

HSV-1 is a pathogen of the human nervous system that uses its own virus-encoded proteins and the normal cellular ESCRT machinery to drive the construction of its envelope. How HSV-1 structural proteins interact with ESCRT components and which subsets of cellular ESCRT proteins are utilized by the virus remain largely unknown. Here, we demonstrate that an essential component of the ESCRT-II complex and two ESCRT-associated Bro1 proteins are dispensable for HSV-1 replication.
KEYWORDS: ESCRT, envelopment, virus assembly, herpes simplex virus
ABSTRACT
Capsid envelopment during assembly of the neurotropic herpesviruses herpes simplex virus 1 (HSV-1) and pseudorabies virus (PRV) in the infected cell cytoplasm is thought to involve the late-acting cellular ESCRT (endosomal sorting complex required for transport) components ESCRT-III and VPS4 (vacuolar protein sorting 4). However, HSV-1, unlike members of many other families of enveloped viruses, does not appear to require the ESCRT-I subunit TSG101 or the Bro1 domain-containing protein ALIX (Alg-2-interacting protein X) to recruit and activate ESCRT-III. Alternative cellular factors that are known to be capable of regulating ESCRT-III function include the ESCRT-II complex and other members of the Bro1 family. We therefore used small interfering RNA (siRNA) to knock down the essential ESCRT-II subunit EAP20/VPS25 (ELL-associated protein 20/vacuolar protein sorting 25) and the Bro1 proteins HD-PTP (His domain-containing protein tyrosine phosphatase) and BROX (Bro1 domain and CAAX motif containing). We demonstrated reductions in levels of the targeted proteins by Western blotting and used quantitative microscopic assays to confirm loss of ESCRT-II and HD-PTP function. We found that in single-step replication experiments, the final yields of HSV-1 were unchanged following loss of EAP20, HD-PTP, or BROX.
IMPORTANCE HSV-1 is a pathogen of the human nervous system that uses its own virus-encoded proteins and the normal cellular ESCRT machinery to drive the construction of its envelope. How HSV-1 structural proteins interact with ESCRT components and which subsets of cellular ESCRT proteins are utilized by the virus remain largely unknown. Here, we demonstrate that an essential component of the ESCRT-II complex and two ESCRT-associated Bro1 proteins are dispensable for HSV-1 replication.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) capsids are assembled and packaged with their DNA genomes in the cell nucleus (1–3). The nucleocapsids subsequently undergo primary envelopment at the inner nuclear membrane (INM), followed by delivery of capsids to the cytoplasm via de-envelopment at the outer nuclear membrane (4–7). The capsids subsequently undergo a final, secondary envelopment by budding into the lumens of organelles in the cytoplasm (8–16). Primary envelopment at the INM and secondary envelopment in the cytoplasm require the activities of multiple virus-encoded proteins (5, 17, 18) in concert with the cellular ESCRT (endosomal sorting complex required for transport) apparatus (19–21).
The ESCRT apparatus is a membrane-remodeling machine that is used to manipulate membrane bilayers during diverse cellular processes, including multivesicular body (MVB) formation, cytokinesis, exosome and microvesicle formation, and repair of plasma and nuclear membranes (22–26). The multisubunit complexes ESCRT-0, -I, and -II, along with additional accessory proteins (24, 25), control the location, timing, and deposition of ESCRT-III (Fig. 1), a spring-like filament assembled by polymerization of CHMP (charged multivesicular body protein) family members on the surface of the lipid bilayer. The polymerized ESCRT-III filament drives the formation of vesicles, tubules, or viral envelopes and then constricts the resulting structure at its neck to catalyze scission, a process accompanied by disassembly and removal of ESCRT-III via the hexameric AAA ATPase VPS4 (vacuolar protein sorting 4) (24, 25, 27).
FIG 1.

Alphaherpesvirus envelopment and candidate ESCRT components. ESCRT complexes are shown on the left, with ESCRT-0 (composed of subunits STAM/HRS) in brown, ESCRT-I (composed of subunits TSG101/MVB12/VPS37/VPS28) in red, and ESCRT-II (EAP30/EAP45/EAP202) in yellow. The ESCRT-III filament is shown as a polymer of CHMP2/3/4 subunits (green) capped by nucleating CHMP6 subunits (cyan). The ESCRT-I and ESCRT-II complex EAP20 subunits can each nucleate filament assembly through recruitment of CHMP6. Members of the Bro1 family of proteins (dark-blue cylinder) are able to directly trigger CHMP4 polymerization. On the right is the generalized structure of an alphaherpesvirus cytoplasmic envelopment intermediate, showing the presumed relationship between the ESCRT-III filament and the lipid bilayer (brown line), the viral capsid (red sphere), the inner tegument (gray layer), and the outer tegument (dark-blue ovals), some of which interact with membrane-embedded envelope glycoproteins (yellow bars). Late in envelopment, ESCRT-III constricts to draw the membrane together, sealing the envelope and pinching the enveloped virus into the organellar lumen (light-blue space). Constriction and ESCRT-III disassembly are catalyzed by the ATPase VPS4 (purple spheres). The positioning of the ESCRT-0, -I, and -II complexes and the Bro1 proteins is not meant to necessarily imply they play roles in alphaherpesvirus envelopment.
The formation and deposition of the ESCRT-III filament are controlled by a number of parallel cellular pathways. ESCRT-I directly binds the ESCRT-III subunit CHMP6 but can also recruit it via association with the complex ESCRT-II (24). In both cases, CHMP6 then nucleates polymerization of the ESCRT-III filament major subunits, principally CHMP4, but also the CHMP2 and CHMP3 subunits (24, 28) (Fig. 1). CHMP polymerization can also be triggered by proteins containing the elongated banana-shaped Bro1 domain (24, 25, 29, 30) (Fig. 1). Humans express five known Bro1 domain family members (described in more detail below), the best characterized of which is ALIX (Alg-2-interacting protein X) (24, 25). ALIX, like ESCRT-II, can be recruited by ESCRT-I, and the ALIX Bro1 domain then binds to CHMP4 (30, 31), activating it and inducing conformational changes that drive its polymerization with itself and other CHMP subunits (24, 25).
Many families of enveloped viruses have evolved means to exploit the ESCRT apparatus in order to gain access to ESCRT-III and VPS4 to drive terminal scission (23, 32, 33). In general, viruses acquire ESCRT-III by recruiting one or more earlier ESCRT components, most commonly ALIX and the ESCRT-I complex (33). The best-understood examples are the retroviral Gag proteins, where sequence motifs termed L domains (late-budding domains) recruit ALIX via the motif YPXnL and the TSG101 subunit of ESCRT-I via the sequence P(T/S)AP (23, 33). ALIX and ESCRT-I are then thought to activate ESCRT-III assembly via their well-characterized direct interactions with (respectively) CHMP4 and CHMP6, though ESCRT-I may also recruit ESCRT-II to the site of HIV-1 budding (34, 35). Other families of enveloped viruses use alternative recruitment mechanisms but commonly target the same subset of ESCRT components (19, 33).
How the Alphaherpesvirinae recruit the ESCRT machinery for cytoplasmic envelopment is poorly understood (19). ESCRT-III and VPS4 appear to be required for completion of cytoplasmic envelopment by HSV-1 (21, 36) and pseudorabies virus (PRV) (21) and play a role during HSV-1 envelopment at the INM (7). However, expression of dominant-negative forms of ALIX and TSG101 have no effect upon HSV-1 replication (20). Similarly, small interfering RNA (siRNA) knockdown of ALIX and TSG101 do not diminish HSV-1 titers or growth rates, even when they are depleted simultaneously to exclude the possibility that they act redundantly (20). As we recently discussed (19), lacking a role for ALIX and ESCRT-I, the most likely remaining cellular proteins that HSV-1 might use to assemble the ESCRT-III apparatus are the ESCRT-II complex and Bro1 family members other than ALIX (Fig. 1).
Although ESCRT-II is most commonly recruited to sites of membrane remodeling via TSG101/ESCRT-I, the core protein of hepatitis B virus has been reported to bind ESCRT-II directly and to utilize it for pre-genomic-RNA trafficking, encapsidation, or stabilization of replication-competent hepatitis B virus nucleocapsids (37). It therefore remains a possibility that structural components of HSV-1 might directly bind ESCRT-II in order to control ESCRT-III assembly (Fig. 1). Regarding Bro1 family members, in addition to ALIX, the human proteome contains four Bro1 domain proteins with broad tissue expression (24, 30, 38): RHPN1 (Rhophilin 1) and RHPN2, HD-PTP (His domain-containing protein tyrosine phosphatase) (39–42), and BROX (Bro1 domain and CAAX motif containing) (29, 30, 43). RHPN1 and RHPN2 are Rho-GTP binding proteins associated with the actin cytoskeleton and have not been clearly linked to the ESCRT pathway (30). In contrast, HD-PTP and BROX both participate in ESCRT-III assembly during endosomal cargo sorting and MVB formation (40, 43), positioning them in an appropriate cellular location for use by enveloping HSV-1 capsids (8, 9, 44). Although the normal physiological role of BROX is unclear (43), its Bro1 domain appears to interact with both CHMP4B and CHMP5 and to recruit CHMP5 to endosomes (29). This is in contrast to ALIX and HD-PTP, both of which are considered to be CHMP4 specific (31, 41, 43), and is interesting, given that dominant-negative alleles of CHMP4B and CHMP5 are two of the most potent inhibitors of HSV-1 replication (20).
In this study, we used siRNA to knock down the essential ESCRT-II subunit EAP20/VPS25 (ELL-associated protein 20/vacuolar protein sorting 25) (19, 24) and the Bro1 proteins HD-PTP and BROX. We found that substantial depletion of these polypeptides could be shown to inhibit the normal cellular processes associated with their functions (for EAP20 and HD-PTP, for which such assays exist) but had no effect upon the final titers of HSV-1 replicating in knockdown cells. We conclude that under our conditions none of these proteins are individually required for ESCRT-III recruitment and envelopment of HSV-1.
RESULTS
Knockdown of ESCRT-II complex function.
ESCRT-II is a Y-shaped complex with a stem consisting of the subunits EAP30 and EAP45 and two arms, each composed of a copy of the protein EAP20/VPS25 (24). It is the EAP20 subunits that are capable of binding and activating CHMP6 (24, 45) to nucleate ESCRT-III polymerization (28) (Fig. 1). We knocked down the EAP20 subunit in HeLa cells using an siRNA directed against exon 5 of the EAP20 transcript. Western blotting confirmed that the EAP20 protein level was substantially reduced after knockdown in HSV-1-infected HeLa cells (Fig. 2B).
FIG 2.

Inhibition of EGFR degradation following knockdown of the ESCRT-II subunit EAP20. HeLa cells were transfected with control siRNA (Ctrl) or an siRNA targeting EAP20. After 24 h, the transfections were repeated, and then the cells were immediately infected with HSV-1. At 13 h postinfection, the cells were serum starved for 5 h; incubated with (+) or without (−) EGF for 2 h, as indicated; and then fixed for DAPI staining and anti-EGFR immunocytochemistry or collected for Western blotting. (A) Fields of cells immunostained for EGFR (green), exhibiting UL25-mCherry fluorescence (red), or in which EGFR and UL25 fluorescence images were merged with DAPI-stained images (Merge). (B) Western blot showing levels of EAP20 or the cell recovery/loading control GAPDH protein at the time of cell recovery. (C) Mean EGFR immunofluorescence intensities from 50 microscopic fields similar to those in panel A were quantitated using NIH ImageJ software (3 to 5 cells were scored per field). Mean EGFR fluorescence intensities and standard deviations from the mean are plotted in arbitrary units (a.u.). ns, not significant (P = 0.59); ****, P < 0.0001.
We next tested whether ESCRT-II was functionally inactivated by examining the ability of EAP20 knockdown cells to degrade the epidermal growth factor receptor (EGFR) in lysosomes, a process known to be partially dependent upon ESCRT-II function (35, 46). HeLa cells were subjected to siRNA-mediated EAP20 knockdown or treated with a control siRNA, infected with HSV-1, and then serum starved for 5 h to deplete epidermal growth factor (EGF) and thus accumulate EGFR at the cell surface. The cells were then incubated with EGF (or with EGF omitted) for 2 h to induce EGFR internalization and targeting to lysosomes for degradation. We then performed anti-EGFR immunocytochemistry to visualize and quantitate EGFR fluorescence at the end of the EGF chase period. As shown in Fig. 2A, when EGF was omitted (−EGF), EGFR was readily detectable on the cell surface regardless of whether cells had been treated with a control (Ctrl) siRNA or with EAP20 siRNA. However, in the presence of EGF (+EGF), EGFR chased from the cell surface into endosomes, and in the presence of control siRNA, some EGFR reached lysosomes, where it was degraded, resulting in a reduction in total anti-EGFR fluorescence intensity (Fig. 2A, left). In contrast, upon EAP20 knockdown, the ESCRT-II-dependent step of delivery to lysosomes was inhibited and EGFR accumulated in bright intracellular puncta (Fig. 2A, right). These data are shown quantitatively in Fig. 2C. In siRNA control cells, approximately 33% of cellular EGFR was internalized and degraded during the time course of the EGF chase (P < 0.0001). In contrast, although EAP20 knockdown cells internalized EGFR (Fig. 2A), no significant EGFR degradation was seen (P = 0.59). These experimental and control data resemble those seen for EGFR turnover following EAP20 knockdown in HEK293 cells (46) and HIV-infected HeLa cells (35).
HSV-1-infected EAP20 knockdown cells similar to those examined above were prepared and then collected immediately after infection or at 20 h postinfection (hpi). The cells were scraped up into their medium, and extracts were prepared and then either Western blotted for EAP20 and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Fig. 3A) or subjected to plaque assay to determine yields of virus. As seen in Fig. 3B, the final yield of HSV-1 in EAP20 knockdown cells at 20 hpi was similar to that in cells treated with a control siRNA, demonstrating that under these conditions, EAP20/VPS25, and therefore the normal function of the ESCRT-II complex, is not necessary for HSV-1 replication.
FIG 3.

siRNA knockdown of EAP20 or PRPF8 and effects on HSV-1 replication. (A and B) Cells were treated with control (Ctrl) siRNA or EAP20 siRNA exactly as for Fig. 2. They were infected with HSV-1 and acid washed to inactivate unpenetrated virus, and cell extracts were prepared immediately (T0) and after 20 h (T20). (A) Cell extracts were subjected to Western blotting for EAP20 and control GAPDH. (B) Plaque assays to determine PFU yields in cell extracts. The plotted values are means and standard deviations from the mean for 6 independent replicates. (C and D) Cells were treated with control (Ctrl) siRNA or PRPF8 siRNA, and the transfections were repeated after 48 h. After a further 24 h, the cells were infected with HSV-1, and extracts were prepared exactly as for the EAP20 knockdown. (C) Cell extracts were subjected to Western blotting for PRPF8 and control GAPDH. (D) Plaque assays to determine PFU yields in cell extracts. The plotted values are means and standard deviations from the mean for four independent replicates.
As a positive control for the effect of siRNA knockdown on HSV-1 replication, we targeted the spliceosomal subunit PRPF8 (pre-mRNA processing factor 8) (47), because its knockdown in HeLa cells is known to inhibit HSV-1 replication without causing cytotoxicity (48). Under infection and replication conditions identical to those used in our EAP20 knockdown experiments, the depletion of PRPF8 (Fig. 3C) resulted in an ∼37-fold reduction in the titer of progeny HSV-1 (Fig. 3D). These results compare well with knockdown of other cellular proteins known to be important for HSV-1 replication in HeLa cells when infected at a multiplicity of infection (MOI) of 1 to 10; HSV-1 progeny titers were reduced 3- to 4-fold by knockdown of nucleolin (49), 2.5- to 5-fold upon knockdown of Rab1a or Rab43 (50), and 4-fold following knockdown of SRSF2 (serine/arginine-rich splicing factor 2) (51).
Knockdown and functional assay of the Bro1 protein HD-PTP.
We next knocked down the Bro1 family member HD-PTP (Fig. 1), using a pool of two siRNAs targeting exons 1 and 2 and exon 8. Western blotting confirmed that HD-PTP levels were substantially reduced in HSV-1-infected HeLa cells (Fig. 4B). HD-PTP is required to efficiently localize the survival motor neuron (SMN) complex to nuclear Cajal bodies (52), so as a functional test for HD-PTP depletion, we examined the accumulation of SMN-positive nuclear puncta. As shown in Fig. 4A, nuclei accumulated substantially fewer SMN-positive puncta in HD-PTP knockdown cells than in controls. Quantitation revealed that loss of HD-PTP resulted in a 57% decrease in the mean number of SMN-positive puncta per nucleus (Fig. 4C, left) (P < 0.0001) and a 49% increase in the number of nuclei that completely lacked such puncta (Fig. 4C, right) (P < 0.0001).
FIG 4.

siRNA knockdown of HD-PTP and its consequences for accumulation of SMN puncta in nuclei. HeLa cells were transfected with control siRNA (Ctrl) or a mixture of two HD-PTP siRNAs, and the transfections were repeated 24 h and 48 h later. Cells were then collected for immunostaining or Western blotting. (A) Fields of cells fixed, stained with DAPI, and immunostained for SMN. (Bottom row) SMN immunostaining alone. The white arrowhead indicates a nuclear SMN punctum. (Top row) Merge of SMN and DAPI channels. (B) Western blot showing levels of HD-PTP or control GAPDH protein in the cells at the time of their recovery. (C) SMN puncta in nuclei were counted and plotted as the mean number of puncta seen per 150 cells (left graph) and the mean number of punctum-free nuclei per 150 cells (right graph). The plotted values are means and standard deviations from the mean for 3 independent experiments, with 150 cells scored per experiment ****, P < 0.0001.
HSV-1-infected HD-PTP knockdown cells similar to those examined above were prepared and then collected immediately after infection or at 20 hpi. Extracts were prepared exactly as for Fig. 3 and then either Western blotted for HD-PTP and GAPDH (Fig. 5A) or subjected to plaque assay to determine the yields of virus. As seen in Fig. 5B, the final yield of HSV-1 in HD-PTP knockdown cells at 20 hpi was similar to that in cells treated with a control siRNA, demonstrating that under these conditions, the Bro1 domain protein HD-PTP is not necessary for HSV-1 replication.
FIG 5.
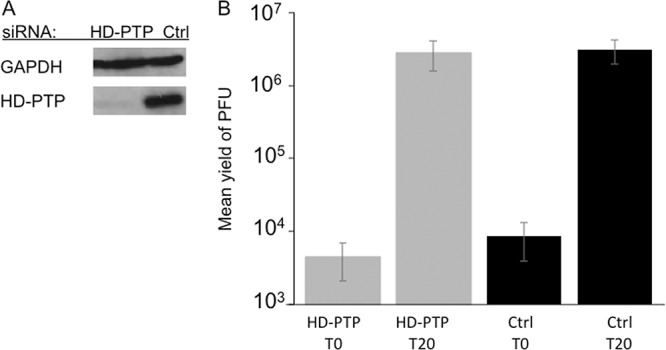
Titers of HSV-1 grown on HD-PTP-depleted HeLa cells. The cells were treated with control siRNA (Ctrl) or HD-PTP siRNAs exactly as for Fig. 4, infected with HSV-1, and then acid washed to inactivate unpenetrated virus. Samples were collected immediately (T0) and after 20 h (T20). (A) Cell extracts were subjected to Western blotting for HD-PTP and control GAPDH. (B) Plaque assays to determine PFU yields in cell extracts. The plotted values are means and standard deviations from the mean for 6 independent replicates.
Knockdown of the Bro1 protein BROX.
Finally, we knocked down the Bro1 protein BROX using a pool of two siRNAs targeting exon 4 and exons 11 and 12. We found this molecule to be rather refractory to siRNA-mediated knockdown, and indeed, to our knowledge, successful knockdown of BROX in mammalian cells has not previously been reported. Nevertheless, following the establishment of a suitable knockdown protocol, we confirmed by Western blotting that BROX levels could be substantially reduced in HSV-1-infected HeLa cells (Fig. 6A). We were unable to independently test the cellular consequences of this BROX knockdown, since there is no known assay to measure BROX function (43).
FIG 6.
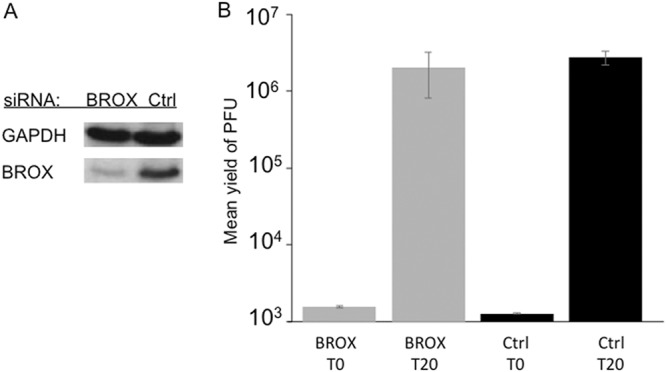
Titers of HSV-1 grown on BROX-depleted HeLa cells. Cells were treated with control siRNA (Ctrl) or a mixture of two BROX siRNAs. The transfection was then repeated 48 h and 96 h after the start of the experiment. After an additional 4 h, the cells were infected with HSV-1 and then acid washed to inactivate unpenetrated virus. Samples were collected immediately (T0) and after 20 h (T20). (A) Cell extracts were subjected to Western blotting for BROX and control GAPDH. (B) Plaque assays to determine PFU yields in cell extracts. The plotted values are means and ranges for 2 independent duplicate experiments.
HSV-1-infected BROX knockdown cells were collected immediately after infection or at 20 hpi, and extracts were prepared as for Fig. 3 and then subjected to plaque assay. As shown in Fig. 6B, the final yield of HSV-1 in BROX knockdown cells at 20 hpi was similar to that in cells treated with a control siRNA, demonstrating that under these conditions, loss of most cellular BROX protein had no effect on HSV-1 replication.
DISCUSSION
The cellular ESCRT apparatus is an ancient and versatile membrane-remodeling machine (24, 25, 32) that has been used by many families of viruses to help construct their envelopes (23, 32, 33). The Alphaherpesvirinae are no exception, as ESCRT-III and VPS4 functions appear to be required for completion of cytoplasmic envelopment by HSV-1 (21, 36) and PRV (21) and for the maturation of these cytoplasmic virions to a form able to traffic along microtubules in vitro (21). However, two major questions remain unresolved: which cellular ESCRT components are utilized by Alphaherpesvirinae during cytoplasmic envelopment, and which viral structural proteins recruit them.
Motifs resembling retroviral YPXnL and P(T/S)AP Gag L domains (known to mediate ALIX and TSG101 binding, respectively) have been identified by sequence inspection of a number of HSV-1 proteins (20). However, siRNA knockdown of ALIX and TSG101, or expression of dominant-negative forms of the proteins, had no effect upon HSV-1 replication (20). Alternatively, the large inner-tegument protein UL36p has long been a strong candidate for ESCRT recruitment due to its essential role in envelopment (53). We showed that although UL36p is not essential for capsid-organelle docking (44), it is required to efficiently couple HSV-1 capsids to sites of VPS4-driven envelopment (54). However, UL36p is required for the assembly of multiple outer-tegument and envelope proteins into the enveloping particle (55–60), and its role in capsid/ESCRT-III/VPS4 assembly may well be indirect (54).
In this study, we tested three cellular candidates that we hypothesized might be used by HSV-1 to recruit and control deposition of the ESCRT-III filament: ESCRT-II and the Bro1 proteins HD-PTP and BROX (Fig. 1). We used siRNA to substantially reduce the levels of the essential ESCRT-II subunit EAP20/VPS25 (Fig. 2 and 3), HD-PTP (Fig. 4 and 5), and BROX (Fig. 6) and confirmed impairment of their normal cellular functions where assays are available (lack of a known role for BROX precluded such a functional assay). In each case, we found that loss of the polypeptide had no significant effect upon the final yields of HSV-1 after 20 h of replication.
In these experiments, we examined only endpoint PFU yields in a single-step growth curve. It is possible that loss of these proteins could result in differences in rates of viral replication earlier in infection or defects that are visible only after multiple cycles of replication. However, it might be difficult to distinguish such modest effects from those arising due to the pleiotropic consequences of ESCRT component knockdown for general cellular health. We also cannot exclude the possibility that HSV-1 might utilize two or more of these polypeptides in a redundant fashion with each other, with the ESCRT-I complex, or with the Bro1 protein ALIX. Given the large number of cellular proteins and complexes that are now known to control ESCRT-III localization and activity, it would be a significant technical challenge to knock down all possible combinations of these molecules, even if the cells were capable of surviving such treatment.
We conclude that either HSV-1 uses multiple cellular ESCRT components in a redundant manner to recruit ESCRT-III or, alternatively, the virus bypasses the need for Bro1 domain proteins, ESCRT-I and ESCRT-II, completely. In the latter case, structural proteins within the HSV-1 particle, perhaps in the inner or outer tegument (18, 19), may have evolved to directly recruit and trigger assembly of ESCRT-III subunits at the site of envelope constriction and scission.
MATERIALS AND METHODS
Cells and viruses.
HeLa cells were maintained in Dulbecco modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin-streptomycin (Gibco Laboratories). HSV-1 GS6807 mimics a previously described HSV-1 strain F recombinant that encodes a UL25/mCherry capsid fusion, HSV1-GS4553 (61), but is derived from an improved self-recombining HSV-1 strain F infectious clone (62). HSV-1 GS6807 was a kind gift from Gregory A. Smith.
RNA interference and HSV-1 infections.
All knockdowns were performed with Ambion Silencer Select siRNAs (ThermoFisher Scientific) transfected into HeLa cells using Lipofectamine RNAiMax (ThermoFisher Scientific) in DMEM containing 10% FCS but lacking antibiotics. For all knockdown experiments, Silencer Select negative control no. 1 siRNA (ThermoFisher Scientific; catalog number AM4611) was used as a control. In none of the siRNA knockdown regimens described below was there any apparent cytopathic effect compared to negative-control siRNA.
For EAP20 knockdown, Silencer Select siRNA s38894 (5′-CGUCUUUACCCUGUAUGAAtt-3′ and 5′-UUCAUACAGGGUAAAGACGga-3′; nucleotides in lowercase correspond to single-stranded overhanging ends [here and in next 3 paragraphs]) targeting EAP20 exon 5 at a concentration of 25 nM was transfected into HeLa cells at the start of the experiment and again 24 h later. Four hours after the second transfection, the cells were infected with HSV-1 GS6807 at an MOI of 10. After 1 h of infection, residual input virus was inactivated by a 30-s wash with glycine-buffered saline (GBS) (136 mM NaCl, 5 mM KCl, 100 mM glycine [pH 2.8]) and then overlaid with fresh antibiotic-free DMEM-10% FCS. Samples were collected immediately (T0) and at 20 hpi (T20) by freezing, thawing, and then scraping cells up into their overlying medium. After two more freeze-thaw cycles, the cell-medium mixture was sonicated and titrated for PFU on Vero cell monolayers.
PRPF8 knockdown used 25 nM Silencer Select siRNA s20798 (5′-GGACAUGAACCAUACGAAUtt-3′ and 5′-AUUCGUAUGGUUCAUGUCCtt-3′) targeting PRPF8 exons 20 and 21. Transfections were performed as described for EAP20, except that cells were transfected at the start of the experiment and again after 48 h. After an additional 24 hours, the cells were infected with HSV-1 strain GS6807 at an MOI of 10, and then samples were collected exactly as described above.
HD-PTP knockdowns made use of a mixture containing 25 nM (each) Silencer Select siRNAs s24775 (5′-AGUUUGUCCUGAAGAAUUAtt-3′ and 5′-UAAUUCUUCAGGACAAACUtc-3′) and s24777 (5′-GGAAGAAACUUGUGCAGAUtt-3′ and 5′-AUCUGCACAAGUUUCUUCCag-3′) targeting HD-PTP exons 1 and 2 and exon 8, respectively. Transfections were performed as described for EAP20 above, except that the cells were transfected at the start of the experiment, after 24 h, and again after 48 h. After an additional 4 h, the cells were infected with HSV-1 strain GS6807 at an MOI of 10, and then samples were collected exactly as described above.
BROX knockdowns were carried out as follows. At the start of the experiment, HeLa cells were transfected with 25 nM (each) Silencer Select siRNAs s45175 (5′-CAAGAAAGCAAGUUACGAUtt-3′ and 5′-AUCGUAACUUGCUUUCUUGgg-3′) and s45176 (5′-CAAGGAUGACAGUACUAAAtt-3′ and 5′-UUUAGUACUGUCAUCCUUGgg-3′) targeting BROX exon 4 and exons 11 and 12, respectively. The transfection was then repeated 48 h and 96 h after the start of the experiment. After an additional 4 h, the cells were infected with HSV-1 strain GS6807 at an MOI of 10, and then samples were collected exactly as described above.
Immunostaining and Western blotting.
When intended for immunostaining experiments, HeLa cells were plated onto glass coverslips precoated with 10 μg/ml poly-l-lysine (Sigma). Following the appropriate experimental treatments, the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) for 15 min at room temperature, and all subsequent steps were performed at room temperature. The cells were washed with PBS, permeabilized with 0.1% Triton X-100 in PBS for 15 min, washed in PBS, and blocked with 20% newborn calf serum (NCS) in PBS for 1 h. They were then incubated for 1 h with the mouse monoclonal antibody anti-EGFR 528 or anti-SMN F-5 (Santa Cruz Biotechnology) as appropriate. After washing in PBS, they were incubated with Alexa Fluor 488-labeled donkey anti-mouse IgG (Invitrogen), washed, and mounted for imaging using ProLong Gold Antifade with DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen).
For Western blotting, whole-cell extracts were subjected to SDS-PAGE on 7.5% gels and then transferred to a polyvinylidene difluoride membrane (Bio-Rad), blocked using a solution of 5% dry milk in Tris-buffered saline-Tween (TBST) (137 mM NaCl, 0.1% [wt/vol] Tween 20, 20 mM Tris-Cl, pH 7.4), and then incubated with the appropriate primary antibody, as follows: anti-EAP20/VPS25, mouse monoclonal antibody B-4 (Santa Cruz Biotechnology); anti-PRPF8, rabbit polyclonal A303-922A (Bethyl Laboratories Inc.); anti-HD-PTP, rabbit polyclonal A304-585A-1 (Bethyl Laboratories Inc.); anti-BROX, rabbit polyclonal PA5-56748 (ThermoFisher Scientific); or anti-GAPDH, mouse monoclonal GA1R (ThermoFisher Scientific). After washing, the membranes were incubated with peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (Millipore Sigma), and bound antibody was detected with an enhanced chemiluminescence (ECL) substrate (PerkinElmer).
EGFR degradation assay.
HeLa cells were seeded onto glass coverslips, and then EAP20 was knocked down and the cells were infected as described above. At 13 hpi, the cells were serum starved for 5 h (to deplete EGF), and then 1 μg/ml EGF was added or omitted for 2 h. The cells were then fixed and processed for immunocytochemistry as described above. EGFR fluorescence intensities were quantified using NIH ImageJ software.
Accumulation of SMN puncta in nuclei.
HeLa cells were seeded onto glass coverslips, and then HD-PTP was knocked down as described above. The cells were fixed 72 h after the beginning of the experiment and immunostained for SMN as described above. SMN accumulations in Cajal bodies (nuclear puncta) were counted in 150 cells per experiment in three independent experiments.
Image recording and analysis.
Imaging was performed in the Analytical Imaging Facility (AIF) of the Albert Einstein College of Medicine. All fluorescence images were collected on a Zeiss Axio Observer CLEM inverted microscope with a 63×, 1.4-numerical-aperture oil immersion objective. All images were saved in Zeiss AxioVision ZVI (Zeiss vision image) format using AxioVision release 4.8.2 image acquisition and management software. NIH ImageJ software was used for image analysis.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grant R01 AI125244 (to D.W.W.). J.B. acknowledges support from the institutional AIDS training grant Training in HIV/AIDS Pathogenesis; Basic and Translational Research T32 AI007501. Microscopy images contained in this study were collected in the AIF of the Albert Einstein College of Medicine, supported by Cancer Center grant NCI P30CA013330.
REFERENCES
- 1.Heming JD, Conway JF, Homa FL. 2017. Herpesvirus capsid assembly and DNA packaging. Adv Anat Embryol Cell Biol 223:119–142. doi: 10.1007/978-3-319-53168-7_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dai X, Zhou ZH. 2018. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 360:eaao7298. doi: 10.1126/science.aao7298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Owen DJ, Crump CM, Graham SC. 2015. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 7:5084–5114. doi: 10.3390/v7092861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mettenleiter TC. 2016. Vesicular nucleo-cytoplasmic transport-herpesviruses as pioneers in cell biology. Viruses 8:266. doi: 10.3390/v8100266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bigalke JM, Heldwein EE. 2017. Have NEC coat, will travel: structural basis of membrane budding during nuclear egress in herpesviruses. Adv Virus Res 97:107–141. doi: 10.1016/bs.aivir.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bigalke JM, Heldwein EE. 2016. Nuclear exodus: herpesviruses lead the way. Annu Rev Virol 3:387–409. doi: 10.1146/annurev-virology-110615-042215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arii J, Watanabe M, Maeda F, Tokai-Nishizumi N, Chihara T, Miura M, Maruzuru Y, Koyanagi N, Kato A, Kawaguchi Y. 2018. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat Commun 9:3379. doi: 10.1038/s41467-018-05889-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollinshead M, Johns HL, Sayers CL, Gonzalez-Lopez C, Smith GL, Elliott G. 2012. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J 31:4204–4220. doi: 10.1038/emboj.2012.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harley CA, Dasgupta A, Wilson DW. 2001. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: role for organelle acidification in assembly of infectious particles. J Virol 75:1236–1251. doi: 10.1128/JVI.75.3.1236-1251.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turcotte S, Letellier J, Lippe R. 2005. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J Virol 79:8847–8860. doi: 10.1128/JVI.79.14.8847-8860.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alwine JC. 2012. The human cytomegalovirus assembly compartment: a masterpiece of viral manipulation of cellular processes that facilitates assembly and egress. PLoS Pathog 8:e1002878. doi: 10.1371/journal.ppat.1002878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Granzow H, Klupp BG, Fuchs W, Veits J, Osterrieder N, Mettenleiter TC. 2001. Egress of alphaherpesviruses: comparative ultrastructural study. J Virol 75:3675–3684. doi: 10.1128/JVI.75.8.3675-3684.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Henaff D, Radtke K, Lippe R. 2012. Herpesviruses exploit several host compartments for envelopment. Traffic 13:1443–1449. doi: 10.1111/j.1600-0854.2012.01399.x. [DOI] [PubMed] [Google Scholar]
- 14.Hambleton S, Gershon MD, Gershon AA. 2004. The role of the trans-Golgi network in varicella zoster virus biology. Cell Mol Life Sci 61:3047–3056. doi: 10.1007/s00018-004-4269-7. [DOI] [PubMed] [Google Scholar]
- 15.Nanbo A, Noda T, Ohba Y. 2018. Epstein-Barr virus acquires its final envelope on intracellular compartments with Golgi markers. Front Microbiol 9:454. doi: 10.3389/fmicb.2018.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cepeda V, Esteban M, Fraile-Ramos A. 2010. Human cytomegalovirus final envelopment on membranes containing both trans-Golgi network and endosomal markers. Cell Microbiol 12:386–404. doi: 10.1111/j.1462-5822.2009.01405.x. [DOI] [PubMed] [Google Scholar]
- 17.Banfield BW. 2019. Beyond the NEC: modulation of herpes simplex virus nuclear egress by viral and cellular components. Curr Clin Microbiol Rep 6:1. doi: 10.1007/s40588-019-0112-7. [DOI] [Google Scholar]
- 18.Crump C. 2018. Virus assembly and egress of HSV. Adv Exp Med Biol 1045:23–44. doi: 10.1007/978-981-10-7230-7_2. [DOI] [PubMed] [Google Scholar]
- 19.Barnes J, Wilson DW. 2019. Seeking closure: how do herpesviruses recruit the cellular ESCRT apparatus? J Virol 93:e00392-19. doi: 10.1128/JVI.00392-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pawliczek T, Crump CM. 2009. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J Virol 83:11254–11264. doi: 10.1128/JVI.00574-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kharkwal H, Smith CG, Wilson DW. 2014. Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J Virol 88:14467–14478. doi: 10.1128/JVI.02777-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alonso YAM, Migliano SM, Teis D. 2016. ESCRT-III and Vps4: a dynamic multipurpose tool for membrane budding and scission. FEBS J 283:3288–3302. doi: 10.1111/febs.13688. [DOI] [PubMed] [Google Scholar]
- 23.Scourfield EJ, Martin-Serrano J. 2017. Growing functions of the ESCRT machinery in cell biology and viral replication. Biochem Soc Trans 45:613–634. doi: 10.1042/BST20160479. [DOI] [PubMed] [Google Scholar]
- 24.Christ L, Raiborg C, Wenzel EM, Campsteijn C, Stenmark H. 2017. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem Sci 42:42–56. doi: 10.1016/j.tibs.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 25.McCullough J, Frost A, Sundquist WI. 2018. Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Annu Rev Cell Dev Biol 34:85–109. doi: 10.1146/annurev-cellbio-100616-060600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Olmos Y, Carlton JG. 2016. The ESCRT machinery: new roles at new holes. Curr Opin Cell Biol 38:1–11. doi: 10.1016/j.ceb.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCullough J, Clippinger AK, Talledge N, Skowyra ML, Saunders MG, Naismith TV, Colf LA, Afonine P, Arthur C, Sundquist WI, Hanson PI, Frost A. 2015. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science 350:1548–1551. doi: 10.1126/science.aad8305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teis D, Saksena S, Emr SD. 2008. Ordered assembly of the ESCRT-III complex on endosomes is required to sequester cargo during MVB formation. Dev Cell 15:578–589. doi: 10.1016/j.devcel.2008.08.013. [DOI] [PubMed] [Google Scholar]
- 29.Mu R, Dussupt V, Jiang J, Sette P, Rudd V, Chuenchor W, Bello NF, Bouamr F, Xiao TS. 2012. Two distinct binding modes define the interaction of Brox with the C-terminal tails of CHMP5 and CHMP4B. Structure 20:887–898. doi: 10.1016/j.str.2012.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhai Q, Landesman MB, Robinson H, Sundquist WI, Hill CP. 2011. Structure of the Bro1 domain protein BROX and functional analyses of the ALIX Bro1 domain in HIV-1 budding. PLoS One 6:e27466. doi: 10.1371/journal.pone.0027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCullough J, Fisher RD, Whitby FG, Sundquist WI, Hill CP. 2008. ALIX-CHMP4 interactions in the human ESCRT pathway. Proc Natl Acad Sci U S A 105:7687–7691. doi: 10.1073/pnas.0801567105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hurley JH. 2015. ESCRTs are everywhere. EMBO J 34:2398–2407. doi: 10.15252/embj.201592484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Votteler J, Sundquist WI. 2013. Virus budding and the ESCRT pathway. Cell Host Microbe 14:232–241. doi: 10.1016/j.chom.2013.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meng B, Ip NC, Prestwood LJ, Abbink TE, Lever AM. 2015. Evidence that the endosomal sorting complex required for transport-II (ESCRT-II) is required for efficient human immunodeficiency virus-1 (HIV-1) production. Retrovirology 12:72. doi: 10.1186/s12977-015-0197-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langelier C, von Schwedler UK, Fisher RD, De Domenico I, White PL, Hill CP, Kaplan J, Ward D, Sundquist WI. 2006. Human ESCRT-II complex and its role in human immunodeficiency virus type 1 release. J Virol 80:9465–9480. doi: 10.1128/JVI.01049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crump CM, Yates C, Minson T. 2007. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J Virol 81:7380–7387. doi: 10.1128/JVI.00222-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stieler JT, Prange R. 2014. Involvement of ESCRT-II in hepatitis B virus morphogenesis. PLoS One 9:e91279. doi: 10.1371/journal.pone.0091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim J, Sitaraman S, Hierro A, Beach BM, Odorizzi G, Hurley JH. 2005. Structural basis for endosomal targeting by the Bro1 domain. Dev Cell 8:937–947. doi: 10.1016/j.devcel.2005.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gahloth D, Heaven G, Jowitt TA, Mould AP, Bella J, Baldock C, Woodman P, Tabernero L. 2017. The open architecture of HD-PTP phosphatase provides new insights into the mechanism of regulation of ESCRT function. Sci Rep 7:9151. doi: 10.1038/s41598-017-09467-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doyotte A, Mironov A, McKenzie E, Woodman P. 2008. The Bro1-related protein HD-PTP/PTPN23 is required for endosomal cargo sorting and multivesicular body morphogenesis. Proc Natl Acad Sci U S A 105:6308–6313. doi: 10.1073/pnas.0707601105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ichioka F, Takaya E, Suzuki H, Kajigaya S, Buchman VL, Shibata H, Maki M. 2007. HD-PTP and Alix share some membrane-traffic related proteins that interact with their Bro1 domains or proline-rich regions. Arch Biochem Biophys 457:142–149. doi: 10.1016/j.abb.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 42.Lee J, Oh KJ, Lee D, Kim BY, Choi JS, Ku B, Kim SJ. 2016. Structural study of the HD-PTP Bro1 domain in a complex with the core region of STAM2, a subunit of ESCRT-0. PLoS One 11:e0149113. doi: 10.1371/journal.pone.0149113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ichioka F, Kobayashi R, Katoh K, Shibata H, Maki M. 2008. Brox, a novel farnesylated Bro1 domain-containing protein that associates with charged multivesicular body protein 4 (CHMP4). FEBS J 275:682–692. doi: 10.1111/j.1742-4658.2007.06230.x. [DOI] [PubMed] [Google Scholar]
- 44.Kharkwal H, Furgiuele SS, Smith CG, Wilson DW. 2015. Herpes simplex virus capsid-organelle association in the absence of the large tegument protein UL36p. J Virol 89:11372–11382. doi: 10.1128/JVI.01893-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yorikawa C, Shibata H, Waguri S, Hatta K, Horii M, Katoh K, Kobayashi T, Uchiyama Y, Maki M. 2005. Human CHMP6, a myristoylated ESCRT-III protein, interacts directly with an ESCRT-II component EAP20 and regulates endosomal cargo sorting. Biochem J 387:17–26. doi: 10.1042/BJ20041227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baldys A, Raymond JR. 2009. Critical role of ESCRT machinery in EGFR recycling. Biochemistry 48:9321–9323. doi: 10.1021/bi900865u. [DOI] [PubMed] [Google Scholar]
- 47.Grainger RJ, Beggs JD. 2005. Prp8 protein: at the heart of the spliceosome. RNA 11:533–557. doi: 10.1261/rna.2220705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Griffiths SJ, Koegl M, Boutell C, Zenner HL, Crump CM, Pica F, Gonzalez O, Friedel CC, Barry G, Martin K, Craigon MH, Chen R, Kaza LN, Fossum E, Fazakerley JK, Efstathiou S, Volpi A, Zimmer R, Ghazal P, Haas J. 2013. A systematic analysis of host factors reveals a Med23-interferon-lambda regulatory axis against herpes simplex virus type 1 replication. PLoS Pathog 9:e1003514. doi: 10.1371/journal.ppat.1003514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Calle A, Ugrinova I, Epstein AL, Bouvet P, Diaz JJ, Greco A. 2008. Nucleolin is required for an efficient herpes simplex virus type 1 infection. J Virol 82:4762–4773. doi: 10.1128/JVI.00077-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zenner HL, Yoshimura S, Barr FA, Crump CM. 2011. Analysis of Rab GTPase-activating proteins indicates that Rab1a/b and Rab43 are important for herpes simplex virus 1 secondary envelopment. J Virol 85:8012–8021. doi: 10.1128/JVI.00500-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang Z, Liu Q, Lu J, Fan P, Xie W, Qiu W, Wang F, Hu G, Zhang Y. 2016. Serine/arginine-rich splicing factor 2 modulates herpes simplex virus type 1 replication via regulating viral gene transcriptional activity and pre-mRNA splicing. J Biol Chem 291:26377–26387. doi: 10.1074/jbc.M116.753046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Husedzinovic A, Neumann B, Reymann J, Draeger-Meurer S, Chari A, Erfle H, Fischer U, Gruss OJ. 2015. The catalytically inactive tyrosine phosphatase HD-PTP/PTPN23 is a novel regulator of SMN complex localization. Mol Biol Cell 26:161–171. doi: 10.1091/mbc.E14-06-1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Desai PJ. 2000. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J Virol 74:11608–11618. doi: 10.1128/jvi.74.24.11608-11618.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kharkwal H, Smith CG, Wilson DW. 2016. HSV capsid localization to ESCRT-VPS4 complexes in the presence and absence of the large tegument protein UL36p. J Virol 90:7257–7267. doi: 10.1128/JVI.00857-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ko DH, Cunningham AL, Diefenbach RJ. 2010. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J Virol 84:1397–1405. doi: 10.1128/JVI.01721-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vittone V, Diefenbach E, Triffett D, Douglas MW, Cunningham AL, Diefenbach RJ. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J Virol 79:9566–9571. doi: 10.1128/JVI.79.15.9566-9571.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mijatov B, Cunningham AL, Diefenbach RJ. 2007. Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 368:26–31. doi: 10.1016/j.virol.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 58.Svobodova S, Bell S, Crump CM. 2012. Analysis of the interaction between the essential herpes simplex virus 1 tegument proteins VP16 and VP1/2. J Virol 86:473–483. doi: 10.1128/JVI.05981-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Koenigsberg AL, Heldwein EE. 2018. The dynamic nature of the conserved tegument protein UL37 of herpesviruses. J Biol Chem 293:15827–15839. doi: 10.1074/jbc.RA118.004481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koenigsberg AL, Heldwein EE. 2017. Crystal structure of the N-terminal half of the traffic controller UL37 from herpes simplex virus 1. J Virol 91:e01244-17. doi: 10.1128/JVI.01244-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Huffmaster NJ, Sollars PJ, Richards AL, Pickard GE, Smith GA. 2015. Dynamic ubiquitination drives herpesvirus neuroinvasion. Proc Natl Acad Sci U S A 112:12818–12823. doi: 10.1073/pnas.1512559112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richards AL, Sollars PJ, Smith GA. 2016. New tools to convert bacterial artificial chromosomes to a self-excising design and their application to a herpes simplex virus type 1 infectious clone. BMC Biotechnol 16:64. doi: 10.1186/s12896-016-0295-4. [DOI] [PMC free article] [PubMed] [Google Scholar]