicted to be the worst-performing library (ΔE = −12 ± 3) and demonstrated the lowest experimental rate of activity (30%). The higher rate of activity among single mutants in this library (67%) than that of double mutants (21%) suggests that the retention of at least one of these two WT residues is necessary for lysin activity. Mutations at either site to the positively charged hydrophilic residues arginine and lysine, or to the structurally disruptive residues proline and tryptophan, resulted in a consistent loss of activity. The remaining seven libraries showed high rates of activity retention (84% to 100%) but no discernible trend in average statistical fitness. Assuming that the results of the halo assay are a monotonic function of the total lysin activity, the relationship between halo-forming variants as a function of the statistical fitness is expected to be sigmoidal in nature (42). The observed similar fractions of active variants for libraries 1 to 7 suggest that the WT lies to the far right of this sigmoid (corresponding to high fitness), such that meaningful differences in the fraction of halo-forming variants would only be seen at considerably lower statistical fitness values, such as the value observed for library 8. Alternatively, or in addition, it is possible that the use of structural information in combination with double mutant ΔE data to constrain site selection led to the construction of libraries with relatively low penalties of mutation and subsequently high observed rates of activity retention.
Constraining the diversity of the libraries using the SwiftLib tool, which is used to specify codon selection based on a known metric, generally did not substantially impact the already high rates of activity retention observed for double mutants in libraries 1, 2, 4, or 5 (94% to 100%). The activity retention of library 3, however, was improved from 82% to 100%. Thus, constraint is a useful tool to design combinatorial libraries based on the effective accuracy of the statistical fitness parameter (the metric in this case).
Sequencing results of 873 unique variants across all libraries showed that the statistical fitness parameter was predictive of the experimental loss or retention of catalytic activity of LysEFm5 variants. Our findings support the previously observed notion that the inclusion of a large set of homologous protein sequences in the starting MSA leads to the best predictive performance of the Potts model (28, 49, 50). Because MSAs aim to create aligned sites that represent related positions in the protein structure, and because all sequences to be used are initially restrained by a relatively strict relevance cutoff, the inclusion of a large number of sequences in the MSA does not “dilute” information, as only relevant portions of these sequences end up being considered, provided that epistatic coupling is taken into account. Conversely, if only a relatively small set of sequences (on the order of hundreds to a few thousand) will be considered in the starting MSA, or if epistatic coupling is not considered, then the inclusion of diverse sequences may worsen predictive performance.
For future protein engineering efforts, utilizing a second-order Potts model to select beneficial sites for mutation can constitute a useful approach, provided that certain criteria are met. The structure of the domain or functional site in the protein of interest must be evolutionarily conserved, allowing for investigation into the dominant sequence constraints acting on familial sequences, and ideally, on the order of tens of thousands of homologous protein sequences must be available for use in the starting MSA. As such, this approach is especially well suited to engineer the catalytic domain of members of the lysin family with N-acetylmuramidase activity, such as LysEFm5, which are known to have distinctly conserved functions and structural features (51).
As the need for alternative or supplemental strategies to treat bacterial infections resistant to conventional antibiotics increases, computationally informed methodologies such as this that allow for the expedited discovery of antimicrobial proteins with improved properties are of great relevance.
MATERIALS AND METHODS
Bacteria used and culture conditions.
Escherichia coli cells were grown either in a liquid culture of lysogeny broth (LB) or on a solid LB agar plate containing 1.5% (wt/vol) agar, and supplemented with 50 g/ml kanamycin (kan). All cultures were grown at 37°C, with liquid cultures shaken at 250 rpm, unless otherwise noted. Enterococcus faecium 8-E9 cells were grown in a liquid culture of brain heart infusion (BHI) medium at 37°C with shaking at 250 rpm.
Inputs for PLMC.
A search in Jackhmmer was performed to determine significant query matches in the UniProtKB database to the WT amidase domain of LysEFm5 (amino acids [aa] 1 to 185) (Fig. 2D) with the taxonomy restricted to E. faecium. The consensus sequence of the top three results was used as the seed sequence in a subsequent search, with the acceptable taxonomy set to either Firmicutes only, Bacteria only, or all, and the minimum expectation value (E value) set to 1.0 × 10−5.
A two-component Gaussian mixture model was constructed to describe the distribution of sequence lengths in the Bacteria and “all” groupings (see Fig. S1 in the supplemental material). Each sequence length was assigned a membership score for two component curves, one describing the main distribution and the other describing the tail of short trailing sequences, presumably outliers. The lax cutoff retained sequences with a component 1 (main distribution) membership score of ≥0.80, and the stringent cutoff required a component 1 membership score of ≥0.95. The same criteria were applied to generate the range of acceptable sequence lengths for the Bacteria and “all” groups; differences between the mean and the spread of each data set resulted in different specific bounding lengths. For the Firmicutes group, there existed a clear outlier length (133 aa), likely due to oversampling. Three Gaussian distributions (components 1 to 3) were fitted such that component 3 represented the set of outliers. A new membership score was calculated for each sequence by weighing scores from component 1 (main distribution) and component 3. These weights were selected such that the outlier length was the lower bound for the lax cutoff. The ranges of acceptable sequence lengths are summarized in Table 1.
PROMALS3D (39) was used to generate an MSA for each of the resulting sets of protein sequences. The PLMC algorithm (28) was run using the recommended regularization parameters for the single-site and pairwise coupling constraints, without the inclusion of gap states. The strength of l2 regularization was set to a λh of 0.01 and λJ of 36.8, and sequences were reweighted to account for redundancy based on an 80% sequence identity cutoff, as recommended.
Design of NNK libraries.
The amidase domain of the major pneumococcal autolysin LytA was identified as a homolog of the amidase domain of LysEFm5 initially via sequence alignment. Thirteen primary ligand-binding residues and ten secondary residues were identified in the putative secondary interaction shell of the LytA crystal structure having the inactivating mutations C60A, H133A, and C136A (37). Twelve structurally analogous residues (one primary and eleven secondary) that occupied the same space in the three-dimensional (3D) structure of the LysEFm5 protein were selected.
The most restrictive MSA (using sequences from group Firmicutesstringent-2k) was used in PLMC to predict changes in the statistical fitness of mutants arising from all possible double mutations at any two of the sites of interest (Fig. 3). This heat map was used to select eight sets of two discrete sites for NNK library creation (Table 2), sampling a range of average ΔE values.
Library 9 was designed using the SwiftLib tool (40). The matrix of predicted change in statistical fitness was discretized into an integer matrix using the following criteria: −10 < ΔE < −8, f(ΔE) = −5; −8 < ΔE < −6, f(ΔE) = −2; −6 < ΔE < −3, f(ΔE) = 0; −3 < ΔE < 0, f(ΔE) = 1; ΔE = 0, f(ΔE) = 2; 0 < ΔE < 1, f(ΔE) = 5; and ΔE > 1, f(ΔE) = 10.
The resulting library, which had a specified maximum size of 252, showed diversity at three positions: 33, 40, and 87. The remaining eight positions of interest encoded the WT residues.
Plasmid creation.
A gBlock gene fragment (Integrated DNA Technologies) encoding the entire 341-aa LysEFm5 gene (18) was amplified via Q5 PCR (New England Biolabs [NEB]), visualized on an agarose gel, and purified. The product was Gibson assembled (41) (NEBuilder HiFi DNA Assembly; NEB) into a pET-24 vector modified to include a C-terminal 6×HIS tag (52) containing a selection marker for kanamycin, which was digested with BamHI and NdeI (NEB). The assembled product was transformed into NEB 5-alpha competent E. coli (NEB), which was cultured at 37°C on an LB agar plate with 50 μg/ml kan for selection overnight. A flask was inoculated with cells from a colony on the plate that harbored the LysEFm5 gene and was left to incubate at 37°C overnight. Plasmid DNA was then isolated from the culture.
Construction of NNK libraries.
The nine NNK libraries were each created by assembling two or three overlap extension PCR products. For each library, the universal forward primer (PRIM01) was used in conjunction with a second primer encoding the randomized codon(s) at one or more of the desired sites. The second primer was designed to anneal to the site immediately downstream of the second codon (in the case of libraries 1, 2, 4, 5, 7, and 8) or the first codon (in the case of libraries 3, 6, and 9). In the second reaction, a reverse primer annealing to the site immediately upstream of the first or second randomized codon, respectively, and the universal reverse primer (PRIM02) were used. For libraries 3, 6, and 9, a third reaction producing a fragment containing the two randomized codons was required, as the distance between the diversified codons did not allow for all to be reasonably encoded by the same primer. Summaries of the reactions performed and primer identities are given in Tables S1 and S2. The expected size of each PCR product was confirmed by running a DNA gel, and the bands were gel purified to yield the final DNA fragments.
Each of the two or three PCR products per library were independently combined using Gibson assembly (NEB). Fragments were combined into a pET-24 vector digested with BamHI and NdeI (NEB). Each reaction was cleaned up using a PCR cleanup kit (Epoch Life Science) and then transformed into 25 μl of MC1061F- electrocompetent cells (Lugicen) quenched with 975 μl of recovery medium. Nine hundred fifty microliters of the transformation product was used to inoculate a 3-ml culture of LB plus kan, which was left to grow at 37°C and 250 rpm overnight. The remaining 50 μl of transformation product was plated onto an LB plus kan agar selection plate. Transformation efficiency was confirmed to be on the order of tens of thousands of transformants for each library. Two to three monoclonal transformants were additionally outgrown and sequenced from each library to confirm the expected diversity.
Halo plate creation.
One hundred milliliters of BHI broth was inoculated with VRE and grown overnight at 250 rpm and 37°C. The flask was autoclaved and centrifuged, and the VRE was washed repeatedly with phosphate-buffered saline (PBS). The final mass concentration of this stock was approximately 0.3 g of cell material per ml of PBS. The stock was stored at 4°C until future use.
The effects of the concentration of IPTG, percent agar, and incubation time on halo radius were investigated in a separate experiment prior to conducting the halo plate assay (Fig. S3). All conditions tested were sufficient to produce results that allowed for the easy identification of halo-forming colonies. It was determined that an IPTG concentration of 0.05 mM, 1.0% (wt/vol) agar, and an incubation time of 19 h were appropriate. Additionally, a saturated culture of E. coli expressing a phage lysin with specific activity against Clostridium perfringens (42) was plated alongside a positive control. At no time did halos form on the plate containing the C. perfringens-specific lysin (Fig. S2).
To create each LB plus kan-VRE-IPTG plate used in the halo plate assay, 2.0% (wt/vol) LB and 1.0% (wt/vol) agar were combined in an Erlenmeyer flask along with enough deionized water to constitute 15 ml of total volume per plate, and the solution was microwaved until it boiled. Then, 50 μg/ml of kan, 0.05% (vol/vol) of the stock autoclaved VRE, and 0.05 mM IPTG (final concentrations) were added to the solution after boiling.
Halo plate assay.
For each library, 0.5 to 2 μl of plasmid DNA isolated from a saturated culture of MC1061F- electrocompetent cells was transformed into 25 μl of T7 Express lysY/Iq competent E. coli (NEB). Nine hundred seventy-five microliters of LB plus kan medium was added, and each transformation product underwent a 1-h outgrowth at 37°C and 250 rpm. Afterwards, the cells were spun down, resuspended in 100 μl of LB plus kan, and plated onto an LB plus kan selection plate. Plates were incubated overnight at 37°C. Between 16 and 18 h later, the lawn of cells on each plate was resuspended in approximately 10 ml LB plus kan, and the cell density was estimated using absorbance measurements taken using a microplate reader (averaged for 1:10, 1:50, and 1:100 dilutions) and an empirically determined coefficient (7.90 × 108 CFU/OD600/ml). This was used to determine the serial dilution scheme needed to obtain 125 CFU/50 μl of cell material. Fifty microliters of cell material for each library was then plated onto LB plus kan-VRE-IPTG agar plates (nine each; three plates per each triplicate). Each plate was incubated for 19 h at 37°C. This procedure was performed for two or three libraries (18 or 27 plates) at a time.
Following incubation, all colonies belonging to one library were designated halo forming if there was visible clearance around the colony or non-halo forming if there was not and then systematically plucked and placed into one of six library-specific bins based on the replicate number (1 to 3) and halo-forming designation. Cell material from each bin was stored at −20°C for between 1 and 4 days. Afterwards, 500 μl of MX1 resuspension buffer (Epoch Life Science) was added to each of the 60 samples. Twenty-microliter aliquots from each sample belonging to the same replicate number and halo-forming designation were pooled across all nine libraries. Plasmid DNA was extracted from each of the six resulting 200-μl samples.
High-throughput sequencing (Illumina MiSeq) sample preparation.
Both sets of five forward (FA1 to -5) and five reverse (RA1 to -5) primers were independently premixed at equal ratios (primer sequence identities are given in Table S3). A 50-μl-volume PCR was performed with a final FA1 to -5 and RA1 to -5 primer concentration of 500 μM and 2 μl of plasmid DNA. Four units of exonuclease I (ExoI) was then added to catalyze the degeneration of single-stranded DNA. The samples were incubated at 37°C for 30 min and then heat inactivated at 80°C for 20 min. Afterwards, 1 μl of the ExoI-digested product was used as a template in a second 50-μl-volume PCR with a final FB and RB1 to -6 sequencing primer concentration of 500 μM. Each product was run on a gel to confirm that it was the expected size. Bands were gel purified, and the concentration of DNA from each was measured using a NanoDrop spectrophotometer (Thermo Fisher). The amount of contributing DNA from groups 1 to 6 was weighted based on the estimated diversity of the group and combined to yield a total of 500 ng of DNA in 100 μl of nuclease-free water. The sample was submitted for a MiSeq 2 × 300-bp paired-end read run with version 3 chemistry.
Production of variants.
During the agar plate assay, eight halo-forming variants were successfully isolated from libraries 1 (×2), 2, 3, 4, 5, 6, 7, and 9 (of 10 plucked), and two non-halo-forming variants were successfully isolated from library 8 (×2) (of eight plucked). These variants were confirmed to encode the full LysEFm5 protein, with diversity at the expected sites.
For each clone, a cell culture tube containing 3 ml of LB plus kan was inoculated with cells and then incubated at 37°C and 250 rpm overnight. The day after, 100 ml of LB was inoculated with 100 μl of confluent culture. The OD600 was monitored using a plate reader spectrophotometer until it was within the range of 0.6 to 0.8, at which point IPTG was added at a final concentration of 0.5 mM and the culture was left to incubate at 30°C and 250 rpm for 6 h. The culture was then spun down, the supernatant was discarded, and 1 ml of lysis buffer (137 mM NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 2 mM PBS, 5% glycerol, 3.1 g/liter 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate [CHAPS], 1.7 g/liter imidazole, with a Pierce Protease Inhibitor Mini Tablet, EDTA free [1 tablet per 10 ml buffer]) was added. Each culture was then supplemented with MgSO4 to a final concentration of 20 mM as well as 2 U of DNase I (New England Biolabs) and 10 μg of RNase A (Thermo Scientific). The cell pellet underwent four freeze-thaw cycles at −80°C and room temperature, respectively. The cell material was then spun down, and the supernatant was filtered and diluted with 1 volume of wash buffer (50 mM sodium phosphate, 300 mM NaCl, 10 mM imidazole, 5% glycerol), applied to 200 μl of HisPur cobalt resin (Thermo Scientific), and rotated end-over-end at room temperature for 30 min. This mixture was then applied progressively to spin columns. Three applications of wash buffer were performed followed by three elutions (with 50 mM sodium phosphate, 300 mM NaCl, 150 mM imidazole, 5% glycerol) to constitute the protein sample in a volume of ∼1,200 μl. Proteins were further purified by application to an ÄKTAprime plus configured with a Superdex 75 Increase 10/300 GL size-exclusion column. Samples were run at 0.2 ml/min with PBS plus 5% glycerol as eluent. Appropriate fractions were collected, mixed, and divided into 100-μl aliquots which were snap-frozen. All subsequent analysis was performed on aliquots thawed on ice immediately before use.
Quantification of variant and WT concentrations.
SDS-PAGE was performed to quantify the produced protein concentration of each variant and the WT; 50 μg/ml of bovine serum albumin (BSA) was used as a standard. Twelve microliters of each variant and the WT, in addition to the BSA standard, was combined with 4 μl of 4× LDS buffer and then denatured at 90°C for 12 min. The samples were loaded onto a NuPAGE bis-Tris 4% to 12% protein gel (Thermo Fisher) along with a PageRuler unstained protein ladder (Thermo Fisher). The gel was run at 200 V for 50 min and then stained with SimplyBlue SafeStain (Thermo Fisher). ImageJ was used to determine the intensity of each band corresponding to the BSA standard and protein variants (having an expected molecular weight of ∼37 kDa). The relative intensity of the BSA standard was used to determine the unknown variant concentrations.
Sypro orange thermal denaturation assay.
Variants were diluted to a concentration of 5 μM, and 45 μl was aliquoted into optically clear PCR tubes. The stock solution of Sypro orange (Thermo Fisher) was diluted to 200× in PBS, 5 μl of which was added to each PCR tube. These solutions were heated from 25°C to 98°C in 0.5°C increments with equilibration time set to 30 s after each temperature elevation in a CFX Connect Real-Time PCR detection system. The fluorescence of the Sypro orange dye was detected via 450- to 490-nm excitation and 560- to 590-nm emission. The maximum change of fluorescence with temperature (defined as the Tm) was determined via smoothing with local second-degree polynomials having widths of 2.5°C using the Savitzsky-Golay filter of the sklearn package in Python.
Quantification of variant and WT activity.
One hundred milliliters of BHI broth was inoculated with a 1,000× dilution of VRE grown overnight at 37°C with agitation. When this culture reached an OD600 of ∼0.5, it was placed on ice and chilled for 15 min. Cells were then pelleted via centrifugation at 6,000 × g for 5 min. The supernatant was removed and resuspended in 1 ml of 50 mM Tris-HCl and then added dropwise to 20 ml of boiling 5% (wt/vol) sodium dodecyl sulfate. This solution was boiled with stirring for 15 min and then allowed to cool to room temperature and centrifuged at 6,000 × g for 5 min. The pellet was resuspended in 1 ml of 1 M NaCl and centrifuged at 17,000 × g for an additional 5 min. This was repeated an additional time with 1 ml of 1 M NaCl and then seven times with pure water, and the pellet was finally resuspended in PBS and stored at 4°C as “crude cell wall.”
In a 96-well plate, 0.5 μg of each variant or blank in 5 μl of PBS with 5% glycerol was combined with 195 μl of crude cell wall diluted to an OD600 of ∼1. Each sample was tested with replication of 4 to 8 with randomized well positions. Measurements of the A600 were taken every 2 min for multiple hours.
Assessment of cell lysis and killing activity.
Enterococcus faecium 8-E9 was streaked onto BHI agar plates and grown overnight at 37°C. The following morning, a colony was used to inoculate 3 ml of BHI broth and was incubated at 37°C with shaking at 250 rpm. When the culture reached mid-exponential phase (OD600 of ∼0.8), cells were washed 2× with sterile PBS with centrifugation of 3,000 × g for 3 min. Cells were then diluted into 3 ml PBS, and 195 μl was applied to 5 μl of 0.5 μg of purified proteins in PBS plus 5% glycerol in a 96-well plate. The plate was then incubated at 37°C with shaking in a spectrophotometer with an OD600 measurement taken every 2 min. After 30 min, the plate was removed, and cell suspensions were serially diluted into BHI broth. CFU were then determined by enumeration of colonies after plating of dilution series onto BHI agar.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a grant from the National Institutes of Health (R01 GM121777).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Barriere SL. 2015. Clinical, economic and societal impact of antibiotic resistance. Expert Opin Pharmacother 16:151–153. doi: 10.1517/14656566.2015.983077. [DOI] [PubMed] [Google Scholar]
- 2.Langdon A, Crook N, Dantas G. 2016. The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med 8:39. doi: 10.1186/s13073-016-0294-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Francino MP. 2015. Antibiotics and the human gut microbiome: dysbioses and accumulation of resistances. Front Microbiol 6:1543. doi: 10.3389/fmicb.2015.01543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Young R. 1992. Bacteriophage lysis: mechanism and regulation. Microbiol Mol Biol Rev 56:430–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.São-José C. 2018. Engineering of phage-derived lytic enzymes: improving their potential as antimicrobials. Antibiotics (Basel) 7:E29. doi: 10.3390/antibiotics7020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loeffler JM, Nelson D, Fischetti VA. 2001. Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science 294:2170–2172. doi: 10.1126/science.1066869. [DOI] [PubMed] [Google Scholar]
- 7.Schuch R, Nelson D, Fischetti VA. 2002. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature 418:884–889. doi: 10.1038/nature01026. [DOI] [PubMed] [Google Scholar]
- 8.Schmelcher M, Donovan DM, Loessner MJ. 2012. Bacteriophage endolysins as novel antimicrobials. Future Microbiol 7:1147–1171. doi: 10.2217/fmb.12.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nelson D, Loomis L, Fischetti VA. 2001. Prevention and elimination of upper respiratory colonization of mice by group A streptococci by using a bacteriophage lytic enzyme. Proc Natl Acad Sci U S A 98:4107–4112. doi: 10.1073/pnas.061038398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Euler CW, Delaune A, Fischetti VA. 2015. Using a novel lysin to help control Clostridium difficile infections. Antimicrob Agents Chemother 59:7447–7457. doi: 10.1128/AAC.01357-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marr AK, Gooderham WJ, Hancock RE. 2006. Antibacterial peptides for therapeutic use: obstacles and realistic outlook. Curr Opin Pharmacol 6:468–472. doi: 10.1016/j.coph.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 12.Fischetti VA. 2008. Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol 11:393–400. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loessner MJ. 2005. Bacteriophage endolysins–current state of research and applications. Curr Opin Microbiol 8:480–487. doi: 10.1016/j.mib.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 14.Leonard E, Ajikumar PK, Thayer K, Xiao WH, Mo JD, Tidor B, Stephanopoulos G, Prather KL. 2010. Combining metabolic and protein engineering of a terpenoid biosynthetic pathway for overproduction and selectivity control. Proc Natl Acad Sci U S A 107:13654–13659. doi: 10.1073/pnas.1006138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vermassen A, Leroy S, Talon R, Provot C, Popowska M, Desvaux M. 2019. Cell wall hydrolases in bacteria: insight on the diversity of cell wall amidases, glycosidases and peptidases toward peptidoglycan. Front Microbiol 10:331. doi: 10.3389/fmicb.2019.00331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Croux C, Ronda C, López R, García JL. 1993. Interchange of functional domains switches enzyme specificity: construction of a chimeric pneumococcal-clostridial cell wall lytic enzyme. Mol Microbiol 9:1019–1025. doi: 10.1111/j.1365-2958.1993.tb01231.x. [DOI] [PubMed] [Google Scholar]
- 17.Díez-Martínez R, De Paz HD, García-Fernández E, Bustamante N, Euler CW, Fischetti VA, Menendez M, García P. 2015. A novel chimeric phage lysin with high in vitro and in vivo bactericidal activity against Streptococcus pneumoniae. J Antimicrob Chemother 70:1763–1773. doi: 10.1093/jac/dkv038. [DOI] [PubMed] [Google Scholar]
- 18.Gong P, Cheng M, Li X, Jiang H, Yu C, Kahaer N, Li J, Zhang L, Xia F, Hu L, Sun C, Feng X, Lei L, Han W, Gu J. 2016. Characterization of Enterococcus faecium bacteriophage IME-EFm5 and its endolysin LysEFm5. Virology 492:11–20. doi: 10.1016/j.virol.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 19.Patterson JE, Sweeney AH, Simms M, Carley N, Mangi R, Sabetta J, Lyons RW. 1995. An analysis of 110 serious enterococcal infections. Epidemiology, antibiotic susceptibility, and outcome. Medicine (Baltimore) 74:191–200. doi: 10.1097/00005792-199507000-00003. [DOI] [PubMed] [Google Scholar]
- 20.DiazGranados CA, Zimmer SM, Klein M, Jernigan JA. 2005. Comparison of mortality associated with vancomycin-resistant and vancomycin-susceptible enterococcal bloodstream infections: a meta-analysis. Clin Infect Dis 41:327–333. doi: 10.1086/430909. [DOI] [PubMed] [Google Scholar]
- 21.Edmond MB, Ober JF, Dawson JD, Weinbaum DL, Wenzel RP. 1996. Vancomycin-resistant enterococcal bacteremia: natural history and attributable mortality. Clin Infect Dis 23:1234–1239. doi: 10.1093/clinids/23.6.1234. [DOI] [PubMed] [Google Scholar]
- 22.Low LY, Yang C, Perego M, Osterman A, Liddington RC. 2005. Structure and lytic activity of a Bacillus anthracis prophage endolysin. J Biol Chem 280:35433–35439. doi: 10.1074/jbc.M502723200. [DOI] [PubMed] [Google Scholar]
- 23.Zoll S, Pätzold B, Schlag M, Götz F, Kalbacher H, Stehle T. 2010. Structural basis of cell wall cleavage by a staphylococcal autolysin. PLoS Pathog 6:e1000807. doi: 10.1371/journal.ppat.1000807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yoong P, Schuch R, Nelson D, Fischetti VA. 2004. Identification of a broadly active phage lytic enzyme with lethal activity against antibiotic-resistant Enterococcus faecalis and Enterococcus faecium. J Bacteriol 186:4808–4812. doi: 10.1128/JB.186.14.4808-4812.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Midelfort KS, Kumar R, Han S, Karmilowicz MJ, McConnell K, Gehlhaar DK, Mistry A, Chang JS, Anderson M, Villalobos A, Minshull J, Govindarajan S, Wong JW. 2013. Redesigning and characterizing the substrate specificity and activity of Vibrio fluvialis aminotransferase for the synthesis of imagabalin. Protein Eng Des Sel 26:25–33. doi: 10.1093/protein/gzs065. [DOI] [PubMed] [Google Scholar]
- 26.Miklos AE, Kluwe C, Der BS, Pai S, Sircar A, Hughes RA, Berrondo M, Xu J, Codrea V, Buckley PE, Calm AM, Welsh HS, Warner CR, Zacharko MA, Carney JP, Gray JJ, Georgiou G, Kuhlman B, Ellington AD. 2012. Structure-based design of supercharged, highly thermoresistant antibodies. Chem Biol 19:449–455. doi: 10.1016/j.chembiol.2012.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cocco S, Feinauer C, Figliuzzi M, Monasson R, Weigt M. 2018. Inverse statistical physics of protein sequences: a key issues review. Rep Prog Phys 81:032601. doi: 10.1088/1361-6633/aa9965. [DOI] [PubMed] [Google Scholar]
- 28.Hopf TA, Ingraham JB, Poelwijk FJ, Schärfe CP, Springer M, Sander C, Marks DS. 2017. Mutation effects predicted from sequence co-variation. Nat Biotechnol 35:128–135. doi: 10.1038/nbt.3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Figliuzzi M, Barrat-Charlaix P, Weigt M. 2018. How pairwise coevolutionary models capture the collective residue variability in proteins? Mol Biol Evol 35:1018–1027. doi: 10.1093/molbev/msy007. [DOI] [PubMed] [Google Scholar]
- 30.Miton CM, Tokuriki N. 2016. How mutational epistasis impairs predictability in protein evolution and design. Protein Sci 25:1260–1272. doi: 10.1002/pro.2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anishchenko I, Ovchinnikov S, Kamisetty H, Baker D. 2017. Origins of coevolution between residues distant in protein 3D structures. Proc Natl Acad Sci U S A 114:9122–9127. doi: 10.1073/pnas.1702664114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levy RM, Haldane A, Flynn WF. 2017. Potts Hamiltonian models of protein co-variation, free energy landscapes, and evolutionary fitness. Curr Opin Struct Biol 43:55–62. doi: 10.1016/j.sbi.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyazawa S. 2017. Selection originating from protein stability/foldability: relationships between protein folding free energy, sequence ensemble, and fitness. J Theor Biol 433:21–38. doi: 10.1016/j.jtbi.2017.08.018. [DOI] [PubMed] [Google Scholar]
- 34.Jacquin H, Gilson A, Shakhnovich E, Cocco S, Monasson R. 2016. Benchmarking inverse statistical approaches for protein structure and design with exactly solvable models. PLoS Comput Biol 12:e1004889. doi: 10.1371/journal.pcbi.1004889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ekeberg M, Lövkvist C, Lan Y, Weigt M, Aurell E. 2013. Improved contact prediction in proteins: using pseudolikelihoods to infer Potts models. Phys Rev E Stat Nonlin Soft Matter Phys 87:012707. doi: 10.1103/PhysRevE.87.012707. [DOI] [PubMed] [Google Scholar]
- 36.Reetz MT, Wang L, Bocola M. 2006. Directed evolution of enantioselective enzymes: iterative cycles of CASTing for probing protein-sequence space. Angew Chem Int Ed Engl 45:1236–1241. doi: 10.1002/anie.200502746. [DOI] [PubMed] [Google Scholar]
- 37.Sandalova T, Lee M, Henriques-Normark B, Hesek D, Mobashery S, Mellroth P, Achour A. 2016. The crystal structure of the major pneumococcal autolysin LytA in complex with a large peptidoglycan fragment reveals the pivotal role of glycans for lytic activity. Mol Microbiol 101:954–967. doi: 10.1111/mmi.13435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finn RD, Clements J, Eddy SR. 2011. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39:W29–W37. doi: 10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pei J, Kim BH, Grishin NV. 2008. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res 36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobs TM, Yumerefendi H, Kuhlman B, Leaver-Fay A. 2015. SwiftLib: rapid degenerate-codon-library optimization through dynamic programming. Nucleic Acids Res 43:e34. doi: 10.1093/nar/gku1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibson D. 2009. One-step enzymatic assembly of DNA molecules up to several hundred kilobases in size. Protocol Exchange doi: 10.1038/nprot.2009.77. [DOI] [PubMed]
- 42.Ritter SC, Hackel BJ. 2019. Validation and stabilization of a prophage lysin of Clostridium perfringens by yeast surface display and coevolutionary models. Appl Environ Microbiol 85:e00054-19. doi: 10.1128/AEM.00054-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clark DP, Pazdernik NJ. 2009. Improving protein secretion, p 312–315. In Clark DP, Pazdernik NJ (ed), Biotechnology: applying the genetic revolution. Elsevier Academic Press, Burlington, MA. [Google Scholar]
- 44.Cheng Q, Fischetti VA. 2007. Mutagenesis of a bacteriophage lytic enzyme PlyGBS significantly increases its antibacterial activity against group B streptococci. Appl Microbiol Biotechnol 74:1284–1291. doi: 10.1007/s00253-006-0771-1. [DOI] [PubMed] [Google Scholar]
- 45.Proença D, Fernandes S, Leandro C, Silva FA, Santos S, Lopes F, Mato R, Cavaco-Silva P, Pimentel M, São-José C. 2012. Phage endolysins with broad antimicrobial activity against Enterococcus faecalis clinical strains. Microb Drug Resist 18:322–332. doi: 10.1089/mdr.2012.0024. [DOI] [PubMed] [Google Scholar]
- 46.Guan R, Roychowdhury A, Ember B, Kumar S, Boons GJ, Mariuzza RA. 2004. Structural basis for peptidoglycan binding by peptidoglycan recognition proteins. Proc Natl Acad Sci U S A 101:17168–17173. doi: 10.1073/pnas.0407856101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang L, Li D, Li X, Hu L, Cheng M, Xia F, Gong P, Wang B, Ge J, Zhang H, Cai R, Wang Y, Sun C, Feng X, Lei L, Han W, Gu J. 2016. LysGH15 kills Staphylococcus aureus without being affected by the humoral immune response or inducing inflammation. Sci Rep 6:29344. doi: 10.1038/srep29344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu Q, Xun G, Feng Y. 2019. The state-of-the-art strategies of protein engineering for enzyme stabilization. Biotechnol Adv 37:530–537. doi: 10.1016/j.biotechadv.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 49.Kosciolek T, Jones DT. 2016. Accurate contact predictions using covariation techniques and machine learning. Proteins 84(Suppl 1):145–151. doi: 10.1002/prot.24863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, Sander C. 2011. Protein 3D structure computed from evolutionary sequence variation. PLoS One 6:e28766. doi: 10.1371/journal.pone.0028766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Broendum SS, Buckle AM, McGowan S. 2018. Catalytic diversity and cell wall binding repeats in the phage-encoded endolysins. Mol Microbiol 110:879–896. doi: 10.1111/mmi.14134. [DOI] [PubMed] [Google Scholar]
- 52.Woldring DR, Holec PV, Stern LA, Du Y, Hackel BJ. 2017. A gradient of sitewise diversity promotes evolutionary fitness for binder discovery in a three-helix bundle protein scaffold. Biochemistry 56:1656–1671. doi: 10.1021/acs.biochem.6b01142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.