

Viruses have evolved a variety of strategies to succeed in a hostile environment. The herpes simplex virus 1 (HSV-1) immediate early protein ICP22 plays several roles in the virus life cycle, including downregulation of cellular gene expression, upregulation of late viral gene expression, inhibition of apoptosis, prevention of aggregation of nonnative proteins, and the recruitment of a cellular heat shock protein, Hsc70, to nuclear domains. We present evidence that ICP22 functionally resembles a cellular J-protein/HSP40 family cochaperone, interacting specifically with Hsc70. We suggest that HSV has taken advantage of the adaptable nature of J proteins to evolve a multifunctional cochaperone that functions with Hsc70 to promote lytic infection.
KEYWORDS: herpes simplex virus, Hsc70, Hsp40, ICP22, J protein, VICE domains, aggresome, heat shock protein, protein chaperone
ABSTRACT
Molecular chaperones and cochaperones are the most abundant cellular effectors of protein homeostasis, assisting protein folding and preventing aggregation of misfolded proteins. We have previously shown that herpes simplex virus 1 (HSV-1) infection results in the drastic spatial reorganization of the cellular chaperone Hsc70 into nuclear domains called VICE (Virus Induced Chaperone Enriched) domains and that this recruitment is dependent on the viral immediate early protein ICP22. Here, we present several lines of evidence supporting the notion that ICP22 functions as a virally encoded cochaperone (J-protein/Hsp40) functioning together with its Hsc70 partner to recognize and manage aggregated and misfolded proteins. We show that ICP22 results in (i) nuclear sequestration of nonnative proteins, (ii) reduction of cytoplasmic aggresomes in cells expressing aggregation-prone proteins, and (iii) thermoprotection against heat inactivation of firefly luciferase, and (iv) sequence homology analysis indicated that ICP22 contains an N-terminal J domain and a C-terminal substrate binding domain, similar to type II cellular J proteins. ICP22 may thus be functionally similar to J-protein/Hsp40 cochaperones that function together with their HSP70 partners to prevent aggregation of nonnative proteins. This is not the first example of a virus hijacking a function of a cellular chaperone, since simian immunodeficiency virus T antigen was previously shown to contain a J domain; however, this the first known example of the acquisition of a functional J-like protein by a virus and suggests that HSV has taken advantage of the adaptable nature of J proteins to evolve a multifunctional cochaperone that functions with Hsc70 to promote lytic infection.
IMPORTANCE Viruses have evolved a variety of strategies to succeed in a hostile environment. The herpes simplex virus 1 (HSV-1) immediate early protein ICP22 plays several roles in the virus life cycle, including downregulation of cellular gene expression, upregulation of late viral gene expression, inhibition of apoptosis, prevention of aggregation of nonnative proteins, and the recruitment of a cellular heat shock protein, Hsc70, to nuclear domains. We present evidence that ICP22 functionally resembles a cellular J-protein/HSP40 family cochaperone, interacting specifically with Hsc70. We suggest that HSV has taken advantage of the adaptable nature of J proteins to evolve a multifunctional cochaperone that functions with Hsc70 to promote lytic infection.
INTRODUCTION
Cells have evolved elaborate homeostatic mechanisms to recognize and respond to environmental and other forms of stress, including viral infections. The cellular protein homeostatic (proteostatic) machinery involves a balance between protein folding carried out by molecular chaperones (heat shock proteins) and protein degradation carried out by the ubiquitin proteasome system (UPS) (1). The proteostatic machinery prevents the accumulation of misfolded proteins (2); however, when this machinery is overwhelmed, misfolded proteins accumulate in cytoplasmic and/or nuclear inclusions, called aggresomes (3, 4). Infection with the alphaherpesvirus HSV-1 results in the drastic spatial reorganization of components of the proteostatic machinery into VICE (Virus Induced Chaperone Enriched) domains that contain Hsc70 and other molecular chaperones, ubiquitin, and components of the UPS (5–7). VICE domains resemble nuclear aggresomes in that they both contain components of the proteostasis machinery (6).
The most well-conserved and ubiquitous heat shock proteins (Hsps) are members of the HSP70 family, and these proteins mediate folding of nascent protein chains, reduce the toxicity of aggregation-prone proteins, and help to assemble of multiprotein complexes (8, 9). The prototype member of the HSP70 family is the heat- and stress-inducible chaperone, Hsp70, while the constitutively expressed version is Hsc70 (10). In addition to their shared roles in proteostasis, Hsc70 and Hsp70 have evolved separate and distinct roles (11). Hsc70 but not Hsp70 is required for chaperone-mediated autophagy (CMA) (12, 13) and clathrin-mediated endocytosis (14, 15). Interestingly, CMA has been implicated in major histocompatibility complex (MHC) class II presentation of viral antigens (16, 17) and clearance of viral pathogens (18), thus implicating Hsc70 in antiviral mechanisms. It is possible that recruitment of Hsc70 into VICE domains during HSV-1 infection may have evolved in part to counteract these antiviral mechanisms.
HSP70 family members work together with obligate partners called J proteins, which are members of the HSP40 family of cochaperones (19). During protein folding, J proteins interact with and stimulate the ATPase activity of their HSP70 partners and provide substrate specificity (20, 21). An important function of HSP70/HSP40 complexes is to reduce toxicity of aggregation-prone proteins such as those implicated in neurodegenerative diseases (22, 23). In addition to their ability to recognize and fold nonnative proteins, HSP40/HSP70 complexes have been shown to play critical roles in a myriad of cellular processes, including the regulation of gene expression and cell cycle control (24, 25). J proteins are highly adaptable, and humans have evolved to encode almost 50 different J proteins that are capable of interacting with a diverse set of client proteins (26, 27).
Cellular chaperones can be commandeered by viruses to facilitate folding of proteins, viral entry, and nuclear transport of viruses and viral proteins (28). In addition to hijacking cellular proteostatic machinery for its own benefit, HSV-1 encodes proteins that resemble cellular chaperones. For instance, UL14 possesses a substrate-binding domain homologous to Hsp70 and has been shown to facilitate nuclear translocation of viral proteins (29). Another viral protein, UL32, acts as a redox-sensitive chaperone that regulates disulfide bond formation during viral assembly (30).
The HSV-1 immediate early protein ICP22 plays several seemingly diverse roles in the virus life cycle and is essential in most but not all cell lines (31, 32). ICP22 is essential for the recruitment of Hsc70 into VICE domains in HSV-infected cells (33). In addition to its ability to influence the host protein quality control machinery (6, 33), ICP22 is required for late viral gene expression in nonpermissive cell lines (31, 32, 34), modification of RNA polymerase (Pol) II (35–39) and cell cycle regulation (40–42). Here, we test the hypothesis that ICP22 plays a chaperone-like function during HSV-1 infection.
(This article was submitted to an online preprint archive [43].)
RESULTS
During HSV infection, ICP22 localizes to discrete nuclear foci that subsequently recruit Hsc70.
In the absence of stress, Hsc70 localizes in a pandiffuse pattern throughout the cell (Fig. 1A, mock); however, following heat shock and other forms of stress, it enters the nucleus and localizes to the nucleolus (44). During HSV infection, Hsc70 relocalizes into VICE domains in an ICP22-dependent fashion (33). In order to determine the localization pattern of ICP22 in relation to Hsc70 as infection progresses, we made use of a plasmid expressing FLAG-tagged ICP22 (FLAG-ICP22) (35) and a viral mutant in which FLAG-tagged ICP22 has been introduced into the HSV genome (TF22) (38). Vero cells were infected with TF22, and Hsc70 and ICP22 localization was monitored as a function of time. In the experiment shown in Fig. 1A, at 2 h postinfection (hpi), ICP22 was pannuclear, and Hsc70 was predominantly localized to the nucleolus, which is consistent with a response to the stress of viral infection. By 4 hpi, ICP22 was localized in numerous brightly staining foci and in replication compartments. On the other hand, at 4 hpi, some Hsc70 localized in domains that resemble replication compartments, some remained in the nucleolus and only a small fraction was colocalized with ICP22 in discrete foci. VICE domains were initially defined as Hsc70-staining foci that could be observed at 6 hpi (6). In this experiment, at 8 hpi ICP22 was localized in discrete foci that resemble VICE domains because they costain with Hsc70 and are adjacent to replication compartments (Fig. 1A). Thus, by 6 to 8 hpi, Hsc70 and ICP22 colocalize in VICE domains. At 16 hpi, the VICE domains were larger and fewer than those seen at 8 hpi. These results indicate that although ICP22 could be observed in discrete foci by 4 hpi, most Hsc70 is not recruited to these domains until 6 to 8 hpi. The ICP22 foci seen at 4 hpi are consistent with previous reports of ICP22-containing nuclear bodies in WT-infected cells (33, 45–47). They also resemble nascent protein domains (NPDs) that result from the rapid accumulation of newly synthesized proteins in HSV-infected cells and progressively recruit Hsc70, 2 h after their initial formation (48).
FIG 1.

Localization of ICP22 and Hsc70 in HSV-infected cells. (A) Vero cells were infected with TF22 (mutant virus with FLAG-tagged ICP22 introduced into the HSV genome) at an MOI of 10. At 2, 4, 8, and 16 h postinfection, the cells were fixed, permeabilized, and labeled with rabbit anti-FLAG and rat anti-Hsc70. Imaging was performed at a magnification of ×2. (B) Vero cells were infected with TF22 (FLAG-tagged ICP22 virus) and d22LacZ (ICP22-null virus) at an MOI of 10. Cells were collected and lysed 10 h postinfection, and immunoprecipitation was performed with mouse anti-FLAG. The cell lysates and pulldown samples were probed with mouse anti-FLAG, rat anti-Hsc70, and mouse anti-ICP4.
Hsc70 coimmunoprecipitates with ICP22.
We next tested whether the progressive recruitment of Hsc70 to ICP22 foci was a result of a physical interaction between ICP22 and Hsc70. Previous studies indicated that while ICP22 deletion constructs were able to pull down Hsc70, full-length ICP22 was not (33). This was surprising since ICP22 was shown to be necessary for Hsc70 recruitment to VICE domains. It has now become clear that detecting relevant protein-protein interactions between chaperones and their cochaperones by coimmunoprecipitation experiments is problematic, due in part to the fact that a misfolded protein can also be recognized by a chaperone as a client rather than a regulatory partner (49). We now suggest that the large deletion constructs in the previous report may have been misfolded and that the IP results reflected client interactions with a misfolded protein rather than biologically relevant interactions. The potentially aberrant interactions may have been strong enough to mask a full-length ICP22 interaction with Hsc70. We have now revisited this question using a lower stringency immunoprecipitation buffer. Vero cells were infected with TF22 expressing a FLAG-tagged version of ICP22 or the null virus, d22LacZ. Analysis of whole-cell lysates by immunoblotting indicated that the infected cells expressed the immediate early protein ICP4 and comparable amounts of Hsc70 (Fig. 1B, lanes 1 and 2). When anti-FLAG monoclonal antibody was used to precipitate whole-cell extracts, FLAG-tagged ICP22 was successfully pulled down in TF22-infected but not d22LacZ-infected cells (Fig. 1B, lanes 3 and 4, respectively). Hsc70 was pulled down from TF22-infected but not d22lacZ-infected cells (Fig. 1C, lanes 3 and 4, respectively), consistent with a physical interaction between ICP22 and Hsc70. Additional protein-protein interaction experiments with purified proteins are needed to validate this conclusion.
Nuclear aggresomes formed in HSV-infected cells recruit Hsc70 in an ICP22-dependent fashion.
Aggregation-prone misfolded proteins have been reported to accumulate in cytoplasmic and/or nuclear aggresomes that also contain chaperones such as Hsp70 (3, 4). This phenomenon has been studied in cells transfected with aggregation-prone mutant proteins such as GFP170* (GFP170*), a truncated version of the Golgi complex protein 170 fused to green fluorescent protein (GFP). In GFP170*-transfected cells, nuclear and cytoplasmic aggresomes were observed that contained the aggregated protein, Hsp70 and a J-protein partner for Hsp70 (HdJ2) (50). Under these conditions, Hsp70 but not Hsc70, was recruited to the nuclear and cytoplasmic aggresomes. Interestingly, when this transfection experiment was performed in the context of HSV infection, we found that Hsc70 was recruited, but only to nuclear aggresomes. In the experiment shown in Fig. 2, mock-infected, WT HSV-infected, or d22lacZ-infected Vero cells were transfected with a plasmid expressing GFP170*. In mock-infected cells, nuclear and cytoplasmic aggregates were observed that contained Hsp70 (Fig. 2, top panel). Under these conditions, Hsc70 staining was diffuse throughout the cell indicating that Hsp70, but not Hsc70, was recruited to aggresomes, a finding consistent with previously reported results (50). In HSV-infected cells transfected with GFP170*, however, the aggregated protein and Hsp70 colocalized in nuclear and cytoplasmic aggresomes. Interestingly, Hsc70 was found to localize in nuclear aggregates (Fig. 2, middle panel), suggesting that in infected cells, Hsc70 is specifically recruited to nuclear aggresomes. On the other hand, in d22LacZ-infected cells transfected with GFP170*, nuclear aggresomes were not observed and cytoplasmic aggresomes were seen that contain Hsp70 but not Hsc70 (Fig. 2, bottom panel). These results indicate that when ICP22 is present, nuclear aggresomes form that can specifically Hsc70. This behavior is reminiscent of cellular J proteins known to recruit their partner chaperones (51–53).
FIG 2.

ICP22 is required for the recruitment of Hsc70 to nuclear aggresomes. Vero cells were mock infected (top panel) or infected at an MOI of 10 with WT HSV (middle panel) or d22LacZ (bottom panel), followed by transfection with plasmid containing model misfolded protein GFP170*. At 12 h postinfection, the cells were fixed, permeabilized, and labeled with rabbit anti-ICP8, mouse anti-Hsp70, and rat anti-Hsc70. GFP was detected by excitation using a 488-nm laser. The contrast was adjusted relative to the nuclear intensity, resulting in saturation of the intensity of cytosolic fluorescence.
ICP22 is sufficient to recruit Hsc70 to nuclear aggresomes in transfected cells.
The experiments shown in Fig. 1 and 2 are consistent with the notion ICP22 may be capable of performing a J-like function during infection. To determine whether ICP22 alone is sufficient to relocalize Hsc70 in the absence of infection, we transfected cells with FLAG-tagged ICP22. Mock-transfected cells exhibited a pandiffuse staining pattern for Hsc70 (Fig. 3A, top panel); however, in cells expressing ICP22, small nuclear inclusions that contain ICP22 and Hsc70 were observed (Fig. 3A, bottom panel). ICP22 and Hsc70 colocalize in these inclusions.
FIG 3.
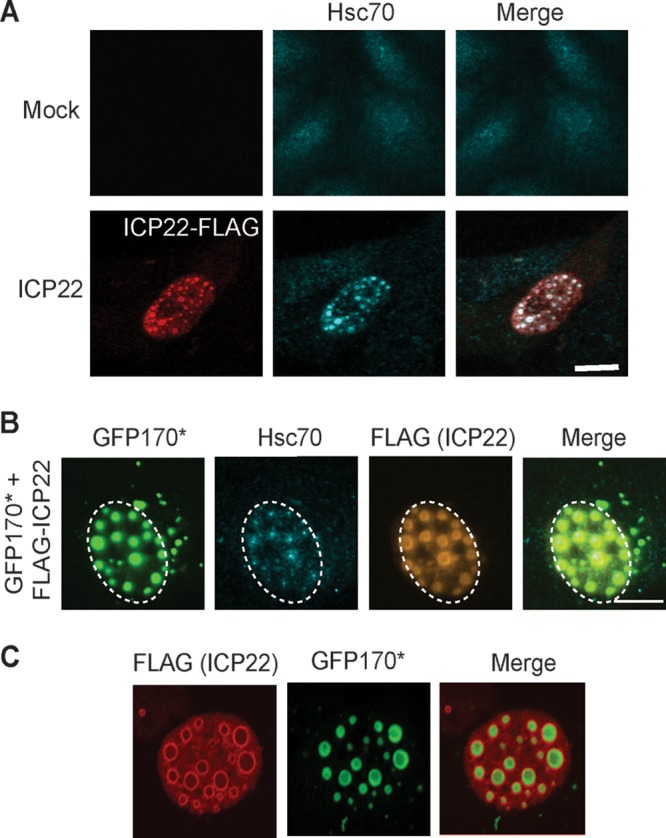
ICP22 is sufficient to recruit Hsc70. (A) Vero cells were transfected with 1 μg of FLAG-ICP22. After 16 h of protein expression, the cells were fixed, permeabilized, and labeled with rabbit anti-FLAG and rat anti-Hsc70 antibodies. (B) Vero cells were transfected with 500 ng of model misfolded protein GFP170* and 500 ng of FLAG-ICP22. After 16 h of protein expression, the cells were fixed, permeabilized, and labeled with rabbit anti-FLAG and rat anti-Hsc70 antibodies. GFP was detected by excitation using a 488-nm laser. (C) Vero cells were transfected as in panel B and labeled with rabbit anti-FLAG antibody. GFP was detected by excitation using a 488-nm laser.
Next, we examined Vero cells transfected with GFP170* and FLAG-ICP22. As shown above in Fig. 2, in cells transfected with GFP170* alone, Hsc70 was present in a pandiffuse staining pattern. On the other hand, in the presence of ICP22, Hsc70 and GFP170* colocalized in large nuclear inclusions that are larger than those seen in the absence of the misfolded protein (compare Fig. 3B and A, bottom panel). Interestingly, ICP22 appeared to form a shell around the misfolded protein and Hsc70 (Fig. 3B and C). In Fig. 3C, the shells of ICP22 surrounding GFP170* were more distinct. We suggest that the ability of ICP22 to induce the formation of inclusions around a misfolded protein reflects an intrinsic J-protein-like property of ICP22. This behavior is reminiscent of nucleophosmin1 (NPM1), a multifunctional chaperone-like protein reported to function in the nucleus to shield the interactive surface of potentially toxic protein aggregates (54).
As mentioned above, HSP40/HSP70 complexes are known to reduce toxic cytoplasmic aggregation in cells transfected with misfolded proteins (53, 55). The J protein DNAJB1 has been shown to facilitate the transport of toxic cytoplasmic aggresomes to the nucleus (51, 52). We next sought to determine whether ICP22 alone is sufficient to reduce cytoplasmic aggregation in a cell transfected with the aggregation-prone protein GFP170*. Cells were transfected with a plasmid encoding GFP170* only or both GFP170* and FLAG-ICP22. A field view of cells transfected with GFP170* alone shows that most of the aggresomes were detected in the cytoplasm, with a few cells containing small nuclear aggresomes marked with white arrows (Fig. 4A, top panel). On the other hand, in cells transfected with GFP170* and ICP22 (Fig. 4A, bottom panel), most of the aggresomes were localized in the nucleus, and these aggresomes were larger than those observed in cells transfected with GFP170* alone in the absence of ICP22. These results were quantified as shown in Fig. 4B by categorizing transfected cells according to presence or absence of robust cytoplasmic aggregates (as depicted in Fig. 3B, top). Approximately 90% of the cells transfected with GFP170* contain robust cytoplasmic aggregates, while only 15% of cells transfected with GFP170* and ICP22 contained robust cytoplasmic aggregates (Fig. 4B). Thus, ICP22 expressed alone by transfection is sufficient to reduce cytoplasmic aggregation. The ability of ICP22 to specifically recruit Hsc70 to nuclear aggresomes is of particular interest in light of recent reports that cytoplasmic aggresomes can be deleterious to cells (51, 54). On the other hand, nuclear aggresomes are thought to be cytoprotective (54).
FIG 4.

Reduction of cytoplasmic aggregates. (A) Vero cells were transfected with model misfolded protein GFP170*, or GFP170* and FLAG-ICP22. After 16 h of protein expression, the cells were fixed, permeabilized, and labeled with chicken anti-ICP22 antibody. A field image of the slide is shown here. (B) Quantification of 100 cells transfected with plasmids encoding model misfolded protein GFP170* or GFP170* and ICP22.
ICP22 confers protection against heat-inactivation of firefly luciferase.
In addition to aggresome formation and reduction of toxic cytoplasmic aggregates, J-protein/HSP70 complexes interact with other chaperones to extend their functionality during proteostasis and often exist within larger chaperone complexes. For instance, a complex containing Hsp40, Hsc70, and Hsp110 (member of the HSP70 family) has been implicated in the protection of cells from heat stress (thermoprotection) (56). In addition, Hsc70 alone has been previously shown to provide protection to proteins against heat inactivation in vitro (57). We made use of a plasmid expressing FlucDM-EGFP, a firefly luciferase mutant that can act as a sensor for heat stress and is specifically dependent on Hsc70 for folding and refolding (58).
In order to determine how ICP22 affects luciferase following heat stress, HEK293T cells were cotransfected with plasmid expressing FlucDM-EGFP alone or with Hsc70, Hsp40 (DNAJB1), or FLAG-ICP22; treated with cycloheximide to inhibit protein synthesis; and treated at 45°C for either 30 min or 1 h (Fig. 5A). Under these conditions, the heat stress would be expected to unfold and inactivate luciferase (58). The luciferase activity was measured after heat shock and normalized to non-heat-shocked samples (representing folded luciferase). The normalized activity was plotted as the percentage of luciferase activity (Fig. 5B). When transfected cells were heat shocked at 45°C for 30 min, the specific activity of luciferase was decreased to 35% in cells transfected with FlucDM alone or in cells transfected with FlucDM and Hsp40. However, in cells transfected with FlucDM and either Hsc70 or ICP22, almost 100% of the specific activity of luciferase was retained, indicating that the expression of Hsc70 or ICP22 provided resistance to damage or unfolding of the luciferase. In cells treated for 1 h at 45°C, transfection with Hsc70 or Hsp40 did not confer significant protection, 15 and 6%, respectively. However, transfection with ICP22 resulted in the retention of 50% of the specific activity of luciferase, indicating that ICP22 was able to significantly protect luciferase from heat-induced inactivation.
FIG 5.
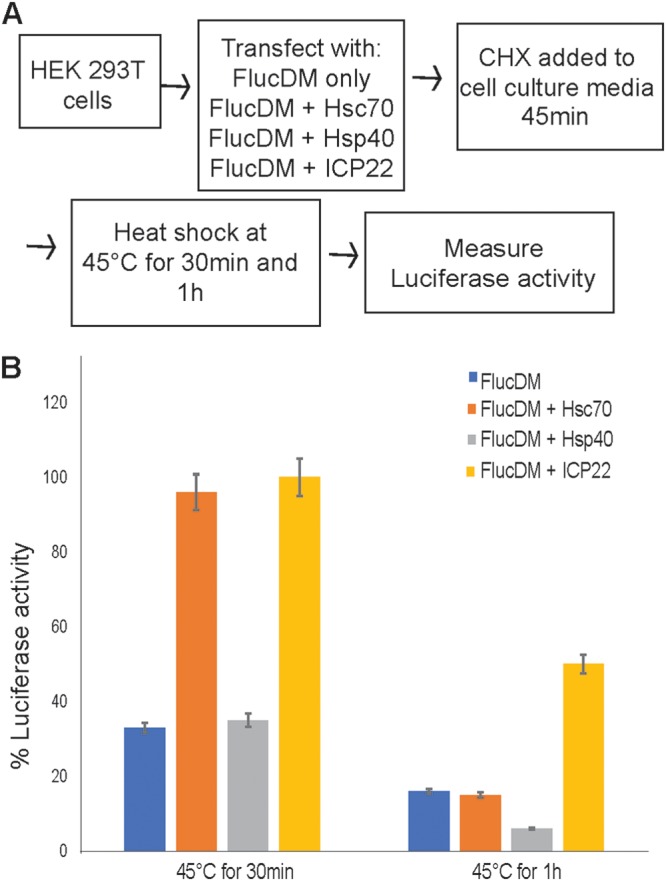
Thermoprotection of luciferase. (A) Flow diagram of experimental procedure. HEK293T cells were used to maximize transfection efficiency. (B) Luciferase activity was measured after heat shock and was normalized to non-heat-shocked samples (representing folded luciferase). The percentage of luciferase activity was plotted for FlucDM alone or for FlucDM with either Hsc70, Hsp40, or ICP22 for cells heat hocked at 45°C for either 30 min or 1 h.
DISCUSSION
J-protein/HSP70 complexes function in a variety of ways to promote protein quality control, including folding and unfolding of nascent proteins, sequestration and degradation of aggregation-prone proteins, and reduction of toxic aggregates from the cytoplasm (51, 53, 55). In addition, it is becoming clear that J proteins can play even more specialized roles in processes such as regulation of gene expression and cell cycle (59). Here, we present several lines of evidence supporting the notion that ICP22 functions as a virally encoded J-like protein that recruits Hsc70. (i) By 4 h postinfection, ICP22 localizes to discrete nuclear foci that subsequently recruit Hsc70. (ii) ICP22 can be immunoprecipitated with Hsc70, suggesting a physical interaction. (iii) In HSV-infected cells transfected with an aggregation-prone protein (GFP170*), Hsc70 was specifically recruited to nuclear aggresomes in an ICP22-dependent fashion. (iv) In cells transfected with ICP22, small nuclear inclusions that contain Hsc70 were observed, suggesting that ICP22 is sufficient to relocalize Hsc70 when expressed alone. If an aggregation-prone protein was also included, both Hsc70 and the aggregation-protein protein were recruited to larger nuclear inclusions in which ICP22 appeared to form a shell around the misfolded protein. (v) Transfection with GFP170* in the absence of ICP22 resulted in the formation of both cytoplasmic and nuclear aggregates; however, when ICP22 was present, a profound decrease in accumulation of cytoplasmic aggregates was observed. (vi) ICP22 was also able to confer protection against heat inactivation of firefly luciferase. These properties are consistent with ICP22 functioning as a virally encoded J-like protein.
The hallmark of cellular J proteins is the presence of a J domain responsible for interacting with an HSP70 family partner (19). Approximately, 50 different human J proteins have been described and are reported to fall into three subclasses (27). A sequence alignment between ICP22 and a prototypical class II J-protein family member, DNAJB1, revealed similarities. Class II J proteins contain an N-terminal J domain, a glycine/phenylalanine-rich domain (G/F-rich linker domain), and a C-terminal substrate binding domain (21). ICP22 contains all three conserved regions: a J domain (residues 1 to 76) that exhibits 49% sequence similarity and 22% identity with the DNAJB1, a G/F linker domain (residues 77 to 180), and a C-terminal substrate binding domain (CTD; residues 180 to 361) that exhibits 72% sequence similarity and 21% identity to the substrate binding domain of DNAJB1 (Fig. 6). However, 43% sequence identity for a small stretch of amino acids 336 to 361 in the C terminus of ICP22 is above what would be expected by chance. Type II J domains generally contain a highly conserved HPD (His-Pro-Asp) motif, critical for stimulation of the ATPase activity of their HSP70 partners (60, 61). Interestingly, ICP22 lacks this conserved motif, suggesting that it may have evolved to recruit Hsc70 but not stimulate its ATPase activity.
FIG 6.

Similarity between ICP22 and J proteins. Sequence alignments between DNAJB1 (top row) and ICP22 (bottom row) are shown. Conserved elements include the J domain (residues 1 to 75), the G/F linker (residues 76 to 180), and two regions of substrate binding domains (residues 180 to 361): CTDI and CTDII. Sequence alignment was performed using Clustal W. For the alignment of ICP22 with CTDI (residues 180 to 256) of DNAJB1, the sequence alignment is displayed for ICP22 residues 180 to 250. For alignment of ICP22 with CTDII (residues 257 to 361), the sequence alignment is displayed for ICP22 residues 257 to 287 and residues 334 to 361. The sequence similarity was consistent all across CTDII, although only residues 257 to 287 and residues 334 to 361 are shown for space limitations. An asterisk (*) indicates positions which have a single, fully conserved residue. A colon (:) indicates conservation between groups of similar properties (scoring > 0.5 in the Gonnet PAM 250 matrix). A period (.) indicates conservation between groups of weakly similar properties (scoring ≤ 0.5 in the Gonnet PAM 250 matrix).
Similar to other J proteins, the CTD of ICP22 contains two conserved regions the putative CTDI (residues 180 to 257) and CTDII (residues 257 to 361) (Fig. 5) (62) that overlap regions of ICP22 required for regulation of gene expression (36, 37). ICP22 residues 193 to 256 have been implicated in inhibition of host gene expression, as well as interaction with CDK9 (39). Residues 240 to 420 are required for promotion of late gene expression (36, 38). These functional domains from residues 193 to 256 and residues 240 to 420 overlap CTDI and CTDII, respectively, suggesting that the ability of ICP22 to regulate gene expression and cell cycle is mediated by chaperone-like functions, perhaps reflecting its ability to interact with a diverse set of client proteins. Taken together with the experimental data presented here, and the sequence conservation support the notion that ICP22 may have evolved as a J-like protein that recruits Hsc70. In fact, this suggests that HSV has taken advantage of the adaptable nature of J proteins to evolve a multifunctional cochaperone that functions with Hsc70 to promote lytic infection.
ICP22 recognizes and sequesters aggregation-prone proteins and provides thermoprotection.
We have previously reported that in HSV-1-infected cells, Hsc70 is recruited to VICE domains by 6 hpi (6). We show here that ICP22 localizes to nuclear bodies at least 2 h before the recruitment of Hsc70. The formation of ICP22 nuclear bodies may reflect the ability of ICP22 to recognize nonnative protein substrates and form nuclear domains that can later recruit Hsc70 (VICE domains). This is reminiscent of the observation by Teo et al. that ICP22 was recruited to nascent protein domains (NPDs) during the first 2 h of infection and that Hsc70 was recruited to these domains approximately 2 h later (48). The ability of ICP22 to recognize nonnative client proteins and subsequently recruit an HSP70 partner supports the notion that ICP22 is a J-like protein (8). Large inclusions were observed in transfected cells that overexpress the aggregation-prone protein GFP170*, along with ICP22. These inclusions do not resemble VICE domains seen in infected cells and are likely the result of overexpression; however, we suggest that the ability of ICP22 to form a shell around an overexpressed misfolded protein reflects an intrinsic property of ICP22 to recognize and sequester nonnative proteins. This behavior is reminiscent of the behavior of nucleophosmin 1 (NPM1), a multifunctional chaperone-like protein that also forms shells around toxic protein aggregates (54).
One of the most important cell protective functions of cellular J-protein/HSP70 complexes involves the sequestration of aggregation-prone proteins into aggresomes, thereby facilitating their removal by the UPS or other clearance mechanisms (63). Failure to degrade or remove misfolded proteins leads to the accumulation of cytotoxic aggregates that can be associated with neurodegenerative diseases (64). As mentioned above, cytoplasmic protein aggregates are reported to be more toxic than those that accumulate in the nucleus by virtue of their ability to interfere with nucleocytoplasmic transport of protein and RNA (51, 54). We report in the present study that ICP22 works in conjunction with Hsc70 to reduce the accumulation of cytoplasmic aggresomes (Fig. 4). Cellular J-protein/HSP70 complexes such as DNAJB6a/Hsc70 are known to reduce the accumulation of toxic poly Q proteins and increase the proportion of aggregated proteins in nuclear aggresomes (53, 65). The J protein DNAJB1 has been implicated in the transport of misfolded proteins from the cytoplasm to the nucleus followed by degradation by the UPS (51, 52). These results are consistent with the suggestion that ICP22 facilitates the intranuclear sequestration of aggregated or misfolded proteins as a protective mechanism consistent with properties of other cellular J proteins.
Some chaperone complexes have the ability to confer protection against heat damage; however, most cellular J-protein/HSP70 complexes do not exhibit this function. The chaperone Hsp110, a member of the HSP70 family, in conjunction with HSP40 and Hsc70, has been implicated in protection of proteins from heat inactivation (56). In the present study, we report that ICP22 alone was able to provide protection against the heat inactivation of firefly luciferase (Fig. 6), and it will be of interest to determine whether ICP22 works in conjunction with Hsc70 or within a larger ICP22/Hsp110/Hsc70 complex to carry out this function. In any case, it appears that ICP22 has evolved the somewhat specialized function of thermoprotection.
Roles of ICP22 in HSV infection.
In addition to interacting with the host protein quality control machinery, ICP22 plays several other roles in HSV infection. It is necessary for efficient viral replication and latency in mouse and guinea pig infection models and is required for viral growth in cell culture in most but not all cell types (31, 32, 40, 66). ICP22 has been implicated in a wide-range of processes, including gene expression, cell cycle control, viral assembly, and nuclear egress (35, 40, 67, 68). ICP22 has been shown to interact with and regulate the activities of several cellular proteins, including RNA Pol II, CDK9, and cyclins (35, 36, 39, 40, 42). In addition, ICP22 is required to recruit the FACT complex and other transcriptional elongation factors to viral genomes (69). Thus, despite its small size (420 residues), ICP22 interacts with a diverse set of cellular and viral proteins and exhibits an impressive array of functions.
The robust program of HSV gene expression during infection would be expected to represent a burden to the cellular protein quality control machinery, and ICP22 may have evolved to help manage the consequences of translation of a large number of nonnative cellular or viral proteins. This suggestion is supported by the observation that ICP22 localizes in the nucleus with nascent and misfolded proteins to form NPDs that recruit Hsc70 (33, 48). In addition, the ability to sequester nascent or misfolded proteins is cytoprotective (6), consistent with previous reports that implicate ICP22 in the prevention of apoptosis (70, 71). It is perhaps not surprising that HSV has evolved to manage robust protein expression by taking advantage of a cellular chaperone such as Hsc70.
We are also intrigued by recent reports suggesting that the protein homeostatic machinery and Hsc70 in particular have been implicated in antiviral defense mechanisms such as aggresome formation, proteolytic degradation of viral proteins, and autophagy (16, 17). In addition to the management of nascent proteins and regulation of gene expression, the ability of ICP22 to relocalize Hsc70 into VICE domains in the nucleus may prevent Hsc70 from carrying out antiviral activities elsewhere in the cell.
MATERIALS AND METHODS
Cells and Viruses.
African green monkey kidney cells (Vero CCl81; American Type Culture Collection, Rockville, MD) were cultured in minimal essential medium (Life Technologies, catalog no. 11095) in 5% fetal bovine serum (Atlanta Biologics, catalog no. S11550) and 1% penicillin-streptomycin (Life Technologies, catalog no. 15070) at 37°C. The KOS strain of HSV-1 was used as the wild-type virus. The ICP22-null virus, d22LacZ, and a virus encoding FLAG-tagged wild-type ICP22 (TF22) were obtained from Stephen Rice (University of Minnesota Medical School, Minneapolis, MN) (33, 38) and propagated on Vero cells.
Plasmids.
Plasmid pcDNA22 encoding N-terminally FLAG-tagged ICP22 was obtained from Stephen Rice (University of Minnesota Medical School, Minneapolis, MN) (35). A plasmid encoding the aggregation-prone protein GFP170* was obtained from Elizabeth Sztul (The University of Alabama at Birmingham, Birmingham, AL) (50). pCIneoFluc-EGFP vector expressing FlucDM-EGFP was obtained from Richard Morimoto and Ulrich Hartl (58). DNAJB1/Hsp40 was cloned into pcDNA3.1(–), and expression and localization were checked using Western blotting and immunofluorescence, respectively. pcDNA3.1 containing Hsc70 was provided by Pramod K. Srivastava lab.
Antibodies. (i) Primary antibodies.
Monoclonal rat anti-Hsc70 (SPA-815) and monoclonal mouse anti-Hsp70 (SPA-810) were purchased from Stressgen (San Diego, CA). Monoclonal mouse anti-ICP8 (11E2, sc-69809) and monoclonal mouse anti-ICP4 (H-943, sc-69809) were purchased from Santa Cruz (Dallas, TX). Polyclonal anti-chicken FLAG (ET-DY100) was purchased from Aves Labs (Tigard, Oregon). Monoclonal mouse anti-FLAG (clone M2, F3165) and polyclonal rabbit anti-FLAG (F7425) were purchased from Sigma Chemical Co. (St. Louis, MO). Polyclonal rabbit anti-ICP8 (clone 367) was obtained from William Ruyechan (State University of New York at Buffalo, Buffalo, NY).
(ii) Secondary antibodies.
Secondary antibodies were purchased from Molecular Probes and include Alexa Fluor 488-conjugated goat anti-mouse, Alexa Fluor 488-conjugated goat anti-chicken, Alexa Fluor 594-conjugated goat anti-rabbit, Alexa Fluor 647-conjugated goat anti-rat, and goat anti-rabbit Pacific Orange antibodies. GFP was detected by excitation using a 488-nm laser.
Transfection.
Vero cells were transfected using Lipofectamine reagent (Invitrogen, catalog no. 18324) according to the manufacturer’s recommended protocol.
Infection.
Infections were performed as previously described (6).
Immunofluorescence experiments.
After infection and/or transfection, the medium was aspirated and coverslips were processed as described previously (11).
Imaging and analysis.
Imaging was performed using Zeiss LSM780/880 confocal microscope with a Zeiss Plan Apochromat 63× objective (numerical aperture, 1.4 oil) at indicated zoom. All images were analyzed using Photoshop CS3 or ImageJ Fiji. All scale bars are drawn to 10 μm. For experiments that required counting of cells, 100 cells were counted from different fields on the slide and are represented as a percentage.
Coimmunoprecipitation.
Vero cells were grown to 90 to 95% confluence in 60-mm plates and infected with TF22 or d22LacZ virus at a multiplicity of infection (MOI) of 10. At 10 h postinfection, the cells were rinsed twice with phosphate-buffered saline, frozen at 80°C, and then lysed on ice for 30 min in lysis buffer (0.1 M potassium acetate, 0.02 M Tris acetate, 10% glycerol, 0.1% NP-40). A protease inhibitor cocktail (1× propidium iodide and 0.1 M phenylmethylsulfonyl fluoride) was added to the lysis buffer prior to use. After lysis, the cell lysates were precleared using normal mouse IgG. To immunoprecipitate FLAG-tagged proteins, mouse anti-FLAG antibody was used. Proteinase A/G-beads were added overnight at 4°C with spinning. The beads were pelleted in a microcentrifuge and washed with the lysis buffer. To elute bound protein, 2× SDS-PAGE sample buffer was added, and the samples were boiled for 5 min. All samples were analyzed by running on SDS-PAGE and immunoblotting. Blots were probed with rat anti-Hsc70, mouse anti-FLAG, and mouse anti-ICP4.
Luciferase activity assay.
HEK293T cells were plated in 24-well plates 20 to 24 h prior to transfection. HEK293T cells are used for this assay to achieve a good transfection efficiency. Transfections were performed using Lipofectamine 2000 reagent (Invitrogen, catalog no. 11668027) according to the manufacturer’s protocol. After 16 to 18 h of transfection, the medium was removed from cells, and 500 μl of heat shock buffer (1× morpholinepropanesulfonic acid and 40 μg/ml cycloheximide) was added per well. The cells were left in heat shock buffer for 45 min at 37°C, followed by heat shock at 45°C for 30 min or 1 h. To collect the samples, 1× passive lysis buffer was added, and samples were frozen on dry ice and stored at –80°C. Luciferase activity was measured in 96-well plates using Steady-Glo reagent (Promega, catalog no. E2510).
ACKNOWLEDGMENTS
We thank members of the Weller laboratory and Jason Gestwicki for helpful discussions and careful readings of the manuscript. We also thank Stephen Rice, Elizabeth Sztul, Richard Morimoto, Ulrich Hartl, Pramod Srivastava, and William Ruyechan for reagents. In addition, we are grateful to Katherine DiScipio for assistance with PyMOL.
The National Institutes of Health provided funding to S.K.W. under grants AI021747 and AI135451.
REFERENCES
- 1.Chen B, Retzlaff M, Roos T, Frydman J. 2011. Cellular strategies of protein quality control. Cold Spring Harb Perspect Biol 3:a004374. doi: 10.1101/cshperspect.a004374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goldberg AL. 2003. Protein degradation and protection against misfolded or damaged proteins. Nature 426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 3.Kopito RR. 2000. Aggresomes, inclusion bodies, and protein aggregation. Trends Cell Biol 10:524–530. doi: 10.1016/s0962-8924(00)01852-3. [DOI] [PubMed] [Google Scholar]
- 4.Johnston JA, Ward CL, Kopito RR. 1998. Aggresomes: a cellular response to misfolded proteins. J Cell Biol 143:1883–1898. doi: 10.1083/jcb.143.7.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Livingston CM, DeLuca NA, Wilkinson DE, Weller SK. 2008. Oligomerization of ICP4 and rearrangement of heat shock proteins may be important for herpes simplex virus type 1 prereplicative site formation. J Virol 82:6324–6336. doi: 10.1128/JVI.00455-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Livingston CM, Ifrim MF, Cowan AE, Weller SK. 2009. Virus-induced chaperone-enriched (VICE) domains function as nuclear protein quality control centers during HSV-1 infection. PLoS Pathog 5:e1000619. doi: 10.1371/journal.ppat.1000619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burch AD, Weller SK. 2004. Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J Virol 78:7175–7185. doi: 10.1128/JVI.78.13.7175-7185.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hendrick JP, Hartl FU. 1995. The role of molecular chaperones in protein folding. FASEB J 9:1559–1569. doi: 10.1096/fasebj.9.15.8529835. [DOI] [PubMed] [Google Scholar]
- 9.Hartl FU, Hayer-Hartl M. 2002. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 10.Clerico EM, Tilitsky JM, Meng W, Gierasch LM. 2015. How Hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol 427:1575–1588. doi: 10.1016/j.jmb.2015.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Daugaard M, Rohde M, Jäättelä M. 2007. The heat shock protein 70 family: highly homologous proteins with overlapping and distinct functions. FEBS Lett 581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- 12.Kaushik S, Cuervo AM. 2012. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol 22:407–417. doi: 10.1016/j.tcb.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiang HL, Terlecky SR, Plant CP, Dice JF. 1989. A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246:382–385. doi: 10.1126/science.2799391. [DOI] [PubMed] [Google Scholar]
- 14.Eisenberg E, Greene LE. 2007. Multiple roles of auxilin and Hsc70 in clathrin-mediated endocytosis. Traffic 8:640–646. doi: 10.1111/j.1600-0854.2007.00568.x. [DOI] [PubMed] [Google Scholar]
- 15.Yu A, Shibata Y, Shah B, Calamini B, Lo DC, Morimoto RI. 2014. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc Natl Acad Sci U S A 111:E1481–E1490. doi: 10.1073/pnas.1321811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou D, Blum JS. 2004. Presentation of cytosolic antigens via MHC class II molecules. Immunol Res 30:279–290. doi: 10.1385/IR:30:3:279. [DOI] [PubMed] [Google Scholar]
- 17.Deffit SN, Blum JS. 2015. A central role for HSC70 in regulating antigen trafficking and MHC class II presentation. Mol Immunol 68:85–88. doi: 10.1016/j.molimm.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Deretic V, Levine B. 2009. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kampinga HH, Craig EA. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Misselwitz B, Staeck O, Rapoport TA. 1998. J proteins catalytically activate Hsp70 molecules to trap a wide range of peptide sequences. Mol Cell 2:593–603. doi: 10.1016/S1097-2765(00)80158-6. [DOI] [PubMed] [Google Scholar]
- 21.Cheetham ME, Caplan AJ. 1998. Structure, function and evolution of DnaJ: conservation and adaptation of chaperone function. Cell Stress Chaperones 3:28–36. doi: 10.1379/1466-1268(1998)003<0028:sfaeod>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jana NR, Tanaka M, Wang G. h, Nukina N. 2000. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet 9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 23.Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. 2000. Chaperones Hsp70 and Hsp40 suppress aggregate formation and apoptosis in cultured neuronal cells expressing truncated androgen receptor protein with expanded polyglutamine tract. J Biol Chem 275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Yang Z, Cao Y, Zhang S, Li H, Huang Y, Ding Y, Liu X. 2008. The Hsp40 family chaperone protein DnaJB6 enhances Schlafen1 nuclear localization which is critical for promotion of cell-cycle arrest in T-cells. Biochem J 413:239–250. doi: 10.1042/BJ20071510. [DOI] [PubMed] [Google Scholar]
- 25.Meng E, Shevde LA, Samant RS. 2016. Emerging roles and underlying molecular mechanisms of DNAJB6 in cancer. Oncotarget 7:53984–53996. doi: 10.18632/oncotarget.9803. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Craig EA, Marszalek J. 2017. How do J Proteins get Hsp70 to do so many different things? Trends Biochem Sci 42:355–368. doi: 10.1016/j.tibs.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. 2009. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones 14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sullivan CS, Pipas JM. 2001. The virus-chaperone connection. Virology 287:1–8. doi: 10.1006/viro.2001.1038. [DOI] [PubMed] [Google Scholar]
- 29.Ohta A, Yamauchi Y, Muto Y, Kimura H, Nishiyama Y. 2011. Herpes simplex virus type 1 UL14 tegument protein regulates intracellular compartmentalization of major tegument protein VP16. Virol J 8:365. doi: 10.1186/1743-422X-8-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albright BS, Kosinski A, Szczepaniak R, Cook EA, Stow ND, Conway JF, Weller SK. 2015. The putative herpes simplex virus 1 chaperone protein UL32 modulates disulfide bond formation during infection. J Virol 89:443–453. doi: 10.1128/JVI.01913-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sears AE, Halliburton IW, Meignier B, Silver S, Roizman B. 1985. Herpes simplex virus 1 mutant deleted in the alpha 22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J Virol 55:338–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poffenberger KL, Raichlen PE, Herman RC. 1993. In vitro characterization of a herpes simplex virus type 1 ICP22 deletion mutant. Virus Genes 7:171–186. doi: 10.1007/bf01702397. [DOI] [PubMed] [Google Scholar]
- 33.Bastian TW, Livingston CM, Weller SK, Rice SA. 2010. Herpes simplex virus type 1 immediate-early protein ICP22 is required for VICE domain formation during productive viral infection. J Virol 84:2384–2394. doi: 10.1128/JVI.01686-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng TI, Chang YE, Roizman B. 1997. Infected cell protein 22 of herpes simplex virus 1 regulates the expression of virion host shutoff gene UL41. Virology 234:226–234. doi: 10.1006/viro.1997.8659. [DOI] [PubMed] [Google Scholar]
- 35.Fraser KA, Rice SA. 2007. Herpes simplex virus immediate-early protein ICP22 triggers loss of serine 2-phosphorylated RNA polymerase II. J Virol 81:5091–5101. doi: 10.1128/JVI.00184-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rice SA, Long MC, Lam V, Schaffer PA, Spencer CA. 1995. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J Virol 69:5550–5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Long MC, Leong V, Schaffer PA, Spencer CA, Rice SA. 1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J Virol 73:5593–5604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bastian TW, Rice SA. 2009. Identification of sequences in herpes simplex virus type 1 ICP22 that influence RNA polymerase II modification and viral late gene expression. J Virol 83:128–139. doi: 10.1128/JVI.01954-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaborowska J, Baumli S, Laitem C, O’Reilly D, Thomas PH, O’Hare P, Murphy S. 2014. Herpes simplex virus 1 (HSV-1) ICP22 protein directly interacts with cyclin-dependent kinase 9 (CDK9) to inhibit RNA polymerase II transcription elongation. PLoS One 9:e107654. doi: 10.1371/journal.pone.0107654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orlando JS, Astor TL, Rundle SA, Schaffer PA. 2006. The products of the herpes simplex virus type 1 immediate-early US1/US1.5 genes downregulate levels of S-phase-specific cyclins and facilitate virus replication in S-phase Vero cells. J Virol 80:4005–4016. doi: 10.1128/JVI.80.8.4005-4016.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bruni R, Roizman B. 1998. Herpes simplex virus 1 regulatory protein ICP22 interacts with a new cell cycle-regulated factor and accumulates in a cell cycle-dependent fashion in infected cells. J Virol 72:8525–8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Advani SJ, Brandimarti R, Weichselbaum RR, Roizman B. 2000. The disappearance of cyclins A and B and the increase in activity of the G2/M-phase cellular kinase cdc2 in herpes simplex virus 1-infected cells require expression of the α22/US1.5 and UL13 viral genes. J Virol 74:8–15. doi: 10.1128/JVI.74.1.8-15.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adlakha M, Livingston CM, Bezsonova I, Weller SK. 2019. The HSV-1 immediate early protein ICP22 is a J-like protein required for Hsc70 reorganization during lytic infection. bioRxiv 10.1101/671412. [DOI]
- 44.Kodiha M, Chu A, Lazrak O, Stochaj U. 2005. Stress inhibits nucleocytoplasmic shuttling of heat shock protein hsc70. Am J Physiol Cell Physiol 289:C1034–C1041. doi: 10.1152/ajpcell.00590.2004. [DOI] [PubMed] [Google Scholar]
- 45.Jahedi S, Markovitz NS, Filatov F, Roizman B. 1999. Colocalization of the herpes simplex virus 1 UL4 protein with infected cell protein 22 in small, dense nuclear structures formed prior to onset of DNA synthesis. J Virol 73:5132–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Markovitz NS, Roizman B. 2000. Small dense nuclear bodies are the site of localization of herpes simplex virus 1 UL3 and UL4 proteins and of ICP22 only when the latter protein is present. J Virol 74:523–528. doi: 10.1128/jvi.74.1.523-528.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xing J, Wang S, Lin F, Pan W, Hu C-D, Zheng C. 2011. Comprehensive characterization of interaction complexes of herpes simplex virus type 1 ICP22, UL3, UL4, and UL20.5. J Virol 85:1881–1886. doi: 10.1128/JVI.01730-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su Hui Teo C, Serwa RA, O’Hare P. 2016. Spatial and temporal resolution of global protein synthesis during HSV infection using bio-orthogonal precursors and click chemistry. PLoS Pathog 12:e1005927. doi: 10.1371/journal.ppat.1005927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sugito K, Yamane M, Hattori H, Hayashi Y, Tohnai I, Ueda M, Tsuchida N, Ohtsuka K. 1995. Interaction between hsp70 and hsp40, eukaryotic homologues of DnaK and DnaJ, in human cells expressing mutant-type p53. FEBS Lett 358:161–164. doi: 10.1016/0014-5793(94)01417-y. [DOI] [PubMed] [Google Scholar]
- 50.Fu L, Gao Y, Tousson A, Shah A, Chen T-L, Vertel BM, Sztul E. 2005. Nuclear aggresomes form by fusion of PML-associated aggregates. Mol Biol Cell 16:4905–4917. doi: 10.1091/mbc.e05-01-0019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hipp MS, Park S-H, Hartl FU. 2014. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol 24:506–514. doi: 10.1016/j.tcb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Park S-H, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, Hayer-Hartl M, Hartl FU. 2013. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell 154:134–145. doi: 10.1016/j.cell.2013.06.003. [DOI] [PubMed] [Google Scholar]
- 53.Gillis J, Schipper-Krom S, Juenemann K, Gruber A, Coolen S, van den Nieuwendijk R, van Veen H, Overkleeft H, Goedhart J, Kampinga HH, Reits EA. 2013. The DNAJB6 and DNAJB8 protein chaperones prevent intracellular aggregation of polyglutamine peptides. J Biol Chem 288:17225–17237. doi: 10.1074/jbc.M112.421685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Woerner AC, Frottin F, Hornburg D, Feng LR, Meissner F, Patra M, Tatzelt J, Mann M, Winklhofer KF, Hartl FU, Hipp MS. 2016. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 351:173–176. doi: 10.1126/science.aad2033. [DOI] [PubMed] [Google Scholar]
- 55.Monsellier E, Redeker V, Ruiz-Arlandis G, Bousset L, Melki R. 2015. Molecular interaction between the chaperone Hsc70 and the N-terminal flank of huntingtin exon 1 modulates aggregation. J Biol Chem 290:2560–2576. doi: 10.1074/jbc.M114.603332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oh HJ, Chen X, Subjeck JR. 1997. Hsp110 protects heat-denatured proteins and confers cellular thermoresistance. J Biol Chem 272:31636–31640. doi: 10.1074/jbc.272.50.31636. [DOI] [PubMed] [Google Scholar]
- 57.Ciavarra RP, Goldman C, Wen KK, Tedeschi B, Castora FJ. 1994. Heat stress induces hsc70/nuclear topoisomerase I complex formation in vivo: evidence for hsc70-mediated, ATP-independent reactivation in vitro. Proc Natl Acad Sci U S A 91:1751–1755. doi: 10.1073/pnas.91.5.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gupta R, Kasturi P, Bracher A, Loew C, Zheng M, Villella A, Garza D, Hartl FU, Raychaudhuri S. 2011. Firefly luciferase mutants as sensors of proteome stress. Nat Methods 8:879–884. doi: 10.1038/nmeth.1697. [DOI] [PubMed] [Google Scholar]
- 59.Ajit Tamadaddi C, Sahi C. 2016. J domain independent functions of J proteins. Cell Stress Chaperones 21:563–570. doi: 10.1007/s12192-016-0697-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kelley WL. 1999. Molecular chaperones: how J domains turn on Hsp70s. Curr Biol 9:R305–R308. doi: 10.1016/s0960-9822(99)80185-7. [DOI] [PubMed] [Google Scholar]
- 61.Tsai J, Douglas MG. 1996. A conserved HPD sequence of the J domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J Biol Chem 271:9347–9354. doi: 10.1074/jbc.271.16.9347. [DOI] [PubMed] [Google Scholar]
- 62.Cuéllar J, Perales-Calvo J, Muga A, Valpuesta JM, Moro F. 2013. Structural insights into the chaperone activity of the 40-kDa heat shock protein DnaJ: binding and remodeling of a native substrate. J Biol Chem 288:15065–15074. doi: 10.1074/jbc.M112.430595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ciechanover A, Kwon YT. 2015. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med 47:e147. doi: 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, Kopito R, McCampbell A, Leblanc G, Venkateswaran A, Nurmi A, Hodgson R. 2017. Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener 6:6. doi: 10.1186/s40035-017-0077-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Månsson C, Kakkar V, Monsellier E, Sourigues Y, Härmark J, Kampinga HH, Melki R, Emanuelsson C. 2014. DNAJB6 is a peptide-binding chaperone which can suppress amyloid fibrillation of polyglutamine peptides at substoichiometric molar ratios. Cell Stress Chaperones 19:227–239. doi: 10.1007/s12192-013-0448-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poffenberger KL, Idowu AD, Fraser-Smith EB, Raichlen PE, Herman RC. 1994. A herpes simplex virus type 1 ICP22 deletion mutant is altered for virulence and latency in vivo. Arch Virol 139:111–119. doi: 10.1007/bf01309458. [DOI] [PubMed] [Google Scholar]
- 67.Maruzuru Y, Shindo K, Liu Z, Oyama M, Kozuka-Hata H, Arii J, Kato A, Kawaguchi Y. 2014. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J Virol 88:7445–7454. doi: 10.1128/JVI.01057-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orlando JS, Balliet JW, Kushnir AS, Astor TL, Kosz-Vnenchak M, Rice SA, Knipe DM, Schaffer PA. 2006. ICP22 is required for wild-type composition and infectivity of herpes simplex virus type 1 virions. J Virol 80:9381–9390. doi: 10.1128/JVI.01061-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fox HL, Dembowski JA, DeLuca NA. 2017. A herpesviral immediate early protein promotes transcription elongation of viral transcripts. mBio 8:e00745-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nguyen ML, Blaho JA. 2006. Apoptosis during herpes simplex virus infection. Adv Virus Res 69:67–97. doi: 10.1016/S0065-3527(06)69002-7. [DOI] [PubMed] [Google Scholar]
- 71.You Y, Cheng A-C, Wang M-S, Jia R-Y, Sun K-F, Yang Q, Wu Y, Zhu D, Chen S, Liu M-F, Zhao X-X, Chen X-Y. 2017. The suppression of apoptosis by α-herpesvirus. Cell Death Dis 8:e2749. doi: 10.1038/cddis.2017.139. [DOI] [PMC free article] [PubMed] [Google Scholar]